
Aislamiento y caracterización de segmentos genómicos...
159
Dirección: Dirección: Biblioteca Central Dr. Luis F. Leloir, Facultad de Ciencias Exactas y Naturales, Universidad de Buenos Aires. Intendente Güiraldes 2160 - C1428EGA - Tel. (++54 +11) 4789-9293 Contacto: Contacto: [email protected] Tesis de Posgrado Aislamiento y caracterización de Aislamiento y caracterización de segmentos genómicos del virus de segmentos genómicos del virus de la Hepatitis B para su utilización la Hepatitis B para su utilización como reactivos biológicos como reactivos biológicos Fernández Cobo, Mariana 1995 Tesis presentada para obtener el grado de Doctor en Ciencias Biológicas de la Universidad de Buenos Aires Este documento forma parte de la colección de tesis doctorales y de maestría de la Biblioteca Central Dr. Luis Federico Leloir, disponible en digital.bl.fcen.uba.ar. Su utilización debe ser acompañada por la cita bibliográfica con reconocimiento de la fuente. This document is part of the doctoral theses collection of the Central Library Dr. Luis Federico Leloir, available in digital.bl.fcen.uba.ar. It should be used accompanied by the corresponding citation acknowledging the source. Cita tipo APA: Fernández Cobo, Mariana. (1995). Aislamiento y caracterización de segmentos genómicos del virus de la Hepatitis B para su utilización como reactivos biológicos. Facultad de Ciencias Exactas y Naturales. Universidad de Buenos Aires. http://digital.bl.fcen.uba.ar/Download/Tesis/Tesis_2734_FernandezCobo.pdf Cita tipo Chicago: Fernández Cobo, Mariana. "Aislamiento y caracterización de segmentos genómicos del virus de la Hepatitis B para su utilización como reactivos biológicos". Tesis de Doctor. Facultad de Ciencias Exactas y Naturales. Universidad de Buenos Aires. 1995. http://digital.bl.fcen.uba.ar/Download/Tesis/Tesis_2734_FernandezCobo.pdf
Transcript of Aislamiento y caracterización de segmentos genómicos...

Di r ecci ó n:Di r ecci ó n: Biblioteca Central Dr. Luis F. Leloir, Facultad de Ciencias Exactas y Naturales, Universidad de Buenos Aires. Intendente Güiraldes 2160 - C1428EGA - Tel. (++54 +11) 4789-9293
Co nta cto :Co nta cto : [email protected]
Tesis de Posgrado
Aislamiento y caracterización deAislamiento y caracterización desegmentos genómicos del virus desegmentos genómicos del virus dela Hepatitis B para su utilizaciónla Hepatitis B para su utilización
como reactivos biológicoscomo reactivos biológicos
Fernández Cobo, Mariana
1995
Tesis presentada para obtener el grado de Doctor en CienciasBiológicas de la Universidad de Buenos Aires
Este documento forma parte de la colección de tesis doctorales y de maestría de la BibliotecaCentral Dr. Luis Federico Leloir, disponible en digital.bl.fcen.uba.ar. Su utilización debe seracompañada por la cita bibliográfica con reconocimiento de la fuente.
This document is part of the doctoral theses collection of the Central Library Dr. Luis FedericoLeloir, available in digital.bl.fcen.uba.ar. It should be used accompanied by the correspondingcitation acknowledging the source.
Cita tipo APA:Fernández Cobo, Mariana. (1995). Aislamiento y caracterización de segmentos genómicos delvirus de la Hepatitis B para su utilización como reactivos biológicos. Facultad de CienciasExactas y Naturales. Universidad de Buenos Aires.http://digital.bl.fcen.uba.ar/Download/Tesis/Tesis_2734_FernandezCobo.pdf
Cita tipo Chicago:Fernández Cobo, Mariana. "Aislamiento y caracterización de segmentos genómicos del virus dela Hepatitis B para su utilización como reactivos biológicos". Tesis de Doctor. Facultad deCiencias Exactas y Naturales. Universidad de Buenos Aires. 1995.http://digital.bl.fcen.uba.ar/Download/Tesis/Tesis_2734_FernandezCobo.pdf

UNIVERSIDAD DE BUENOS AIRESFACULTAD DE CIENCIAS EXACTAS Y NATURALES
Tema de Tesis
AISLAMIENTOY CARACTERIZACION DE SEGMENTOS GENOMICOSDEL VIRUS DE LA HEPATITIS B PARA SU UTILIZACION
COMO REACTIVOS BIOLOGICOS
AutorMariana Fernández Cobo
Director de TesisDr. Ramón De Torres
Lugar de TrabajoInstituto Nacional de Microbiología "Carlos G. Malbran"
Tesis presentada para optar al título de Doctor en Ciencias BiológicasAño 1995

AGRADECIMIENTOS
A mi director, Dr. Ramónde Torres por su aliento y asesora
miento permanente.
Al Instituto Nacional de Microbiología "Carlos G. Malbrán" por
su apoyopara la realización y presentación de esta tesis.
A todos mis compañeros de laboratorio por su colaboración, sus
valiosas sugerencias, apoyo, crítica y estimulo
A mi familia y a todos mis amigos por su estímulo constante.


ABREVIATURAS...............................................iiiABREVIATURASY GLOSARIODE LOS GENOTIPOS................I. INTRODUCCION................................ ..............1l Hepatitisvirales................................ ................l
1.1. Patogenia . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . ..11.2. Agentes causales . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . ..2
2. HBV................................. ............................52.1. Estructura . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . ..5
2.2 Biología . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . ..302.3 Diagnóstico . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . ..372.4. Tratamiento . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . ..39
2.5 Epidemiología . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . ..40
II. OBJETIVO....................................... . . . . . . . ..42III. MATERIALESYMETODOS........... . . . .....................431. ProductosQuímicos.............................................432. MaterialRadioactivo... ........................................433. HBV............. . . . . . . . . . . . . . . . . . . . . ... . . . . . . . . . . . . . . . . . . . . . . ..43
3.1. Purificación de partículas de DANE. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . ..433.2. Purificación de HBV-DNA. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . ..44
4. Cepashuéspedy vectores.......................................444.1. Cepas Bacterianas . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . ..444.2. Vectores . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . ..45
4.3. Extracción y purificación de plásmidos . . . . . . . . . . . . . . . . . . . . . . . . . . ..464.4. Inducción de la expresión bacteriana . . . . . . . . . . . . . . . . . . . . . . . . . . . . ..48
5. Reacciones enzimáticas...... . . . . . . . . . . . . . ....... . . . . . . ... . . . . ..495.1. Restricción . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . ..49
5.2. Ligación . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . ..495.3. Defosforilación . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . ..505.4. Fill-in . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . ..50
6. Transformación.................................................506.1. Preparación de células competentes . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . ..506.2. Introducción de plásmidos en E. coli . . . . . . . . . . . . . . . . . . . . . . . . . . . . ..526.3. Preparación de placas . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . ..52
7. Marcaciónde DNAin vitro . . . . . . . . . .................. . . . . . . . . . ..538. Reacciónde hibridación................. . . . . . . . . . . . . . . . . . . . . . ..54
8.1. De colonias . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . ..548.2. Dots . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . ..54
8.3. Southern . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . ..548.4. Prehibridación e hibridación . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . ..548.5 Autoradiografïa . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . ..568.6 Quimioluminiscente . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . ..56
9. Electroforesis en gel............. . . . . . . . . . . . . . . . . . . . . . . . . . . . ..579.1. Geles de agarosa . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . ..579.2. Geles de poliacrilamida . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . ..SB
10.Secuencia.....................................................58

IV.RESULTADOS.............................................. 601. Construcciónde plásmidosrecombinantes........................60
1.1. Estrategia de clonado . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . ..601.2 Purificación de partículas de Dane . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . ..601.3. Extracción de DNA. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . ..621 4 Restricción del DNA. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . ..621.5 Elección del vector . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . ..631.6. Transformación (eficiencia) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . ..63
2. Seleccióny análisis de recombinantes..........................642.1. Selección por hibridación de colonias . . . . . . . . . . . . . . . . . . . . . . . . . . . ..642.2. Verificación de la presencia de plásmidos . . . . . . . . . . . . . . . . . . . . . . . ..662.3. Análisis, de los plásmidos seleccionados, por restricción ehibridación. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . ..662.4. Determinación de la zona viral presente en los recombinantes . . . . ..692.5. Secuencia . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . ..76
3. Utilización de uno de los recombinantescomosonda............1043.1. Determinación de la especificidad de la sonda . . . . . . . . . . . . . . . . . . ..1043.2. Determinación de la sensibilidad . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . ..105
4. Subclonadoenvectoresde expresión...........................1094.1. Estrategia de clonado en vectores de expresión . . . . . . . . . . . . . . . . . ..1094.2. Transformación . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . ..112
5. Seleccióny análisis de expresión. . . . .........................1l4V. CONCLUSIONESGENERALES......... . . . . . ....................116VI.BIBLIOGRAFIA...........................................123Anexo......................................................140

ABREVIATURAS
aaATP
Ci
cpm
dATP
dCTP
dGMP
dGTP
DNA
DO
DTT
dTTP
EDTA
HBV
kDa
kpb
log
NC
nm
ntNTP
ORF
pbPIPES
PM
PyRFLP
aminoácidotrifosfato de adenosina7mGppp= 7-metil trifosfato de guanosinaCuriecuentas por minutoDalton: unidad de peso moleculartrifosfato de desoxiadenosinatrifosfato de desoxicitosinamonofosfato de desoxiguanosinatrifosfato de desoxiguanosinaácido desoxirribonucleicodensidad ópticadoble cadenaditiotreitoltrifosfato de desoxitimidinaetilendiaminotetraacetato de sodioaceleración de la gravedadvirus de la hepatitis BkiloDalton (1000 Da)kilopares de bases (1000 pares de bases)litrologaritmo base 10molarnitrocelulosananometronucleótidotrifosfato de nucleósido (sin especificar)open reading frame = marco de lectura abiertopares de bases(ácido piperazino-N,N'-bis[2-etanosulfónico]; ácido1,4-piperazino dietanosulfónico)peso molecularpurinapirimidinapolimorfismo del largo de los fragmentos derestricción

RNasa
rpmSDS
ssSSC
SSCP
Tris
X-gal
ácido ribonucleicoribonucleasarevoluciones por minutododecil sulfato de sodiosimple cadenabuffer citrato salino standardpolimorfismo dependiente de la conformación de lasimple cadenaTris(hidroximetil)amino metanotrifosfato de uridinaultravioleta5-bromo-4-cloro—3-indoli1 B-d-galactopiranósido
Abreviaturas de los aminoácidos:Alanina Ala AArginina Arg RAspargina Asn NAspartato Asp DCisteína Cys CFenilalanina Phe FGlicina Gly GGlutamato Glu EGlutamina Gln QHistidina His HIsoleucina Ile ILeucina Leu LLysina Lys KMetionina Met MProlina Pro PSerina Ser STirosina Tyr YTreonina Thr TTriptofano Trp WValina Val V

ABREVIATURAS Y GLOSARIO DE LOS GENOTIPOS
deo R
end A1
FI
gyr A96
hst
hst
lacZAMlS
AU169
recArelA
supEthi-l080ÓBOdlachS
mutación en gen regulador que permite la expresiónconstitutiva de genes involucrados en la síntesis dedeoxiribosa.Permite la entrada de plásmidos grandes.mutación que suprime la actividad inespecïfica de laendonucleasa 1.F episomal.mutación en DNAgirasa. Confiere resistencia alácido nalidíxico.actividad endógenade restricción abolida peromantiene actividad de modificación (metilación)ambasactividades (restricción y modificación)abolidas.deleción parcial del gen de B-D-galactosidasa.Expresa el fragmento carboxi-terminal (frag. omega)deleción del operón lac completo más una cantidadvariable de DNAflanqueante.suprime la recombinación homóloga.mutación que elimina el factor “stringent” queregula procesos biosintéticos celulares. Permite lasíntesis de RNAen ausencia de sintesis de proteínastRNAsupresor ámbar (UAG)que inserta una glutamina.requiere tiamina.lleva el profago ÓBO.
profago defectivo que lleva la deleción lacM15.

GLOSARIO :
buffer solución de sustancias químicas que mantienenconstante el pH.
primer oligonucleótido iniciadorpolylinker zona en el DNAde un plásmido que posee varios
sitios únicos de restricción, disponibles parael clonado molecular
Nota: En este trabajo se utilizan términos en inglés, de usocorriente en la literatura científica, cuandoéstos son de difícil traducción o carecen de un equivalente castellano ampliamente difundido.


I. INTRODUCCION
1 Hepatitis virales
1.1. Patogenia
Las manifestaciones clínicas de la inflamación hepática
(hepatitis) integran un síndrome complejo. Es muydifícil que
el conjunto de síntomas permita predecir la etiología, que
puede tener origen en toxicidad química o puede resultar de
infecciones causadas por parásitos, bacterias y una variedadde virus evolutivamente diferentes.
El cuadro clínico de la infección viral es diverso y
puede presentarse comouna infección subclínica o asintomá
tica, comouna forma leve anictérica, comoenfermedad aguda o
comohepatitis fulminante. El signo más evidente de la disfun
ción hepática en las formas agudas es la coloración amarilla
de la piel y de la esclerótica ocular lo que refleja la presencia de niveles elevados de bilirrubina circulante, signo alque se llama ictericia. En algunos casos, especialmente en las
infecciones producidas por virus transmitidos por vía parenteral, se establece un estado de portador persistente (crónico), en el cual puede haber un equilibrio que se expresa en
un estado oligosintomático, o evoluciona a cirrosis, carcinoma
hepatocelular, etc. con síntomas variados y signos claves deenfermedad.
En el caso particular de las infecciones por virus HBV,
objeto de este trabajo, el estado de persistencia del genomaviral es prácticamente una constante.
Dicha persistencia se instala por la capacidad del genoma

de HBVde integrarse al DNAcromosómico.
Respecto a la evolución de la patología en el individuo
infectado, el resultado final depende de la relación que se
establezca entre los mecanismosbioquímicos inducidos por el
virus en el hepatocito y la respuesta inmuneresultante en el
individuo. Esta relación interdependiente es la que establece
el grado y tipo de la enfermedadclínica y el equilibrio posterior.
1.2. Agentes causales
Comoya se mencionó existe una variedad de virus que pue
den producir un cuadro clínico de hepatitis.
Puede reconocerse un grupo de agentes virales en los que
no siempre el daño hepático directo es la patología más impor
tante que se produce en el curso de la infección. Entre éstos
hay que mencionar virus causantes de fiebres hemorrágicas
(fiebre amarilla, dengue, etc.), rubeola, sarampión, Epstein
Barr (EBV), citomegalovirus (CMV),Herpes Simplex (HSV), etc.
(Fagan, 1992).
Sin embargo los virus humanosreconocidamente hepatotro
pos más importantes son los llamados “virus de las hepatitis”:
A (HAV), B (HBV), C (HCV)antes incluida en la nomenclatura
hepatitis NoANoBpost-transfusional, D (HDV), E (HEV)antes
hepatitis NoANoBentérica, y otros, reconocidos clínicamente
aunque aún no bien descriptos como por ejemplo el F y G, que
continúan englobados en el nombre genérico de NoANoBNoC
(NANBNCV).(Chooet al, 1989; Fagan et al, 1989; Phillips et
a1, 1991; Reyes et al, 1990; Fagan, 1992).

La situación actual de los estudios virológicos en estegrupo, toma en consideración, por sobre los estudios clásicos
de caracterización convencionales, el criterio del estudio genómico del virus, especialmente si ha sido clonado y/o secuenciado.
Debe aclararse que este "estado actual" es muyfluido, ya
que existen publicaciones que presentan información incompa
tible. Este es el caso del HFV,en el que Fagan et al. (1989),
Seelig et al. (1992) y Deka et al. (1994) postulan agentes virales totalmente distintos.
Las características generales de los principales virus
hepatotropos citados se muestran en la Tabla 1.

FamiliaGénero Genoma Virión
Envoltura
Transmisión
HEPATITISA
Picornaviridae
Hepatovirus
ssRNA+ 7,5kb 27nm
icosahédrico
no
entérica
Tabla1:Cuadrocomparativo
HEPATITISB
Hepadnaviridae
Hepadnavirus
dsDNAincomp.
3,2kpb42nm
esférico
Sl
parenteral
HEPATITISCFlaviviridae
Hepacavirus
ssRNA+ 9-10kb50-60nm
desconocido
Sl
parenteral
HEPATITISD sinclasif. DeltavirusssRNAcirc.
1,7kb 35nm
esférico
Sl
parenteral
HEPATITISE
Caliciviridae
Hepevirus
ssRNA+7,2kb
27-34nm
icosahédrico
no
entérica
delosprincipalesvirushepatotropos.

2. HBV
2.1. Estructura
El HBVes un virus pequeño, complejo, con características
únicas e inusuales que lo distinguen de otros virus. Sin em
bargo ha sido filogenéticamente relacionado con los retrovi
rus, ya que utiliza un paso de transcripción inversa para la
replicación de su DNA(Summers & Mason, 1982) y posee cierta
homología genómica, especialmente con los retrovirus tipo C de
las linfosarcomatosis murinas como MoMLV(Moloney murine leu
kemia virus) y MoMSV(Moloney murine sarcoma virus) (Miller &
Robinson, 1986).
El virión o partícula de Dane tiene forma esférica y mide
aproximadamente 42nm de diámetro. Posee una envoltura de 7nm
de espesor y un core interno o nucleocápside hexagonal de 27nm
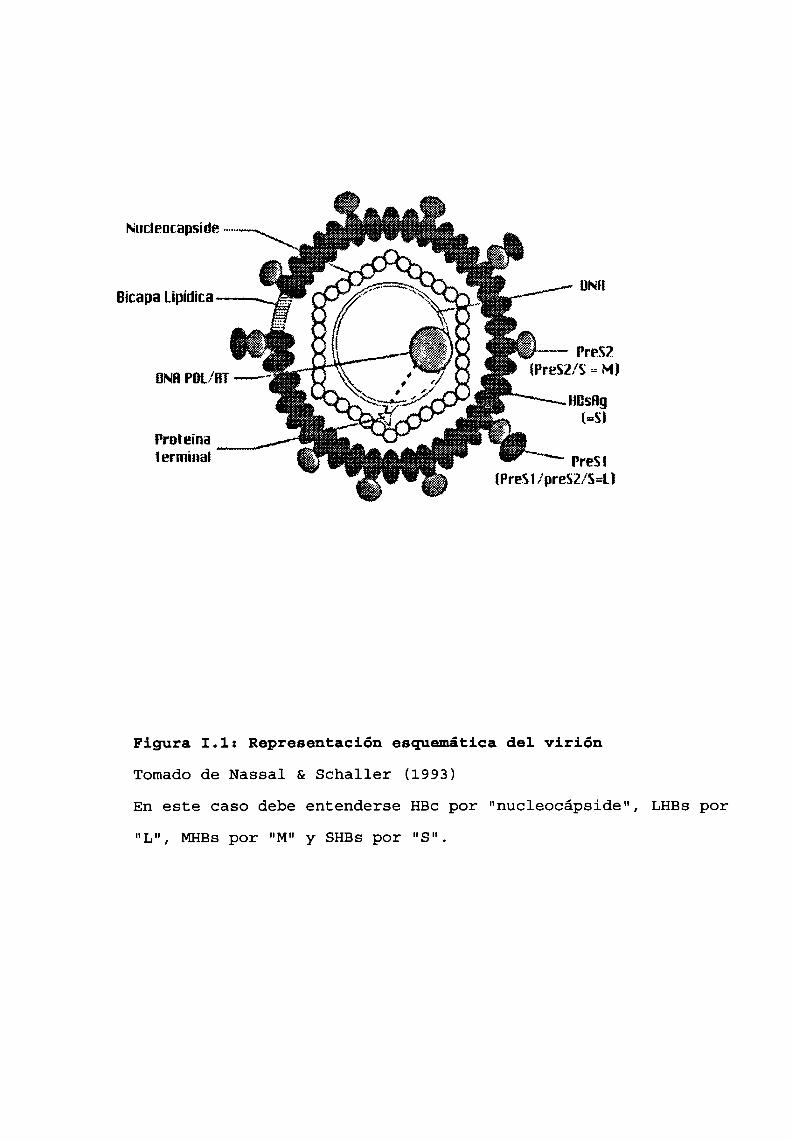
de diámetro (Daneet al., 1970). (Fig. I.1).
La envoltura viral está constituida por lípidos presentesen la membranacitoplasmática, carbohidratos también de origen
celular y una "proteína de superficie" (HBs), esta última es
equivalente al llamado "antígeno de superficie (HBsAg)",que
inicialmente fue denominadoantígeno australiano en virtud de
hallazgos inmunoquímicosrelacionados a estudios de polimor
fismos de proteínas séricas en humanos en Nueva Guinea
(Blumberget al., 1965). Este “antígeno”, en realidad, es un
complejo molecular constituido básicamente por tres proteínas
específicas, relacionadas, que se denominan grande (LHBs), me
dia (MHBs)y pequeña (SHBs). Las moléculas que integran el
complejo HBsse sintetizan en exceso durante la expresión gé
nica viral y se autoensamblan formandopartículas esféricas de
Nuclencapside
' - ¡JM!Bicapa Lipldlca
PreSZ
[INRPOL/HT----" I ÍPI’ESZ/S M]
llBsflgl=Sl
Pro! einaterminal x PreS!
{PreSl¡mew/5:0
Figura 1.1: Representación esquemática del viriónTomado de Nassal & Schaller (1993)
En este caso debe entenderse HBcpor "nucleocápside", LHBspor
"Lu, MHBSpor "Mu SHBS por "S".Y

22nmde diámetro y agregados de estas últimas que forman fila
mentos de igual diámetro y 200nmde longitud. Todas estas for
mas, acompañadas o no de partículas de Dane, se encuentran en
el plasma de ciertos individuos infectados, estado evolutivo
de la infección al que se llama "antigenemia".
La nucleocápside está formada por una proteína llamada
comúnmente core (HBc) o antígeno core (HBcAg)y contiene una
molécula de DNAque constituye el genoma del HBV. Este DNA
tiene una cadena circular completa y la complementaria es más
corta, por lo que la molécula entera es parcialmente de doble
cadena; mide aproximadamente 3,2 kpb, y presenta una proteína
unida al extremo 5' de la cadena negativa y una DNApolimerasa
(Pol).
Genoma: Consiste en una pequeña molécula de DNAcircular,
sólo parcialmente de doble cadena, de una longitud promedio de
0,78um (Robinson et al., 1974) y cuyo número real de nucleóti
dos varía ligeramente entre los 3182 y 3221 pb debido a pequeñas inserciones o deleciones. Este tamaño lo ubica entre las
moléculas informacionales más pequeñas que poseen independen
cia genética y están asociadas a mecanismosde infección“convencionales”.
La cadena completa, de polaridad negativa, no es un cír
culo cerrado covalentemente, sino que está interrumpida cerca
del extremo 5' de la cadena corta (Sattler &Robinson, 1979).
La estructura circular del genomaestá mantenida por el apareo
de bases de los extremos 5' de ambas cadenas que se extiende
aproximadamente por 200 nucleótidos. La cadena corta,

3200/1
UAA 333
Figura 1.2: Organización genómica

positiva, tiene una longitud variable que va desde el 15 al
6096de la cadena larga (Delius et al., 1983). (Fig. I.2).
Este pequeño genoma es muy compacto ya que todo el DNA
está integrado por secuencias codificantes, parcialmente su
perpuestas. Esto requiere un complejo mecanismode control de
la transcripción y expresión que asegura la síntesis ordenada
de al menossiete productos virales a partir de cuatro marcos
de lectura abiertos (ORFs). Respecto a los elementos regulado
res, estos se encuentran, superpuestos en las regiones codifi
cantes, distribuidos por todo el genomaa diferencia de lo queocurre en retrovirus, en los cuales las secuencias de control
están concentradas en una región determinada del genoma.
La cadena negativa tiene cuatro ORFsllamados: core
(preC/C), superficie (preS/S), polimerasa (P) y X (Fig. 1.2).
Además, todos los HBVexaminados tienen dos ORFs adicionales:
ORFSlocalizado en la misma cadena y con un tamaño de 70-100
codones; y ORF6localizado en la cadena complementaria
(positiva) con un largo aproximadamente de 210 codones (Miller
et al., 1989); aunque hasta ahora no se ha demostrado la exis
tencia de las proteínas predichas y, en el virus de la marmota
(WHV)parecería que estos ORFsno representan genes auténticos
(Chen et al., 1993).
La estructura de la cadena positiva varía en distintos
virus emparentados que infectan otras especies. Mediante un
programa de predicción de secuencias, se demostró en la cadena
positiva del virus de pato (DHBV),un ORFen el área comple
mentaria al extremo 3' del ORFde la polimerasa y 5' del pre
core; este ORFestá conservado en tres cepas de DHBVy la pro

teïna predicha tiene aproximadamente 80 aa (Tagawaet a1.,
1990). Debe tenerse en cuenta que en el virus del pato la ca
dena positiva esta casi completa.
Tanto el ORFpreC/C como el preS/S poseen más de un codón
de iniciación (AUG)en fase y un codón de terminación común,
pudiéndose así traducir más de un polipéptido de cada una de
estas regiones. El ORFpreS/S tiene tres AUG,uno al comienzo
de cada zona: pre-Sl, pre-82 y S, produciendo así las tres
proteínas LHBs, MHBsy SHBs respectivamente. El ORFpreC/C
tiene dos AUG,si la traducción se inicia en el primero se
forma el producto de pre-C/C que es el precursor del antígeno
e (HBeAgo HBe); en cambio si comienza en el segundo AUGda
origen a la proteína HBc.
Secuencias regulatorias: La regulación, tanto de la
transcripción comode la replicación viral, es un proceso com
plejo en el que, no sólo están involucrados un gran número de
factores celulares para los cuales se han identificado sitiosde unión en el genomadel virus, sino que también parece estar
influido de modotejido-específico.
La transcripción en HBVestaría controlada por cuatro
promotores, dos “aumentadores” (enhancers), un “silenciador”
(silencer) y una sola señal de poliadenilación. (Fig. I.3).Ademásse han identificado sitios de unión para un gran número
de factores moduladores de la transcripción mediante ensayos
de movilidad diferencial en geles (gel mobility shift assay) e
improntas de protección a la DNasaI (DNasaI footprinting)
(Schaller &Fischer, 1991).

Ipr‘eC/C) Ipr‘e S/S ) lpr‘eC/C
I PIJL )
l] [l Ü I]
UDD3200/1
I l I ll l l
1500 2000 2500 3000 300 500 13601'500 edoo
Ü SiLenciQolor‘:IPY‘OMOÏOY‘ C
¡:Enh I E3 Promotor" XÍ::|Enh II EEpsiLon
I DRI I DRII
[:18PIÍ:]SP II
Figura 1.3: Principales secuencias regulatoriasAdaptado de Schaller & Fisher (1991)
No se representan los sitios de unión de los diversos factores
de regulación que han sido identificados.

De los cuatro promotores, llamados: core, X, SPI y SPII,
solamente SPI presenta una típica TATAbox consenso.
El promotor core no sólo dirige la sintesis de los mRNA
que codifican para HBc, HBey Pol sino también del RNAprege
nómico, el cual es el templado para la síntesis del DNAviral.La secuencia nucleotídica 1074-1234 tiene una función de
enhancer (EnhI) que activa los promotores SPI, SPII, core y X.
Un segundo enhancer (EnhII) que mapea entre los nucleótidos
1636-1741 dentro del ORFX, e inmediatamente anterior al ORF
preC/C , estaría compuesto por dos elementos II-A (nt 1636
1690) y II-B (nt 1704-1741) y estimularía la actividad trans
cripcional de los promotores de los genes superficie, core y X
( Yuh & Ting, 1991).
Respecto al "silenciador", este elemento, que actúa en
cis, sólo regularía negativamente al promotor core (Gerlach &
Schloemer, 1992).
Otras regiones importantes son: las secuencias DR1y DR2
que son repeticiones directas de 11 pb localizados a cada lado
de los extremos cohesivos del genomay son importantes en la
síntesis del DNAviral, y, 1a señal de encapsidación (e) cuyasfunciones veremos más adelante.
Variantes genómicas: Las primeras “variantes” en ser des
criptas fueron los subtipos o serotipos, los cuales se definenpor criterios inmunoquímicos(inmunodifusión), por la presen
cia de pares de determinantes antigénicos, de este modoun
aislamiento es d o y (Le Bouvier, 1971) y w o r (Bancroft et
a1., 1972). Posteriormente se definieron los subdeterminantes

de w (Couroucé et al., 1976), el determinante q (Magnius et
al., 1975; Couroucé-Pauty et al., 1978) y el par i/t (Ohnuma
et al., 1993). Esta diversidad antigénica se debe, en varios
casos, a 1a sustitución de un sólo nucleótido, generando así,
polipéptidos con estructuras primarias muysimilares pero po
sibles conformaciones inmunológicas distintas. En otros casos,
la situación es más compleja ya que más de una posición nu
cleotïdica parece estar involucrada.( Norder et al., 1992a).Tabla compendiode posiciones aparentemente claves:
posición aa determinante124-147 a
122 lis d
arg Y
160 lis w
arg r
127 pro w1/w2
thr w3
leu w4
126/7 ile/pro ithr/pro t
177/8 val/pro q+
ala/pro adrqhval/gln adw4q

Por otra parte la clasificación genética, usando la se
cuencia nucleotídica del genomacompleto o de la región S, ba
sada en la divergencia del @%o más, permitió la clasificación
en 6 genotipos designados de A a F (Okamotoet al., 1988;
Norder et al., 1992b). Si bien en algunos casos existe alguna
relación, estos agrupamientos genotïpicos no se corresponden
exactamente con los grupos serológicos.
genotipo serotipo
A adwz aywl
B adwg aywi
C adw ayr adr adrq'
D ang ayw3
E aywÁ
F adw4
Dada la completa utilización de las secuencias genómicas
para la codificación de proteínas, se podría suponer que el
HBVha evolucionado hasta un punto en que, pocas de las muta
ciones que se producen en un ciclo normal de replicación po
drían mantenerse sin ser letales. La comparaciónde las se
cuencias publicadas muestra, sin embargo, que varias regiones
difieren en el númerode secuencias conservadas y estas dife
rencias se dan, no sólo entre genomascorrespondientes a los
distintos genotipos, sino tambiénentre distintos aislamientosdel mismotipo; aunque, en general, la divergencia intra-sub
tipo es menor que la inter-subtipo (Okamotoet al., 1988; Lin
et al, 1991a; Kremsdorf et al., 1991).

El genomade HBVtiene una tasa de sustitución de nucleó
tidos estimada de 1,4 x 10‘5 a 3,2 x 10'5 por sitio por año,
la cual es algo menor que la demostrada para algunos retrovi
rus (que también usan transcripción inversa) y varias veces
mayor(aproximadamente104) que la tasa de sustitución de la
mayoría de los virus con genomas a DNA.
Entre las zonas más conservadas se encuentran 1a región
pre-C y S; y entre las altamente variables las secuencias que
van desde el final de C y se continúan a través de los pre-S
hasta el S, y, entre S y X. En particular la región que com
prende los nt 2500 a 3200 es hipervariable.
Es importante mencionar que dentro de estas regiones con
servadas, se producen dos tipos de mutaciones que tendrían una
relación relevante con la evolución de la patología; son las
llamadas mutantes e‘ y las mutantes de escape (“escape
mutants”). Las primeras, aparentemente ligadas al cuadro más
severo (hepatitis fulminante), presentan, en la mayorparte de
los casos, una mutación (Gmgsa A) que incorpora un codón de
terminación en la región preC no permitiendo así la biosínte
sis de HBe. Las segundas, en general, también se deben a una
mutación puntual, que en este caso se da en SHBs; la más fre
cuente de estas mutaciones cambia G97 a A, lo que implica un
cambio de Glst a Arg, en el determinante antigénico a contra
el cual esta dirigida la inmunidad inducida por la mayoría de
los antígenos vacunales actualmente en uso, dándole así al vi
rus la capacidad de escapar a los anticuerpos neutralizantes.

HBx: El ORFX, presente en todos los hepadnavirus de ma
míferos (orthohepadnavirus) y ausente en los aviarios
(avihepadnavirus), está altamente conservado en todos los sub
tipos de HBV(Lo et al., 1988) y codifica una proteína (HBx)
de 154 aa (17 kDa) (Fig. 1.4).
Se detectaron anticuerpos contra HBxen individuos infec
tados ( Kay et al., 1985; Levrero et al., 1990) lo que demues
tra que proteínas originadas en la expresión de este gen sesintetizan in vivo.
A diferencia de lo que ocurre con los otros genes del
HBV,la secuencia nucleotïdica del ORFX indica que su prefe
rencia en el uso de codones es similar a la de los genes presentes en células eucariotas. Esto también ocurre con los on
cogenes de retrovirus, por lo que se ha sugerido que este gen
sería de origen celular. Esta hipótesis, sin embargo, no pa
rece posible ya que su secuencia no revela homología signifi
cativa con otros oncogenesretrovirales ni celulares (Miller &
Robinson, 1986).
HBxes un importante trans-activador de secuencias regu
latorias, estimulando la expresión de una variedad de genes
autólogos y heterólogos, entre ellos el gen B-interferón
humano, c-myc, c-fos, HIV-1 y HIV-2 LTR, promotor temprano de
SV40, HTLV-l LTR, RSV LTR, etc..
Es de notar que proteínas HBxtruncadas mantienen sus
propiedades transactivadoras. Esto se explica porque el extremo carboxi-terminal no es necesario para esta función. La
presencia del primer codón AUGpor el contrario, es muy impor
tante para la síntesis de una proteína activa, lo mismoque

[ERE preC/C
ORF P
ATG TAAnt1374 1836
‘ I l II I III IV ‘ V
NH2 ' I ¡ COOH¿o 100 150 aa
I: región conservada, mayormentea hélice o plieguehidrofóbico
II: región variable, con predominancia de residuos Pro y Ser
III: región compleja, de secuencia conservada interrumpida por
una zona hipervariable y carga distribuida en una zonaacídica y otra básica
IV: región muyconservada, semejante a "Leu-zipper" quecontiene 6 residuos Leu con intervalos de 4 a 6 aa
V: cola hidrofóbica de secuencia poco conservada
Figura 1.4: HBx
Adaptado de Colgrove et al. (1989), Arii et al. (1992) y Kimet al. (1993).

varias secuencias internas para su acción (Balsano et al.,1991; Arii et al., 1992).
Se han propuesto varios mecanismos de acción: mientras
algunos autores sugieren que HBxse une a algún factor de
transcripción secuencia-específico, estableciendo una interac
ción con el complejo iniciador de la transcripción (Colgrove
et al., 1989; Yen et al., 1991); otros proponen que regula la
renovación (turnover) de diversos factores de transcripción, a
través de la inhibición de una serin-proteasa involucrada en
el clivaje proteolítico de proteínas celulares (Takada&Koike, 1990; Takada et al., 1994), o, activando estos factores
a través de la quinasa C (PKC) (Kekulé el al., 1993).
Se ha demostrado que HBxtiene actividad de quinasa y
puede catalizar la fosforilación tanto de sí misma, comode
otras proteínas, en los residuos serina y treonina, aunque no
en tirosina (Wuet al., 1990). Es posible entonces que esta
proteína puedaactivar la transcripción regulada por distintassecuencias, catalizando la fosforilación de factores celularesinvolucrados en 1a regulación de la transcripción (Robinson et
al. , 1991).
En suma, está bien demostrado el rol transactivador de
HBxaunque existe controversia acerca de los mecanismos invo
lucrados mediante los cuales HBxejerce su acción trans-acti
vadora (Rossner, 1992; Murakamiet al., 1994).
HBc: Es una proteína de 183 o 185 aa, dependiendo del
subtipo, y un tamaño aparente de 21-22 kDa. Estos monómeros se
organizarían formandouna estructura icosahédrica de 180

subunidades por nucleocápside, que se produce en ausencia de
otras proteínas virales, por lo cual muestra una alta capacidad de autoensamble.
Esta proteína presenta dos regiones estructural y funcionalmente distintas: una amino-terminal de aproximadamente 144
aa que forma un dominio estable e independiente con una fuerte
tendencia a formar dímeros y suficiente por sí mismapara au
toensamblarse; esto por lo menosen los sistemas de expresión
heterólogos, en que se sintetizan y acumulan grandes concen
traciones de estas proteínas. La otra parte, correspondiente
al carboxi-terminal, está cargada positivamente y muestra ca
pacidad para unirse a RNA(Fig. 1.5).
HBcposee cuatro Cys en las posiciones 48, 61, 107 y 183,
estos residuos parecen estar evolutivamente conservados ya que
están presentes, en posiciones equivalentes, en la proteína
core de todos los hepadnavirus de mamíferos estudiados, pese a
que estos virus exhiben una variación de secuencia promedio
del 3096. Se ha sugerido que al menos los dos primeros de estos
residuos podrían estar implicados, a través de puentes disulfuro, sino en la formación, por lo menos en el mantenimiento
de los dímeros, y la CyslB3 en la de polímeros (Nassal et al,
1992).
Respecto a los 39aa carboxi-terminales, éstos contienen
16 residuos Arg, 14 de los cuales se presentan en 4 agrupa
mientos (clusters), tres de ellos presentando el motivo
SerProArgArgArg(Arg), cuyo prototipo (SerProLysLys) esta pre
sente en varias proteínas que se unen al DNA(DNA-binding).
Estas proteínas, comopor ejemplo: extremo carboxi-terminal de

20
ORF P
ATG TAG
1901 ' 245o nt
NH2 J C%É
rnllvv “¿‘“¿L uuuuu .ZonnnnanSRESQc l
región carboxi-terminal con los agrupamientos de Arg y lasseñales de localización nuclear
x Cys en posición 48, 61, 107 y 183
Figura 1.5: HBc
Adaptado de Nassal (1992) y Nassal et a1. (1992).

21
la histona H1, c-myc murino, etc., interaccionan con el surco
menor de la doble hélice del DNA,preferencialmente en zonas
ricas en AT (Churchill &Travers, 1991).
La expresión en E.coli de un gen de HBccon una deleción
carboxi-terminal permite el autoensamblede la proteína sinte
tizada y, por ende, la formación de nucleocápsides, pero éstas
ya no contienen RNA.Esto indica que el ensamble de las partí
culas de HBcpuede concretarse sin el requerimiento de una
unión previa a ácidos nucleicos, y que la eliminación de estos
agrupamientos del sector carboxi-terminal previene la unión
con el RNA(Gallina et al., 1989).
En cuanto a experimentos de expresión transitoria de ge
nomas de HBVcompletos en células animales, sugieren que la
primera mitad de la parte carboxi-terminal, hasta el aa 164,
es necesaria y suficiente para encapsidar el RNApregenómico y
que los aa desde el 163 al 173 actuarían como componentes au
xiliares en la replicación, en particular durante la síntesisde la segunda cadena de DNA(Nassal, 1992).
La cantidad de RNAempaquetado corresponde a 18 bases por
HBc, es decir 3240 bases por partícula de core, lo cual está
de acuerdo con el tamaño del pregenoma que es de aproximada
mente 3300 pb, más la cola de poly-A (Melegari et al., 1991);
aunque la alta especificidad de secuencia observada en el em
paquetamiento in Vivo probablemente es debida a la cooperación
entre el pregenoma, HBcy la polimerasa (Lavine et al., 1989;
Hirsch et al., 1990). Es de importancia tener en cuenta que,
la proteína HBcaislada de viriones purificados a partir de
suero de pacientes, no se une a RNAaunque posee los agrupa

22
mientos de Arg. Esto indicaría que se ha producido una modifi
cación en esta región, posterior al ensamblado; un mecanismo
posible para generar esta modificación sería la fosforilación
de los residuos serina que están dispersos entre las argininas; esta actividad podría ser debida, tal vez a HBxo, a la
protein-quinasa C (PKC)del huésped la cual sería encapsidada
dentro de 1a nucleocápside en una proporción de dos moléculas
de PKCpor partícula (Kann & Gerlich, 1994).
Esta fosforilación del extremo carboxi-terminal también
podría desempeñarun rol importante en la regulación del
transporte nuclear de HBc. Un mecanismosimilar ocurre en el
transporte del antígeno T de SV40, que se ve facilitado por 1a
fosforilación de los residuos serina que flanquean la señal delocalización nuclear (Rihs & Peters, 1989). En HBcesta señal
estaría conformadapor: dos repeticiones directas (Pro Arg Arg
Arg Arg Ser Gln Ser) que comprenden los dos últimos agrupa
mientos de Arg (Yeh et al, 1990); o por dos regiones que com
prenden el primer y cuarto cluster y representarían secuenciasde localización nuclear distintas e independientes, aunque
para evitar la acumulación de HBcen el núcleo ambas señales
deben estar ausentes o mutadas (Eckhardt et al, 1991).
HBe: Es el producto de la traducción de la región pre-C/C
que resulta en una proteína de 212 aa, es decir 29 aa más que
HBc, de los cuales los primeros 19 aa amino-terminales consti
tuyen el péptido señal para la inserción del polipéptido naciente en la membranadel reticulo endoplasmático (R.E.). El
péptido señal es clivado por una proteasa específica y el po

23
ORF P
ATGATG G TAG
1814 2450 nt
x Cys
péptido señal
agrupamientos de Arg
l <:> sitio de clivaje fijo
sitio de clivaje variable
Figura 1.6: HBe
Adaptado de Carman & Thomas (1992)

24
lipéptido restante es translocado al lumen. Durante el trans
porte en el aparato de Golgi es modificado por proteólisis delextremo carboxi-terminal, eliminándose un númerovariable de
agrupamientos de Arg. La proteína resultante, es secretada sin
ningún proceso de ensamble o formación de partículas, y aún
contiene 10 aa de la región pre-C en su extremo amino. (Fig.
I.6)
El análisis de esos 10 aa pre-core residuales reveló que
éstos inhiben la formación de partículas y la Cys en posición
-7 juega un rol crítico en prevenir el ensamble. Así cuando la
Cys -7 es sustituida por glutamina (Gln) se restablece el en
samble típico de HBC. (Schodel et al., 1993)
Aún no se ha asignado una función específica a esta pro
teína en el aspecto funcional de la replicación viral, sin em
bargo su detección en plasma es un indicador clínico impor
tante. La presencia de antígeno e es interpretada comouna re
plicación viral activa en el huésped y es indicador de un ma
yor riesgo de transmisibilidad y mal pronóstico de la evolución.
Pol: El producto del ORF-P (Pol) es una proteína con ac
tividad de transcriptasa inversa/DNApolimerasa (Bavandet
al., 1989). Está contenida dentro de la nucleocápside del vi
rión infeccioso, y en presencia de los dNTPrepara la cadena
incompleta del genoma generando una molécula de DNAcircular
de doble cadena y ademásparticipa en varios pasos del ciclode vida viral.
En realidad sería una proteína multifuncional de unos 95

25
kDa aproximadamente, dentro de 1a cual, y en sentido 5’-3’, se
pueden distinguir cuatro dominios que funcionan independiente
mente: una primasa o proteína terminal (TP), una secuencia es
paciadora no esencial, una transcriptasa inversa/DNApolime
rasa (RT) y una RNasaH(Radziwill et al., 1990) (Fig. 1.7);
además es requerida para el empaquetamiento del RNApregenó
mico, estando esta última función separada de la actividad de
polimerasa (Hirsch et al, 1990).
Esta proteína polifuncional puede ser sintetizada a par
tir del mRNApregenómico de 3,5 kb por iniciación de la tra
ducción a partir del codón AUGcorrespondiente al ORF-P (Chang
et al., 1989). Este mecanismodifiere del que se desarrolla en
retrovirus donde el gen pol es expresado en forma de una pro
teína de fusión gag-pol por un mecanismode desplazamiento del
marco de lectura (frameshifting) ribosomal y luego la polime
rasa es liberada por procesamientoproteolïtico post-traduccional. En el sistema HBVlas proteínas core y pol pueden ser
sintetizadas independientemente usando el RNApregenómico como
templado y un mecanismo de “leaky scanning” (Kawamotoet al,
1990; Lin & Lo, 1992). Además la traducción de pol no es inhi
bida por el análogo del cap m7GpppG.Estos resultados indican
que la traducción ocurre de una forma nueva que no requiere la
presencia del cap (cap-independiente).
El dominio amino-terminal (TP) es idéntico a la proteína
unida a1 extremo de la cadena negativa del DNAviral
(Bartenschlager &Schaller, 1988) y actuaría como “primer” en
la transcripción inversa (Seeger &Maragos, 1989) posiblemente
interactuando con la señal para la transcripción inversa

26
ORF X
ORF preCZE] I ‘ ORF’preS/S
ATGTGAnt
2310 1621
! x TP ‘ Spacer ¡ RT , RNasaH ‘
NH2 f l 1 COOH200 4%“) 600 800 aa
4; Tyr63
Figura 1.7: Pol
Adaptado de Radziwill et al. (1990) y Roychoudhury et al.
(1991)

27
(SRT), que se encuentra en la región e, a través de su Tyr63
(Weberet al., 1994).
La detección de una proteína de 65-70 kDa en viriones hu
manos mediante antisuero P-especïfico (Macket al., 1988) ha
sugerido un procesamiento proteolïtico después del empaqueta
miento; y ya que esta proteína reacciona con antisuero contra
el extremo carboxi-terminal de P se podría presumir que la
proteólisis ocurriría dentro del dominioespaciador, y que
este producto de P, unos 600 aa, contendría la transcriptasa
inversa y la RNasaH (Gerlich & Heermann, 1991). Sin embargo
otras experiencias indicarïan que la proteína Pol actúa sin
ser procesada, al menosdurante los pasos iniciales de ensam
blado de la nucleocápside y transcripción inversa
(Bartenschlager et al, 1992).
Un antisuero preparado contra 1a región conservada de la
transcriptasa inversa, reacciona con la HBV-polimerasay con
transcriptasas inversas de varios retrovirus (Macket al.,
1988). La inactivación por mutaciones del dominio de la RNasaH
permite la síntesis de 1a cadena de DNAnegativa, pero anula
la síntesis de la cadena positiva (Radziwill et al., 1990).
HBs: Es un grupo de glicoproteinas relacionadas, cotermi
nales, producto de la iniciación en tres AUGsdiferentes si
tuados en fase (Fig. 1.8).
La iniciación en el AUG3genera la principal proteína de
superficie SHBsde 226 aa (p24 y su forma glicosilada gp27);
la iniciación en el AUG2agrega entre 44 y 56 aa a1 extremo
amino-terminal generando MHBs(p30 y sus formas mono y digli

28
ORF P
ATG ATG ATG TAAA A2848 \J k1 833 nt
LHBS l
MHBs 1
SHBs I
|N lI f IV l III v IIIII c |
I, II y III: a hélices hidrofóbicasI y II: señales de translocaciónIV y V: regiones moderadamentehidrofïlicas
V: región de determinantes antigénicos
N y C: regiones amino y carboxi-terminal respectivamente
Figura 1.8: HBs
Adaptado de Carman & Thomas (1992).

29
cosilada gp33 y gp36) y la iniciación en el AUGlagrega de 108
a 119 aa a MHBspara dar LHBs (p39 y gp41).
El sitio de glicosilación de las tres proteínas es elmismo y está localizado en la región S; MHBs,además, tiene
otro oligosacárido, también unido vía N-acetilglucosamina (N
linked), en el dominio específico de preSZ.
LHBstiene otra modificación post-traduccional con la
adición de un grupo ácido mirïstico a su residuo Gli amino
terminal (Persing et al., 1987).
Comoya se mencionóentre estas tres proteínas, no existe
relación precursor-producto sino que provienen de distintos
transcriptos iniciados a partir de los dos promotores corres
pondientes, por lo tanto cualquier mutación en las zonas preS,
ya sea que cambie el marco de lectura o introduzca codones de
terminación, no afecta la síntesis de SHBsque es el principal
componentede las partículas de Dane y también de las partículas subvirales.
Las partículas de Dane se producen cuando las nucleocáp
sides preformadas brotan dentro de las membranasdel reticulo
endoplásmico (ER) que contiene a SHBs, MHBsy LHBs, y el re
sultado de análisis genéticos ha demostrado que SHBs, LHBs, y
probablemente MHBstambién, son requeridas para una correcta
morfogénesis de la partícula de Dane. Otros experimentos, con
proteínas quimera (mezclando dominios del HBVy WHV)y coex
presión de las distintas proteínas de membranade ambosvirus,
sugieren que el proceso de ensambladorequiere interacciones
moleculares precisa entre estos componentes; siendo importan
tes para la secreción eficiente de SHBslos residuos cisteína

en posición 48, 65, 69, 107, 138 y 149 (Gerhardt & Bruss,
1995).
Los receptores celulares deben jugar un rol importante en
la patogénesis viral. El dominiopreSl contiene un sitio de
unión a la membranadel hepatocito y otras células de origen
extrahepático (Neurath et a1., 1990). Este sitio de unión, lo
calizado entre los aa 21 y 47 de la secuencia preSl, es muy
semejante a una secuencia presente en la región constante de
la cadena pesada de IgA (Neurath & Strick, 1990).
2.2. Biología
El principal obstáculo para dilucidar los procesos de
adsorción viral, penetración y pérdida de la envoltura(uncoating) ha sido la dificultad de infectar líneas celularesestablecidas con viriones infecciosos. A pesar que se han he
cho avances al identificar a1 receptor celular putativo y elsitio de unión del virion, los pasos subsiguientes de penetra
ción y uncoating están mayormentesin definir. Se ha sugerido
que la nucleocápside es liberada de la envoltura y transportada al núcleo celular, en virtud de la señal de translocación
nuclear de HBc, donde se realizaría el uncoating del genoma
viral y su conversión, de la forma de DNAparcialmente de do
ble cadena, a la forma de DNAcircular covalentemente cerrado
(ccc-DNA).
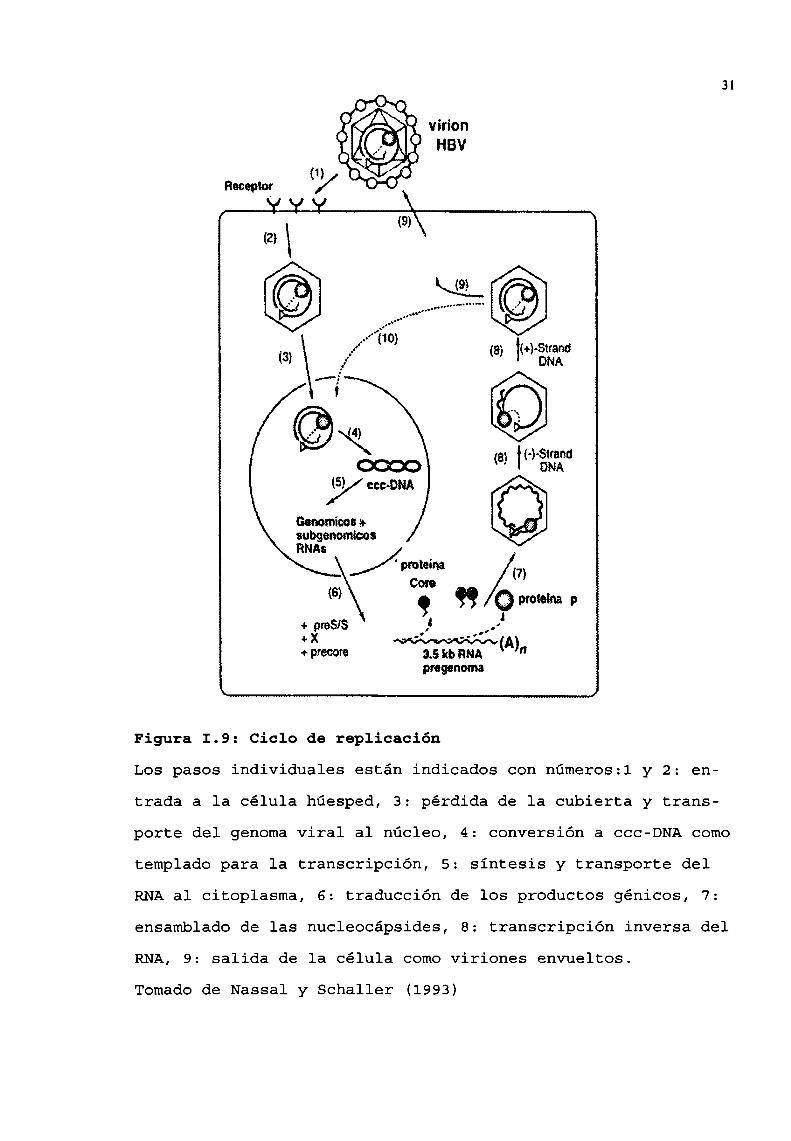
En la figura I.9 se presenta el esquemageneral
3l
Genomicos ersubgenomicoa
RNAS/ 'pmnma(7)c
(6) o,” 9? e proteinapl ,‘
+Ímfis . H.* ' ' A+precore 3.5 kb RNA ( )"
pmguwnm
Figura 1.9: Ciclo de replicación
Los pasos individuales están indicados con números:l y 2: en
trada a 1a célula húesped, 3: pérdida de la cubierta y trans
porte del genomaviral al núcleo, 4: conversión a ccc-DNAcomo
templadopara la transcripción, 5: síntesis y transporte delRNAal citoplasma, 6: traducción de los productos génicos, 7:
ensambladode las nucleocápsides, 8: transcripción inversa delRNA,9: salida de la célula comoviriones envueltos.
Tomadode Nassal y Schaller (1993)

32
Adsorción y entrada: A pesar de los esfuerzos realizados,
a causa de la ya mencionada falta de un sistema celular apro
piado para propagar el virus, se conoce muypoco de los pasos
iniciales de la infección. La mayoría de los datos disponibles
provienen de experimentos utilizando células transfectadas con
DNAviral, generalmente clonado; y estos métodos sólo permiten
un ciclo único de producción viral.
La unión del virus al hepatocito estaría mediada por un
segmentoparticular de la región preSl.
Respecto a la fusión de la membranacelular y la envol
tura viral, que permitiría la entrada de la nucleocápside al
citoplasma, se ha postulado un rol fusogénico a los veintitrés
aa amino-terminales de SHB(Rodríguez Crespo et al., 1994).
Formación de ecc-DNA: La formación de esta especie mole
cular requiere una serie de pasos cuyo orden cronológico, en
zimas involucradas (celulares y/o virales) y si requieren o no
la integridad de la nucleocápside, no están perfectamente ca
racterizados. Estos pasos serían: a) terminación de la cadena
positiva del DNA;b) disociación de TP del extremo 5' de la
cadena negativa; c) eliminación de la redundancia terminal de
la cadena negativa; d) eliminación del primer de oligoRNAdel
extremo 5' de la cadena positiva y e) ligación de los extremos
5' y 3’. La precisión de este último punto es crítica para la
integridad del genomaviral, ya que la cadena negativa es el
templado para la síntesis del RNApregenómico.
Transcriptos: La transcripción se lleva a cabo, a partir

33
de DNAcircular covalentemente cerrado, utilizando la maquina
ria de transcripción nuclear de la célula huésped (RNApolII)
y produce un grupo de transcriptos, unidireccionales y super
puestos, ya que dicha transcripción se inicia en varios promo
tores distribuidos a lo largo del genomay termina en un solo
sitio de poliadenilación comúna todos localizado aproximada
mente 20 nt después del hexanucleótido conservado TATAAA(Fig.
I.3).En los hepatocitos infectados se producen varias clases
de transcriptos: los transcriptos genómicos (que son terminal
mente redundantes) de aproximadamente 3,5 kb y comprenden: a)
el RNApregenómico, cuyo extremo 5’ es único y esta localizado
5 nucleótidos después del AUGdel precore, y que es el tem
plado para la transcripción inversa que produce el DNAviral y
además mensajero para la síntesis de las proteínas (HBcy Pol)
que formaran las nucleocápsides competentes para la replica
ción y b) otro menos abundante y ligeramente más largo, que
incluye el sitio de iniciación de traducción del precore(comienza 20-30 nt antes) y sirve comomRNApara la síntesis
de un producto de fusión precore/core que luego de ser procesado dará a HBe.
De los transcriptos subgenómicos, los más abundantes son
los RNAde 2,1 kb utilizados para la síntesis de MHBsy SHBs
(transcriptos desde el promotor SPII) y también se sintetizan
un transcripto de 2,4 kb para la síntesis de LHBsdesde SPI y
otro de aproximadamente 0,8 kb para HBx.
Todos estos transcriptos presentan cap, una cola de polyA
y no son procesados (unspliced).

34
longitud función/producto extremo 5' promotor
Transcriptos genómicos3,30 kb —pregenoma único, 5 nt posterior C
- C mRNA/HBc al preC —AUG
- P mRNA/P
3,35 kb preC mRNA/HBe variable, 20-30ntantes del preC-AUG
Transcriptos subgenómicos2,4 kb preSl mRNA/LHBs único (z nt 2807) SPI
2,1 kb - presz mRNA/MHBS z nt 3157/8 SPII- S mRNA/SHBS nt 3174/5, 5/6, 113
0.9 kb X mRNA/HBx nt 1226, 1242 X
Además de estos mRNAno procesados, se han detectado
transcriptos procesados, en tejidos infectados por el HBVy
células transfectadas, aunque aún no se ha demostrado cuál
pueda ser su rol biológico in vivo (Suzuki et al., 1989;
Schaller & Fischer, 1991; Wuet al., 1991).
Comoya se indicó el RNApregenómico tiene extremos ter
minalmente redundantes que exceden el largo de su templado de
DNA,por lo que tanto las secuencias DRl como la señal de en
capsidación (8) están localizadas tanto en el extremo 5’ como
cerca del extremo 3' de la molécula; una tercera copia de la
repetición directa (DR2)queda también en el extremo 3' peroantes de DR1*.

35
mSijlllemyDR1 DR2 DRl'
Ensamblado: El elemento (8) en cis del RNApregenómico
responsable por el empaquetado, ha sido mapeado en una estre
cha región cerca de su extremo 5’. Esta secuencia es requerida
para el ensamblado de esta molécula en los cores inmaduros ya
que un corto segmento de 85 pb de este elemento es suficiente
para la encapsidación de RNAheterólogo no viral (Junker
Niepmannet al, 1990). La estructura secundaria predicha para
este elemento preveé la formación de una estructura en horqui
lla compuesta por dos "stem", un "loop" y un "bulge".
Es interesante notar que esta señal de empaquetado ha
sido mapeadacerca de la unión (junction) preC-C, esta región
del genoma también corresponde a una secuencia de 60-70 pb al
tamente conservada con un alto grado de homologïa con la re
gión U5 del LTRde retrovirus. En los sistemas retrovirales la
región U5 juega un rol en el empaquetado del genoma viral
(Moloney murine leukemia virus). Ya que el más largo de los
RNAtranscriptos de 3,5 kb, que también contiene esta señal 8,
no es encapsidado, se supone que el desarrollo de estructuras
secundarias y/o procesos transcripcionales o traduccionalesadicionales deben jugar un rol en un mecanismode encapsida

ción tan altamente selectivo
Comoya se mencionó HBc es capaz de autoensamblarse a
través de dímeros intermediarios, pero la correcta encapsida
ción del RNAapropiado también requiere la presencia de Pol,
por lo que se ha sugerido el siguiente modelo: el RNApregenó
mico actúa como mRNApara HBc y eventualmente para Pol, en
este último caso esta proteína se uniría inmediatamente (en
cis) a la señal 8 del extremo 5’ del RNA;esta unión inhibiría
la traducción de HBca partir de esa molécula de RNA, “saca”
los ribosomas y estimularïa la adición de dímeros de HBc (en
trans) que estabilizarïan el complejo. Este mecanismoasegura
la coencapsidación eficiente del pregenomay de Pol previ
niendo la transcripción inversa de RNAscelulares y también
explica el bajo número (probablemente sólo una) de moléculas
de Pol por partícula.
Transcripción inversa: El RNApregenómico encapsidado se
convierte en el genomaDNAcircular parcialmente de doble ca
dena mediante un proceso complejo.
Se había postulado que el sitio de iniciación para la ca
dena negativa del DNAera el DR1*, pero nueva evidencia (Wang
& Seeger, 1993) indica que la reacción que produce el primer
para la síntesis de dicha cadena utiliza comotemplado una se
cuencia presente en una de las zonas de simple cadena de 8. De
este modo la Tyr63 actúa comoprimer para la formación de una
unión covalente (que ya no se romperá hasta el próximo ciclo)
entre TP y dGMP(complementario a la C del RNA)para sintetiAzar una cadena corta (de 3 o 4 nt) de DNAque será el verda

37
dero primer para la formación de la cadena negativa del DNA
genómico. Este primer (proteína + nt) luego se transpone y
aparea con la secuencia complementaria en DR1*desde donde
continúa la síntesis del DNAhacia el extremo 5' del RNApre
genómico, mientras éste es degradado por acción de la activi
dad de RNAsaHde Pol, excepto los últimos 15-18 nt que inclu
yen a DR1y sirven como primer para la cadena positiva del DNA
luego de ser transpuestos a DR2;esta cadena se elonga hacia
el extremo 5' de la cadena negativa, donde el salto hacia el
extremo 3' se ve facilitado por la redundancia terminal.
El mecanismopor el cual Pol lleva a cabo las transposi
ciones de los primers para la síntesis de ambas cadenas de DNA
no esta resuelto, pero parece evidente que el complejo ácido
nucleico-Pol puede organizarse de modotal de mantener en una
gran proximidad y/o yuxtaponer los distintos elementos involucrados.
2.3. Diagnóstico
La mayoría de las infecciones son subclïnicas y conducen
a una recuperación aparentemente completa con desarrollo de
anticuerpos virus-específicos. Sin embargouna proporción
significativa (probablemente 1-596) pueden evolucionar a dese
quilibrios de la función celular y generar estados de hepatopatías crónicas de varios tipos, cirrosis y cáncer primario dehígado.
Unporcentaje muydifícil de establecer desarrolla una
patología aguda (hepatitis fulminante) que produce la muerte,con una evolución de treinta a sesenta días.

38
La definición de viremia en infecciones persistentes con
HBVha ido avanzando desde las primeras detecciones de partí
culas de Dane por inmunomicroscopía electrónica (Daneet al.,
1970), y la correlación indirecta con HBeAg(Shikata et al.,
1977) hasta las técnicas de hibridación molecular incluyendo
el uso reciente de PCR. La sangre de pacientes HBeAgpositivos
contienen concentraciones relativamente altas de HBV-DNA,in
dicando un alto nivel de replicación de HBVen el hígado. Pa
cientes que son HBeAgnegativos y anti-HBe positivos general
mente tienen cantidades moderadas o pequeñas de HBV-DNA.Con
técnicas de hibridación en puntos (dot blot), el HBV-DNApuede
ser detectado en el 5-60% de los pacientes anti-HBe, depen
diendo de su origen racial (Karayiannis et al., 1985) y algunos autores los consideran infectados con HBVmutantes. Usando
PCRy detección de los productos amplificados por hibridación
en Southern blot, HBV-DNAfue detectado en el 8996de los pa
cientes anti-HBe positivos con hepatitis crónica (Kanekoet
al., 1989); esta combinaciónde técnicas permitiría detectar
104 pg de HBV-DNA,o sea tres genomas virales, por muestra de
20 pl, lo que se aproxima a la sensibilidad de los ensayos de
infectividad en chimpancés (Wanget al., 1991).
La detección serológica de HBsAgo anti-HBc es aceptada
comoevidencia diagnóstica de infección por HBVactual o pa
sada, dada la consistente presencia de estos dos marcadores eninfecciones crónicas.
A pesar de que el virus ha sido identificado en tejidos
extrahepáticos, comoel endotelio vascular, epitelio de losductos biliares, médulaósea y linfocitos, preferentemente del

39
tipo B, de sangre periférica (Blumet al., 1983), el sitio de
infección principal y más importante es el hepatocito.
La disponibilidad de sondas para detectar el ácido nucleico de
HBVes muy importante en aquellos casos en que las pruebas se
rológicas no permiten establecer un resultado definitivo o en
casos de terapia antiviral a fin de monitorear y determinar lareal efectividad de la misma.
Aún no se ha demostrado un mecanismo oncogénico específi
camente viral para HBV,sin embargola infección persistenteha sido fuertemente asociada con el desarrollo del carcinoma
hepatocelular (HCC). En pacientes portadores de HBV,la mayoría de las células de los HCCcontienen secuencias virales
integradas en el DNAcelular y esto planteó la cuestión de si
tales integraciones contribuyen, alguna vez, a la oncogénesis.
Sin embargo no se ha observado un patrón de integración espe
cífico, sugiriendo que HBVtendría un rol en el desarrollo de
neoplasias a través de la acción en trans de uno de sus pro
ductos génicos.
2.4. Tratamiento
Hasta el presente no existe ningún tratamiento antiviral,
específico contra la patología inducida por el virus de la he
patitis B, sin embargo,existen series de pacientes tratados
con diferentes protocolos, en los que fundamentalmente se han
utilizado distintos tipos de interferones, aunque es demasiado
prematuro aventurar conclusiones sobre estos estudios.

40
2.5. Epidemiología
El HBVtiene una distribución mundial si bien pueden re
conocerse zonas endémicas y otras de menor prevalencia. Asi
mismolos genotipos y/o subtipos principales también tienen
una distribución geográfica diferente (Couroucé-Pautyet al.,
1983; Norder et al., 1994). Sin embargoel carácter distintivo
es que el mecanismomás importante de transmisión varía en
distintas regiones del mundo.
Así, existen regiones en las que la historia natural de
la enfermedad mantiene la endemia por transmisión perinatal,
propia de áreas con alta endemicidad, que contrastan con
aquellas de baja endemicidad en las que 1a transmisión se man
tiene por transfusiones y/u otro métodobiomédico y por trans
misión sexual.
El este y sudeste asiáticos y las comunidadesque viven
en las regiones africanas al sur del desierto del Sahara, son
las de mayor incidencia y prevalencia del HBVen el mundo. Al
gunos otros focos tienen el mismoperfil; es el caso de algu
nas islas del Caribe comopor ejemplo la República Dominicana.
En cambio los registros más bajos se presentan en el
oeste europeo, América del Norte y algunos países del cono sur
de América del Sur.
Este tipo de clasificación en países con distinto tipo de
riesgos es aceptado internacionalmente, y se basa en estudios
serológicos efectuados en adultos, en muchoscasos derivados
de resultados obtenidos en el estudio de dadores de sangre:

41
a) baja prevalencia:
menos del 196 de individuos con HBsAgen sangre
menos del 1596de individuos con evidencia de haber su
frido una infección previa con el HBV.
b) moderada prevalencia:
individuos con HBsAgen sangre entre el 2 y el 796
individuos con evidencia de infección previa con el HBV
entre el 15 y el 40%
c) alta prevalencia:
individuos con HBsAgen sangre mayor al 896
individuos con marcadores positivos indicadores de infec
ción previa con el HBV, igual o mayor del 45%u


42
II. OBJETIVO
En el Instituto Nacional de Microbiologia (INM)funciona
el Centro Nacional de Referencia de Hepatitis Virales. La apa
rición de mutantes que complican el diagnóstico a nivel se
rológico, justifica explorar técnicas basadas en la detección
genómicadel virus de la hepatitis B.
La metodología a desarrollar intenta aportar resultados
en la búsqueda de posibles variantes de genomasvirales y diseñar una o más sondas destinadas a detección de DNAviral en
estudios diagnósticos, preferentemente en seguimiento de casos
particulares de patología crónica.Los aspectos propuestos son:
A.—Aportar información sobre secuencias del DNAviral, en es
pecial de las regiones vinculadas con el core y la región
preS/S, y ver su inserción en colecciones de genomasde dis
tintas regiones geográficas ya analizadas.
B.- Generar materiales para el desarrollo de reactivos diagnósticos, confiables y de baja complejidad, apuntando funda
mentalmentea la detección de posibles variantes virales asociadas a procesos evolutivos poco frecuentes

ODO

43
III. MATERIALES Y METODOS
1. Productos Químicos
Los reactivos y enzimas utilizados fueron de grado analí
tico o grado biología molecular de los siguientes laborato
rios: Boehringer Mannheim (Mannheim, Alemania), Fluka (Buchs,
Suiza), BRL (Gaithersburg, EE.UU), Mallinckrodt (Nueva York,
EE.UU), Merck (Darmstadt, Alemania), Sigma (St.Louis, EE.UU),
Bio-Rad (Hercules, EE.UU).
Los medios de cultivo bacteriano fueron de Difco
(Detroit, EE.UU) o Gibco (Gaithersburg, EE.UU).
2. Material Radioactivo
El material utilizado, aÜÏHdCTP> 3000 Ci/mmol para las
marcaciones de DNAin vitro y P%fldATP> 1000 Ci/mmol para las
reacciones de secuencia, fue de NewEngland Nuclear (Boston,
EE.UU) .
3. HBV
3.1. Purificación de partículas de DANE
Los viriones se purificaron parcialmente a partir de un
pool de plasma humano.
El plasma fue clarificado en un Rotor 19 de Beckman
Instruments Inc. (NewJersey, USA)a 5.000 rpm durante 30 mi
nutos a 4°C. El sobrenadante se sometió a una ultracentrifuga
ción a 160.000xg durante 6 horas a 4°C.
El pellet resultante se resuspendió en 0,01MTris (pH
7,2) con Pronasa al 196incubándose 1 hora a 37°C, y se peleteó
a través de un colchón de sacarosa al 3096en el mismobuffer

44
ultracentrifugándose otras 6 hs a 210.000xg. Este paso de pu
rificación se repitió una segunda vez en las mismascondiciones.
El último pellet se resuspendió en 0,01MTris (pH 7,2)
para ser aplicado a un gradiente lineal de sacarosa 15-6096que
se corrió en un rotor SW41a 40.000 rpm por 6 hs. El gradiente
se recogió en alícuotas de lml por punción del tubo.
3.2. Purificación de HBV-DNA
Las partículas de Dane se llevaron a una concentraciónfinal de 1%)SDS
10 mM EDTA
100 pg/ml Proteinasa K
incubándose durante 30 minutos a 45°C. Posteriormente se rea
lizaron dos extracciones con fenol-cloroformo-alcohol isoamï
lico (25:24:1) y otras dos con cloroformo-alcohol isoamïlico
(24:1). El DNAde la fase acuosa final se precipitó con ace
tato de amonioy etanol. El pellet de DNAresultante se resus
pendió en agua tridestilada conservándose a —20°C.
4. Cepas huésped y vectores
4.1. Cepas Bacterianas
4.1.1. Características y genotipos
Se utilizaron las siguientes cepas de E. coli:
4.1.1.1. DHSaF'.
Huéspedpara vectores que tienen la información genética
para el péptido a de la B-galactosidasa
Genotipo: F' ÓBOdlacZAMlS A(1acZYA-argF)U169 endAl

45
recAl hst17(r{nm+) deoR thi-l supE44 l‘ gyrA96 relAl
(Liss,L.R. 1987; Woodcocket al. 1989)
4.1.1.2. POP2136
Cepa que contiene el Xcl ts857 para la propagación y ex
presión controlada de vectores pEX.
Genotipo: F' endA thi hsdk malT malPQ lc1857 lacZ'
(Bernard et al. 1979).
4.1.2. Condiciones de crecimiento
Las cepas bacterianas fueron crecidas en medio LBy el
mismomedio en presencia del antibiótico ampicilina, en una
concentración final de 100 pg/ml, cuando correspondía.
La incubación se realizó, durante 16-18 hs, con agitación
a 37°C excepto la cepa POP2136que se incubó a 30°C.
Medio de cultivo LB (Luria Bertani)
Para un litro: triptona 10 g
extracto de levadura 5 g
NaCl 10 g
Se ajustó el pH a 7,5 con HONa.
4.2. Vectores
4.2.1. Características
4.2.1.1. pUC
Vectores de clonado derivados: del plásmido pBR322, del
que conservan el origen de replicación y el gen de la B-lacta
masa; y del vector M13mp(modificación del bacteriófago M13),
que aportó a la construcción el péptido-a del gen de la B-ga
lactosidasa y el sitio múltiple de clonado (polylinker).

46
4.2.1.2. pEX
Es una familia de vectores de expresión construidos a
partir de otros plásmidos: pUC8, pLK91y pCL19que a partir de
una fusión génica l cro-E.coli lacZ expresan una proteína de
fusión bajo el control del promotor PRdel fago l. Poseen un
polylinker en el extremo 3’ del gen lacZ en cada uno de los
tres marcos de lectura, permitiendo que cualquier ORFse ex
prese comouna proteína B-galactosidasa híbrida. Al final del
polylinker posee los terminadores de transcripción del fago fd
y señales de fin de traducción sintéticas.( Stanley &Luzio,
1984 ; Haymerle et al, 1986).
4.3. Extracción y purificación de plásmidos
Se utilizó la técnica de Birboim y Doly (1979) con modificaciones:
Para las minipreparaciones uno a tres ml de cultivo de
células se centrifugaron en tubo eppendorf durante 2 minutos
en microcentrïfuga. El pellet se resuspendió en 100 pl de so
lución de lisis con 10 ug/ml de RNAsay se incubó en hielo du
rante 5-10 minutos. Se agregaron luego a la suspensión 200 ul
de solución alcalina. Se mezcló por inversión y se incubó por
otros 5 minutos en hielo. Se agregaron 150 pl de acetato de Na
3 MpH 4,6-5,2 y se incubó durante 15 minutos a la misma tem
peratura.La suspensión se centrifugó 5 minutos en microcentrífuga
recuperándose el sobrenadante al que se agregaron 2-2,5 vol.
de etanol absoluto. Luego se centrifugó 10 minutos a 13000xg.

47
El pellet obtenido se resuspendió en un volumen adecuado
de agua tridestilada, evaluándose la cantidad y calidad del
DNAallí presente, por electroforesis, de una alícuota, en gelde agarosa al 0,8%.
Solución de lisis: Tris-Cl pH 8 25 mM
EDTA 10 mM
Glucosa 9 mg/ml
Solución alcalina: HONa 0,2 mMSDS l 96
Cuandofue necesario reprecipitar el DNAse agregaron 0,5
volúmenes de acetato de amonio 7,5 MpH=7,3 y la mezcla se
dejó en hielo 10 minutos. Luego se centrifugó 10 minutos a
13000xg; se recuperó el sobrenadante y se descartó el precipi
tado proteico. Se precipitó luego el DNAcon dos volúmenes de
etanol durante 15 minutos a 13000xg; se dejó secar el pellet y
luego se resuspendió en agua tridestilada guardándose las alí
cuotas a -20°C.
En el caso de maxipreparaciones se partió de un cultivo
de 100-150 ml ajustándose proporcionalmente los volúmenes de
las soluciones utilizadas. Este DNAse purificó a través de un
gradiente de cloruro de cesio para lo cual se resuspendió en
3,6 ml de buffer TE. Se agregaron 4,2 g de CsCl y 0,5 m1 de
bromuro de etidio 5 mg/ml.
Se centrifugó a 10.000 rpm, durante 10 minutos, transfi
riéndose el sobrenadante a un tubo de rotor 65 Ti para centri
fugar a 45000 rpm por 16 horas, al cabo de las cuales, la
banda de DNAplasmídico se tomó con jeringa de 1 ml.

48
Se extrajo el bromurode etidio con isopropanol saturado
en CsCl, descartándose la fase orgánica hasta que ésta quedó
incolora; luego se dializó contra TE, con 3 cambios de buffer
por lo menos.
Se evaluó la cantidad y calidad del DNApor lectura de DO
en un espectrofotómetro y corrida en gel de agarosa.
buffer TE: Tris 10 mM pH:8
EDTA l mM
4.4. Inducción de la expresión bacterianaLas bacterias se crecieron en medio LBcon ampicilina
(100 ug/ml) a 30°Ccon agitación. Cuando el cultivo alcanzó
una DO = 0,5 a 550 nm se indujo a 42°C durante 90 minutos.
Los cultivos se centrifugaron 15 minutos a 800-1000 xg a
4°C, descartando luego el sobrenadante, y se resuspendió el
pellet conteniendo las bacterias en medio de lavado. Se sonicó
durante 3 minutos manteniendo la muestra sobre agua-hielo. Se
centrifugó en microcentrïfuga durante 10 minutos a velocidad
máximaen frío. El sobrenadante se descartó y el pellet se re
suspendió en buffer de muestra para proteinas.
Medio de lavado de bacterias Pex: Tris pH:8 50mM
EDTA lmM
ClNa 50mM
En el momentode usar se agregan los inhibidores de proteasas:
PMSFy TLCKen una concentración de lmMfinal.

49
5. Reacciones enzimáticas
5.1. Restricción
Las condiciones de reacción, concentración salina y tem
peratura, para las distintas enzimasde restricción utilizadas
fueron las recomendadaspor los productores. En general, para
las restricciones totales, el tiempo de incubación fue de dos
horas, luego de las cuales se agregaron 0,5 pl más de enzima y
se extendió la incubación por otras dos horas o toda la noche.
En caso de la restricción parcial sólo se realizó la primera
incubación y por un lapso de una hora. La efectividad de la
reacción se monitoreó corriendo una alícuota en minigel de
agarosa .
5.2. Ligación
La reacción se realizó durante 2 hs a temperatura am
biente, en un volumen de 10 o 20 pl.
La relación molar inserto-vector utilizada fue de 1:1.
La enzima utilizada fue la T4 DNA-ligasa que cataliza la
formación de un enlace fosfodiester entre terminales yuxta
puestos 5' fosfato y 3' hidroxilo de DNAdoble cadena.
En todas las ligaciones se cuidó que la concentración del
DNApor ml de volumen final de reacción no superara los Bug/ml
Buffer de ligación: Tris-HCl pH= 7,6 25 mM
MgClz 10 mM
Dithiothreitol (DTT) 10 mM
ATP 0,4mM

50
5.3. Defoaforilación
Cinco microgramos del vector, digerido con la enzima de
restricción, fueron tratados con 1 unidad de la fosfatasa al
calina bacteriana (BAP), en el buffer adecuado, e incubados 1
h a 65 °C; luego fueron tratados con proteinasa K, en una con
centración final de 100 pg/ml, durante 30 minutos a 37 °C y ex
traídos con fenol-cloroformo-alcohol isoamílico. El DNAse
precipitó con acetato de amonioy etanol.
Buffer: Tris-HCl pH= 8 10 mM
NaCl 10 mM
5.4. Fill-in
A aproximadamente 2 pg del DNAelectroeluido se agregó
una solución que contenía los cuatro dNPTs (para concentración
final 0,1 mM)en el buffer apropiado y dos unidades del frag
mento Klenow de la DNApolimerasa I de E.coli. Se incubó 20
minutos a temperatura ambiente y luego de terminada la reac
ción, agregando EDTAhasta una concentración final de 10 mM,
se hizo una extracción fenol-c10roformo-alcohol isoamïlico. El
DNAse precipitó con acetato de amonio y etanol.
6. Transformación
6.1. Preparación de células competentes
Inducción de la competencia bacteriana:Un m1 de un cultivo fresco de E. coli (DHSaF') se sembró
en 100 m1 de caldo psiB. Se incubó el cultivo durante 2 hs a

fi
37°C con agitación controlándose periódicamente la absorbancia
hasta que esta alcanzó un valor aproximado de 0,4-0,5 a 600
nm. Las bacterias se recolectaron luego con centrifugación a
5000 rpm a 4°C. El pellet se resuspendió en 40 m1 be I mante
niendo siempre el material en frío y se volvió a centrifugar 5
minutos a 5000 rpm. El nuevo pellet se resuspendió en 4 ml de
be II y se mantuvoen hielo durante 5 minutos.
La suspensión de bacterias así tratadas se fraccionó en
tubos eppendorf en alícuotas de 200 pl las que se congelaron a
-70° C hasta su transformación. (Hanahan,1983; Hanahan et a1,
1991).
Mediopsi-B: Extracto de levadura Sg/litro
Triptona ZOg/litro
M9804 Sg/litro
Se ajustó el pH a 7,6 con KOH.Se filtró a través de un filtro
de 45 micrones y se autoclavó.
beI: KAC 30 mM
RbClz o KCl 100 mM
CaClz 10 mM
MnClz 50 mM
Se agregó glicerol hasta 15%nSe ajustó el pH a 5,8 con ácido
acético 0,2 M. Se esterilizó por filtración.
beII: MOPSo PIPES 10 mM
CaClz 75 mM
RbClz o KCl 10 mM
Glicerol 15‘%
Se ajustó el pH a 6,5 con KOHy se esterilizó por filtración.

52
6.2. Introducción de plásmidos en E. coli
Luego de descongelarlas y mantenerlas en hielo durante 10
minutos, 50 pl de células competentes se incubaron con 1 a 5
ng de DNAplasmïdico durante 30' (en hielo). Luego se incubó
la suspensión a 42 °C, 90 segundos y se la enfrió en hielo in
mediatamente por 2 minutos.
A cada tubo de suspensión sometido a los tratamientos
térmicos descriptos se agregaron 200 ul de caldo Luria o SOCy
la mezcla se incubó a 37° C con agitación durante una hora.
Comocontroles de transformación se incluyeron siempre un
tubo sin DNAplasmídico y otro conteniendo 1 ng de vector.
Medio SOC: Triptona 2 96
Extracto de levadura 0,5 96
NaCl 10 mM
KCl 2,5 mM
MgCl2 10 mM
MgSO4 10 mM
Glucosa 20 mM
6.3. Preparación de placas
Se prepararon placas de agar Luria. Las placas contenían
100 ug/ml de ampicilina y, cuando se trabajó con pUCcomo vec
tor, 40 pg/ml de X-gal.
Cincuenta ul de las bacterias transformadas fueron sem
brados con espátula de Drigalsky en las placas que se incuba
ron a la temperatura requerida.La eficiencia de transformación se calculó como:
E: (unidades formadoras de colonias/pg de DNA)
E= N1_de_colonias x factor de dilución x factor demasa DNA conversión de unidad

53
En este caso E = N:_de_gglgnias x 5 x 103masa DNA
Agar L: se prepara igual que el medio LB y se agrega 1,2 96 de
agar (1,2 g/lOO ml de medio LB). Se ajusta a pH: 7,2 con HONa
4N.
7. Marcación de DNAin vitro
Para realizar el marcadose siguieron las instrucciones
de Bethesda Research Laboratory (BRL), fabricante de los
equipos de nick translation y randomprimer utilizados.
Para cada DNAa marcar radioactivamente se agregó: a[nP]
dATP o dCTP 10 pci/¡.11 (400 Ci/mmol)o 0L[32P]dCTP 10 pCi/pl
(>3000 Ci/mmol).
En el caso de utilizarse quimioluminiscencia se marcó con
el mismo equipo de random primer utilizando DIG-dUTPy, para
revelar, el equipo para detección luminiscente de ácidos
nucleicos de Boehringer Mannheim,utilizado según sus
instrucciones.
Para eliminar los nucleótidos no incorporados durante la
reacción se precipitó el DNAcon los volúmenes correspondien
tes de acetato de amonio y etanol centrifugando durante 15 mi
nutos a 13000xg.
Se resuspendió el DNAen TE pH 7,6 o en agua destilada
estéril, se midió la radioactividad incorporada con un conta
dor y se calentó 5 minutos a 100°C para desnaturalizar el DNA

54
y luego se dejó 5 minutos en hielo previamente a su utiliza
ción.
8. Reacción de hibridación
8.1. De colonias
Se realizó de acuerdo a la técnica descripta por Maniatis
et al. (1982).
8.2. Dots
Los sueros probados (100 pl) fueron clarificados por
centrifugación a 13.000 rpm durante 10 minutos en una
microcentrífuga. Luego se les agregó HONapara concentración
final lN y se calentó 10 minutos a 65°C.
Los sueros así tratados se filtraron con vacío suave a
través de un dispositivo comercial para dots sobre una
membranade nitrocelulosa o nylon. Los filtros se secaron al
aire y posteriormente 1 hora a 80°C.
8.3. Southern
Transferencia de geles: Se realizó de acuerdo a la técnica de
Southern modificada según Maniatis et al. (1982).
8.4. Prehibridación e hibridación
Luego de las 2 horas de prehibridación se agregó la sonda
marcada y se dejó agitando durante toda la noche a la tempera
tura deseada. Luego se lavaron los filtros con solución de 1a

55
vado.
Se varió la temperatura y la concentración de las sales,
de las soluciones de prehibridación y de lavado, para conse
guir el nivel de homologïa buscado.
La temperatura de fusión, Tm, es una medida de la estabi
lidad térmica de los híbridos. Es la temperatura a la cual, la
mitad de las moléculas están asociadas y la otra mitad sin
asociarse. Es característica de cada especie de DNAo RNA.La
ecuación general para determinarla es:
Tm = 81,5 + 16,6 (log M) + 0,41 U%G+C) —0,72 (96 formamida)
donde M = molaridad del catión monovalente (Na+) y
Tm - temperatura de trabajo = 96 de mal apareamiento permitido
(Hames & Higgins, 1985).
En nuestro caso se tomó comoporcentaje promedio de todo
el genoma del HBV: 47 96 GC lo cual nos da los siguientes
valores de Tm : 1x SSC :9 Tm = 88°C y 0,1x SSC :9 Tm = 71°C
Solución de prehibridación:
SSCde acuerdo a la severidad del ensayo
SDS 0,5 96
leche en polvo 0,3 %
En algunos casos se usó también: DNAde esperma de arenque so
nicado 100 ug/ml, calentado 5 minutos a 100° C y dejando en
hielo otros 5 minutos.
Solución de lavado:

56
SSCde acuerdo con la severidad del ensayo
SDS 0,5 a 1 %
1x SSC: NaCl 0,15 M
Citrato de sodio 0,015 M
8.5. AutoradiografíaLuego de lavados, los filtros fueron colocados sobre una
placa radiográfica en un recipiente metálico apropiado
(holder) y se dejó toda la noche o durante un tiempo variable,
de acuerdo a cada caso, a -70°C o a temperatura ambiente.
Reactivos
Placa radiográfica XARKodak
Revelador para autoradiografía Kodak
Lavador: ácido acético al 5‘%
Fijador Kodak
8.6. Quimioluminiscente
Los filtros equilibrados en el buffer adecuado se incuba
ron con anti-digoxigenina Fab conjugada con fosfatasa alcalina
y se lavaron de acuerdo a las indicaciones del fabricante del
equipo (Boehringer Mannheim). Los híbridos se detectaron por
reacción con el sustrato quimioluminiscente formulado como
Lumi-Phos 530 (Boehringer Mannheim) sobre una hoja de acetato
y se expuso a una placa radiográfica XARa 37°C durante 1 h.

57
9. Electroforesis en gel
9.1. Geles de agarosa
Se realizaron corridas electroforéticas en geles de agarosa de diversa concentración: 0,8, 1 y 1,2 96. El buffer del
gel y el buffer de corrida utilizado fue TBE0,5x.Las condiciones de corrida fueron variables: durante toda
la noche a 25 volts o bien aproximadamente 4 hs a 100 volts.
Los geles se colorearon con Bromurode Etidio 0,5 pg/ml,
en agua destilada después de la corrida electroforética. Seobservaron a la luz ultravioleta (300 mm)con un transilumina
dor y se fotografiaron con un filtro adecuado.
Buffer TBE. 1x: Tris 0,089 M
Acido bórico 0,089 M
EDTA 0,002 M
pH= 8,3
Buffer de siembra 6X: Azul de bromofenol 0,3 96
Xylene Cyanol FF 0,3 96
Glicerol 30 96
SDS 6 96
Electroelución de fragmentos de DNA:
Se digirió el DNAcon endonucleasas de restricción. Se
realizó una corrida electroforética en un gel de agarosa en
función de separar los fragmentos de DNAde acuerdo a su peso
molecular. Se tiñó el gel con bromuro de etidio y se recortó
la banda correspondiente al fragmento que se deseaba electroe
luir. La electroelución se llevó a cabo según Maniatis et al.(1982).

58
9.2. Geles de poliacrilamida
Las proteínas se prepararon por lisis de las células en
el buffer de muestra y se separaron en geles de poliacrilamida
discontinuos (Laemmli, 1970).
Buffer de muestra: Tris pH:7,S SOmM
SDS 1%
B-mercaptoetanol 1%
Urea 6M
azul de bromofenol 0,1%
glicerol 10%
10. Secuencia
Se siguió la metodología de secuenciación de Sanger
(Sanger et al. 1977), la cual se basa en el uso de terminado
res específicos (ddNTP)de la elongación de la cadena de DNA.
Se utilizaron los siguientes primers universales para el sis
tema M13/pUC
primer forward 22 mer (-47) : 5'-GCCAGGGTTTTCCCAGTCACGA-3’
primer reverse 23 mer (-48) 5’-AGCGGATAACAATTTCACACAGG-3'
la DNApolimerasa del fago T7 (Pharmacia) o alternativamente
la Ampliïaqu DNApolimerasa (Perkin Elmer Cetus) y como nu
cleótido radioactivo PSSkLNTP.
Las cadenas resultantes se separaron en geles de poliacrilamida en condiciones desnaturalizantes (696acrilamida
(19:1)—7Murea).
El gel fue expuesto a una placa radiográfica y la se
cuencia se obtuvo por la lectura directa de la placa.

Las secuencias nucleotídicas fueron estudiadas por los
siguientes programas de computación:
- DNASIS(Hitachi Software Engineering Co.)
- PC-GENE(IntelliGenetics, Inc.)
- GCG (Genetics Computer Group)
59


60
IV. RESULTADOS
1. Construcción de plásmidos recombinantes
1.1. Estrategia de clonado
El esquema general seguido para la obtención de recombi
nantes fue el siguiente: (Fig. IV.1).
1.2. Purificación de partículas de Dane
Un requisito importante para la obtención de un HBV-DNA
para clonar es poder disponer de una masa adecuada de virus
purificado. En la mayoría de los virus esto se logra mediante
1a infección de líneas celulares susceptibles. En el caso deHBVel desarrollo de un sistema de cultivo celular adecuado,
es decir, permisivo para todos los pasos del ciclo de replica
ción no ha sido sencillo. Los cultivos primarios de hepatoci
tos son capaces de soportar algunas etapas de la replicación
viral (Gripon et al, 1988; Ochiya et al, 1989) pero no de pro
ducir partículas virales. Envarios laboratorios se identificaron líneas celulares derivadas de hepatomas (comolas HepG2)
que son competentes para la expresión de genes de HBVluego de
ser transfectadas con DNAviral clonado (Sureau et al, 1986;
Tsurimoto et al, 1987; Sells et al, 1987; Yaginumaet al,
1987).
Por otra parte el suero de ciertos pacientes es infectivo
y también la fuente natural más importante para obtener virus
purificado.Cuandose encaró este proyecto, en nuestro laboratorio
disponïamos de cantidades adecuadas de plasma de individuos
infectados. Los valores de un primer estudio nos informaron
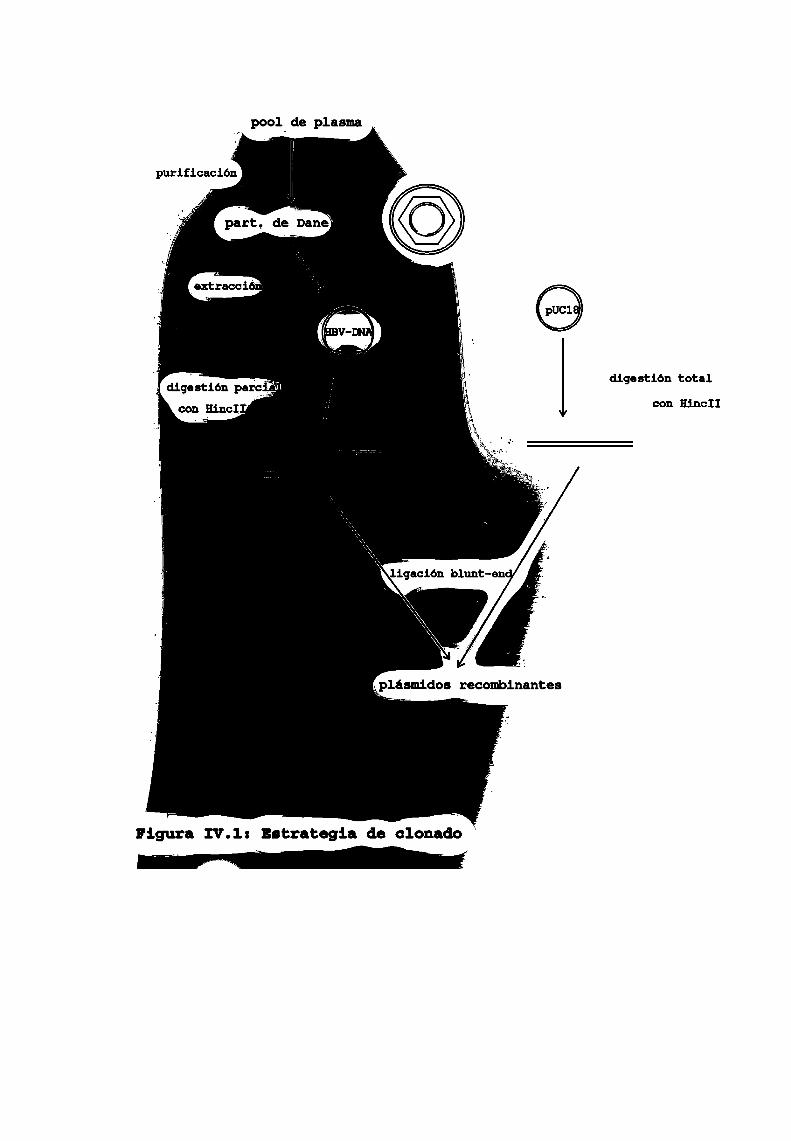
pool de plasma H
purificación
digestión totalcon HincII
Kplásmidos recombinantes
Figura IV.1: Estrategia de clonado '
M

62
sobre la posibilidad de manejar cantidades aceptables de antí
geno comopara evitar las dificultades del cultivo celular.
Se partió de 3,6 l de un pool de plasma HBs+ HBe+y se
purificaron parcialmente las partículas de Dane, por ultracen
trifugación, según protocolo en III.3.1.
1.3. Extracción de DNA
La extracción del DNAdel HBV(parcialmente de doble ca
dena) se realizó comose detalla en III.3.2, obteniéndose
aproximadamente 1,2 ug de DNAsegún se determinó por corrida
en gel de agarosa en comparación con un standard. Teniendo en
cuenta que, nuestro objetivo nunca fue clonar genomasvirales
completos; consideramos innecesario llevar a cabo el protocolo
usual de reparación de la cadena incompleta del DNAviral, me
diante la actividad de la DNA-polimerasaendógena.
1.4. Restricción del DNA
Dado que se partió de un pool de plasma obtenido de va
rios pacientes y, comoya se indicó, las distintas secuencias
publicadas del genomadel HBVindican cierto grado de variabi
lidad en los mapasde restricción, se decidió realizar una di
gestión parcial del DNAviral con la enzima HincII, la cual,
en los mapas publicados del serotipo adw, que sería el más
frecuente en la Argentina (Couroucé-Pauty et al., 1983), no
presenta sitios de corte dentro del ORFde la proteína core.
Esta enzima reconoce un hexanucleótido cuya secuencia es
5'..GTPy¿PuAC..3' y al cortar deja extremos romos.

63
1.5. Elección del vector
Actualmente se dispone de una gran variedad de vectores,
la mayoría de los cuales tienen una región que concentra un
númeroimportante de sitios únicos de restricción (polylinker)
y marcadores que facilitan la identificación de los recombi
nantes; de este modola elección del vector depende de la na
turaleza del experimento.
En este caso se eligió un vector de la línea pUCya que
posee las siguientes ventajas:a) un sitio HincII en el polylinker
b) el polylinker está dentro del gen que codifica
para el péptido a de la B-galactosidasa, por lo que la inser
ción de un DNAen dicha zona inactiva dicho gen y el recombi
nante aparece comouna colonia blanca en una placa que con
tenga al indicador.c) la disponibilidad de primers comerciales que per
miten la secuenciación del DNAinsertado sin necesidad de sub
clonados en vectores especiales.
El vector utilizado fue el pUC18que se sometió a unarestricción total con la enzimaHincII.
La ligación se llevó a cabo según el protocolo para ex
tremos romos que se detalla en materiales y métodos (III.5.2).
1.6. Transformación (eficiencia)
Se utilizó la cepa DHSaF' de E. coli , que se hizo compe
tente por el método de Hanahany presentó una eficiencia de
transformación de 1,2 x 106 colonias por pg de DNAtransfor
mante .

Las bacterias transformadas con la mezcla de ligación, se
sembraron en placas de agar LBcon ampicilina y X-gal, dife
renciando así las colonias recombinantes (blancas) de las que
portaban el plásmido vector religado (azules) y las recombi
nantes que por tener insertado un fragmento muy pequeño de DNA
no llegan a inactivar totalmente la B-galactosidasa (celestes)
Según catálogo, las eficiencias usuales para ensayos como
el nuestro, es decir, ligaciones de extremos romos, con el
vector sin defosforilar, son: 5 x 104 transformantes/ ug de
DNAcon una frecuencia de recombinación del 1096, o, en otros
casos, 5 x 105 transformantes/ ug de DNAcon frecuencias 8596
parental y 1596recombinante.
En nuestro caso obtuvimos una eficiencia de 1,5 x 105 con
frecuencias: 90%parental y 10%)recombinante, valores éstos
que se encuentran dentro de los parámetros esperados.
Algunas de las colonias recombinantes (35) junto con dos
colonias control (correspondientes a transformantes con pUC),
se repicaron por duplicado, en placas de agar LB con ampici
lina, para su análisis.
2. Selección y análisis de recombinantes
2.1. Selección por hibridación de colonias
A partir de una de estas placas, se transfirieron las colonias a un filtro de nitrocelulosa donde se detectaron los
clones portadores de secuencias de DNAviral por hibridación,
utilizando como sonda el mismo HBV-DNAobtenido de las partí
culas de Dane purificadas, marcado con bzPL
El resultado de esta hibridación se muestra en la figura

65
lt \á L6 2| zz ¿3 ¿q 25
‘¿6 21 za zq 3° J! 31 33
3‘1 35 3: 31. 31 3Q
42' "¿Qr
Figura IV.2 : Hibridación de las colonias recombinantesA y esquema, B : autorradiografïa- Las colonias transferidas al filtro de N.C. se hibridaroncon [”EflHBV—DNAen condiciones de alta exigencia (Tm - 5).
Tiempo de exposición : 24 hs.—Las posiciones 38 y 39 corresponden a los controles pUC.e La diferencia en intensidad se debe a diferencias en eltamaño de las colonias.—No se observa reacción cruzada con el vector.

66
IV.2 donde puede observarse que la mayoría de las colonias re
combinantestiene secuencias específicas del virus.
2.2. Verificación de la presencia de plásmidos
Los clones recombinantes se amplificaron, a partir de la
segunda placa, en cultivo líquido y se hizo una miniprepara
ción de plásmidos para comparar su tamaño respecto del vector
pUC.
En la figura IV.3 (a y b) se ve el resultado de analizar
la preparación en gel de agarosa. Las calles marcadas con x
corresponden a pUC. Se observa que los plásmidos obtenidos
presentan un tamaño aparente mayor que el del vector pUC, y
claramente distinguibles entre si, lo que indica que son re
combinantesportadores de insertos de distinta longitud, lo
que era de esperarse dada la estrategia de clonado.
2.3. Análisis, de los plásmidos seleccionados, por restricción
e hibridación.
Se seleccionaron algunos de los clones anteriores, representativos de los distintos recombinantes, y se sometieron a
una digestión total con la enzima HincII con el objeto de li
berar el fragmento insertado. Comose observa en la figura
IV.4 se comprobóla presencia de varios clones diferentes,
como los correspondientes a las calles N° 2, 3, 7, 9 y 10. Al

67
Figura IV.3 : Contenido plasmídico de los recombinantes—Los DNAplasmídicos obtenidos de las minipreparaciones dealgunos de los clones recombinantes se analizaron por electroforesis en gel de agarosa a1 0,8%L—En las calles marcadas x se sembró el vector pUCpara lacomparación del tamaño aparente.

68
1234567891011
Figura IV.4 : Análisis de los plásmidos recombinantesobtenidos- Algunos de los clones de la figura anterior se eligieron,por su tamaño aparente, comorepresentantes de los distintosrecombinantes, y se cortaron con la enzima de restricciónHincII. Los fragmentos de DNAresultantes se corrieron en ungel de agarosa al 1%“ para determinar el tamaño del fragmentoinsertado, utilizando comocontroles el vector pUC18cortadocon la misma endonucleasa (calles 1 y 11) y el fago k digeridocon HindIII (calle 6).

69
gunos de ellos, por ejemplo los presentados en las calles N° 2
y 4, portadores de un inserto clivable por la mismaenzima, lo
que se explica por la restricción parcial que los originó.
Para corroborar que se trataba de fragmentos específicos
de HBV,el gel de la figura IV.5a, que muestra el resultado
del análisis de algunos de los clones, se transfirió a papel
de nitrocelulosa y se hibridó con el mismo HBV-DNAdel punto
2.1 (Fig. IV.5b).
El resultado obtenido demuestra claramente que los frag
mentos, que miden aproximadamente 1700 pb, 500 pb y 350 pb,
corresponden a HBVy que el vector no da hibridación cruzada
comoya se había visto en la hibridación en colonias.
2.4. Determinación de 1a zona viral presente en los recombi
nantes
Comoresultado de los experimentos precedentes se eligie
ron, en base al tamaño del inserto, tres clones (pHB4, pHB7y
pHB20)comorepresentantes de los principales fragmentos genó
micos clonados. Estos clones corresponden a las calles 2, 3 y
5 respectivamente, de la figura anterior.A estos recombinantes se los continuó analizando para de
terminar qué zona del genomaviral estaba comprendida en cada
uno de ellos. Para esto se digirieron con varias enzimas de
restricción, comparándoseluego los mapas así obtenidos con
uno de los disponibles en un banco de datos (IBI/Pustell ver
1.80 HPBADW,que corresponde a V000866 del Anexo I).
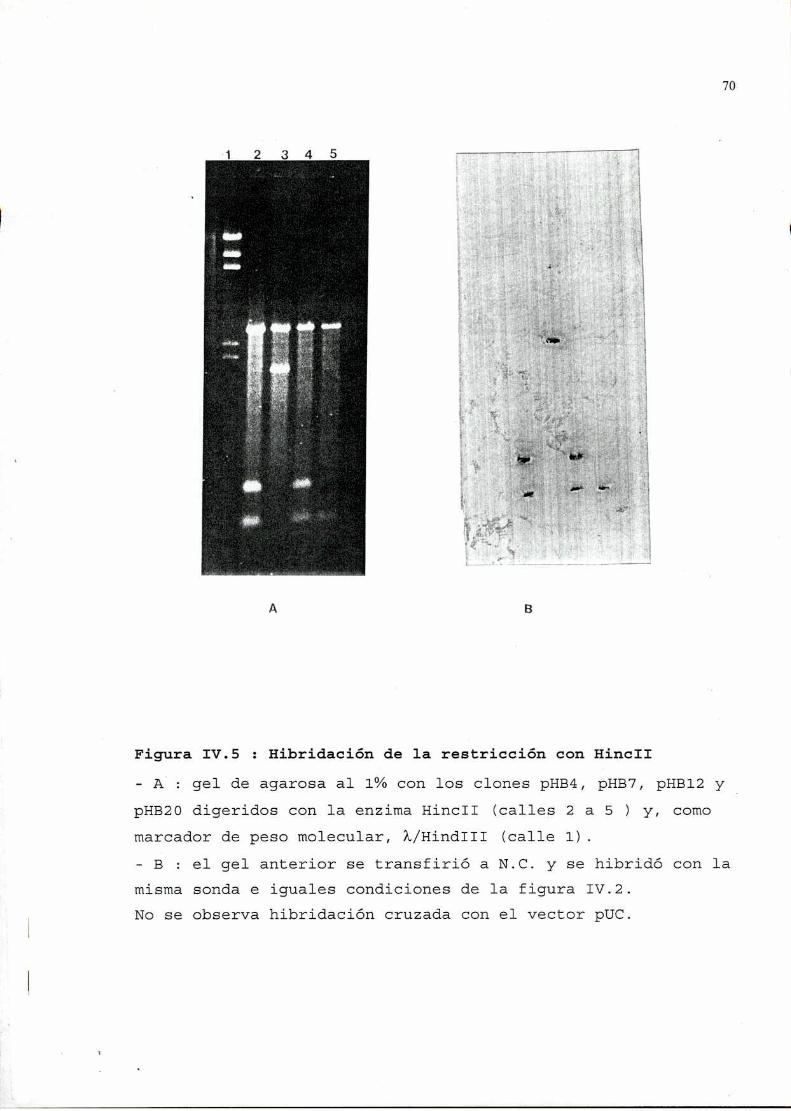
70
Figura IV.5 : Hibridación de la restricción con HincII—A : gel de agarosa al 1%)con los clones pHB4, pHB7, pHB12 y
pHB20digeridos con la enzima HincII (calles 2 a 5 ) y, comomarcador de peso molecular, k/HindIII (calle 1).—B : el gel anterior se transfirió a N.C. y se hibridó con lamisma sonda e iguales condiciones de la figura IV.2.No se observa hibridación cruzada con el vector pUC.

71
Las figuras siguientes muestran algunos de los geles de
agarosa en los que se analizó el resultado de digerir los
clones ya mencionados con las endonucleasas que se mencionan
en las figuras respectivas y en los mapasresultantes.
El clon pHB4 (Fig. IV.6a restricción y IV.6b mapa) abarca
la zona del genomaviral entre los nt 2589-220 comprendiendo
las regiones preSl, preS2 y los primeros 22 aa de S.
El clon pHB7 (Fig. IV.7a restricción y IV.7b mapa) com
prende el fragmento entre los nt 1684-220 ,desde los 50 aa delextremo carboxi-terminal de HBxhasta los 22 aa del amino-ter
minal de SHBs, incluyendo el ORFdel core.
Por último, el clon pHB2O(Fig. IV 8a restricción y IV Bb
mapa) va del nt 3119 al 220 cubriendo desde los últimos 30 aa
de preSl hasta los primeros 22 aa de SHBs.
En la Fig. IV.9 se diagrama la posición relativa de estos
clones respecto del genomaviral, observándose que los tres se
superponen en la región preS/S y se extienden en distinta me
dida hacia el ORFpreC/C, siendo el clon pHB7el único de los
tres que lo incluye.
Es evidente que, en el pool de plasma del cual se partió,
había representados más de un tipo genómico, ya sea que fueran
variaciones presentes en un mismodador o provenientes de los
distintos componentesdel pool. De este modo, insertos que re
presentan una misma zona del genomaviral, en alguno de los
clones, carecen de determinados sitios de restricción para las
enzimas ensayadas como es el caso de pHB7para las enzimas
BamHI,HincII y PstI en la región correspondiente a preS.

1 2 3 4 5 6 7 8 9 10 11 12
4363_
_26862319_
18103568
j118
Figura IV.6 : Perfil de restricción del clon pHB4- A : gel de agarosa al F% del Clon pHB4digerido con variasenzimas de restriccióncalle l: pBR322linealizado con BamHI+ digerido con BglI;calles 2 a 10: pHB4digerido con: 2:BstEII, 3: BstEII/HindIII,4: BstEII/BglI, 5: BstEII/ECORI, 6: EcoRI, 7: BamHI, 8: BglI,9: BglI/EcoRI, 10: AvaII, calle 11: pHB4sin digerir; calle12: pUClinealizado con BamHI+ digerido con BglI.- B : mapa del inserto linealizado con HincII (H).Enzimas: B = BamHI, Bs = BstEII, E = EcoRI, P = PstI, S = StyINopresenta sitios de corte para: Aval, BglI, HindIII, BglII.
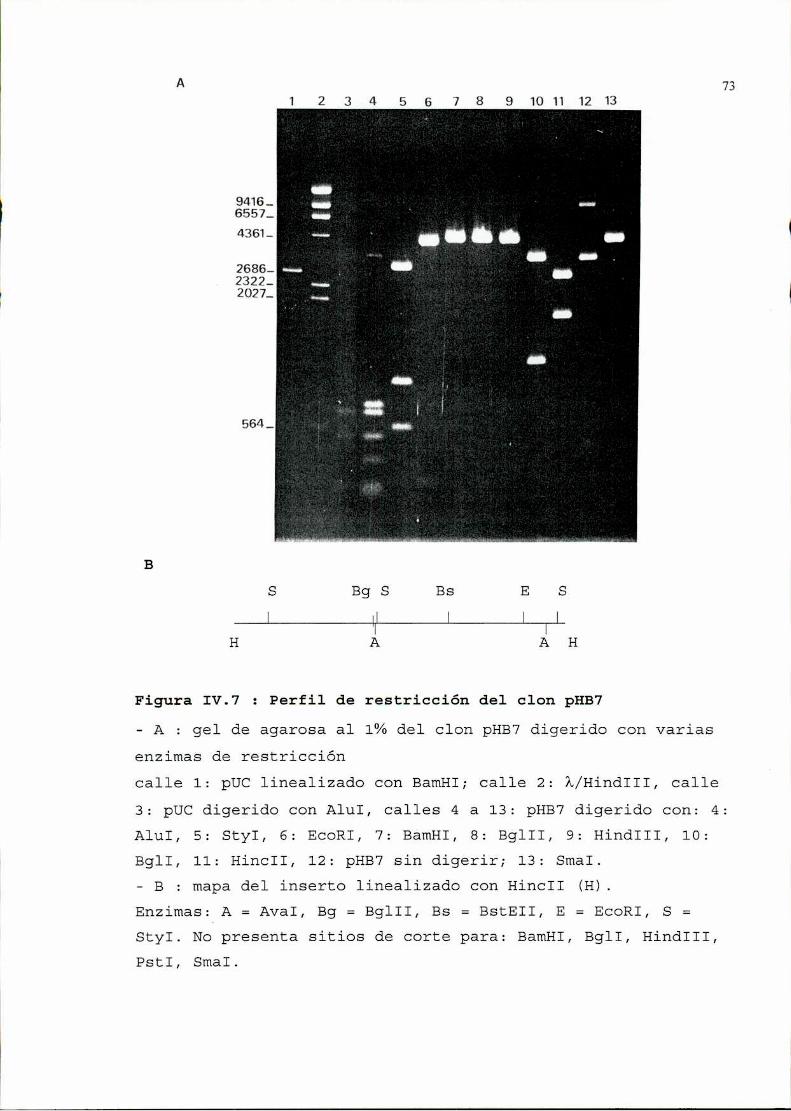
12345678910111213
S Bg S Bs E S
Figura IV.7 : Perfil de restricción del clon pHB7—A : gel de agarosa a1 1% del clon pHB7digerido con variasenzimas de restriccióncalle 1: pUClinealizado con BamHI;calle 2: K/HindIII, calle3: pUCdigerido con AluI, calles 4 a 13: pHB7digerido con: 4:AluI, 5: StyI, 6: ECORI, 7: BamHI, 8: BglII, 9: HindIII, lO:BglI, 11: HinCII, 12: pHB7sin digerir; 13: SmaI.- B : mapa del inserto linealizado con HincII (H).Enzimas: A = Aval, Bg = BglII, Bs = BstEII, E = ECORI, S =StyI. Nopresenta sitios de corte para: BamHI,BglI, HindIII,PstI, SmaI.
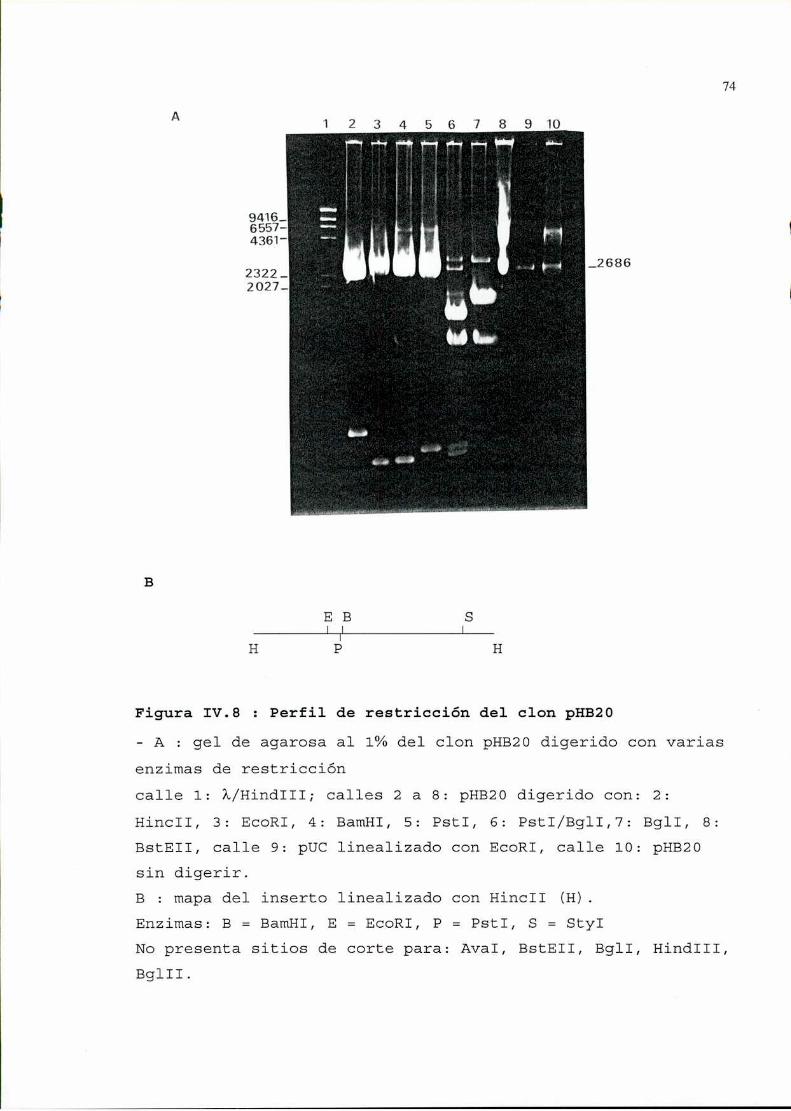
74
Figura IV.8 : Perfil de restricción del clon pHB20- A : gel de agarosa al l%>de1 clon pHB20digerido con variasenzimas de restriccióncalle 1: X/HindIII; calles 2 a 8: pHB20digerido con: 2:HincII, 3: EcoRI, 4: BamHI,5: PstI, 6: PstI/BglI,7: BglI, 8:BstEII, calle 9: pUClinealizado con EcoRI, calle 10: pHB20sin digerir.B : mapa del inserto linealizado con HincII (H).Enzimas: B = BamHI, E = EcoRI, P = PstI, S = StyINopresenta sitios de corte para: Aval, BstEII, BglI, HindIII,BglII.
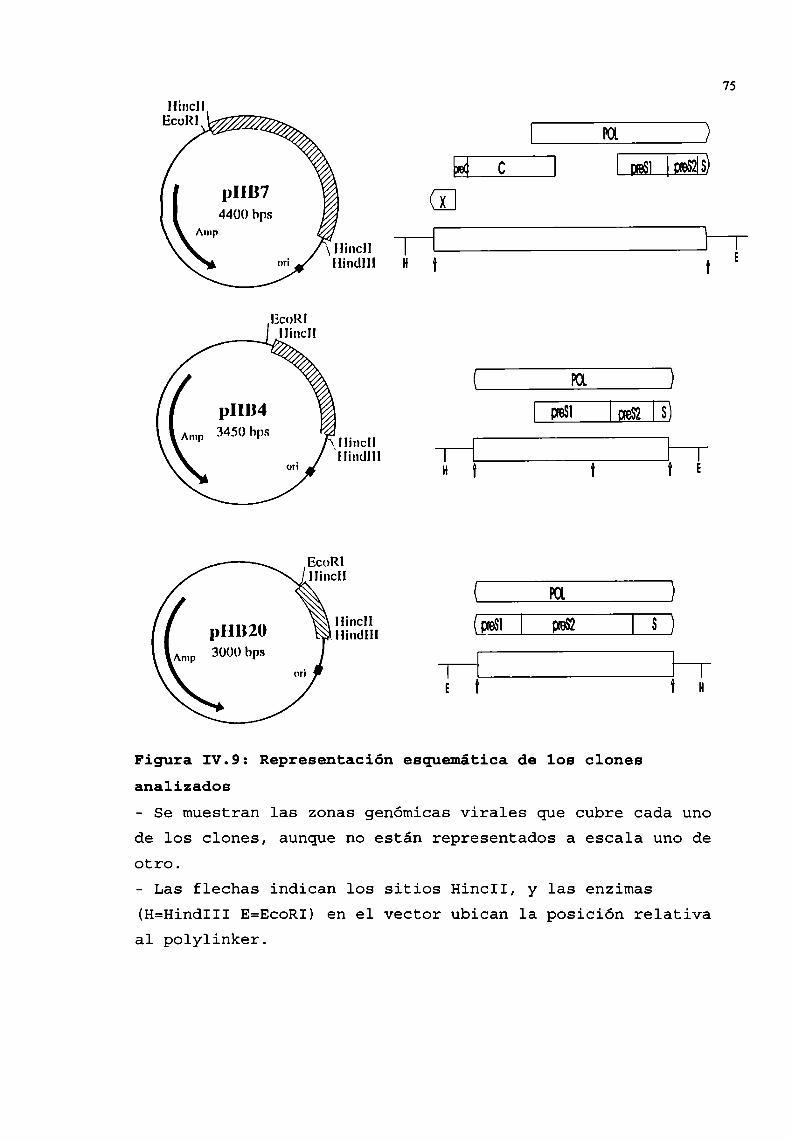
¡in
4400 bpsAmp
'\ Hincllllhldlll
ECORIllincll
I
3450 hpsAmp
EcoRll Ilincll
pI-IBZO3000 bpsAmp
Figura IV.9: Representación esquemática de los clonesanalizados- Se muestran las zonas genómicas virales que cubre cada unode los clones, aunque no están representados a escala uno deotro.- Las flechas indican los sitios HincII, y las enzimas(H=HindIII E=EcoRI)en el vector ubican la posición relativaal polylinker.
75

76
2.5. Secuencia
Se secuenciaron, al menos dos veces en experimentos inde
pendientes, total o parcialmente, los tres clones en ambos
sentidos (primers "forward" y "reverse")
El clon pHBZOse secuenció en su totalidad, mientras que
los clones pHB7y pHB4sólo en sus extremos, utilizándose la
nomenclatura "pS" y "p3" para los extremos 5' y 3' correspondientes.
Las secuencias nucleotídicas obtenidas y su correspon
diente traducción a aa, mediante el programa DNASIS,se pre
sentan en las figuras IV.10, 11 y 12
Comopuede observarse en la parte que se superponen (Fig.
IV.13), los tres clones analizados no son idénticos en sus se
cuencias. A excepción de la mutación única (anyáT) que pre
senta pHB4, esta secuencia y pHB20se corresponden exactamente
en la zona de superposición lo que indica que están muyrela
cionadas entre sí; siendo pHB7la que representa una variante
con mayor grado de diferencias, confirmando así la disparidad
vista en los mapasde restricción.
La divergencia, calculada en esta zona, de pHB7respecto
a los otros dos clones es de 3,596 con pHB20y 3,896 con pHB4.

FILE NAME PHB7P5.SEQSEQUENCE : 404 BP; 88 A; 101 C; 95 G; 120 T.
CAA CGA CCG ACC TTG AGG CCT ACT TCA AAG ACT GTG TGT TTA AAG ACT GGG AGG
* I I OI. I I ll. III IIIAsp Cys Val Phe Lys Asp Trp Glu Glu# Thr Thr Asp Leu Glu Ala Tyr Phe Lys
AGT TGG GGG AGG AGA TTA GGT TAA AGG TCT TTG TAT TAG GAG GCT GTA GGC ATA
Leu Gly Glu Glu Ile Arg Leu Lys Val Phe Val Leu Gly Gly Cys Arg His Lys
AAT TGG TCT GCG CAC CAT CAT CAT GCA ACT TTT TCA CCT CTG CCT AAT CAT CTC
... ... ... ... ... ... ... Met Gln Leu Phe His Leu Cys Leu Ile Ile SerLeu Val Cys Ala Pro Ser Ser Cys Asn Phe Phe Thr Ser Ala *** ... ... ...
TTG TAC ATG TCC CAC TTT TCA AGC CTC CAA GCT GTG CCT TGG ATG GCT TTG GGG
Cys Thr Cys Pro Thr Phe Gln Ala Ser Lys Leu Cys Leu Gly Trp Leu Trp Gly
CAT GGA CAT TGA CCC TTA TAA AGA ATT TGG AGC TAC TGT GGA GTT ACT CTC GTT
Met Asp Ile Asp Pro Tyr Lys Glu Phe Gly Ala Thr Val Glu Leu Leu Ser Phe
TTT GCC TTC TGA.CTT CTT TCC TTC CGT CCG GGA TCT ACT AGA CAC AGC CTC AGC
Leu Pro Ser Asp Phe Phe Pro Ser Val Arg Asp Leu Leu Asp Thr Ala Ser Ala
TCT GTA TCG GGA AGC CTT AGA GTC TCC TGA GCA TTG CTC ACC TCA CCA TAC AGC
Leu Tyr Arg Glu Ala Leu Glu Ser Pro Glu His Cys Ser Pro His His Thr Ala
ACT CAG GCA AGC CAT TCT CTG CTG GG /////
Leu Arg Gln Ala Ile Leu Cys Trp
* : ORFpreC/C Se indica el comienzo de preC y C.# : ORF X
FILE NAME : PHB7P3.SEQSEQUENCE 316 BP; 68 A; 103 C; 68 G; 77 T.
/////TCC TCC TCC TGC CTC CAC CAA TCG GCA GTC AGG AAG GCA GCC TAC TCC CAT CTC
* Ser Ser Ser Cys Leu His Gln Ser Ala Val Arg Lys Ala Ala Tyr Ser His Leu# Pro Pro Pro Ala Ser Thr Asn Arg Gln Ser Gly Arg Gln Pro Thr Pro Ile Ser
TCC ACC TCT AAG AGA CAG TCA TCC TCA GGC CAT GCA GTG GAA TTC CAC AGC TTT
Ser Thr Ser Lys Arg Gln Ser Ser Ser Gly His Ala Val Glu Phe His Ser PhePro Pro Leu Arg Asp Ser His Pro Gln Ala Met Gln Trp Asn Ser Thr Ala Phe
77

1k
#
CCA CCA AGC TCT GCA AGA TCC CAG AGT CAG GGG CCT GTA TTT TCC TGC TGG TGG
Pro Pro Ser Ser Ala Arg Ser Gln Ser Gln Gly Pro Val Phe Ser Cys Trp TrpHis Gln Ala Leu Gln Asp Pro Arg Val Arg Gly Leu Tyr Phe Pro Ala Gly Gly
CTC CAG TTC AGG AAC ACT CAA CCC TGT TCC AAA TAT TGC CTC TCA CAT CTC GTC
Leu Gln Phe Arg Asn Thr Gln Pro Cys Ser Lys Tyr Cys Leu Ser His Leu ValSer Ser Ser Gly Thr Leu Asn Pro Val Pro Asn Ile Ala Ser His Ile Ser Ser
AAT CTC CTC GAG GAC TGG GGA CCC TGC GAC GAA CAT GGA GAA CAT CAC ATC AGG
Asn Leu Leu Glu Asp Trp Gly Pro Cys Asp Glu His Gly Glu His His Ile ArgIle Ser Ser Arg Thr Gly Asp Pro Ala Thr Asn Met Glu Asn Ile Thr Ser Gly
ATT CCT AGG ACC CCT GCT CGT GTT ACA GGC GGG GTT TTT CTT GTT G
Ile Pro Arg Thr Pro Ala Arg Val Thr Gly Gly Val Phe Leu ValPhe Leu Gly Pro Leu Leu Val Leu Gln Ala Gly Phe Phe Leu Leu
ORF P
: ORFpreS/S. Se indica el comienzo de preSZ y S.
Fig. IV.10 : pHB7:secuencia nucleotídica y traducción a aa.
"forward") y 3'El clon pHB7se secuenció en su extremo 5' (primer
(primer "reverse") utilizando la técnica deSanger.
La secuencia de aminoácidos se determinó a partir dela secuencia nucleotídica mediante el programa DNASIS.
78

79FILE NAMESEQUENCE :
PHB4P5.SEQ332 BP; 104 A; 73 C; 63 G; 92 T.
CAA CAA TTT GTG GGC CCT CTC ACT GTA AAT GAA AAG AGA AGA TTG AAA TTA ATT
Gln Gln Phe Val Gly Pro Leu Thr Val Asn Glu Lys Arg Arg Leu Lys Leu Ile*
# ... ... ... ... ... ... ... ... ... ... .. ... ... ... ... .. ... ...ATG CCT GCT AGA TTC TAT CCT ACC CAC ACT AAA TAT TTG CCC TTA GAC AAA GGA
Met Pro Ala Arg Phe Tyr Pro Thr His Thr Lys Tyr Leu Pro Leu Asp Lys Gly
ATT AAA CCT TAT TAT CCA GAT CAG GTA GTT AAT CAT TAC TTC CAA ACC AGA CAT
Ile Lys Pro Tyr Tyr Pro Asp Gln Val Val Asn His Tyr Phe Gln Thr Arg His
TAT TTA CAT ACT CTT TGG AAG GCT GGT ATT CTA TAT AAG AGG GAA ACC ACA CGT
Tyr Leu His Thr Leu Trp Lys Ala Gly Ile Leu Tyr Lys Arg Glu Thr Thr Arg
AGC GCA TCA TTT TGC GGG TCA CCA TAT TCT TGG GAA CAA GAG CTA CAG CAT GGG
Ser Ala Ser Phe Cys Gly Ser Pro Tyr Ser Trp Glu Gln Glu Leu Gln His Gly... ... ... ... ... ... ... ... ... ... ... ... ... ... ... ... MetGlyAGG TTG GTC ATC AAA ACC TCG CAA AGG CAT GGG GAC GAA TCT TTC TGT TCC CAA
Arg Leu Val Ile Lys Thr Ser Gln Arg His Gly Asp Glu Ser Phe Cys Ser GlnGly Trp Ser Ser Lys Pro Arg Lys Gly Met Gly Thr Asn Leu Ser val Pro Asn
CCC TCT GG /////
Pro SerPro Leu
* t ORF P
# : ORFpreS/S Se indica el comienzo de preSl
FILE NAME : PHB4P3.SEQSEQUENCE : 356 BP; 78 A; 111 c; 81 G; 86 T.
/////GAC CTC AGG CTC AGG GCA TAT TGA CCA CAG TGT CAA CAA TTC CTC CTC CTG CCT
* Thr Ser Gly Ser Gly His Ile Asp His Ser Val Asn Asn Ser Ser Ser Cys LeuPro Gln Ala Gln Gly Ile Leu Thr Thr Val Ser Thr Ile Pro Pro Pro Ala SerZu:
CCA.CCA ATC GGC AGT CAG GAA GGC AGC CTA CTC CCA TCT CTC TAC CTC TAA GAG
His Gln Ser Ala Val Arg Lys Ala Ala Tyr Ser His Leu Ser Thr Ser Lys ArgThr Asn Arg Gln Ser Gly Arg Gln Pro Thr Pro Ile Ser Leu Pro Leu Arg Asp
ACA GTC ATC CTC AGG CCA TGC AGT GGA ATT CCA CTG CCT TCC ACC AAG CTC TGC
Gln Ser Ser Ser Gly His Ala Val Glu Phe His Cys Leu Pro Pro Ser Ser AlaSer His Pro Gln Ala Met Gln Trp Asn Ser Thr Ala Phe His Gln Ala Leu Gln

80
AGG ATC CCA GAG TCA GGG GTC TGT ATT TTC CTG CTG GTG GCT CCA GTT CAG GAA
Gly Ser Gln Ser Gln Gly Ser Val Phe Ser Cys Trp Trp Leu Gln Phe Arg AsnAsp Pro Arg Val Arg Gly Leu Tyr Phe Pro Ala Gly Gly Ser Ser Ser Gly Thr
CAG TAA ACC CTG CTC CGA ATA TTG CCT CTC ACA TCT CGT CAA TCT CCG CGA GGA
Ser Lys Pro Cys Ser Glu Tyr Cys Leu Ser His Leu Val Asn Leu Arg Glu AspVal Asn Pro Ala Pro Asn Ile Ala Ser His Ile Ser Ser Ile Ser Ala Arg Thr
CTG GGG ACC CTG TGA CGA ACA TGG AGA ACA TCA CAT CAG GAT TCC TAG GAC CCC
Trp Gly Pro Cys Asp Glu His Gly Glu His His Ile Arg Ile Pro Arg Thr ProGly Asp Pro Val Thr Asn Met Glu Asn Ile Thr Ser Gly Phe Leu Gly Pro Leu
TGC TCG TGT TAC AGG CGG GGT TTT TCT TGT TG
Ala Arg Val Thr Gly Gly Val Phe Leu ValLeu Val Leu Gln Ala Gly Phe Phe Leu Leu
* ORF P
# : ORFpreS/S. Se indica el comienzo de preSZ y S
Fig. IV.11 pHB4secuencia nucleotídica y traducción a aa.
"forward") y 3'f-‘ooE clon pHB4 se secuenció en su extremo 5'
(primer "reverse")(primer
utilizando la técnica deSanger.
la secuenciaLa secuencia de aminoácidos se determinó a partir de
nucleotídica mediante el programa DNASIS.

81
FILE NAME : PHBZO.SEQSEQUENCE : 323 BP; 70 A; 103 C; 72 G; 78 T.
CAA CAA TTC CTC CTC CTG CCT CCA CCA ATC GGC AGT CAG GAA GGC AGC CTA CTC
* Asn Asn Ser Ser Ser Cys Leu His Gln Ser Ala Val Arg Lys Ala Ala Tyr SerThr Ile Pro Pro Pro Ala Ser Thr Asn Arg Gln Ser Gly Arg Gln Pro Thr ProId:
CCA TCT CTC CAC CTC TAA GAG ACA GTC ATC CTC AGG CCA TGC AGT GGA ATT CCA
His Leu Ser Thr Ser Lys Arg Gln Ser Ser Ser Gly His Ala Val Glu Phe HisIle Ser Pro Pro Leu Arg Asp Ser His Pro Gln Ala Met Gln Trp Asn Ser Thr
CTG CCT TCC ACC AAG CTC TGC AGG ATC CCA GAG TCA GGG GTC TGT ATT TTC CTG
Cys Leu Pro Pro Ser Ser Ala Gly Ser Gln Ser Gln Gly Ser Val Phe Ser CysAla Phe His Gln Ala Leu Gln Asp Pro Arg Val Arg Gly Leu Tyr Phe Pro Ala
CTG GTG GCT CCA GTT CAG GAA CAG TAA ACC CTG CTC CGA ATA TTG CCT CTC ACA
Trp Trp Leu Gln Phe Arg Asn Ser Lys Pro Cys Ser Glu Tyr Cys Leu Ser HisGly Gly Ser Ser Ser Gly Thr Val Asn Pro Ala Pro Asn Ile Ala Ser His Ile
TCT CGT CAA TCT CCG CGA GGA CTG GGG ACC CTG TGA CGA ACA TGG AGA ACA TCA
Leu Val Asn Leu Arg Glu Asp Trp Gly Pro Cys Asp Glu His Gly Glu His HisSer Ser Ile Ser Ala Arg Thr Gly Asp Pro Val Thr Asn Met Glu Asn Ile Thr
CAT CAG GAT TCC TAG GAC CCC TGC TCG TGT TAC AGG CGG GGT TTT TCT TGT TG
Ile Arg Ile Pro Arg Thr Pro Ala Arg Val Thr Gly Gly val Phe Leu ValSer Gly Phe Leu Gly Pro Leu Leu Val Leu Gln Ala Gly Phe Phe Leu Leu
* : ORF P
# : ORFpreS/S. Se indica el comienzo de preSZ y S.
Fig. IV.12 : pHB20:secuencia nucleotídica y traducción a aa.
El clon pHB20fue secuenciado totalmente a partir de suextremo 5' (primer "reverse") y del 3' (primer "forward").
La secuencia de aminoácidos se determinó a partir de lasecuencia nucleotídica mediante el programa DNASIS.

PHB20 ------------------------------- --CAACAATTCCTCCTCCT17PHB4P3 GACCTCAGGCTCAGGGCATATTGACCACAGTGTCAACAATTCCTCCTCCT 50PHB7P3 -------------------------------------- —-TCCTCCTCCT10
**********
PHBZO GCCTCCACCAATCGGCAGTCAGGAAGGCAGCCTACTCCCATCTCTCCACC 67PHB4P3 GCCTCCACCAATCGGCAGTCAGGAAGGCAGCCTACTCCCATCTCTCTACC 100PHB7P3 GCCTCCACCAATCGGCAGTCAGGAAGGCAGCCTACTCCCATCTCTCCACC 60
"k********************************************* i'k'k
PHB20 TCTAAGAGACAGTCATCCTCAGGCCATGCAGTGGAATTCCACTGCCTTCC 117PHB4P3 TCTAAGAGACAGTCATCCTCAGGCCATGCAGTGGAATTCCACTGCCTTCC 150PHB7P3 TCTAAGAGACAGTCATCCTCAGGCCATGCAGTGGAATTCCACAGCTTTCC 110
****ti***************************i******** ** ****
PHBZO ACCAAGCTCTGCAGGATCCCAGAGTCAVUUULLLULHLLLlblelebl 167PHB4P3 ACCAAGCTCTGCAGGATCCCAGAGTCAUUUU1LLULALlllblelebï 200PHB7P3 ACCAAGCTCTGCAAGATCCCAGAGTCAUUUULLlULAl1llblelebl 160
************* ***************** ******************
PHB20 GGCTCCAGTTCAGGAACAGTAAACCCTGCTCCGAATATTGCCTCTCACAT 217PHB4P3 GGCTCCAGTTCAGGAACAGTAAALLLlbLlLLbAATALLULLLLILACAT 250PHB7P3 GGCTCCAGTTCAGGAACACTCAALLLLei¿LLAAATAL¿utLLLLLACAT 210
****************** * ******* *** *****************
PHB20 CTCGTCAATCTCCGCGAGGACTGGGGACCCTGTGACGAACATGGAGAACA 267PHB4PB CTCGTCAATCTCCGCGAGGACTGGGGACCCTGTGACGAACATGGAGAACA 300PHB7P3 CTCGTCAATCTCCTCGAGGACTGGGGACCCTGCGACGAACATGGAGAACA 260
************* ****************** *****************
PHB20 TCACATCAGGATTCCTAGGACCCCTGCTCGTGTTACAGGCGGGGTTTTTC 317PHB4P3 TCACATCAGGATTCCTAGGACCCCTGCTCGTGTTACAGGCGGGGTTTTTC 350PHB7P3 TCACATCAGGATTCCTAGGACCCCTGCTCGTGTTACAGGCGGGGTTTTTC 310
**************************************************
PHBZO TTGTTG 323PHB4P3 TTGTTG 356PHB7P3 TTGTTG 316
******
'*' posición del alineamiento perfectamente conservada
Figura IV.13: Alineamiento de los tres clonesLos tres clones secuenciados fueron alineados en la zona de
superposición mediante el programa CLUSTALincluido en PC/GENE

83
Con el fin de comparar las secuencias obtenidas con otras
depositadas en los bancos de datos (GeneBanky EMBL),primera
mente se realizó una búsqueda de secuencias homólogas en el
Centro Nacional de Información Biotecnológica (NCBI= National
Center for Biotechnology Information) de la Biblioteca Nacio
nal de Medicina (USA)mediante el algoritmo Blast (Altschul et
al., 1990). Dicho programa encontró entre 125 y 62 entradas,
dependiendodel clon probado, que satisfacían el criterio de
corresponder a secuencias con un grado de conservación mayor
al 6596; de éstas sólo se tomaron en cuenta las 50 primeras. Deentre estas últimas se seleccionaron veintiséis en base a los
siguientes criterios:—que fueran genomas completos, con el fin de poder comparar
las mismas con todos nuestros clones,
- que estuvieran representados todos los genotipos y serotipos
En la figura IV.14 se comparan las secuencias obtenidas
de los tres clones correspondientes a la zona preS/S con estas
secuencias depositadas en los bancos de datos y cuyas princi
pales características están resumidas en el AnexoI. La compa
ración se realizó mediante los programas PC/GENEy GCG.
Según el dendogramaobtenido es evidente que las tres
pertenecen al genotipo A definido por Okamotoet al. en 1988.
Sin embargo, es de notar, que el clon pHB7junto con la se
cuencia M57663forman un subgrupo dentro de la rama del geno
tipo A; las diferencias en secuencia nucleotídica que lo ocasionan ya se habian visto en el perfil de restricción. Estehecho se da sólo en esta región, ya que en el dendograma co
rrespondiente a la zona preC/C (fig. IV.15) no se presenta

84
esta subdivisión.
Por otra parte puede observarse que la mutación T3u2, en
la zona preSl, de pHB4, que involucra un cambio de Pro a Leu,
no es compartida por ninguna otra de las secuencias analizadas
ni las obtenidas con el programa BLAST.En cambio la falta del
sitio BamHIde pHB7, confirmada por el cambio G” -+ A, parece
estar más difundida, sin que la presencia de este sitio de
restricción sea característica de ningún grupo, comoalguna
vez fue sugerido.
En la figura IV.15 se compara la zona preC/C del clon
pHB7con las secuencias antes mencionadas. Este clon presenta
un par de mutaciones, ambas compartidas con M57663, en la re
gión e. Una de ellas Gugg-+ A, que al aparearse con Uïm1, en
el RNApregenómico, convierte el par "imperfecto" U:G, ubicado
en el segundo stem, en el par U=Adando mayor estabilidad a la
estructura. La otra Gm;2-+T, esta localizada en el "bulge" y
sería el templado para el cuarto nucleótido que se incorpora
al primer, que luego se translocará a DR1*,al iniciar la
transcripción inversa (Fig. IV.16).En la figura IV.17 se observa el alineamiento correspon
diente al extremo 5' del clon pHB4que comprende el extremo
terminal de TP y comienzo del separador (Spacer) de Pol y el
principio de preSl.Puede observarse que, aún siendo esta región, la zona del
genomaviral que presenta mayorvariación, nuestro clon coin
cide con Z35717, cosa que no ocurría cuando se analizó el ex
tremo 3' del mismoclon, que por el contrario parecía más re
lacionado con otras secuencias del mismogenotipo. Este hecho

Figura IV.14
X75658
D00329Consenso
X75658X75663X69798X75657X75664J02203
Consenso
086. . . . . . . . .. A..A TGT c.A.....CT .......A. . . . . . . . .. A.....TG ..A.....CT .........A. . . . . ..A.. A. A .TGG ..A.....CT .........A. . . . . . . . . . . . . .. c.A.A....T .........A. . . . . . . ... . A.... G c.A.A. .T . . . . . . . ..A
. . . . . . . . . . . . . . . . . .. C.ACA.. TT .. . ...AA. . . . . . . . . . . . . . ..... A.ACA.. CT ...... .AA. . . . . ..... .......... c.ACAc.C. ........AA
.... ... . . . . . . . . .. .G.C. C. . . . . . ..GAT
. . . . . . . . . . . . . . . . . . . . . . . ..C... T .A..AT--- .A..AT
.A.. ..... ... . . . . . .. ....C. .. T..A..AT
. . . . . . ... ...... . .....C. . .T..A..AT
. . . . . . . . . . . . . . . . . . . . . . . ..C. T..A..AT
........ . . . . . . . . . . . . . . ..C. . T..A..AT
. . . . . . . . . . . . . . . . . . . . . . . ..C.... T..A..AT
. . . . . . . . . . . . . . . . . ..G .....Cc .. T..A..AT. . . . . .. A..A . . . . .. .... . . . . . . . . . . . . . ... . . . .. .. .. c...... .. .......... ..........
. . . . . . . . . . . . . . . . . ..C .......... .........., . . . . . . . . ..... .. . ....C ........
........ . . . . . . . . . . . . . . . . . . . . . . . . ..T. .
. . . . . . . . . . . . . . ..c.. c.c.....T. . . . . . . . . ... . . . . ..A. .......... C.C.....T. .....A... . . . . . . . . . . . . . . . . . .. C.C...T.T. ..........GCCCTCAGGC TCAGGGCATA TTGACAACAG TGCCAGCAGC
3136.... .. . .T. c..G...A. .A..C..AG..T....... . ..T. . c..G...A.. ..A..C.AAG.T. . . . . . . . . . . ..G. C..g..AA.. ..A..C..AG
... . . . . . .. ... . . . . . .. . . . . . . . . . . . . . ..C..A.
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . ..C..A...... ...C . . . . . . . . . . . . . . . . . . ..C..GC
.....T. . .C..... .. . . . . . . . . . . . ..C..GC....T. . ..C...A . . . . . . . ..A .. .C..TC
..A . . . . . . . . . . .... . . . . . . . . . . . . . . . . . . ..
. .....G. .......... .......... ....... .
. . . . . . . . . . . . . . . . . . . . . . . . ..A... .....G. ..
. . . . . . . . . . . . . . . . . . . . . . . . ..A.. .. .G. .
. . . . . . . . . . . . . . . . . . . . . . . . ..A.. . . . . . . . . ..
.TT. . . . . . . . . . . . . . . . . . . . . ..A... ... ......
....... . . . . . . . . . . . . . . . ..A.. . . . . . . . . ..
. . . . . . . . . . . . . . . . . . . . . . . . ..A... ........... . . . . . . . .. ........ ......A.. ..........
.....T. . . . . . . . . . . . . . . ..A... . . . . . . . . ... . . . . . . . . . . . . ... . . .......... .........T
. . . . . . . . . . . . . . . . . ..T .......A.. .........C. .. .... . . . . . . . ..C
AGGAAGGCAG CCTACTCCCA
...G......
. o o o o o o o o o
o o o o o o o o oo
o o o o o a o o o.
o a o o . o o a o o
o o n a a o o o o o
o o o c o o o o o o
n n o n - . o n o o
a o o o o . o o o o
o y o o o o o o o o
o g o c o o o o o o
TCTCTCCACC

X75658X75663X69798X75657X75664J02203Z35716X65258M57663pHB7p3L13994
pHBZOpHB4p3X70185Z357l7X02763V00866X5197OX75656X75665D12980M12906XO4615M38454VOO867DOO63ODOO33OD00331DOO329
Consenso
X75658X75663X69798X75657X75664J02203235716X65258M57663pHB7p3L13994
Consenso
a o n ' o o o o o o
o o . o . o . . o o
. . o o . o g o a o
o o o o n o o o o o
o o o c c o o o o o
o o o o o a a o o o
o o o o o o o o o .
o ' o o c o o o o o
o o o u o . a . 0 .
o c o u o a u o o o
o o o o o o o o o o
c o o o o o o o o o...-oTCTAAGAGAC
o o a o a a o n o o
o o o c o o . o o o
v o o o o u - o o o
o o o o o o o o oo
o . o o o o o o o.
o o a o o o o o o o
o o u o o o o o c o
oooovonooACCAAGCTCT
u o . o o o o o o o
o o c o ' o o o o o
o o o o o o o o o o
. o g c o o o o a .
o o g o o o o o . .
u . . . o o o o o o
u . c o . o o c o o
o o o o c o o o o o
o o o o n n o . o o
a o o . o o o o o .
n o . . n o o o oo
o o o o o o o o o.
o o o u o o o o c a
o o o o a o o o o o
. . . . . . . . ..
. . . . . . . . ..
. . . . . . . . ..
...G n a o o o....G.........G o o o o . o
GCAAGATCCC
o o o o o n o o o o
o o o o o o a o o o
v o o o o o c o o o
o c o o o o o o a o
o . o o o o o o o o
o o o o o o a o o o
. o o o o a o c o o
o o o o a . o o o o
o o . o o o o o o o
o . o o o o c . o o
o . . . . o c o o o
. o o o o c . o o o
o o o o a o o o o o
o a o o o o o c . o
o o c o o o o c a o
' o o o o o o . o o
o o a . g . o o o o
o c o o o o o o . o
n o c o o u o o o a
o o o o o o o o o o
no...
o a o o o a o o o.
o o o o o o o o o.
o n o o o a o g o o
o o o o o o o o o o
o o o o n o a o o.
o o o a . o o o o.
AGAGTCAGGG
o u o o o o o o o o
o o o o o o a o o o
o o o o o o o o o g
o . o o o o o . o o
o o o o . . o o . o
o o . o c o o o o o
o o o o o o a o o .
. o . o o o o o o o
o o o o c c o o . c
a o a o n n o o o.
o c a o o o n o oo
o o o o o o o o o o
o o o o o o c o o o
o o o o o c o o o o
o o o o o e o o o o
C .....C..GCCTGTATTT
o o o a o o o o o o
o o . o a o o o o o
o .C. ¡Go o
... ......c . a . . a . ...C.. u o q o n o.C . . . . . . . ...... . . ........... . . . . . . . ... . . . . . . . ............ . . . . . . . ... . . . . . . . ............ . . . . . . . ... . . . . . . . ........ .... . . . . . . . ... . ........... ..... . . . . . . . ... . . . . . . . ... . . . . . . . ... . . . . . . . ... . . . . . . . ........ ... . . . . . . . ..TCCTGCTGGT
86

X75658X75663X69798X75657X75664J02203235716X65258M57663
D00329Consenso
X75658X75663X69798X75657X75664J02203Z35716X65258M57663pHB7p3L13994
pHBZOpHB4p3X70185Z35717X02763V00866X5197OX75656X75665D12980M12906XO4615M38454V00867D00630DOO33OD00331D00329
Consenso
o . . o o o.
o o . s o - o . o.
o . a o . o con
oo o o o . oo.oo... o.o o o o 00.000o . . . o o o . o.
o o o. onooo-oo...c o o . c . c c ..
. o o o . o ....a o o . o .oooo o . o . . o . .......o.oc . . . o o o . o.0.000....o o . . . . ooo00.00.0000o... o o.o o . o o o . c oo
. o . o o o . . ..
o v o o o o . o.
o a o o o . oo
a o o . o . . g ..
. o . . o o o . ..
ooo o o . o o a.ooo o.o o o o o o o o o .
o o v o o o
o o n o o o o o o o
o o o o o o u o . o
o a o o . n o o o o
. o o o o o o n o o
o o o o o o o o o o
ATCGTCAATC TTCTCGAGGA CTGGGGACCC
o o o o o o o o o o
. . o o o o o . o.
u o o o o o o o o.
.o o a o . o o oo
a o o . a o o o o.
ooo o o a o oo
o o o . o o o oo
o o o o . o a o.
v u o o o a o o o a
c o o o a c o o o .
o a o o o o o o o o
o o o o o o o o o o
o - o o o o o o a o
o c o o o o o a a o
a o o o o o o o o o
o o o o o o o ' v .
o a o . o o o o o o
o o o o o o o o o o
o o c o o n o o o .
o o o a c . o n n o
Ao...
o o o o y o o o o o
o o o o o o o n ¡o
a o o o o o o o oo
o c o o o c o o o c
a o o o o o a o o o
ooGaooooCo
n o o o n n o o o o
o o o a o o o a o o
o o o o o o o o o g
o o o o o o o o o o
o o o o o o o o . o
o o o c o o o a o o
H
c o o n o o o o o o
n o o o o o c o o o
o o o u o o a o o o
o . o o o o o o a o
CCGACTACTG
oooToToooooooToToosc
o ACooooo
ooToooo..T.....
o o o o a o o
o o o . o o o
..T.Coo.oo..T.c.....TGCGACGAAC
o o . o o o o u - o
o o . o o u o . o o
o o o o o o o g o o
. o o o o o o o o o
o . o o . o . o o a
o o o o o o n o o o
o o o o o o o o n o
o o o o o o o n o .
o o o a n o o o o o
. o o a o o o a c o
o o a o c . o o o o
o o o o o o o o o
o o o o o o o o vo
o o o o o . o o o a
ATGGAGAACA
87

X75658 . . . . . . . . .. .C . . . . . . .. ... . . . . . . . . . . . . . . . .. ..T..G....x75663 . . . . . . . . .. .C. . . . . . . . . . . . . ..G.. .......... ..T..G....X69798.T........ .C.. . . . . .. .......... .. . . . . . . .. ..T..G....X75657 .......... . . . . o. . . . . . . . . . . o. . . . . . . . . . . .. ......o...X75664 ... . . . . . .. .... . . . . . . . . . . . . . . . . . . . . . . . . .. ...-......J02203 . . . . . . . . . . . . . . . . . . .. .....T.... . . . . . . . . .. ..........Z35716 .......... o. . . . . . . . . . . o....... . . . . . . . . .. ... . . . . . ..X65258 . . . . . . . ... . . . . . ..... ... . . . . . .. .......... . . . . . . . . ..M57663 .......... .......... . . . . . . . . .. .. . . . . . . . . . . . . . . . . ..pHB7p3 ... . oo. . .. ... oo. . . . o o. . . . ....o o. oo. . ooo. ......o...L13994 . . . . . . .... . . . . . . . . . . . . . . o . . . . . . . . . . . . . . . o o . . . . . . ..
o o o o o o o o o. oocooooooo connooocov ooo o a o a o o o o o o o o a o o .pHB4p3 . . . . . . . . .. .... . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . ..X70185 . . . . . . . . . o . . . . . . . . . . . . . . o . o . . . . . . . . . . . .. ......o...Z35717 ... . . . . . . . . . . . . . oo. . . . . . . . .... oooooocooc ..........X02763 ... . . . . . . . . . . ....... . . . . . . . . .. ...-...... o. o. . . . . ..V00866 . . . . . . . . . . . . . ....... . . . . . . . . . . . . . . . ..... . . . . . .....X51970 . . . . . . . . . . . . . . . . . . . . . . . . . ..C.. . . . . . . . . . . . . . . . . . . ..X75656 CA........ . . . . . . . . .. .......... . . . . . . . . . . . . . . . . . . ..X75665 CA........ . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . ..D12980 CA........ . . . . . . . . . . . . . . ...... ..... . . . . . . . . . . . ....M12906 CA. . . . . . . . . . . ....... . . . . . . . . . . . . . . . . . . . . . . . . . . . . ..XO4615 CA. . . . . . .. .... . o . . . . . . . . . . . . . . . . . . . . .... . . . . . . . . ..M38454 CA. . . . . . . . . . . . ...... ... o o . . o . . . . . . . . . . . . . . . . . . . . ..V00867 CA........ .......... . . . . . . . . . . . . . . . . . . . . . . . . . .....DOO63O CA. . . . . . . . . . . ....... .......... . . . . . . . . . o . . . . . . . . ..D00330 ..G....... .C.. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . ..D0033l ..G . . . . ... .C . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . ..D00329 ..G . . . . . .. .C . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . ..
Consenso TCACATCAGG ATTCCTAGGA CCCCTGCTCG TGTTACAGGC GGGGTTTTTC
X75658 o. . . -X75663 - - - - -X69798 o - - - -X75657 - - - - oX75664 - - - - -J02203 - - - - -‘235716 .-o--.
M57663 - - ' - -' Comparación de la secuencia de pHBZO
L13994 - - - - -- y del extremo 3' de los insertos en lospHB4p3 -----° clones pHB7 y pHB4 con las secuencias235717 - o»- -- descriptas en el Anexo 1.
V00866 - - - - -- Se destacan las posiciones corresponX75656 - - ' oo- dientes a los ATG preSZ y S.
Consenso TTGTTG
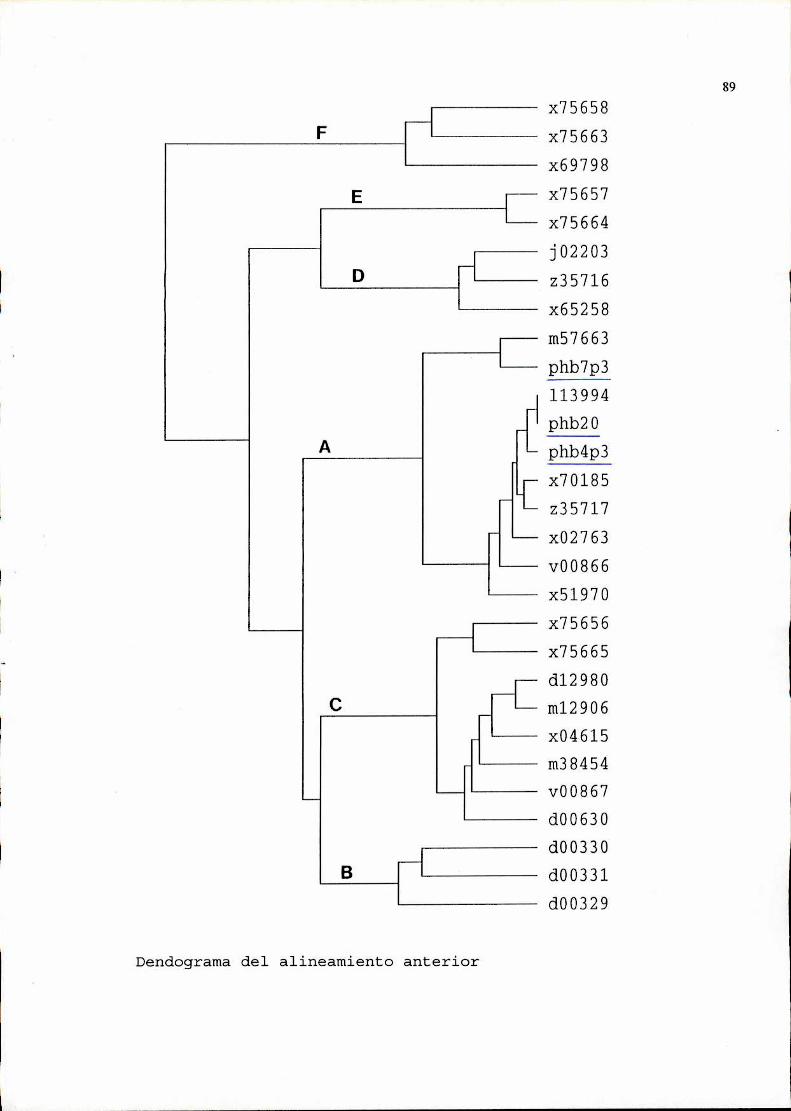
F C|___
D | F3333;
——[:
EDendogramadel alineamiento anterior
X75658
x75663
X69798
X75657
X75664
j02203235716
X65258
m57663
phb7p3113994
phb20
phb4p3X70185
235717
x02763
V00866
X5197O
X75656
x75665
d12980
m12906
X04615
m38454
v00867
d00630
d00330
d00331
d00329
89

Figura IV. 15
X75658X75663X69798X75657X75664J02203Z35716X65258X70185Z35717L13994X02763
Consenso
X75658X75663X69798X75657X75664J02203235716X65258X70185Z35717L13994X02763V00866X5197O
Consenso
1684ooTIoo
oooTooooTG GA.-°°T°°'-TG GA
n . o o a o o o o o
a o o o o o o o o o
o n o a n a o o o o
a o o o n o a c o o
c o o o o o o o o o
o a . o o a o o o o
o o o o o o o o n o
o o o a o a o a y o
u o o o a o o o 0 o
o o a a o o n o o o
o o a a o a o . o o
o o o 0 o c c c 0 a
a o o o o o n c o a
n o n o o o o o a .
o o a o o o o . o o
o a a a o o a o o o
o o c o o o o o o c
o o o o o o o o c o
o
90
1733
...G . . . . ....G . . . . ..
. .G . . . . ...G . . . . ...G . . . . ..
. .G . . . . ...G . . . . ...G . . . . ..
..G . . . . ..
c...... .
..G . . . . ..
..T..G. .
.CT..G....T..G...TAAAGACTG
1783
.G.. . ..
o o o o o o o o v o
aooo.0C00 0000.0000. 000000.... ¡000000... 0Co'ocoochGAGGAGTTGG GGGAGGAGAT TAGGTTAAAG GTCTTTGTAC TAGGAGGCTG

X75658 o. o o o o o o o c . o . ¡00.0.0 o . c o o o o o n o a . . o o o a o o o o o . . o o o . oocooo. c cooo .ocoaooaTT ..... ooooc . oo.....o. . ooo. . ooa.. o . . o o . c o . o o o . u o ..TT o u . o o o .... a a o . . o . . o. o o o o . - o a o.
X75657 o o c o u . o o o . o o o . g . .... o . o o o o c . oo ......oooo ono-00000.X75664 c . o o o o o o o o o c . o o ....o 00 . . o o . u o o o o o c c o . o o o a o o . o . o . noJ02203 . o . . o o a a u a o n o o o o .... .aouooc... o . . c c o o o o o o o o . o ....o235716 o o . o o . . o o o . o o o o o o .oo o... . a - a o c o o o . . o o o . . . c o o . . o o a.X65258 o o o o o o . . . o o o . o . o o o o. ..oo-oooo. o o o o o o o o . . o o o . a o o u ooX70185 ......oooa o . . o o . o ... ¡0.00.0009 o o o . . o o o o o . . o . o . o o c.235717 oncoo o n - o o . c . o o o .... u o . . o . o o u o o o o . . o o u o o . o o o s o a c o.Ll3994 o o a o o . . o o o o u c o o o nooo o o . . u . . c o o o o o ...-ooo o o o o ......X02763 o o o o ..Coo. tooo-coto. o o o o . o o o u o o . . o o o o . o o o o . . . no...V00866 o... o o . o o o . . o o o o a o .. o a o o s o o a o o o o o . . o . . o o o . o o o o . s o.
X51970 . o o o o . o o o. ¡tooo-ooo. o o o o . o o . o o c o ' o o o o . . . o u o o . o o . ¡o
pHB7p5 o o o o o oo... o a o o . . . o a o o o o o-TooTc a o a o o o o u o c . o g o o o o o ooM57663 o . o o o o o . .. o. . a o o c c . . o o o ..TT.T. o o o o o o . . o . o . o o . o . o o.X75656 . . o o o . . o .o o a o o . o o o o. .o o o . o . o o . c o o o . u o o . o . o c a o o - o c.
X75665 o... . o o . o- ... a o n o o o o . o o o o o o... o. . o o o o . o o o . o o . . o o o.D1298O o o a a ...... o u u ' a a ..TT o. o o . c u o o n u . o o . . o o c o . o o o o c o o o.M12906 .....o-oo- 00.0000vTT o o u c o o o o o . o o g o o o o o o a o a o o . c o . ooX04615 o o . . . . . s . o . . . o o . ..TT g o o o . . o o . o . o o o . . o o .‘ ...C o o o e ..M38454 o o o o . o o . o. ....ooooTT o o o . o o o o o o c . o . . o o o o o o o o o o c o . o.V00867 .......___ ____...... ... . . . . . ..DOO63O oooooosoo. ooooooooTT o . c u o o o o o o o o o o o . o o o o . o o o o o o o o.DOO33O 40000000... ...oosooTT o o o s o o o o o . . o o a - . o o o c o o o o . o . o o.DOO331 . g a o o o o . o o o . . o o o ..TT o o o o o o o o g o o o o . o o o . o o . o o o . a o . ..
D00329 o o . . . uo a o c o o . o o o ooTT 00.000.... ......c... o o o o c . o o o.Consenso TAGGCATAAA TTGGTCTGCG CACCAGCACC ATGCAACTTT TTCACCTCTG
L*
X75658 . . . . . . . . . . . . . . . ..T.. ....C..... . . . . . . . . . . . . . . . . . . ..X75663 . . . . . . . . .. . . . . . . . . .. ....C . . . . . . . . . . . . . . . . . . . . . . . ..X69798 . . . . . . . . .. .T . . . . . . .. ....C . . . . . . . . . . . . . . . . . . . . . . . ..X75657 . . . . . . . . . . . . . . . . . ... ... . . . . . . . . . . . . . . . .. ..........X75664 .......... .. . . . . . . . . . . . . . . . . .. .......... . . . . . . . . ..J02203 . . . . . . . . . . . . . . . . . . . . . . . . ...... .......... ..........Z35716 . . . . . . . . .. ..A . . . . . . . . . . . . . .... .. . . . . . . .. ..........X65258 . . . . . . . . . . . . . . ..A... ....C..... .. . . . . . . .. ...-......X70185 . . . . . . . . . . . . . . ..A... ....C . . . . . . . . . . . . . . . . . . . . . . . ..Z35717 . . . . . . . . . . . . . . ..A... ....C..... . . . . . . . . .. . . . . . . . . ..Ll3994 . . . . . . . . . . . . . . ..A... ....C . . . . . . . . . . . . . . . . . . . . . . . ..X02763 . . . . . . . . . . . . . . ..A... ....C . . . . . . . . . . . . . .. . . . . . .....V00866 . . . . . . . . . . . . . ...A... ....C . . . . . . . . . . . . . .. .. . . . . . . ..X51970 . . . . . . . . . . . . . . ..A... ....C . . . . . . . . . . . . . .. . . . . . .....pHB7p5 . . . . . . . . . . . . . . ..A... ....C...T. . . . . . . . . . . . . . . . . . . ..M57663 . . . . . . . . . . . . . . ..A... ....C...T. .......... . . . . . . . . ..X75656 . . . . . . . . .. ..A . . . . . . . . . . . . . . . . . . . . . . ..... . . . . ......X75665 . . . . . . . . .. ..A . . . . . . . . . . . . . . . .. ... . . . . . .. .. . . . . . . ..D12980 . . . . . . . . .. ..A . . . . . .. ... . . . . . . . . . . o . . . . . . o . . . o . . . ..M12906 . . . . . . . . .. ..A . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . ..X04615 . . . . . . .... ..A . . . . . . . . . . . . . .... . . . . . . . . .. . . . . . . . . ..M38454 . . . . . . . . .. ..A . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . ..vo0867 . . . . . . . . .. ..A ‘ . . . . . . . . . . . . . . .. . . . . . . . . . . . . . . . . . . ..D00630 .. . . . . . . .. ..C . . . . . . . . . . . . . . . .. ..... . . . . . . . . . . . . . ..D00330 . . . . . . . . .. ..A . . . . . . . . . . . . . .... . . . . . . . . . . . . . . . . . . ..D00331 . . . . . . . . .. ..A . o . . . o . . . . . . . . . .. .... . . o o . . . . . . . . . . ..D00329 ....G . . . . . . . . . . . . . .. .......... .......... . . . . . . . . ..
Consenso CCTAATCATC TCTTGTTCAT GTCCTACTGT TCAAGCCTCC AAGCTGTGCCl n
G

X75658X75663X69798X75657X75664J02203Z35716X65258X70185Z35717Ll3994X02763V00866X51970pHB7p5M57663X75656X75665D12980M12906X04615M38454V00867DOO63OD0033OD00331D00329
Consenso
X75658X75663X69798X75657X75664J02203235716X65258X70185235717L13994X02763V00866X51970pHB7p5M57663X75656X75665D12980M12906X04615M38454V00867D0063OD0033OD00331D00329
Consenso
1884 1933o o o o . . o o o o o o o o u o o o a o o . . . g . o o o . o o . o a o o g o o o a q . o . o o a.
n o o o o c o o o o o o o o . o o . o o . . . ¡ooo .......... oca-ooo...o o o o . . o o . c c o o o a . . c o o o a o a o . . c o . o c . c . o o . o . o o o o c o c . ..
0.0000 oo ooo-so . . o o o o o o o u o o - o o o u . o o o o a ooo o..ooo.aaa.. o ...... .......... .......... a........ooooooo-o 00000.00 oocooCo oooO-00000 ¡tooo-noooo. aac. oa .. oo .o. .....C.o. 00-0000000 nooo-00000oo u u c . n o o . o o o u a ooo . o o . o o c o c o o n . . o o o o o o o o o o u o o...
o. oooo. . . . o. q. go. o0| . . g. ........ . ........o.. g. . . ... .....ooooo 0000.00.00 00.000 o. con... oo o o o o o o o o o o . o . . o o o o o a . o o o o o . . o c . o o o . o o o a o o . o o . o o .o
c. . oo. . o.o . coo... ¡00000000. o...-..... 00000000..o o o o o o . o o o s o o . o . o . o o o . o o o o o . o . o - o o o o . o c o o - o o o o a o o
c v . . o o . o o . o c o o o o c o o o o o o o o o . o c o o . o o o . o o o o o o o c o o n - a.
.A o o c . o o o o o . o c o . o o . o a o o o o u c o . o o . o c o o c . o o o o o c o o.
00A a . . . . o . o o o . . o o o . . . o o o o o o . . . . o o . . o o a o o o o _ . o o .oo o o o . o o c o o o o o . o ¡no o o o .. .co... ooo-000.00. oo. c oogoc . ooooo . .o. .o. .. .o. cocos-ooo. 00.000....
o o o o ooo oaoocooooo oGoooo o . o o o o o c . o o.oooooooooo ooo. gnoooo .. ....... .G........ 0.00.0000.oooooooo.. tooo-oo... ¡0.000.000 oG....ooao ccoo-00000ao. o. ooo. . . . . . ... .......... .G........ ooo-coo.... a . o a . . o o o o . . o... . . . . . ... .G.o o o g o o . o o a 00A. oooooooo. vooocoooo cooooooooA .G.ooc.o ooo-cuco.o o o o o . o a o o n o o . o o o o o o o c o o o o o . o. .Gooooooo- oc........o o o o o a a o o o o o o o . . a o o o o o o o o o . o o. oGuo . . coa-ooo...
o 0C... o o o o a o o o o . . o o oo
1934 1983oooonovo «GT.. To. ooo-ooo... ooo-noo... choAA Co.
ToooooooAI oGTcoooTno . .G a . o o . o . o o . o o o o. .Gooooo- oT c o o o o o o u . . u o o - ocho ooo... ooo o T. oooCo .AooGoooo.oo. . oc nooo ooc uooaGoo ooo-ooo... .o.....oo ...A..AA.ooo. oooo. o . ooooocGO. noooovooc- o.......o. .vooAAo. . ooooooo. ooo oG-o .o.....oo. 000000.0. o. .Aovoooo. oa oa ou o oo c Goo oo...... o 00.000.. ooAavo. . . . . . c c uo a . . .o. G.. . o . 000.00.00. o. 0C00Aoo
. u o o a o n . c o o ooGoo o . o . q o o . o o . . o o o o o . oo . C..CA.o o o o o o o o o o o o u o o ooGov o o... .......... o .C .CA.oooooooooo . o oooooGoo oooooooc. .......... oooocooAAo. . . . o . ooa o . a . ooooG- ooo... 0.00000... ocooc .CA.oo ¡o o o . cooG-o oo o. o .. ooo .ooocovoo. uoaoaco. . och. ooo... 0.0.0.000. ooooc .CAou. o. . cocaa ccoooooGoo ooooooo-oc oo........ .o..c .C... o o o c o . o . o o c c o ...A.. . o o o c o o o o o o o o o o o o o o. ooooC .Co.T o o o o o a . o a o . . c o ooTo. o o . . o o o o o. ..T o o c o o o. .A. .A....T o o o o o o o o y a o o o c .uT. a . . o o o o o o. T o o o o o o. .A.o.Aooo.T o o u . . o o o o u . . v o aoTao o o o . . o o o o u . o n . o o o o o o o o o .vo...
T o o o o o n . no oo ooToo ooo . o o o o c o . o o c o o o o c . o o o ocAcooT. . . . . o a .. g n o o o acTo o ooo o o o u o ’ o o o o o n o o ovooooT o . o . o o c a c o c o o o ooTo o o o u o o . o o o o o o o o o o o . . o . o ..A.o..
T o o o o o o c o o o o o o o ooTo o o o o a o o o o o o o c c . . o o oo .G .voooo
T o c o c c a o o u o c o o oooTo c . o . o o o c u o u . o o o o o . a . o o a ovoo.
T... o c o a o o o o o o . ooTo o o o o o o o o o o . . a o o o n o o. oGnoGooGooT o . o o o o c o o o o c . o ooTo o o o . o a o o o o ' o - c o o o o o o o u o ooA-ooo
¡000000009 o o a . u .oTug ooo-occ... a o c o a . o o oo . Go 0G. .Go .ACTGTGGAGT TACTCTC-TT TTTGCCTTCT GACTTCTTTC CTTCTGTTCG

84 2032x75658 .C..T C . _ . . . . . . . . . . . . . . . . . . . . . . . ..T.. G. . . . ....AX75663 G .C.....C _... . . . . . . . . ..C .C.....T . . . . .....AX69798 G C..A. C _... T . . T c.....T . T. . . . . . ..AX75657 . . . . ..T.. _.T . . . . . . . . . . . . . . . . . . . . . ..T . . . . . . . . ..X75664 . . . . ..T.. _ T. . . . . . . . . ... .. . . . . . ..T . . . . ..A...J02203 . . . . ..T.. _.T . . . . . . . . . . . . . . . . . . . . . . . . . ......Z35716 . . . . ..T.. ._ .. . . . . . . ..C. . . . . . . . . . . . . . . . . . . ..
. . . . . . . . .. _.... . . . . . . . . . . . . . ... . . . . . . . . ..X70185 . . . . . . . . .. ._ ...... . . . . . . .. A. . . . . . . . . . . . . ..Z35717 . . . . . . . . .. _ .. . . . . . . . . . . . . . .. .. A . . . . . . . . . . . . . ..L13994 . . . . . . . . . . . _ . . . . . . . . . . . . . . . ... ..A . . . . . . . . . . . . ‘.GXO2763 . . . . . . . . .. _ . . . . . . . . . . . . . . .. A . . . . . . . . . . . . . ..vo0866 . . . . . . . . .. _ . . ...... ...A . . . . . . . . . . . . . ..X51970 . . . . . . . . . . . _ . . . . . . .. G . . .. ...... .. . . . . . . ..
Gasco-A ._ ..oA. oco. . . . . .. .. . . . . . . . . . . . . . . ..M57663 Q.....A A T .A . . .A . . . . . . . . . . . . . . . . . . ..X75656 ...C.....C . _ . . . . . . . . . . . . . . . . . . . . . . . ..G . . . . . . . ..X75665 c.....C . _ . . . . . . . . . . . . . . . . . . . . . . . ..G . . ......Dlzggo . . . . . . . ..C _ ....... T... . ... g. . . . . . . . . ..M12906 . . . . . . . ..C . _ . . . . . . .. ..T . . . . . . . . . . . . ..G.. . . . . . . . . ..xo4615 . . . . . . . ..C . _. . . . . . .. . T....... ......G. . . . . . . . . ..M38454 . .. ...c ._ . .... . . . . . . ... .. . .G . . . . . . . . ..vo0867 . . . . . . . ..T _ . . ..T.. . . ...... g.. . . . . . . . . ..D00630 . ..C . _ . . . . . . .. TT. . . . ..C . . . . ...G . ........ .D0033O . . . . . . . ..C . _ . . . . . . .. T T. . . . . . ..G ......Doo331 . . . . ..T C . _ . . . . . . .. .T . . . . . . . . . . . . ..G.. .... . . . . ..D00329g..c . . . . .. .-.T....T. ..T....... . . . . . . . . .. ....A.A...
Consenso AGATCTCCTA G-ACACCGCC TCAGCTCTGT ATCGGGAAGC CTTAGAGTCT
2033 2082X75658 ..G..A.... .CA.C..CA.T.....C..T ..... . . . . . . . . ..T....X75663 ..G..A.... .CA.C..CA.T.....C..T . . . . ...... .....T....X69798 .....A.... .CA.T..CA. ... . . . . ..T . . . . . . . . . . . . . ..T....X75657 . . . . . . . . . . . . . . . . . . .. ...C . . . . . . . . . . . . . . .. .C.....T..X75664 .....A.... . . . . . ..G.. ...C . . . . . . . . . . . . . . .. .C.....T..
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .. .A.....T..Z35716 . . . . . . . ’ . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .. .A.....T..
. . . . . . . . . . . . . . . . . . .. ... . . . . . . . . . . . . . .... .A.....T..X70185 ... . . . . . .. .C.. . . . . .. ... . . . . . . . . . . . . . . ... .C.....C..235717 .. . . . . . . .. .C . . . . . . . . . . . . . . . ... . . . . . . . . .. .C.....C..L13994 ..C . . . . . .. .C . . . . . . . . . . . . . . . . . . . . . . . . . . .. .C.....C..XO2763 . . . . . . . . .. .C . . . . . . . . . . . . . . . . . . . . . . . . . . .. .C.....C..vo0866 . . . . . . . . .. .C . . . . . . . . . . . . . . . . . . . . . . . . . . .. .C.....C..X51970 . . . . . . . . .. .C. . . . . . . . . . . . ..c... . . . . . . . . .. .c.....C..pHB7p5 . . . . . . . . .. .C . . . . . . . . . . . . ..A... . . . . . . . . .. .C.....C..M57663 .. . . . . . . .. .C . . . . . . . . . . . . ..A... . . . . . . . . .. .C.....C..X75656 ..G..A.... ... . . . . . . . . . . . ..A... . . . . . . . . .. ..G . . . . . ..X75665 ..G . . . . . . . . . . . . . . . . . . . . . ..A... . . . . . . . . .. ..G . . . . . ..D12980 ..G..A.... . . . . . . . . .. ......A... . . . . . . . . .. ....C.....M12906 ..G..A.... . . . . . . . . . . . . . . ..A... . . . . . . . . . . . . . . . . . . ..x04615 ..G..A.. . . . . . . . . . . . . . ..A... . . . . . . . . . . . . . . . . . . ..M38454 ..G..A.. C. . . . . . . . . . . . ..C.. . . . . . . . . . . . . . . . . . . ..V00867 ..G..A.. ....... . . . . ..A. . . . . . . . . . . . . . . . . . . ..D00630 ..G..A.. . . . . . . . . . . . . . . . ..A. . ... .....G...Doo33o ..G..A.... . . . . . . . . .. ......G... . . . . . . . . . . . . . . . . . . ..D00331 ..G..A.... . . . . . . . . . . . . . ...G... . . . . . . . . . . . . . . . . . . ..D00329 . . . . . . . . .. .C . . . . . . .. ...c..A... . . . . . . . . . . . . . . . . . . ..
Consenso CCTGAGCATT GTTCACCTCA CCATACTGCA CTCAGGCAAG CTATTCTGTG

X75658X75663X69798X75657X75664J02203Z35716X65258X70185235717L13994X02763V00866X51970pHB7p5M57663X75656X75665D1298OM12906X04615M38454V00867D00630D0033ODOO331D00329
Consenso
Comparación de la secuencia del extremo 5'
94
ooooo
oo...oo...es...oc...ooo.o.....o....c...o.....o........oo...ha...
del inserto enel clon pHB7con las secuencias descriptas en el Anexo 1.
Se destacan las posiciones correspondientes a los ATGprecore y core,
(—-—)8, DR1el comienzo del RNApregenómico (*), la señal
<——-).y el sitio de poliadenilación

mX75658
X75663
X69798
r—— X756571%¡— X75664
j02203235716
X65258
X70185
235717
113994
X02763
v00866
X51970>
mmm
Dendogramadel alineamiento anterior
m57663
X75656
x75665
d12980
m12906
XO4615
m38454
v00867
d00630
dOO33O
d00329
phb7p5
95

96
Length: 90 Energy: -27.4
90H >
Figura IV.16: Estructura secundaria de e- Se presenta la conformación que adoptarïa, en el RNApregenómico, la señal de encapsidación (e) del virus clonadoen el recombinante pHB7.- Los nucleótidos se numeraron a partir del primero del RNApregenómico que corresponde al nt 1819.- La transición G -+ A corresponde a la Amde la figura. Sedestaca el AUGde iniciación del core

Figura IV.17
X75658X75663X69798X75657X75664J02203235716
X75665D12980M12906X04615M38454V00867DOO630D00330D00331D00329
consenso
X75658X75663X69798X75657X75664J02203Z35716X65258pHB4p5Z35717X70185Ll3994X02763X51970V00866M57663X75656X75665D12980M12906XO4615M38454V00867DOO630D00330D00331D00329
Consenso
GoaoG.o oG-cocacoooG. o-G. g.
o o o o o o o o o o
o n o o o o o o o o
o o o o o a o o o o
o . . o g o o a o o
A.g.......CAACAATTTG
o o o o o o o o o o
. a o ..........
. o g . . . . a ..o o o o o o ooo
o o o o o a n o .o
o 0000- occoo... oo o u o o c o . o.
o o o o o o o o .o
... o o
. . . . o o o . o.o . . o . o . o o.
o c o o . o o.
u . . o o . o . oo
o o o o o . . c o...........
oooA c o o o o.AATTATGCCT
.
o . o
o. . . . o . . ........o . . o o o . oo
. o o . . e o . .
. . . o o o o o oo..........
.A o . o o o . ..ooo . o . o o oo
. . . . o . ..o . o o s o o o o .
n o o o o o o o o o
o o c . o . o o o .
o o . o o o o o o o
o o o o . o c a o o
oooIvoooo..C.voo.o
o o . o o o o o o.
o o u o o o - o o.
o u o o o o o a o .
¡ooooAaaToGCTAGGTTCT
o o o o c o
o o o o o o o o o o
o o o a a o o o n o
o o o o o o o o o o
c c o o o o o c o.
g o o a c o o o oo
o o c o o . n a o o
....C..Tg.ATCCTAACCT
o. .G...ooo oGo...
oo .GooooooocoGo o.oooonGooooccoo-Go o..ooooGooooo o o o . oo...
o. . . . o . o.o. . . . o o o .0
oooT o - a . noooocoo oo.ccCo. o.noGoooo otvo-oooo
o..T . oo . ..oooC c o o . o.TAC-AAATAT
n o o o o a o o o o
ooCoGC0000ooooGCoooo
o o o . o o a o o o
occ o o o n lo.GATTAAAATT
2688
o o a o o o u o o c
..o .oco o.o........
. o . o a o o o .o
o . . . o o o o .. o - o o oo...
o o c a o . c o o n
c o o o o a o c o o
TTGCCCTTAG
97

X75658X75663X69798X75657X75664J02203Z35716X65258pHB4p5Z35717X70185L13994X02763X5197OV00866M57663X75656X75665D12980M12906XO4615M38454V00867DOO63ODOO33ODOO331DOO329
Consenso
X75658X75663X69798X75657
DOO63ODOO33ODOO331DOO329
Consenso
G oA-o . o o o o o o o o e . . o o . g ..G. .C . . . . oo.G...o.G. . o o o coco o o o o o o aoTo Gac. o ..T c o o . u o .- Coooooco oooooTooGo oC o o o o o.¡(S-a...ch A o o o o o o o o o c o c o ¡no o o o o o c o y .’.GoooooTo- A . o o o ' o o . o o c o o o o .oTA . o o o o . u a c.oTooGooTcl o o . o o o a o o o o o o o o g ocA. ¡oC o a . a . oooTcoGooToo o g o o u o o . . o o o o o o o .vo noc o o o o o o.oTooG-oToo o o o u o u o c o . o o o . . o ooA- occ o o o c o o.oooooooAco cocacoooao nooo-aooTo .G........o. ogoovoo «cono-ooo. oaoooaocT. .G. ooooooc-Aoo 0.00000... ooo-ooooTo .Gaooooco.cooooovoo o......... oo-oooTo oGoo-ooooooooooooA. 0000000000 e. o .T. oG-ooo-.oa. oooovo. coo-ooo... ¡choco-otro ¡Gone-¡octoooooovo .ooooocoao oooaooooT. .Gouaooaou. . oonooGoo vooooo oooooTc-To o......’-o.T . o . . a . o u o o o o u c . . . o o o c oaTonA. .coC o o o a oco o o o o o o o o o o o c u a o o o o c y o o o o u 00A. ¡GAC-oooao
.T . . o o g a o . o o o . c c o . o o o o o ooTovo ¡oC o o o c o.
.T o o o o o o o o o o o o o . o a o o o . o coTooAa oC o o o o oo
c . c o o c o a o o o o o oooGoo oooctT oAT ooCooooooo o o o o o o o o o o o o o cvo- cooocTovo o 0Cooooooo o a a o o u o n o n . o o ooGooo oooooTovo 0CooloooTu-o-oTo occ-oo . o. .T..A. .oc oc. o..T.....Go CoooooG. o o c n o o ooGT o a o o o o o o o.
oTooiovoo a o o o a o o . o o o o o a o o ooG. o o o o o . o o o.cTcooocToo C o c o o a o . o . o o o o o . ooGo o o o a o o o o aoACAAAGGCAT TAAACCTTAT TATCCAGA-C ATGTAGTTAA
2739o o o o o o o a o . a o o o o ¡.G. ooTnAo oooooGovoo o o o o o o o o o o o o o o oaGo ocTvooo oooooGooAa
A u o o u o o n o. o o o o o ouGo ooTvoo oooooGooAva. aoan. oo. .C.....-.. .CooA. ..oo.G..C.. o. . . oo.o. .C........ o. c..A-. ........GConoooToo o oCo . o o o oo. C... 0A. oooooGooooa o a a o o o o a o o o o o o o o o no CoooooAaoo oooooGo o.nooooToo coo-ccoo. Co 0A. oooO-Go...
o a a o o o o o o o o a o o o o o o o o o o o o o o . o o o o o o ooGoaoo
ooooooCooo ¡tooo-.00. 0.000000 o oouoooooGoA. ¡AouG o o o o . o o . o o o o c g ooG. o . n o o o o ..CoAoo oTooGo o o o o o osG. o o o u ooGo o o n . . o o o.A. .TooG o o o o a ooo-o oGo G. o . o o o . occ.Ao. .T-oG a o o o o o o o o . o o o o ooGo o . . o a . oocoA. cTooGo . o o o o o . . . o o c v c ooGo o o o o o a occ.A. .oTooG oo ooooo a. oaoocoGo oooooGooC.A oT-oGo o o o o o n ‘ o o o o o c o ooGo o o . . o o oocoA....T..G o . o . o o o o y o o o o o ooGo . o o o o ooocu
oGaoGC... o o a a a o o. C o q o a o o o . o o o . ooGooGooGooGoo o o o o o o o o o o o o o o o o o o o o . o o .oGo. ..G..T.... . . . . . ..G. G
CAAACCAGAC ATTATTTACA TACTCTTTGG AAGGCTGGTA
2738. . . . ..T..T. . . . ..T..T. . . . . . . ..T
. . . . . . . ..T
. . . . ..T.
TCATTACTTC
2788
..T.. . ..
.CT . . . . . ..A . . . . . . . ...AT . . . . . ..CT . . . . . ..
.AT . . . . . ..
.CT . . . . . ..G . . . . . . . ..
CT . . . . . ...CT . . . . . ...CT . . . . . ..TTCTATATAA

X75658X75663X69798X75657X75664J02203235716X65258pHB4p5Z35717X70185L13994X02763X5197OVOO866M57663X75656X75665D1298OM12906XO4615M38454V00867D00630DOO33ODOO331DOO329
Consenso
X75658X75663X69798X75657
M38454V00867D00630D0033ODOO331DOO329
Consenso
789 2838. .oc...T.C.o........ .......... .o........ .........a. oooaoooC ..-...... .o...... o.......A u...C o........T.C . . o . a o a . . . . . o . o o . o . . . o a o . . . o . o . . .oc....
A . . o o o o .aT . . o o o . . . . . . . . . . . . a . . o . o o u o . .oT . . o . . . . a .oA . . . . . o ooT o . . . a o o . . o . . o o o . o . .. .... a o . o . . . a a . ... ..o. o-ooooA .A . . . . . . o o a .... o .. . . o o o o . o o . o c . . . a.. . . . .o. ..A .A..... ........-. ......... ........... .ooo...A .oA . . o . o o . a . u . . . . o . o . .... . s . . . o . . ... .Goo ..C an. .A. o . C. . . . a . a .o o . o n a o . o ..
.Goaoooc o o . o o . o . o. .A.. . . Co. . . . o o . o . . o o .o....¡aoGon .C . . . a o . . o .. .A . . . o . o .- C . . . c . . o .. o . . o n . o o o....G.....C c. . o. o.o. .A.. .... Co. o. . . . . . . . . oooo...C.G.....C o a . . o . .o. .A . o . o . . .. C . . . o . . . o. .. o . a a . o.
.. .o....C o o o o . o n . .. .A . . . . o . .. C . u . . . o .o. . a . . u . a o o.000G... oc a . . . . . . . o. .A . o . o o . .. C o o . . . . . o. . o u . o . .....G.....T . . o . . o . . . . . . . . o . o o .. C . a a . . . o o . o . . . . o . . ..A. . o . . . .oT o....C. T. ... ..... . . oo . . . . o. o. oo. . . o..coco. . . ..T o..--C.... .g. .a.... .o........ .o........A. o . ooo.oT .....C.... .T.... . . . . . oo . . oo oo oo. . . .o.A o o . a . . .oT -.C. .. .T. o o c c . . o o o a . . o a . . . o o o a . o............T c....C.... a . . oc a . ooo o. . . . o. o. o . s oo. . o. o.o. . . . ooooT G..C.... .......... .o........ .o........o.oo.....T o... Co... ....... .......... .o.........o......-T a . c . . . ..T. o. . oo.a... ... . . . oo. o . . oc . . . . ..A. o. C o oo. o. . .o..... C.... o. . . o . o. uo. ooooA.....G.. o o c . o . . . o . o . . o . . a . o. C . c a . o . a . . . o a . o o . o o.A.....GT.A a... . . . . . . c . . oo . a oo C. . . o . . .- -.. oa o . . .GAGAGAAAC- ACACGTAGCG CCTCATTTTG TGGGTCACCA TATTCTTGGG
2839 2888. o. o. . o . . . oooo. . . o . o . . .CGA.AA oo .GG. . . oo
o. . . o . o . . o . o . . ooo. o o . o .CGAoAA . . .GG. a . . oo o o o o o o o o o a o . o . o . o o . .CGAoA. . .G o o . . o .
.o. onnoo. .oTo. o. o. .CT.GG A.GGTC.... .Ta___. oooo. . . o. .T. .... .CToGG A.GGTC.....T.___o. n. . ..T o. . .. oooo. .... ...-..... o. . o.oT. .o......... . . . . . o o. . o o o . . o oo ... o .aA o o o o a o o o o. ¡C o . o o o o .o. . . .u. . ... .o....... .........A .....-.-.. .C. oa...oo. oooo. . o . o. oooo... o .o. .A .......... .C......... . . . . . . . . . . . . . o..... .c.......A .......... .C.......
. o o o o a . o . . . . o o o . . . . o o o a ... .A u a . . . o . o o. ¡C o . o . . o no. oooo. o. oa o. . oao. . . . a . o.... ¡A .......... .C.......ao. . . aaao. .o. ___ o. .C........noa. oo. . o. .T...... o o... A .......... .C......... . . o. o. oo. oooo. o . oo. . o. ... ... .C....... .G.. o.... o . . o a a . . . . . . o . . . o . . . . o . a . o . . . . o . . . . o o o. .G........
c o o oo o o . o o . . o . o o. o. occ... ..C oo o o o .. ..C..-..... o. oooooo. .G..- o. ........ a .oco...... .ocoo....... o . o . . . o o o o o . o . . . . o o . . . . o occ ..C o . o o o o. .C . . . a . o.. . . o o o a o o . . o o . a o o o o o o . . . . a . . .. ..C....... .Coooooo.o o o o . o . o o . . o o o a . . o . o o o . o o o o... ..C....... .ocoo...... o o o a . o o o o . o . . . . . a . . . . . o o . o o .. ..C. o... .Cn......o.o.. ..T.. .o........ .o........ ..C...-... ........... . ooo. o. o. ......... .........c .ocoo..... .C.......
ooc... . o . . . oo . . . .. . . . o.aT.. .o........ o......a..AACAAGAGCT ACAGCATGGG AGGTTGGTCT TCAAAACCTC GAAAAGGCAT
99

1002889 2920X75658 ...ACA.... c.....G. .... .A.. .X75663 ...ACA.... ........G. . ..A. ..X69798 ...ACA.... ..C.....G. . . . . . . . . .. .X75657 .....A.... A....CACCA. . . . . ..... ..X75664 .....A.... A....CACCA . . . . . . . . .. ..J02203 ——___A.... .....CACCA G. . . . . . . .. ..Z35716 ——___A.... .....CACCA G. . . . . . . .. ..X65258 ——___A......A..CACCAg......... ..
0000000000.o o. oooooo -...C..... o.Z35717.......... .. . . . . . . .. ....C..... ..X70185 . . . . . . . . . . . . . . . . . . .. ....C..... ..L13994 . . . . . . . . . . . . . . . . . . .. ....C..... ..X02763 . . . . . . . . . . . . . . . . . . . . . . . . . . . . .. ..X5197o . . . . . . . . . . . . . . . . . . .. ....C..... ..vo0866 .......... . . . . . . . . .. ....C..... ..M57663 . . . . . . . ..C . . . . . . . . .. ....C..... ..X75656 .......... . . . . . . . . . . . . . . . . . . .. ..X75665 . . . . . . . . . . . . . ..G..A. . . . . . . . . .. ..D12980 .......... . . . . . . . . . . . . . . . . . . .. ..M12906 . . . . . . . . . . . . . . . . . . .. .......... ..X04615 . . . . . . . . . . . . . . . . . . . . . . . . . . . . .. ..M38454 .......... . . . . . . . . . . . . . . . . . . .. ..V00867 ... . . . . . .. .. o . . . . . . . . . . . . . . . .. ..D00630 . . . . . . . . . . . . . . . ..... .......... ..Doo330 . . . . ..A... . . . . . . ..C. .......... ..D00331 . . . . ..A... .....C..C. . . . . . . . . .. ..D00329......A... ......C ,.
Consenso GGGGACGAATCTTTCTGTTC CCAATCCTCT GG
Comparación de la secuencia del extremo 5' del inserto en el _“clon pHB4con las secuencias descriptas en el Anexo 1. ' cSe destaca la posición correspondiente al ATGde la zona preSl. {'x‘

m¿a
firm¡#1L-EDendogramadel alineamiento anterior
X75658
X75663
x69798
X75657
x75664
j02203235716
x65258
phb4p5
235717
X70185
113994
x02763
x51970
V00866
m57663
x75656
x75665
d12980
m12906
x04615
m38454
V00867
d00630
d00330
d00331
d00329
10]

102
es importante ya que nos alerta sobre la posibilidad de sacar
conclusiones apresuradas analizando sólo una parte del genoma.
Dadoque la identificación de regiones de secuencia alta
mente conservada facilita el diseño de primers, tanto para la
detección del HBVcomopara el secuenciamiento de zonas de in
terés; en la figura IV.18 se muestra el gráfico de puntaje de
similitud de los alineamientos vistos en las figuras anteriores.
Estas gráficas confirman que la zona correspondiente al
extremo carboxi-terminal de TP y al espaciador de Pol es la
más variable (puntaje promedio = 0,845), aunque con fragmentos
de alta conservación de secuencia, comopor ejemplo, entre los
nt 2820 y 2850, región útil para diseñar un primer que permita
el secuenciamiento de preSl a partir de cualquier genotipo.

ms;I
Isso 1150 |89° ‘9°° ¡ono
SimilarityScore
O a,
lll
O O
SinilaricyScore
O ¡b
l
3190 '0 ‘ "°Position
UO8C
Figura IV.18: Gráficos de puntaje de similitud
A: correspode al alineamiento de la fig. IV.15; B: al de la
fig. IV.17 y C: al de la fig. IV.14.Nótese la diferencia de escala.

104
3. Utilización de uno de los recombinantes comosonda
El estudio de los marcadores virales convencionales en el
suero, genera información valiosa para conocer el estado ac
tual, predecir la evolución y establecer un pronóstico. Inclu
sive, en forma indirecta, permite conocer la actividad replicativa del virus en el huésped.
La posibilidad de disponer de sondas que permitan detec
tar el ácido nucleico de HBVen suero, otros líquidos biológi
cos o células es muyimportante en cuadros crónicos (persis
tentes), y/o el control de intentos terapéuticos donde laspruebas serológicas no permiten establecer una interpretacióndefinitiva.
En nuestro caso decidimos utilizar el clon portador del
inserto mayor (pHB7)como sonda para monitorear un panel de
sueros de pacientes crónicos, otro de pacientes en fase aguda
y aguda reciente, y sueros control, en todos los cuales se ha
bían realizado las determinaciones serológicas clásicas, a fin
de determinar la especificidad, sensibilidad y las mejorescondiciones para el ensayo.
3.1. Determinación de la especificidad de 1a sonda
Se utilizó la técnica de hibridación en puntos (dot-blot)
usando comocontroles negativos sueros de personas sanas, de
pacientes con otras patologías (HAV,HCV,CMV)y buffer; y
comocontroles positivos los provistos por un ensayo comercial
(Genosticm de ABBOTT)utilizado como indica el productor, es
decir 100 pl de una solución de 103 i 10 pg/ml y sueros de
pacientes en etapa aguda.

m5
Las condiciones de hibridación utilizadas fueron: 1x SSC
a una temperatura de 65°C, y los lavados se realizaron con
0,1x SSCa 62°C; lo cual equivale a trabajar aproximadamente aTm-lO.
3.2. Determinación de la sensibilidad
Para determinar la sensibilidad, también mediante la téc
nica de dot-blot, se utilizaron diluciones de cantidades cono
cidas de pHB7en sueros normales y controles comerciales
(Genosticm de ABBOTT) con concentraciones conocidas de DNA.
Las condiciones de hibridación utilizadas fueron las mis
mas que para el ensayo anterior.
En la figura IV.19 se muestra el resultado de la hibri
dación del filtro, donde se ve que la sonda no da señales
inespecïficas con ninguno de los sueros negativos. Ademásse
observa que la cantidad mínima detectada fue la menor de las
concentraciones probadas, o sea, 1 pg lo que equivale a 3 x
10S genomas virales en los 100 ul de suero probados.
Esto está dentro del rango comunmentedetectado por
equipos comerciales y de la concentración estimada de 104-109
viriones/ 100 ul de suero, medida por microscopía electrónica(Robinson, 1986).
Una mayor sensibilidad se logra mediante el uso de la
técnica de PCR. Sin embargo, según un programa de control de
calidad establecido por el European Group for Rapid Viral
Diagnosis and European Expert Group on Viral Hepatitis, con el
objeto de monitorear los métodos de detección para el HBV,de
los laboratorios participantes, que utilizaron la PCR,sólo un

106
12345A.Ba
Figura IV.19: Utilización de pHB7como sonda
Los sueros tratados según materiales y métodos, fueron fijados
a la membrana de NCsegún el siguiente diagrama:
A1: control comercial positivo, lOpg
B1: dilución 1/10 del anteriorA2 a A5: sueros de individuos sanos
B2 a B4: sueros de individuos con otras patologías
B5: control comercial negativo
C1 a C5: dilución de pHB7 en suero, 1, 10, 20, 50 y lOOpg
D1 a D3: sueros de individuos en etapa aguda
D4: buffer
D5: pHB7 en buffer
La hibridación se realizó a Tm-lOcon la sonda marcada con
[nEfldCTP. Tiempo de exposición : 36 hs.

107
2396mostraron resultados correctos en todo el panel de mues
tras, mientras que el resto falló en especificidad y/o sensibilidad. Concluyendoasí que la interpretación de los resulta
dos de la PCRdebe hacerse cautelosamente y no es un test
aplicable por todos los laboratorios (Quint et al., 1995).
Dentro del marco de desarrollo de la sonda, aplicable a
equipos diagnósticos de detección de DNAdel HBV,se estudió
un panel de 22 sueros, provenientes de pacientes con distintos
tipos de patologías; fundamentalmenteindividuos en etapa
temprana de la enfermedad o que cursaban evoluciones crónicas;
y que cubrían un amplio margen de concentraciones de HBV-DNA.
En la tabla IV.1, ademásde los resultados obtenidos, se
explicitan datos de otros marcadores clásicos. Todos los
pacientes eran antiHBc-IgG +.
El resultado comparativo de la sonda pHB7con el reactivo
de Abbott, que también es el utilizado por Quint et al. como
standard, reveló una coincidencia del 100%u
Datos integrales correspondientes a los estudios en los
que se incluyó el control de calidad y uso de la sonda para
diagnóstico han sido comunicadospreviamente (González et al.,
1990; Fernández Cobo et al., 1990).
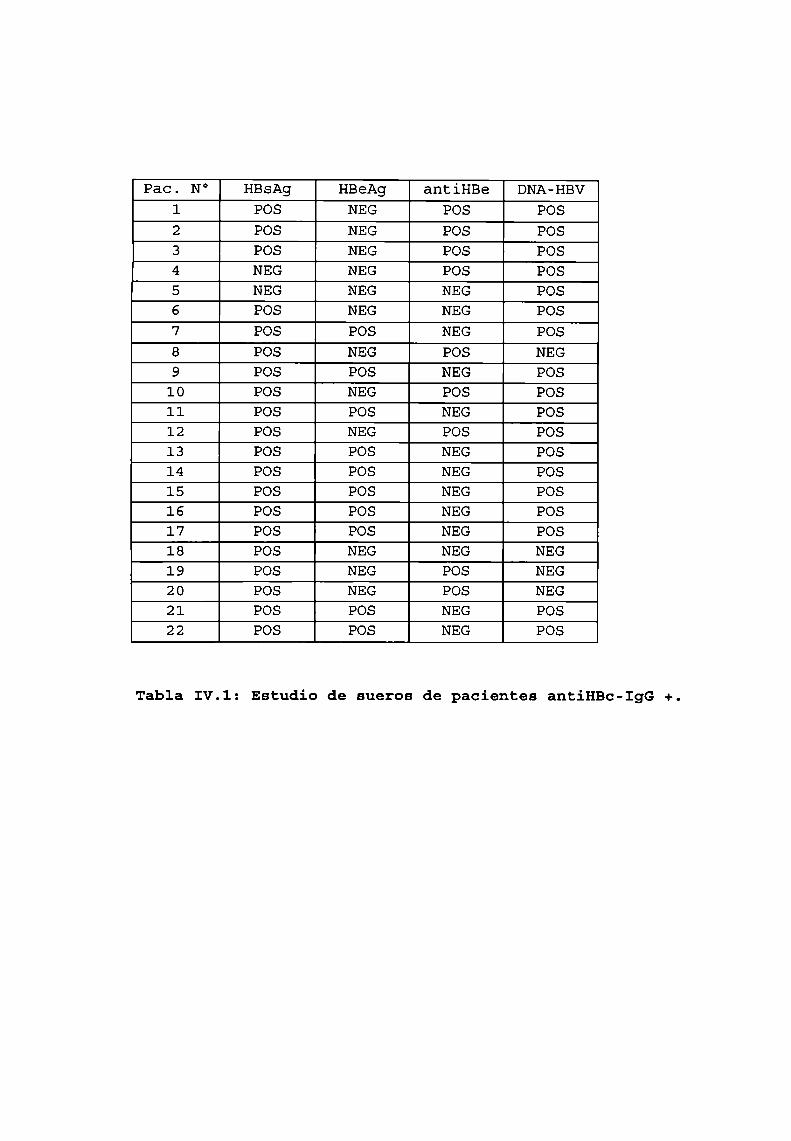
Pac. N° HBsAg HBeAg antiHBe DNA-HBV1 POS NEG POS POS
2 POS NEG POS POS3 POS NEG POS POS
4 NEG NEG POS POS
5 NEG NEG NEG POS
6 POS NEG NEG POS
7 POS POS NEG POS
8 POS NEG POS NEG
9 POS POS NEG POS10 POS NEG POS POS11 POS POS NEG POS12 POS NEG POS POS13 POS POS NEG POS14 POS POS NEG POS15 POS POS NEG POS16 POS POS NEG POS17 POS POS NEG POS18 POS NEG NEG NEG19 POS NEG POS NEG20 POS NEG POS NEG21 POS POS NEG POS22 POS POS NEG POS
Tabla IV.1: Estudio de sueros de pacientes antiHBe-IgG +.

m9
4. Subclonado en vectores de expresión
La obtención de HBcrecombinante es de suma utilidad para
el desarrollo de reactivos para el diagnóstico de la infección
por HBV,al utilizarlo comoantígeno de captura en técnicas de
enzimoinmunoensayopara la detección de anticuerpos anti-HBc.
4.1. Estrategia de clonado en vectores de expresión
El esquema general seguido para la obtención de recombi
nantes fue el siguiente: (Fig.IV.20).
Se decidió aislar el gen core a partir del clon pHB7uti
lizando para ello la enzima StyI que reconoce la secuencia
CiCWWGG(W= A o T). Al cortar este clon con dicha enzima uno
de los fragmentos que se obtiene, como se ve en la calle N°5
de la figura IV.7, tiene 581 pb (del nt 1884 al 2465) y com
prende el gen core completo más los 18 nt previos y los 9 nt
posteriores. Este fragmentose aisló por electroelución delgel.
Los extremos 3’ recesivos pueden ser completados por la
actividad de polimerasa del fragmento Klenow de la DNApolime
rasa I de E. coli en presencia de los dNTPsapropiados; de
este modo, mediante la reacción de "fill in" o rellenado se
obtuvo el fragmento de interés con los extremos romos.
A su vez, el vector de expresión pEX3se cortó con la en
zima SmaI, que deja extremos romos y tiene un único sitio de
reconocimiento en el polylinker. Posteriormente, se trató conla fosfatasa alcalina bacteriana (BAP)con el fin de remover
el fosfato en 5', y así, optimizar la ligación con el inserto

110
restricción
con StyI
restricción
P SP con maIP
P
P
Pel V
g 1>
electroelución P
P
EAPP
fill-in
P
P
ligasa
plásmido recombinante
Figura IV.20: Estrategia de clonado para expresión

pHB7
StyI
pEX3..C»LCTTGGA.
..GGAACTCT.
SmaI
.GAATTAATTCCCJ,GGGGATCCGI
CTTAATTAAGGGCCCCTAGGCfill-inI GluLeuIleProGlyAspPro.i
TTGCTTGGA.
CGAACCT.
Proteínadefusión
core
....GAATTAATTCCCCTTGGATGGCTTTGGGGCfATG......
CTTAATTAAGGGGAACCTACCGAAACCCCGTAC...... GluLeuIleProLeuGlyTLpLeuTrpGly.Mét..
TGTTAGTATTCCTTGGGGGATCCG...... ACAATCATAAGGAACCCCCTAGGC......
...Lisend
FiguraIV.21:Proteínadefusiónobtenida
H1

112
evitando la recircularización del vector; ya que, en este sistema el único marcadorpara seleccionar es la resistencia al
antibiótico ampicilina, y en la placa no se distinguen las colonias portadoras de recombinantes de las portadoras del vec
tor religado.La ligación se llevó a cabo según el protocolo para ex
tremos romos que se detalla en materiales y métodos (III.5.2).
En la figura IV.21 se ve la secuencia y la proteína de
fusión que se generaría, observándose que se regenera el sitio
de restricción con StyI y, que la proteína resultante mantiene
los últimos seis aminoácidos correspondientes al precore
4.2. Transformación
La cepa de E. coli utilizada fue la pop2136 que presentó
una eficiencia de 2 x 105 transformantes por pg de DNA.
La estrategia para la confirmación de las colonias recom
binantes, fue comoen el caso del clonado, la hibridación de
colonias utilizando como sonda una fracción del mismo DNApu
rificado que se pretende clonar. Para ello las recombinantes
más una colonia control portadora del vector, se repicaron por
duplicado a placas de agar con ampicilina.
Unade las placas se transfirió a un filtro que se hi
bridó con el mismo fragmento de pHB7electroeluido, marcado
con digoxigenina (DIG-dUTP)(Fig. IV.22). Comopuede obser
varse, excepto dos de las colonias (N°5 y 50), todas las de
más eran portadoras del inserto requerido.

113
17. BJ! 5-g|12qj1o unsal-¡1415m!" 18mm
4 i py l
24‘22 2a 29‘26 ae ’¡mlm27 50 31 31 33 ¿a ‘35 iJC
aviu'm ¿o ü! 42 m 44.
Í 45 ha 41 4€ ¡#9 50‘ 57 YI ¿255 54:15.;¡56 54
l ¿Í ¿9160 lei‘ Y I
A
Figura IV.22: Detección de recombinantes
A : esquema, B autorradiografïa
- Las colonias transferidas al filtro se hibridaron con el
fragmento de DNAmarcado con digoxigenina en condiciones de
alta exigencia (Tm —5).
- No se observa reacción cruzada con el vector que corresponde
a las posiciones 59 y 60.

114
5. Selección y análisis de expresión
Algunas de estas colonias recombinantes, y el control, se
repicaron a caldo, para realizar una minipreparación, y ana
lizarlas por restricción y expresión.El resultado del gel de agarosa con la restricción de la
minipreparación de plásmidos (datos no mostrados); demostró
que la enzima StyI libera, en el recombinante, un fragmento de
aproximadamente 580 pb como esperábamos.
Unode los clones recombinantes y el control, fueron in
cubados a 30 °C hasta alcanzar la fase logaritmica (DOSW=
0,4-0,5) y luego a 42 °C durante 90 minutos a fin de inducir
la expresión de la proteína de fusión. En la figura IV.23 seobserva el resultado del análisis, medianteelectroforesis en
gel desnaturalizante de poliacrilamida, de los lisados bacterianos inducidos. Se incluyeron varios controles a fin de com
parar los niveles basales de proteínas en estas condiciones(bacterias sin plásmidos) con la producción de B-galactosidasa
(bacteria portadora del plásmido vector) y la proteína híbrida
core-B-galactosidasa (bacteria portadora del plásmido recombinante). Este análisis demostró que nuestro recombinante pro
duce una proteína del tamaño esperado, aproximadamente 140
kDa.

115
Figura IV.23: Expresión de una proteína core recombinante
Las bacterias recombinantes y las controles fueron
inducidas comose describe en materiales y métodos; y las
proteínas analizadas mediante un gel desnaturalizante de
poliacrilamida al 7.5%)teñido con azul de Coomassie.
Calle 1: clon portador del plásmido recombinante; calle 2:
clon sin ningún plásmido; calle 3: clon portador de plásmidovector sin inserto.


H6
V. CONCLUSIONES GENERALES
Se ha presentado en esta tesis el análisis molecular de
DNAdel virus de la hepatitis B, aislado a partir de plasma de
pacientes.Se purificó DNAa partir de partículas de Dane y clonaron
fragmentos genómicoscorrespondientes al virus de la hepatitisB.
De los clones obtenidos, se seleccionaron tres. Los virus
representados en ellos resultaron pertenecer al genotipo A, lo
que está de acuerdo con algunas de las previsiones para el
área geográfica.
De la comparación de nuestras secuencias con los
"prototipos" de este genotipo A, se desprende que, de las dos
variantes reconocidas por nosotros, una de ellas está más re
lacionada con los aislamientos de USA,mientras que la otra
parece más cercana al virus aislado en Filipinas.De acuerdo a los objetivos planteados se desarrolló la
metodología, y se obtuvo una sonda diagnóstica, que utilizada
adecuadamente, demostró poseer una confiabilidad comparable a
las aprobadas para su comercialización en nuestro país.
En el Dep. de virus del INMfunciona la unidad de hepato
patïas virales y el servicio de Biología Molecular. El grado
de complejidad de los problemas a resolver motivó la necesidad
de encarar los objetivos de esta tesis.Pese a la disponibilidad de equipos comerciales y aunque
no se defina un programa de producción inmediato, existe la

117
necesidad de contar, en el INM,con las bases operativas nece
sarias destinadas a asegurar la realización de estudios genómicos.
El estudio de la factibilidad de producir reactivos paradetección de HBV-DNA,acompañado de un acabado cálculo de cos
tos y de asegurar su continuidad, excede el marco de los obje
tivos de esta tesis, pero del resultado de la mismase generan
todos los elementos para su planteo.
En cuanto a la contribución de este trabajo al conoci
miento de los perfiles genómicosde virus circulantes en el
área geográfica, se puede integrar en una serie de conocimientos actuales
Es evidente, que a pesar de lo altamente evolucionado que
parece estar el HBV,aún tiene la capacidad de seguir diversi
ficándose a través de nuevas mutaciones que no afectan la via
bilidad viral. Algunasde estas variaciones son clínicamente
relevantes, mientras que el resto parecería que sólo revisteimportancia a nivel epidemiológico.
Dentro de las variaciones asociadas a aspectos clínicos
determinados, las más importantes son las ya mencionadas mu
tantes e' y “escape mutants”; las primeras incapaces de sinte
tizar el antígeno e , y las segundas que generan proteínas que
pueden escapar al efecto protector de la inmunidad inducida
por ciertas vacunas.En pacientes crónicos se han detectado mutantes con dele
ciones amino-terminales de presz, acompañadas o no con dele
ciones carboxi-terminales de presl, algunas de las cuales eli

118
minan la región del promotor SPII y sitios de reconocimiento
de células B y T, pero no el sitio de unión de pre81¡ por loque, sin afectar la penetración viral a las células hepáticas,implicarïan una desventaja tanto para la eliminación viral del
organismo (“clearance”) comopara la eficacia de las nuevas
vacunas que contienen las proteínas preS. (Gerkenet al.,
1991; Santantonio et al., 1992; Yamamotoet al., 1994).
En cuanto a las mutantes e-, al no producir el antígeno
HBemodifican la respuesta inmune. La supresión de la expre
sión de esta proteína se produce a través: de mutaciones en el
promotor core que inhiben la transcripción del preC mRNA,o,
de mutaciones en el codón de iniciación, corrimiento del marco
de lectura o la introducción de codones de terminación en la
región precore; dentro de estos últimos ya se mencionó que el
más frecuente es el que transforma el triptofano del codón 28
de preC. A pesar de que aún no se han establecido diferencias,
en las características biológicas, asociadas a los distintosgenotipos o serotipos; estas últimas mutantes HBe-serían
menos frecuentes en los genotipos A (Li et al, 1993) y F ya
que ambos tienen C153que al aparearse con Gun; estabilizarían
a la señal e (Loket al., 1994); por lo tanto en el caso
particular de estos dos genotipos, se especula que la
principal causa de este fenotipo serían las mutaciones en el
promotor (Okamotoet al., 1994).
Estos HBe- se encuentran principalmente en Asia y la zona
del Mediterráneo, áreas en las que predominan los genotipos B,
C y D. En cambio, los genotipos A y F serían predominantes en
América y norte y centro de Europa.

ll9
En nuestro caso el clon secuenciado en esta zona genó
mica, perteneciente al genotipo A, no presenta ningún impedi
mento para una correcta expresión de este antígeno, ya que las
particularidades que presenta no afectarían al promotor coreni a una correcta síntesis de HBe.
Respecto a la vigilancia epidemiológica, su objetivo fun
damental es determinar el patrón de diseminación del virus,
para así, mejorar las estrategias diseñadas para interrumpirla transmisión. En este contexto, las cuestiones más críticas
están basadas en: la relación entre casos y brotes que ocurren
en determinadas regiones, y los patrones de transmisión
locales, regionales y mundiales.
Antes de la aplicación de los métodos moleculares, muchas
de estas cuestiones eran difíciles de abordar. La precisión y
velocidad, con las cuales cualquier métodomolecular puede di
ferenciar cepas, y por lo tanto monitorear la transmisión vi
ral, nos permiten, además, analizar en un marco de contempora
neidad, la evolución genómicadel virus durante la replicación
en un huésped determinado.
Ya que la mayoría de las mutaciones se fijan acumulativa
mente en el genoma, la cadena de transmisión puede ser infe
rida por la localización y tipo de mutación encontrada en los
aislamientos clínicos. Sin embargo, cabe aclarar, que la evo
lución vertiginosa de las metodologías aplicables a los estu
dios genómicos, genera una seria discusión acerca de la elección de una técnica determinada cuando se llevan a cabo estos
estudios; al respecto podemosdecir que existen tres líneas
principales:

l20
a) clonado directo: el DNAviral se clona directamente, para
ello se necesita una cantidad considerable de virus presente
en el material de partida. Esta fue la técnica elegida por no
sotros, ya que, en nuestras condiciones esta metodología era
posible y disponïamos del plasma necesario. La ventaja del
método es que no hay ningún tipo de selección previa, generán
dose así clones de todas las variantes genómicas presentes en
la muestra; si bien en este trabajo no se analizaron todos los
clones obtenidos (sólo tres de algo más de veinte clones
establecidos), hemospodido detectar diferencias genómicas que
muestran la presencia de, por lo menos, dos tipos diferentes.
b) PCRy secuenciado y/o restricción directa del producto:
este métodotiene la ventaja de su sensibilidad que le permite
amplificar cantidades mínimas de HBV-DNA.Sin embargo, en los
casos estudiados exhaustivamente, se ha detectado un número
considerable de variantes genómicascocirculando, en distinta
proporción, en la misma muestra (Yamanakaet al., 1990; Liang
et al., 1994); por lo tanto, es muyprobable que la PCRdi
recta, estaría favoreciendo la amplificación de sólo la va
riante predominante. En el caso de aplicarla a un estudio de
cadena de transmisión, funcionará bien si la variante genómica
predominante es realmente la transferida; en caso contrario,
no se pueden sacar conclusiones.
c) PCRy clonado: la alternativa ideal es amplificar por PCR
para aprovechar la sensibilidad de la técnica y clonar el DNA
amplificado, ya que este método, luego de secuenciar un número
significativo de clones, permite tener una visión más exactade todas las variantes presentes en la muestra original.

121
El último paso de las tres estrategias mencionadas, es el
secuenciamiento genómico, ya que éste es el método definitivo
para asignar relación genética. Aunquepara simplificar el
análisis de la variabilidad, también se han propuesto otras
técnicas, comopor ejemplo el estudio del polimorfismo de los
fragmentos de restricción (RFLP) (Shih et al., 1991) o el aná
lisis del polimorfismo de 1a conformación de las cadenas sim
ples (SSCP) (Yusof et al., 1994).
La comparación de regiones genómicas seleccionadas (como
por ejemplo zonas reconocidamente polimórficas), que represen
tan tan solo el 396del DNAtotal, aporta información sobre la
relación genética entre virus (Lin et al., 1991b; Uyet al.,
1992), aunque debe tenerse en cuenta que identidad en una re
gión tan pequeña no implica identidad en todo el genoma. Como
ejemplo, nuestra secuencia pHB4p5, que comprende aproximada
mente un 1096del genomaviral en la zona hipervariable (nt
2590 al 2920), resulta coincidente con otra secuencia deposi
tada en los bancos de datos (fig. IV.17), mientras que el otro
extremo del mismo clon, la secuencia pHB4p3, que también in
cluye parte de dicha zona hipervariable, se diferencia clara
mente de la misma secuencia (fig. IV.14).
La hibridación con sondas de DNAes un método rápido para
determinar homologïa de secuencias. Así es posible preparar
dos clases de sondas: a) aquellas que se aparean con secuen
cias genómicas conservadas y por lo tanto hibridan con todas
las cepas de la especie viral, y b) las que se unen a regiones
variables, características de cepas particulares.Los dos métodos (hibridación y secuenciación) son comple

122
mentarios en muchosaspectos. Los “aislamientos” pueden ser
rápidamente tamizados por homología de secuencias utilizando
las sondas mientras que, el secuenciamiento revela la relaciónevolutiva.
La información de secuencia también sirve para el diseño
de sondas para hibridar, primers para PCRy elección de zonas
adecuadas para la expresión de antígenos útiles, para el diag
nóstico y/o inmunización, de acuerdo a las características locales.
/l EHTO DE TOR ES
no ¡son TITULAR
cneon o: MICROBIOLOGIA

OGRA

H3
VI. BIBLIOGRAFIA
Altschul,S.F., Gish,w., Miller,W., Myers,E.W. &Lipman,D.J.(1990) Basic local alignment search tool. J. Mol. Biol. 215403-410.
Arii,M., Takada,S. &Koike,K. (1992) Identification of threeessential regions of hepatitis B virus Xprotein for trans-activation function. Oncogene7 : 397-403.
Balsano,C., Avantaggiati,M.L., Natoli,G., De Marzio,E.,Elfassi,E., Will,H. &Levrero,M. (1991) Transactivation of cfos and c-myc protooncogenes by both full-length and truncatedversions of the HBV-Xprotein. En Viral Hepatitis and LiverDisease, pp 572. Eds. F.B.Hollinger, S.M.Lemon& H.Margolis.Williams &Wilkins. Baltimore.
Bancroft,W.H., Mundon,F.K. &Russel,P.K. (1972) Detection ofadditional antigenic determinants of hepatitis B antigen. J.Immunol. 109 842-848.
Bartenschlager,R. &Schaller,H. (1988) The amino-terminal domain of the hepadnaviral P-gene encodes the terminal protein(genome-linkedprotein) believe to prime reverse transcription. EMBOJ. 7 : 4185-4192.
Bartenschlager,R., Kuhn,C. &Schaller,H. (1992) Expression ofthe P-protein of the humanhepatitis B virus in a vaccinia virus system and detection of the nucleocapsid-associated P-geneproduct by radiolabelling at newly introduced phosphorylationsites. Nucleic Acids Res. 20 195-202.
Bavand,M., Feitelson,M. &Laub,O. (1989) The hepatitis B virus-associated reverse transcriptase is encodedby the viral

124
pol gene. J. Virol. 63 1019-1021.
Bernard,H.U., Remaut,E., Hershfield,M.V., Das,H.K., Helinski,D.R., Yanofsky,C. & Franklin,N. (1979) Construction of plasmidcloning vehicles that promote gene expression from bacteriophage lambda pL promoter. Gene 5 : 59-76.
Birboim,H.C. &Doly,J.A. (1979) A rapid alcaline extractionprocedure for screening recombinant plasmids. Nucl. Acid Res.7 : 1513-1523.
Blum,H.E., Stowring,L., Figus,A., Montgomery,C.K., Haase,A.T.,Vyas,G.N. (1983) Detection of hepatitis B DNAin hepatocytes,bile duct epithelium, and vascular elements by in situ hybridization. Proc. Natl. Acad. Sci. (USA)80 6685-6688.
B1umberg,B,S., Alter,H.J. &Visnich,S. (1965) A “new” antigenin leukemia sera. J. Am. Med. Assoc. 191 541-546.
Carman,W.F. & Thomas,H.C. (1992) Genetic variation in hepatitis B virus. Gastroenterology 102 711-719.
Colgrove,R., Simon,G. &Ganem,D. (1989) Trans-activation ofhomologous and heterologous genes by hepatitis B virus X protein in cells permissive for viral replication. J. Virol. 634019-4026.
Couroucé,A.M., Holland,P.V., Muller,J.Y. &Soulier,J.P. (1976)HBsantigen subtypes. Bibliotheca Haematologica 42 1.
Couroucé-Pauty,A.M., Lemaire,J.M. & Roux,J.F. (1978) Newhepatitis B surface antigen subtypes inside the ad category. VoxSanguinis 35 304-308.

D5
Couroucé-Pauty,A.M., Plancon,A. &Soulier,J.P. (1983) Distribution of HBsAgsubtypes in the world. Vox Sang. 44 : 197-211.
Chang,L.J., Pryciak,P., Ganem,D. &Varmus,H.E. (1989) Biosynthesis of the reverse transcriptase of hepatitis B virus involves de novo translational initiation not ribosomalframeshifting. Nature 337 : 364-368.
Chen,H.S., Kaneko,S, Girones,R., Anderson,R.W., Hornbuckle,W.E., Tennant,B.C., Cote,P.J., Gerin,J.L., Purcell,R.H. &Miller,R.H. (1993) The woodchuckhepatitis virus X gene isimportant for establishment of virus infection in woodchucks.J. Virol. 67 : 1218-1226.
Choo,Q-L., Kuo,G., Weiner,A.J., Overby,L.R., Bradley,D.w. &Houghton,M. (1989) Isolation of a cDNAclone derived fromblood-borne nonA-nonBviral hepatitis genome. Science 244359-362.
Churchill,M.E.A. &Travers,A.A. (1991) Protein motifs thatrecognize structural features of DNA.Trends Biochem. Sci. 16
92-97.
Dane,D.S., Cameron,C.H. and Briggs,M. (1970) Virus-like particles in serumof patient with Australia-antigen associatedhepatitis. Lancet 1 697-698.
Deka,N., Sharma,M.D. &Mukerjee,R. (1994) Isolation of thenovel agent from humanstool samples that is associated withsporadic Non-A, Non-Bhepatitis. J. Virol. 68 7810-7815.
Delius,H., Gough,N.M., Cameron,C.H. & Murray,K. (1983) Structure of the hepatitis B virus genome. J. Virol. 47 337-343.

D6
Eckhardt,S.G., Milich,D.R. &McLachlan,A. (1991) Hepatitis Bvirus core antigen has two nuclear localization sequences inthe arginine-rich carboxyl terminus. J. Virol. 65 575-582.
Fagan,E.A. (1992) Hepatitis A to G and beyond. Br. J. Hosp.Med. 47 127-131.
Fagan,E.A., Ellis,D.S., Tovey,G.M., Portmann,B., Williams,R. &Zuckerman,A.J. (1989) Toga-like virus as a cause of fulminanthepatitis attributed to sporadic non-A, non-Bhepatitis. J.Med. Virol. 28 150-155.
Fernández Cobo,M., González,J. & Hosokawa,R. (1990) Obtenciónde una sonda molecular de HBVy su aplicación en la determinación de infectividad. ResumenN° 48 del III Congreso Argentino de Virología.
Gallina,A., Benelli,F., Zentilin,L., Rindi,G., Muttini,M. &Milanesi,G. (1989) A recombinant hepatitis B core antigenpolypeptide with the protamine-like domaindeleted self-assembles into capsid particles but fails to bind nucleic acids. J.Virol. 63 : 4645-4652.
Gerhardt,E. &Bruss,V. (1995) Phenotypic mixing of rodent butnot avian hepadnavirus surface proteins into humanhepatitis Bvirus particles. J. Virol. 69 1201-1208.
Gerken,G., Kremsdorf,D., Capel,F., Petit,M.A., Dauguet,C.,Manns,M.P., Meyer zum Buschenfelde,K.H. & Brechot,C. (1991)Hepatitis B defective virus with rearrangements in the preSgene during chronic HBVinfection. Virology 183 555-565.
Gerlach,K.K. & Schloemer,R.H. (1992) Hepatitis B virus C genepromoter is under negative regulation. Virology 189 59-66.

H7
Gerlich,W.H. &Heermann,K.H. (1991) Functions of hepatitis Bvirus proteins and virus assembly. En Viral Hepatitis andLiver Disease, pp 121. Eds. F.B.Hollinger, S.M.Lemon&H.Margolis. Williams &Wilkins. Baltimore.
González,J., Banchero,A., Fernández Cobo,M., Alonso,A.,Otegui,L. &Calviño,M. (1990) Marcador serológico alternativoen la determinación de la replicación viral en pacientes conhepatitis B crónica. ResumenN° 15 del III Congreso Argentinode Virología.
Gripon,P., Diot,C., Theze,N., Fourel,I., Loreal,O., Brechot,C.&Guguen-Guillouzo,C. (1988) Hepatitis B virus infection ofadult humanhepatocytes cultured in the presence of dimethylsulfoxide. J. Virol. 62 : 4136-4143.
Hames,B.D. &Higgins,S.J. eds. (1985) Nucleic acid hybridisation: a practical approach. IRL Press. Oxford
Hanahan,D. (1983) Studies on transformation of Escherichiacoli with plasmids. J. Mol. Biol. 166 557-580.
Hanahan,D., Jessee,J. &Bloom,F. (1991) Plasmid transformationof E.coli and other bacteria. En Methods in enzimology Vol.204 : 63-113.
Haymerle,H., Herz,J., Bressan,G.M., Frank,R. &Stanley,K.K.(1986) Efficient construction of a DNAlibrary in plasmid expression vectors using an adaptor strategy. Nucl. Acids Res.14 8615
Hirsch,R.C., Lavine,J.E., Chang,L.J., Varmus,H.E. &Ganem,D.(1990) Polymerase gene products of hepatitis B viruses are required for genomic RNApackaging as well as for reverse tran

ns
scription. Nature 344 552-555.
Junker-Niepmann,M., Bartenschlager,R. & Schaller H. (1990) Ashort cis-acting sequence is required for hepatitis B viruspregenomeencapsidation and sufficient for packaging of foreing RNA. EMBOJ. 9 3389-3396.
Kaneko,S., Miller,R.H., Feinstone,S.M., Unoura,M., Kobayashi,K., Hattori,N. &Purcell,R.H. (1989) Detection of serumhepatitis B virus DNAin patients with chronic hepatitis usingthe polymerase chain reaction assay. Proc. Natl. Acad. Sci.(USA) 86 312-316.
Kann,M. & Gerlich,W.H. (1994) Effect of core protein phosphorylation by protein kinase C on encapsidation of RNAwithincore particles of hepatitis B virus. J. Virol. 68 : 7993-8000.
Karayiannis,P., Fowler,M.J.F., Lok,A.S.F., Greenfield,C.,Monjardino,J. & Thomas,H.C. (1985) Detection of serum HBVDNAby molecular hybridization: Correlation with HBeAg/anti-HBestatus, racial origin, liver histology and hepatocellular carcinoma. J. Hepatol. 1 99-106.
Kawamoto,S., Yamamoto,S., Ueda,K., Nagahata,T., Chisaka,0. &Matsubara,K. (1990) Translation of hepatitis B virus DNApolymerase from the internal AUGcodon, not from the upstreamAUGcodon for the core protein. Biochem. Biophys. Res. Commun.171 1130-1136.
Kay,A., Mandart,E., Trepo,C. & Galibert,F (1985) The HBVHBxgene expressed in E. coli is recognized by sera from hepatitispatients. EMBOJ. 4 1287-1292.
Kekulé,A.S., Lauer,U., Weiss,L., Luber,B. &Hofschneider,P.H.(1993) Hepatitis B virus transactivador HBxuses a tumor pro

129
moter signalling pathway. Nature 361 742-745.
Kim,Y-H, Kang,S-K & Lee,Y.I. (1993) Functional anlysis ofhepatitis B virus transactivator X : implication of theleucine zipper-like region and C-terminal seven conservedamino acids in functional regions. Biochem. Biophys. Res.Commun. 197 : 894-903.
Kremsdorf,D., Thiers,V., Garreau,F., Nakajima,E., Chappey,C.,Schellekens,H., Wands,J.R., Sninsky,J., Tiollais,P. &Brechot,C. (1991) Nucleotide sequence analysis of hepatitis B virusgenomesisolated from serologically negative patients. En Viral Hepatitis and Liver Disease, pp 222. Eds. F.B.Hollinger,S.M.Lemon & H.Margolis. Williams & Wilkins. Baltimore.
Laemmli, U.K. (1970). Cleavage of structural proteins duringthe assembly of the head of bacteriophage T4. Nature 227680-685.
Lavine,J.E., Hirsch,R.C. &Ganem,D. (1989) A system for studying the selective encapsidation of hepadnavirus RNA.J. Virol.63 : 4257-4263.
Le Bouvier,G.L. (1971) The heterogeneity of Australia antigen.J. Infec. Dis. 123 : 671-675.
Levrero,M., Jean-Jean,0., Balsano,C., Will,H. &Perricaudet,M.(1990) Hepatitis B virus (HBV)X gene expression in humancells and anti-HBx antibodies detection in chronic HBVinfection. Virology 174 : 299-304.
Li,J-S., Tong,S-P., Wen,Y-M.,Vitvitski,L., Zhang,Q. &Trépo,C. (1993) Hepatitis B virus genotype A rarely circulatesas an Hbe-minusmutant: possible contribution of a single nu

B0
cleotide in the precore region. J. Virol. 67 5402-5410.
Liang,T.J., Hasegawa,K., Muñoz,S.J., Shapiro,C.N., Yoffe,B.,McMahon,B.J., Feng,C., Bei,H., Alter,M.J. &Dienstag,J.L.(1994) Hepatitis B virus precore mutation and fulminant hepatitis in the United States. A polymerase chain reaction-basedassay for the detection of specific mutation. J. Clin. Invest.93 550-555.
Lin,C.G. &Lo,S.J. (1992) Evidence for involvement of a ribosomal leaky scanning mechanismin the translation of the hepatitis B virus pol gene from the viral pregenome RNA.Virology188 342-352.
Lin,H.J., Lai,C.L., Lau,J.Y.N., Chung,H.T., Lauder,I.J. &Fong,M.W. (1991a) DNAanalysis of a mutant hepatitis B virus(HBV)in four membersof a chinese family: Evidence for intrafamilial spread of infection. En Viral Hepatitis and LiverDisease, pp 207. Eds. F.B.Hollinger, S.M.Lemon&H.Margolis.Williams &Wilkins. Baltimore.
Lin,H.J., Lai,C.L., Lauder,I.J., Wu,P.C., Lau,T.K. & Fong,M.W.(1991b) Application of hepatitis B virus (HBV)DNAsequencepolymorphisms to the study of HBVtransmission. J. Infec. Dis.164 284-288.
Liss,L.R.(1987) NewM13host: DHSaF' competent cells. Focus9(3) 13.
Lo,S.J., Chien,M.L. &Leet,Y.W. (1988) Characteristics of theX gene of hepatitis B virus. Virology 167 289-292.
Lok,A.S.F., Akarca,U. &Greene,S. (1994) Mutations in the precore region of hepatitis B virus serve to enhance the stability of the secondary structure of the pre-genomeencapsidation

B1
signal. Proc. Natl. Acad. Sci. (USA)91 : 4077-4081.
Mack,D.H., Block,W., Nath,N. & Sninsky,J.J. (1988) Hepatitis Bvirus particles contain a polypeptide encodedby the largestopen reading frame: a putative reverse transcriptase. J.Virol.62 z 4786-4790.
Magnius,L.O., Kaplan,L., Vyas,G.N. & Perkins,H.A. (1975) A newvirus specified determinant of hepatitis B surface antigen.Acta Pathol. Microbiol. Scand. 83B 295-297.
Maniatis,T., Fritsch,E.F. &Sambrook,J. (1982) Molecular cloning: a laboratory manual. Ed. Cold Spring Harbor LaboratoryPress, Cold Spring Harbor, NewYork.
Melegari,M., Bruss,V. &Gerlich,W.H. (1991) The arginine-richcarboxy-terminal domain is necessary for RNApackaging byhepatitis B core protein. En Viral Hepatitis and Liver Disease, pp 164. Eds. F.B.Hollinger, S.M.Lemon&H.Margolis.williams &Wilkins. Baltimore.
Miller,R.H. & Robinson,W.S. (1986) Commonevolutionary originof hepatitis B virus and retroviruses. Proc. Natl. Acad. Sci.(USA) 83 : 2531-2535.
Miller,R.H., Kaneko,S., Chung,C.T., Girones,R. &Purcell,R.H.(1989) Compactorganization of the hepatitis B virus genome.Hepatology 9 : 322-327.
Murakami,S., Cheong,J.H., Ohno,S., Matsushima,K. & Kaneko,S.(1994) Transactivation of humanhepatitis B virus X protein,HBx, operates through a mechanismdistinct from protein kinaseC and okadaic acid activation pathways. Virology 199: 243-246.

B2
Nassal,M. &Schaller,H. (1993) Hepatitis B virus replication.Trends Microbiol. 1 : 221-228.
Nassal,M. (1992) The arginine-rich domain of the hepatitis Bvirus core protein is required for pregenomeencapsidation andproductive viral positive-strand DNAsynthesis but not for virus assembly. J. Virol. 66 : 4107-4116.
Nassal,M., Rieger,A. &Steinau,0. (1992) Topological analysisof the hepatitis B virus core particle by cysteine-cysteinecross-linking. J. Mol. Biol. 255 : 1013-1025
Neurath,A.R. & Strick,N. (1990) Antigenic mimicry of an immunoglobulin A epitope by a hepatitis B virus cell attachmentsite. Virology 178 : 631-634.
Neurath,A.R., Strick,N., Sproul,P., Ralph,H.E. &Valinsky,Y.(1990) Detection of receptors for hepatitis B virus on cellsof extrahepatic origin. Virology 176 : 448-457.
Norder,H., Couroucé,A.M. &Magnius,L.O. (1992a) Molecular basis of hepatitis B virus serotype variations within the fourmajor subtypes. J. Gen. Virol. 73 : 3141-3145.
Norder,H., Couroucé,A.M. & Magnius,L.O. (1994) Completegenomes,phylogenetic relatedness, and structural proteins ofsix strains of the hepatitis B virus, four of which representtwo new genotypes. Virology 198 : 489-503.
Norder,H., Hammas,B., Lófdahl,S., Couroucé,A.M. & Magnius,L.O.(1992b) Comparison of the amino acid sequences of nine different serotypes of hepatitis B surface antigen and genomic classification of the correspondinghepatitis B virus strains. J.Gen. Virol. 73 : 1201-1208.

B3
Ochiya,T., Tsurimoto,T., Ueda,K., Okubo,K., Shiozawa,M. &Matsubara,K. (1989) An in vitro system for infection withhepatitis B virus that uses primary humanfetal hepatocytes.Proc. Natl. Acad. Sci. USA86 : 1875-1879.
Ohnuma,H., Machida,A., Okamoto,H., Tsuda,F., Sakamoto,M.,Tanaka,T., Miyakawa,Y. &Mayumi,M. (1993) Allelic subtypic determinants of hepatitis B surface antigen (i and t) that aredistinct from d/y or w/r. J. Virol. 67 927-932.
Okamoto,H., Tsuda,F., Akahane,Y, Sugai,Y, Yoshiba,M.,Moriyama,K., Tanaka,T., Miyakawa,Y. & Mayumi,M. (1994) Hepatitis B virus with mutations in the core promoter for an e antigen-negative phenotype in carriers with antibody to e antigen.J. Virol. 68 8102-8110.
Okamoto,H., Tsuda,F., Sakugawa,H., Sastrosoewignjo,R.,Imai,M., Miyakawa,Y. & Mayumi,M. (1988) Typing hepatitis B virus by homologyin nucleotide sequence: comparison of surfaceantigen subtypes. J. Gen. Virol. 69 2575-2583.
Persing,D.H., Varmus,H.E. & Ganem,D. (1987) The preSl proteinof hepatitis B virus is acylated at its aminoterminus withmyristic acid. J. Virol. 61 1672-1677.
Phillips,M.J., Blendis,L.M., Poucell,S., Patterson,J.,Petric,M., Roberts,E., Levy,G.A., Superina,R.A., Greig,P.D.,Cameron,R., Langer,B. &Purcell,R.H. (1991) Syncytial giantcell hepatitis. Sporadichepatitis with distinctive pathological features, a severe clinical course, and paramyxoviral features. N. Engl. J. Med. 324 : 455-460
Quint,W.G., Heijtink,R.H., Schirm,J., Gerlich,w.H. &Niesters,H.G.M. (1995) Reliability of methods for hepatitis B virus DNA

B4
detection. J. Clin. Microbiol. 33 : 225-228.
Radziwill,G., Tucker,W. &Schaller,H. (1990) Mutational analysis of the hepatitis B virus P gene product: Domainstructureand RNaseHactivity. J. Virol. 64 613-620.
Reyes,G.R., Purdy,M.A., Kim,J.P., Luk,K-C., Young,L.M., Fry,K.E. & Bradley,D.W. (1990) Isolation of a cDNAfrom the virusresponsible for enterically transmitted non-A, non-Bhepatitis. Science 247 1335-1339.
Rihs,H-P. &Peters,R. (1989) Nuclear transport kinetics dependon phosphorylation-site-containing sequences flanking thekaryophilic signal of the simian virus 40 T-antigen. EMBOJ. 8
1479-1484.
Robinson,W.S. (1986) Hepatitis B virus. En Fundamental Virology, pp 657. Ed.B.N.Fields & D.M.Knipe. NewYork, Raven Press
Robinson,W.S., Clayton,D.N. & Greenman,R.L. (1974) DNAof ahumanhepatitis B virus candidate. J. Virol. 14 : 384-391.
Robinson,W.S., Wu,J. &Twu,J. (1991) Transcriptional activation of homologousand heterologous genes by hepatitis B virus. En Viral Hepatitis and Liver Disease, pp 149. Eds. F.B.Hollinger, S.M.Lemon&H.Margolis. Williams &Wilkins.Baltimore.
Rodríguez Crespo,I., GómezGutiérrez,J., Nieto,M., Peterson,D.L. &Gavilanes,F. (1994) Prediction of a putative fusionpeptide in the S protein of hepatitis B virus. J. Gen. Virol.75 : 637-639.
Rossner,M.T. (1992) Review: Hepatitis B virus X-gene product:A promiscuous transcriptional activador. J. Med. Virol. 36

BS
101-117.
Roychoudhury,S., Faruqi,A.F. & Shih,C. (1991) Pregenomic RNAencapsidation analysis of eleven missense and nonsense polymerase mutants of humanhepatitis B virus. J. Virol. 653617-3624.
Sanger,F., Nicklen,S. & Coulson,A.R. (1977) DNAsequencingwith chain-terminating inhibitors. Proc. Natl. Acad. Sci.(USA) 74 5463-5467.
Santantonio,T., Jung,M.C., Schneider,R., Fernholz,D., Milella,M., Monno,L., Pastore,G., Pape,G.R. &Will,H. (1992) HepatitisB genomesthat cannot synthesize pre-S2 proteins occur frequently and as dominantvirus populations in chronic carriersin Italy. Virology 188 948-952.
Sattler,F. &Robinson,W.S. (1979) Hepatitis B viral DNAmolecules have cohesive ends. J. Virol. 32 : 226-233.
Schaller,H. &Fischer,M. (1991) Transcriptional control ofhepadnavirus gene expression. Curr. Top. Microbiol. Immunol.168 21-39.
Schodel,F., Peterson,D., Zheng,J., Jones,J.E., Hughes,J.L. &Milich,D.R. (1993) Structure of hepatitis B virus core and eantigen. A single precore amino acid prevents nucleocapsid assembly. J. Biol. Chem. 268 1332-1337.
Seeger,C. &Maragos,J. (1989) Molecular analysis of the function of direct repeats and a polypurine tract for plus-strandDNApriming in woodchuckhepatitis virus.J.Virol.6321907-1915.
Seelig,R., Weisser,C.F., Zentraf,H.W., Bottner,C., Seelig,H.P.& Renz,M. (1992) The use of PCRfor epidemiological studies of

B6
HNANBviruses in arthropod vectors. En PCR: clinical diagnostics and research. pp A32. Ed. A.Rolss, I.Schuller, U.Sinckh &I.Weber-Rolss. Springer Verlag. Germany.
Sells,M.A., Chen,M&Acs,G. (1987) Production of hepatitis Bvirus particles in HepG2cells transfected with cloned hepatitis B virus DNA.Proc. Natl. Acad. Sci. USA84 1005-1009.
Shih,J.W., Cheung,L.C., Alter,H.J., Lee,L.M. &Gu,J.R. (1991)Strain analysis of hepatitis B virus on the basis of restriction endonuclease analysis of polymerase chain reaction products. J. Clin. Microbiol. 28 1640-1644.
Shikata,T., Karasawa,T., Abe,K., Uzawa,T., Suzuki,H., Oda,T.,Imai,M., Mayumi,M.&Mortisugu,Y. (1977) Hepatitis B e antigenand infectivity of hepatitis B virus.J.Infec.Dis. 1362571-576.
Stanley,K.K., Luzio,J.P. (1984) Construction of a new familyof high efficiency bacterial expression vectors: identification of cDNAclones coding for human liver proteins. EMBOJ. 3
1429-1434.
Summers,J. & Mason,W.S. (1982) Replication of the genome of ahepatitis B-like virus by reverse transcription of an RNAintermediate. Cell 29 : 403-415.
Sureau,C., Romet-Lemonne,J., Mullins,J.I. &Essex,M. (1986)Production of hepatitis B virus by a differentiated humanhepatoma cell line after transfection with cloned circular HBVDNA. Cell 47 : 37-47.
Suzuki,T., Masui,N., Kajino,K., Saito,I. &Miyamura,T. (1989)Detection and mapping of spliced RNAfrom a human hepatomacell line transfected with the hepatitis B virus genome.Proc.

B7
Natl. Acad. Sci. (USA) 86 8422-8426.
Tagawa,M., Omata,M. & Marion,P.L. (1990) Open reading frameson plus strand genomeof duck hepatitis B virus. Gastroenterol-Jpn 25 Suppl.2 : 20-22.
Takada,S. &Koike,K. (1990) X protein of hepatitis B virus resembles a serine protease inhibitor. Jpn. J. Cancer Res. 811191-1194.
Takada,S., Kido,H., Fukutomi,A., Mori,T. & Koike,K. (1994) Interaction of hepatitis B virus Xprotein with a serine protease, tryptase TL2as an inhibitor. Oncogene 9 341-348.
Tsurimoto,T., Fujiyama,A. &Matsubara,K. (1987) Stable expression and replication of hepatitis B virus genomein an integrated state in a humanhepatomacell line transfected withthe cloned viral DNA.Proc. Natl. Acad. Sci. USA84 444-448.
Uy,A., Wunderlich,G., Olsen,D.B., Heermann,K-H., Gerlich,W.H.& Thomssen,R. (1992) Genomicvariability in the preSl regionand determination of routes of transmission of hepatitis B virus. J. Virol. 73 3005-3009.
Wang,G-H& Seeger,C. (1993) Novel mechanism for reverse transcription in hepatitis B viruses. J.Virol. 67 : 6507-6512.
Wang,J.T., Wang,T.H., Sheu,J.C., Shih,L.N., Lin,J.T. &Chen,D.S. (1991) Detection of hepatitis B virus DNAby polymerasechain reaction in plasma of volunteer blood donors negativefor hepatitis B surface antigen. J. Infec. Dis. 163 397-399.
Weber,M., Bronsema,V., Bartos,H., Bosserhoff,A.,Bartenschlager,R. &Schaller,H. (1994) Hepadnavirus P proteinutilizes a tyrosine residue in the TP domainto prime reverse

BS
transcription. J. Virol. 68 : 2994-2999.
Woodcock,D.M.,Crowther,P.J., Doherty,J., Jefferson,S., DeCruz,E., Noyer-Weidner,M., Smith,S.S., Michael,M.Z. & Graham,M.W.(1989) Quantitative evaluation of Escherichia coli hoststrains for tolerance to cytosine methylation in plasmid andphage recombinants. Nucleic Acid Res. 17 3469-3478.
Wu,H.L., Chen,P.J., Tu,S.J., Lin,M.H., Lai,M.Y. & Chen,D.S.(1991) Characterization and genetic analysis of alternativelySpliced transcripts of hepatitis B virus in infected humanliver tissues and transfected HepG2cells. J. Virol. 651680-1686.
Wu,J-Y., Zhou Z.Y., Judd,A., Cartwright,C. & Robinson W.S.(1990) The hepatitis B virus encoded transcriptional transactivation hbx is a novel protein serine/threonine kinase. Cell63 : 687-695.
Yaginuma,K., Shirakata,Y., Kobayashi,M. & Koike,K. (1987)Hepatitis B virus (HBV)particles are produced in a cell culture system by transient expression of transfected HBVDNA.Proc. Natl. Acad. Sci. USA84 : 2678-2682.
Yamamoto,K.,Horikita,M., Tsuda,F., Itoh,K., Akahane,Y.,Yotsumoto,S., Okamoto,H., Miyakawa,Y. & Mayumi,M. (1994) Naturally occurring escape mutants of hepatitis B virus with various mutations in the S gene in carriers seropositive for antibody to hepatitis B surface antigen. J. Virol. 68 2671-2676.
Yamanaka,T., Akahane,Y., Susuki,H., Okamoto,H., Tsuda,F.,Miyakawa,Y &Mayumi,M. (1990) Hepatitis B surface antigen particles with all four subtypic determinants: point mutations ofhepatitis B virus DNAinducing phenotypic changes or doubleinfection with viruses of different subtypes. Mol. Immunol.27

B9
443-449.
Yeh,C-T., Liaw,Y-F. &Ou,J-H. (1990) The arginine-rich domainof hepatitis B virus precore and core proteins contains a signal for nuclear transport. J. Virol. 64 6141-6147
Yen,T.S.B., Mitchell,P.J. &Seto,E. (1991) Mechanismof transcriptional trans-activation by the hepatitis B virus Xprotein. En Viral Hepatitis and Liver Disease, pp 186. Eds.F.B.Hollinger, S.M.Lemon&H.Margolis. Williams &Wilkins.Baltimore.
Yuh,C-H. & Ting,L-P. (1991) Cooperation of two elements withinthe second enhancer of the humanhepatitis B viral genome. EnViral Hepatitis and Liver Disease, pp 176. Eds. F.B.Hollinger,S.M.Lemon &H.Margolis. Williams &Wilkins. Baltimore.
Yusof,J.H.M., Flower,A.J.E. &Teo,C.G. (1994) Transmission ofhepatitis B virus analysed by conformation-dependent polymorphisms of single-stranded viral DNA.J. Infec. Dis. 169:62—67.


N°acceso
M57663 V00866
02763 00331 00329 00330 12906 00867 75656 75665 04615 02203 75657 75664 75658
N O O D Z > N x N b N N N x75663
serotipo
adwadwz adw2
adwadwz adwz
adr adradrq adrq’
ayraYW3 aYW4 aYW4
adW4q‘ adw4q'
genotipo
A A m m U U U U U D LI]
N°nt.3221 3200 3221 3215 3215 3215 3215 3188 3215 3215 3215 3182 3212 3212 3215 3215
origen
Filipinas
USA USA
Indonesia
Japón Japón Japón Japón
Polinesia
Nva.Caledonia
Japón
Francia
W.Africa
Senegal FranciaColombia
locus
HPBADWZCG
HBVADW
HBVADW2 HPBADW3 HPBADWl HPBADW2 HPBADRA
HBVADR
HHVCCHA
HHVBC
HEHBVAYR
HPBAYW
HHVBBAS
HHVBE4
HHVBFFOU
HHVBF
TABLAconlasprincipalescaracterísticasdelassecuenciasutilizadas
clon
pFDW294 pHBV933
pHBV3200
pIDW420 pJDW233 pODW282 pHBVl-l pHBr330
CHA HMA
pYRB259
ECOHBVDNA
BAS KOU FOU9203
cita
7 8
Anexo
MO

Q EI O p-"l N >< N NN.E.:noespecificado
12980 38454 00630 13994 51970 70185 35717 69798 65258 35716
adr adr adw adw adwadw2 adw2 adW4
aYWaYW4
Estacioetal.
(1988)J.
Ono(1983)NucleicAcids Valenzuelaetal. Okamotoetal.
(1980)
(1988)J.
Kobayashi&Koike(1984) Norderetal.
3215N.E.HPBCGSRADR9 3221N.E.HPBADRlCG10 3215N.E.HPBADRC5 3221N.E.HUMPRECX11 3221N.E.HVHEPBHBV99112 3221N.E.HBVXCPS13 3221PoloniaHBVGEN2PHB61414 3215BrasilHBVADW4A15 3182N.E.HBVAYWCI16 3182PoloniaHBVGENlPHB32114
Gastroenterol.Hepatol.3215-222 Res.111747-1757 enAnimalVirusGenetics57-70,AcademicPress,N.Y. Gen.Virol.692575-2583 Gene30:227-232
(1994)Virology198:489-503
Ml

7.-Okamotoetal.(1986)J.Gen.Virol.67:2305-2314 8.-Galibertetal.(1979)Nature281:646-650 9.-Mukaideetal.(1992)NucleicAcidsRes.20:6105 10.-Renbaoetal.(1987)Sci.Sin.30:507-521 11.-Meiseleta1.(1993)sinpublicar 12.-Koechel.H(1990)sinpublicar 13.-Preisler-Adamsetal.(1993)NucleicAcidsRes.21:2258 14.-Plucienniczak(1994)sinpublicar 15.-Naumannetal.(1993)J.Gen.Virol.74:1627-1632 16.-Laietal.(1992)sinpublicar


















