
Alzheimer gaixotasunarekin erlazionaturiko proteinen ultraegitura … · 2015-11-09 ·...
206
Alzheimer gaixotasunarekin erlazionaturiko proteinen ultraegitura mailako kokapena baldintza fisiologikotan: APP eta PS1 arratoi helduen hipokanpoan Jakintza-arloa: Medikuntza Egilea: NAROA ANABITARTE GONZALEZ Urtea: 2009 Zuzendariak: IZASKUN ELEZGARAI GABANTXO, PEDRO R. GRANDES MORENO Unibertsitatea: UPV-EHU ISBN: 978-84-8438-326-0
Transcript of Alzheimer gaixotasunarekin erlazionaturiko proteinen ultraegitura … · 2015-11-09 ·...

Alzheimer gaixotasunarekin erlazionaturiko proteinen ultraegitura mailako kokapena baldintza fisiologikotan: APP eta PS1 arratoi helduen hipokanpoan
Jakintza-arloa: Medikuntza
Egilea: NAROA ANABITARTE GONZALEZ Urtea: 2009 Zuzendariak: IZASKUN ELEZGARAI GABANTXO, PEDRO R. GRANDES MORENO Unibertsitatea: UPV-EHU ISBN: 978-84-8438-326-0

Hitzaurrea Tesi hau 2003ko urritik 2009ko ekainera bitartean egin zen UPV/EHUko neurozientzien sailean. 2009ko ekainaren 19an izan zen tesiaren defentsa. Biokimikako ikasketen azken urtea Norvegiako laborategi batean eman nuen, eta han piztu zitzaidan hain zuzen ere neurozientziekiko interesa. Horren ondorioz, Euskal Herrian gai horretan zebiltzanekin harremanetan jarri, eta haiek aholkatu zidaten tesia egitea. Horrela, bereziki mikroskopio elektronikoaz baliatuz, eta beste teknika batzuekin ikerketa osatuz, APP eta PS1 izeneko proteinen ultraegitura mailako kokapena ikertu genuen arratoi helduen hipokanpoko neuronetan. Bi proteina horiek Alzheimer gaixotasunean zeresan handia dutenez, garrantzitsua iruditu zitzaigun baldintza fisiologikoetan haien kokapenaren berri izatea. Interesgarria izan daiteke lortutako emaitza horiei jarraipen bat ematea aztertutako kokapen hori animali gaixoen neuronetako kokapenarekin alderatu eta ondorioak atereaz; alegia, Alzheimer gaixotasunaren modeloa betetzen duten animalien neuronetan eta baldintza fisiologikoetan proteina horien kokapena berdina den edo ez begiratu daiteke. Beste aukera bat, APPren proteolisian parte hartzen duten beste entzimen kokapena teknika berdinekin aztertzea izango litzateke. Hauek adibide batzuk besterik ez dira, tesi ororekin gertatzen den bezala, bide baten zati txiki bat besterik ez da izan, eta oraindik bide asko daude ikerketa lerroarekin jarraitu ahal izateko. Hala ere, tesiaren bukaera iritsi zenetik, tesia egin zen laborategian beste proiektu batzuek zuten lehentasuna zela eta denboraldi batez etenaldia izan zuen ikerketa lerro horrek. Gainera, beste lan eta ikasketa batzuetan murgildu naiz ordutik aurrera, eta beraz, ez dut gai horren jarraipen zuzena egin ordutik.
Naroa Anabitarte
2010

Alzheimer gaixotasunarekin erlazionatutako proteinen
ultraegitura mailako kokapena baldintza fisiologikoetan:
APP eta PS1 arratoi helduen hipokanpoko neuronetan
Naroa Anabitarte Gonzalez
Leioa, 2009ko ekainaren 19a
Doktore-tesi honen zuzendariak
Pedro R. Grandes Moreno
Izaskun Elezgarai Gabantxo
Neurozientzien Saila
Medikuntza eta Odontologia Fakultatea
Euskal Herriko Unibertsitatea


Tesiaren amaiera
ailegatu zaigu berez
urte hauen laburpena
ezin da azaldu errez
Gazteluan ta familian
bete naute esker onez
ezin ahaztu inolaz
zuzendari ta lankideez
Eskerrak gehiegi dira
hori nola esan hitzez
bertsotan saiatuko naiz
beste modu batzuen ordez
zoriontsu izan naiz eta
nahi dizuet esan zinez:
ondoan egon zaretenoi
milesker bihotz-bihotzez
Aita eta amari, emandako guztiagatik.


Anai-arrebak, entzun ene aho-otsa: izaite bat ez daike hezur hutsez osa;
herria da gorputza, hizkuntza bihotza; bertzetik berextean bitarik bakotxa, izaite horrendako segurra hil hotza.
Xalbador
Bizitza herriaren oroimena da, historiaren katearen jarraipena izatearen talde-kontzientzia, pentsatzeko eta bizitzeko modua.
Milan Kundera


LABURDUREN ZERRENDA
ABC: abidina biotina konplexua
ACh: azetilkolina
ADAM: disintegrina eta
metaloproteasa domeinudun proteinen
sendia
AFS: Izozte bidezko ordezkapen
automatikoa
AICD: APPren zelula barneko
domeinua (ingelesezko APP
intracellular domain)
AMPA: azido α-amino-3-hydroxy-5-
methyl-4-isoxazolepropioniko
hartzailea
APH-1: Aph-1 fenotipoarekin
erlazionatutako proteina (ingelesezko
Anterior Pharnyx defective 1)
APP: amiloide peptidoaren proteina
aitzindaria (Ingelesezko Amyloid
Precursor Protein)
APLP: APPren antzeko proteinak
APLP1: APPren antzeko 1 proteina
APLP2: APPren antzeko 2 proteina
βA: β-amiloide peptidoa
BACE1: β-gunean APP mozten duen 1
entzima
BACE2: β-gunean APP mozten duen 2
entzima
BSA: behi sero-albumina
C83: α-sekretasaren mozketatik
eratorritako APPren mintzeko zatia
C99: β-sekretasaren mozketatik
eratorritako APPren mintzeko zatia
CA1, CA2, CA3: ammon-en adarraren
guneak
CAAT: zenbait generen transkripzio-
gunearen aurreko nukleotidoen sekuentzia
CaMK: kalmodulina kinasa
cAMP: AMP ziklikoa
cdk-5: ziklinaren menpeko kinasa
C/EBP: CAAT kutxarekin elkarrekintzak
dituen transkripzio faktore sendia
cGMP: GMP ziklikoa
CREB: cAMPren erantzunerako
elementuak kodetzen dituen DNA zatiari
lotzen zaion transkripzio faktorea
CTF: karboxi muturreko zatia
CTFα: α-sekretasaren mozketatik
eratorritako APPren karboxi muturreko
zatia
CTFβ: β-sekretasaren mozketatik
eratorritako APPren karboxi muturreko
zatia
CTFγ: γ-sekretasaren mozketatik
eratorritako APPren karboxi muturreko
zatia
DAB: 3,3´diaminobentzidina
DAG: diazil glizerol
DISLL: dileuzina-talde domeinu azidoa
DPX: ehuna portetan montatzeko medioa
DTT: ditiotreitol
ECL: kimioluminiszentzia-areagotzailea
EE: erretikulu endoplasmatikoa
EGF: hazkuntza-faktore epidermala
ERK MAPK: seinale transdukzio bide bat

EPSC: korronte postsinaptiko
kitzikatzailea
Fe65: proteina adaptatzaileen sendia
GABA: azido gamma-amino butirikoa
GABAA: GABAren hartzaile
ionotropikoa
GABAB: GABAren hartzaile
metabotropikoa
GC: guanilato ziklasa
GFAPε:
GGA: Golgin kokatzen diren eta γ-
“belarria” duten ADP erribosilazio-
faktoreari lotzen zaizkion proteinen
familia
GluR1: AMPA hartzailearen
azpiunitatea
GluR2: AMPA hartzailearen
azpiunitatea
GPCR:
GTP: guanosina 5'-trifosfatoa
5-HT2a: serotoninaren hartzailea
5-HT2c: serotoninaren hartzailea
HRP: errefau peroxidasa (ingelesezko
horseradish peroxidase)
HS: heparan sulfatoa
HZ: hortz-zirkunboluzioa (gyrus
dentatus)
I-CLiP: mintz barruan mozten duten
proteasak (ingelesezko Intramembrane
Cleaving Proteases)
IDE: intsulina suntsitzen duen entzima
(ingelesezko insulin degrading enzyme)
IP3: inositol tri-fosfatoa
LRP: dentsitate baxuko lipoproteinen
hartzailearekin erlazionatutako proteina
LTD: epe luzeko depresioa
LTP: epe luzeko potentziazioa
(ingelesezko long term potentiation)
mACh: azetilkolinaren hartzaile
muskarinikoak
MAPK: MAP kinasa
ME: mikroskopio elektronikoa
MENA: zelulen mugikortasunean
eragiteko gai diren proteina-sendia
mGluR1: glutamatoaren 1 motako
hartzaile metabotropikoa
mRNA (edo RNAm): RNA mezularia
MVB: xixku anitzdun gorputzak
nACh: azetilkolinaren hartzaile
nikotinikoak
NCT: nikastrina
NGS: ahuntzaren sero normala
NHS: zaldiaren sero normala
NMDA: azido N-metil D-aspartikoa
NO: oxido nitrikoa
NR1: NMDA hartzaileen azpiunitatea
NTF: amino muturreko zatia
NY: Y neuropeptidoa
p3: α- eta γ-sekretasaen mozketatik
eratorritako APPren peptidoa
p38 MAPK: MAP kinasa
PAGE: poliakrilamidazko gel
elektroforesia
PAR-4: prostatako apoptosirako 4
erantzun-proteina (ingelesezko prostate
apoptosis response-4)
PB: fosfato indargetzailea

PBS: fosfato/gatz-indargetzailea
PEN-2: presenilinaren areagotzailea 2
PIV: hesteetako polipeptido
basoaktiboa
PKA: cAMParen menpeko proteina
kinasa
PKC: C kinasa proteina
PKG: G kinasa proteina
PLC: C fosfolipasa
PPSE: potentzial postsinaptiko
kitzikatzailea
PS1: presenilina-1
PS2: presenilina-2
PSD95: dentsitate postsinaptikoko 95
proteina
PVDF: polibinilideno difluoroa
RIP: mintz barneko proteolisi
erregulatua (ingelesezko Regulated
Intramembrane Proteolysis)
RNA: azido erribonukleikoa
S2P: I-CLiP mota bat
SAP97: sinapsiarekin lotutako 97
proteina
sAPPα: α-sekretasaren mozketatik
eratorritako APPren zati disolbagarria
sAPPβ: β-sekretasaren mozketatik
eratorritako APPren zati disolbagarria
SDS: sodio dodezil sulfatoa
SNAP25: sinaptosomekin lotutako
proteina
sorLA/LR11: sortilinarekin
erlazionatutako hartzailea (ingelesezko
sortilin-related receptor)
SPP: seinale peptidoen peptidasa
(ingelesezko signal peptide peptodases)
STM: epe laburreko oroimena
(ingelesezko short term memory)
TACE: TNFα eraldatze-entzima
TBS: tris/gatz-indargetzailea
TBST: tween-a duen tris/gatz
indargetzailea
TEMED: N,N,N,N'-
tetrametiletilenodiamina
TGS: trans golgi sarea
TMD: Transmintz domeinua
WB: western blot


AURKIBIDEA


AURKIBIDEA Orrialdea
1. Sarrera 1
1.1. Oroimena 1
1.2. Hipokanpo egitura 2
1.2.1. Orokortasunak 2 1.2.2. Hipokanpo egitura eta konexioak 4 1.2.3. Hipokanpo egiturako neuronen anatomia kimikoa 6 1.2.4. Hipokanpoko CA1 gunearen konexioak 7 1.2.5. Hipokanpo egiturako azpiguneen berezitasunak oroimena gordetzeko 7
1.3. Sinapsia eta plastizitate sinaptikoa 9
1.3.1. Sinapsi kimikoak 10 1.3.2. Neurotransmisoreak eta hartzaileak 11 1.3.3. Plastizitatea eta oroimena 13
1.3.3.1. Oroimen inplizitua edo ez-deklaratiboa 13 1.3.3.2. Oroimen esplizitua edo deklaratiboa 13
1.4. Amiloide Peptidoaren Proteina Aitzindaria (APP: Amyloid Precursor Protein) 19
1.4.1. Orokortasunak 19 1.4.2. APPren egitura 20 1.4.3. APPren kokapena zelula barruan 21 1.4.4. APP eta sinapsia 21 1.4.5. APPren proteolisia 22
1.4.5.1. Bide ez-amiloidogenikoa 23 1.4.5.2. Bide amiloidogenikoa 24
1.4.6. APP osoa 25 1.4.7. APP eta bere zatien funtzioak sinapsian eta plastizitate sinaptikoan 27
1.4.7.1. sAPPα 27 1.4.7.2. β-amiloide peptidoak 29 1.4.7.3. APPren zelula barruko domeinua (AICD) 35
1.5. APPren proteolisian parte hartzen duten sekretasak 37 1.5.1. α-Sekretasa 37
1.5.1.1. ADAM sendia 38 1.5.1.2. ADAM proteinen egitura 38 1.5.1.3. ADAM proteinak nerbio-sisteman 39 1.5.1.4. ADAM proteinak eta APP 40

1.5.1.5. Substratuak ezagutzeko modua 41 1.5.1.6. α-sekretasa aktibitatearen erregulazioa 41
1.5.2. β-Sekretasa 43
1.5.2.1. Orokortasunak 43 1.5.2.2. Sendia eta egitura 43 1.5.2.3. BACE 1en garapena zelula barruan 45 1.5.2.4. BACE1en funtzioak eta substratuak 46 1.5.2.5. Substratuak Ezagutzeko Modua 47 1.5.2.6.BACE 1en aktibitatearen erregulazioa 47
1.5.3. γ-Sekretasa 48 1.5.3.1. Mintz-Barneko Proteolisi Erregulatuta 48 1.5.3.2. γ-sekretasaren osagaiak 51 1.5.3.3. γ-sekretasa konplexuaren eraketa 57 1.5.3.4. γ-sekretasaren substratuak 57 1.5.3.5. γ-sekretasaren mozketa guneak APPren barnean 60 1.5.3.6. γ-sekretasaren zelula barneko kokapena 61 1.5.3.7. γ-sekretasa eta sinapsia 61
1.6. APP eta bere sekretasen zelula barneko garraioa 63
1.6.1. Sekretasen garraioa eta erregulazioa 63 1.6.1.1. γ-sekretasa 63 1.6.1.2. α-sekretasa 65 1.6.1.3. β-sekretasa 67
1.6.2. APPren zelula barneko garraioa 69
2. Helburuak 73
3. Materialak eta Metodoak 77
3.1. Argi-mikroskopiorako teknika immunozitokimikoak 79
3.1.1. DABren metodoa 79
3.2. Mikroskopio elektronikorako teknika immunozitokimikoak 83
3.2.1. Erretxinan murgildu aurreko immunozitokimika: nanourrearen protokoloa 83 3.2.2. Erretxinan murgildu ondorengo immunozitokimika 89
3.3. Western Blot teknika 97

3.3.1. Hipokanpoaren homogeneizazioa eta proteina kontzentrazioaren neurketa 97 3.3.2. Western Blot (WB): elektroforesia (SDS-PAGE),
transferentzia eta inmunodetekzioa 100
3.4. Sinaptosomak prestatzea 107
3.5. Kontrolak 110
3.5.1. Sagu knockout-ekin kontrolak 110 3.5.2. Bigarren antigorputzaren kontrolak 110
3.6. Kuantifikazioa eta analisi estatistikoa 110
4. Emaitzak 115
4.1. Antigorputzen kontrolak 115
4.1.1. Antigorputz primarioen kontrolak 115 4.1.2. Bigarren antigorputzen kontrolak 120
4.2. Emaitzak 121
4.2.1. Western Blot teknikarekin lortutako emaitzak 121 4.2.2. Argi-mikroskopiaz egindako esperimentuen emaitzak 123 4.2.3. Mikroskopio elektronikoaz eta sinaptosometan egindako esperimentuen emaitzak 124
5. Eztabaida 145
5.1. APP eta PS1 sinapsian kokatzen dira nagusiki 145
5.2. APPren kokapena 149
5.3. PS1en kokapena 152
5.4. APPren proteolisia ematen den eskualdea 153
5.5. Laburbilduz 159
6. Ondorioak 163
7. Bibliografia 167


1. SARRERA


Sarrera
1. SARRERA 1.1. OROIMENA Oroimena da pertsona bat bera dena izatea bermatzen duen mekanismoa. Oroimena
galtzea, norberaren nortasuna galtzea da, eta, azken batean hori, hiltzea bezala da; izan
ere, norberak bere nortasuna galtzean, bere bizitzaren berri ez izateak, ez dio zer den
antzematen ere uzten.
Oroimena garunaren funtzioa da; garunaren funtzio harrigarrienetako bat, gainera. Izan
ere, esperientzia bidez ikasitako informazioa gorde eta informazio hori borondatez
berreskuratu eta erabiltzeko gaitasuna izatea da oroimena. Nerbio-sistemak informazio
berria bereganatzeari ikasketa-prozesu deritzo, eta oroimena, ikasitako informazio hori
kodetu, gorde, eta berreskuratzeari deitzen zaio. Aldi berean, informazioa ahazteko
gaitasuna ere (modu osasuntsuan; ez gaixotasun-egoera batean) garrantzitsua da.
Oroimena deskribatzeko irizpide ezberdinak erabili dituzten sailkapenak daude.
Oroimena gordetzeko denbora irizpidetzat hartuz, adibidez, denbora labur eta luzeko
oroimenak daude. Gogoratzen dugunaren izaerari begiratuz egindako sailkapen
orokorrean, gizakietan, bi oroimen mota deskribatu dira: oroimen deklaratiboa, eta
oroimen ez-deklaratiboa. Lehenengoa kontzientzian gordeta dagoena da, eta hitzez
adieraz daiteke (norberaren iraganeko pasarte bat, adibidez), eta bigarrena, berriz, ez da
kontzienteki gordetzen (gaitasun motorrak adibidez) (Kandel eta lank., 2001).
Oroimena zer den eta nola jarduten duen jakiteko, ikerketa asko egin izan dira garunean.
Horrela, ikusi da edozein oroimen mota gordetzeko mekanismoa oinarri berdinean
finkatzen dela; nerbio-sistemaren plastizitatean. Nerbio-sistema inguruneko estimuluen
arabera egokitzen da erantzunak emateko, eta egokitze-prozesu hori konexio
sinaptikoen kopuru eta indarren aldaketen bidez egiten da. Horri plastizitate sinaptikoa
deritzo, eta edozein oroimen gordetzeko oinarria dela uste da.
Garuneko gune ezberdinetan, hainbat oroimen mota gordetzen dira (hizkuntza baten
ezagutza, ibiltzen jakiteko gaitasuna, norberaren iraganeko pasadizo bat gogoratzeko
gaitasuna….). Gainera, oroimen mota guztiak ez dira garunean zehar edonola gordetzen,
eta gune bakoitzak nolabaiteko “espezializazioa” du. Gizakia ez den beste animalia eta
ugaztunetan, ikusi da hipokanpoa eta beste zenbait egitura (lobulu temporal-medial eta
berarekin loturiko egituretan daudenak) beharrezkoak direla denbora-espazioan
oroimenak gorde eta trinkotzeko. Aldi berean, gizakiak, egitura horiek erabiliko ditu
1
Sarrera
hasiera batean oroimen deklaratiboak kodetzeko, eta, ondoren oroimen horiek
gordetzeko. Epe luzean gordetzen den informazio deklaratiboa badirudi garun
kortexeko bestelako gune espezializatuetan gordetzen dela.
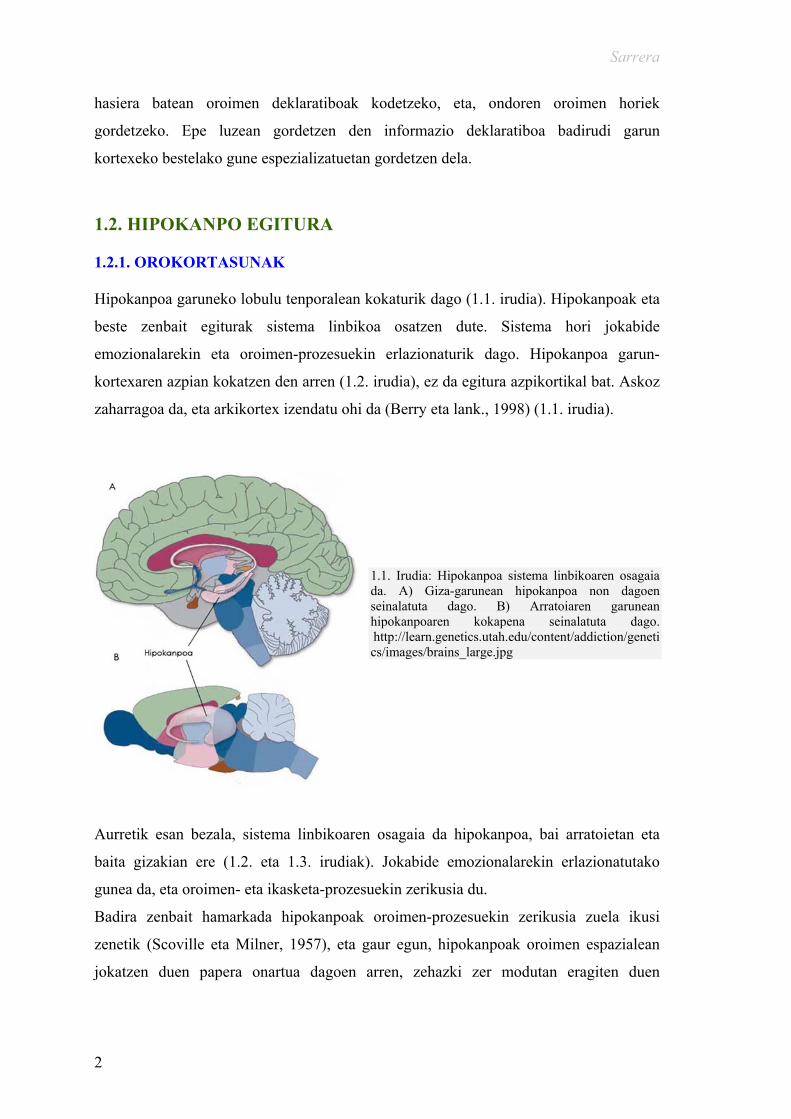
1.2. HIPOKANPO EGITURA 1.2.1. OROKORTASUNAK Hipokanpoa garuneko lobulu tenporalean kokaturik dago (1.1. irudia). Hipokanpoak eta
beste zenbait egiturak sistema linbikoa osatzen dute. Sistema hori jokabide
emozionalarekin eta oroimen-prozesuekin erlazionaturik dago. Hipokanpoa garun-
kortexaren azpian kokatzen den arren (1.2. irudia), ez da egitura azpikortikal bat. Askoz
zaharragoa da, eta arkikortex izendatu ohi da (Berry eta lank., 1998) (1.1. irudia).
1.1. Irudia: Hipokanpoa sistema linbikoaren osagaia da. A) Giza-garunean hipokanpoa non dagoen seinalatuta dago. B) Arratoiaren garunean hipokanpoaren kokapena seinalatuta dago. http://learn.genetics.utah.edu/content/addiction/genetics/images/brains_large.jpg
Aurretik esan bezala, sistema linbikoaren osagaia da hipokanpoa, bai arratoietan eta
baita gizakian ere (1.2. eta 1.3. irudiak). Jokabide emozionalarekin erlazionatutako
gunea da, eta oroimen- eta ikasketa-prozesuekin zerikusia du.
Badira zenbait hamarkada hipokanpoak oroimen-prozesuekin zerikusia zuela ikusi
zenetik (Scoville eta Milner, 1957), eta gaur egun, hipokanpoak oroimen espazialean
jokatzen duen papera onartua dagoen arren, zehazki zer modutan eragiten duen
2

Sarrera
eztabaidagai da oraindik (Rolls eta Kesner, 2006; Smith eta Mizumori, 2006; Diana eta
lank., 2007;).
1.2. Irudia: Arratoiaren eta gizakiaren garunaren ebaketa koronala. Hipokanpoak seinalaturik daude kasu guztietan.
Argi dago, behintzat, hipokanpoa oroimen- eta ikasketa-prozesuen sistema baten zati
garrantzitsua dela. Sistema horren barruan beste hainbat egituraren artean, hipokanpoa
bera inguratzen duten kortexek parte hartzen dute (entorrinala, perirrinala eta
parahipokanpala), baita hipokanpotik proiekzioak jasotzen dituzten zenbait gunek ere
(subikulua, kortex infralinbikoa eta kortex prefrontaleko beste zenbait gune) (Bunsey
eta Eichenbaum, 1993). Gainera, aipatzekoa da Alzheimer gaixotasunean pixkanaka-
pixkanaka nerbio-sistemako hainbat egitura kaltetuak agertzen direla; kaltetutako
egitura horien artean, hipokanpoa hasieratik kaltetua agertzen den gunea da.
1.3. Irudia: Ezkerrean arratoiaren garunaren ebaketa koronala. Bertan, karratuan sartuta, hipokanpo egitura ikusten da. Eskubian, hipokanpo egitura zehaztasun handiagoz. CA1, CA2 eta CA3 guneak markatuta daude, baita hortz-zirkunboluzioa (HZ) ere. Paxinos eta lank., 1999.
3

Sarrera
1.2.2. HIPOKANPO EGITURA ETA BERAREN KONEXIOAK Egitura hau definitzeko irizpide bat baino gehiago erabili dira, eta ondorioz, zenbait
sailkapen mota aurkitu daitezke. Amaral eta Witter-ek erabilitako definizioaren arabera,
hipokanpo formazioa lau egituraz osatuta dago; hipokanpoa, hortz-zirkunboluzioa (HZ),
konplexu subikularra (subikulua, presubikulua eta parasubikulua), eta kortex entorrinala
(Amaral eta Witter, 1995).
Definizio hori hipokanpo formazioa osatzen duten egituren arteko konexioetan
oinarrituta dago. Egitura horiek bata bestearekin konektatuta daude, norantza bakarrean
doazen konexioen zirkuitu bat osatuz (1.4. irudia).
1.4. Irudia: Diagraman hipokanpo formazioko konexio intrintseko nagusiak erakusten dira. Ikusten den bezala, haietako asko (bide zulatzailea (kortex entorrinala→hortz-zirkunboluzioa), zuntz goroldiotsuak (Hortz-
zirkunboluzioa→CA3), Schaffer-en albokideak (CA3→CA1)) norantza bakarreko konexioak dira.
Hortz-zirkunboluzioko zelula nagusiak pikor geruza osatzen duten pikor zelulak dira.
Zelula horiek geruza molekularrean zehar zabaltzen diren dendrita unipolarrak dituzte,
eta dendrita horiek jasotzen dituzte hortz-zirkunboluziora iristen diren proiekzio
aferente gehienak (kortex entorrinaletik batez ere). Pikor zelula horien geruzak
hipokanpoko zelula piramidalen geruzaren zati bat inguratzen du.
Hipokanpoan, zelula piramidalak dira zelula nagusiak eta CA1, CA2 eta CA3 guneetan
banatzen dira, “Ammon-en adarra” deritzona osatuz. Bertan, zenbait geruza bereizten
dira. Geruza nagusia zelula piramidalez osatuta dagoen estratu piramidala izango da.
Geruza piramidalarekiko azal aldera, stratum oriens kokatzen da, bereziki zelula
piramidalen dendrita basalez eta zenbait interneuronen dendritez osatuta dagoena.
Zelula piramidal eta subikuluko axoiek osatzen duten Alveus-a estratu radiatuaren
goialdetik pasatzen dira finbriatik ateratzeko. Estratu luzidoa zuntz goroldiotsuak
CA3ko dendritekin kontaktatzeko estratu piramidalaren eta estratu erradiatuaren artetik
pasatzean sortzen da (gizakian estratu hori ez da hain nabaria, eta CA1 eta CA2
4

Sarrera
guneetan ez da existitzen). Eta geruzarik sakonena estratu erradiatua da. Zelula
piramidalen dendrita apikalak estratu erradiatuan zabaltzen dira. (Berry eta lank., 1998)
(1.5. irudia). Estratu erradiatu horren behealdetik estratu lakunar-molekularra pasatzen
da. Azken horretatik kortex entorrinaletik hortz zirkunbuluzioara doazen zuntzak
pasatzen dira zelula piramidalen dendrita apikal distalekin kontaktua egiten duten
albokideak utziz.
CA3ko zelula piramidalak dira hipokanpoko zelularik handienak. Bestalde, CA3-
CA2ren arteko muga ez da oso muga zehatza. CA2ko zelula piramidalek oso geruza
trinkoa osatzen dute. Eta gune horretara ez dira zuntz goroldiotsuak iristen hortz-
zirkunboluziotik. CA2ra iristen den aferentzia garrantzitsu bat hipotalamoaren gune
supralaminarretik eta tuberomamilarretik dator. Esaten da CA1 gunea hipokanpoko
azpiatalen artean konplexuena dela. CA1-CA2 muga ere ez da oso argia. Zelula
piramidalen geruzaren zabalera 10-30 zelulakoa edo zelula gehiagokoa izaten da.
Geruza horretako zelulen % 10, gutxi gorabehera, interneuronak izaten dira (Berry eta
lank., 1998).
1.5. Irudia: Hipokanpo formazioko zelula motak eta haien arteko konexioak. Geruza bakoitzeko zelulek beste geruzetako zelulekin egiten dituzten konexioak ikusten dira. Gorriz zelula piramidalak CA1, CA2 eta CA3 guneak osatuz. Urdinez pikor geruza. Kahle eta Frotscher, 2008tik moldatua. Kortex entorrinaletik hortz-zirkunboluzioko pikor zeluletara proiekzioak iristen dira, eta
bide horri bide zulatzailea deritzo. Hortz-zirkunboluzioko pikor zelulak, zuntz
goroldiotsuen bidez, CA3ra bidaltzen dituzte proiekzioak; CA3ko zelula piramidaletara.
Proiekzioak CA3tik CA1eko zelula piramidaletara (Schaffer-en albokideak) eta
CA1etik subikulura doaz (1.6. irudia) (Lorente de No, 1934; Amaral, 1978; Amaral eta
Witter 1989; Kahle eta Frotscher, 2008) eta horrek zirkuitua isteko kortex entorrinalera
proiektatzen du. Norantza bakarreko proiekzio horiek ez dira oso ohikoak konexio
kortiko-kortikaletan; normalena konexio erreziprokoak aurkitzea da.
5

Sarrera
Dena dela, aipatutako “sinapsi-zirkuitu hirukoitza” (Hortz-zirkunboluzioa-CA3-CA1)
baino askoz elkarrekintza konplexuagoak daude hipokanpoaren barnean. Alde batetik,
hipokanpoko zelula kitzikatzaileetan efektu inhibitzailea gauzatzen duten interneuronak
daude, eta bestetik, hipokanpoaren aktibitatean, bestelako proiekzio estrinsekoek ere
eragiten dute.
1.6. Irudia: Hipokanpo egituraren “sinapsi-zirkuitu hirukoitza”. Bide zulatzaileak, zuntz goroldiotsuek, Schaffer-en albokideek eta CA1etik kortex entorrinalera itzultzen diren konexioek osatzen dute norantza bakarreko zirkuitu hau. Kandel, 2001. 1.2.3. HIPOKANPO EGITURAKO NEURONEN ANATOMIA KIMIKOA Anatomia kimikoari dagokionez, ikusi da hipokanpo formazioko hiru bideetan
glutamatoa eta/edo aspartatoa direla neurotransmisore kitzikatzaile nagusiak: bide
zulatzailean, zuntz goroldiotsuetan eta Schaffer-en albokideetan. Hortz-zirkunboluzioko
geruza molekularrean, GABA-neurona ugari agertzen dira. Hipokanpoari dagokionez,
stratum oriens geruzan aurkitu ditzakegu GABA-neurona gehienak, nahiz eta zelula
piramidalen geruzan eta geruza erradiatuan ere badauden (Berry eta lank., 1998).
Hipokanpo formazioan, peptidoak dituzten neurona ugari daude. Horrela, hortz-
zirkunboluzioko pikor zelulek dinorfina peptidoa izaten dute, eta baita CA3ko zuntz
goroldiotsuek ere. Geruza entorrinaletik datozen zuntzetan, berriz, entzefalina aurkitu
izan da. Hortz-zirkunboluzioko geruza molekularrean, eta hipokanpoko geruza lakunar-
molekularrean, somatostatina duten zuntz ugari daude. Baita “Ammon-en adarra”-ren
stratum oriens geruzan ere. GABArekiko immunoerreaktibitatea duten neurona askok
badute somatostatinarekiko inmunoerreaktibitatea ere, baita Y neuropeptidoarekiko
(NY) ere. Heste-polipeptido basoaktiboa (PIV) duten neuronak ere asko dira hipokanpo
formazioan (Berry eta lank., 1998).
6

Sarrera
1.2.4. HIPOKANPOKO CA1 GUNEAREN KONEXIOAK CA1 gunearen konexioez arituko gara; izan ere, gune horretako sinapsiak aztertuko
ditugu lan honetan. CA1 gunera sartzen diren proiekzio nagusiak, aurretik aipatu bezala,
CA3tik datozen Schaffer-en albokideak (sinapsi-zirkuitu hirukoitza) eta kortex
entorrinaletik zuzenean (bide zulatzailea) iristen direnak dira.
CA1 guneko konexio intrintsekoen artean, CA1eko axoiek subikulura bidean egiten
dituzten kontaktuak daude, bai CA1 guneko beste zelula piramidalen dendritekin, eta
baita geruza piramidal eta oriens-eko interneuronekin egiten dituztenak ere.
Konexio estrinsekoen artean, nukleo septaletik iristen zaio aferentzia bat (ez da CA3ra
iristen dena bezain bestekoa). Locus coeruleus-etik ere nolabaiteko aferentzia xume bat
iristen zaio (noradrenergikoa) (Swanson eta lank., 1987), eta errafe nukleoetatik
proiekzio serotoninergiko bat (Freund eta lank., 1990). Amigdala-konplexuko nukleo
basolateraletik ateratzen diren axoiak CA1eko estratu lakunoso molekularrera iristen
dira, hipokanpoko fisuratik gertu (Krettek eta Price, 1977). Estratu horretara, proiekzio
handi bat bidaltzen du reuniens nukleoak (Wouterlood eta lank., 1990; Dolleman-Van
der Weel eta Witter, 1992). Gainera, hipokanpo kontralateraleko CA1 gunerantz doan
proiekzio komisural txiki bat ere badago (Van Groen eta Wyss, 1990).
Eferentzien artean, berriz, CA1etik kortex entorrinalaren geruza sakonenetara bidaltzen
dira proiekzio batzuk; hala, zirkuitu bat isten da. Bestalde, subikulu ipsilateralerantz
topografikoki ordenatua doa beste eferentzia bat; CA1eko zelula piramidalen axoiak,
stratum oriens-a zeharkatuz, subikulura iristen dira, eta han adarkatze-prozesu handia
jasaten dute (Amaral eta lank., 1991). CA1etik, CA2 eta CA3tik, nukleo septal
lateralerantz beste proiekzio bat bidaltzen da, baita kortex retrosplenial eta perirrinalera,
Brocaren banda diagonaleko nukleorantz, taenia tecta-rantz, kortex medial
frontalerantz, usaimeneko nukleo lateralerantz, usaimen erraboilerantz, nukleo
accumbenserantz, amigdalako nukleo basaletarantz, eta hipotalamorantz (Jay eta lank.,
1989; Van Groen eta Wyss, 1990; Jay eta Witter, 1991).
1.2.5. HIPOKANPO EGITURAREN AZPIGUNEEN BEREZITASUNAK OROIMENA GORDETZEKO Hipokanpoko gune bakoitza (CA1, CA2, CA3), eta baita hortz-zirkunboluzioa (HZ) ere,
hainbat modutan aritzen da oroimen espazialaren prozesu horietan. Hortz-
zirkunboluzioan (HZ), zenbait lekuren errepresentazioa modu bereizian irudikatzen
7

Sarrera
(gordetzen) da bertako zeluletan. Antzeko lekuei buruzko informazioa gorde behar den
kasuetan, garrantzi handikoa da hortz-zirkunboluzioa. Mekanismo hori ezinbestekoa da
informazioa CA3ra pasatzeko.
CA3 guneko zelula piramidalen sinapsi gehienak gune bereko beste zelulen axoien
albokideen bidez sortutakoak dira. Horregatik, CA3 gunera hainbat lekutatik iristen
diren informazioak elkarrekin lot daitezke. Beraz, hortz-zirkunboluzioko zeluletan
modu bereizian gordetako informazioa CA3ra pasatzen da, eta bertan, informazio hori
beste guneetatik datorkion informazioarekin integratzen da. CA3 gunea oso
garrantzitsua da espazioarekin erlazionatuta dauden oroimen berriak sortzeko. Eratu
berri diren oroimen horiek, hala ere, denbora labur batean besterik ez dira gordetzen
CA3n.
CA1 gunea, berriz, oroimenaren sekuentziak eta denbora parametroa duten oroimenak
elkartzeaz arduratzen da. CA1era iristen den informazioa denboran kokatzeko gaitasuna
du gune horrek, eta gainera, oroimenak trinkotzeko gunea ere bada. CA3tik datorren
informazioa sendotu, eta CA3 gunean baino denbora luzeagoan gordetzen dira
oroimenak bertan.”Erdibideko oroimena” delakoa izango da hori (1.7. irudia).
Horrela, hipokanpoan, bi motatako denbora laburreko oroimen (STM) gordetzen dira;
bata berehalakoa (CA3an), eta bestea laburra (CA1en). CA1 gunetik informazioa
neokortexera itzuliko da berriro ere; beraz, esan liteke hipokanpoa egitura beharrezkoa
dela neokortexeko memoriak sendotzeko. Neokortexean, luzaroago gordetzen dira
oroimenak, eta hipokanpoarekiko menpekotasuna galtzen dute.
Hipokanpoak oroimena prozesatzeko duen modu hori da, hain zuzen, Rolls eta Kesner-
ek proposatzen dutena (Rolls eta Kesner, 2006), eta badira horri buruzko beste zenbait
teoria. Argi dago, dena den, hipokanpoko gune bakoitzaren papera ezberdina dela.
Gainera, CA1 gunearen kasuan, garrantzitsua da jakitea bertatik ateratzen direla
neokortexerantz hipokanpotik irteten diren konexio nagusiak, eta, aurretik aipatu bezala,
oroimen-prozesuetan garrantzizko papera jokatzen dute bertako zelulek; bai informazioa
CA3 gunean baino luzaroago gordez, eta baita neokortexean oroimena sendotzeko
orduan ere.
Hipokanpoa eta neokortexa oroimen-sistema osagarriak dira, eta zirkuitu bat eratzen
dute. Hau da, neokortexeko egitura ugarietatik hipokanpora iristen den informazioa
garatu egiten da hipokanpoan. Hipokanpoko guneetan, informazioa batetik bestera
pasaz, eta gune bakoitzean informazio hori osatuz, hipokanpoan denbora mugatu batez
gorde eta trinkotutako oroimenak neokortexean modu garatu eta sendotuan gordeko
8

Sarrera
dira. Beraz, denborarekin, hipokanpoarekiko menpekotasuna galtzen dute, eta, horrela,
zirkuitua isten da (Rolls eta Kesner, 2006).
1.7. Irudia: Hipokanpo formazioko azpigune bakoitzak oroimena “gordetzeko” dituen berezitasunak. Oroimen mota batzuk trinkotzeko egitura garrantzitsua da hipokanpo egitura; ondoren, neokortexera bidaliko du informazio hori bertan epe oso luzerako gordeta gera dadin. Kandel, 2001. 1.3. SINAPSIA ETA PLASTIZITATE SINAPTIKOA Aurretik aipatu bezala, edozein oroimen mota gordetzeko mekanismoa nerbio-
sistemaren plastizitatean oinarrituta dago. Nerbio-sistema egokitu egiten da inguruneko
estimuluen arabera erantzunak emateko, eta egokitze-prozesu hori konexio sinaptikoen
kopuru eta indarraren aldaketen bidez egiten da. Horri plastizitate sinaptikoa deritzo, eta
uste da edozein oroimen gordetzeko oinarrian dagoela (Purves eta lank., 2008a).
Sinapsiak informazioa neurona batetik bestera pasatzeko guneak dira; ondorioz,
informazioaren transmisioaren oinarria dira. Neuronek, batez beste, 1000 konexio
sinaptiko sortzen dituzte, eta 10000 konexio sinaptiko jaso ditzakete. Kasu batzuetan,
100000raino ere iristen da jaso ditzaketen konexio sinaptikoen kopurua (Kandel eta
Siegelbaum, 2001). Sinapsi horiek elektriko edo kimikoak izan daitezke. Bi kasuetan,
transmisio sinaptikoaren intentsitatea areagotu edo murriztu egin daiteke. Hori da
plastizitate sinaptikoa eta ezinbestekoa da garunak goi-mailako funtzioak betetzeko.
Sinapsi kimikoak elektrikoak baino konplexuagoak dira, eta zelulen erreakzio
konplexuagoak bidera ditzakete. Mota horretako sinapsiak garrantzi handikoak dira
garunaren funtzioak ulertu ahal izateko (De Camilli eta lank., 2001; Purves eta lank.,
2008b).
9

Sarrera
1.3.1. SINAPSI KIMIKOAK Sinapsi kimikoetan, neurona presinaptiko eta postsinaptikoaren artean tarte bat dago,
tarte sinaptikoa (synaptic cleft). Neurona presinaptikoan, normalean axoiaren amaieran
(terminal presinaptikoa), neurotransmisoreak dituzten xixkuak daude. Gune aktibo
deritzen eremuetan pilatzen dira xixku horiek, eta bakoitzak milaka transmisore
molekula ditu bere barnean. Alde presinaptikoan ekintza potentzial bat gertatzen
denean, Ca2+ ioiak sartzen dira zelula presinaptikora, gune aktiboan dauden kaltzio-
kanaletan barrena. Zelula barneko Ca2+ kontzentrazioaren igoera dela eta, xixkuak mintz
presinaptikoarekin lotzen dira, eta neurotransmisorea tarte sinaptikoan askatzen da.
Neurotransmisore horiek zelula postsinaptikoan dauden hartzaile espezifikoekin lotzen
dira. Horrela, zelula postsinaptikoan ioi-kanalak ireki edo itxiko dira, eta zelula
postsinaptikoaren mintz potentzialean aldaketak gertatuko dira. Transmisio sinaptiko
kimikoak “anplifikazio” deritzon ezaugarri garrantzitsua du; izan ere, xixku bakar batek
askatzean, milaka neurotransmisore molekula askatzen dituenez, horrek zelula
postsinaptikoan milaka ioi-kanalen irekiera bultza dezake. Horrela, korronte elektriko
ahul bat besterik sortzerik ez duen terminal txiki bat gai izan daiteke zelula
postsinaptiko handi bat despolarizatzeko (1.8. irudia) (Kandel eta Siegelbaum, 2001;
Purves eta lank., 2008b).
Neurotransmisoreen hartzaileek modu zuzenean (hartzaile ionotropikoak) edo
zeharkakoan (hartzaile metabotropikoak) bete ditzakete euren funtzioak (1.9. irudia).
Gainera, neurotransmisorearen eragina hartzailearen araberakoa izango da, hau da,
neurotransmisore berdinak kontrako efektuak eragin ditzake lotzen den hartzailearen
arabera (Kandel eta Siegelbaum, 2001).
Nerbio-sistema zentraleko neurona batek, ehunka neuronaren konexioak jasotzen ditu.
Gainera, seinale kitzikatzaile eta inhibitzaileak jaso ditzake, eta zenbait
neurotransmisorek eragin dezakete neurona berdinean, eta, hala, zenbait ioi-kanalen
aktibitatean eragin. Aurretik esan bezala, horietako kanal batzuetan neurotransmisoreak
zuzenean eragiten du eta beste batzuetan, berriz, zeharka. Ondorioz, nerbio-sistema
zentraleko neuronek zenbait aferentzia koordinatu eta erantzunak emango dituzte.
Adibide gisa, neurona sentsitibo batek, neurona motorrean, potentzial postsinaptiko
kitzikatzaile (PPSE) txiki bat sor dezake, baina horrek bakarrik ez du intentsitate
nahikoa izango neurona motorrean ekintza potentzial bat sortzeko, ez baita nahikoa
despolarizatuko. Baina zenbait PPSE batera gertatzen badira, neurona motorrean ekintza
10

Sarrera
potentzial bat hasiko da. Gainera, potentzial sinaptiko inhibitzaileak ere kontuan hartu
behar dira. Horiek efektu kitzikatzaile horren aurka egingo dute (Kandel eta
Siegelbaum, 2001; De Camilli eta lank., 2001).
1.8. Irudia: Sinapsi kimikoetan transmisio sinaptikoa zenbait pausotan gertatzen da. Axoi terminalera iristen den ekintza potentzialaren ondorioz, potentzial-diferentziaren menpekotasuna duten Ca2+ kanalak irekitzen dira. Ca2+ axoiaren barrura sartuko da, eta neurotransmisoredun xixkuak mintz presinaptikoarekin lotu, eta beren edukia askatuko dute. Askatutako neurotransmisorea mintz postsinaptikoan dauden hartzaileekin lotuko da. Hori dela eta, kanal postsinaptikoen irekiera edo itxiera gertatuko da. Korronte postsinaptikoaren ondorioz, potentzial kitzikatzaile ala inhibitzaile bat sortuko da mintz postsinaptikoan. Purves eta lank, 2008b. 1.3.2. NEUROTRANSMISORE ETA HARTZAILEAK Modu orokor batean, garrantzitsua da aipatzea glutamatoa dela garunean kitzikatzailea
den neurotransmisore nagusia. Zenbait hartzaile ionotropiko (NMDA, AMPA eta
Kainato) eta metabotropiko (mGluRak) ditu glutamatoak. Hartzaile ionotropikoen
kasuan, NMDA ez diren hartzaileek, Na+ eta K+ ioien fluxuak bideratzen dituzte, eta
potentzial kitzikatzaile postsinaptiko (PPSE) goiztiarrak sortzen dituzte. NMDA
11

Sarrera
hartzaileen kasuan berriz, Ca2+, Na+ eta K+ ioiak igaro daitezke kanal horietatik, eta
potentzial-diferentziaren mendekotasuna ere badute hartzaile horiek. Hau da, mintza
atseden-egoeran dagoenean, hartzaile horiek Mg2+ ioiez blokeaturik daude. Baina
blokeo hori zelula despolarizatzean desagertzen da. Beraz, NMDA hartzaileek
glutamatoaren eta zelularen despolarizazioaren beharra dute ireki ahal izateko. Hartzaile
metabotropikoen kasuan, zeharkako bide baten bidez, bigarren mezulariak erabiliz
eragiten dute ioi-kanaletan (Purves eta lank., 2008c).
.
1.9. Irudia: Neurotransmisoreek zuzenean (aktibatze zuzena) edo zeharka (zeharkako aktibatzea) eragiten dute ioi-kanaletan. Zuzenean aktibatzen dituenean , hartzaile ionotropikoek hartzen dute parte. Zeharkako aktibatzean, berriz, hartzaile metabotropikoek. Kandel eta Siegelbaum,2001. GABA eta glizina dira nerbio-sistema zentralean hartzaile inhibitzaileak aktibatzen
dituzten neurotransmisore nagusiak. GABAk hartzaile ionotropikoak (GABAA) aktiba
ditzake, eta horiek, Cl- ioia pasarazten duten kanalak eratzen dituzte. Hartzaile
metabotropikoak ere aktibatzen ditu (GABAB), eta modu horretan, K+-aren
kanporanzko iragazkortasuna handitu, edota tentsioaren mendeko Ca2+ kanalak
inhibitzen dituzte (Purves eta lank., 2008c).
Transmisio kolinergikoa ere hipokanpo eta memoriarekin erlazionatua izan da.
Bideratzen duen azetilkolina (ACh) neurotransmisorea, halaber, hartzaile ionotropikoei
eta metabotropikoei lotzen zaie. Gehien ikertu diren hartzaileak hartzaile nikotinikoak
12

Sarrera
(ionotropikoak) dira (nAChR). Katioi ez-selektiboen kanalak osatzen dituzte, eta
erantzun postsinaptiko kitzikatzaileak eragiten dituzte. Metabotropikoen kasuan,
AChren hartzaile muskarinikoak daude (mAChR). Garunean, AChren efektu gehienak
hartzaile horien bidez bideratzen dira (Purves eta lank., 2008c).
Oinarrian, beraz, sinapsi kimiko batean gertatzen dena ioi-fluxuen gorabeherekin azaldu
daiteke. Neurona presinaptikoan ekintza potentzial batek, Ca2+ ioien barneratzea dela
eta, neurotransmisorea askatzea bultzatuko du. Aldi berean, neurotransmisore hori
neurona postsinaptikoko hartzaileei lotuko zaie. Hartzailearen arabera, ioi-en fluxu bat
edo beste gertatuko da neurona postsinaptikoan, eta, modu horretan, neurona horretan
ekintza potentziala gertatuko da edo ez da gertatuko. Horrela, informazioa transmititzen
joango da neuronaz neurona (Purves eta lank., 2008c).
1.3.3. PLASTIZITATEA ETA OROIMENA Baldintza zehatz batzuetan, neuronen arteko sinapsi horiek aldaketak jasan ditzakete,
indartu edo ahuldu egin daitezke, eta konexio sinaptiko gehiago ere sor daitezke. Hori
guztia da aurretik aipatu dugun plastizitate sinaptikoa, eta ikasketa eta oroimen-
prozesuak modu horretan bideratzen direla uste da (Kandel, 2001; Purves eta lank.,
2008a). Sinapsietan gertatzen diren aldaketa horiek denbora mugatu batez gerta
daitezke, edota iraunkorrak izan daitezke.
1.3.3.1. Oroimen Inplizitua edo ez-deklaratiboa
Oroimen ez-deklaratiboaren kasuan (oroimen inplizitua), zenbait prozesu gertatzen dira:
ohitze-prozesua, sentsibilizazio-prozesua, eta baldintzapen klasikoa. Horietan guztietan,
ikusi da, epe luzerako oroimenak gordetzeko orduan, cAMP-PKA-MAPK-CREB
bideak zerikusia duela (1.10. irudia).
1.3.3.2. Oroimen esplizitu edo deklaratiboa
Oroimen deklaratiboa edota esplizituaren kasuan, hipokanpoko epe luzeko potentziazioa
(LTP) inplikatuta dago ugaztunetan. Lehenago ere esan dugu hipokanpoa oroimen mota
hori prozesatzeko egitura garrantzitsua dela. Eta, hipokanpoan, hiru bide nagusi ditugu:
bide zulatzailea (kortex entorrinaletik hortz-zirkunboluzioko pikor zeluletara), zuntz
goroldiotsuen bidea (hortz-zirkunboluzioko pikor zeluletatik CA3ko zelula
13

Sarrera
piramidaletara), eta Schaffer-en albokideak (CA3ko zelula piramidaletatik CA1eko
zelula piramidaletara). Bide horietan guztietan, neuronek aurretik duten aktibitateak
zerikusi handia du ondorengo erantzunean, hau da, maiztasun altuko kitzikapenek,
neurona dianetan, potentzial postsinaptikoen anplitudea handitzen dute. Horri LTP
deritzo. Dena dela, LTP hori jazo dadin gertatzen diren mekanismoak ez dira berdinak
hiru bide horietan.
1.10. Irudia: Oroimen inplizituaren kasuan, epe luzerako oroimenean, bi aldaketa garrantzitsu gertatzen dira neuronetan: PKAren etengabeko aktibitatea, eta konexio sinaptiko berriak sortuko dituzten egitura mailako aldaketak. Horretarako, proteina berrien ekoizpena beharrezkoa da, eta hori PKAren bidez hasiko den mekanismoa da. PKA MAPKrekin batera, nukleora sartuko da, eta bertan CREBen fosforilazioa bideratuko du PKAk. Horrela, zenbait generen transkripzioa gertatuko da. Hori gerta dadin, hau da, CREB-1 aktibatzeko, beharrezkoa da CREB-2 inhibitzea, eta hori, MAPKren bidez gertatuko da. CREBen bidez aktibatuko den geneetako batek ubikitina hidrolasa kodetzen du, eta, horri esker, PKAren etengabeko aktibitatea mantenduko da. CREBek aktibatuko duen beste gene batek C/EBP transkripzio-faktorea kodetzen du, eta hori, CAATrekin lotuko da. Azken horrek, konexio berriak sortzeko beharrezkoak diren proteina garrantzitsuen geneak aktibatzen ditu. Kandel,. 2001.
Zuntz goroldiotsuen kasuan hortz-zirkunboluzioko pikor zelulen axoiek glutamatoa
askatzen dute, eta glutamato hori CA3ko neurona postsinaptikoen hartzaileei lotzen
zaie. Dena dela, badirudi LTP hori ez dagoela neurona postsinaptikoetako NMDA
hartzaileekin, ezta Ca2+ ioiak zelula postsinaptikoan sartzearekin lotuta; maiztasun
altuko estimuluen ondoren, zelula presinaptikoan sartzen diren Ca2+ ioiekin lotuta
baizik. Ioi horiek Ca2+/kalmodulina menpeko adenilziklasa aktibatzen dute, horrela,
cAMP mailak igotzen dira, eta PKA aktibatzen da neurona presinaptikoan. Prozesu
horretan, LTP eragiteko, zuntz goroldiotsuetara iristen diren beste aferentzia batzuen
14

Sarrera
bidez (aferentzia noradrenergikoak), β-adrenergikoak diren hartzaileak inplikatzen dira,
eta guztien eraginaz LTP gertatzen da.
Schaffer-en albokideen kasuan ere glutamatoa da erabiltzen den transmisorea, baina,
kasu horretan, LTP gerta dadin, NMDA hartzaileen aktibitatea beharrezkoa da.
Ondorioz, CA1eko LTPak CA3ko LTPak ez dituen bi ezaugarri berezi ditu, NMDA
hartzaileen izaeratik eratortzen diren ezaugarriak alegia.
Alde batetik, Schaffer-en albokideetako LTPan, zenbait axoi aferenteren aldi bereko
aktibazioa behar da. Horren arrazoia, esan bezala, NMDA hartzaileen izaeran aurkitzen
dugu; hau da, hartzaile hori funtzional bihurtzeko, glutamatoa hartzaile postsinaptikoari
lotu behar zaio, eta, bestalde, zelula postsinaptikoak nahikoa despolarizatua egon behar
du zenbait axoi aferenteren deskarga kooperatiboa dela eta. Horren ondorioz, NMDA
hartzaileen ahotik, Mg2+ askatzen da. Mg2+ askatzean, Ca2+ ioien fluxua gerta daiteke
kanal horietatik barrena, eta Ca2+ ioiak zelula postsinaptikora sartzen dira. Kaltzioa
zelula postsinaptikoan sartzean, zenbait proteinkinasaren aktibatzea gertatzen da
(Ca2+/kalmodulina-ren mendeko proteinkinasa, proteinkinasa C, proteinkinasa A, eta
tirosina-proteinkinasa fyn). Horrela, transmisio sinaptikoaren erraztea gertatzen da.
Bestalde, Schaffer-en albokideen bidean, zelula presinaptiko eta postsinaptikoen
aktibitate bateratua behar da, zelula postsinaptikoa modu egokian despolarizatzeko.
Horrela, ba, CA1 guneko LTP gertatzeko, zenbait faktore postsinaptiko beharrezkoak
dira: zelula postsinaptikoaren despolarizazioa, NMDA hartzaileen aktibatzea, Ca2+ ioien
barneratzea eta Ca2+ ioien bidezko zenbait bigarren mezulariren sistemen aktibatzea.
Ca2+ ioien mendekotasuna duten zenbait kinasa aktibatuko dira, eta, horren ondorioz,
ez-NMDA hartzaileen fosforilazioa gertatuko da (Ca2+/kalmodulin kinasaren bidez) eta
hartzaile horien glutamatoarekiko sentikortasuna areagotuko da. Gainera, badirudi
zelula postsinaptikoak atzeranzko mezulariak ere askatzen dituela (haietako bat oxido
nitrikoa dela uste da (NO)). Mezulari horiek zelula presinaptikoan dauden kinasa
proteinetan eragingo dute, neurotransmisore gehiago askatu dadin. (1.11. irudia).
LTPak, lehenago aipatutako oroimenaren biltegiratze-prozesuak bezala, faseak ditu.
Alde batetik, LTP goiztiarra dugu (epe motzekoa; 1-3 h; eta ez du proteina berrien
sintesiaren beharrik), eta bestalde, estimulu gehiagorekin, LTP berantiarra dugu (24 h-
ko iraupena du, gutxienez, eta proteina berrien eta RNAren sintesia gertatzen da) (1.12.
irudia). Aipatu dugun bezala, epe motzeko fase goiztiarreko mekanismoak nahiko
ezberdinak dira Schaffer-en albokideen eta zuntz goroldiotsuen bidean. Fase
berantiarreko mekanismoak aldiz, hau da, epe luzekoak, antzekoak dira bi bideetan.
15

Sarrera
Bietan, LTPn, proteina eta RNA mezulariaren (RNAm) sintesia gertatzen da, eta,
seinaleak transmititzeko, cAMP-PKA-MAPK-CREB bidea erabiltzen da.
Gainera, badirudi fase goiztiarrean ez dela sinapsi gehiagorik sortzen; LTPren fase
berantiarrean, berriz, gune presinaptiko eta postsinaptiko aktiboen kopurua handitzen da
(1.13. irudia).
1.11. Irudia: LTPren indukzioaren fase goiztiarraren modeloa. Transmisio sinaptiko arruntean, hau da, frekuentzia baxuko estimuluen kasuan, glutamatoa askatzen da (Glu) terminaletik. Horrek, NMDA hartzaileetan eta ez-NMDA hartzaileetan eragiten du. Baina ioiak ez-NMDA hartzaileen kanaletan barrena igaroko dira; izan ere, NMDA kanalak Mg2+ ioien bidez blokeatuta daude. Epe luzerako potentziazioaren kasuan, berriz, NMDA hartzaileak ez diren hartzaileen ekintza dela eta, mintz postsinaptikoa despolarizatu egiten da. Hori frekuentzia altuko estimulu tetanikoen ondorioz gerta daiteke. Hori dela eta, Mg2+ ioiak askatu egiten dira, NMDA hartzaileak desblokeatuz, eta, horrela, ioiak kanal horietan zehar ere igaro daitezke. Ondorioz, Ca2+ ioien gorakada bat gertatuko da dendrita-arantzan, eta Ca2+-aren mendeko kinasek (Ca2+/kalmodulin kinasa eta C kinasa proteina (PKC)) eta Fyn tirosina kinasak, elkarrekin, LTP eragingo dute. Kandel, 2001.
16

Sarrera
1.12. Irudia: LTPren fase goiztiar eta berantiarraren modeloa. LTP goiztiarra ekintza potentzial multzo baten ondorioz sortzen da. Baina, ekintza potentzial multzo horiek behin eta berriro errepikatzen badira, Ca2+ ioien barneratzeak adenilziklasa ere sartzen du jokoan. Horrek AMP ziklikoaren (cAMP) mendekotasuna duen kinasa proteina aktibatzen du (cAMP kinasa). Nukleora translokatzen da, eta CREB proteina fosforilatzen du. CREBek, egitura mailako aldaketak egin ditzaketen eragileak aktibatuko ditu, eta sinapsi berriak eratu daitezke. Kandel,2001. Hori guztia ikusita, oroimen espazialean hipokanpoko LTPk garrantzi handia duela
ikusi dugu. Esan dugun bezala, LTPren indukzioan eta mantentzean hainbat molekulak
hartzen dute parte, eta zenbait seinale-bide aritzen dira horretan. Molekula ugarik
eragiten dute LTP hori, plastizitate sinaptikoa areagotuz edo murriztuz. Ondoren
17

Sarrera
ikusiko dugun moduan, APP (amiloide peptidoaren proteina aitzindaria) eta beraren
proteolisiaren ondorioz eratzen diren zatiek zerikusi handia dute horretan guztian.
1.13. Irudia: CA3 guneko zelula baten eta CA1 guneko beste zelula baten arteko transmisio sinaptikoa aztertuz, LTP ren fase goiztiarra eta berantiarra bereizi daitezke. CA3 guneko zelula bakar bat kitzikatuz, eta CA1 guneko zelula batean sortuko duen potentzial sinaptikoa neurtuz, ikus daiteke kontrolen kasuan, gune aktiboan “dena ala ezer ez” moduan neurotransmisoredun xixkuak askatzen direla. LTP goiztiarraren kasuan, neurotransmisoreen askatze-probabilitatea handitu egiten dela ikusten da, baina oraindik ere, gune aktibo bakarra dago. LTPren fase berantiarrean berriz, erantzuna gune aktibo gehiagoren bidez gertatzen da, sinapsi berriak sortu direlako. Kandel, 2001.
18

Sarrera
1.4. AMILOIDE PEPTIDOAREN PROTEINA AITZINDARIA (APP)
1.4.1. OROKORTASUNAK
APP, izenak adierazten duen bezala, β-amiloide peptidoaren (βA) proteina aitzindaria
da.
Alzheimer gaixotasuna dutenen garunetan, ezaugarri bereziak dira amiloide-xaflak eta
zelula barruko tau proteina hiperfosforilatuz osatutako mikrozuntzak. Amiloide-xaflen
osagai nagusia 42aa-ko βA peptidoa da, eta, aurretik aipatu bezala, peptido hori APP
izeneko proteinatik eratortzen da, aitzindari horren proteolisiaren ondorioz.
APPk, ondoren aipatuko dugun bezala, funtzio garrantzitsu eta positibo ugari ditu, baina
ez zen horregatik ezagutzera eman. Alzheimer gaixoen garunetan agertzen ziren
xafletako β-amiloide peptidoa identifikatu ondoren (Glenner eta Wong, 1984; Masters
eta lank., 1985), peptido horren sekuentziatik abiatuz, beraren aitzindaria klonatzea
lortu zuten: amiloide peptidoaren proteina aitzindaria; APP (Kang eta lank., 1987;
Gralle eta Ferreira, 2007). Neuronetan nagusia den APPren isoforma klonatu zuten, 695
aminoazidokoa hain zuzen ere, eta hurrengo urteetan, beste zelula mota askotan nagusi
ziren APPren beste isoforma batzuk ezagutu zituzten: APP751 eta APP770 (Kitaguchi
eta lank., 1988; Ponte eta lank., 1988; Tanzi eta lank., 1988). APP 21. kromosoman
kodetzen da (21q21.2-3) eta beraren pre-mRNAren aukerazko splicing-a dela eta,
zenbait isoforma sortzen dira. APPren isoforma ugari dauden arren, aipatutako hirurak
dira APPren isoforma nagusiak.
APP ez da garunean bakarrik adierazten, gorputzean zehar leku askotan adierazten da,
eta isoformen proportzioa ezberdina da organoaren arabera (Selkoe eta lank.,1988).
Horregatik, APPk bizitzeko oinarrizkoak diren funtzioak zituela pentsatzen zen, eta
ezustean harrapatu zituen ikerlariak APP knockout saguak bizi zitezkeela ikusteak
(Zheng eta lank., 1995).
Dena dela, gaur egun, jakina da APP gene-sendi txiki bateko kidea dela. Sendi
horretakoak dira APLP1 eta APLP2, eta bestalde, beste zenbait espezietan ere aurkitu
dira gene horren homologoak. Horrela, APP knockout saguetan, APLP1 eta APLP2k
beraren funtzioak nolabait ordezkatzen dituzte (Heber eta lank., 2000; Turner eta lank.,
2003). Hala ere, sendiaren barnean, APP da βA peptidoaren sekuentzia duen bakarra
(Thinakaran eta Koo, 2008).
19

Sarrera
1.4.2. APPren EGITURA
APP, I motako transmintz proteina da. Zatirik luzeena zelularen kanpoaldean edo
organuluen lumenean kokatzen den ektodomeinua da. Ondoren, transmintz domeinua
du, eta, azkenik, zitoplasmako C mutur laburra.
1.14. Irudia: APP770 isoformaren eskema. Zelularen kanpoko aldean, zenbait azpidomeinu azaltzen dira. 1) Zisteinatan aberatsa den domeinua. 2) Domeinu anionikoa. 3A) 7 exoia: Kunitz proteasa inhibitzailea den domeinua, APP695 isoforman ez da azpidomeinu hau agertzen. 3B) 8 exoia. Domeinu hau ez da APP 695 isoforman agertzen, ezta APP751 isoforman ere. 4) Funtzio neurobabeslea egotzi zaion domeinua. Irudian, heparina, kolagenoa, zink-a eta kobrea lotzeko guneak markaturik daude. 4. domeinuan, hexagono bidez agertzen da N-glikosilazio gunea. Neurotrofikoa den RERMS sekuentzia eta itsaste-funtzioekin zerikusia duen RHDS sekuentzia, biribil handiagoekin irudikatuta daude. Disulfuro zubiak ere agertzen dira. Turner eta lank., 2003.
Zelulaz kanpoko domeinuan E1 eta E2 domeinu kontserbatuak daude. Domeinu
horietan zenbait azpidomeinu daude, eta horrek APPk dituen funtzio ugariak azaltzen
ditu (1.14. irudia) (Ohsawa eta lank., 2001).
20

Sarrera
1.4.3. APPren KOKAPENA ZELULA BARRUAN
Nerbio-sisteman modu zabalean kokatzen dela esan izan da, eta kontzentrazio handitan
agertzen da usaimen-erraboileko, garun-kortexeko, septum diagonal-aren bandako,
globus pallidus-eko, zerebeloko eta hipokanpoko neuronetan (Card eta lank., 1988) eta
glian ere adierazten dela ikusi da (Ouimet eta lank., 1994).
APP zelula barneko mintz askotan kokatzen da; erretikulu endoplasmatikoaren (EE) eta
Golgi aparatuaren mintzetan ez ezik, mintz plasmatikoan ere aurkitzen da (Caporaso eta
lank, 1994). Baita gorputz multibesikular (MVB) (Tomimoto eta lank., 1995) eta
lisosometan ere (Haass eta lank., 1992). Sinapsiari dagokionez, APP, mintz
sinaptikoetan kokatzen dela ikusi da (Kirazov eta lank., 2001; Shigematsu eta lank.,
1992; Schubert eta lank., 1991; Caporaso eta lank., 1994).
1.4.4. APP ETA SINAPSIA
Ikerketa talde batzuek adierazi dute APP sinapsian kokatzen dela (Schubert eta lank.,
1991; Shigematsu eta lank., 1992; Caporaso eta lank., 1994; Kirazov eta lank., 2001).
APP eta sinaptofisinaren kolokalizazioaz ere hitz egin izan da sinapsian (Schubert eta
lank., 1991).
APPren adierazpen-maila aztertu duten taldeen arabera, adierazpen hori garapenean
zehar handitzen doa, eta maila gorena lortzen du sinaptogenesian (Kirazov eta lank.,
2001). Badirudi sinapsien eraketa eta mantenuan garrantzi handia duela (Kirazov eta
lank., 2001), sinaptogenesian, dendriten arantzak sortzeko orduan, eta plastizitate
sinaptikoan eraginez (Sabo eta lank., 2003). Gainera, garun helduan, aldaketa sinaptiko
intentsoa jasaten duten guneetan maila altuan adierazten da APP (Ouimet eta lank.,
1994).
APPren adierazpen-mailaren arabera, glutamatoarekiko erantzuna aldatzen dela ikusi
izan da (Tominaga-Yoshino eta lank., 2001). Beste zenbait ikerketaren arabera, APPren
gabezia dagoenean, elektrofisiologia mailan ondorioak daude. Ondorio horiek
kaltegarriak izaten dira; hala, hazkuntzetan, ikusi da sinapsi funtzionalen kopurua
jaisten dela, hau da, APP falta denean, sinapsien eraketan eta transmisioan galerak
gertatzen direla (Priller eta lank., 2006). APPren proteolisia ere transmisio
sinaptikoarekin lotuta dago (Kamenetz eta lank., 2003), eta, aurretik aipatu dugun
21

Sarrera
bezala, proteolisitik eratorritako zatiek transmisio sinaptikoan eta plastizitate
sinaptikoan eragiten dute.
APPri egozten zaizkion funtzioak direla eta, proposatu izan da isoformaren arabera
funtzio ezberdinak izan ditzakeela; hala, APP695ek neuronen konexio helduen
garapenean parte hartuko luke, eta APP751 isoformak sinaptogenesian eragingo luke
(Apelt eta lank., 1997).
In vivo egindako ikerketetan ere, APPk sinapsien garapenean, bai morfologikoki eta
baita elektrofisiologikoki ere, garrantzi handia duela ikusi da. Izan ere, APP/APLP2
knockout bikoitzetan, ikusi da motoneuronen axoiek ez dituztela sinapsiak leku egokian
eratzen. Horren ondorioz, jaiotzean, sinapsien transmisioan galera handiak izaten
dituzte sagu horiek, eta handik egun gutxira hil egiten dira (Heber eta lank., 2000;
Herms eta lank., 2004; Wang eta lank, 2005; Yang eta lank., 2005). Enbrioien zelula
ametatik eratorritako neuronetan ere, APP/APLP2 knockout bikoitzetan, ikusi da
transmisio sinaptikoan galerak egoten direla (Schrenk-Siemens eta lank., 2008).
1.4.5. APPren PROTEOLISIA
APPk prozesu proteolitikoa jasaten du, hau da, sekretasa batzuek moztu egiten dute,
APPren tamaina ezberdinetako zatiei bide emanez. α-, β-, eta γ-sekretasa deritze
APPren prozesu proteolitikoa gauzatzen duten entzimei. α- eta β-sekretasak APPren
zelulaz kanpoko domeinuan mozten dute. γ-sekretasak, berriz, transmintz domeinua
mozten du, mintz barneko proteolisi erregulatua (RIP) deritzon prozesuaren bidez.
Bakoitzaren ezaugarri zehatzak aurrerago aipatuko ditugun arren, modu orokorrean
esateko, parte hartzen duten sekretasen arabera, APPren zenbait zati sortuko dira, eta
sortzen diren zatien funtzioak bata bestetik oso ezberdinak dira. Gainera, batzuek
ondorio kaltegarriak eragin ditzakete, eta beste batzuen eraginak, berriz, fisiologikoak
dira. Ondorioz, proteolisia zein bidetatik gertatzen den garrantzi handikoa da, zerikusi
zuzena baitu Alzheimer gaixotasunarekin.
Bi mozketa bide nagusi daude: bide ez-amiloidogenikoa eta bide amiloidogenikoa.
Izenek adierazten duten bezala, lehenengoan mozketen ondoren ez da beta-amiloide
peptidoa (βA) sortzen; bigarrenean, aldiz, bai (1.15. irudia).
22

Sarrera
1.15. Irudia: APPren proteolisia. Bide ez amiloidogenikoan (ezkerrean) α-sekretasak β-amiloide peptidoaren sekuentziaren barnean mozten du. Bide amiloidogenikoan (eskuinean), aldiz, β-sekretasa bidezko mozketa gertatzen da APPren zelulaz kanpoko zatian, eta ondoren γ-sekretasak mintzean geratu den zatia mozten du. Mozketa horien ondorioz, β-amiloide peptidoa sortzen da, eta peptido horrek oligomeroak eratu ditzake. Oligomero horiek eta haiekin osatutako xaflek sinapsien arteko komunikazioa oztopa dezakete. β-amiloidez osatutako xaflak Alzheimer gaixoen garunetan agertzen dira. Friesen eta Woolridge, 2008.
1.4.5.1. Bide ez-amiloidogenikoa
Prozesu proteolitiko honetan, α- eta γ-sekretasek hartzen dute parte. Lehenengo, α-
sekretasak APPren zelulaz kanpoko domeinua mozten du, transmintz domeinutik gertu
(lys613-leu614 aminoazidoen artean APP695 isoformaren kasuan) (Turner eta lank.,
2003). Mozketa β-amiloidearen (βA) sekuentziaren barnean gertatzen denez, ezinezkoa
da bide horretatik peptido hori askatzea. Horrela, APPren zati disolbagarri luzea
askatzen da (sAPPα) eta mintzari lotuta C83 deritzon APPren zatia gelditzen da. C83
zatia izango da ondoren γ-sekretasak moztuko duena. Mozketa hori, aurretik esan
bezala, transmintz domeinuan gertatzen da; alde batetik, p3 peptidoa askatzen da, eta,
bestetik, C muturreko zelula barneko zatia (AICD) (1.16. irudia) (Turner eta lank.,
2003).
23

Sarrera
1.16. Irudia: APPren mozketa ez-amiloidogenikoa. Irudian ikusten dugu lehenengo α-sekretasa bidezko mozketa gertatzen dela. Mozketa horren ondorioz, sAPPα zati disolbagarri luzea askatzen da, eta c83 deritzon zatia gelditzen da mintzean. Ondoren, c83 zatia, γ-sekretasak moztuko du. Mozketa horren ondorioz, p3 zatia sortuko da, eta baita zelula barneko zati laburra ere (AICD). Hartman, 2005. http://www.alzforum.org
1.4.5.2. Bide amiloidogenikoa
Beste bide honetan, lehenengo, APPren β-sekretasa bidezko mozketa gertatzen da.
APPren zelulaz kanpoko domeinuan hau ere, baina APP695en 596-597 aminoazidoen
artean kasu gehienetan, nahiz eta, aurrerago aipatuko dugun bezala, beste aminoazido
batzuen artean ere mozketak egin ditzakeen. Hori β-amiloide peptidoaren N muturra da.
Mozketa hori dela eta, zati disolbagarri luzea askatzen da, sAPPβ. Zati hori, hala ere,
sAPPα baino pixka bat laburragoa da. Mintzari lotuta, C99 deritzon zatia gelditzen da,
eta hori γ-sekretasa bidez moztuko da transmintz domeinuan. Azken mozketa horren
ondorioz, βA peptidoa askatzen da, eta AICD zelula barneko zatia. γ-sekretasa bidezko
mozketa transmintz domeinuan zenbait lekutan gerta daiteke; horrela, tamaina
ezberdinetako β-amiloide peptidoak sortuko dira (1.17. irudia). βA42k βA40k baino
hauspeatzeko joera handiagoa du (Turner eta lank., 2003). Peptido horiek, zenbait
entzimaren bidez “garbituak” izan daitezke (Turner eta lank., 2004), eta peptidoen
ekoizte/garbiketa oreka apurtzean efektu kaltegarriak gerta daitezke, Alzheimer
gaixotasunean gertatzen den bezala (Pearson eta Peers, 2006)
24

Sarrera
1.17. Irudia: APPren mozketa amiloidogenikoa. β-sekretasaren eta, jarraian, γ-sekretasaren bidezko mozketen ondorioz, β-amiloide (βA) peptidoa sortuko da. Lehenengo mozketan (β-sekretasa), sAPPβ zati disolbagarri luzea askatzen da, eta c99 deritzon zatia gelditzen da mintzean. Zati hori izango da ondoren γ-sekretasaren bidez moztuko dena, eta, mozketa horretatik, βA peptidoa eta zelula barneko AICD zati laburra sortuko dira. Hartman, 2005. http://www.alzforum.org
1.4.6. APPren FUNTZIO OROKORRAK
Aurretik aipatu dugun bezala, APPk azpidomeinu ugari ditu eta horrek funtzio-
aniztasuna ematen dio. Gainera, beraren proteolisiaren ondorioz sortutako zatikiek
zenbait modutan eragin dezakete, eta horrek guztiak areagotu egiten du molekula eta
beraren funtzioa ulertzeko konplexutasuna. Beraz, eta funtzioa oraindik ere zehatz
ezagutzen ez dela jakin arren, zenbait funtzio egotzi zaizkio. Hartzaile gisa aritu
daitekeela pentsatu da (Rossjohn eta lank., 1999). Hazkuntza-faktore funtzioa ere eman
zaio (Sheng eta lank., 2008). Zelulen ugalkortasunean, itsasketan, eta migrazioan ere
zerikusia duela uste da (Sheng eta lank., 2008). Glutamatoaren hartzaileen erregulazioan
aritzen dela ere esan izan da (Tominaga-Yoshino, eta lank., 2001). Ikusi da APP
axoietan zehar garraiatzeko mekanismoarekin eralzionaturik dagoela (Kamal eta lank.,
2000; Kamal eta lank., 2001) eta uste da neuronen egituran eta seinaleen transdukzioan
eragina duela (Marcello eta lank., 2008).
APP knockout saguetan, neuronen egitura eta funtzioetan alterazio ugari ikusi dira, hala
nola gliosia, sinaptofisinaren maila-jetsiera, dendriten luzera txikitzea hipokanpoko
neuronetan, hazkuntzetako neuronen bizi-iraupen laburragoa eta LTP galera, besteak
beste (1.1. taula) (Seabrook eta lank., 1999; Dawson eta lank., 1999; Seabrook eta
Rosahl, 1999). APP, lehen esan bezala, proteina-sendi bateko kide da, eta beste kideek
25

Sarrera
APP falta denean beraren funtzioa nolabait betetzen dutenez, zaila da knockout saguen
bidez modu zuzenean beraren funtzioa ezagutzea (Heber eta lank., 2000).
26
1.1.Taula APP, APLP1 eta APLP2 knockout konbinaketa ezberdinen ezaugarriak.
Genotipoa Fenotipoa Erreferentzia
Knockout bakunak
APP -/- Bizi dira. Ugaltzeko gaitasuna
dute
Zheng eta lank., 1995
Gorputzaren masa murriztua Müller eta lank, 1994
Aktibitate lokomotorra murriztuta Li eta lank., 1996
Aldaketak garapen
sensorimotorrean
Tremml eta lank., 1998
Epilepsiarako hipersentsibilitatea Steinbach eta lank., 1998
APLP1 -/- Bizi dira. Ugaltzeko gaitasuna
dute
Heber eta lank., 2000
Gorputzaren masa
murriztua
Heber eta lank., 2000
APLP2 -/- Bizi dira. Ugaltzeko gaitasuna
dute
Von Koch eta lank., 1997
Itxuraz arruntak Von Koch eta lank., 1997
Knockout bikoitzak
APLP2-/- APP-/- Jaio ondorengo heriotza Heber eta lank., 2000
Histopatologikoki arruntak Von Koch eta lank., 1997
APLP2-/- APLP1-/- Jaio ondorengo heriotza Heber eta lank., 2000
Histopatologikoki arruntak Heber eta lank., 2000
APLP1-/- APP-/- Bizi dira. Ugaltzeko gaitasuna
dute
Heber eta lank., 2000
Gorputzaren masa murriztua Heber eta lank., 2000
APLP1-/-, APP-/-, APLP2+/- Jaio ondorengo heriotza Heber eta lank., 2000
Histopatologikoki arruntak Heber eta lank., 2000

Sarrera
1.4.7. APP ETA BERAREN ZATIEN FUNTZIOAK SINAPSIAN ETA
PLASTIZITATE SINAPTIKOAN
APPren proteolisiaren ondorioz sortutako peptidoen funtzioei buruz oraindik asko dago
jakiteko, baina ezagutzen dira haiei egozten zaizkien hainbat funtzio. Gehien ikertu
diren peptidoak sAPPα, βA peptidoa eta AICDak dira (1.2. taula). Sinapsiarekin eta
plastizitate sinaptikoarekin lotuta, sAPPα eta βA peptidoek garrantzi handia dute, eta
AICDren kasuan, berriz, ikusi da zenbait generen transkripzioan eragin dezakeela.
1.2. Taula: APPren proteolisitik eratorritako zatiki ezberdinen funtzioak.
APPren proteolisitik
eratorritako zatia
Funtzioa
sAPPα
Neurobabeslea. Neuronen gehiegizko kitzikatzea ekiditen du.
Ca2+-aren homeostasia erregulatzen du.
Oro har, LTP bultzatzen du. Kontzentrazio handiegitan LTPren indukziorako
kaltegarria izan daiteke.
βA
Zenbait sistematan eragiten du (glutamatergikoa, kolinergikoa..).
Sistema horietan oro har, LTP oztopatzen du zenbait mekanismoren bidez, baina,
kontzentrazioaren arabera, LTPren indukzioan lagun dezake.
AICD
Seinaleen transdukzioan, apoptosian eta zitoplasmako dinamiketan eragiten du.
Zenbait generen adierazpena bultzatzen du.
Plastizitate sinaptikoan eragin dezake, zenbait proteinarekin konplexuak eratuz, eta neuriten
garapenean eraginez.
1.4.7.1. sAPPα
sAPPα zatia modu konstitutiboan askatzen da APP molekula osotik, baina baita modu
erregulatuan ere. Sistema kolinergikoko eta baita glutamatergikoko zenbait hartzaile
aktibatzeak zerikusia du zati disolbagarri hori sortzearekin. Azetilkolinaren hartzaile
muskarinikoetatik, m1 eta m3 aktibatzean, APPren zati disolbagarri hori askatzea
bultzatzen da; baita glutamatoaren zenbait hartzaile aktibatzean ere, bai ionotropikoak,
bai metabotropikoak (Turner eta lank., 2003). M1 eta m3 hartzaile muskarinikoek, eta
baita mGlu1 hartzaile metabotropikoek ere, DAG bidezko seinale-bideen bitartez
eragiten dute. Bide horretan DAGk PKC aktibatzen du, eta hori garrantzi handikoa da
sAPPα zatia askatu dadin (Turner eta lank., 2003).
Bide ez-amiloidogenikoan askatzen den sAPPα zati disolbagarriaren funtzio ugari ikusi
dira horren inguruan egin diren ikerketetan. Besteak beste, neuronen zelula
27

Sarrera
aitzindarietan, eta baita bestelako zelulen aitzindarietan ere, sAPPα zatia ugaltze-faktore
gisa aritzen da (Pietrzik eta lank., 1998; Caillé eta lank., 2004); efektu neurobabesle eta
neurotrofikoak dituela ere ikusi da zenbait zelulatan (Mucke eta lank., 1996); eta
neuriten hazkuntza ere areagotzen duela esan izan da (Milward eta lank., 1992).
SAPPα zelulen mintzen gune zehatzetan lotzen da, eta, gainera, lotura hori asegarria da;
beraz, sAPPα-rentzat hartzaileak daudela pentsatzen da (Hoffman eta lank., 1999).
Gainera, APPren zatiki disolbagarri horrek hartzaileei lotutako guanilato-ziklasa
aktibatzen du (1.18. irudia) (Barger eta Mattson, 1995). Badirudi bide horren bidez
bideratzen direla sAPPα-k plastizitate sinaptikoan dituen eraginak (Turner eta lank.,
2003).
1.18. Irudia: zelula barneko cGMPren bidearen eskema. Guanilato ziklasari (GC) lotutako hartzailea aktibatzean hari lotutako GC entzimak GTP cGMP bihurtzea bideratzen du. cGMPk PKG aktibatu eta kinasa horrek zenbait proteinaren fosforilazioa eragingo du, eta fosfatasek, berriz, proteina horien desfosforilazioa bideratuko dute. Mekanismo horien bidez, zelulak maila funtzionalean erantzunak emango ditu. http://www.sigmaaldrich.com
Zelulen kitzikatze eta transmisio sinaptikoari dagokionez, APPtik eratorritako zati
disolbagarri horrek mintzaren hiperpolarizazioa eragiten du, eta neuronen kitzikatzea
ekidin. Hiperpolarizazio hori, gutxienez, potasio-kanal baten bidezko korronteen
aktibatzearen ondorioz gertatzen da (Furukawa eta lank., 1996a). Seinale-transdukzio
bide horretan, ikusi da cGMPrekiko menpekotasuna duen PKG kinasak parte hartzen
duela. PKGz gain, zenbait fosfatasak ere erregulatzen dituzte sAPPα-ren eraginak
(Turner eta lank., 2003).
28

Sarrera
Mintzaren kitzikapenean duen paperaz gain, kaltzioaren homeostasia ere erregulatzen
du sAPPαk (Mattson eta lank., 1993). Neuronen soman kaltzio askearen mailak jaisten
dira, potasio-kanalen aktibatzea dela eta. Potasio kanal horiek blokeatzean sAPPαk
eragiten duen kaltzioaren erregulazioa ez da gertatzen (Turner eta lank., 2003). sAPPαk
NMDA hartzaileen bidezko korronteak ere erregulatzen ditu (Turner eta lank., 2003;
Taylor eta lank., 2008). PKG eta fosfatasen inhibitzaileek ere bide hori mozten dutenez,
kasu honetan ere, kinasa eta fosfatasa horien parte-hartzea nabarmen geratzen da
(Furukawa eta Mattson, 1998).
Aurretik aipatutako horrekin guztiarekin kontraesanean, esan da sAPPα zatiak LTPren
indukzioan laguntzen duela eta epe iraunkorreko depresioa LTD, aldiz, oztopatu egiten
duela (Ishida eta lank., 1997; Taylor eta lank., 2008). NMDA korronteak areagotu eta
modu horretan LTPa bideratzen duela esan dute ikerketa horietan (Taylor eta lank.,
2008). Hala ere, kasu horretan ere sAPPαren dosi handiek LTP galarazi zuten (Taylor
eta lank., 2008).
Laburbilduz, sAPPα zatiak plastizitate sinaptikoan eragiten du. Sinapsiaren
testuinguruak ere zerikusia izango du efektu bat edo bestea lortzeko orduan (sinapsiaren
inguruneak eta historiak); halaber, sAPPα-ren kontzentrazioak ere zerikusia izango du.
Bestalde, plastizitate sinaptikoa erregulatzeko beste modu bat alde morfologikoari
dagokio. Badirudi, sAPPα-ren N muturraren bidez terminal presinaptikoen kopuruaren
igoera gertatzen dela, eta axoien adarkatze-prozesua ere areagotzen dela (Turner eta
lank., 2003).
Gai honekin lotutako orain arteko ikerketen arabera, argi dago sAPPα plastizitate
sinaptikoan eta hipokanpoko oroimen-prozesuetan garrantzi handiko erregulatzailea
dela, nahiz eta hipokanpoan, oroimen-prozesuen hasierako pausoetan edo memoriaren
trinkotze-prozesuetan eragiten duen eztabaidagai den (Turner eta lank., 2003; Taylor eta
lank., 2008).
1.4.7.2. β-Amiloide peptidoak
β-amiloide peptidoek Alzheimer gaixotasunean zeregin kaltegarria duten arren, peptido
horiek badituzte beste hainbat funtzio fisiologiko (Pearson eta Peers, 2006). Haiek
suntsitzeko edota “garbitzeko” makineria existitzeak berak (neprilysin, IDE eta abar)
funtzio fisiologikoak dituztela berresten digu (1.19. irudia).
29

Sarrera
1.19. Irudia: APPren proteolisia. Bi bideak ikusten dira irudian: bide ez-amiloidogenikoa (α-sekretasa bidea), eta bide amiloidogenikoa (β-sekretasa bidea). Bide amiloidogenikoan βA peptidoa sortzen da. Peptido horrek funtzio fisiologikoak ditu bere ekoizpen-maila eta suntsipen edo garbiketa orekatuak daudenean. Suntsipen-garbiketa hori neprilisina, intsulina suntsitzen duen entzima (IDE) eta dentsitate baxuko lipoproteinen hartzailearekin erlazionatutako proteinak (LRP) gauzatzen dute, besteak beste. Ekoizpen-suntsipen oreka galtzean, eta βA peptido gehiegi dagoenean, oligomeroak eratu daitezke eta oligomero horiek kaltegarriak dira, neuronen funtzio fisiologikoa oztopatzen baitute.
Sinapsien osaketan zeregin garrantzitsua dutela uste da (Priller eta lank., 2006). β-
amiloide peptidoek sinapsiaren aktibitatea kontrolatzen dute. Peptido horiek sinapsiaren
aktibitatea murrizten dute, aktibitate hori gehiegizkoa bihur ez dadin, eta beraz,
eszitotoxizitatea gerta ez dadin (Kamenetz eta lank., 2003; Lesné eta lank., 2005;
Selkoe, 2006). Norabide berdinean, βA peptidorik ez dagoenean, hazkuntzetan egindako
ikerketen arabera, neuronen heriotza gertatzen da (Plant eta lank., 2003), eta horrek
pentsarazten du neuronen biziiraupenean βA peptidoa garrantzitsua dela.
SAPPα zatiarekin gertatzen den bezala, litekeena da funtzio hori potasio-kanalekin
lotutako mekanismoren baten bidez gauzatzea. Potasio-kanal batzuen aktibitatea
garrantzitsua da neuronen iraupen-prozesuan, zelularen kitzikapenean zerikusia
dutelako; gainera, zelula barneko K+ kontzentrazioak zerikusi zuzena du apoptosiarekin
(Yu, 2003; Pearson eta Peers, 2006). Aldi berean, kaltzioaren homeostasiarekin ere
zerikusia dutela uste da (LaFerla, 2002; Reinhard eta lank., 2005; Pearson eta Peers,
2006).
Peptido horien ondorio fisiologikoekin konparatuz, kaltegarriak luze eta zabal ikertuak
eta eztabaidatuak izan dira. Aurretik esan bezala, βA42 peptidoa da Alzheimer gaixoen
garun-xafletako osagai nagusia. Dena dela, gaur egun esaten da peptido horiek osa
ditzaketen oligomero disolbagarriak direla toxikoenak (Walsh eta Selkoe, 2007). Ez
30

Sarrera
zelula kanpoan bakarrik, baita zelula barruan ere (Billings eta lank., 2005; LaFerla eta
lank., 2007).
Ikerketa ugari egiten ari dira β-amiloide peptidoen oligomeroen eta sinapsiaren galerak
lotuz. Postsinapsiko PSD95ekin elkarrekintzak izan ditzakete oligomeroek, eta baita
NMDA hartzaileekin ere. Arantza dendritikoen murrizte bat eragiten dute oligomero
horiek, eta baita fisiologikoki aktiboak diren sinapsien galera ere hipokanpoko neurona
piramidaletan (Marcello eta lank., 2008).
Plastizitate sinaptikoari dagokionez, βA peptidoek LTP eta LTDan eragina dute, eta
zenbait seinalizazio-bidetan eragiten dute. Seinalizazio-bide horietako batzuk dira p38
MAPK eta ERK MAPKen bideak (Dineley eta lank., 2001; Turner eta lank., 2003), eta
bestalde, PKAren aktibitatean eragiteko gaitasuna ere badute (Turner eta lank., 2003).
βA peptidoek, beste zenbait proteinaz gain, sistema kolinergikoko eta glutamatergikoko
hainbat hartzailerekin lotzeko gaitasuna dute, sistema horietan eraginez (Verdier eta
Penke, 2004). Plastizitate sinaptikoan, beraz, βA peptidoek efektu kontrajarriak lor
ditzakete; izan ere, zenbait sistematan (glutamatergikoa eta kolinergikoa, adibidez),
seinalizazio-bide ezberdinen bidez eragiten dute, eta, gainera, azken ikerketa batzuen
arabera, plastizitate sinaptikoa murriztu edo areagotu egin daiteke βA peptidoen
kontzentrazioaren arabera ere. Kontzentrazio txikitan, areagotu egingo dute, eta
kontzentrazio handitan, LTP galarazteko gai izango dira (Puzzo eta lank., 2008).
Sistema glutamatergikoa eta kolinergikoa Alzheimer gaixoen garunetan modu
goiztiarrean kaltetzen diren sistemak dira. Haietan βA peptidoek duten eragina ikusiko
dugu.
1.4.7.2.A. Sistema kolinergikoa
Zenbait hartzaile izan dira proposatuak βA peptidoentzat. Horien artean, nACh
hartzaileekiko afinitate handia dutela ikusten da, nACh azpimota batzuekiko bereziki
(Chin eta lank., 2007). Beraz, neurotransmisio kolinergikoaren erregulazioan garrantzia
du, eta bai aktibitate presinaptikoan, bai postsinaptikoan eragiten du. Sistema
kolinergikoak kalte handiak izaten ditu Alzheimer gaixotasunean, eta hipokanpoaren
funtzioan, nAChR hartzaileak dituzten neuronak garrantzi handikoak dira. Hartzaile
nikotinikoak, bai hipokanpoko zelula piramidaletan, eta baita GABA-neuronak diren
interneurona inhibitzaileetan ere kokatzen dira eta pisu handia hartzen dute
hipokanpoaren aktibitatean (Pettit eta lank., 2001).
31

Sarrera
βA peptidoak, sistema kolinergikoan duen eragina peptidoa zuzenean hartzaileari lotuz
burutzen da (Pettit eta lank., 2001). Lotura hori dela eta, kaltzioaren korronteak aktibatu
eta ERK2 MAPKren bidea aktibatzen da. Seinalizazio-bide horretan, CREBen
fosforilazioa ere gertatzen da. Ezaguna da ERK2 MAPK kaskadak paper garrantzitsua
jokatzen duela hipokanpoko plastizitate sinaptikoan; izan ere, LTPren indukziorako
neurotransmisoreek erabiltzen duten bide bat da (Dineley eta lank., 2001). βA peptidoek
denbora luzean nACh hartzaileak aktibatuz gero, haien gainerregulazioa gertatzen da.
Horrekin batera, ERK2 MAPKen hiperaktibazioa, eta jarraian, azken horien (ERK2
MAPK) azpierregulazioa gertatzen da. Alzheimer gaixoen oroimen-galeren arrazoia edo
arrazoietako bat gutxienez, horretan aurki liteke, β-amiloide peptido asko egotean,
hartzaile horien gainerregulazioa eta ERK MAPK kaskadaren disfuntzioan alegia,
CREBen fosforilazio-maila ere modu berean erregulatzen baita (1.20. irudia) (Dineley
eta lank., 2001). βA peptidoei antagonista papera eman dioten beste ikerkuntza batzuk
ere badira, hauek nACh hartzaileen aktibitatea blokeatzen dutela adierazten dute (Liu
eta Wu, 2006). Hala ere, badirudi mekanismo guzti horietan, aurretik aipatu bezala, βA
peptidoen kontzentrazioaren araberakoa izango da efektu bat ala bestea (Puzzo, eta
lank., 2008).
1.20. Irudia: ERK/MAPK bidea zenbait modutan aktiba daiteke. Ezkerrean eta goian, nACh hartzailea βA peptidoen bidez aktibatuko da. Horren ondorioz, Ca2+ zelula barrura sartuko da. Zelula barruko Ca2+ ioien gorakadak zenbait mekanismo aktibatuko ditu, eta, haien artean ERK/MAPK bidea. Bide horretan, CREBen fosforilazioa ere gerta daiteke, eta, horren ondorioz, zenbait gene aktibatuko dira. Sweatt, 2001
1.4.7.2.B. Sistema glutamatergikoa
• AMPA
Β-amilode peptidoak badu sistema glutamatergikoan ere eraginik, bai hartzaile
ionotropikoen bidez, eta baita metabotropikoen bidez ere. AMPA hartzaileen kasuan,
hartzaileei zuzenean lotu eta haien funtzioan eragin dezakete βA peptidoek. Ikusi da,
hipokanpoko CA1 guneko neuronetan, peptido horiek AMPA bidezko korronteen
32

Sarrera
jatsiera bat eragiten dutela. Horren inguruan talde batzuek emaitza kontraesankorrak
lortu dituzte; horren azalpena hau izan liteke: peptidoen kontzentrazioaren, peptidoen
agregazio-mailaren eta zelula motaren arabera erantzun bat edo beste lortzen dela
(Parameshwaran eta lank., 2008). βA peptidoek, hartzaile horiek mintz postsinaptikoan
maila baxuagoan adieraztea eragiten dute (Parameshwaran eta lank., 2008). Izan ere,
AMPA hartzaileen azpiunitateen entzima bidezko suntsipenean eragin dezakete eta
modu horretan, ez dira egoera normalean mintz postsinaptikoan egoten diren beste
hartzaile egongo. βA peptidoaren ekoizpen maila altuaren ondorioz, AMPA hartzaileen
bidezko korronte postsinaptiko kitzikatzaileen (EPSC) anplitudea jaisten da, esan
bezala. Jaitsiera hori AMPA hartzaileen sinapsietako kokapenean jatsiera bat gertatzen
delako izaten da. Kasu horretan, jaitsiera hori βA peptidoaren ekoizpen-maila altuak
GluR2 azpiunitatearen S880 fosforilazioa eragiten duelako da. Ondorioz, AMPA
hartzaileen endozitosia gertatzen da (Parameshwaran eta lank., 2008). Hasieran
Alzheimer gaixoen garunetan gertatzen diren sinapsien funtzioen galeretan, horrek
guztiak zerikusia izan dezake (Parameshwaran eta lank., 2008).
Frekuentzia altuko estimuluen bitartez, βA 1-42 peptidoaren aplikazio akutuak LTPren
galera dakar, eta Ca2+/calmodulina menpeko CAMKIIren autofosforilazioa eta azken
horrek fosforilatutako GluR1en fosforilazioa eragozten du (1.21. irudia)
(Parameshwaran eta lank., 2008).
1.21. Irudia: βA peptidoek AMPA hartzaileen mintzeko adierazpenean eta funtzioan eragiten dute. Irudian, gezi ez-jarraituek prozesu inhibitzaileak adierazten dituzte. Gezi jarraituek, aldiz, errazte-prozesuak. βA peptidoek kaspasa bidezko AMPA hartzaileen mozketa bultzatzen dute. CaMKIIren fosforilazioa inhibitzen du, eta baita CaMKIIren bidezko AMPA hartzaileen fosforilazioa ere. βA peptidoek Glu azpiunitateen (GluR2) fosforilazioa bultzatzen dute, eta, horrela, AMPA hartzaileen endozitosia bideratzen dute. Bestalde, peptido horiek PSD-95 eta GluR2 azpiunitatearen mailak jaisten dituzte NMDA hartzaileen eta cdk 5-en mendeko prozesuen bidez. Parameshwaran eta lank., 2008.
33

Sarrera
• NMDA
NMDA hartzaileen kasuan, βA peptidoek, hartzaile horien bidezko neurotoxizitatea
bultza dezakete. Hartzaile horiek aktibatu eta mezulari erretrogadoen askapenaren
ondorioz, zelularen kanpoaldean glutamatoa pilatzen da. Gainera, NMDA hartzaileen
aktibatze ahulak peptido horien sintesia areagotzen du, eta β-sekretasa bidezko mozketa
bultzatzen da, α-sekretasa bidezko mozketaren ordez. Beraz, hartzaile horien
erregulazio zuzena beharrezkoa da βA peptidoen sintesia maila egokian gerta dadin.
Ondorioz, ziklo antzeko bat gertatzen da; NMDA hartzaileen gainaktibatzeak β-
amiloide peptidoaren sintesi-maila igoko du eta, ondorioz, glutamato gehiegi pilatuko
da, zeinak NMDA hartzaileak aktibatuz jarraituko duen, eta horrela neurona hil arte
(Parameshwaran eta lank., 2008).
Neurotoxizitatean eragiteaz gain, badirudi βA peptidoek kontrako eraginak izan
ditzaketela Alzheimer gaixotasunaren hasieran, NMDA hartzaileen funtzioa
azpierregulatuz. Hartzaile horiek zelularen azalean duten adierazpena jaisten dute, eta
bere funtzioa galarazi; hala, glutamato bidezko transmisio sinaptikoa jaisten dute (1.22.
irudia) (Parameshwaran eta lank., 2008).
1.22. Irudia: βA peptidoak NMDA hartzailearen funtzioa eta mintzeko adierazpena oztopatzen ditu zenbait mekanismoren bidez. Gezi ez-jarraituek prozesu inhibitzaileak adierazten dituzte; gezi zurrunek, berriz, errazte-prozesuak. βA peptidoak NMDA hartzaile bidezko LTD, βA peptidoen barneratzea eta NMDA hartzaileen endozitosia bideratzen ditu. Bestalde, βA peptidoek Ca2+ ioien fluxua areagotzen dute NMDA hartzaileetan barrena, eta, zelularen heriotzera eramango duen eszitotoxizitatea bultzatzen dute. NMDA hartzaileen aktibitatearen jaitsierak βA peptidoak sortzea bultzatzen du. Peptido horiek PKC bidezko mGlu hartzaileen (mGluR) bidezko NMDA eta GABA hartzaileen aktibatzea eragozten dute, eta horrek garuneko zenbait gunetan ikaste- eta oroimen-prozesuen galera ekarriko du. Parameshwaran eta lank., 2008.
34

Sarrera
II. taldeko hartzaile metabotropikoen aktibatzeak, PKCren menpeko mekanismo baten
bidez, NMDA hartzaileen korronteak areagotzen ditu. βA peptidoek potentziazio hori
jaisteko ahalmena dute. Horrela, ikusten da peptido horiek plastizitate sinaptikoan
eragiten dutela. Esate baterako, hipokanpoko CA1 gunean, NMDA bidezko LTD
bultzatzen dute (Parameshwaran eta lank., 2008).
• Glutamatoaren hartzaile metabotropikoak
β-amiloide peptidoek glutamatoaren hartzaile metabotropikoetan duten eragiteko
modua, askotan, seinalizazio-bide ezberdinen bidez izaten denez, beste hartzaile
batzuetan ere eragiten dute. Adibidez, kortex prefrontaleko neuronetan, mGluR5en
aktibazioak, PKCren menpeko bide batez, GABA-transmisioa bultzatzen du. β-amiloide
peptidoek bide hori galarazten dute (Tyszkiewicz eta Yan, 2005). Lehenago ere aipatu
dugu II. taldeko hartzaile metabotropikoen aktibatzeak dakarren PKC menpeko NMDA
hartzaileen korronteen areagotzea ere galarazten dutela peptido horiek (Tyszkiewicz eta
Yan, 2005). Gainera, esan behar da APPren proteolisi ez-amiloidogenikoa areagotu
egiten dela mGluR hartzaileen bidezko PKCren aktibatzearen bidez, eta, alderantziz, βA
peptidoen ekoizpena bultzatuko da mGluR hartzaileen bidezko PKCren aktibatzea
eragoztean.
1.4.7.3. APPren C muturreko zelula barneko domeinua (AICD)
Aurretik aipatu bezala, APPren bi bide proteolitikoen ondorioz askatutako APPren zati
bat da AICD, 5 KDa-eko zatia, edo APPren zelula barneko domeinua. AICD
domeinuak ere Alzheimer gaixotasunean eragina izan dezakeela pentsatzen da; gainera,
domeinu horiek zelula barnean askatzen direnez, haien efektua β-amiloide peptidoena
baino gogorragoa izan liteke, nahiz eta eragin hori aztertzen ari diren (LaFerla eta lank.,
2007; Müller eta lank., 2008).
Sortzen diren AICD horiek aminoazido kopuru ezberdinak izaten dituzte. Horrela,
AICD50, AICD48, AICD51, AICD31 zatien existentzia deskribatu da (Müller eta lank.,
2008). Isoforma horiek guztiek YENPTY motiboa dute beren barnean, eta motibo hori
da AICDk zenbait proteina adaptatzailerekin elkarrekintzak izateko erabiltzen duen
zatia. Aminoazido horiek dira klatrina bidezko endozitosiaren arduradunak. Domeinu
horretan, fosforilazioa gertatzen da; fosforilazioaren ondorioz lotura peptidikoen
35

Sarrera
angeluetan aldaketa batzuk gertatzen dira, eta horrek proteina adaptatzaileekin lotzeko
afinitatea baldintzatzen du. Adibidez, FE65 proteina adaptatzailea, T668ren
fosforilazioa dela-eta lotzen zaio APPri (Müller eta lank., 2008).
1.23. Irudia: APPren C muturrak proteina askorekin elkarrekintzak izan ditzake. APP 695 isoformaren C muturra ikus daiteke irudian. Gezi ez-jarraituek proteina-proteina elkarrekintzak erakusten dituzte. Gezi zurrunek elkarrekintza horien ustezko ondorio funtzionalak adierazten dituzte. Marra jarraituetan ikusten da APPren C muturraren zein sekuentziak dituen elkarrekintzak proteina horiekin. Turner eta lank., 2003.
AICDrekin elkarrekintzak dituzten 20 proteina baino gehiago deskribatu dira.
Elkarrekintza horien ondorioz, AICDk seinaleen transdukzioan, apoptosian eta
zitoeskeletoko dinamiketan eragina izango du (1.23. irudia) (Müller eta lank., 2008).
Seinaleen transdukzioak nola gauzatzen dituen azaltzeko zenbait modelo daude,
oraindik argi ez dagoen gaia da eta (Müller eta lank., 2008). AICDk zenbait generen
adierazpena erregulatzen duela ere ikusi da (Müller eta lank., 2008).
36

Sarrera
AICD /FE65 konplexuak neuriten garapenean eragina du (Müller eta lank., 2008).
Konplexu hori MENA proteinei lotzen zaie (zelulen mugikortasunean eragiteko gai
diren proteina batzuk dira MENA proteinak), eta MENAk profilin biltzen du
konplexura, eta azken horrek G-aktinarekin elkarrekintzak ditu. Elkarrekintza horien
ondorioz, aktinaren polimerizazioa gertatzen da (Müller eta lank., 2008).
1.5. APPren PROTEOLISIAN PARTE HARTZEN DUTEN
SEKRETASAK
Aurreko atalean ikusi dugun bezala, APPk proteolisia jasaten du. Proteolisi hori gainera,
bi bidetatik gerta daiteke: bide ez-amiloidogenikoa eta bide amiloidogenikoa.
Bakoitzetik, APPtik eratorritako zenbait zatiki sortuko dira, eta zati bakoitzak hainbat
funtzio beteko ditu zelulan. Zenbait entzima arduratzen dira APPren proteolisiaz. Bide
ez-amiloidogenikoan, α-sekretasa eta γ-sekretasak; bide amiloidogenikoan, berriz, β-
sekretasa eta γ-sekretasak. Sekretasa horien zenbait ezaugarri aipatuko ditugu jarraian.
1.5.1. α-SEKRETASA:
APPren proteolisia nagusiki, bide honetatik gertatzen da (Postina, 2008); lehenengo α-
sekretasak mozketa egin ondoren, γ-bidezko mozketa gertatzen da. Aurreko atalean esan
dugun bezala, mozketa horien ondorioz APPren zati ugari sortuko dira; sortutako zati
bakoitzak bere ibilbideari jarraituko dio eta zenbait funtzio beteko ditu. Lehenago esan
bezala, α-sekretasak β-amiloidearen sekuentzian eragiten duenez, peptido hori sortzea
ekiditen du; horregatik, hasiera batean, APPren proteolisi bide osasuntsutzat zeukaten
bide hori eta β-amiloide peptidoa sortzen zen bidea, aldiz, patogenikotzat hartzen zen.
Hala ere, gaur egun badakigu β-amiloidea sortzeko proteolisi bidea ere bide fisiologiko
bat dela, hau da, baldintza normaletan ere gertatzen dela, eta peptido horrek funtzio
fisiologiko ugari dituela (Pearson eta Peers, 2006).
Ikertzaile ugari α-sekretasa aktibitatea gauzatzen zuen entzimaren nortasunaren bila ibili
dira azken urteotan. Kontutan harturik α-sekretasa bidezko mozketa aminoazido-
sekuentzian ez, baizik eta mintzetik mozketa gunerako distantzian eta α-helize
37

Sarrera
konformazioan oinarritzen dela (Sisodia, 1992), proteinasa inhibitzaileak erabiliz, α-
sekretasa zink-metaloproteinasa zela ikusi zuten (Roberts eta lank., 1994). Horrela,
disintegrina eta metaloproteasa domeinudun proteinen sendiko (ADAM) zenbait
entzimak horretan parte hartzen zutela ikusi zuten.
1.5.1.1. ADAM sendia
ADAM sendia disintegrina eta metaloproteasa domeinuak dituzten proteinen sendia da
(Wolfsberg eta lank., 1995a). Proteina horiek, beraz, onak zein kaltegarriak diren
prozesu askotarako garrantzi handikoak dira, proteolisi eta itsaste-funtzioak betetzeko
gai direlako (Wolfsberg eta lank., 1995b). Zenbait ehunetan adierazten direla ikusi izan
da (barrabilak, obarioak, giltzurrunak……) eta Wolfsberg eta lankideak izan ziren
garunean lehenengo aldiz ADAM sendiko partaide batzuen presentzia deskribatu
zutenak (ADAM 1 edo fertilin α eta ADAM 5). ADAM proteinak oraindik ere ez dira
guztiz ezagutzen neurozientzien arloan, baina argi dago horietako proteina batzuek
izugarrizko garrantzia dutela neuronen garapenean (Yang eta lank., 2006). Esate
baterako, ADAM 10, Alzheimer modeloa diren saguetan gainadieraziz gero, β-
amiloidearen biltegien jaitsiera eta ezagutza kognitiboen galeren jaitsiera ere gertatzen
da (Postina eta lank., 2004). Era berean, ADAM 10 neurogenesian eta axoien
hazkuntzan erregulatzaile garrantzitsua da enbrioiaren garapenean, eta ADAM 21-ek
funtzio bera betetzen du jaio-ondorengo eta ugaztun helduetan (Yang eta lank., 2005).
Horrela, ADAM proteinak neuronen konponketarekin lotuak egon daitezkeen proteina
gisa ezagutarazten ari dira (Yang eta lank., 2006).
1.5.1.2. ADAM proteinen egitura
ADAM proteinak I motako transmintz proteinak dira, eta beren egituran domeinu hauek
errepikatzen dira: propeptido domeinua, transmintz domeinu bat, metaloproteasa
domeinua, disintegrin domeinua, zisteinatan aberatsa den gunea, hazkuntza-faktore
epidermalaren sekuentzia antzekoa (EGF), eta isats zitoplasmatikoa (1.24. irudia).
Metaloproteasa domeinua aktibatzeko, endoproteasa baten bidez propeptido domeinua
kendu behar da; kasu batzuetan, funtzio hori betetzen duena furinaren moduko
endoproteasa bat da (Loechel eta lank., 1998). ADAM gehienek proteasa domeinu
katalitiko hau izaten dute: HEXGHXXGXXHD, eta beren aktibitatea zinkaren
presentziaren araberakoa izaten da (1.25. irudia).
38

Sarrera
1.24. Irudia: ADAM proteinen eskema-egitura. Koloreek domeinu ezberdinak adierazten dituzte. Urdinez, mintza zeharkatzen duen domeinua.
1.5.1.3. ADAM proteinak nerbio-sisteman
Ugaztunetan ezagutzen diren 33 ADAM proteinetatik 17 gutxienez ikusi izan dira
nerbio-sisteman (Alfandari eta lank, 2001). 17 horietatik, 10 (1, 8-10, 12, 15, 17, 19, 21,
33) katalitikoki metaloproteasa aktiboak dira, eta integrina bidezko lotura eta proteinen
mozketa funtzio bikoitzak betetzeko gai dira (Primakoff eta Myles, 2000). Gainerako
7ek metaloproteasa domeinuan katalisirako motibo klasiko baten gabezia dute; horrek
pentsarazten du zelulen arteko seinalizazioan eragiten dutela, disintegrina domeinuaren
bidez adibidez (Yong eta lank., 2001). Horrela, ADAM sendiko proteinak, integrinen
bidez, zelulen arteko seinalizazioan eta itsasketan parte hartzeaz gain, zenbait proteinen
ektodomeinuak aska ditzaketen transmintz proteina bakarrak dira. Funtzio horiek
guztiak, garrantzitsuak dira neurogenesian, neuronen migrazioan, axoien hazkuntzan eta
plastizitatean (Yang eta lank., 2006).
39

Sarrera
1.25. Irudia: ADAM proteinen domeinuak, zelularen prozesuak koordinatuz. A) ADAM proteinen eskema-irudia. Metaloproteasa domeinuari esker (MP), substratua moztu dezakete (1). Disintegrin domeinuaren bidez (DI), integrina heterodimero hartzailearekin lot daitezke. Zisteinatan aberatsa den domeinuaren bidez (ZA), askotan, zenbait proteinekin elkarrekintzak izan ditzakete. Zitoplasmako domeinuaren bidez (Z), zelula barneko seinalizazio-prozesuak bidera ditzakete. B) ADAM proteinen 3D-ko egitura. Domeinuak markatuta daude irudian. Yang eta lank., 2006.
1.5.1.4. ADAM proteinak eta APP
Zenbait ikerketaren arabera, APPren α-sekretasa guneko mozketa, ADAM sendiko hiru
proteinak egin dezakete: ADAM17 (edo TACE), ADAM 10 eta ADAM 9 (Buxbaum
eta lank., 1998a ; Koike eta lank., 1999; Lammich eta lank., 1999; Asai eta lank., 2003).
Aurreko atalean aipatu dugu APPren α-sekretasa bidezko mozketa modu konstitutiboan
edo erregulatuan gerta daitekeela. ADAM10ek mozketa konstitutiboa gauzaten du, eta
besteek erregulatua (Sannerud eta Annaert, 2008).
ADAM batzuen sagu knockout-ak erabiliz egindako lanetan, ikusi nahi izan da ea APP
mozteko ezinbestekoak diren ala ez.
Horrela, ADAM 10 -/- saguak enbrioiak bederatzi egun t´erdi dituenean hiltzen dira,
gorputzaren garapenean izaten dituzten akatsak direla eta (nerbio-sisteman, sistema
kardiobaskularrean…). Euren garunek akats morfologiko ugari dituzte. Baina APPren
mozketari dagokionez, ADAM 10 gabeko saguetatik eratorritako fibroblasto zelula-
lerroetan ikusi denez, zelula-lerro guztietan izan ez arren, gehienetan APPren α-
sekretasa bidezko mozketa gertatzen da (Hartmann eta lank., 2002).
ADAM 17 -/- saguak jaio bezain pronto hiltzen dira birika eta sistema
kardiobaskularrean izaten dituzten akatsen ondorioz (Peschon eta lank., 1998; Zhao eta
lank., 2001; Shi eta lank., 2003). Baina, kasu horretan ere, katalitikoki aktiboa den
40

Sarrera
ADAM 17 gabeko fibroblastoetan, ikusten da APP disolbagarria askatzen dela
(Buxbaum eta lank., 1998a).
ADAM 9 -/- saguen hipokanpoko hazkuntzetan egindako ikerketen arabera, APPren
prozesu proteolitikoan ez da aldaketarik ikusten (Weskamp eta lank, 2002). Ez dute
agertzen nerbio-sistemarekin lotutako portaera edota akats morfologiko esanguratsurik.
Horrekin pentsa daiteke, nahiz eta neuronetan modu zabalean adierazten den, beraren
funtzioa ez dagoela nerbio-sistemaren garapenarekin lotuta edota beste ADAM batzuk
beraren funtzioa betetzeaz arduratzen direla bera falta denean (Yang eta lank, 2006).
Aipatutako guztiarekin, agerian gelditzen da APPren α-sekretasa mozketaz ADAM
sendiko proteina ugari arduratzen direla baldintza fisiologikoetan (Postina, 2008). Hala
ere, badirudi ADAM sendiko proteina asko APPren proteolisian antzeko moduan
inplikatuta daudela, eta haien artean funtzio-erredundantzia dagoela (Zhang eta Xu,
2007).
1.5.1.5. Substratuak ezagutzeko modua
Lehenago esan bezala, α-sekretasa bidezko mozketa ez da aminoazido-sekuentziaren
arabera gertatzen, mintzetik mozketa gunerako distantzia eta proteinak hartzen duen
konformazioaren arabera baizik (Sisodia, 1992). α-sekretasa bidezko mozketa β-
amiloide peptidoaren 16 (lys) eta 17 (leu) aminoazidoen artean gertatzen da (Esch eta
lank., 1990; Anderson eta lank, 1991; Wang eta lank., 1991), hau da, mintzetik 12-13
aminoazidoko distantziara, eta badirudi, α-helize konformazioa mozketaren
eraginkortasunarekin zuzenki erlazionatuta dagoela (Sisodia, 1992).
1.5.1.6. α-Sekretasaren aktibitatearen erregulazioa
G proteinei lotutako zenbait hartzailek (GPCRs) APPren proteolisi ez-amiloidogenikoa
bultzatzen dute. Azetilkolinaren hartzaile muskarinikoak dira horietako batzuk
(mAChR). Hala ere, hartzaile horien artean, azpimotaren arabera ezberdintasunak
daude. Horrela, m1 eta m3 motako mAChRak, APPren α-sekretasa bidezko mozketa
bultzatzen dute; m2 eta m4 motakoek, berriz, ez dute efektu hori lortzen. Hartzaile
azpimota horien arteko ezberdintasuna bigarren mezulari-sistema da. M1 eta m3
azpimotak fosfolipasa C-ri lotuta daude (PLC), eta m2 eta m4 azpimotak berriz, adenil-
ziklasari lotuak. M1 eta m3 motak PLC-ri lotuak daudenez, DAGz (diazilglizerol)
erregulatutako PKCren aktibazioak α-sekretasa bidezko mozketarekin zerikusia du
(Nitsch eta lank., 1993) . Fosfatidilinositolen hidrolisitik sortzen diren DAG eta inositol
41

Sarrera
tri-fosfatoaren (IP3) ekintzen ondorioz, PKC aktibatzen da; DAGak PKC aktibatzen du,
edota, IP3k zelula barruan metatutako kaltzio-erreserbetatik kaltzioa askatzea bideratzen
du, eta horrek PKC aktibatzen du. Aipatzekoa da zelula barneko kaltzio-mailen igoerak
bakarrik ez duela APPren α-sekretasa bidezko mozketa bultzatzen (Nitsch eta lank.,
1992); gainera, forbol-ester bidezko PKC-ren aktibazio zuzenak α-sekretasa bidezko
mozketa bultzatzen du, APP disolbagarriaren mailak handituz, eta βA-ren mailak
txikituz, bai APP arrunta adierazten duten zelulen hazkuntzetan, eta baita Alzheimer
gaixotasunarekin zerikusia duten APP ren mutazio mota batzuk dituzten zeluletan ere
(Hung eta lank., 1993).
PLCri lotutako beste GPCR batzuen kitzikapenak ere, APP ren zati disolbagarriaren
mailak igotzen ditu; adibidez, 1α-motako glutamatoaren hartzaile metabotropikoak
(mGluR1α) (Lee eta lank., 1995; Ulus eta lank., 1997), eta baita serotoninaren 5-HT2a
eta 5-HT2c hartzaileak ere. mGluR hartzaileen kasuan, APPren zati disolbagarrien
askatze hori, PKCren aktibazioarekin lotuta dagoen arren (Lee eta lank., 1995), ez da
gauza bera gertatzen serotoninaren hartzaileen kasuan. 5-HT2a-ren kasuan, PKC ez da
beharrezkoa APPren zati disolbagarrien askatzea kitzikatzeko. PKC zuzenean
kitzikatuz, APPren zati disolbagarrien maila igotzen den arren, PKCren blokeoa
eragitean ez da 5-HT bidezko sAPPren igoera hori blokeatzen. Bestalde, PLA2ren
inhibitzaileak erabiliz, oinarrizko sAPPren sekrezio-maila jaisten ez den arren, 5-HT
bidezko sAPPren igoera inhibitzen zen (Nitsch eta lank., 1996). Beraz, 5-HT2a
hartzaileen aktibatzeak, sAPPren askatzea azelera dezake, PLA2-ren bidez. 5- HT2c-ren
kasuan, berriz, PKCren azpierregulazioa eta PLA2ren inhibitzea konbinatuz lortzen da
5-HT bidezko sAPPren sekrezioa inhibitzea. Beraz, badirudi, 5-HT2c-ren kasuan, PKC
eta PLA2ren aktibitateek zerikusia dutela APPren sekrezioa estimulatzeko orduan
(Nitsch eta lank., 1996).
Serotoninaren 5-HT4 hartzailea estimulatzeak ere APP ren α-sekretasa bidezko
mozketarekin erlazioa du, AMP ziklikoaren menpeko, baina, PKArekiko (A proteina
kinasa) independientea den mekanismo baten bidez (Robert eta lank., 2001).
Beste zelula seinale-bide batzuek ere APPren α-sekretasa bidezko proteolisia areagotzen
dute. Haien artean, MAP kinasak (Kojro eta lank., 2006) daude. Horrela, APPren
proteolisiaren erregulazioan zerikusia duten hartzaile ezagunen artean, G-proteinei
lotutako hartzaileak eta tirosina-kinasari lotutako hartzaileak ditugu. Sistema efektore
arduraduna, PKC-menpeko edo PKC ez-menpeko moduan erregulatua izan daiteke, eta
42

Sarrera
tirosina kinasen aktibazioa ekar lezake. MEK/ERK, PKC eta tirosina kinasa hartzaileen
bidezko APPren erregulazioan inplikatua egon daiteke (Mills eta lank., 1997).
1.5.2. β-SEKRETASA
APPren proteolisian, bide amiloidogenikoaren kasuan, β-sekretasa eta γ-sekretasa
bidezko mozketak gertatzen dira, eta horrela, tamaina bat baino gehiagoko βA
peptidoak eratzen dira.
1.5.2.1. Orokortasunak
β-sekretasa mozketaren arduraduna den entzima, duela hamarkada bat identifikatu
zuten, eta pepsinen barruko aspartil-proteasa azpifamiliako kide bat zela ohartu ziren
(Vassar eta lank., 1999; Lin eta lank., 1999; Yan eta lank., 1999; Hussain eta lank.,
1999); β-gunean APP mozten duen 1 entzima (BACE1), memapsin 2 eta Asp2 ere
deitua.
1.5.2.2. Sendia eta egitura
BACE1 eta BACE2 (beraren homologo bat) pepsin sendiko aspartil-proteasen
azpifamiliakoak dira. Horrela izan arren, beren domeinu katalitikoan, disulfuro zubiak
eratzeko moduan eta beste hainbat gauzetan ezaugarri bereziak dituztenez, beste sendi
bat eratzen dutela esan izan da (Bennet eta lank., 2000a).
BACE1 11. kromosoman kodetzen den (11q23.2) transmintz proteina bat da, I motako
transmintz proteina hain zuzen ere. 501 aminoazido ditu. N muturrean 21 aminoazidoko
seinale peptidoa du, eta haren ondoren, propeptido domeinua dago, 22. aminoazidotik
45. aminoazidoraino. 46. aminoazidotik 460. eraino, proteinaren alde luminala/zelulaz
kanpokoa dago. Ondoren, transmintz domeinu bakarra du, 17 aminoazidokoa, eta
zitoplasman –COOH mutur laburra, 24 aminoazidokoa (1.26. irudia) (Vassar eta lank.,
1999).
Domeinu luminalean, bi gune aktibo ditu, 93-96 aminoazidoen artean bat, eta 289-292
aminoazidoen artean bestea. Gune horietako bakoitzak ondo kontserbatutako aspartil-
proteasen sekuentzia du: D t/s G t/s. Gune horietan, aspartato aminoazidoa ezinbestekoa
da entzimaren aktibitate katalitikorako (Vassar eta lank., 1999). Domeinu luminalean,
43

Sarrera
halaber, sei zisteina ditu, molekula barneko disulfuro-zubiak eratzen dituztenak, eta
glikosilazio guneak ere baditu (Cole eta Vassar, 2007).
1.26. Irudia: BACE1en eskema-egitura. Koloreen bidez domeinu ezberdinak adierazten dira. Urdinez, mintza zeharkatzen duen domeinua agertzen da, eta zelulaz kanpoko (edo organuluen lumeneko) aldean, proteolisia gauzatzeko beharrezkoak diren bi aspartatoak (D) markatuta daude.
Haren homologoa den BACE2 proteina 21. kromosoman kodetzen da (21q22.3). Bi
proteasa horiek ehunaren araberako adierazpen-ezberdintasunak dituzte. BACE1
garunean ugari agertzen da; BACE2, berriz, ehun periferikoetan adierazten da, eta
garunean adierazpen txikia du (Vassar eta lank., 1999; Lin eta lank., 1999; Bennett eta
lank., 2000a). Garunean, BACE1en mRNA leku guztietan agertzen da; gehienbat
hipokanpoan, kortexean eta zerebeloan agertzen da (Vassar eta lank., 1999). Gainera,
BACE1 maila handiagoan adierazten da neuronetan glian baino (Vassar eta lank.,
1999). Badirudi APPren β-sekretasa aktibitatea BACE1ek gauzatzen duela (Vassar eta
lank., 1999; Yan eta lank., 1999; Sinha eta lank.,1999; Bennett eta lank., 2000a).
Ikerketa batzuen arabera, BACE2 beste α-sekretasa baten moduan aritzen da, BACE1en
antagonista gisa (Yan eta lank., 2001). Badirudi BACE2k gehien eragiten duen APPren
mozketa gunea Phe19 eta Phe20ren artean dagoela, α-sekretasa gunetik behera, eta
APPren mozketa gune berri bat erabiltzen du. Mozketa horrekin ere, βA peptidoa
sortzea ekiditen da (Sun eta lank., 2006).
BACE1en modu aktiboaren kristal-egitura proposatu dute orain dela gutxi (Shimizu eta
lank., 2008), eta, horren arabera, BACE1en gune aktiboa estalita agertzen da, eta
substratuaren gune aktiborako sarbidea kontrolaturik dago.
Bestalde, aspartil proteasek pH-aren menpekotasuna izaten duten konformazio-
aldaketak izaten dituzte, eta BACE1en aktibitatea aldaketa horien araberakoa da.
44

Sarrera
Konformazio-aldaketa horiek eta aspartil proteasek izaten dituzten bi ur-molekula
kontserbatuen portaeraren araberakoa izango da BACE1en aktibitatea (1.27. irudia)
(Shimizu eta lank., 2008).
1.27. Irudia: BACE1en aktibitatearen eskema zenbait baldintzetan (aldagaiak pH-a eta eta ur-molekulen presentzia dira). pH azidoa eta ur molekulen presentzia beharrezkoak dira beraren aktibitaterako. Shimizu eta lank., 2008.
Beraren aktibitate gorena, pH 4,5eko ingurunean izaten da, eta zenbait ikerketak hori
agerian utzi dute beraren aktibitatea bide sekretorioko konpartimendu azidoetan ikusi
dutenez gero (Vassar eta lank., 1999).
1.5.2.3. BACE1en garapena zelula barruan
BACE1 erretikulu endoplasmatikoan sintetizatzen da; ~60 kDa-ko pisu molekularra du
hasieran, eta dagoeneko N-glikosilatua agertzen da. Garatzen doan heinean, azukre
gehiago jasoko ditu, eta azkenik, modu garatuan ~70 kDa-ko pisura iristen da (Cole eta
Vassar, 2007). Badirudi glikosilazioak erretikulu endoplasmatikotik ateratzean gertatzen
direla (Capell eta lank., 2000; Cole eta Vassar, 2008). Beste proteasa asko bezala, pro-
entzima gisa sortzen da hasiera batean, eta erretikulu endoplasmatikoa uztean,
propeptidoaren mozketa gertatzen da, BACE1en garapenean beste pauso bat emanez.
Badirudi mozketa hori Golgi aparatuan gertatzen dela (Bennett eta lank., 2000b). N-
glikosilatutako BACE1 sulfatua izaten da, eta beraren zitoplasmako muturrean
palmitoilazioa gertatzen da (Benjannet eta lank., 2001). Horrela, bide sekretorioan
gertatzen diren aldaketa horien bidez, BACE1 modu garatura iristen da (Haniu eta lank.
2000; Cole eta Vassar, 2008). Ez ohikoa den arren, propeptido domeinuak ez dio pro-
BACE1i proteasa aktibitatea kentzen, eta gai da APP mozteko, potentzialki toxikoa den
45

Sarrera
βA peptido andana bat zelula barnean sortuz (Creemers eta lank., 2001; Cole eta Vassar,
2007; Cole eta Vassar, 2008;). Glikosilazioak, berriz, proteasa aktibitatean eragiten du,
eta BACE1en C-muturraren palmitoilazioak haren kokapenean eta garraioan eragiten du
(Cole eta Vassar, 2008), nahiz eta azken ikerketa batzuen arabera hori zalantzan jarri
den, kokapen azpizelularrean eraginik ez duela esanez, baina bai lipid raft-ekin
asoziatzeko orduan (Vetrivel eta lank., 2008).
Nahiko proteina egonkorra da BACE1. ~9 orduko bizi-itxaropena du. Endoproteolisi
bidez suntsitzen da lisosometan eta ubikitin-proteasoma bidean (Cole eta Vassar, 2007).
1.5.2.4. BACE1en funtzioak eta substratuak
BACE1en funtzioari dagokionez, aurretik aipatu dugu beharrezkoa dela βA peptidoak
APPtik askatzeko. BACE1-/- saguek funtzio kognitibo eta emozionaletan gabeziak
izaten dituzte eta oroimen espaziala ere kaltetua izaten dute besteak beste (Cole eta
Vassar, 2007).
Neuronetan APPren zati bat BACE1en bidez mozten da, baina bestelako ehunetan,
APPk nagusiki α-sekretasa bidezko mozketa jasaten du. Gainera, BACE1ek APPz gain
beste substratu batzuk ere badituela ikusi da, eta horrekin lotuta, BACE1ek izan
ditzakeen funtzioak ezagutaraztera ematen ari dira (Cole eta Vassar, 2007). Horietako
substratu batzuk dira APLP eta dentsitate baxuko lipoproteinen hartzailearekin
erlazionatutako proteina (LRP). Bestalde, neuregulin-1 eta Sodio kanalen β-azpiunitatea
ere BACE1en substratuak direla ikusi da, adibidez. Horrekin, BACE1ek mielinazioan
eta neuronen aktibitatean zerikusia duela erakutsi da (Wong eta lank., 2005; Willem eta
lank., 2006; Cole eta Vassar 2008). BACE1en substratuetako asko estres edota lesioen
erantzunetan zerikusia duten substratuak dira. Gainera, BACE1en maila handitua
agertzen da estres baldintzetan; horrela, neuroinflamazioarekin zerikusia duten zenbait
substratu ditu BACE1ek (Cole eta Vassar, 2007). Estres-egoeretan erantzunerako
beharrezkoak diren funtzioak izan ditzakeen arren, uste da estres kronikoaren kasuan
BACE1en maila altuegiak kaltegarri eta patogeniko bihurtzen direla (Cole eta Vassar,
2007).
BACE1en substratu batzuk axoi terminaletan kokaturik egon daitezkeela uste da eta
sinapsiaren funtziorako BACE1en bidezko mozketa beharrezkoa da. APPren mozketa
β-amiloidea sortzeko, axoi terminaletan gerta daitekeela ere esan da (Lazarov eta lank.,
2002; Cole eta Vassar, 2007)
46

Sarrera
1.5.2.5. Substratuak ezagutzeko modua
Substratuak ezagutzeko modua sekuentziaren menpekoa da; izan ere, β-sekretasaren
mozketa gunean mutazioak gertatuz gero, β-sekretasa bidezko APPren proteolisia igo
edota jaitsi egiten da mutazioaren arabera (Vassar eta lank., 1999). Horrela, APPren
mozketatik eratorritako βA peptidoak, nagusiki, Asp1 aminoazidoan hasten dira.
Badaude beste aminoazidoetan hasten direnak ere (Val 3, Ile 6 eta Glu 11), baina askoz
gutxiago dira; gainera, Val 3 eta Ile 6ren kasuetan, beste β-sekretasa baten mozketaren
ondorioz sortzen direla uste da (Citron eta lank., 1996). β-sekretasak mintzari lotutako
substratuak bakarrik mozten ditu; gainera, beraren aktibitate gorena pH azidoan denez,
bide sekretorioko konpartimenduetan (Trans Golgi Sarean (TGS) eta endosometan)
egiten du bere lana bertako mintz-proteinak moztuz (Cole eta Vassar, 2007).
1.5.2.6. BACE1en aktibitatearen erregulazioa
BACE1en aktibitatea zenbait modutan erregulatzen da. Esate baterako, BACE1 heldua
lipid raft-etan kokatzen da, eta baita APP eta presenilina-1 ere. APPren proteolisi
amiloidogenikoa lipid raft horietan gerta daiteke eta zenbait lipidok BACE1en
aktibitatea areagotzen dute (Kalvodova eta lank., 2005). Lipidoek zerikusi handia dute
APPren proteolisi amiloidogenikoan eta β-sekretasa aktibitatean. Badirudi lipid raft-
etatik at kokatzen den APP α-sekretasa bidetik prozesatua izango dela, hau da, bide ez
amiloidogenikoa jarraituko duela. Lipid raft-etan kokatzen den APPk, berriz, bide
amiloidogenikoa jarraituko du (Riddell eta lank., 2001; Ehehalt eta lank., 2003).
Lipidoez gain, beste zenbait molekulak ere BACE1en aktibitatean eragina dute.
BACE1ekin elkarrekintzak izaten dituzte eta beraren aktibitatea areagotu edo murrizten
dute. Horietako molekula batzuk dira prostatako apoptosirako 4 erantzun-proteina
(PAR-4), sortilinarekin erlazionatutako hartzailea (sorLA/LR11) eta HS (heparan
sulfatoa) besteak beste (Scholefield eta lank., 2003; He eta lank., 2004; Xie eta Guo,
2005; Angeletti eta lank., 2005; Wickham eta lank., 2005; Spoelgen eta lank., 2006).
Horietako molekula batzuk sinapsiarekin erlazio estua dute, bai euren kokapenean eta
baita sinaptogenesian ere (Duan eta lank., 1999; Yamaguchi, 2001).
Bestalde, Golgin kokatzen diren eta γ-“belarria” duten ADP erribosilazio-faktoreari
lotzen zaizkion proteinen familiak (GGA) zerikusia du BACE1en TGS-endosoma
arteko garraioan; kokapen horrek, halaber, BACE1 bidezko proteolisian eragina du, eta
maila horretan ere aktibitate horren erregulazioa gertatzen da aurrerago ikusiko dugun
bezala (Cole eta Vassar, 2007).
47

Sarrera
1.5.3. γ-SEKRETASA
APPren proteolisian bigarren mozketa gauzatzen duena da γ-sekretasa entzima. Lehenik
eta behin, aurretik aipatu bezala, APPren N muturrean α- edo β-sekretasa bidezko
mozketa gertatzen da, eta mintzean sartuta APPren C muturra geratzen da. C mutur
horrek γ-sekretasa bidezko mozketa jasan dezake. Mozketa horren ondorioz zenbait
peptido (p3 α-sekretasaren kasuan; eta βA peptidoa β-sekretasaren kasuan), eta karboxi
muturreko APPren zelula barneko domeinuak (AICD) askatzen dira.
γ-sekretasa zenbait osagai dituen konplexu bat da eta mintzean kokatzen da. Presenilina
da konplexu horren osagai katalitikoa. Konplexu horrek zelula barnean hainbat kokapen
erakutsi ditu. Bestalde, proteolisi mota hori ez da ohikoena, izan ere, APPren C muturra,
mintz barruan mozteko ahalmena du. Horri, “Mintz barneko proteolisi erregulatua”
(RIP) deritzo.
1.5.3.1. Mintz-barneko proteolisi erregulatua (RIP)
Esan dugu γ-sekretasa ez dela ohiko aspartil proteasa bat; izan ere, mintz barruan
gauzatzen du substratuen mozketa.
APP identifikatu zenarekin batera (Kang eta lank, 1987), gero eta argiago ikusten zen
βA peptidoa mintzetik askatzen zela, ziurrenik mintz barneko mozketa baten ondorioz.
Baina hasiera batean ezinezkotzat jotzen zen mozketa hori modu fisiologikoan
gertatzea; izan ere, mintz barneko ingurune hidrofobikoan ur molekulak egotea ez da
batere gertaera arrunta. Ondorioz, ikerlariek, denbora luzean zehar, gertaera hori
patologikotzat zuten, eta uste zuten βA peptidoa askatzeko modu bakarra mintza
suntsitzea zela. Mintza puskatu eta peptidoaren aitzindaria askatzean proteasa ohikoek
mozketa eragiten zutela pentsatzen zen (Steiner eta lank, 2008). Horrela, hiru
laborategitan, modu independentean, ikusi zuten βA modu fisiologikoan ere sortzen
zela, eta horrek mintz barneko proteolisi bat gertatzea zekarren berekin (Haass eta
lank.,1992; Shoji eta lank., 1992; Busciglio eta lank., 1993)
APPren proteolisiak beste zenbait proteinaren proteolisiarekin batera (Brown eta
Goldstein, 1997; Wolfe eta lank.,1999c), mintz-barneko proteolisia ezagutzeko bidea
irekitzeaz gain, zelula barneko bide berri bat ezagutzeko ere balio izan zuen, RIP hain
zuzen ere.
48

Sarrera
Mintz barneko proteolisi erregulatua mintza behin zeharkatzen duten zenbait transmintz
proteinaren prozesatze sekuentziatua da. Bigarren mozketa bikapa lipidiko baten
barruko giro hidrofobikoan gertatzen da (Steiner eta lank., 2008).
Denbora askoan ezagutu izan diren proteasak disolbagarriak ziren arren, azken urteetan,
bikapa lipidikoaren barneko giro hidrofobikoan eragiten duten proteasak ezagutzera
eman dira. Haien substratuak ere ez dira oso ohikoak; normalki, α-helize bat osatzen
dute; eta eraso nukleofilikoa lotura peptidikora iristea eragozten dute. Horrela,
proteolisi mota hori egin behar duten entzimek katalisirako urarentzat eta aminoazido
hidrofilikoentzat ezinbestekoa den mikroingurua sortu behar dute; gainera, substratuek
biribilketak eta desbiribilektak jasan behar dituzte eta lotura peptidikoak hidrolisirako
prest utzi. Badirudi, substratu horien transmintz domeinuetan, lotura peptidikoa
apurtuko den lekuaren inguruan, α-helize konformaziorako joera txikia duten
aminoazidoak kokatzen direla; hala, desbiribilketa errazago gerta daiteke (Ye eta lank,
2000; Lemberg eta Martoglio, 2002; Lemberg eta Martoglio, 2004). Aurrerago aipatuko
dugun bezala, badaude salbuespenak hala ere.
Mintz barnean eragiten duten ez ohiko proteasa horiek, zentzu horretan ezberdinak eta
bereziak izan arren, katalisirako erabiltzen dituzten aminoazidoen konbinazioak
proteasa disolbagarriek erabiltzen dituztenen berdinak dira (Wolfe eta Kopan, 2004).
Ez ohiko proteolisi hau orain arte ezagutzen diren lau proteasa moten bidez gauzatzen
da. Proteasa horiei “mintz barnean mozten duten proteasak” (I-CliP) deitzen zaie.
Ondokoak dira ezagutzen diren I-CliP-ak: S2P (zink metaloproteasa bat dela uste da)
(Beel eta Sanders, 2008), seinale peptidoen peptidasa (SPP) (aspartil proteasa),
rhomboideak (serine proteasak) eta presenilinak (PS) (aspartil proteasak) (Spasic eta
Annaert, 2007; Beel eta Sanders, 2008) (1.28. irudia). Bakoitzak substratu anitz ezagut
ditzakeenez, RIP zelularen prozesu askotan gertatzen den proteolisia da, eta gainera,
bizidun askotan kontserbaturik dagoen kontrol-mekanismoa da (Brown eta lank, 2000).
RIPean, bi pauso ematen dira. Lehenengoan, substratuaren ektodomeinuaren zati handi
bat askatzen da sheddasa baten mozketaren eraginez. Kasu askotan, sheddasa horiek
ADAM familikoak izaten dira. Beste sheddasa bat da, adibidez, BACE1, baina askoz
substratu gutxiago mozten ditu horrek (Cole eta Vassar, 2008). Bigarren pausoan, I-
CLiP proteinek geratu den zatia mozten dute, eta mozketa bikapa lipidikoaren barruan
49

Sarrera
gertatzen da. Orokorrean bi pauso horiek errepikatzen diren arren, rhomboideen kasuan
ez da ezinbestekoa lehenengo mozketa gertatzea bigarrena gerta dadin (Wolfe eta
Kopan, 2004).
1.28. Irudia: Ezagutzen diren I-CliP-ak: S2P, SPP, presenilina eta rhomboideak. Irudian haien egitura, eta moztu ditzaketen proteina motak ikusten dira. Baita mozteguneak ere. Wolfe eta Kopan, 2004
Proteolisi mota hori jasaten duten gero eta proteina gehiago ezagutzen dira, eta
horietako adibide batzuk dira APP eta Notch, besteak beste. Biak presenilinen
substratuak dira, eta presenilinak dira lau I-CliP proteasen artean substratu aniztasun
handiena dutenak. Orain arte, 30 proteina baino gehiago ezagutu zaizkie haien
substratutzat, eta guztiak I motako transmintz proteinak dira. Horien artean, APP izan
zen lehenengo ezagutu zen substratua (Spasic eta Annaert, 2007). Presenilinek badituzte
beste berezitasunak gainerako I-CLiP-ekin alderatzean; alde batetik, haien substratuek
ez dute desbiribiltzeko beharrezkoak den aminoazidorik izaten (Wolfe eta Kopan,
2004), nahiz eta berezitasun hori ez dagoen gaur egun oso argi (Beel eta Sanders,
2008), eta bestetik, ezinbestekoa dute zenbait kofaktoreren laguntza beren aktibitatea
egiteko. Nikastrina (NCT), Aph1 fenotipoarekin eralzionatutako proteina (APH1) eta
PSren areagotzailea (PEN2) dira kofaktore horiek. Lau mintz proteina horiek elkartzean
γ-sekretasa konplexua osatzen dute zeinean presenilinak jartzen baititu bi aspartil
katalitikoak (De Strooper eta lank. 1998; Wolfe eta lank. 1999b)
50

Sarrera
1.5.3.2. γ-Sekretasaren osagaiak
Gaur egun argi dago γ-sekretasaren aktibitatea esan bezala mintz-proteina konplexu
batek gauzatzen duela; konplexu horretako oinarrizko osagaiak presenilina (PS1 edo
PS2), nikastrina (NCT), APH-1 (APH-1aL, APH-1aS, edo APH-1b) eta PEN-2 dira
(1.29. irudia) (Shah eta lank., 2005; Wolfe, 2008). Bai PSk eta baita APH-1ek ere
zenbait isoforma dituzte, eta isoforma horiek γ-sekretasa konplexuan sartu
daitezkeenez, zenbait γ-sekretasa mota daude. Horrela, presenilinaren kasuan bi
isoforma (PS1 eta PS2; PS1 isoforma nagusia da), eta APH-1en kasuan hiru isoforma
ezagutu dira gizakian (APH-1aS, APH-1aL, APH-1b). Hirurek γ-sekretasa konplexuan
parte hartzen dute, bai PS1-ekin batera, eta baita PS2-rekin batera ere. Ondorioz, sei γ-
sekretasa mota daudela pentsatzen da .
.
1.29. Irudia: γ-sekretasa proteina-konplexu bat da. Presenilina konplexu horren osagai bat da. Beste osagaiak APH-1, PEN-2 eta Nikastrina dira. Parks eta Curtis, 2007
γ-Sekretasa konplexuaren pisu molekularra ez dago argi; izan ere, hori lortzeko egin
diren saiakeretan, emaitza ezberdinak lortu dira, eta horrek eztabaida piztu du. 200-
250kDa izan da eman den zifrarik txikiena (Kimberly eta Wolfe, 2003), baina 440kDa
eta gehiago ere eman izan dira (Edbauer eta lank., 2002).
γ-sekretasa konplexuaren oinarrizko lau osagaietako bakoitzaren ezaugarri orokorrak eta
konplexuan betetzen dituzten funtzioak aipatuko ditugu:
51

Sarrera
1.5.3.2.A. Presenilina
1.5.3.2.A.a Presenilina
γ-sekretasa aspartil proteasa bat da (Wolfe eta lank., 1999a; Tsai eta lank., 2002).
Presenilina γ-sekretasa konplexuaren osagai katalitikoa da, eta bi aspartato kontserbatu
ditu bere sekuentzian, seigarren eta zazpigarren transmintz domeinuetan (TMD6 eta
TMD7) hain zuzen ere (hau da, bata presenilinaren NTF zatian eta bestea CTF zatian)
(1.30. irudia) (Esler eta lank., 2000; Li eta lank., 2000). γ-sekretasaren substratuak
mozteko ez ezik, presenilinaren endoproteolisirako ezinbestekoak dira bi aspartatoak
(Wolfe eta lank., 1999b). γ-sekretasaren gune aktiboa, beraz, presenilinaren bi zati
horien arteko interfasean kokatzen da (Esler eta lank., 2000; Li eta lank., 2000).
Presenilinaren endoproteolisian, 30 kDa-eko N muturra eta 20 kDa-eko C muturra
sortzen dira (Thinakaran eta lank., 1996a). Mozketa TMD6 eta TMD7ren artean dagoen
domeinu labur hidrofobiko batean gertatzen da (1.30. irudia). Endoproteolisi hori ez da
behar-beharrezkoa γ-sekretasa aktibitatea gerta dadin nahiz eta hasiera batean horrela
pentsatu zen (Wolfe eta lank., 1999b), baina TMD6 eta TMD7ren bi aspartato gune
aktiboen konformazio egonkorra maximizatzeko balio du (Steiner eta lank., 2008).
1.30 Irudia: Presenilinaren egitura. Irudian biribil horiz irudikatzen dira seigarren eta zazpigarren transmintz domeinuetan dauden aspartatoak. Guztiz beharrezkoak dira presenilinak gauzatzen duen proteolisia egiteko. Guztira hamar domeinu hidrofobiko ditu proteinak, baina badirudi denek ez dituztela transmintz domeinuak eratzen. Irudian bederatzi transmintz domeinu agertzen dira, eta hamargarrena zelularen zitoplasman ikusten da. Domeinu horretan gertatzen da, hain zuzen ere, presenilinaren endoproteolisia (laranjaz markatuta). Laudon eta lank., 2005.
52

Sarrera
1.5.3.2.A.b Presenilinaren egitura. Mintz- topologia
Presenilinen geneek gutxi gorabehera 50 kDa-eko proteinak kodetzen dituzte, eta
mintza zenbait alditan zeharkatzen duten mintz-proteinak dira (Wolfe, 2008).
Presenilinen mintz-topologiaren inguruan talde batzuek lortutako emaitza ezberdinen
artean eztabaida dago (Wolfe, 2008). Sekuentzia primarioaren arabera, hamar domeinu
hidrofobiko dituen arren, eta guztiak teorikoki transmintz domeinuak osa ditzaketen
arren, talde batzuek sei, zazpi eta zortzi transmintz domeinu zituztela proposatu izan
dute, eta N muturra organuluen lumenean/zelularen kanpoaldean edota zitoplasman
kokatzen zela esaten zuten (Kim eta Schekman, 2004). Hala ere, gehienbat defendatu
izan den hipotesiaren arabera, zortzi transmintz domeinuz osatuak daude, eta bai N
muturra eta baita C muturra ere, zitoplasman kokatzen dira, kate hidrofiliko luzearekin
batera (Li eta Greenwald, 1996; Doan eta lank., 1996; Li eta Greenwald, 1998).
Presenilinaren topologiari buruzko azken ikerketen arabera, bederatzi transmintz
domeinu ditu eta N muturra kate hidrofiliko luzearekin batera zitoplasman, eta C
muturra lumenean/espazio estrazelularrean kokatzen dira (1.31. irudia) (Laudon eta
lank., 2005; Oh eta Turner, 2005; Jozwiak eta lank., 2008).
1.31. Irudia: Presenilinaren egitura ezberdinak proposatu izan dira. Irudian, zortzi (Li eta Greenwald, 1996) eta bederatzi (Laudon eta lank., 2005) transmintz domeinuko modeloak.
53

Sarrera
1.5.3.2.A. c Presenilinaren zelula barneko garapena
Presenilina proteina aitzindari gisa sintetizatzen da, eta, egonkortasuna lortzeko,
garapena jasan behar du. Horretarako, konplexu baten barruan integratu behar du.
Integratzen ez diren presenilinak proteasoman suntsitzen dira (De Strooper, 2003).
Garapen-prozesu horretan presenilinaren endoproteolisia ere gertatzen da. Presenilina bi
zatitan banatzen da endoproteolisiaren ondorioz; hala, N muturreko zatia (NTF) eta C
muturreko zatia (CTF) sortzen dira. Bi zatiak banatuta baina elkarturik mantentzen dira
denbora askoan (Thinakaran eta lank., 1996a; Ratovitsky eta lank., 1997; Thinakaran
eta lank., 1997). Heterodimero horrek 1:1 estekiometria mantentzen duela uste da, eta
holoproteinak baino batazbesteko bizitza luzeagoa du; gainera, presenilinaren forma
biologiko aktiboa dela uste da (Capell eta lank., 1998).
1.5.3.2.A.d Mutazioak presenilinaren geneetan
Hasiera batean, PS Alzheimer gaixotasunarekin erlazionatutako gene gisa ezagutu zen;
izan ere, gizakian agertzen diren PSren bi homologoetan (PS1 eta PS2), mutazioak,
FAD (Familial Alzheimer´s Disease) delakoarekin lotuta agertu ziren (Giliberto eta
lank., 2009). Mutazio horiek βA42ren igoera bat eragiten zuten, APPren transmintz
domeinuko (TMD) C muturrean agertzen diren mutazioek eragiten duten bezala (Herl
eta lank., 2009). Ondorioz, bai proteasa mailan eta baita substratu mailan gerta
daitezkeen mutaketak FADrekin lotura estua dute. Kasurik gehienetan, PS1 genean
gertatzen dira mutazioak; gaur egun, 150 mutazio baino gehiago ezagutzen dira gene
horretan, eta, PS2ren genean, berriz, gutxi batzuk besterik ez (Ikus:
http://www.alzforum.org/res/com/mut/; http://www.molgen.ua.ac.be/ADMutations/ )
1.5.3.2.A.e Presenilinaren zenbait funtzio
γ-sekretasa aktibitateaz gain, beste funtzio batzuk ere egotzi zaizkio presenilinari.
Horrela, APPren garraioarekin ere zerikusia duela esan izan da (Kaether eta lank., 2002)
eta funtzio fisiologiko garrantzitsu batzuetan ere zeresana duela esan da, hala nola
kaltzioaren homeostasian, neuronen seinalizazioan, proteinen garraioan, sistema
immunean, neuriten hazkuntzan, apoptosian, oroimenean eta plastizitate sinaptikoan
(Wang eta lank., 2007; Dewachter eta lank., 2008).
54

Sarrera
1.5.3.2.B. Nikastrina (NCT)
Nikastrina γ-sekretasa konplexuaren beste oinarrizko osagai bat da. Gizakian 1.
kromosoman kodetzen den mintz-proteina integrala da, I motako mintz-proteina
integrala hain zuzen ere. ∼100 kDa-eko pisu molekularra duen glikoproteina bat da (Yu
eta lank., 2000; Wolfe, 2008). 709 aminoazido ditu; aminoazido horiek ektodomeinu
luze bat eta zitoplasmako domeinu laburra osatzen dute. Ia proteina osoa organuluen
lumenean/zelularen zati estrazelularrean kokatzen da (1.32. irudia) (Wolfe, 2008).
1.32. Irudia: Nikastrinaren egituraren eskema. Zenbait kolore erabili dira domeinu ezberdinak bereizteko. Transmintz domeinua urdinez adierazita dago, eta, ikusten da ektodomeinu luzea eta zitoplasmako isats laburra dituela. Glikosilazio-guneak ere markatuta daude (Yu eta lank., 2000).
Badirudi γ-sekretasaren substratuak hasiera batean nikastrinak ezagutzen dituela. Uste
da substratu horien N muturra ezagutzen duela (Struhl eta Adachi, 2000; Shah eta lank.,
2005; Chávez-Gutiérrez eta lank., 2008).
1.5.3.2.C. APH-1 fenotipoarekin erlazionatutako proteina (APH1) eta PSren
areagotzailea 2 (PEN2)
Presenilina eta nikastrina bikotea bakarrik ez dira gai, ez presenilinaren endoproteolisia
gauzatzeko, eta ezta γ-sekretasa aktibitatea egiteko ere. Konplexuaren beste bi osagaiak
APH-1 eta PEN2 dira (Goutte eta lank., 2002; Francis eta lank., 2002).
55

Sarrera
1.5.3.2.C.a APH-1
APH-1 genea lehenengo kromosoman kokatzen da, eta mintza zenbait aldiz zeharkatzen
duen mintz-proteina bat kodetzen du, 308 aminoazidokoa (1.33. irudia) (Francis eta
lank., 2002). 30kDa inguruko pisu molekularra du (De Strooper, 2003) eta beharrezkoa
da nikastrina zelularen mintzean kokatzeko (Goutte, eta lank., 2002). APH-1-i egotzi
zaizkio γ-sekretasa konplexuaren osagaien elkartzea, egonkortasuna mantentzea, eta
zelula barnean garraiatzea (Dries eta lank., 2008).
1.33. Irudia: APH-1 proteinaren egitura. Zazpi transmintz domeinu ditu, eta amino muturra zelularen kanpoaldean (edota organuluen lumenean) kokatzen da. Karboxi muturra, berriz, zelularen zitoplasman dago.
1.5.3.2.C.b PEN2
γ-sekretasaren oinarrizko laugarren osagaia da PEN2 (Presenilin enhancer 2) (1.34.
irudia) (Francis eta lank., 2002). PEN2ren genea hirugarren kromosoman dago.
Proteinak gutxi gorabehera, 12 kDa-eko pisu molekularra du (De Strooper, 2003) eta
101 aminoazido dituela aurreikusten da (Francis eta lank., 2002). Presenilinaren
endoproteolisian zerikusia duela uste da (Luo eta lank., 2003).
1.34. Irudia: PEN-2 γ-sekretasa konplexuaren oinarrizko laugarren osagaia da. Bi transmintz domeinu ditu, eta irudian ikusten den bezala, amino eta karboxi muturrak zelularen kanpoaldean (organuluen lumenean) kokatzen dira.
56

Sarrera
1.5.3.3. γ-Sekretasa konplexuaren eraketa
Konplexuaren estekiometria molekularra Sato eta lankideek zehaztu dute orain dela
gutxi, eta, haien arabera, 1:1:1:1 proportzioa betetzen da PS:Nct:Aph-1:Pen-2 lau
oinarrizko osagaientzat. Oinarrizkoak ez diren bestelako kofaktoreen parte-hartzea ez
dago argi. Espezie batzuetan egindako ikerketetan, ikusi da γ-sekretasaren oinarrizko
lau osagaietako bakoitzak beste hiru osagaien egonkortasun eta garapenean eragina
duela (Sato eta lank., 2007).
Konplexuaren eraketa eta garapenak lau osagaiak behar ditu, eta elkartze-prozesua
zehatz-mehatz nola eta non gertatzen den jakiteke dagoen arren, badirudi bide
sekretorioan modu goiztiarrean gertatzen dela (Kim eta lank., 2004; Capell eta lank.,
2005). Lehenengo, Nikastrina eta APH-1 elkartzen dira eta azpikonplexu bat eratzen
dute (LaVoie eta lank., 2003; Beel eta Sanders, 2008). Ondoren, presenilina elkartzen
zaio eratu den Nct-APH-1 konplexuari (Gu eta lank., 2003; Takasugi eta lank., 2003).
Azkenik, PEN-2 elkartzen da, eta horri esker presenilinaren autoendoproteolisia
gertatuko da, eta sortuko diren presenilinaren NTF/CTF heterodimeroak egonkor
mantenduko dira PEN-2ren bidez (Prokop eta lank., 2004; Watanabe eta lank., 2005).
Badago hala ere, bestelako hipotesi bat; eta, aurrekoarekin bat dator hasierako
Nikastrina-APH-1 elkarrekintzak gertatzen direlako puntuan. Baina, aldi berean, esaten
du presenilina eta PEN-2k ere beste azpikonplexu bat eratu eta ondoren elkartzen direla
lehen konplexuarekin (Spasic eta Annaert, 2008).
Esan bezala, γ-sekretasa konplexuaren eraketa bide sekretorioan modu goiztiarrean
gertatzen da. Hala, elkarturik eta modu funtzionalean dauden konplexuak bakarrik
garraiatuko dira mintz plasmatikora, edota biologikoki aktiboa den bide sekretorioko eta
endozitosi bideko konpartimentutara (Capell eta lank, 2005; Steiner eta lank., 2008).
1.5.3.4. γ-Sekretasaren substratuak
1.5.3.4.A. γ-Sekretasaren substratuen ektodomeinuaren askatzea
γ-sekretasaren substratuak, oro har, proteina aitzindari handiago baten eratorriak dira,
eta sekretasa horrek bere ekintza betetzeko, lehenengo aurrebaldintza bat bete behar
izaten da: ektodomeinuaren askatzea (Struhl eta Adachi, 2000). Beraz, presenilinak
egiten duen proteolisia, proteolisi-jarraiera baten bigarren pausoa da; lehenengoan zati
estrazelular luzea askatzen da. Lehenengo proteolisia mintzetik gertu gertatzen da, eta,
57

Sarrera
mozketaren ondoren, 30 aminoazido edo gutxiago dituen ektodomeinu txikia gelditzen
da. Modu horretan, γ-sekretasak bere substratu moduan ezagutzen du (Struhl eta
Adachi, 2000).
Zenbait faktorek eragiten dute γ-sekretasaren substratuen ektodomeinuaren askatzea
gertazeko; ligando baten loturak, substratuaren dimerizazio/oligomerizazioak,
substratuaren glikosilazio mailan gerta daitezkeen aldaketak…. (Beel eta Sanders,
2008). Ikusi da ektodomeinuaren askatze aurreko substratuaren dimerizazio edo
oligomerizazioa zenbait substratutan gertatzen dela; horien artean, APP, APLP-1 eta
APLP-2 (Scheuermann eta lank., 2001).
1.5.3.4.B. γ-Sekretasa bidezko substratuaren harrapatzea
Nikastrinaren funtzio horren inguruan gaur egun eztabaidak dauden arren (Chávez-
Gutiérrez eta lank., 2008), substratuaren ezagutzan beharrezkoa delako modeloa nahiko
zabaldua dago (Shah eta lank., 2005). Modelo horren arabera, behin substratuaren
ektodomeinua askatu ondoren, gelditzen diren aminoazido horiek ezagutzen ditu
Nikastrinaren ektodomeinuak, eta euren artean lotzen dira.
Elkarrekintza horien ondorioz, substratua hasierako “substratuaren lotura gunean”
kokatuko da (docking site) (1.35. irudia). Gune hori ez da gune katalitikoa bera. Bi gune
horiek espazialki banaturik daude zenbait aminoazidoko distantziara (3 aminoazido edo
gutxiago) (Wolfe eta Kopan, 2004).
1.35. Irudia: γ-sekretasak substratuak ezagutzeko mekanismoaren modeloa. I motako transmintz proteinak (1) sheddasa baten bidez mozten dira, eta haien ektodomeinua askatu egiten da (2). Nikastrinak gelditu den N-mutur laburrarekin elkarrekintzak ditu (3), eta, horri esker, substratua hasierako lotura gunean kokatzen da (4). Hasierako lotura gunea, mozketa gunetik nahiko gertu dago, eta hurrengo pausoan, mozketa gunera pasako da substratua. Bertan, mozketa gertatuko da (5). Shah eta lank., 2005
58

Sarrera
Modelo horren arabera beraz, hasiera batean, Nikastrinaren eta substratuaren N
muturraren arteko elkarrekintzak gertatuko dira; horrela, substratua “hasierako lotura
gunean” kokatzen da. Gune hori presenilinaren NTF eta CTFren artean kokatzen da.
Ondoren, translokazio fisiko baten bidez, edota substratuaren transmintz domeinuaren
tolesketa baten ondorioz, substratua gune katalitikoan kokatuko da, eta bertan gertatuko
da mozketa (1.36. irudia).
1.36. Irudia: γ-sekretasa bidezko mozketa. Hasieran, sustratua “hasierako lotura gunean” kokatzen da (A). Ondoren, “tolesketa” baten ondorioz, gune katalitikoan kokatzen da, eta bertan gertatuko da substratuaren mozketa. Beel eta Sanders, 2008.
1.5.3.4.C. γ-Sekretasak substratuarekiko duen zehaztasun maila
γ-sekretasa substratu asko dituen proteasa da, eta dagoeneko, jakina da I motako
transmintz proteina ugari mozten dituela (60 inguru gutxi gora behera). Substratu
horietako askok, mozketaren ondoren nukleora translokatzen diren domeinuak dituzte,
eta modu horretan zenbait generen transkipzioan eragiten dute (Beel eta Sanders, 2008).
Beste batzuen mozketa-produktuek, aldiz, ez dute horrelako eragin jakinik. Hori dela
eta, “mintz proteasoma” papera ere eman zaio γ-sekretasari (Beel eta Sanders, 2008).
γ-sekretasaren substratuek izan zitzaketen ezaugarri komunen bila ibili dira ikerlariak,
eta, besteak beste, substratuaren ektodomeinu laburra (mozketaren ondorioz lortutakoa)
eta I motako transmintz proteinak izatea (proteinaren transmintz orientazioan, N
muturra zelularen kanpoalderantz) nahiko ezaugarri errepikatuak zirela ikusi zen arren,
bi ezaugarrien aurkako adibideak agertu dira (Beel eta Sanders, 2008) eta horrek agerian
59

Sarrera
utzi du beste faktore batzuek eragina izan behar dutela. Substratuen lehenengo, bigarren
eta maila altuagoko egituren ezaugarriak aztertu dira γ-sekretasa bidezko mozketaren
substratu-baldintzak aurkitzeko bidean (Beel eta Sanders, 2008). Ez dago batere argi γ-
sekretasak substratuak zein faktorerengatik ezagutzen dituen. Badirudi aminoazidoen
sekuentziak eta substratuen bigarren mailako egiturak ez duela horretan zeresan
handirik. γ-sekretasaren zenbait substratuk haien transmintz domeinuen bidezko
elkarrekintzen ondorioz dimerizatzeko ahalmena dute. Horietako adibide bat da APP,
eta badirudi beraren homodimerizazio-prozesuak zerikusia izan dezakeela γ-sekretasa
bidezko mozketaren erregulazioan (Scheuermann eta lank., 2001).
1.5.3.5. γ-Sekretasaren mozketa guneak APPren barnean
Beste zenbait I-CLiP bezala, γ-sekretasa gai da bere substratuak gune ezberdinetan
mozteko. APPren kasuan ere horrela da eta zenbait mozketa gune ditu γ-
sekretasarentzat; γ- (APPren transmintz domeinuaren erdian kokatuta, 40-42
aminoazidoen ondoan, βAren aminoazidoen zenbaketaren arabera), ζ,- (46.
aminoazidoaren ondoan kokatuta) eta ε (49. aminoazidoaren ondoan kokatuta,
transmintz domeinuaren C muturrean) guneak (Weidemann eta lank., 2002; Zhao eta
lank., 2005). Mozketa gune horien inguruan egin diren ikerketen arabera, βA49 peptidoa
βA46ren aitzindaria izango da eta azken hori, aldi berean, βA40/42 lortzeko erdibideko
aitzindari bat da. Mozketa jarraituen modelo bat proposatzen da; lehenengo mozketa
mintza-zitoplasma interfasearen ondoan gertatzen da, eta, ondoren, C muturra moztu
zaion lekutik ~3 aminoazidoko mozketa egingo da aldi bakoitzean (1.37 .irudia).
1.37. Irudia: γ-sekretasak APPren zenbait aminoazidoren artean egin dezake mozketa. Irudian ikusten da β-sekretasa bidezko mozketaren ondoren γ-sekretasak zenbait mozketa jarraitu egin ditzakeela (ε, ζ, γ). Irudian ikusten dena mozketa jarraituen modelo bat da; azken mozketatik βA42 peptidoa askatzen da. Beel eta Sanders, 2008.
60

Sarrera
1.5.3.6. γ-Sekretasaren zelula barneko kokapena
γ-sekretasaren zelula barneko kokapena eztabaidagai da gaur egun ere, zenbait
ikerketaren arabera, leku anitzetan kokatzen dela uste baita. Horrela, erretikulu
endoplasmatikoan (Walter eta lank., 1996; Annaert eta lank., 1999; Cupers eta lank.,
2001; Kim eta lank., 2004), Golgi/TGS aparatuan (Baulac eta lank., 2003),
endosometan (Lah eta Levey, 2000), lisosometan (Pasternak eta lank., 2003),
fagosometan (Jutras eta lank., 2005), bakuola autofagikoetan (Yu eta lank., 2004; Yu
eta lank., 2005), mintz plasmatikoan (Ray eta lank., 1999; Kaether eta lank., 2002;
Chyung eta lank., 2005) eta sinapsietan (Georgakopoulos eta lank., 1999) kokatzen
dela esan izan da.
Erretikulu endoplasmatikoan (EE) kontzentrazio handian agertzen den arren, zenbait
ikerketaren arabera γ-sekretasa modu aktiboan erretikulu endoplasmatikoaren
ondorengo konpartimentuetan agertzen zaigu, post-EE konpartimentuetan alegia. Horri
paradoxa espaziala deitu izan zaio (Cupers eta lank., 2001). Ez da zehazki ezagutzen
kokapen horietako bakoitzean γ-sekretasaren aktibitate proteolitikoa zenbaterainokoa
den, baina uste da proteolisiaren gune nagusiak mintz plasmatikoa eta endosoma
goiztiarrak direla (Kaether eta lank., 2006).
1.5.3.7. γ-Sekretasa eta sinapsia
γ-sekretasa sinapsietan kokatzen dela esan izan da (Georgakopoulos eta lank., 1999).
Gainera, presenilina sinapsietan eta zelula epitelialen zelula-zelula kontaktuetan
kontzentratzen da, eta kaderina, β-katenina eta plakoglobinari lotzen zaie; hala,
kaderina-katenina konplexuaren egonkortasuna sustatzen du eta horrek aktinazko
zitoeskeletoarekiko duen lotura bultzatzen du.
PS1 knockout saguekin egindako ikerketa batean, presenilinak sinapsien osaketarekin
erlazio estua duela ikusi da. Hala, hartzaile bidezko zelula barneko seinalizazioa
moteldua gertatzen da sagu horien sinapsi glutamatergikoetan (Parent eta lank., 2005).
γ-sekretasaren substratuen artean, asko dira zelulen arteko itsasketa-molekulak.
Adibidez, nektina 1α, γ-protokaderinak, E-kaderinak eta N-kaderinak. Substratu horiek
agerian uzten dute γ-sekretasak zelulen itsasketan eta funtzio sinaptikoan garrantzi
handia duela (Parks eta Curtis, 2007). Nektina 1α neuronen arteko atxiki-loturetan
(adherens junctions) kokatzen da, eta sinapsiaren eraketan zerikusia dauka. γ-sekretasa
61

Sarrera
bidezko nektina 1αren mozketa inhibituz gero, zelulen arteko itsasketa inhibitu eta
migrazioa bultzatzen da. γ-protokaderinen kasuan, garunean espezifikoak diren
kaderinekin erlazionatutako proteinak dira, eta sinapsietako konexioetan parte hartzen
dute. γ-protokaderinaren mozketaren emaitza den γ-ICDak γ-protokaderinaren zenbait
gene sustatzailetan transkripzio-aktibitatea du. γ-protokaderinaren geneak ez dituzten
saguetan, sinapsiaren morfologian akatsak eta neuronen hiltzea izaten dute ondorio gisa.
γ-sekretasaren aktibitatearen galerak protokaderinaren mozketan izan ditzakeen
ondorioak neuronen endekatze gisa agertzen dira. Kaderinek zerikusia dute kaltzioaren
menpeko zelula-zelula itsasketan eta sinapsiaren funtzio eta plastizitate sinaptikoan.
Presenilinak, N-kaderinaren heltzea eta erretikulu endoplasmatikotik mintzerako
garraioa bideratzen du.
62

Sarrera
1.6. APP ETA BERAREN SEKRETASEN ZELULA BARNEKO
GARRAIOA
Aurretik aipatu dugu mintz barneko proteolisi erregulatua (RIP) dela APPren proteolisi
modua. Erregulazio hori zenbait modutan egiten da. Alde batetik, aurretik ere esan dugu
rhomboideen kasuan izan ezik beste kasu guztietan substratuen ektodomeinua moztuko
duten sheddasek hartzen dutela parte. Ondorioz, hasierako mozketa horretan parte
hartuko duten entzimek mugatuko dute bigarrenaren mozketa-maila; beraz, horiek
erregulazio faktore bat izango dira. Bestalde, RIP proteolisietan, bai substratuaren eta
baita entzimen (sheddasak eta iCLiP-ak) garraioa izango da prozesu horiek
erregulatzeko modu garrantzitsua; izan ere, substratu eta entzimen garraioa izango da
beren arteko topaketa bideratu edo desbideratuko duena. Hainbat talde dabiltza APPren
eta bere sekretasen zelula barneko garraioaren nondik norakoak aztertzen (Sannerud eta
Annaert, 2008).
1.6.1. SEKRETASEN GARRAIOA ETA ERREGULAZIOA
1.6.1.1. γ-Sekretasa
PS1/γ-sekretasa konplexua nagusiki erretikulu endoplasmatikoan (EE), Golgi
goiztiarrean, trans-Golgi sarean (TGS) eta endozitosi xixkuetan kokatzen da. APPren γ-
sekretasa bidezko mozketa βA peptidoa emateko nagusiki trans-Golgi sarean eta
endosometan gertatzen da (Xu eta lank., 1997; Greenfield eta lank., 1999; Tarassishin
eta lank., 2004). PS1ren beste zenbait substratuk (Notch adibidez) mintz plasmatikoan
jasaten dute mozketa hori (Tarassishin eta lank., 2004). PS1ren zelula barneko garraioa
1.38.irudian ikus daiteke (1.38. irudia).
γ-sekretsaren aktibitateaz gain, PS1ek zenbait proteinaren zelula barneko garraioan
parte hartzen du. Horietako batzuk dira γ-sekretasa konplexuaren beste osagaiak
(nikastrina, APH-1 eta PEN-2), eta baita APP molekula ere (Vetrivel eta lank., 2006;
Liu eta lank., 2009). Era berean, APPk ere proteinen axoietan zeharreko garraio
anterogrado azkarrean parte hartzen du, eta kinesinari lotuta zenbait proteina axoi-
muturrera garraiatzen ditu. Horietako batzuk dira PS1 eta BACE1 (Kamal eta lank.,
2000; Kamal eta lank., 2001). Bide horretan elkarrekin doazela eta gainera β- eta γ-
63

Sarrera
bidezko proteolisia bertan gertatu daitekeela eztabaidan dagoen arren (Lazarov eta lank.,
2005), axoietan barrena doan garraio anterogrado azkarra egiten dute hirurek, esan
bezala (Beher eta lank., 1999; Kasa eta lank., 2001; Papp eta lank., 2002; Capell eta
lank., 2002). PS1/γ-sekretasa axoietan barrena garriatua izateaz gain, kontrako alderantz
ere (soma-dendritetarantz) ateratzen da (1.39. irudia) (Capell eta lank., 2002).
1.38. Irudia: PS1/γ-sekretasa konplexua erretikulu endoplasmatikoan, Golgi goiztiar eta trans-Golgi sarean, mintz plasmatikoan, endosometan, fagosometan, mitokondrietan eta lisosometan ikusi izan da (Vetrivel eta lank., 2006). APPren γ-sekretasa bidezko mozketa nagusiki trans-Golgi sarean eta endosometan gertatzen dela uste da (Liu eta lank., 2009).
γ-sekretasaren kokapena baldintzatuko duten zenbait faktore daude. Proteina ugarirekin
izan ditzake elkarrekintzak, eta, elkarrekintza horien ondorioz, beraren kokapena
baldintzatu daiteke. Ondorioz, beraren proteolisi-funtzioa ere bai. Presenilina edota γ-
sekretasa konplexuarekin elkarrekintzak dituzten 40 proteina baino gehiago deskribatu
izan dira (Van Gassen eta lank., 2000; Parks eta Curtis, 2007). Horien artean,
garraioarekin zerikusia duten zenbait proteinarekin zuzenean elkarrekintzak dituela
ikusi da, hala nola zitoeskeletoari lotutako proteinekin (restin, GFAPε, filamin..), eta
endosoma eta xixkuei lotutako proteinekin (Rab11, Syntaxin 1A, Syntaxin 5, LRP1).
Badirudi restin, filamin eta syntaxin 5ek presenilinaren funtzio eta kokapenean eragiten
dutela, eta beste proteinekin dituen elkarrekintzek, berriz, presenilinaren garraioan edota
berak garraiatzen dituen proteinen garraioan eragiten dutela (Parks eta Curtis, 2007).
64

Sarrera
1.39. Irudia: PS1 garraio anterogradoz axoietan barrena garraiatuko da somatik axoi muturrera iritsi arte. Alderantzizko norantzan ere, soma-dendritetarantz aterako da.
Elkarrekintza horietako askok sortuko diren βA peptidoen tamaina baldintzatu, eta,
peptido horien proportzioan eragiten dutela ikusi den arren, ez dago argi eragin hori γ-
sekretasaren aktibitatearen gain zuzenean den ala konplexuaren osaketan, garraioan eta
ondorioz kokapenean eragiten duten (Spasic eta Annaert, 2008). Adibidez, Rer1p
proteinak zelulan gertatzen den γ-sekretasaren aktibitatea erregula dezake, eta
konplexuaren osaketan eragiten du (Spasic eta lank., 2007; Kaether eta lank. 2007;
Spasic eta Annaert, 2008).
Beste faktore batzuek endozitosian eragiten dute; adibidez, β2-hartzaile adrenergikoaren
kasuan, presenilinarekin elkarrekintza zuzenak izanik, beraren barneratzean eta
endosometarako garraioan eragiten du (Ni eta lank., 2006).
Bestalde, eta γ-sekretasa konplexuan 19 transmintz domeinuk parte hartzen dutela
jakinik, begibistakoa da lipidoek sortuko duten mikroingurunea garrantzi handikoa dela,
eta horrek γ-sekretasaren kokapenean eta aktibitatean eragingo duela (Runz eta lank.,
2002; Urano eta lank., 2005; Osenkowski eta lank., 2008; Hur eta lank., 2008)
1.6.1.2 α-Sekretasa
ADAM9, ADAM10 eta ADAM17 α-sekretasaren aktibitatea egiten duten entzimak
direla uste da. Guztiak I motako transmintz proteinak dira, eta aldaketak jasaten dituzte
Golgi aparatuan mintz plasmatikora garraiatzen diren arte (Lammich eta lank., 1999).
Ez da asko ezagutzen haien kokapen eta garraioari buruz, baina APPren α-sekretasa
bidezko mozketa erretikulu endoplasmatikoan, trans-Golgi aparatuan eta mintz
plasmatikoan deskribatu izan da (Parvathy eta lank., 1999; Nunan eta Small, 2000).
65

Sarrera
1.40. Irudia: α-sekretasa aktibitatea erretikulu endoplasmatikoan, trans-Golgi aparatuan eta mintz plasmatikoan deskribatu izan da.
APPren α-sekretasa bidezko mozketa aipatutako lekuetan deskribatu den arren, ADAM
proteinak preproproteina gisa sintetizatzen dira, eta prodomeinuaren askatzea Golgi
aparatuan gertatzen denez (Lammich eta lank., 1999), APP nagusiki Golgitik mintzera
bidean eta mintz plasmatikoan moztuko dute (Turner eta lank., 2003). Aktibitate
gehiena mintzean dutela uste den arren, zelula barneko aktibitatea ere deskribatu izan da
(1.40. irudia) (Huovilla, 2005).
Zelula polarizatuetan α-sekretasa basolateralki/soma-dendritetarantz ateratzen dela ikusi
da (Capell eta lank., 2002; Cole eta Vassar, 2007). Neuronetan sinapsiko alde
postsinaptikora garraiatua dela ikusi da (1.41.irudia) (Marcello eta lank., 2007).
1.41. Irudia: α-sekretasa basolateralki/soma-dendritetarantz ateratzen da zelula polarizatuetan.
66

Sarrera
1.6.1.3. β-Sekretasa
BACE1 zikloka ibiltzen da bide sekretorioan, zelularen mintzean eta endosoma-
sisteman zehar zenbait alditan, eta ziklo horietan aktibatua izan daiteke.
Bide sekretorioan barrena doanean, BACE1ek garapen prozesu bat jasaten du, N-
glikosilazio konplexu bat eta propeptidoaren askatzea, besteak beste (Capell eta lank.,
2000). Behin garatuta, BACE1 zelularen azalera garraiatua izango da, eta bertatik
berriro ere barneratua izan daiteke endosometara (Huse eta lank., 2000) (1.42. irudia).
Bere zelula barneko kokapenarekin bat dator APPren β-sekretasa bidezko mozketa ere,
trans Golgi sarean, endosometan eta mintz plasmatikoan ikusi izan direla (Vassar eta
Citron, 2000; Walter eta lank., 2001a). Hala ere, nagusiki β-sekretasa bidezko APPren
mozketa endosoma/lisosometan gertatzen da (Koo eta Squazo, 1994; Haass eta lank.,
1995, Thinakaran eta lank., 1996b).
1.42. Irudia: β-sekretasa erretikulu endoplasmatikotik Golgi aparatura garraiatzen da. Handik mintz plasmatikora, eta hortik, endozitosi bidez, endosometara garraiatua izan daiteke. Endosometatik Golgi aparatura edo mintz plasmatikora birziklatua izan daiteke. Bestela, lisosometara joango da, eta han suntsitua izango da. APP β-sekretasa bidez trans-Golgi sarean, endosometan eta mintz plasmatikoan moztua izango da. Nagusiki, endosoma/lisosometan.
BACE1en garraioa, beraren zitoplasmako domeinuaren fosforilazioaren bidez
erregulatzen da (Walter eta lank. 2001b; Pastorino eta lank 2002). Fosforilazio-egoera
horiek GGA proteinekiko (Golgi kokatzen diren eta γ-“belarria” duten, ADP
erribosilazio faktorera lotzen diren proteinak) elkarrekintzak baldintzatzen dituzte; izan
ere, proteina horiek, beren VHS domeinuaren bidez, garraiatu behar dituzten proteinen
sekuentzia zehatz batzuekin lotzen dira (DXXLL). BACE1en kasuan, DISLL (Acid
Cluster-dileucine motif edo ACDL ere deitzen zaio) sekuentzia du bere karboxi-
67

Sarrera
muturretik gertu. GGA proteinek, “kargo proteinak” garraiatzen dituzte TGS eta
endosomen artean (Wahle eta lank., 2006).
GGA proteinen artean, GGA1 nagusiki neuronetan agertzen da giza garunean. GGA1ek
BACE1en zitoplasmako domeinuarekin dituen elkarrekintzak ditu. Elkarrekintza horien
ondorioz, BACE1en garraioa (TGS eta endosomen artean eta baita endosomak eta
lisosomen artean (Cole eta Vassar, 2007)) baldintzatzen du, eta, ondorioz, β-sekretasa
bidezko APPren mozketan eragiten du. Izan ere, APP eta BACE1ek bide antzekoa
jarraitzen dute; Golgitik zelularen mintzera, eta handik endosometara barneratuak izan
daitezke biak. GGA1ek endosometatik TGSera eraman dezake BACE1, eta alderantziz,
Golgitik endosometara. Modu horretan, APPrekin konpartimendu horietan aurkitzeko
aukerak aldatzen ditu (1.43. irudia) (Wahle eta lank., 2006).
1.43. Irudia: BACE1 erretikulu endoplasmatikotik (EE), trans-Golgi sarera pasatzen da (TGS), eta hortik mintz plasmatikora. Han, BACE1en endozitosia gerta daiteke, eta endosometan (E), GGArekin dituen elkarrekintzak direla eta, Golgira garraiatua izan daiteke berriro. Bestalde, lisosometara (L) ere pasa daiteke, eta han suntsitzen da. Proteasoman (PR) ere gerta daiteke BACE1en suntsipena. Cole eta Vassar, 2007.
GGA proteinek BACE1ekiko menpekotasunik gabeko beste bide batzuen bidez ere
eragin dezakete APPren metabolismoan; izan ere, APPren kokapenean eragina duen
SorLa/LR11 GGA proteinen “kargo” proteina dela ere ikusi da, eta berarekin izan
ditzakeen elkarrekintzen bidez, APPren kokapenean eta proteolisian eragin dezakete
(Jacobsen eta lank., 2002).
In vivo BACE1 axoietan zehar garraiatua izango da, bide zulatzailean barrena (Cole eta
Vassar, 2008). Ikusi da APP eta presenilina ere, bide horretan zehar, kortex
entorrinaletik hortz-zirkunboluziorako bidean axoietan barrena garraiatzen direla
(Sheng eta lank., 2003), eta, BACE1 markatzaile presinaptikoekin batera kokatzen
dela. Zelula polarizatuetan axoietarantz/alde apikalerantz garraiatua izaten da BACE1
(1.44. irudia) (Capell eta lank, 2002; Sheng eta lank., 2003; Cole eta Vassar, 2007; Cole
eta Vassar, 2008).
68

Sarrera
1.44. Irudia: BACE1 zelula polarizatuetan alde apikalerantz/neuronen axoietarantz garraiatua izaten da modu anterogradoan.
1.6.2. APPren ZELULA BARNEKO GARRAIOA
APPk, erretikulu endoplasmatikoan sortu ondoren, mintz plasmatikora bidean, itzulpen
ondorengo aldaketak jasaten ditu; N- eta O-glikosilazioak, ektodomeinu eta
zitoplasmako fosforilazioak, sulfatazioa…..
APPren zelula barneko garraioa faktore garrantzitsua da beraren proteolisiari begira.
Izan ere, APPk, kokapenaren arabera, bide amiloidogenikoa edo ez-amiloidogenikoa
jarraituko du. Adibide gisa aipa daiteke sorLA/LR11 proteina, zeina APPri lotzen zaion
eta beraren garraioan eragiten duen. Proteina horren adierazpen-mailak APPren
proteolisian eragiten du, beraren kokapenarekin jokatuz (Shah eta Yu, 2006).
Sortutako APP molekula berrien oso proportzio txikia egongo da mintz plasmatikoan
(~% 10 jotzen da); APP molekula gehienak atseden-egoeran Golgi aparatuan eta TGSn
egongo dira. Mintzera iristean, bere zitoplasmako muturrean duen YENPTY sekuentzia
dela eta, barneratua izango da endozitosi bidez. Horrela, APP endosometara garraiatzen
da, eta handik, berriro mintzera birziklatua izan daiteke. Endosometatik jarrai dezakeen
beste bide bat suntsitze-bidea da, lisosometan alegia (1.45. irudia) (Thinakaran eta Koo,
2008).
Bestalde, neuronetan, APP modu anterogradoan garraiatua izaten da erdiko nerbio-
sistemako eta nerbio-sistema periferikoko axoietan zehar. Adibidez, kortex
entorrinaletik hipokanpoko formaziora doan bide zulatzailean barrena, axoietan zehar
garraiatua izaten da (Buxbaum eta lank., 1998b). Axoietan zehar azkar garraiatzen da
presinapsira, eta, aurretik esan bezala, denbora gutxi irauten du mintzean, barneratua
69

Sarrera
izaten baita (Koo eta lank., 1990; Lyckman eta lank., 1998). Trans Golgi Saretik
ateratzean, APP axoietan zehar modu azkarrean garraiatzen diren xixkuetan sartzen da
(Koo eta lank., 1990; Sisodia eta lank., 1993; Gralle eta Ferreira, 2007).
1.45. Irudia: Sortzen ari diren APP molekula berriak (marratxo laranjak), bide sekretorioan barrena garatzen joango dira; mintz plasmatikora iristean, berriro ere zelulan barneratuak izan daitezke endozitosiaren bidez eta, ondoren, berriro mintzera birziklatuak izan daitezke, edota lisosometara garraiatuak izan, eta han suntsipena jasan dezakete.
APP berak besikula kinesinari lotzen diola esaten duten hipotesiak badauden arren,
eztabaida dago gai horren inguruan (Kamal eta lank, 2000; Lazarov eta lank., 2005).
Modu horretan axoietan barrena sinpasira garraiatzen da. Behin xixkuak axoiaren edo
sinapsiko mintzarekin elkartuta, neuronaren gainazalean agertzen da APP, baina
agertzen diren molekula guztietatik batzuek proteolisia jasaten dute, eta beste batzuk
klatrinaren menpeko endozitosi bidez barneratuak izango dira, zelularen somara eta
dendritetara (1.46. irudia) (Yamazaki eta lank., 1995; Marquez-Sterling eta lank, 1997).
1.46. Irudia: APP, garraio anterogradoz, axoietan zehar axoi-muturrera iritsiko da. Kontrako norantzan ateratzen dela ere ikusi da, hau da, alde somato-dendritikorantz.
70

2. HELBURUAK


Helburuak
73
2. HELBURUAK Helburu orokorra:
Alzheimer gaixotasunarekin erlazionatutako proteinen ultraegitura mailako
kokapena aztertzea baldintza fisiologikoetan. Amiloide peptidoaren proteina
aitzindaria (APP), eta bere sekretasaren (PS1: presenilina-1) kokapenaren azterketa
arratoi helduen hipokanpoko CA1 guneko neuronetan.
Helburu zehatzak:
1. Amiloide peptidoaren proteina aitzindariaren (APP) kokapenaren ultraegitura
mailako azterketa arratoi helduen hipokanpoko CA1 guneko sinapsietan.
2. Presenilina-1-en (PS1) kokapenaren ultraegitura mailako azterketa arratoi
helduen hipokanpoko CA1 guneko sinapsietan.
3. Amiloide peptidoaren proteina aitzindariaren (APP) molekula osoaren eta bere
proteolisitik eratorritako zatikien kokapenaren azterketa mintz domeinu
ezberdinetan.
4. Presenilina-1 en (PS1) kokapenaren azterketa mintz domeinu ezberdinetan.


3. MATERIAL ETA METODOAK


Materialak eta metodoak
3. MATERIALAK ETA METODOAK Doktore-tesi hau egiteko, zenbait teknika erabili ditugu:
1.-Argi-mikroskopiorako teknika immunozitokimikoak:
1.1. DABren metodoa
2. Mikroskopio elektronikorako immunodetekzioa:
2.1.-Mikroskopio elektronikorako erretxinan murgildu aurreko
immunozitokimika
2.2.-Mikroskopio elektronikorako erretxinan murgildu ondorengo
immunozitokimika
3.-Western Blot teknika:
3.1.-Ehunaren homogeneizazioa eta proteinen kuantifikazioa
3.2.-Elektroforesia, transferentzia eta immunodetekzioa
4.-Sinaptosomak prestatzea
Teknika horietan guztietan dagozkien kontrolak egin dira, eta mikroskopio
elektronikoaren kasuan urre partikulen kuantifikazioa eta analisi estatistikoa ere gauzatu
dira.
Ikerketa animalien garunetan egin da; arratoi, jatorrizko sagu eta APP knockout saguen
hipokanpoetan zehazki. Arratoien kasuan, arratoi helduak izan dira denak, Sprague
Dawley arratoiak hain zuzen ere. Erabilitako saguak, berriz, C57 sagu helduak izan dira,
eta azkenik, APP knockout xaguak SN 4133, B6.129S7-Apptm1Dbo/J zepako saguak
izan dira, Charles River Laboratories-en erositakoak hain zuzen ere. Guztiekin egin
genituen esperimentuak, Europar Batasuneko Zuzendaritzak 2003ko uztailaren 22an
animaliekin ikertzeko erabakitako arauen arabera (2003/65/CE) eta Espainiako arauen
arabera (Real Decreto 1201/2005, BOE 21-10-2005) diseinatu genituen.
ERABILITAKO ANTIGORPUTZ PRIMARIOAK
Erabilitako antigorputz primarioak hauek izan ziren: APP N muturraren aurka, saguan
egindako antigorputz monoklonala (Cat: MAB 348; 22C11 (Chemicon)); APP C
muturraren aurka untxian ekoiztutako bi antigorputz poliklonal erabili ziren (Cat:
77

Materialak eta metodoak
127002 (Synaptic Systems) eta A8717; 026K4844 (Sigma-Aldrich); eta PS1en N
muturraren aurkako antigorputz poliklonala erabili zen (B19.3 (Bart De Strooper-en
oparia)). Sinaptosomekin egindako Western Blotentzat aipatutako APPren eta PS1en
aurkako antigorputzez gain, saguan ekoiztutako sinaptofisinaren aurkako antigorputz
monoklonala (Tebu, Le-Perray-en Yvelines; France), SNAP25en aurkako antigorputz
poliklonala (Synaptic Systems), eta NR1en aurkako antigorputz poliklonala (Chemicon)
erabili ziren.
Bigarren antigorputz gisa zenbait antigorputz erabili genituen, teknikaren arabera. Argi-
mikroskopiorako teknika immunozitokimikoen kasuan biotinilatutako antigorputzak
erabili ziren, zaldian ekoiztutako eta saguaren aurkako IgG-a eta ahuntzean ekoiztutako
eta untxiaren aurkako IgG-a (Vector laboratories). Mikroskopio elektronikorako
erretxinan murgildu aurreko teknika immunozitokimikoetan, nanourrez markatutako
antigorputzak erabili genituen; ahuntzean ekoiztutako eta untxiaren aurkako Fab´ zatia,
eta ahuntzean ekoiztutako eta saguaren aurkako Fab´ zatia (nanoprobes Inc. Stony
Brook, NY, USA). Mikroskopio elektronikorako erretxinan murgildu ondorengo
immunozitokimika esperimentuetan tamaina ezberdinetako urre partikulez markatutako
antigorputzak eraibli ziren; 30 nm-ko urrez markatutako ahuntzean ekoiztutako eta
saguaren aurkako antigorputza (BBInternational. EM GAM 30), eta 15 nm-ko urrez
markatutako ahuntzean egindako untxiaren aurkako antigorputza (BBInternational. EM
GAR 15). Western Blot teknikarako erabilitako bigarren antgorputzak errefau
peroxidasari lotutako Fab´ zatiak izan dira, ardian (NA 9310V. GE HealthCare UK) eta
astoan (NA 9340V. GE HealthCare UK) ekoiztuak.
78

Materialak eta metodoak
3.1. ARGI-MIKROSKOPIORAKO TEKNIKA IMMUNOZITO_
KIMIKOAK
3.1.1. DABren METODOA
Hipokanpoan, APPren N muturra, C muturra eta PS1en argi-mikroskopia mailako
kokapena aztertzeko, Avidina-Biotina Peroxidasa (ABC) metodoa erabili genuen (Hsu
eta Raine, 1981). Horretarako, arratoiak, jatorrizko saguak eta APP knockout saguak
erabili genituen.
Teknikaren oinarria:
Teknika honetan, biotinaz markatutako 2. antigorputzak erabiltzen dira. Antigorputz
horiek 1. antigorputza sortu den animalia ostalariaren aurka ekoiztu dira, eta beraz, 1.
antigorputza ezagutzen dute, eta berarekin lotzen dira. Aldi berean, 2. antigorputzaren
biotina (bitamina bat da), peroxidasa biotinilatuari konjugatutako abidina konplexuari
lotuko zaio; izan ere, biotinarentzat leku libre bat dauka konplexu horrek.
3.1. Irudia: Argi-mikroskopiorako proteinak DABz markatzeko teknikaren eskema. 1. antigorputzak antigenoa ezagutzen du. Bigarren antigorputza lehenengoari lotzen zaio, eta, aldi berean, abidina-biotina peroxidasa konplexuaz markatua dago. Hidrogeno peroxidoaren presentzian, erreakzioaren ondorioz, DABk kolore marroiko hauspeakina sortzen du.
Ondoren, DAB kromogenoa gehitzen dugu (3,3´diaminobentzidina), eta hori oxidatu
egiten da abidina-biotina konplexuaren peroxidasa eta hidrogeno peroxidoaren
presentzian; erreakzioaren ondorioz, argi-mikroskopioan ikus daitekeen marroi
koloreko produktu bat sortzen da. Peroxidasak hidrogeno peroxidoaren deskonposizio-
erreakzioa katalizatzen du, eta askatzen den oxigenoak DAB oxidatzen du.
Diaminobentzidina oxido hori disolbagaitza denez, hauspeakin marroi-gorrixka bat
sortzen da, eta argi-mikroskopioan ikus daiteke. Mikroskopio elektronikorako erabiliko
79

Materialak eta metodoak
bagenu, teknika horrekin gure proteina kokatzen den konpartimendu neuronala ikusiko
genuke, baina ez urrearekin ikus dezakegun zehaztasun-mailarekin, DAB difunditu
egiten baita. (3.1. irudia).
Materialak:
-Gailuak:
-Kriotomoa (Leica, CM 1325)
-Argi-mikroskopioa (Zeiss, Axiostar)
-Soluzioak:
-Kloral hidratoa % 4 H2O ultrapuruan:
- Chloral Hydrate FLUKA 23100
- 4 g 100 ml H2Odd-tan
-Fosfato/gatz-indargetzailea (Phosphate buffered saline; PBS):
-PBS 10X: dH2Otan. pH-a 7,4 ra egokitu.
-KH2PO4 14,7 mM
-Na2HPO4 81 mM
-NaCl 1369 mM
-KCl 26,82 mM
-PBS 1X: PBS10X hamar aldiz diluitu, eta pH-a 7,4ra
egokitu.
-Finkatze-disoluzioa : % 4 formaldehidoa 0,1 M NaPi-
tan, pH = 7,4 eta glutaraldehidoa % 0,1.
-Blokeo-soluzioak: NGS (Ahuntz sero normala) edo NHS
(zaldi sero normala) PBS1Xetan.
-ABC kit komertziala ( ABC, Elite, Vector, Burlingame,
CA).
-DAB % 0,05 PB 0,1 M-etan.
80

Materialak eta metodoak
Pausoak:
Teknika hau erabiltzean pauso hauek jarraitu genituen:
1.-ANESTESIA:
Ultraegitura mantentzeko perfusioari ekin aurretik, animaliak anestesiatu genituen,
injekzio intraperitoneal baten bidez kloral hidratoa sartuz. Erabili beharreko kantitatea
animaliaren pisuaren arabera aukeratzen genuen, eta 40 mg kloral hidrato erabiltzen
genituen animaliaren 100 g-ko.
2.-PERFUSIOA:
Lehenengo fosfato/gatz-indargetzailea (PBS=Phosphate buffered saline) (pH 7,4)
pasarazten genuen 20 segunduan gutxi gorabehera, eta ondoren finkatze-disoluzioa
pasarazten genuen.
3.-KRIOBABESA:
Behin ehuna finkatuta genuelarik, animalien garunak atera eta % 15eko sakarosa-
disoluzio kriobabeslean sartzen genituen, erabat murgiltzen ziren arte. Jarraian, %
30eko sakarosa-disoluzioan sartzen genituen, hor ere erabat murgiltzen ziren arte.
4. KRIOTOMOA:
30 µm-ko hipokanpoaren ebakin koronalak egiten genituen.
5.-IMMUNO-MARKAKETA:
A.-Lotura ez-zehatzen blokeoa:
NGS (Ahuntz sero normala) edo NHS (zaldi sero normala) erabiltzen genituen blokeoa
egiteko. Ordubete inguru izaten genituen ebakinak % 1,5eko kontzentrazioa zuen
blokeo-soluzioarekin inkubatzen, mugimenduan eta giro-tenperaturan.
B.- 1. Antigorputzarekin egindako inkubaketak:
Bi egunez egiten genituen inkubaketak, 4 ºC-an, eta mugimenduan. Antigorputzen
kontzentrazio hauek erabili genituen (3.1. taula):
81

Materialak eta metodoak
3.1. taula: Erabilitako 1. antigorputzak eta kontzentraizoak
ANTIGORPUTZA KONTZENTRAZIOAAPP Nter (Chemicon) 1:100
APP Cter (Sigma) 1:10000
APP Cter (Synaptic Systems) 1:50
PS1 (Bart De Strooper; B19.3) 1:5000
C.-Garbiketak:
PBS edo fosfato/gatz-indargetzailea erabiltzen genuen, ordubetean, mugimenduan eta
giro-tenperaturan, garbiketak egiteko.
D.- 2. Antigorputzarekin egindako inkubaketak:
Ordubeteko inkubaketa egiten genuen giro-tenperaturan eta mugimenduan. 1:200
kontzentrazioan jartzen genituen antigorputzak.
E.-Garbiketak:
PBS edo fosfato/gatz-indargetzailea erabiltzen genuen, ordubetean, mugimenduan eta
giro-tenperaturan, garbiketak egiteko.
F.-Abidina-biotina-peroxidasa konplexuarekin egindako inkubaketak:
ABC kit komertziala erabiliz, kit horretako A eta B osagaiak aurretik nahasten genituen,
eta ondoren ordubeteko inkubaketa egiten genuen giro-tenperaturan.
G.- Garbiketak:
PBS edo fosfato/gatz-indargetzailea erabiltzen genuen, ordubetean, mugimenduan, eta
giro-tenperaturan garbiketak egiteko.
H.-DABrekin egindako inkubaketak:
% 0,05eko kontzentrazioan zegoen DAB, eta % 0,01ean hidrogeno peroxidoa PB 0,1
M-etan. 5 min edo gutxiagoko inkubaketa egiten zen, ebakinek kolorea hartzen zutela
ikusten zen arte.
82

Materialak eta metodoak
I.-Garbiketak:
PB 0,1 M-etan garbiketak 30 minutuan .
6.- MUNTAIA:
Porta gelatinizatuetan jartzen genituen ebakinak.
7.-LEHORKETA:
Portaren gainean ebakinak lehortzen uzten genituen, gau bat normalean.
8.-DESHIDRATAZIOA:
Goranzko graduazioa zuten alkoholetan murgilduz, ebakinak deshidratatzen ziren: 50º,
70º, 96º, 100º, eta Xilol-etan.
9.-EBAKINEN BABESTEA:
DPX erabiliz, kristaltxo babesle bat itsasten genuen ebakinen gainean.
10.-BEHAKETA:
Argi-mikroskopioa erabiliz, gure ebakinak aztertzen genituen eta argazkiak ateratzen
genituen.
3.2. MIKROSKOPIO ELEKTRONIKORAKO TEKNIKA
IMMUNOZITOKIMIKOAK
3.2.1. ERRETXINAN MURGILDU AURREKO IMMUNOZITOKIMIKA:
NANOURREAREN PROTOKOLOA
Teknika hau erabili genuen APPren zati ezberdinen (N eta C muturrak) eta PS1en zelula
barneko kokapena aztertzeko. Horretarako, arratoiak, jatorrizko saguak eta APP
knockout saguak erabili genituen.
83

Materialak eta metodoak
Teknikaren oinarria:
Teknika immunozitokimiko honetan, 1,4 nm-ko urre partikla loturik duen 2.
antigorputzaren Fab´zatia erabiltzen dugu. Urrearen tamaina txikiari esker, ehunean
sartzeko aukera handiagoa izaten dute antigorputzek eta horrela, metodoaren
sentsibilitatea handitzen da. Behin antigorputza antigenoarekin elkartzen denean, urre
partikulak zilarrarekin intentsifikatzen dira, beren tamaina handitu eta mikroskopio
elektronikoan ikusi ahal izateko (3.2. irudia).
3.2. Irudia: Erretxinan murgildu aurreko immunozitokimikaren oinarria. 2. antigorputzaren F(ab´) zatiak lehenengo antigorputza ezagutzen du. Aldi berean, F(ab´) zatia 1,4 nm-ko urre partikula batez markatua dago. Mikroskopio elektronikoan ikusi ahal izateko, zilar partikulekin egiten da seinalearen intentsifikazioa.
Materialak:
-Gailuak:
-Bibratomoa (Energy Beam Sciences)
-Ultramikrotomoa (Reichert-Jung)
-Mikroskopio Elektronikoa (Phillips EM 208S; Jeol X-100, Peabody,
MA)
-Soluzioak:
-Kloral hidratoa % 4 H2O ultrapuruan:
- Chloral Hydrate FLUKA 23100
- 4 g 100 ml H2Odd-tan
-Fosfato/gatz-indargetzailea (Phosphate buffered saline; PBS):
-PBS 10X: dH2Otan. pH-a 7,4 ra egokitu.
-KH2PO4 14,7 mM
-Na2HPO4 81 mM
-NaCl 1369 mM
84

Materialak eta metodoak
-KCl 26,82 mM
-PBS 1X: PBS10X hamar aldiz diluitu, eta pH-a 7,4ra
egokitu.
-Blokeo-soluzioa: (% 20 NHS edo % 20 NGS PBS
1Xetan).
-Finkatze-disoluzioa : % 4 formaldehidoa 0,1 M NaPi-tan,
pH = 7,4 eta glutaraldehidoa % 0,1
-Postfinkatze-disoluzioa : % 1 Glutaraldehidoa , PBS 0,1
Metan.
-Fosfato indargetzailea (Phosphate buffer; PB 0,2 M edo
NaPi 0,2 M) pH=7,4:
- NaH2PO4 · H2O 0,04 M
- Na2HPO4 0,16 M
-Fosfato indargetzailea (PB 0,1 M)
-PB 0,2 M dH2O-tan 1:1 disolbatu
-Erretxina-propileno oxido disoluzioa (1:1; bol:bol)
-Berun zitratoa:
-Berun nitratoa 0,08 M
-Sodio zitratoa 0,012 M
-NaOH 1 N
-ddH2O-tan
-HQ Silver izeneko kit-a: HQ silver kit, Nanoprobes Inc.Stony
Brook, NY, USA
Pausoak:
Teknika hau erabiltzean, pauso hauek jarraitu genituen:
1.-ANESTESIA:
Ultraegitura mantentzeko perfusioari ekin aurretik, animaliak anestesiatu genituen
injekzio intraperitoneal baten bidez kloral hidratoa sartuz. Erabili beharreko kantitatea
85

Materialak eta metodoak
animaliaren pisuaren arabera aukeratzen genuen eta 40 mg kloral hidrato erabiltzen
genituen animaliaren 100g-ko.
2.-PERFUSIOA:
Aurretik deskribatuta zegoen bezala (Luján eta lank, 1997) transkardiakoki egin
genituen perfusioak, lehenengo fosfato/gatz-indargetzailea (PBS=Phosphate buffered
saline) (pH 7,4) pasarazten genuen 20 segunduan gutxi gorabehera, eta ondoren
finkatze-disoluzioa pasarazten genuen.
3.-BIBRATOMOA:
Behin ehuna finkatuta genuelarik, garezurretik animaliaren garuna atera, eta ehuna
bibratomoan mozteari ekiten genion. 50 µm-ko ebakinak ateratzen genituen.
4.- IMMUNO-MARKAKETA
A.-Lotura ez-zehatzen blokeoa:
Preinkubaketa egiteko NGS-an edo NHS-tan ordu batez egiten genuen
inkubaketa, aukeratutako seroan, % 20-ko kontzentrazioan fosfato/gatz-indargetzailean
(PBS 1X), eta giro-tenperaturan.
B.-1. Antigorputzaren markaketa:
Erabiltzen genuen antigorputzaren arabera, erabili beharreko kontzentrazioa
aukeratzen genuen. Antigorputza, % 1,5 seroa PBS 1X-an duen disoluzioan diluitzen
genuen zegokion kontzentrazioan. Horrela, kontzentrazio hauek erabili genituen (3.2.
taula):
ANTIGORPUTZA KONTZENTRAZIOA
APP Nter (Chemicon) 1:100
APP Cter (Sigma) 1:10000
APP Cter (Synaptic Systems) 1:50
PS1 (Bart De Strooper; B19.3) 1:5000
3.2. taula: Erabilitako antigorputzak eta kontzentrazioak
86

Materialak eta metodoak
2 egunetan 4 ºC-an eta mugimenduan mantentzen genituen ebakinak,
antigorputza ehunean sar zedin.
C.-Garbiketak:
PBS-tan eta 5-10 minuz behin likido garbitzailea aldatzen genion. 1-2 orduz
egiten genituen garbiketak.
D.- 2. Antigorputzarekin markaketa:
Bigarren antigorputza, % 1,5 seroa PBS 1X-tan duen disoluzioan diluitzen
genuen, 1:100 kontzentrazioan. 3-4 orduko inkubaketa egiten genuen, giro-tenperaturan
eta mugimenduan.
E.-Garbiketak:
PBS-tan garbiketak egiten genituen. Aurreko garbiketen antzera, 5-10 minutuz behin
likido garbitzailea aldatzen genuen, baina, kasu honetan, gau osoan uzten genuen PBS-
tan, 4 ºC-an eta mugimenduan.
5.-POSTFIXAPENA:
Ebakinak % 1 glutaraldehido PBS 1X-tan duen disoluzioan inkubatzen genituen 10
minutuz.
6.-GARBIKETA:
Ur ultrapuruz garbitzen genituen ebakinak 30 minutuan eta 5 minutuz behin ura
aldatzen genuen.
7.-ZILARREZ AREAGOTU:
Zilar areagotzailearen HQ Silver izeneko kit-a (HQ silver kit, Nanoprobes Inc. Stony
Brook, NY, USA) erabiltzen genuen, 12 minutuz. Hori gela ilunean egiten genuen, argi
gorriaren laguntzaz.
8.-GARBIKETAK:
Ur ultrapuruz garbiketak egiten genituen, oraindik ere gela ilunean, bizpahiru alditan.
Egun-argitara atera, eta garbiketa gehiago egiten genituen 30 minutuz; eta 5-10 minutuz
87

Materialak eta metodoak
behin ur ultrapurua aldatzen genuen. Jarraian, 0,1 M fosfato indargetzaileaz (PB 0,1 M),
garbiketa gehiago egiten genituen 30 minutuz.
9.- OSMIFIKAZIOA:
Osmifikatzeko, kanpaian eta 20 minutuz inkubatzen genituen ebakinak fosfato
indargetzailean disolbatutako % 1 osmio tetroxidorekin. (Proportzioa: OsO4 1 mL; PB
0,2 M 2 mL; ddH2O 1 mL; osmioa % 4ean dagoenez, azkenean % 1ean geratuko
zaigu). Horrekin, zelulen mintzak kontrastatzeaz gain, zilarra eta urrearen markaketa
finkatua geratzen zitzaigun.
10.- GARBIKETAK:
Garbiketak fosfato indargetzaile 0,1 Mean. 30minutuz.
11.-ALKOHOLEN BIDEZKO DESHIDRATAZIOA:
Gero eta gradu altuagoa zuten alkoholetan inkubatzen genituen ebakinak. % 50eko
alkoholetik hasten ginen, ondoren % 70ekoa, % 96koa, eta % 100ekoa. Bakoitzean
5minutu inkubatzen genituen, eta % 100ekoan 3 alditan eta 5 minutuz aldi bakoitzean.
Azkenik, propileno oxidotan inkubatzen genituen ebakinak, 3 alditan, 5 minutuz aldi
bakoitzean.
12.-ERRETXINAN MURGILTZEA:
EPON erretxina eta propileno oxidozko nahastea 1:1 proportzioan erabiliz, ebakinak
bertan jartzen genituen erretxinan murgil zitezen. Mugimendu ez oso arinean uzten
genituen gau osoan eta giro-tenperaturan, propileno oxido guztia lurrundu zedin.
Gau guztiaren ondoren, erretxina purua gehitzen genien, eta beste bizpahiru orduz uzten
genituen mugimenduan.
13.-ERRETXINAREN POLIMERIZAZIOA:
Xafletan murgiltzen genituen ebakinak (Flat embedding), eta estufan sartzen genituen
60 ºC-an gutxienez 2 egunean.
14.- INTERESATZEN ZAIGUN EHUNAREN ZONA AUKERATU:
Mikroskopio fotonikoa (argi ikuskorrekoa) erabiliz, gure intereseko zona topatzen
genuen (hipokanpoko CA1 gunea) erretxinan murgilduta genituen ebakinetan eta zatitxo
88

Materialak eta metodoak
hori mozten genuen kontu handiz laban batekin. Zatitxoa erretxinazko kapsula baten
puntan itsasten genuen, kola erabiliz. Behin horrela, ehunaren zati ultrafinak mozten
genituen ultramikrotomoan.
15.-ULTRAMIKROTOMOA
Zati semifinak mozten genituen lehenengo, diamantezko labanatxoa erabiliz. 0,99 µm-
ko zatiak mozten genituen. Richarson tintzioa erabiliz, immunomarkaketa guk
mikroskopio elektronikoan ikusi nahi genuen eskualdean ikusten genuenean, ebakin
ultrafinak mozteari ekiten genion. Diamantezko labanatxoa hartu, eta 60-80 nm
bitarteko mozketak egiten genituen, eta lortutako ebakinak nikelezko saretxoetan
biltzen genituen.
16.-KONTRASTEA BERUN ZITRATOAZ (Mintzak kontrastatzeko):
% 2,5ean zegoen berun zitratoaz, 15-30 minutuz kontrastatzen genituen gure ebakin
ultrafinak.
Ondoren, ur ultrapuruz garbitzen genituen.
17.- BEHAKETA:
Mikroskopio elektronikoan materiala aztertu, eta argazkiak ateratzen genituen.
3.2.2.-ERRETXINAN MURGILDU-ONDORENGO IMMUNOZITOKIMIKA
Teknika hau erabili genuen APPren N muturraren, C muturraren eta PS1en zelula
barneko kokapena ezagutzeko. Zehazki, sinapsian non kokatzen ziren ikusteko erabili
genuen metodo hau. Horretarako, arratoiak erabili genituen.
Teknikaren oinarria:
Izenak berak adierazten duen bezala, metodo honekin, immunozitokimika esperimentua
ehuna erretxinan murgildu ondoren egiten da. Ehuna erretxinan murgildu ondoren, zati
ultrafinak moztu eta ebakin horietan egiten da immunozitokimika. Horrela, teknika
honetan, laginak lehenengo finkatu, erretxinan murgildu, moztu, eta saretxoetan
muntatzen dira immunomarkaketari ekin aurretik. Ondorioz, eta hau da teknikaren
abantaila nagusia, epitopo guztiak agerian daudenez, markatuak izateko probabilitate
89

Materialak eta metodoak
bera dute. Laginek erretxinan murgiltzeko, disolbatzaile organikoen bidezko (azetona
edo alkoholak) deshidratazio bat jasan behar dute; horrek antigenoen eraldaketak ekar
ditzake eta antigenizitatea jaisten da. Gainera, galera hori areagotu egiten da normalean
erabiltzen diren erretxinek polimerizatzeko behar dituzten tenperatura altuak direla eta
(Araldita eta Epoxy) (60 ºC). Horri aurre egiteko aukera bat lagina metakrilato antzeko
plastikoetan murgiltzea da (Lowicryl K4M eta HM20); izan ere, kasu horretan
deshidratazioa tenperatura baxuetan egiten da alkoholen bidez, eta antigenoak gutxiago
eraldatzen dira. Gainera, polimerizazioa, argi ultramoreei esker, 45 ºC-an gerta daiteke.
Horregatik, modu horretan murgildutako laginek abantailak dituzte ondoren egingo
diren mikroskopio elektronikoarentzako teknika immunozitokimikoei begira. Hori da
guk erabili dugun modua gure laginen murgilketa egiteko.
Horrela bada, teknika honetan, zati ultrafinen immunomarkaketa egiten da tamaina
ezberdinetako urrez markatuta dauden 2. antigorputzekin. Kasu honetan ez da
zilarrarekin intentsifikatu behar, urre partikulek mikroskopio elektronikoan ikusi ahal
izateko tamaina nahikoa izaten baitute. Gainera, immuno bat baino gehiago egin
daitezke batera tamaina ezberdineko urreak erabiliz (3.3. irudia).
3.3. Irudia: Erretxinan murgildu ondorengo immunozitokimikaren oinarria. Ebakin ultrafinak saretxoetan bildu, eta lehenengo antigorputzak interesatzen zaigun proteina ezagutzen du. 2. antigorputza lehenengoari lotzen zaio, eta, aldi berean, 2. antigorputz horrek tamaina ezberdinetako urre partikulak ditu lotuak. Mikroskopio elektronikoan urre partikula horiek ikusten ditugu.
Oso baliagarria da teknika hau kuantifikazioak egiteko; gure kasuan, helburua sinapsiko
partikulen ikerketa bat egitea zenez, erretxinan murgildu aurreko teknika
immunozitokimikoek antigorputzarentzat sor ditzaketen sarketarako oztopoak
gainditzeko ere oso baliagarria da teknika hau. Tauletan ikusten dugun bezala (3.3. eta
3.4. taulak), teknika bakoitzak bere abantaila eta desabantailak ditu, eta horrela bata
bestearen osagarri bihur daiteke.
90

Materialak eta metodoak
Erretxinan murgildu aurreko immunozitokimika (Preembedding):
3.3. taula: erretxinan murgildu aurreko immunozitokimikaren abantaila eta desabantailak
ABANTAILAK DESABANTAILAK
Sentikortasun handia Immunorreaktiboak ehunean difunditzen
dira
Argi-mikroskopio eta mikroskopio
elektronikoko teknikak konbinagarriak
Ez da kuantitatiboa
Ez da aparatu berezirik behar Urrez markatutako immunomarkaketa
bikoitzak ezinezkoak
Ehuna MEko irudi konbentzionalen
moduan ikusten da: ondo bereizten dira
zelulen elementu ezberdinak
Ez du aukerarik ematen kokapen zehatz
batzuetako proteinak ikusteko (sinapsiko
proteina batzuk, adibidez)
Erretxinan murgildu ondorengo immunozitokimika (Postembedding):
. 3.4. taula: erretxinan murgildu odorengo immunozitokimikaren abantaila eta desabantailak.
ABANTAILAK DESABANTAILAK
Proteinen epitopoak eskuragarri ikusi ahal
izateko (sinapsian adibidez)
Murgildu aurreko teknikak baino
sentikortasun txikiagoa
Immunorreaktiboak ez dira ehunean
difunditzen
Aparatu berezi eta garestien beharra
Metodo kuantitatiboa da Ehunaren antigenizitatea murrizten da
Tamaina ezberdinetako urreak erabiliz,
askotariko markaketak egin daitezke (3
markaketa ezberdin immuno batean)
Ezin da argi-mikroskopiarekin konbinatu
Ehunaren ultraegitura ez da murgildu
aurreko teknika bezain argia, jasan duen
murgiltze-prozesua gogorragoa baita.
91

Materialak eta metodoak
Materialak:
-Gailuak:
-Bibratomoa (Energy Beam Sciences)
-Kriofinkatze Sistema Unibertsala (KF80; Reichert-Jung, Wien, Austria)
-AFS (Automatic freeze substitution, (Reichert-Jung, Wien, Austria)
-Argi-mikroskopioa (Zeiss. Axiostar)
-Ultramikrotomoa (Reichert-Jung)
-Mikroskopio elektronikoa (Phillips EM 208S; Jeol X-100, Peabody,
MA)
-Soluzioak:
-Kloral hidratoa (Fluka, Chemika)
-Fosfato/gatz-indargetzailea (Phosphate buffered saline; PBS)
-Finkatze-disoluzioa: % 4 formaldehidoa 0,1 M NaPi-tan, pH = 7,4 eta
glutaraldehidoa % 0,1
-HNO3 % 10 disoluzioa dH2O-tan
-Etanol % 70 disoluzioa dH2O-tan
-TBST eta NaBH4 eta Glizina: TBST % 0,1 NaBH4 eta 50 mM Glizina
-TBST: dH2O-tan
-Tris-HCl: 50 mM pH 7,4
-NaCl: 0,15 M
-NaN3: % 0,02
-Tritón X-100: % 0,1. Bukaeran gehitzen zaio.
-Blokeo soluzioa. % 3 BSA TBST-tan
-Uranilo azetatoa
-Berun zitratoa:
-Berun nitratoa 0,08 M
-Sodio zitratoa 0,012 M
-NaOH 1 N
-ddH2O-tan
92

Materialak eta metodoak
Pausoak:
Teknika hau erabiltzean, pauso hauek jarraitu genituen:
1.-ANESTESIA:
Ultraegitura mantentzeko perfusioari ekin aurretik, animaliak anestesiatu genituen,
injekzio intraperitoneal baten bidez kloral hidratoa sartuz. Erabili beharreko kantitatea
animaliaren pisuaren arabera aukeratzen genuen, eta 40 mg kloral hidrato erabiltzen
genituen animaliaren 100 g-ko.
2.-PERFUSIOA:
Lehenengo fosfato/gatz-indargetzailea (PBS=Phosphate buffered saline) (pH 7,4)
pasarazten genuen 20 segunduan gutxi gorabehera, eta ondoren finkatze-disoluzioa
(NaPi) pasarazten genuen.
3.-BIBRATOMOA:
Behin ehuna finkatua genuelarik, animaliaren garuna atera eta ehuna bibratomoan
mozteari ekiten genion jarraian. Bibratomoan 1 mm-ko ebakinak mozten genituen, eta
ondoren, guri interesatzen zitzaigun eskualdea mozten genuen (CA1) (1 mm x 1 mm x 1
mm), karratu-itxurako lagin bat lortuz. Hauxe izango zen erretxinan murgilduko genuen
zatia. Murgildu aurretik egun batzuk itxaron behar bazituen, erabilitako likido
finkatzailearen (NaPi) 1:10 disoluzioan sartzen genuen.
4.-TENPERATURA BAXUAN ERRETXINAN MURGILDU ETA IZOZTE
BIDEZKO ORDEZKAPENA:
A.-Lehenik eta behin, ehuna kriobabestu behar zen modu honetan:
-% 10 (w/v)glizerol NaPi-tan 30 minutu.
-% 20 (w/v)glizerol NaPi-tan 30 minutu.
-% 30 (w/v)glizerol NaPi-tan gau guztia 4 ºC-an.
B.- KF80 “Sistema Criofijación Universal” erabiliaz, eta -190 ºC-an dagoen nitrogeno
likidoa erabiliaz hoztutako propanol likido hotzean murgiltzen zen ehuna.
C.- AFS (Automatic freeze substitution) aparatura eramaten zen ehuna; horretarako,
hoztutako pintzak erabiltzen ziren. Aparatu hori hoztu egiten zen lehenengo.
93

Materialak eta metodoak
D.-Izozte bidezko ordezkapenerako, ehuna metanol anhidroa eta % 0,5 azetato uraniloa
zuen soluzio batean murgiltzen zen gau guztian zehar. Hasieran -90 ºC-an egoten zen,
eta orduro 4 ºC-ko igoera bat izaten zuen; azkenean 45 ºC-raino iristen zen.
E.- Ehuna zenbait alditan metanol anhidroz garbitzen zen, ur-soberakina eta uranilo
azetatoa kentzeko.
F.- Lowicryl HM20an murgiltzeko pausoa pixkanaka egiten zen, pausoz pauso;
Lowicryl proportzioa handitzen zen bitartean, metanolarena jaisten zen. Azkenean,
Lowicryl purua izatera iristen zen:
-Lowicryl:Metanol 1:2 proportzioan ordubete.
-Lowicryl:Metanol 1:1 proportzioan ordubete.
-Lowicryl:Metanol 2:1 proportzioan ordubete.
-Lowicryl puruan gau guztian.
G.- Polimerizazioa 360 nm-ko uhin-luzerako argi ultramorez katalizatzen zen.
-45 ºC-an bi egun egoten zen eta, jarraian, giro-tenperaturan egun bat.
5.-ULTRAMIKROTOMOA:
Zati semifinak mozten genituen lehenengo, diamantezko labanatxoa erabiliz. 0,99 µm-
ko zatiak mozten genituen. Richarson tintzioa erabiliz, ehunera iritsi ginela ikustean,
zati ultrafinak mozteari ekiten genion. Diamantezko labanatxoa hartu, eta 60-80 nm
bitarteko mozketak egiten genituen, eta formvar deritzon pelikula zuten nikelezko
saretxoetan biltzen genituen zati ultrafin horiek, ondoren bertan immunomarkaketa egin
ahal izateko.
6.-ZATI ULTRAFINEN IMMUNOMARKAKETA:
Tantatxoetan egiten zen immunomarkaketa, 30 µl-ko tantak gutxi gorabehera.
Garbiketak egiteko, nahi izanez gero mintz osoa zegokion garbiketa disoluzioan
murgiltzen genuen, baina bestelako pausoak tantetan egiten genituen.
Mintzaren garbiketa
1.-Mintza hartu, eta % 10 HNO3 ur ultrapuruan sartzen genuen 30 minutuz eta giro-
tenperaturan. Horrela, saretxoak kokatzen genituen zulotxoak garbitzen genituen.
2.-Ur ultrapuruan sartzen genuen mintza, minutu bat prezipitatu-ontzi batean, eta beste
minutu bat beste prezipitatu-ontzi batean.
3.-% 70 Etanol disoluzioan sartu-irten bat egiten genion.
94

Materialak eta metodoak
Immunomarkaketa
Saretxoak mintzean kokatzen genituen kontu handiz, pintzekin formvar filma puskatu
ez genezan.
A.-Ehunen garbiketa:
% 0,1 NaBH4 eta 50 mM glizina dituen TBST soluzioaz garbitzen genituen ehunak 10
minutuan.
B.-Garbiketak:
Ehunak TBST-tan garbitzen genituen, 3x1 minutu, eta azken garbiketa bat 5-10
minutukoa.
C.-Lotura ez-zehatzen blokeoa:
10 minutuan % 3 BSAan blokeatzen eduki genituen gure ehunak antigorputzaren lotura
ez-zehatzak ekiditzeko, blokeo-soluziozko tantatxoetan.
D.- 1. Antigorputzarekin egindako inkubazioa:
Antigorputz bakoitza, zegokion kontzentrazioan diluitzen genuen erabilitako blokeo-
soluzioan (% 3 BSA) (3.5. taula). Gau osoan eta giro-tenperaturan egiten genituen
tantatxoetan inkubaketak.
3.5. taula: Erabilitako antigorputzak eta kontzentrazioakANTIGORPUTZA KONTZENTRAZIOA
APP Nter (Chemicon) 1:100
APP Cter (Sigma) 1:1000
APP Cter (Synaptic Systems) 1:50
PS1 (Bart De Strooper; B19.3) 1:500
E.-Garbiketak:
TBSTtan. 3x1 minutuz, eta 5-10 minutuko azken garbiketa bat.
95

Materialak eta metodoak
F.-Lotura ez-zehatzen blokeoa:
Aurretik erabilitako blokeo-soluzio bera erabiliz (% 3 BSA), 10 minutuko beste blokeo
bat egiten genuen.
G.- 2. Antigorputzarekin egindako inkubazioa:
Blokeo-soluzioan (% 3 BSA TBST-tan) 1:20 kontzentrazioan diluitzen genuen urreaz
markatutako antigorputza. Bi orduan edukitzen genuen inkubaketa tantatxoetan eta giro-
tenperaturan. Erabilitako antigorputzak:
H.-Garbiketak:
3x1 minutuan ur ultrapuruan, eta berriro ere azken garbiketa bat 5-10minutuan
7.-KONTRASTEA URANILO AZETATOZ:
10 mg/mL-ko kontzentrazioko disoluzioa erabiliz, tantatxoetan inkubaketa egiten
genuen 20 minutuz.
8.-GARBIKETAK:
3x1 minutuz ur ultrapuruan, eta berriro ere azken garbiketa bat 5-10 minutuz.
9.- KONTRASTEA BERUN ZITRATOZ:
% 2,5ean zegoen berun zitratoaz, 15-30 minutuz kontrastatzen genituen gure zati
ultrafinak.
10.-GARBIKETAK:
Ur ultrapuruzko tantatxoetan egiten genituen garbiketak.
11.- BEHAKETA:
Mikroskopio elektronikoan materiala aztertu, eta argazkiak ateratzen genituen.
96

Materialak eta metodoak
3.3. WESTERN BLOT TEKNIKA
3.3.1. HIPOKANPOAREN HOMOGENEIZAZIOA ETA PROTEINA
KONTZENTRAZIOAREN NEURKETA:
Arratoi, jatorrizko sagu, eta APP sagu knockout-en hipokanpoak homoegneizatu
genituen, eta haien proteina kantitatea neurtu genuen, ondoren Western Blot teknikaren
bidez APP N muturra, C muturra eta PS1en immunomarkaketari ekiteko.
• HOMOGENEIZAZIOA:
Teknikaren oinarria:
Zelulak apurtzeko, lisi soluzio indargetzailea erabiltzen da. Ondoren, abiadura
ezberdinetan zentrifugazioak eginez, zelulen osagaiak abiaduraren arabera hauspeatu
edota gainjalkinean geldituko dira (López de Jesús M eta lank., 2006) (3.4. irudia).
Horretan oinarrituz, interesatzen zaizkigun zelula-osagaiak banatu ditzakegu, ondoren,
proteina kantitatea neurtu eta Western Blot-a egin ahal izateko (3.5. irudia).
Materialak:
-Gailuak:
-Homogeneizagailua (Polytron Aggregate. KINEMATIKA.
Luzern)
-Ultrazentrifuga. Kontron Centrikon T2070
-Soluzioak:
-Homogenizazio-soluzio indargetzailea
10mM Tris-HCl, pH 7,4
%1 (b/b) Proteasen inhibitzaileen koktela
H2O bolumena bete arte
97

Materialak eta metodoak
Pausoak:
1. 100-400 mg ehun + Homogeneizazio soluzio indargetzailearen (10 mM Tris-
HCl indargetzaile pH 7,4 + proteasen inhibitzaileak) 15 bolumenetan.
2. Homogeneizatu potter-ean (2´ / 800 rpm)
3. Zentrifugatu: 4´ / 3.500 rpm / 4 ºC-an
4. P1 eta S1 zatikiak eskuratu
P1 (Nukleoak eta organuluen mintzak)
S1
- Soberakina
- Ultrazentrifugatu: 35´ / 35.000 rpm (100.000 g) / 4 ºC-an
- P2 eta S2 zatikiak eskuratu
P2 (mintz plasmatikoak)
- Hauspeakina (garbitu buffer-aerkin) + 150 µl soluzio indargetzailea
- Proteina neurtu (Bradfford)
S2 (zitosol)
- Soberakina
- Proteina neurtu (Bradfford)
Zentrifugatu1500 g / 10 min / 4 ºC
Ultrazentrifugatu100000 g / 35 min / 4 ºC
Mintzzelularrak
GainjalkinaHomogeneizazio-bufferra gehitu
Homogeneizatu800 rpm / 1 min / 4 ºC2 43500
Zentrifugatu1500 g / 10 min / 4 ºC
Ultrazentrifugatu100000 g / 35 min / 4 ºC
Mintzzelularrak
GainjalkinaHomogeneizazio-bufferra gehitu
Homogeneizatu800 rpm / 1 min / 4 ºC2
Zentrifugatu1500 g / 10 min / 4 ºC
Ultrazentrifugatu100000 g / 35 min / 4 ºC
Mintzzelularrak
GainjalkinaHomogeneizazio-bufferra gehitu
Homogeneizatu800 rpm / 1 min / 4 ºC2 43500
3.4. Irudia: Ehunak homogeneizatzeko orduan jarraitzen genituen pausoen eskema.
98

Materialak eta metodoak
3.500 / 4´ S2 S1 35.000 / 35´
P2 P1 3.5. Irudia: Abiadura ezberdinetako zentrifugazioekin zelulen osagaiak jasotzen genituen. Irudian, koloretan agertzen zaizkigu P1 (nukleoak), S2 (zitoplasma), eta P2 (mintzak)
• PROTEINA-KONTZENTRAZIOAREN NEURKETA (BRADFORD
METODOA):
Teknikaren oinarria:
Teknika hau, Coomasie urdin disdiratsuak polipeptidoekiko duen afinitatean oinarritzen
da (Bradford, 1976). Teknika kolorimetrikoa da, hau da, zenbait konposatu kimikok
argia xurgatzeko duten gaitasunean oinarritzen da. Koloratzailea proteinetara lotzean,
gai kimiko horren xurgapenaren uhin-luzeran aldaketa sortzen da: 465 nm-tik 595 nm-ra
pasatzen da. Koloratzailea asetzailea denez, xurgapenaren gehikuntza proteina-
kontzentrazioarekiko zuzenki proportzionala da. Hala, xurgapenaren gehikuntza
espektrofotometroan neurtuz, proteina kantitate ezagunekin egindako xurgapenaren
patroi-zuzen batean interpola daiteke.
Materialak:
-Gailuak:
-Espektrofotometroa
-Soluzioak:
-BSA soluzioa (1 mg/mL)
-Bradford errreaktiboa (BioRad 5000006)
-Erreaktiboak:
-BSA (behi sero-albumina, A8022, SIGMA)
99

Materialak eta metodoak
Pausoak:
1.-Patroi-zuzena prestatzea:
BSA soluzioaren bolumen bat (taula) eta Bradford erreaktiboa nahasten ziren (3.6.
taula).
3.6. taula: Patroi-zuzena prestatzeko erabilitako osagai eta kantitateak
Ur destilatua
(µL)
BSA soluzioa
(µL)
Bradford
erreaktiboa (µL)
800 0 200
796 4 200
792 8 200
788 12 200
784 16 200
780 20 200
Espektrofotometroan, 595 nm-tan absorbantzia neurtu eta patroi-zuzena ateratzen
genuen; hala, absorbantzia-emaitzak kontzentrazioarekiko proportzionaltasun zuzena
azaltzen zutela ziurtatzen da.
2.-Laginak prestatu:
Gure laginen homogeneizatuak hartu, eta hiru erreplika Bradford erreaktiboarekin
nahasten ziren. (800 µL dH2O, 1 µL lagin, 200 µL Bradford erreaktibo erreplika
bakoitzean).
Ondoren, espektrofotometroan absorbantzia neurtu, eta patroi-zuzenean interpolatuz,
proteina-kontzentrazioa ateratzen zen.
3.3.2. WESTERN BLOT (WB): ELEKTROFORESIA (SDS-PAGE),
TRANSFERENTZIA ETA INMUNODETEKZIOA.
Teknika hau erabili genuen APPren N muturra eta C muturra ezagutzen zuten
antigorputzek APPren zein zati zehatz ezagutzen zuten ikusteko, markatzen zuten pisu
molekularrean oinarrituz. Horretarako, arratoiak, jatorrizko saguak, eta APP knockout
saguak erabili genituen.
100

Materialak eta metodoak
Teknikaren oinarria:
Elektroforesia proteinen nahasketa konplexuak banatzeko erabili ohi da, eta helburua,
banaketaren ondoren proteinak identifikatzea edota purifikatzea izaten da. SDSren
presentzian egindako dimentsio bakarreko poliakrilamidazko gel-elektroforesian (SDS-
PAGE), proteinak, eremu elektriko bati esker, geleko poroetan zehar mugitzen dira.
Akrilamida-kontzentrazioa handitzean poroen tamaina txikitzen denez, gelaren poroen
tamainak eta proteinaren karga, neurri eta itxurak mugatuko dute migrazio-abiadura.
Gelean korriarazitako proteinak nitrozelulosa, nylon edo PVDFzko (Polyvinylidene
difluoride) mintz baten azalari itsasteari transferentzia deritzo. Prozesu hori
elektroforesiz egiten da, eta behin prozesua bukatuta, proteinen tindaketa itzulgarria
egiteko aukera dago.
Immunodetekzioa, antigorputz monoklonal edo poliklonal baten bidez, guk detektatu
nahi dugun proteina bilatzeko erabiltzen da. Lehenengo, antigorputza gure proteinari
lotuko zaio. Ondoren, bigarren antigorputzarekin inkubatuko dugu. 2. antigorputz hori
entzima bati lotua egongo da (errefau peroxidasa), eta kimioluminiszentziako
osagaiekin konjugatzean, ECL substratua erreaktibo sentikor bihurtzea katalitzatzen du
HRP konplexuak. Erreakzioaren ondorioz, daukagun proteina-kontzentrazioaren
araberako luminiszentzia emango digu; izan ere, hidrogeno peroxidasa bidezko
oxidazioaren ondorioz, karbonilo hirukoitz kitzikatua sortzen da, zeinak argia botatzen
duen karbonilo bakar bihurtzean. Argazki-film batean errebelatuko dugu.
Materialak:
-Gailuak:
-Elektroforesi eta transferentzia-euskarriak. Mini Trans-Blot III.
Bio-Rad
-Soluzioak:
-Tris-HCl/3M/pH 8,8 Trizma base 3 M ddH2Otan.
-Tris-HCl/0,5 M/pH 6,8 Trizma base 0,5 M ddH2Otan.
-Sodio dodezil sulfatoa (SDS) % 10 ddH2Otan.
-Persulfato amonikoa % 10 dH2Otan.
-Proteinentzako karga indargetzailea:
-Urea 10 M
-SDS 0.35 M
101

Materialak eta metodoak
-DTT 1,5 M
-Tris-HCl 1 M pH=8 (0,1 M bukaeran)
-dH2Otan
-bromophenol urdina
-Elektroforesi indargetzailea 10X:
-Trizma base 0,25 M
-Glizina 1,92 M
-SDS % 20
-dH2Otan
-Elektroforesi indargetzailea 1X:
-10X Elektroforesi indargetzailea 10 aldiz diluituta.
-Transferentzia indargetzailea 10X:
-Trizma base 0,25 M
-Glizina 1,92 M
-dH2Otan.
-Transferentzia indargetzailea 1X:
-% 20 Etanol
-% 8 Transferentzia indargetzailea10X
-% 72 dH2O
-Blokeo soluzioa: % 5 Hauts-esne esnegaingabetua
-Garbiketa-soluzioak:
-TBS10X: 1,5 M NaCl; 200 mM Tris-HCl; pH 7,4
-TBS 1X: TBS10X 100 mL; 1 L-raino dH2O
-TBST: TBS1X; % 0,1 Tween-20
-Stripping indargetzailea: guanidina-tiozianatoa (6 M) dH2Otan.
-Erreaktiboak:
-Persulfato amonikoa: APS; A9464, Sigma
-Proteasa inhibitzaileak: Complete protease inhibitor coctail
tablets. ROCHE.
-TEMED: N,N,N´,N´, Tetramethylethylene-diamine. T9281.
Sigma.
-Sec-butanol: 2-Butanol B8.591-9 Aldrich.
102

Materialak eta metodoak
-Tween 20, P1379. Sigma-Aldrich
-SDS: Sodium Dodecyl Sulfate L3771. Sigma
-Akrilamida: Acrylamide/Bis % 40 ,29:1. 161-0146. Biorad
-Kimioluminiszentzia-areagotzailea: Enhanced
chemiluminesence; ECL eta ECL plus. Westerm blotting detection reagents and
analysis system. Amersham biosciences.
Pausoak:
1.-GELAK PRESTATZEA:
Guk detektatu nahi genuen proteinaren pisu molekularrean oinarrituz zenez, taula
teoriko batez baliatuz, prestatu beharreko gelaren kontzentrazioa erabakiten genuen.
Gure kasuan % 7,5 akrilamida-kontzentrazioa zuten gelak prestatzen genituen
gehienetan, baina pisu molekular baxuko APPren C muturra detektatu nahi genuenean,
% 18 akrilamida-kontzentrazioko gelak prestatu genituen (3.6.A irudia).
3.6. Irudia: A) Gelen akrilamida portzentaiaren arabera, pisu molekularrak ze altueratan gelditzen diren erakusten duen taula. B) Transferentzia hezea gauzatzeko egiten den muntaia.
103

Materialak eta metodoak
Tauletan agertzen dira bi gel prestatzeko erabiltzen genituen kantitateak:
Gel korriarazlea (Running gel):
3.7 taula: Gel korriarazlea prestatzeko erabilitako osagaiak eta kantitateak OSAGAIA BOLUMENA
Tris HCl 3M pH=8,8 2,8 mL
Acrylamida %40 4,2 mL
SDS %20 112,5 µL
APS %20 175 µL
TEMED 15 µL
DH2O 15,23 mL
Gel korriarazlea prestatzean, sec-butanol gehitzen genion, gelaren azala mailakatua
geldi zedin. Polimerizatzen zenean, sec-butanola kendu eta dH2O-z ondo garbitzen
genuen gel kontzentratzailea gehitu aurretik.
Gel kontzentratzailea (Stacking gel):
3.8. taula: Gel kontzentratzailea prestatzeko erabilitako osagaiak eta kantitateak OSAGAIA BOLUMENA
Tris HCl 0,5M pH=6,8 3,75 mL
Acrylamida %40 1,45 mL
SDS %20 75 µL
APS %20 90 µL
TEMED 20 µL
DH2O 10 mL
2.-LAGINAK PRESTATU ETA GELEAN KORRIARAZTEA:
Hipokanpo homogeneizatuak prest genituelarik, eta haien proteina-kontzentrazioa
neurtuta genuelarik, kale bakoitzean 30-50 µg proteina kargatzen genituen. Bai,
hipokanpoko mintzen homogeneizatua, bai zati disolbagarria kargatzen genituen.
104

Materialak eta metodoak
Horretarako, lehenengo, homogeneizatuak, proteinentzako karga-indargetzailearekin
berreseki edo diluitzen ziren, eta 3-5 minutuan irakiten jartzen ziren. Jarraian, gelean
kargatzen genituen.
Proteinak, 126,127 V-ean korriarrazten ziren, bromofenol urdina gelaren bukaerara iritsi
zela ikusten genuen arte.
3.-TRANSFERENTZIA- METODO HEZEA:
Irudiko muntaia erabiliz, (6B irudia) 100V-eko eremu elektrikopean jartzen genuen 1h-
1h 30´-an.
4.-IMMUNO-MARKAKETA:
A.-Lotura ez-zehatzen blokeoa:
Lotura ez-zehatzak ekiditeko, mintza ordubetean blokeo-soluzioan murgiltzen zen, giro-
tenperaturan, eta mugimenduan.
B.- 1. Antigorputzarekin egindako inkubaketa:
Gau osoan, eta 4 ºC-an egiten zen inkubaketa,mugimenduan. Antigorputzen
kontzentrazio hauek erabili ziren TBS 1Xetan (3.9. taula):
ANTIGORPUTZA KONTZENTRAZIOA
APP Nter (Chemicon) 1:500
APP Cter (Sigma) 1:4000
APP Cter (Synaptic Systems) 1:500
PS1 (Bart De Strooper) 1:5000
3.9. taula: Erabilitako antigorputzak eta kontzentrazioak
C.-Garbiketak:
Garbiketak egiteko, TBST soluzioa erabiltzen zen. 5-15 minutuko hiru garbiketa egiten
ziren.
105

Materialak eta metodoak
D.- 2. Antigorputzarekin egindako inkubaketa:
Antigorputz hauek 1:3000 kontzentrazioan, TBS 1X etan erabili genituen. Bi orduko
inkubaketa egiten zen, giro-tenperaturan eta mugimenduan.
E.-Garbiketak:
Garbiketak egiteko, TBST soluzioa erabiltzen zen. 5-15 minutuko hiru garbiketa egiten
ziren.
5.-ERREBELAKETA:
Mintza ECL edo ECL plus kit-ak erabiliz, kit-en osagaiekin murgiltzen zen. Jarraian,
gela ilunean, argazki-filma gainean jarriz, eta errebelatzeko likidoez baliatuz,
antigorputzen bandak errebelatzen ziren.
6.-STRIPPING-A:
Mintzak berrerabiltzen genituenean, stripping deituriko teknika erabiltzen zen. Mintza,
guanidina-tiozianatoan (6M), 5-10 segundoan murgiltzen zen; gero, garbiketak egiten
ziren TBSTrekin, eta berriro, lotura ez-zehatzen blokeotik hasiz, beste proteina baten
immunodetekziorako erabil zitekeen.
106

Materialak eta metodoak
3.4. SINAPTOSOMAK PRESTATZEA
Sinaptosomak prestatu genituen, sinapsiko mintzak isolatu eta lortutako mintz horietan
Western Blot teknika erabiliz, APPren zatiak eta PS1 kokatu ahal izateko. Horretarako,
arratoiak erabili genituen.
Teknikaren oinarria:
Zelula barneko zenbait osagaik dituzten dentsitate isopikniko ezagunetan oinarrituz,
pauso bakarreko prozedura bat erabiliz prestatzen dira sinaptosomak (Cohen RS eta
lank. 1977). Ultrazentrifugadora abiadura ezberdinetan erabiliz, sakarosa-gradienteekin
eta pH-arekin jokatuz, sinaptosomak isolatzea eta ondoren beraren mintzak banatzea
lortzen genuen.
Pausoak:
Sinaptosomak prestatzea:
1.- Lau arratoi helduren hipokanpoak 3,75 mL-ko disoluzioan homogeneizatzen
genituen (0.32 M sakarosa, 0.1 mM CaCl2, 1 mM MgCl2, 1X proteasa inhibitzaile
koktail-a, Roche). Hori 4 ºC-an egiten genuen, Teflon-glass homogeneizatzailea
erabiliz.
2.-Homogeneizatu bakoitza, 1,25 M sakarosa-kontzentraziora eramaten genuen, 18 mL
2 M sakarosa, 7,5 mL 0.1 mM CaCl2 + 3X Complete protease inhibitor cocktail (Roche)
gehituz. Ondoren, 14 x 95 mm-ko hiru Ultraclear ultrazentrifugaren hodietan banatzen
genituen homogeneizatuak.
3.- Hodi bakoitzean, 2,5 mL 1.0 M sakarosa, 0,1 mM CaCl2 gehitzen ziren, eta
hodiaren goialdea, homogeneizazio-soluzioz betetzen genuen. Ondoren, SW40 Ti
errotorean (Beckman) zetrifugazioa egiten genuen, 100000 x g-tan, hiru orduz, eta 4 ºC-
an.
4.- Zentrifugazioaren ondoren, sinaptosomak sakarosa 1,25 M /1 M interfasean jasotzen
genituen.
107

Materialak eta metodoak
5.-Jasotako sinaptosomak, homogeneizazio-disoluzioan (40 mL) berresekitzen genituen;
ondoren, zentrifugazio bidez kontzentratzen genituen (15000 x g, 30 min, 4 ºC) .
6.-Pellet-ean gelditzen ziren sinaptosomak, homogeneizazio-soluzioan (1 mL)
berresekitzen genituen, eta -80 ºC-an gordetzen genituen, aurrerago prozesatu ahal
izateko.
Sinaptosomen mintzen banaketa:
Hori egiteko, Phillips eta lankideek (Phillips GR eta lank., 2001) deskribatutako
metodoa jarraitu genuen.
1.- Lehendik prestatutako sinaptosomak (1 mL), 0,1 mM CaCl2-tan 1:10 diluziora
eraman genituen. Lortutako esekidura, 20 mM Tris, pH 6.0 eta % 1 TritonX100
bukaerako kontzentraziora eraman genuen.
2.- 30 minutuan, eta 4 ºC-an, mintzak askatu ziren (estrakzioa), eta lortutako disoluzioa,
14 x95 mm-ko bi Ultraclear ultracentrifugadora hoditan (Beckman) banatu ziren. SW40
Ti (Beckman) errotorea erabiliz (40000 x g, 30 min, 4 ºC), disolbagaitza zen materiala
pellet-ean gelditzen zen. Lortutako gainjalkina (zati estrasinaptikoari zegokiona)
izotzetan gordetzen genuen ondorengo pausoetarako.
3.- Pellet bakoitza 5 mL-ko disoluzioan (20 mM Tris, pH 8,0, % 1 TritonX100)
berresekitzen zen. 30 minutuz estrakzioa egiten zen, eta aurreko estrakzio-pausoan
bezala zentrifugazioa egiten zen beriro ere. Urrats horretan lortutako gainjalkina zati
presinaptikoari zegokion.
4.- Aurreko bi pausoetan lortutako gainjalkinak (zati estrasinaptikoa eta zati
presinaptikoa) hartu, eta azetona erabiliz (3xbolumena), 2 orduz eta -20 ºC-an, zati
horietan zeuden proteinen hauspeatzea eragiten genuen. Ondoren, 18000 x g abiaduran,
30 minutuan eta 15 ºC-an, zentrifugazioz berreskuratzen genituen.
5.- 3. pausoan lortutako pellet-a (zati postsinaptikoa) eta 4. pausoan lortutako proteinen
hauspeakina, % 5 SDS-tan disolbatzen ziren.
108

Materialak eta metodoak
6.- Hasierako homogeneizatuaren 100 µL eta aurreko prozeduran lortutako
sinaptosomak ere, % 5 SDS kontzentraziora diluitzen genituen proteinen
kontzentrazioa neurtu aurretik.
Sinaptosoemekin eta haietatik eratorritako zatikiekin Western Blot-ak:
Sinaptosomak eta haietatik eratorritako zatikiak (mintz estrasinaptikoa, mintz
presinaptikoa eta mintz postsinaptikoa) erabiliz, Western Blot esperimentuak aurretik
deskribatu dugun bezala egin ziren. Antigorputz bakoitzarekin zatiki bakoitzean
lortutako bandak elkarren artean alderatu genituen, horrela kasu bakoitzean proteinaren
presentzia nagusiki presinaptikoa, postsinaptikoa edo estrasinaptikoa zen ikusi zen.
Kimioluminiszentziaz lortutako bandak zirenez, banden detekzioa eta banden arteko
intentsitatearen alderaketa Chemi Genius2 analisi sistemaren bidez egin zen, GeneSnap
eta GeneTools software-ak (Syngene) erabiliz. Modu horretan egindako 3 esperimentu
erabili ziren gutxienez histogramak egiteko eta % ± SEM moduan adierazi ziren.
Proteina batentzat zenbait banda lortzen zirenean kolore ezberdinetako gezi-muturren
bidez seinalatzen dira eta histograman gezi bakoitzari dagokion banda kolore berdina
erabiliz adierazten da. Histogramako datuen analisi estatistikoa “bide bakarreko
ANOVA”-ren bidez (one way ANOVA) egin da, eta ezberdintasunak esanguratsuak
direnean hizki ezberdinen bidez adierazita daude (p<0,05).
Sinaptosomak eta bere zatikiak ondo banatuta zeudela jakiteko erabilitako antigorputzak
eta kontzentrazioa (urdinez) 3.10. taulan agertzen dira:
1. ANTIGORPUTZA 2. ANTIGORPUTZA
mAb anti-synaptophysin
(clone SY38; 1:2500; Tebu, Le-Perray-en
Yvelines; France)
Errefau peroxidasari lotutako F(ab´).
Ardian ekoiztua saguaren aurkka (GE
Healthcare UK)
pAb anti-SNAP25 (1:10000; Synaptic
Systems)
Errefau peroxidasari lotutako F(ab´).
Astoan ekoiztua untxiaren aurka (GE
Healthcare UK).
pAb anti-NR1 (1µg/mL; Chemicon) Errefau peroxidasari lotutako F(ab´).
Astoan ekoiztua untxiaren aurka (GE
Healthcare UK).
3.10. taula: Sinaptosomen kontroletarako erabilitako antigorputzak.
109

Materialak eta metodoak
3.5. KONTROLAK
Esperimentuen fidagarritasunaz ziurtatzeko, eta bereziki, antigorputz komertzialek
zehaztasun arazoak ematen dituztenez, zenbait kontrol egin genituen.
3.5.1. APP KNOCK OUT SAGUekin KONTROLAK
Aurretik deskribatu dugun bezala, argi-mikroskopioa, mikroskopio elektronikoa eta
Western blot metodoak erabili genituen sagu knockout-ekin kontrolak egiteko. APPrik
ez zutenez, 1. antigorputzaren eta 2. antigorputzaren kontrolak egiten genituen sagu
horiek erabiliz.
3.5.2. 2. ANTIGORPUTZAREN KONTROLAK
Argi-mikroskopioa, mikroskopio elektronikoa eta Western blot metodoak erabiliz egin
genituen kontrolak. 1. antigorputza bota gabe eta beste pauso guztiak berdin mantenduz,
immunomarkaketa gertatzen ote zen begiratzen genuen. Markaketa gertatzen bazen, 2.
antigorputza ez-espezikfikoa zen zerbaiti lotzen zitzaiola ondorioztatzen genuen.
3.6. KUANTIFIKAZIOA ETA ANALISI ESTATISTIKOA
Erretxinan murgildu aurreko eta ondorengo immunozitokimika-esperimentuak n=3
arratoiekin egin genituen antigorputz bakoitzeko. Arratoi bakoitzeko hiru esperimentu
egin genituen, eta ziurtatu genuen ez zegoela taldeen arteko ezberdintasun
esanguratsurik.
Urre partikulen kuantifikazioa egiteko, Imaje J programa erabili genuen. Horrela,
sinapsiaren alde presinaptikoan eta postsinaptikoan zeuden urre partikulak kontatzen
genituen. Partikula kopuruak kontatzen genituen sinapsietan eta mintz
estrasinaptikoetan. Imaje J programarekin, mintz horien luzera neur genezakeen, eta
beraz, urre partikulak/mikra kontatzen genituen. Kasu guztietan sinapsitik 30 nm-ko
distantziara zeuden urre partikulak sinapsitik kanpo zeudela kontsideratu genuen.
110

Materialak eta metodoak
Erretxinan murgildu aurreko immunozitokimika bidez lortutako emaitzetan, APPren N
muturraren aurkako antigorputza erabili genuen kasurako n=79 sinapsitako urre
partikulak kontatu genituen, eta baita sinapsi horiei zegokien mintz estrasinaptikoko
urre partikulak ere (terminal eta dendritetan). APPren C muturraren aurkako
antigorputzarekin lortutako emaitzetan, n=84 sinapsitan egin genuen zenbaketa. Eta
PS1en kasuan, berriz, n=85 sinapsitan.
Erretxinan murgildu ondorengo immunozitokimika bidez lortutako emaitzetan, APPren
N muturraren aurkako antigorputza erabiliz lortutako emaitzetan, n=141 sinapsi erabili
genituen kontaketa egiteko. APPren C muturraren aurkako antigorputza erabiliz, n=139
izan zen, eta PS1en kasuan, n=135. Kasu honetan ere, sinapsi horiei zegokien mintz
estrasinaptikoetako urre partikulak ere kuantifikatu ziren, bai terminalen mintzetako
partikulak eta baita dendritetakoak ere.
Behin datu guztiak bilduta, analisi estatistikoari ekiten genion. Emaitzen atalean
erakusten diren balioak kuantifikazioaren ondoren lortutako batez besteko balioak ±
SEM gisa emanda daude. Batez besteko balioen arteko analisi estatistikoa eta
ezberdintasunak esanguratsuak ziren ala ez Graph Pad PRISM (version 4.0; Graph Pad
Software Inc. San Diego, CA, USA) erabiliz lortu genituen. “Bide bakarreko” ANOVA
(ez-parametrikoentzat zuzenketekin) eta Bonferroniren post test-a erabiliz
ezberdintasunak esanguratsuak ziren ala ez ikusten genuen.
111


4. EMAITZAK


Emaitzak
4. EMAITZAK
4.1. ANTIGORPUTZEN KONTROLAK
4.1.1. ANTIGORPUTZ PRIMARIOEN KONTROLAK
APPren kontrako antigorputzen kasuan, APPren generik gabeko sagu knockout-ak
erabili dira antigorputz primarioen kontrolak egiteko. Sagu horiek erabiliz eta
genetikoki manipulaturik ez zeuden saguekin alderatuz, hau da, jatorrizko saguekin
alderatuz, APP modu zehatzean ezagutzeko erabilitako antigorputzen gaitasuna aztertu
da. Teknika bat baino gehiago erabili dira kontrolak egiteko; izan ere, batzuetan,
antigorputz bat teknika baterako baliagarria izateak ez du esan nahi beste teknika
guztietan ere modu egokian aritzen denik. Beraz, Western Blot teknika, argi-
mikroskopioa eta mikroskopio elektronikoa erabili dira antigorputzen fidagarritasun
maila aztertzeko.
PS1en kontrako antigorputz primarioa Bart De Strooperrek emandako antigorputza da;
lehenago karakterizatua izan dena (De Strooper eta lank., 1997; Hébert eta lank., 2004).
Hona hemen lortutako emaitzak:
4.1.1.1. Western Blot
4.1.1.1.1. APPren C muturraren kontrako Sigma antigorputza
Antigorputz honek APPren C muturra ezagutzen du. APP695 isoformaren 676-
695 aminoazidoei dagokien peptido sintetikoaren kontra ekoiztu da
(KMQQNGYENPTYKFFEQMQN).
Western Blot esperimentuetan 110 kDa inguruko banda bat lortu zen mintzari
zegokion kalean jatorrizko saguen kasuan. Banda hori desagertu egiten da sagu
knockout-en kaleetan, eta baita jatorrizko saguen zati disolbagarrian ere. Pisu molekular
hori bat dator APPrentzat teorikoki deskribatutako pisuarekin. (4.1.A irudia).
4.1.1.1.2. APP ren C muturraren kontrako Synaptic Systems antigorputza
Antigorputz honek APPren C muturreko azken hamabost aminoazidoen
GYENPTYKFFEQMQN sekuentzia ezagutzen du; izan ere, sekuentzia hori duen
peptido baten aurka sintetizatutako antigorputza da. Egindako esperimentuetan, 110
kDa inguruko banda bat ikus daiteke jatorrizko saguen mintzari dagokion kalean. Banda
115

Emaitzak
hori sagu knockout-ei dagokien kaleetan desagertu egiten da, eta baita jatorrizko saguen
zati disolbagarriari dagokion kalean ere (4.1.B irudia).
4.1. Irudia: Sagu arrunten eta sagu knockout-en hipokanpoetan APPren C muturraren kontrako antigorputz komertzialak erabiliz egindako Western Blot-a. Kasu bakoitzean, hipokanpoen mintzak eta zati disolbagarriak bereizi dira zentrifugazio-abiadura ezberdinak erabiliz. Kale guztietan 50 µg proteina kargatu dira. A) Sigma antigorputz komertzialarekin egindako esperimentuak. B) Synaptic Systems antigorputzarekin egindako esperimentuak.
4.1.1.1.3. APPren N muturraren kontrako Chemicon antigorputza
Antigorputz honek APPren amino muturreko 66-81 aminoazidoen sekuentzia ezagutzen
du (KEGILQYCQEVYPELQ). Egindako esperimentuetan lortutako emaitzak 4.2.
irudian ikus ditzakegu. 110 kDa inguruan, banda bat agertzen da jatorrizko saguen
hipokanpoei dagokien kaleetan bai mintzari dagokion kalean, bai zati disolbagarriari
dagokionean. Banda horiek desagertu egiten dira erabat sagu knockout-en hipokanpoei
dagokien kaleetan.
4.2. Irudia: APPren N muturra ezagutzen duen Chemicon antigorputz komertzialaz, jatorrizko saguen eta sagu knockout-en hipokanpoen zati disolbagarri eta mintz plasmatikoaren zatiak erabiliz lortutako immunoblot-a. Kale bakoitzean 30 µg proteina sartu dira.
116

Emaitzak
4.1.1.2. Argi-mikroskopioa
Teknika honekin ere APPren aurka erabilitako hiru antigorputzen zehaztasun-maila
ikusi nahi izan genuen. Horretarako, sagu knockout-en eta jatorrizko saguen
hipokanpoetan, DABz errebelatutako immunozitokimikaren bidez, markaketak alderatu
ditugu. Erabilitako hiru antigorputzen kasuan jatorrizko saguen hipokanpoko CA1
guneko markaketa patroia (4.3. irudiko A, C eta E argazkiak) desagertu egiten da sagu
knockout-en kasuan (4.3. irudiko B, D eta F argazkiak).
4.3. Irudia: APPren kontrako zenbait antigorputzekin egindako immunomarkaketaren argi-mikroskopioko irudiak. A, C eta E irudietan jatorrizko saguen hipokanpoko CA1 guneko neurona piramidalen geruzaren immunomarkaketa ikusten da. A eta C irudietan APPren C muturraren aurkako bi antigorputz erabili dira, eta E irudian N muturraren aurkako antigorputza erabiliz lortutako immunomarkaketa ikusten da. B, D eta F irudietan ikusi sagu knockout-ekin egindako esperimentuetan lortutako markaketa eza. Hiru kasuetan ikus daiteke immunomarkaketa desagertu egiten dela. Irudi guztietan eskalak 50µm ditu.
117

Emaitzak
4.1.1.3. Mikroskopio elektronikoa
Mikroskopio elektronikorako egin den immunomarkaketaren kontrol moduan,
erretxinan murgildu aurreko immunozitokimika erabili dugu. Kontrolen kasuan, APPren
sagu knockout-ak erabili ditugu antigorputz primarioen zehaztasun-maila neurtzeko.
Sagu knockout horietan immunomarkaketa egin ondoren, jatorrizko saguekin alderatu
ditugu erabilitako antigorputzak fidagarriak diren ala ez erabakitzeko. Horrela, aurreko
teknikekin egin dugun bezala, APPren karboxi muturra ezagutzen duten bi
antigorputzen, eta APPren amino muturrra ezagutzen duen antigorputzaren kontrolak
egin ditugu. Emaitza hauek lortu ditugu:
4.1.1.3.1. APP ren C muturra ezagutzen duen Sigma antigorputza
Lehenengo antigorputz gisa, KMQQNGYENPTYKFFEQMQN sekuentzia ezagutzen
duen Sigma antigorputza erabili dugu. Antigorputz hori ezagutzen duen bigarren
antigorputza 1´4 nm-ko urre partikulaz markatuta dago, zeinaren seinalea zilarraz
areagotu dugun mikroskopio elektronikoan ikusteko (4.4. irudia). Irudian ikusten da
sagu knockout-ekin egindako esperimentuetan ez dela immunomarkaketarik agertzen.
4.4. Irudia: APP ren C muturra ezagutzen duen Sigma antigorputzarekin egindako immunozitokimika, sagu knockout-en hipokanpoko CA1 geruzako neuronetan. Bigarren antigorputza urrez markatuta dago. A) Nukleo bat agertzen zaigu (n) urre partikularik gabe. B) Urre partikularen bat ikusten da (gezia) eta lotura ez-zehatz bat adierazten du. Gainerakoa garbi dago. C) Sinapsiak (gezia), mitokondriak (m) eta bestelako organulu intrazelular guztiak urre partikularik gabe agertzen ziren. D) Nukleo bat (n), eta ondoan terminal bat (t) dendrita batekin (d) sinapsia egiten, mielinaz inguratutako axoi batekin (a) ondoan. Azterturiko eremuetan ez zen markaketa esanguratsurik agertu. Argazki guztietan, eskalak 400 nm ditu.
118

Emaitzak
4.1.1.3.2. APP ren C muturra ezagutzen duen Synaptic Systems antigorputza
Aurretik aipatu dugun bezala, antigorputz honek APPren C muturreko azken hamabost
aminoazidoak ezagutzen ditu. Antigorputz honekin egindako esperimentuetan ere,
mikroskopio elektronikorako egindako esperimentuetan, sagu knockout-ak erabiliz,
immunourre partikulak agertzen diren ala ez ikusi nahi izan dugu antigorputzaren
zehaztasun maila aztertzeko. Ikus daiteke sagu knockout-ekin egindako esperimentuetan
ez dela immunomarkaketarik agertzen (4.5. irudia).
4.5. Irudia: APPren C muturraren aurkako Synaptic Systems etxe komertzialeko antigorputza erabiliz egindako imunozitokimika. Irudietan ikus daitekeenez, ez da agertzen APP proteina edo beraren zatikiak seinalatzen dituen urre partikularik. Terminal eta dendriten arteko sinapsiak urrerik gabe agertzen dira. Oso kasu gutxitan agertzen diren urre partikulak geziz seinalatuak daude A eta C irudietan. Mitokondriak: m; mielinadun axoiak: a; termialak: t; dendritak: d. Eskalak 400 nm ditu irudi guztietan.
4.1.1.3.3. APPren N muturra ezagutzen duen Chemicon antigorputza
Antigorputz honekin ere, nahiz eta kasuren batean immunomarkaketa ez-zehatz bat
erakusten duen urre partikularen bat ikus daitekeen, oro har sagu knockout-en ehunean
egindako esperimentuetan ez da urre partikularik agertzen. (4.6. irudia).
119

Emaitzak
4.6. Irudia: APPren N muturra ezagutzen duen Chemicon antigorputzarekin APP sagu knockout-etan egindako immunozitokimika. Irudietan ikusten da terminaletan (t), dendritetan (d), mitokondrietan (m), sinapsietan (A, B geziak) eta mielinadun axoietan (a) ez dela urre partikularik agertzen. Kasu batzuetan lotura ez-zehatzak adieraziz urre partikula batzuk agertu diren arren (C geziak), oro har sagu knockout-etan urre partikularik agertzen ez dela ikus daiteke irudietan. Argazki guztietan eskalak 400 nm ditu.
4.1.2. BIGARREN ANTIGORPUTZEN KONTROLAK
Antigorputz hauen baliagarritasuna aztertzeko, material eta metodoetan aipatu bezala,
erabilitako tekniketan, esperimentuak antigorputz primarioarekin inkubatu gabe eta
beste pauso guztiak berdin utziz egin ditugu. Horrela, bigarren antigorputzen
zehaztasun-maila ikus daiteke. Era horretan, ikusi dugu immunomarkaketa ez-zehatzik
ez dela agertzen. Irudiak ez ditugu jarri.
120

Emaitzak
4.2. EMAITZAK
Jatorrizko saguak sagu knockout-ekin alderatuz, antigorputzen zehaztasun-maila ikusi
ondoren, APPren kasuan antigorputz horiek erabiliz, Sprague Dawley arratoiekin egin
dira esperimentuak. Bestalde, PS1en kokapena ere aztertu nahi izan dugu. Emaitza
hauek lortu ditugu:
4.2.1. WESTERN BLOT TEKNIKAREKIN LORTUTAKO EMAITZAK
Western Blot esperimentuak arratoien hipokanpoen homogenatuekin egin dira.
Homogenatuak zati disolbagarrien eta mintz plasmatikoaren zatienak dira, zeinak
zenbait zentrifugazio-abiadura erabiliz lortu ditugun, material eta metodoen atalean
azaldu dugun bezala.
4.2.1.1. APPren kontrako antigorputzak erabiliz lortutako emaitzak
APP proteina osoa eta proteolisia jasan ondoren proteina horretatik eratortzen diren
zatiak pisu molekularren arabera ezagututa, APPren aurkako (eta aurretik aipatutako)
hiru antigorputzak erabiliz detektatu nahi izan ditugu. Horretarako, arratoien
hipokanpoko homogenatua, bi zatitan banatu dugu, zenbait zentrifugazio-abiadura
erabiliz, eta horrela, APP molekula oso gisa, mintz plasmatikoan edo zati disolbagarrian
dagoen ikusi dugu, eta gauza bera proteolisiaren ondoren APPtik eratortzen diren
zatiekin. APPren C muturraren kontra erabilitako bi antigorputzekin mintzei dagokien
zatietan, 110 kDa inguruko banda bat lortu dugu. Zati disolbagarrian banda hori
desagertu egiten da (4.7. irudia). APPren N muturra ezagutzen duen antigorputza
erabiliz, bai mintzeko homogenatu zatietan, eta baita zati disolbagarrietan ere, 110 kDa
inguruko banda lortu dugu, eta hori bat dator, lehenago esan bezala, APPrentzat
deskribatutako pisu molekular teorikoarekin (4.7. irudia). N muturraren antigorputzak
estrazelularki askatzen den APParen zatikia ezagutzen du zati disolbagarrian agertzen
denean. Bestalde, APPren C muturraren aurkako antigorputzak erabiliz mintzaren zatian
17 kDa-etik beherako bandak lortu dira. Banda horiek APPren proteolisitik eratorritako
CTFei dagozkie (4.7. irudia). Banda horiek zati disolbagarrian desagertu egiten dira
beti; bai mintz plasmatiko zein zelula barneko mintzekin erlazionaturik baitaude.
121

Emaitzak
4.7. Irudia: Arratoien hipokanpoen zati disolbagarriak eta mintzak bereizi ondoren egindako Western Blot-en emaitzak. A) APPren C muturra ezagutzen duen antigorputzekin bandak mintz plasmatikoaren zatian lortzen genituen. Banda horiek APP osoari dagozkie (Goran eta erdian, APP C) APPren N muturraren aurkako antigorputza erabiliz APP osoari edo N muturra daraman zatikiari dagokion banda zati disolbagarrian eta mintzaren zatian lortzen da. B) APPren C muturraren aurkako antigorputzekin (Sigma goran eta Synaptic Systems beheran), 17 kDa-en azpitik APPren proteolisitik eratorritako CTFei dagozkien bandak lortu ditugu mintzei dagokien kaleetan.
4.2.1.2. PS1en N muturraren kontra erabilitako antigorputzarekin lortutako emaitzak
Presenilinaren immunodetekzioa egiteko ere, arratoien hipokanpoak hartu eta
homogeneizatu ondoren zentrifugazio-abiadura ezberdinak erabiliz mintz plasmatikoa
eta zati disolbagarria banatu ditugu. Bi zati horietan Western Blot-a egin dugu PS1en
aurkako B19.3 antigorputza erabiliz. Antigorputz hori presenilinaren N muturraren
aurkako antigorputza da. Egindako esperimentuetan, mintz plasmatikoari dagokion
zatian 50 kDa inguruan banda bat agertzen da. Zati disolbagarrian, ordea, ez da bandarik
agertzen. Pisu molekular hori, PS1 osoari dagokion pisu molekular teorikoarekin bat
dator (4.8. irudia).
4.8. Irudia: PS1en N muturraren aurkako antigorputza erabiliz egindako Western Blot-a. Banda mintz plasmatikoan agertzen da 50kDa inguruan. Zati disolbagarrian ez da bandarik agertzen.
122

Emaitzak
4.2.2. ARGI-MIKROSKOPIAZ EGINDAKO ESPERIMENTUEN EMAITZAK
APPren eta PS1en kokapena aztertu da argi-mikroskopia mailan (4.9. irudia).
4.9. Irudia: Arratoien hipokanpoan APPren aurkako hiru antigorputzekin (N muturraren aurkakoa (Chemicon), C muturraren aurkakoa (Sigma eta Synaptic Systems), eta PS1en aurkako antigorputzarekin (B19.3) egindako immunomarkaketa. Ezkerreko zutabean (A, D, G, J irudiak) hipokanpoaren ikuspegi orokorra agertzen da. Irudi horietan eskalak 200 µm ditu. Erdiko eta eskubiko zutabeetan, berriz, CA1 guneko zelulak handipen gehiagorekin ikusten dira. Zelula piramidalek immunomarkaketa erakusten dute (gezi beltzak). Baita zenbait interneuronak ere (gezi urdinak C, I eta L irudietan). Kasu batzutan dendrita apikalek ere APPren C muturraren antigorputzarekin markaketa erakusten dute (F irudia gezi urdinak). Eskala-barra B, C, E, F, H, I, K eta L irudietan: 50 µm.
Immunomarkaketa egiteko, zenbait antigorputz erabili dira (Western Blot-etan erabili
direnak): APPren N muturra ezagutzen duena, APPren C muturra ezagutzen dutenak eta
PS1en aurkako antigorputza. DABrekin errebelatu ondoren, ikusi dugu
immunomarkaketaren hauspeakinak kasu guztietan, hipokanpoko zelula piramidalen
geruza markatzen duela. APPren kasuan, hiru antigorputzekin geruza piramidaleko
zelulen mintzak markatuta ikusten dira. Argi-mikroskopio mailan ezinezkoa da
123

Emaitzak
konfiantza osoz markaketa hori terminaletako markaketa presinaptikoa ala markaketa
postsinaptikoa den jakitea. APPren C muturraren aurkako antigorputzekin kasu
batzuetan zelula piramidalen dendrita apikalak ere markatuta agertzen dira (4.9.F irudia
gezi urdinak) stratum radiatum-ean Baita zenbait interneurona ere stratum oriens-ean
(4.9.C irudia gezi urdinak). N muturraren aurkako antigorputzarekin ere, geruza
piramidaleko zelulez gain, stratum oriens-eko zenbait interneurona ere markatuak
agertzen dira (4.9.I irudia gezi urdina).
PS1en kasuan, zelula piramidalak dira gehien markatzen direnak. Kasu horretan ere,
stratum oriens-eko zenbait interneurona markatuta agertzen dira. PS1en kasuan, DAB
bidezko markaketa hori, hala ere, ez da mintzera mugatua ikusten, APPren kasuan
bezala. Presenilinaren kasuan, zelula piramidalen somak markatzen direla ikusten da.
4.2.3. MIKROSKOPIO ELEKTRONIKOAZ eta SINAPTOSOMETAN
EGINDAKO ESPERIMENTUEN EMAITZAK
4.2.3.1. APP eta PS1 sinapsian eta mintz estrasinaptikoan
Zenbait esperimentu egin ditugu APP eta PS1 sinapsian aberastuta agertzen diren ala ez
jakiteko. APPren kasuan, N muturra eta C muturraren kontrako antigorputzak erabili
ditugu, lehenago aipatu dugun bezala. APPk prozesu proteolitikoa jasaten du, eta,
horren ondorioz, APPtik eratorritako zatikiak ager daitezke zelularen barruan.
Horregatik, APPren zenbait domeinu ezagutzen dituzten antigorputzak erabili ditugu.
Mikroskopio elektronikoarekin lortutako emaitzetan zein zatiki ikusten ari garen
bereiztezina den arren, sinaptosomen Western Blot esperimentuekin pisu
molekularraren inguruko informazioa lortzen dugunez, informazioa osa dezakegu.
Erretxinan murgildu aurreko immunozitokimika erabiliz, urre partikula ugari ikusi
ditugu neuronen mintzetan. Urre partikula horiek mintz sinaptikoan eta estrasinaptikoan
kokatzen dira, bai APPren kasuan eta baita PS1en kasuan ere. Urre partikulen
kuantifikazioa egitean datu hauek lortu ditugu: APPren N muturraren aurkako
antigorputza erabiliz, sinapsian batez beste 1,558 ± 0,1982 partikula/mikra, eta mintz
estrasinaptikoan, berriz, 0,095 ± 0,0166 partikula/mikra dentsitatea lortu dugu. APPren
C muturraren kontrako antigorputza erabiliz, sinapsian 1,552 ± 0,1933 partikula/mikra
eta mintz estrasinaptikoan 0,228 ± 0,0358 partikula/mikra kontatu ditugu batez beste.
124

Emaitzak
Presenilinaren kasuan ere, urre partikulak kuantifikatu ditugu eta, batez beste, emaitza
hauek atera zaizkigu: sinapsian 1,689 ± 0,1887 partikula/mikra eta mintz
estrasinaptikoan 0,173 ± 0,0236 partikula/mikra (4.10.c irudia). Kuantifikazio hori
egiteko irizpide hau erabili da: sinapsitik 30 nm-ko distantzia baino urrutiago dauden
partikulak estrasinaptikotzat hartu ditugu. Irizpide bera erabili dugu ikerketa guztian
egin ditugun urre-kuantifikazio guztietan. APP eta PS1 sinapsian aberastuta daudela
ikusi dugu. Hiru kasuetan, estatistikaren arabera, ezberdintasunak esanguratsuak direla
ikusi dugu (p<0,0001) (4.10.b irudia).
4.10. Irudia: a) APP eta PS1 sinapsian agertzen dira. Geziz seinalaturik daude APP eta PS1 markatzen dituzten urre partikulak. Eskalak 400 nm ditu kasu guztietan. b) Urre partikulen kuantifikazioa egin ondoren, kasu bakoitzean lortutako grafikoak (ezkerrean APP C muturra, erdian APP N muturra eta eskubian PS1). Hiru kasuetan urre partikulak nagusiki sinapsian kokatzen dira mintz estrasinaptikoarekin alderatuz. c) Batez beste kasu bakoitzean kontatutako urre partikulak taulan agertzen dira.
Material eta metodoetan azaldu dugun bezala, erretxinan murgildu ondorengo
immunozitokimikak abantaila batzuk ditu erretxinan murgildu aurreko
immunozitokimikarekin alderatuz; antigorputzek ez dute ehunera sartu beharrik epitopo
125

Emaitzak
guztiak eskuragarri daudelako. Horregatik, teknika horrekin ere APP eta PS1en
kokapena sinapsian eta mintz estrasinaptikoan ikusi nahi izan dugu.
Horrela egindako esperimentuetan ere, urre partikula ugari ikusi ditugu neuronen
mintzetan. Bai mintz plasmatikoan, eta baita zelularen barruko organuluen zenbait
mintzetan. Sinapsiari dagokionez, urre partikula ugari ikusi ditugu bai APPren kasuan bi
muturren aurkako antigorputzak erabiliz eta baita PS1en aurkako antigorputza erabiliz
ere. Urre partikula horiek sinapsiko mintzetan agertzen dira; bai mintz presinaptikoan,
eta baita mintz postsinaptikoan ere kasu guztietan (4.11. irudia).
4.11. Irudia: Erretxinan murgildu ondorengo immunozitokimika erabiliz APP eta PS1 sinapsian aberasturik daudela ikusi da. a) Urre partikulak geziz seinalaturik daude sinapsian. Eskalak 400 nm ditu hiru kasuetan. b) Grafiketan urre partikulen kuantifikazioa egin ondoren ateratako datuak adierazten dira. Ezkerrean, APP C muturrari dagokion grafikoa; erdian, APP N muturrari dagokiona; eta eskuinean, PS1i dagokiona. c) Urre partikulen kuantifikazioaren bidez lortutako datuak taulan agertzen dira.
Sinapsiko urre partikulen kuantifikazioa egitean, emaitza hauek lortu ditugu: APPren N
muturraren aurkako antigorputza erabiliz, batez beste 2,388 ± 0,1653 partikula/mikra
eta APPren C muturraren aurkako antigorputza erabiliz 3,273 ± 0,2444 partikula/mikra
126

Emaitzak
dentsitatea lortu dugu. Presenilina-1en kontrako antigorputzarekin, berriz, 3,127 ±
0,2297 partikula/mikra zenbatu ditugu batez beste. Mintz estrasinaptikoko urre
partikulen kuantifikazioa egitean, emaitza hauek atera zaizkigu: APP N muturraren
aurkako antigorputza erabiliz, batez beste 0,011 ± 0,0037 partikula/mikra, APP C
muturraren aurkako antigorputza erabiliz, batez beste 0,135 ± 0,0193 partikula/mikra
eta PS1en aurkako antigorputzarekin, batez beste 0,109 ± 0,0198 partikula/mikra (4.11.c
irudia).
Berriro ere lortutako datuekin egindako analisi estatistikoaren arabera, datuen arteko
aldea esanguratsua dela ikusi dugu (p<0,001) (4.11.b irudia).
Mikroskopio elektronikoarekin lortutako emaitza horiek, Western Blot teknikarekin
egindako immunomarkaketarekin alderatu ditugu. Horretarako, arratoien hipokanpoko
homogenatuetan eta hipokanpo horietatik eratorritako sinaptosometan egin ditugu
esperimentuak (4.12. irudia).
4.12. Irudia: Arratoien hipokanpoko homogenatuetan eta haietatik eratorritako sinaptosometan egindako Western Blot-ak. a) Sinaptosomak ondo banatuta daudela ziurtatzeko sinapsi-markatzaileekin egindako esperimentuetan, hiru kasuetan sinaptosomei dagokien banda askoz biziagoa da, eta horrek adierazten du sinaptosomak ondo banatuta daudela b) APP eta PS1en aurkako antigorputzak erabiliz biak sinaptosometan aberastuak daudela ikusi da. APP osoaren kasuan, N muturraren eta C muturraren aurkako antigorputzak erabili dira, eta guztietan patroi bera errepikatzen da. Gainera, APP C muturraren aurka egindako esperimentuetan, APP osoaz gain, proteolisitik eratorritako zatikiak ere sinaptosometan aberastuak agertzen dira (b erdian).
127

Emaitzak
Hipokanpo osoaren homogenatuan (H) lortutako bandak, sinaptosometan (S)
lortutakoekin alderatu ditugu. Sinaptosomak azukre-gradiente desberdinetan
oinarritutako prozeduraren bidez lortu ditugu. Metodo horrek mintz espezifikoak
banatzeko gaitasuna du. Gure esperimentuetan lortu ditugun laginetan sinapsiko
mintzak ondo bananduak zeuden frogatzeko, sinapsian espresatzen diren zenbait
markatzaile erabili ditugu: sinaptofisina, SNAP25 eta NR1. Western Blot-ak egin ditugu
hipokanpoko homogenatu osoan eta sinaptosometan. Markatzaile horiek
sinaptosometan aberastuak agertzeak prozedura ondo gauzatu dela adierazten du
(4.12.a irudia).
Horrela lortutako sinaptosometan, APPren N muturraren, C muturraren eta PS1en
aurkako antigorputzak erabiliz, Western Blot-ak egin ditugu. APPren aurkako
antigorputzak erabiliz (N eta C muturraren aurkako antigorputzak), banda intentsoagoa
lortu dugu kasu guztietan sinaptosomei dagokien zatian, eta gauza bera gertatzen da
PS1en bandarekin ere (4.12. irudia). Gainera, APPren C muturraren aurkako
antigorputza erabiliz, zenbait banda lortu ditugu. Antigorputz horrek APPren
proteolisiaren ondorioz eratzen diren zati batzuk ezagut ditzake. Alde batetik, APP
molekula osoari dagokion banda (110 kDa inguruan) eta, bestetik, proteolisiaren
ondoren mintzari lotuta geratzen diren zatien bandak (CTFen bandak; 10-15 kDa
artean), sinaptosometan aberastuak agertzen dira homogenatu osoarekin alderatuta
(4.12.b irudia). PS1en kasuan, 37 kDa-etik behera lortzen genuen banda, eta hori ere bat
dator PS1en N zatiari dagokion pisuarekin.
Beraz, teknika honekin ere, ikusi da bai APP eta baita PS1 ere sinapsian aberastuta
agertzen direla; gainera, ikusi dugu pisu molekularrari begiratuz, APPren proteolisi
produktu diren CTFak ere sinapsian aberastuta agertzen direla.
4.2.3.2. APPren N muturraren aurkako antigorputza
Mintz estrasinaptikoa eta sinapsia alderatuz lortutako emaitzetan APP eta PS1 sinapsian
aberastuta agertzen direla ikusi ondoren, sinapsiaren bi aldeen artean (presinaptikoa eta
postsinaptikoa) ezberdintasunak agertzen diren ala ez ikusi nahi izan dugu. Mintz
estrasinaptikoaren kasuan ere, terminalen eta dendriten mintzen arteko ezberdintasunak
aztertu ditugu.
Mikroskopio elektronikorako, erretxinan murgildu aurreko immunozitokimika erabiliz
lortutako emaitzen arabera, ikusi dugu APP N muturra nagusiki sinapsiaren alde
presinaptikoan agertzen dela. Urre partikulak mintz estrasinaptikoaren eta sinapsiaren bi
128

Emaitzak
aldeetan banatuta daude, baina, nagusiki, mintz presinaptikoan kokatzen dira (4.13.
irudia).
Urre partikula horien kuantifikazioa egin dugu eta datu hauek lortu ditugu: sinapsiaren
kasuan, mintz presinaptikoan batez beste 1,766 ± 0,3183 partikula/mikra eta mintz
postsinaptikoan 1,351 ± 0,2361 partikula/mikra. Mintz estrasinaptikoaren kasuan,
berriz, terminalaren mintzean batez beste 0,094 ± 0,0226 partikula/mikra eta dendritaren
mintzean 0,096 ± 0,0246 partikula/mikra zenbatu ditugu (4.13.c irudia).
Urre partikulen kuantifikazioaren arabera, APP presinaptikoki kokatzen da nagusiki,
baina analisi estatistikoa egiterakoan, ikusi dugu ezberdintasunak esanguratsuak ez
direla (4.13.b irudia).
Mikroskopio elektronikoa erabiliz, azterketa zehatzagoa egiteko, erretxinan murgildu
ondorengo teknikaren bidez errepikatu dira esperimentuak, APP N muturraren kontrako
antigorputza erabiliz.
Teknika horren bidez ere antzeko datuak lortu ditugu, hau da, APP N muturra nagusiki
sinapsiaren alde presinaptikoan agertzen da. Berriro ere ikusi dugu urre partikulak
sinapsiaren mintzetan (mintz presinaptiko eta postsinaptikoan) eta sinapsitik kanpoko
mintzetan (mintz estrasinaptikoan) kokatzen direla. Azken kasu horretan, bai terminalen
mintz estrasinaptikoetan eta baita dendritetan ere (4.14. irudia).
Mintz horietan APPk duen kokapen-maila aztertzeko, urre partikulen kuantifikazioa
egin dugu. Lehenago ere esan dugun bezala, erretxinan murgildu ondorengo
teknikarekin lortzen diren kuantifikazio-datuak zehatzagoak dira erretxinan murgildu
aurreko immunozitokimikaren kasuan baino. Izan ere, antigorputzak eskuragarri ditu
antigenoak lotura gerta dadin, eta gainera, zilar bidezko intentsifikazioaren beharrik ez
dagoenez, urre partikulak zuzenean zenbatuz kuantifikazio zehatzagoa lortzen da.
Horrela, urre partikulen kuantifikazioa egin dugu eta datu hauek lortu ditugu:
sinapsiaren kasuan, mintz presinaptikoan 3,705 ± 0,2376 partikula/mikra, eta mintz
postsinaptikoan 1,071 ± 0,1687 partikula/mikra. Mintz estrasinaptikoen kasuan, berriz,
terminalen mintzean 0,010 ± 0,0049 partikula/mikra eta dendriten kasuan 0,012 ±
0,0055 partikula/mikra zenbatu ditugu (4.14.c. irudia).
Ondorioz, erretxinan murgildu ondorengo teknikarekin ere, APPren kokapen
presinaptikoa nagusitzen da, N muturraren aurkako antigorputza erabiliz (4.14. irudia).
Baina, erretxinan murgildu aurreko datuekin gertatzen ez den bezala, analisi estatistikoa
129

Emaitzak
egitean, kasu honetan ezberdintasunak esanguratsuak direla ikusi dugu (p<0,0001)
(4.14.b irudia).
4.13. Irudia: Erretxinan murgildu aurreko teknikarekin eta APP N muturraren aurkako antigorputza erabiliz lortutako kokapen eta kuantifikazioa. a) Urre partikulak presinaptikoki kokatuta agertzen zaizkigu lau argazkietan, urre partikulak gezien bidez seinalaturik daude. Eskalak 400 nm ditu kasu guztietan. Alde presinaptikoan kokatzen da nagusiki, baina irudiko grafikan (b) ikusten dugu datuen arabera ezberdintasunak ez direla esanguratsuak. Grafikoan sinapsiko datuak mintz estrasinaptikoko datuen ondoan jarri ditugu, baina mintz estrasinaptikoa osotasun gisa hartu beharrean, terminalen eta dendriten mintzen artean ezberdindu dugu. Urre partikulek antzeko banaketa daukate mintz estrasinaptikoetan. (Sin Pre: sinapsiaren alde presinaptikoa; Sin Post: sinapsiaren alde postsinaptikoa; MtEx Ter: Terminalaren mintz estrasinaptikoa; MtEx Den: Dendritaren mintz estrasinaptikoa). Datu guztiak zenbakitan taulan ikus ditzakegu (c). Bertan, batez besteko urre partikula kopurua mikrako ikus dezakegu.
130
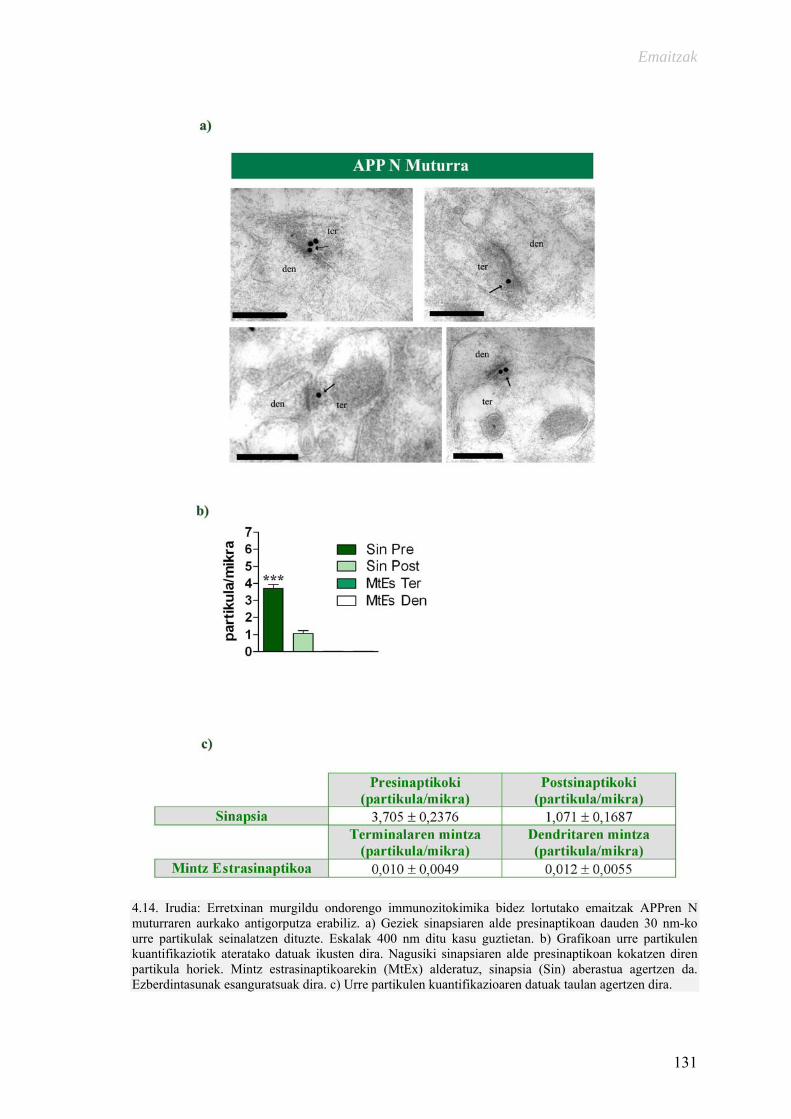
Emaitzak
4.14. Irudia: Erretxinan murgildu ondorengo immunozitokimika bidez lortutako emaitzak APPren N muturraren aurkako antigorputza erabiliz. a) Geziek sinapsiaren alde presinaptikoan dauden 30 nm-ko urre partikulak seinalatzen dituzte. Eskalak 400 nm ditu kasu guztietan. b) Grafikoan urre partikulen kuantifikaziotik ateratako datuak ikusten dira. Nagusiki sinapsiaren alde presinaptikoan kokatzen diren partikula horiek. Mintz estrasinaptikoarekin (MtEx) alderatuz, sinapsia (Sin) aberastua agertzen da. Ezberdintasunak esanguratsuak dira. c) Urre partikulen kuantifikazioaren datuak taulan agertzen dira.
131

Emaitzak
Western Blot teknika erabili da APPren sinapsi barneko kokapen zehatza aztertu eta
mikroskopio elektronikoaren bidez lortutako emaitzekin bat datorren ikusteko. Gainera,
teknika horrekin detektatzen ari garen proteinaren pisu molekularra ikus dezakegu eta
horrekin datu osagarri bat lortzen dugu. Horretarako, sinaptosomen zatiketatik lortutako
zatikiak erabili ditugu (mintz presinaptikoa, mintz postsinaptikoa eta mintz
estrasinaptikoa). Lortutako emaitzak bat datoz APPren N muturraren aurkako
antigorputzarekin beste tekniketan lortutako kokapen presinaptikoarekin (4.15. irudia).
4.15. Irudia: Sinaptosometatik eratorritako zatikietan egindako Western Blot-en emaitzak APPren N- muturraren aurkako antigorputza erabiliz. a) Sinaptosometatik mintz-zatikietarako banaketa ondo egin dela ziurtatzeko kasu bakoitzeko markatzaileekin egindako Western Blot-a. b) APPren N muturraren aurkako antigorputzarekin lortutako emaitzetan, ikusten da nagusiki mintz presinaptikoan agertzen direla bandak. Grafikoan, banden intentsitate totalaren portzentaiak adierazita daude. Hizki ezberdinekin zutabeen arteko ezberdintasun esanguratsuak adierazten dira (p<0,05); hizki berdinekin, aldiz, ezberdintasun ez-esanguratsuak.
Sinaptosomen banaketa, zati presinaptikoa, postsinaptikoa eta estrasinaptikoa lortzeko,
modu egokian egin dela ziurtatzeko, markatzaile batzuk erabili ditugu Western Blot
esperimentuetan. Markatzaile estrasinaptiko gisan, sinaptofisina erabili da, markatzaile
presinaptiko gisa, SNAP25, eta markatzaile postsinaptiko gisa, NR1 (4.15.a irudia).
Mintzen arteko banaketa egokitzat eman ondoren, APPren N muturraren kontrako
antigorputza erabiliz Western Blot-ak egin dira sinaptosomen zati horiek erabiliz. 100
kDa inguruan agertzen diren bandak askoz biziagoak dira mintz presinaptikoan, beste
mintzekin alderatuta. Banda horien pisu molekularra bat dator APP osoaren pisu
132

Emaitzak
molekular teorikoarekin. Mintz postsinaptikoan ere banda ahulago bat agertu arren,
askoz intentsoagoa da presinaptikoki lortutako banda (4.15.b irudia).
Beraz, teknika horrekin, mikroskopio elektronikoarekin lortutako datuak berresten dira,
hau da, APPren sinapsiko kokapena nagusiki presinaptikoa dela ikusi dugu, APPren N
muturraren aurkako antigorputza erabiliz.
4.2.3.3. APPren C muturra sinapsian
APPren karboxi muturraren aurkako antigorputza erabiliz ere, APPk sinapsian agertzen
duen adierazpen-patroia aztertu nahi izan dugu. Mintz estrasinaptikoen kasuan ere,
terminalen edo dendriten mintzen artean bereizketa egin dugu. Kasu honetan, berriro
ere, mikroskopio elektronikorako erretxinan murgildu aurreko eta ondorengo
immunozitokimika erabili ditugu, eta baita sinaptosometatik eratorritako zatikien
Western Blot-a ere. Azken horrekin, C muturraren aurkako antigorputzak ezagutzen
dituen APPren proteolisi zatikiak bereiz ditzakegu.
Mikroskopio elektronikorako erretxinan murgildu aurreko immunozitokimikaren bidez,
APPren C muturra nagusiki sinapsiaren alde postsinaptikoan kokatzen dela ikusi dugu
(4.16. irudia). Urre partikulak bai mintz sinaptikoan eta baita estrasinaptikoan ere
agertzen zaizkigu. Aurretik ikusi dugu APP aberastua agertzen dela mintz sinapikoetan
mintz estrasinaptikoekin alderatuz gero. Eta gainera, lortutako ezberdintasunak
esanguratsuak direla ikusi da. Hala ere, aurretik ikusitako emaitza horietan ez genuen
sinapsiko mintzen artean (mintz presinaptiko eta postsinaptiko) bereizketarik egin; ezta
mintz estrasinaptikoen (terminalen eta dendriten mintza) artean ere. Kasu honetan,
bereizketa hori egin nahi izan dugu, eta mintzen arteko kokapenean APPk dituen
ezberdintasunak ikusi nahi izan ditugu.
Urre partikulen kuantifikazioa egitean, emaitza hauek lortu dira: sinapsian, mintz
presinaptikoan batez beste 0,698 ± 0,2046 partikula/mikra eta mintz postsinaptikoan,
berriz, 2,405 ± 0,3016 partikula/mikra. Mintz estrasinaptikoen kasuan terminalen
mintzean 0,177 ± 0,0370 partikula/mikra eta dendriten mintz estrasinaptikoan 0,278 ±
0,061 partikula/mikra zenbatu dira (4.16.c irudia).
Datu horiekin, ikusi dugu C muturraren aurkako antigorputza erabiliz, APPren
kokapena sinapsian nagusiki postsinaptikoa dela (4.16.b irudia). Datu horien analisi
estatistikoen arabera, ezberdintasunak esanguratsuak direla ikusi dugu gainera
(p<0,0001) (4.16.b irudia).
133

Emaitzak
4.16. Irudia: Erretxinan murgildu aurreko teknikarekin eta APP C muturraren aurkako antigorputza erabiliz lortutako kokapen eta kuantifikazioa. Urre partikulak, sinapsien alde postsinaptikoan ikus ditzakegu a irudian agertzen diren argazkietan. Eskalak 400 nm ditu kasu guztietan. Irudiko grafikoetan (b) ikus dezakegun bezala, mintz presinaptiko eta postsinaptikoan kontatutako urre partikulen arteko aldea esanguratsua da. Nagusiki alde postsinaptikoan kokatzen da (p<0,0001). Mintz estrasinaptikoari dagokionez, terminaletan eta dendritetan kontatutako urre partikulak alderatuz, ikusi dugu gehienbat dendriten mintzean kokatzen direla (b irudian grafika). Datu horiek guztiak, zenbakitan, taulan (c) agertzen dira eta kasu guztietan kontatutako batez besteko urre partikulak mikrako adierazi ditugu.
134

Emaitzak
Erretxinan murgildu aurreko immunozitokimika erabiliz datu horiek lortu ondoren,
azterketa bera errepikatu dugu erretxinan murgildu ondorengo immunozitokimika
erabiliz. Hau da, APPren C muturraren aurkako antigorputza erabiliz mintz sinaptiko eta
estrasinaptikoetan duen kokapena ikusi nahi izan dugu (4.17. irudia).
Metodo horrekin ere, urre partikula ugari ikusi dira sinapsien mintzetan. Bai mintz
presinaptikoan eta baita postsinaptikoan ere. Lehenago ere ikusita geneukan APP
sinapsian aberastuta agertzen zela APPren aurkako antigorputz bera erabiliz, mintz
estrasinaptikoarekin alderatuz. Hala ere, mintz estrasinaptikoetan ere zenbait urre
partikula kokatzen zirela ikusi genuen.
Urre partikula horiek nagusiki zein mintzetan kokatzen diren jakiteko, kuantifikazioa
egin dugu. Emaitza hauek lortu ditugu: sinapsian, 1,734 ± 0,2768 partikula/mikra mintz
presinaptikoan. Mintz postsinaptikoan, berriz, 4,813 ± 0,3589 partikula/mikra. Mintz
estrasinaptikoen kasuan, urre partikulak kuantifikatu ditugu bai terminalen eta baita
dendriten mintzetan ere. Datu hauek lortu ditugu: terminalen mintz estrasinaptikoan
0,145 ± 0,0283 partikula/mikra eta dendriten mintz estrasinaptikoan, berriz, 0,124 ±
0,0264 partikula/mikra. (4.17. irudia)
Beraz, erretxinan murgildu ondorengo immunozitokimikarekin ere, erretxinan murgildu
aurreko immunozitokimikarekin lortutako datu berak lortu ditugu, hau da, APPren C
muturraren aurkako antigorputza erabiliz kokapena nagusiki postsinaptikoa dela ikusi
dugu. (4.17. irudia).
Ezberdintasunak esanguratsuak diren ala ez ikusteko, analisi estatistikoa egin dugu eta
ezberdintasunak adierazgarriak direla ikusi da kasu honetan ere (p<0,0001) (4.17.b
irudia).
Mikroskopio elektronikoaren bidez lortutako emaitzak sinaptosometatik eratorritako
zatietan (mintz presinaptikoa, mintz postsinaptikoa eta mintz estrasinaptikoa) egindako
Western Blot-ekin alderatu ditugu.
APPren C muturra ezagutzen duen antigorputzarekin mikroskopio elektronikorako
lortutako markaketa (APPren kokapen postsinaptikoa) ez dator bat APPren N
muturraren aurkako antigorputza erabiltzean lortutako emaitzekin (APPren kokapen
presinaptikoa), beraz, C muturraren aurkako antigorputzarekin sinaptosometatik
eratorritako mintz-zatiki bakoitzean antigorputz horrekin zer pisu molekularretako
APPren zatia ikusten ari garen jakin nahi izan dugu.
135
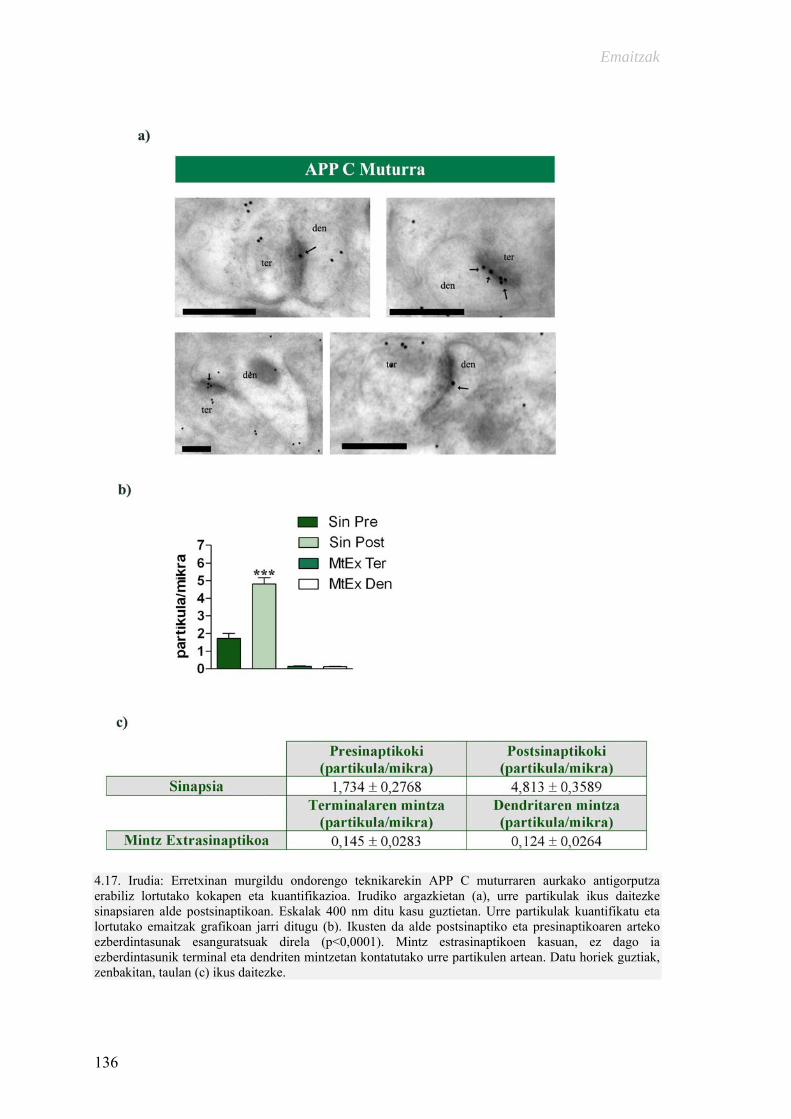
Emaitzak
4.17. Irudia: Erretxinan murgildu ondorengo teknikarekin APP C muturraren aurkako antigorputza erabiliz lortutako kokapen eta kuantifikazioa. Irudiko argazkietan (a), urre partikulak ikus daitezke sinapsiaren alde postsinaptikoan. Eskalak 400 nm ditu kasu guztietan. Urre partikulak kuantifikatu eta lortutako emaitzak grafikoan jarri ditugu (b). Ikusten da alde postsinaptiko eta presinaptikoaren arteko ezberdintasunak esanguratsuak direla (p<0,0001). Mintz estrasinaptikoen kasuan, ez dago ia ezberdintasunik terminal eta dendriten mintzetan kontatutako urre partikulen artean. Datu horiek guztiak, zenbakitan, taulan (c) ikus daitezke.
136

Emaitzak
Western Blot-ekin lortutako emaitzak 4.18. irudian agertzen dira. Markatzaileen bidez
mintzen banaketa modu egokian gauzatu dela ziurtatu ondoren (4.18.a irudia), C
muturraren aurkako antigorputza erabiliz, Western Blot-ak egin dira zati horietan.
APP osoari dagokion banda (∼100 kDa), sinapsian alde presinaptikoan kokatzen da
nagusiki. Mintz postsinaptikoan ere kokatzen den arren, maila altuagoan kokatzen da
alde presinaptikoan (4.18.b irudia).
4.18. Irudia: Sinaptosometatik eratorritako mintz-zatikietan egindako Western Blot-a, APPren C muturraren aurkako antigorputza erabiliz. a) Markatzaileen bidez sinaptosometatik eratorritako mintzak ondo banatuta dauden jakiteko egindako kontrola. b) APP osoari dagokion banda nagusiki mintz presinaptikoan agertzen da. Proteolisitik eratorritako zatikien kasuan, 15 kDa-en inguruko CTFα eta CTFβri dagozkien bandak estrasinaptikoki kokatzen dira nagusiki, baina sinapsiaren barruan presinaptikoki); 10 kDa-etik beherako banda (CTFγri dagokiona) nagusiki mintz presinaptikoan agertzen da. Grafikoetan banden intentsitate totalaren portzentaia adierazten da. Goran, APP osoaren bandei dagokien grafikoa. APP osoari dagokion banda bat baino gehiago daudenez, grafikoan zenbait koloretan adierazten da bakoitzaren intentsitatea. Behean, APPren proteolisiaren ondoren sortzen diren CTFen bandei dagokien grafika. Grafikoko zutabeetan hizkiak daude. Hizki ezberdinek ezberdintasun esanguratsuak adierazten dituzte (p<0,05); hizki berdinek, aldiz, ezberdintasun ez-esanguratsuak
15 kDa-etik behera banda bat agertzen da, bai mintz estrasinaptikoari dagokion kalean,
eta baita mintz presinaptikoari dagokion kalean ere. Banda hori CTFα eta CTFβren pisu
molekularrarekin bat dator. Mintz postsinaptikoaren kasuan agertzen den banda askoz
ahulagoa da (4.18.b irudia). Pisu molekular hori bat dator teorikoki APPren
proteolisiaren ondoren sortzen diren C muturrei dagokien pisuarekin. Ondorioz, APPren
prozesatu ondoren lortutako CTFak nagusiki mintz estrasinaptikoan kokatzen dira, eta
137

Emaitzak
sinapsiaren barruan, berriz, alde presinaptikoan. Bestalde, APP osoa ere, mintz
presinaptikoan kokatzen da eta 10 kDa-etik behera ere, beste banda bat ikusten da,
mintz presinaptikoaren kasuan (4.18.b irudia). Banda horren pisu molekularra bat dator
CTFγren pisu molekularrarekin.
4.2.3.4. PS1 sinapsian
PS1 ere sinapsian eta mintz estrasinaptikoetan kokatzen dela ikusi ondoren, proteina
hori sinapsiaren alde presinaptiko eta postsinaptikoan zer mailatan kokatzen den jakin
nahi izan dugu. Mintz estrasinaptikoen kasuan ere, bereizketa egin da terminalen eta
dendriten mintzetan kokatzen diren urre partikulen artean, euren arteko ezberdintasunak
nolakoak diren ikusteko asmotan.
Lehenago ere ikusi genuen PS1 sinapsian aberastua zegoela mintz estrasinaptikoekin
alderatuz gero. Sinapsian bertan, erretxinan murgildu aurreko immunozitokimika
erabiliz, PS1 nagusiki alde postsinaptikoan kokatzen dela ikusi dugu (4.19. irudia). Urre
partikula asko ikusi ditugu mintz postsinaptikoan. Sinapsiaren alde presinaptikoan ere
agertzen dira partikulak, eta baita mintz estrasinaptikoetan ere, baina maila txikiagoan.
Mintz estrasinaptikoen kasuan, bai terminalen eta baita dendriten mintzetan ere ikusi
ditugu partikulak.
Mintz horietako bakoitzean zer mailatan kokatzen den jakiteko, urre partikulak
kuantifikatu ditugu. PS1en kasuan ere, mintz presinaptiko eta postsinaptikoko
partikulak kontatu ditugu alde batetik, eta bestetik, mintz estrasinaptikoetan,
terminaletan eta dendritetan agertzen diren urre partikulak. Horrela egindako
kuantifikazioan, emaitza hauek lortu dira: sinapsiaren alde presinaptikoan 0,944 ±
0,2151 partikula/mikra. Alde postsinaptikoan, berriz, 2,434 ± 0,2895 partikula/mikra.
Mintz estrasinaptikoen kasuan, 0,147 ± 0,0337 partikula/mikra kontatu ditugu
terminalen mintzetan, eta 0,199 ± 0,0329 partikula/mikra dendriten kasuan (4.19.c
irudia).
Datu horiekin, beraz, presenilinaren kasuan, sinapsiko mintz postsinaptikoko kokapena
nagusitzen dela ikusi dugu. Mintz horretako kokapen-mailak beste mintzetako kokapen-
mailarekin ezberdintasunik duen ikusteko, analisi estatistikoa egin dugu, eta
ezberdintasunak esanguratsuak direla ikusi dugu (p<0,0001) (4.19.b irudia).
138

Emaitzak
4.19. Irudia: Erretxinan murgildu aurreko teknikarekin PS1en aurkako antigorputza erabiliz lortutako kokapen eta kuantifikazioa. Irudiko argazkietan (a), urre partikulak agertzen dira sinapsiaren alde postsinaptikoan. Eskalak 400 nm ditu kasu guztietan. Urre partikulen kuantifikazioa egin ondoren lortutako grafikoak agertzen dira (b). Taulan (c), berriz, zenbakitan agertzen dira kuantifikatutako balioak. Ikusten da, mintz estrasinaptikoaren kasuan, ez dagoela ezberdintasun esanguratsurik terminal eta dendriten mintzentzat lortutako balioen artean.
139

Emaitzak
PS1en kasuan ere, azterketa bera errepikatu nahi izan dugu erretxinan murgildu
ondorengo immunozitokimika erabiliz, teknika bakoitzak dituen abantaila eta
desabantailak direla-eta emaitza berdinak ala ezberdinak lortuko genituen ikusteko
asmotan. Kasu honetan ere, bat egiten dute emaitzek erretxinan murgildu aurreko
immunozitokimikaren bidez lortutako emaitzekin.
Urre partikulak bai sinapsian eta baita mintz estrasinaptikoetan banatuta ikusi ditugu.
Gainera, sinapsiaren kasuan, mintz postsinaptiko eta presinaptikoan agertzen dira, eta
mintz estrasinaptikoen kasuan terminal eta dendriten mintzetan. Mintz postsinaptikoari
dagokionez, mintz presinaptikoan baino urre partikula gehiago ikusten dira (4.20.
irudia).
Kuantifikazioa egin dugu, aurreko kasuetan bezala, mintz presinaptiko eta
postsinaptikoan eta terminal eta dendriten mintz estrasinaptikoetan. Datu hauek lortu
ditugu: sinapsiko mintz presinaptikoan 1,178 ± 0,1950 partikula/mikra, eta mintz
postsinaptikoan 5,076 ± 0,3423 partikula/mikra. Mintz estrasinaptikoaren kasuan,
terminalen mintzetan 0,096 ± 0,0254 partikula/mikra eta dendriten kasuan 0,122 ±
0,0303 partikula/mikra zenbatu dira (4.20.c irudia).
Datu horiek, erretxinan murgildu aurreko immunozitokimikako datuekin batera, PS1en
kokapena nagusiki postsinaptikoa dela adierazi digute. Presinaptikoki ere kokatzen da
maila txikiagoan, eta mintz estrasinaptikoetan, berriz, askoz maila txikiagoan.
Kokapen-maila horien arteko ezberdintasunak ikusteko analisi estatistikoa egin
genuenean, ezberdintasunak esanguratsuak direla ikusi dugu (p<0,0001) (4.20.b irudia).
Lortutako emaitzak Western Blot teknikarekin egindako immunomarkaketekin alderatu
ditugu. Berriro ere, horretarako, sinaptosometatik abiatuta lortutako zatikiak erabili dira
(mintz presinaptikoa, mintz postsinaptikoa eta mintz estrasinaptikoa). Lehenago egin
genuen bezala, sinapsiko alde bakoitzarentzat markatzaileak erabiliz, banaketa modu
egokian egin dela ziurtatu dugu (4.21.a irudia). Ondoren, PS1en kontrako antigorputza
erabiliz egindako Western Blot.-en arabera, alde presinaptikoan ere berari dagokion
pisu molekularrean (4.21.b irudia) banda bat agertzen den arren, mintz postsinaptikoan
agertzen den banda intentsoagoa zen (4.21.b irudia). Emaitza horiek bat datoz
mikroskopio elektronikoaren bidez lortutako emaitzekin.
140

Emaitzak
4.20. Irudia: Erretxinan murgildu ondorengo immunozitokimika bidez lortutako emaitzak PS1en aurkako antigorputza erabiliz. Irudian, teknika hau erabiliz, ateratako zenbait argazki agertzen dira (a), eta, haietan, urre partikulak sinapsiaren alde postsinaptikoan erakusten dira. Eskalak 400 nm ditu kasu guztietan. Urre partikulen kuantifikaziotik lortutako grafikoa ikus daiteke (b), eta alde postsinaptiko eta presinaptikoaren arteko ezberdintasun esanguratsuak erakusten ditu (p<0,0001). Datuak, zenbakitan, taulan agertzen dira (c).
141

Emaitzak
4.21. Irudia: a) Sinaptosomen banaketa modu egokian egin dela ziurtatzeko, sinapsiko eta mintz estrasinaptikoko markatzaileekin egindako Western Blot-a. b) PS1en kontrako antigorputzarekin, sinaptosomen banaketatik eratorritako mintz-zatikietan egindako Western Blot-a. 37 kDa-etik behera banda intentsoena mintz postsinaptikoan agertzen zaigu; presinaptikoki kokatzen dela ere ikusten den arren. Grafikoan, banden intentsitate totalaren portzentatiak adierazita daude. Hizki ezberdinak dituzten zutabeen artean ezberdintasunak esanguratsuak dira (p<0,05); hizki berdinak dituztenen artean, aldiz, ezberdintasunak ez dira esanguratsuak.
142

5. EZTABAIDA


Eztabaida
5. EZTABAIDA
Gure emaitzek APP eta bere sekretasen sinapsiko aberastasuna erakusten dute.
Presinapsian azaltzen den dentsitateak iradokitzen du APPk gune horretan funtzio
espezifikoa eduki behar duela. Bestalde molekula osoaren eta beraren zatikien kokapen
presinaptiko horrek, presenilina eta bestelako sekretasen kokapen presinaptikoarekin
batera APPren metabolismo presinaptikoaren aldeko argudio esanguratsuak ematen
ditu.
5.1. APP eta PS1 sinapsian kokatzen dira nagusiki Mikroskopio elektronikoak ematen duen zehaztasun-mailaz, mintz plasmatikoaren
eskualdeetan Alzheimer gaixotasunarekin erlazionaturiko APP eta PS1 proteinen
kokapena aztertu nahi izan genuen. Horretarako, APP proteinaren bi atal desberdin
ezagutzen dituzten antigorputzak erabili genituen, N muturra eta C muturra ezagutzen
dituzten antigorputzak hain zuzen ere. Halaber, PS1en kokapena aztertzeko beste
antigorputz bat erabili genuen. Gure emaitzen arabera, bata zein bestea, mintz
estrasinaptikoarekin alderatuz, maila altuagoan kokatzen dira sinapsiko mintzetan.
Mikroskopio elektronikoarentzat egindako immunourre protokoloaren ondoren, urre
partikulen kuantifikazioak ezberdintasun esanguratsuak zeudela erakutsi zuen, bai
APPren N muturraren kasuan, C muturraren kasuan eta baita PS1ren kasuan ere.
Gainera, bai erretxinan murgildu aurreko eta baita murgildu ondorengo
immunozitokimika erabiliz ere emaitza horiek lortu genituen. Emaitza horiek bat datoz
aurretik beste talde batzuek iradoki dutenarekin; izan ere, esan izan da APP eta PS1
sinapsian kokatzen direla eta gune horietan garrantzi handikoak direla (Georgakopoulos
eta lank., 1999; Ribaut-Barassin eta lank., 2000; Kirazov eta lank., 2001; Soba eta
lank., 2005; Gralle eta Ferrerira, 2007).
Erabiltzen ari ginen antigorputzak fidagarriak ziren ala ez jakiteko, APP knockout
saguekin esperimentuak egin genituen, bai Western Blot teknika erabiliz, eta baita argi-
mikroskopio eta mikroskopio elektronikorako inmunozitokimika erabiliz ere. Argi-
mikroskopioaren kasuan, bai N zein C muturraren aurka erabili genituen antigorputzak
fidagarriak zirela ondorioztatu genuen; izan ere, jatorrizko saguen kasuan agertzen
zitzaigun inmunomarkaketa desagertu egiten zen sagu knockout-en kasuan.
145

Eztabaida
Mikroskopio elektronikorako immunourre protokoloa erabiliz, antigorputz guztien
kasuan, urre partikula gehienak desagertzen ziren sagu knockout-ak erabiltzen
genituenean. Agertzen ziren urre partikula urriak, APPren sendiko APLP proteinen
immunomarkaketa izan zitezkeen. Izan ere, sendi horretako proteinek antz handia dute
aminoazidoen sekuentzian, eta posible da erabilitako antigorputzek beste proteina
horiek ezagutzea (Pardossi-Piquard eta lank., 2005). Dena dela, markaketa inespezifiko
hori ez zen sinapsietan agertzen ia kasu guztietan, eta gure ikerketa, nagusiki, gune
horietan izan da. Gainera, APLP eta APPren kokapen mailako ezberdintasunez aritu
izan dira lehenago zenbait talde, eta, ikerketa horien arabera, APP mintz sinaptikoetan
kokatzen da eta APLP, berriz, dentsitate postsinaptikoan ikusi da baina mintz
sinaptikoetatik kanpo (Kim eta lank., 1995). Horrekin guztiarekin, ondorioztatu genuen
antigorputzak fidagarritasun handikoak zirela mintz sinaptikoetan kuantifikazioa
egiteko. Gainera, Western Blot teknika erabiliz, APPren aurka erabili genituen hiru
antigorputzekin, APPren banda (∼110kDa) desagertu egiten zen sagu knockout-en
kasuan.
Mikroskopia elektronikoaz gain, bestelako informazio gehigarria ematen zuen teknika
erabili genuen; sinaptosomekin egindako Western Blot teknika, alegia. Lehenengo,
sinaptosomak lortzen ziren. Modu horretan dentsitate ezberdina duten mintz-eremuak
banatzea lortzen da. Ondoren, immunoblotak egiten genituen sinaptosoma horiekin. Era
horretara, proteina zehatz batentzat mintz espezifiko bakoitzak duen aberastasuna
neurtzeaz gain, proteina horren pisu molekularra adierazten da aldi berean. Informazio
hori garrantzi handikoa da APP proteinaren kasuan, hainbat prozesu proteolitiko jasaten
baititu. Teknika hori erabilita lortutako emaitzek eta mikroskopio elektronikoarekin
lortutakoek ondorio berdinetara garamatzate. Sinaptosomekin egindako Western Blot
esperimentuetan APP eta PS1 sinapsian aberastuta ote zeuden ikusi nahi izan genuen.
Horretarako, APPren N eta C muturren aurkako antigorputzak erabili ziren, eta PS1ren
aurkako antigorputz bat, mikroskopio elektronikoarekin egindako esperimentuetan
bezala. Horrela egindako esperimentuetan, bai APP eta baita PS1 ere sinapsian
aberastuta zeudela ikusi genuen; izan ere, hipokanpoko ehunaren homogenatu osoarekin
alderaturik, banda intentsoagoak lortzen ziren sinaptosomen zatian. Bestalde, APPren C
muturraren aurkako antigorputza erabiliz, zenbait banda lortzen ziren, eta ikusi genuen
CTFri (CTFα eta CTFβ) zegokiona (10-15kDa artean) ere sinaptosometan intentsoagoa
zela ehunaren homogenatu osoan baino.
146

Eztabaida
Nahiz eta APP eta beraren proteolisi bide eta garraioari buruz ikerketa asko egin diren,
gaur egun oraindik kontraesanak handiak dira eta ez dira asko proteina horien kokapen
fisiologikoa aztertu duten ikerketak, ez ultraegitura mailan, eta are gutxiago
immunourreak ematen duen zehaztasun-mailaz. Hori dela eta, beharrezkoa iruditu
zitzaigun gaur egun dauden teoria guztiei, in situ, ultraegitura mailako substratu
morfologiko bat ematen saiatzea .
Gurea ez da izan APP eta PS1 sinapsian kokatzen direla esan duen ikerketa bakarra.
Horren inguruan, gero eta ikerketa gehiago aurki daitezke teknika desberdinen bitartez
APP eta sinapsia erlazionatzen dituztenak.
5.1.1. Aurrekariak
APP
APP zelula barneko mintz askotan kokatzen da. Erretikulu endoplasmatikoaren eta
Golgi aparatuaren mintzetan ez ezik, mintz plasmatikoan ere aurkitzen da (Caporaso eta
lank., 1994). Baita gorputz multibesikular (MVB) (Tomimoto eta lank., 1995) eta
lisosometan ere (Haass eta lank., 1992).
Sinapsiari dagokionez, APP bertan kokatu izan duten ikerketa ugari egon dira (Ikin eta
lank., 1996; Buxbaum eta lank., 1998b; Hoe eta lank., 2009; Kirazov eta lank., 2001;
Shigematsu eta lank., 1992).
APPren funtzioari dagokionez, hazkuntzekin egindako ikerketa ugarik APPri itsaste-
funtzioak egotzi dizkiote, bai zelulen arteko itsasketa-funtzioak, eta baita zelula-matrize
estrazelularreko osagaiekin itsasteko funtzioak ere (Gralle eta Ferreira, 2007; Soba eta
lank., 2005). APP eta beraren sendiko kideek dimeroak eratu ditzakete; zelula
ezberdinetan kokaturik daudelarik zelulen arteko elkarrekintza trans-zelularrak bidera
ditzakete, eta mota horretako konplexuak sinapsietan ere ikusi izan dira (Soba eta lank.,
2005). APPk dituen funtzio horiek guztiak ikusiz, ez da harritzekoa garunaren
garapenean garrantzi handia izatea, neuronek konexio egokiak egin ditzaten bideratzen
baititu. Gainera, APP garrantzi handikoa da neuronen morfogenesian sinapsi
funtzionalak eratzeko orduan.
Hipokanpoko neuronen hazkuntzetan ikusi da, APPren adierazpenak neurona horien
glutamatoarekiko erantzuna areagotzen duela hartzaile funtzionalen kopuruan gertatzen
den igoera dela eta (Tominaga-Yoshino eta lank., 2001). Beste ikerketa batzuek erakutsi
dutenez, APPren gabeziak ondorioak ditu elektrofisiologia mailan (Priller eta lank.,
147

Eztabaida
2006). APP knockout saguekin egindako ikerketek erakutsi dutenez, proteina horren
gabeziak sinapsi mailako eraginak ekartzen ditu (Wang eta lank., 2005; Yang eta lank.,
2005; Priller eta lank., 2006).
APPren adierazpen mailari dagokionez, ikusi da sinaptogenesian haren adierazpenaren
gorakada bat gertatzen dela; horrekin, agerian gelditzen da neuriten hazkuntzan, zelulen
targeting-ean, eta garun helduan sinapsien mantenuan APPk izan dezakeen garrantzia
(Kirazov eta lank., 2001). Gainera, garun helduan, sinapsiek aldaketa bortitzak izaten
dituzten guneetan maila altuan adierazten da APP (Gralle eta Ferreira, 2007; Löffler eta
Huber, 1992).
Bestalde, APPren proteolisiaren ondorioz sortzen diren zatikiek zerikusia dute
sinapsiaren aktibitatearekin (Mattson, 1997). Adibidez, esan izan da neuronen
aktibitateak beta amiloide peptidoaren askatzea bideratzen duela eta peptido horrek, aldi
berean, transmisio sinaptiko kitzikatzailea murrizten du (Kamenetz eta lank., 2003).
PS1
Gure emaitzak adierazten dutenarekin bat eginaz PS1 sinapsian deskribatu izan da
(Uemura eta lank., 2009; Kasa eta lank., 2001; Beher eta lank., 1999). Beste kokapen
batzuk ere egotzi zaizkio (Busciglio eta lank., 1997) PS1en zelula barneko kokapenaz
aritu izan direnean; esan izan da nagusiki erretikulu endoplasmatikoan, Golgi aparatuan,
eta endosometan kokatzen dela (Cook eta lank., 1996; De Strooper eta lank., 1997;
Walter eta lank., 1996; Kovacs eta lank., 1996; Kasa eta lank., 2001; Lah eta Levey,
2000; Cupers eta lank., 2001).
PS1en funtzioekin lotuta, PS1 kaderinan oinarritutako atxiki-lotuneen (“adherens
junctions”) osagaia dela ikusi da. Garunean ere, kaderina/katenina sistema ikusi izan da
sinapsietan, mintz pre- eta postsinaptikoa elkartuz (Uemura,1998; Uemura eta lank.,
2007; Uemura eta lank., 2009) eta PS1 sinapsietan sistema horren osagaia da
(Georgakopoulos eta lank., 1999). Gainera, sinapsiaren plastizitatearekin ere zerikusia
du; izan ere, PS1en geneetan mutaketak daudenean, sistema kolinergikoko plastizitate
sinaptikoa eragozten da (Wang eta lank., 2007; Knight eta lank., 2007).
Bestalde, PS1ek zenbait substratu ditu, eta horietako batzuk sinapsian kokatzen dira
(Kim eta lank., 2002; Haapasalo eta lank., 2007). Gainera, aldi berean, bai PS1 eta baita
APP ere, kaspasen bidez moztuak izan daitezke, eta kaspasen zenbait substratu ere
sinapsian kokatzen direla ikusi da (Chan eta Mattson, 1999).
148

Eztabaida
5.2. APPren kokapena Esan bezala APPren bi domeinu desberdin ezagutzen dituzten antigorputzak erabili
genituen APPren kokapena aztertzeko. Lehenengoak N muturraren sekuentzia
ezagutzen du; horrela, APP molekula osoa edota α− eta β−sekretasaren eraginez
askaturiko eremu estrazelularra ezagutzen ditu. Erabili genituen beste antigorputzek C
muturreko sekuentzia ezagutzen dute. Kasu horretan antigorputzak molekula osoa edota
γ−sekretasaren eraginez banaturiko sekuentziak seinalatuko ditu. Baita α- eta β-
sekretasen eraginez mintzari lotuta gelditzen diren APPren CTF zatiak ere. Era horretan
eta sinaptosomen Western Blotetatik eratorritako informazioaz baliatuz APP osoaren eta
proteolisiaren ondoren sortutako molekulen ultraegitura mailako jarraipena egiten saiatu
ginen.
5.2.1. APPren N muturraren aurkako antigorputza
Gure emaitzen arabera, APPren N muturraren aurkako antigorputza erabiliz, mintz
presinaptikoan kokatzen da nagusiki APP, mintz postsinaptikoarekin alderatuz gero.
Emaitza horiek lortu genituen, bai mikroskopio elektronikorako egindako
esperimentuetan (erretxinan murgildu aurretik eta erretxinan murgildu ondoren), eta
baita sinaptosometatik eratorritako mintz zatikietan egindako Western Blot
esperimentuetan ere. Mikroskopio elektronikorako egindako esperimentuetan,
erretxinan murgildu aurretik egindako immunozitokimikako datuen kuantifikazioa egin
ondoren, ikusi genuen sinapsiko mintz presinaptiko eta postsinaptikoaren arteko aldea
ez zela esanguratsua. Erretxinan murgildu ondoren egindako immunozitokimikako
datuen kuantifikazioa egitean, berriz, ezberdintasunak esanguratsuak zirela ondorioztatu
genuen. Bi teknikekin lortutako emaitzen aldea antigorputzak ehunean antigenoarekin
lotzeko duen erraztasunarekin azaldu ditzakegu. Material eta metodoen atalean ikusi
dugu, erretxinan murgildu ondorengo immunozitokimika egitean, antigorputzak epitopo
guztiak agerian dituela eta beraz, erretxinan murgildu aurreko immunozitokimikan
baino fidagarraitasun handiagoa du alde horretatik. Gainera, erretxinan murgildu
ondorengo immunozitokimikan egindako kuantifikazioa zehatzagoa da; izan ere, urre
partikulak zuzenean kontatzen dira, ez erretxinan murgildu aurreko immunozitokimikan
bezala. Kasu horretan urre partikulak zilarrarekin intentsifikatzen dira, eta
kuantifikazioa ez da hain zehatza.
149

Eztabaida
Sinaptosometatik eratorritako mintzen zatikietan ere, APPren N muturraren aurkako
antigorputza erabiliz, mintz presinaptikoan ∼110kDa-ko pisu molekularraren inguruan
bandak agertzen zaizkigu. Mintz postsinaptikoan banda ahulak agertzen diren arren,
sinapsiko bi mintzak elkarren artean alderatuz gero, mintz presinaptikotan aberastua
agertzen da. Pisu molekular hori APPren molekula osoari dagokion pisua izango da.
Aurkeztu izan dira APPren presentzia sinapsiaren alde presinaptikoan deskribatu izan
duten ikerketak. Izan ere, haren zelula barneko garraioaren inguruko lanetan ikusi da
APP axoietan barrena garraiatua izaten dela, eta alde presinaptikoan sinaptofisinarekin
batera kokatzen dela (Koo eta lank., 1990 , Massone eta lank., 2007, Kamal eta lank.,
2000, Sisodia eta lank., 1993; Morin eta lank., 1993). Hipokanpoan, ikusi da APP bide
zulatzailetik barrena axoietan modu anterogradoan garraiatu ondoren, hipokanpo
formazioko neurona entorrinalen terminalen presinapsian kokatzen dela (Buxbaum eta
lank., 1998b). Mikroskopio elektronikoaz egindako ikerketa batean (Ribaut-Barassin eta
lank., 2000), esan zen APPren kokapena hipokanpoan, nagusiki postsinaptikoa dela eta
bertan PS1ekin batera kokatzen dela. Presinaptikoki ia agertzen ez dela deskribatu zuten
ikertzaile horiek. Haiek erabilitako metodoa immunoperoxidasarena izan zen. APPren
aurka erabili zituzten antigorputzek N muturra ezagutzen zuten (Ribaut-Barassin eta
lank., 2000). Gure emaitzekiko ezberdintasunen azalpena erabilitako teknika
desberdinen sentsibilitatean egon daiteke. Immunoperoxidasaren metodoa erretxinan
murgildu aurreko metodo bat da; antigorputzentzat metodo horrek sarkortasun arazoak
dakartza, eta, horrez gainera, teknika horren markaketaren difusio maila dela eta, eta
APPren zitoplasmako presentzia ugaria kontutan hartuz, ezinezkoa da kasu askotan
mintz edo zitoplasmako markaketa den zehaztea.
5.2.2. APPren C muturraren aurkako antigorputza
Sinaptosometatik eratorritako mintz zatikiekin egindako Western Bloten emaitzen
arabera, APPren C muturrak (CTFα eta CTFβ) sinapsiaren alde presinaptikoan eta mintz
estrasinaptikoan kokatzen dira. APPren CTFγ (10kDa-etik beherako zatia)
presinaptikoki kokatzen dela ikusi dugu. Zenbait ikerketaren arabera, esan izan da APP
eta beraren proteolisitik eratorritako C99 alde presinaptikoan kokatzen direla bide
zulatzailean,, eta han gertatu litekeela C99tik beta amiloide peptidorako proteolisia
(Buxbaum eta lank., 1998b). Antigorputz horrekin APP molekula osoari dagokion
banda alde presinaptikoan ikusi dugu, eta alde postsinaptikoan ere agertzen zen arren,
nagusiki alde presinaptikoan ikusten zen. Hau da, N muturraren aurkako
150

Eztabaida
antigorputzarekin ikusitako kokapen-patroi bera errepikatu zaigu C muturraren aurkako
antigorputza erabiliz.
Mikroskopio elektronikoaren kasuan, berriz, bai erretxinan murgildu aurreko eta baita
ondorengo immunozitokimikan lortutako emaitzen arabera, nahiz eta zenbaitetan
presinapsian kokatu APPren C muturra sinapsiaren alde postsinaptikoan kokatzen da,
batez ere; gainera, alde presinaptikoarekin alderaturik, ezberdintasun esanguratsuak
lortzen genituen erretxinan murgildu aurreko eta ondorengo inmunozitokimikaren
bidez.
APP osoa, dentsitate postsinaptikoan ikusi izan da, sinapsi kitzikatzaileetan (Hoe eta
lank., 2009). Ikerketa horien arabera, APPren kokapena, presinaptikoa eta
postsinaptikoa da (Hoe eta lank., 2009). Sinaptosomekin egin dituzten ikerketa horietan,
APPren kokapen presinaptiko eta postsinaptikoaz ari direnean, APP osoaz ari dira eta
bandak 110 kDa inguruan agertzen zaizkie. Gure kasuan, mikroskopio elektronikoaren
kasuan, ezin dezakegu jakin urre partikulek markatzen duten proteina APPren C
muturra bakarrik den, ala APP osoa den. Beraz, izan liteke, gu mikroskopioaz
postsinaptikoki ikusten ari garen markaketa APP osoari dagokion markaketa izatea; izan
ere, gure sinaptosomen arabera, pisu molekularrari kasu eginaz, C mutur askeak (CTF
zatikiak; CTFα, CTFβ eta CTFγ) sinapsian daudenean presinaptikoak dira, baina,
molekula osoa nagusiki presinaptikoki agertzen bada ere, maila txikiago batean
postsinaptikoki ere aurkezten da.
Esan dugu azken ikerketa batzuen arabera APP alde presinaptikoan eta postsinaptikoan
kokatzen dela (Hoe eta lank., 2009). Gainera, APPren garraioari buruzko ikerketa ugari
egin dira, eta, lan horien arabera, sortu berria den APPa, axoi-muturrera garraiatua
izaten da, garraio anterogrado azkarraren bitartez, kinesinaren menpeko mekanismoaren
bidez (Koo eta lank., 1990; Buxbaum eta lank., 1998b); ondoren, transzitosia gerta
daiteke eta axoietik gune somatodendritikora garraiatzen da (Simons eta lank., 1995).
Hori dela eta, APP dendrita eta terminaletan kokatzen da (Kaether eta lank., 2000) gure
emaitzekin bat eginez.
Mikroskopian lortu ditugun emaitzen arabera, APP sinapsiaren bi aldeetan kokatzen
dela argi dagoela dirudien arren, alde batean eta bestean molekula fase desberdinetan
egon daitekeela edota prozesamendu desberdinak jasan ondoren kokapen espezifiko bat
har dezakeela adierazten dute. N muturra nagusiki presinaptikoa da; C muturraren
aurkako antigorputzak, berriz, APP edo beraren zatiki bat batez ere postsinapsian
151

Eztabaida
aurkitzen du. Sinaptosomen esperimentuetan lortutako emaitzen arabera, berriz, C
muturraren kokapena presinaptikoa izango litzateke. Baliteke kontraesan hori tekniken
arteko aldean egotea. Mikroskopia elektronikoaren kasuan, molekulak bere egitura
tertziarioan mantenduko dira, interakzio molekularrak mantenduz. Mikroskopio
elektronikorako erabilitako gure tekniken mugak kontutan hartuz, litekeena da APPk
alde presinaptikoan bere karboxi muturraren bidez izan ditzakeen elkarrekintza ugariak
direla eta (Turner eta lank, 2003), C muturraren aurka erabilitako antigorputzak
sekuentzia aurkitzeko arazoak izatea (Katona eta lank., 2006). Hori dela eta, APPren C
muturra ezagutzen duen antigorputz kantitatea murriztu egingo da alde presinaptikoan.
Bestalde, postsinaptikoki kokatzen den APP modu ahul xamarrean egon liteke mintz
postsinaptikoari lotuta. Hori horrela izanik, sinaptosometatik zatikiak banatzeko
prozeduran, ahulki lotutako proteina horiek solubilizatuak izan daitezke lehen pausoan
eta, ondorioz, mintz postsinaptikoan ikusi beharrean mintz estrasinaptikoan ikusten dira
CTF-ei dagozkien banda horiek.
5.3. PS1en kokapena Mikroskopio elektronikoan lortutako emaitzen arabera, presenilina-1 sinapsian kokatzen
da, modu aberastuan mintz estrasinaptikoarekin alderatuz gero, eta gainera, sinapsian
zehazki, alde postsinaptikoan agertzen da nagusiki. Mikroskopio elektronikoarentzat
egindako esperimentuetan, bai erretxinan murgildu aurreko immunozitokimika erabiliz
eta baita erretxinan murgildu ondorengo immunozitokimika erabiliz ere, presenilina-1
nagusiki alde postsinaptikoan kokatzen dela ikusi dugu. Mintz estrasinaptikoan oso
maila baxuan agertzen zitzaigun sinapsiarekin alderatuz.
Gainera, sinaptosometatik eratorritako mintz zatikietan egindako Western Blot-ekin ere,
emaitza berberak lortu ditugu. Hau da, presenilina, bai sinapsiaren alde presinaptikoan
eta baita postsinaptikoan ere agertzen zaigu, baina alde postsinaptikoan nagusiki. Mintz
estrasinaptikoan ez zen detektatu.
Presenilina-1 axoietan kokatzen den ala ez eztabaidagai izan da. Zenbait taldek kokapen
hori ukatu izan dute; beste batzuek, berriz, bertan koka zitekeela defendatu dute (Cook
eta lank., 1996; Lah eta lank., 1997; Elder eta lank., 1996, Kovacs eta lank., 1996;
Moussaoui eta lank., 1996). Horrela, PS1 axoietan barrena garraiatua izaten dela
deskribatu da, eta sinapsian lokalizatu izan da (Beher eta lank., 1999; Lah eta lank.,
152

Eztabaida
1997). Garraio hori modu anterogradoan (zelularen somatik terminaletarantz) eta
retrogradoan (terminaletatik somarantz) egiten du, APPk egiten duen modu berean
(Kasa eta lank., 2001; Papp eta lank., 2002).
Aurretik aipatu bezala, zenbait ikerketak PS1 axoietan kokatzen ez dela proposatu duten
arren (Cook eta lank., 1996) PS1, bai axoietan eta baita dendritetan ere ikusi izan da
(Busciglio eta lank., 1997; Capell eta lank., 1997). Ikerketa horietan, gure kasuan
gertatzen den moduan, axoietan agertu dela esan arren, axoiekin alderatuz gero
somatodendritikoki maila altuagoan adierazten dela esan izan dute (Busciglio eta lank.,
1997; Capell eta lank., 1997). Bai mintz presinaptikoan eta baita postsinaptikoan agertu
zaigun arren, mintz postsinaptikoa izan da kokapen nagusia. Gurekin bat datoz
immunoperoxidasa erabiliz mikroskopio elektroniko mailan egindako ikerketeta bitan;
presenilina nagusiki dendrita eta arantza dendritiko mailan agertu izan da, eta, askoz
gutxiago zen arren, (Lah eta lank., 1997, Ribaut-Barassin eta lank., 2000) presinaptikoki
ere ikusi izan dute (Lah eta lank., 1997).
5.4. APPren Proteolisia gertatzen den eskualdea APP garrantzizko proteina da sinaptogenesian eta, garun helduaren kasuan, sinapsien
eraldaketa-prozesuetan (Ribaut-Barassin eta lank., 2000). Lehenago esan dugun bezala,
APP modu anterogradoan garraiatzen da axoietan barrena, eta axoi-muturretara iristen
da (Simons eta lank, 1995; Kins eta lank., 2006). Axoi-terminaletik, transzitosi-prozesu
baten bidez, dendrita postsinaptikoetara hel daiteke (Simons eta lank., 1995). APPren
garraio bide horretan, bere sekretasekin elkarrekintzak izan ditzake gune batzuetan, eta
beraren proteolisia gerta daiteke hala nola, mintz plasmatikoan, erretikulu
endoplasmatikoan, Golgi aparatuan eta endosometan (Marcello eta lank., 2007; Kovacs
eta lank., 1996; Haass eta Selkoe, 1998; Ni eta lank., 2006).
Sarrerako atalean aipatu dugu γ-sekretasa bidezko mozketa Mintz Barneko Proteolisi
Erregulatuaren (RIP) bitartez gertatzen dela; beraz, aurretik beste sekretasa baten
bidezko mozketa gertatzen da (α- edo β-sekretasaren bidezkoa). Ondoren, mozketa
horretatik eratorritako zatiaren bigarren mozketa gertatuko da,; γ−sekretasaren bidezko
mozketa. Garrantzitsua da mozketa horietako bakoitza zelula barnean non gertatzen den
jakitea, bai bide ez-amiloidogenikoaren kasuan, eta baita bide amiloidogenikoaren
kasuan ere. Sekretasa horien zelula barneko garraioan, ezberdintasunak deskribatu izan
153

Eztabaida
dira, eta beraz, sekretasa bakoitza APPrekin zelularen gune zehatzetan elkartuko da.
APPren proteolisian, parte-hartzaileen kokapena garrantzitsua izan daiteke APPk bide
amiloidogenikoa ala ez amiloidogenikoa jarraituko duen jakiteko (Cole eta Vassar,
2007; Capell eta lank., 2002; Sheng eta lank., 2003).
APPren proteolisi konstitutiboa sinapsitik kanpo gertatzen dela dirudien arren, orain arte
hurbildu ditugun hainbat eta hainbat ikerketek APP osoak eta bere zatikiek
sinapsiarekin duten erlazio estua adierazten dute. Horrekin batera, bere sekretasek ere
kokapen sinaptikoa izateak, egoera jakin batzutan APPren proteolisia sinapsi mailan ere
gerta daitekeela pentsarazten du.
5.4.1. Bide ez-amiloidogenikoa
α-sekretasa bidezko mozketa, ustez, Trans Golgi Sarean eta nagusiki mintz
plasmatikoan gertatzen da (Marcello eta lank., 2007). Sinapsiari dagokionez, hala ere,
azken urteetako ikerketa batzuek ADAM10en sinapsiko kokapenaz hitz egin dute
(Marcello eta lank., 2007). Gainera, SAP97 proteinarekin elkarrekintzak dituela esan da
(proteina hori, beste zenbait proteina sinapsi kitzikatzaileetara garraiatzeaz arduratzen
da). Ikerketa horren arabera, ADAM10 postsinapsira garraiatu ondoren, elkarrekintza
horiek eragina dute ADAM10en α-sekretasa aktibitatean hipokanpoko sinapsi
glutamatergikoetan (Marcello eta lank., 2007).
Hala ere, sinaptosomekin egindako gure esperimentuetan, ADAM10 sinapsian kokatzen
zen arren, nagusiki sinapsitik kanpo kokatzen zela ikusi zen. Ez da hori gertatzen
BACE1 eta PS1 proteinekin. Azken horiek, sinapsian kokatzen ziren nagusiki, mintz
estrasinaptikoekin alderatuz gero (erakutsi gabeko datuak).
Horregatik guztiagatik eta, gainera, aurretik deskribatua izan den moduan (Marcello eta
lank., 2007), APPren proteolisi ez-amiloidogenikoa sinapsitik kanpoko egituretan
gertatzen dela uste dugu. Dena dela, ADAM10en sinapsiko kokapenaz hitz egin denez
(Marcello eta lank., 2007), eta, gure emaitzetan ere sinapsian (maila baxuagoan bada
ere) agertzen zenez, ez dugu erabat baztertzen sinapsian kasuren batean APPren
proteolisi ez-amiloidogenikoa gerta daitekeela; gainera, horretarako jarraitu beharko
lituzkeen mekanismoak deskribatuak izan dira (Blobel, 2000, 2005; Reiss eta lank.,
2005; Muresan eta lank., 2009; Janes eta lank., 2005; Dewji eta Singer, 1996). Baina
hala ere, ADAM10ek, APPz gain, beste substratu batzuk ere baditu. Horietako batzuk
sinapsian kokatzen dira, eta hori izan daiteke ADAM10 sinapsian kokatzearen arrazoia
(Reiss eta lank., 2005). Datu horiek guztiek, aurretik esan bezala, APPren sinapsitik
154

Eztabaida
kanpoko proteolisi bide bati buruz pentsatzera garamatzate bide ez-amiloidogenikoaren
kasuan.
5.4.2. Bide amiloidogenikoa
Beta sekretasaren kasuan, behin garatuta, BACE1 zelularen azalera garraiatua izango da
eta bertatik berriro ere barneratua izan daiteke endosometara (Huse eta lank., 2000).
BACE1en eta APPren zelula barneko kokapenarekin bat datoz APPren beta sekretasa
bidezko mozketa Trans Golgi Sarean, endosometan eta mintz plasmatikoan ikusi izan
duten ikerketak (Vassar eta Citron, 2000; Walter eta lank., 2001a). Hala ere, β-sekretasa
bidezko APPren mozketa endosoma/lisosometan deskribatu da nagusiki (Koo eta
Squazo, 1994; Haass eta lank., 1995, Thinakaran eta lank., 1996b).
BACE1, eta aurretik esan dugun bezala APP eta presenilina, axoietan barrena
garraiatuak izaten dira axoien mutur presinaptikoraino. Xixku berdinetan elkarrekin
garraiatuak diren ala ez eta bide horretan prozesu proteolitiko amiloidogenikoa
gertatzen den ala ez eztabaidagai den arren, axoietan barrena presinapsirainoko garraio
anterogradoaz luze eta zabal hitz egin izan da (Kamal eta lank., 2000; Kamal eta lank.,
2001; Kins eta lank., 2006; Papp eta lank., 2002; Lazarov eta lank., 2005; Capell eta
lank., 2002; Sheng eta lank., 2003).
β-amiloidea askatzen den gune nagusia neuronen alde presinaptikoa dela ikusi da (Kins
eta lank., 2006). Gainera, bide zulatzailean, APP eta beraren proteolisitik eratorritako
C99, alde presinaptikoan kokatzen direla, , eta bertan gerta litekeela C99tik β−amiloide
peptidorako proteolisia (Buxbaum eta lank., 1998b) iradoki da. Hori guztia bat dator
guk lortutako emaitzekin non APPren kokapen presinaptikoa eta sinaptosometatik
eratorritako mintz zatikiekin egindako Western Blot-etan APPren CTFak alde
presinaptikoan kokatzen zirela ere ikusi dugun. Izan ere, gure emaitzen arabera CTFα
eta CTFβ mintz extrasinaptikotan eta sinapsiaren alde presinaptikoan kokatzen dira.
CTFγ berriz, mintz presinaptikoan aberasturik agertzen da.
Gure sinaptosomekin eginiko esperimentuetan BACE1 batez ere presinaptikoki
kokatzen den proteina da (erakutsi gabeko datuak). BACE1en aktibitate optimoa pH
azidotan izaten da, eta pH hori ez da normalki mintz plasmatikoan aurkitzen dena. Hala
ere, gerta liteke, xixku sinaptikoek askatu ditzaketen eduki azidoen ondorioz, sinapsian,
une batez, pH azido hori lortzea. Horrela, BACE1ek bere proteolisia gauzatuko luke,
eta, ondoren, presenilinak eragingo luke, bide amiloidogenikoaren bigarren pausoa
155

Eztabaida
emanaz, eta β-amiloide peptidoa sortuz. Presenilina postsinaptikoki kokatzen dela ikusi
dugu gure emaitzetan. Hori horrela izan arren, presinaptikoki ere agertzen zen.
Presinaptikoki kokatzen den hori izango litzateke modu horretan mozketa gauzatuko
lukeena (5.1.A irudia). Aipatzekoa da, presenilina γ-sekretasa konplexuan sartuta egoten
dela mozketa egiteko orduan, eta konplexuaren gainerako osagaiak ere sinapsian ikusi
direla; horietako batzuk (Pen-2 eta Nct), gainera, presinapsian aberasturik (Siman eta
Salidas, 2004; erakutsi gabeko datuak). Bide hori gertatzen bada, alde presinaptikoa
APPren proteolisi amiloidogeniko erregulatutako gunea izango litzateke, hau da,
sinapsiko aktibitatearen araberakoa. Hori bat dator sinapsiko aktibitatearen gorakadaren
ondorioz β-sekretasa aktibitatearen gorakada bat ere gertatzen dela esan duten zenbait
ikerketekin (Kamenetz eta lank., 2003). Egon dira sinapsi aktibitatearen ondorioz
CTFβren ekoizpenean igoerarik ikusi ez dituzten taldeak ere (Cirrito eta lank., 2005),
baina haiek ere bat datoz β-amiloide peptidoaren ekoizpenaren eta sinapsiaren
aktibitatearen artean erlazio zuzena dagoela dioen hipotesiarekin. Esan izan da, halaber,
xixku sinaptikoen zikloa bera β-amiloidea neuronetatik askatzeko nahikoa dela; hau da,
neuronaren despolarizazioa ez dela behar horretarako (Cirrito eta lank., 2005). Kontutan
hartu behar da ikerketa horretan, hala ere, garun guztiko CTFβ mailak aztertu zituztela,
eta ez zehazki sinapsi mailan zeuden CTFβ mailak.
Proposatutako APPren proteolisi bide horretan ez da aintzat hartzen postsinapsian
kokaturik dagoen PS1. Gainera,gure emaitzen kasuan, PS1en kokapena nagusiki
postsinaptikoa da. Sinapsiaren alde postsinaptikoan kokatzen diren zenbait substratu
ditu PS1ek, eta bertan horien proteolisian aritu daitekeela pentsa daiteke (Mizoguchi eta
lank., 2002; May eta lank., 2004; Eugenin eta lank., 2007; Krivosheya eta lank., 2008).
Hala ere, PS1en kokapen postsinaptikoa kontutan hartzen duen β-/γ-sekretasa bidea
gertatzeko beste modu bat ere proposatu izan da; gure emaitzekin guztiz bat datorrena.
APPren proteolisi bide horretan, eta baita beste proteina batzuen kasuan ere, trans-
sinapsi bidezko mozketa mota bat gertatzen da (5.1.B irudia). Presenilinaren eta APPren
arteko elkarrekintzak gertatzen dira; bata sinapsi bateko alde presinaptikoan kokaturik
dagoelarik, eta bestea berriz alde postsinaptikoan (Dewji eta Singer, 1996; Dewji,
2006). Badirudi, APP presinapsian egonik eta PS1 postsinapsian, elkarrekintzak izango
dituztela haien artean, eta, ondorioz, elkarrekin endozitosi bidez barneratuak izango
direla neurona presinaptikora. Bertan, gorputz multibesikularretara bideratuko dira, eta
hor daudenean izango da β-/γ-sekretasa mozketak jasango dituzten unea. Ondoren,
156

Eztabaida
sortutako β-amiloide peptidoa neurona presinaptikotik askatua izango da (Dewji eta
Singer, 1996; Dewji eta Singer, 1998; Dewji, 2006). Gainera, β-amiloidea sortzeko
APPren endozitosiaren beharra deskribatua izan da (Cirrito eta lank, 2008). Gure
emaitzak kontutan izanik, hipotesi hori guztiz zilegia izango litzateke β-amiloidea
sortzen den APPren proteolisi bidea deskribatzeko.
5.1. Irudia: A) APPren proteolisi amiloidogenikoa sinapsi mailan. BACE1ek APP moztuko du mintz presinaptikoan; xixku sinaptikoek eduki azidoa askatzean inguruneko pH azidoak beraren aktibitatea bermatzen du. Ondoren, mintz presinaptikoan gelditzen den CTFβ muturra presinaptikoki kokatzen den γ-sekretasa konplexuak moztuko du. Sinapsiaren bi aldetan kokatzen diren proteinak nagusiki zein aldetan kokaturik dauden “marratxoen” bidez adierazita dago.
5.1. Irudia: B) APPren proteolisi amiloidogenikoa sinapsi mailan gertatzeko beste aukera bat. Mintz postsinaptikoan kokaturik dagoen γ-sekretasak, PS1en bidez mintz presinaptikoan kokaturik dagoen APPrekin trans-elkarrekintzak izango ditu. Elkarrekintza horien ondorioz biak endozitosi bidez zelula presinaptikora sartuko dira. Gorputz multibesikularretan BACE1en eta γ-sekretasaren aktibitatearen ondorioz βA peptidoa sortuko da, eta peptido hori presinapsitik kanporatua izango da.
Orain arte plazaratuak izan diren hainbat ikerketak eta gure emaitzek adierazten
dutenez, APPren proteolisia sinapsian gerta daiteke (Buxbaum eta lank., 1998b; Ribaut-
Barassin eta lank., 2000). Horrekin lotuta, zenbait ikerketak APPren proteolisiaren eta
157

Eztabaida
lipid raft-en arteko erlazio estua adierazi dute. Bai APP, BACE1 eta γ-sekretasa
konplexua ere lipid raft-etan kokaturik ikusi izan dira, eta baita β-sekretasa bidezko
mozketaren ondoren lortzen den APPren C mutur laburra ere (CTFβ) (Ehehalt eta lank.,
2003; Wahrle eta lank., 2002; Riddell eta lank., 2001). Bestalde, beste ikerketa batzuen
arabera, ADAM10, detergente bidezko estrakzioaren ondoren, disolbagarria zen, raft-
ekin lotuta ez dagoela erakutsiz, eta gauza bera gertatzen da CTFα-rekin ere (Ehehalt
eta lank., 2003; Riddell eta lank., 2001; Kojro eta lank., 2001).
Ikerketa batek baino gehiagok hitz egin dute sinapsiko lipid raft-en presentziari buruz
(Suzuki eta lank., 2007;. Pfrieger, 2003). Kolesterolak sinapsiaren garapen eta
egonkortasunean garrantzi handia du (Pfrieger, 2003), eta gainera, lipid raft-etan,
zenbait hartzaile, seinalizazio-molekula, zenbait tirosina kinasa eta zitoeskeletoko
proteinak kokatzen direla esan izan dute (Matsuura eta lank., 2007; Delint-Ramírez eta
lank., 2008; Besshoh eta lank., 2005). Alde presinaptikoari buruz zehazki ere hitz egin
izan da lipid raft-en inguruan egindako lanetan, eta, ikusi denez, terminal
presinaptikoak kolesteroletan aberastuta daude (Pfrieger, 2003). Gainera, sinapsiaren
garapenean, esan izan da BDNFk zenbait proteina presinaptikoren maila areagotzen
duela lipid-raft domeinuetan, eta, erdiko nerbio-sisteman, funtzio presinaptikoen
garapenerako, kolesterolaren biosintesian ere eragina du (Suzuki eta lank., 2007).
Talde askok esan dute APPren bi proteolisi bideak zelularen gune ezberdinetan
gertatzen direla (Marcello eta lank., 2007). Hau da, α-sekretasa aktibitatea Trans-Golgi
Sarean eta mintz plasmatikoan deskribatu da (Lammich eta lank., 1999); β-sekretasa
aktibitatea, berriz, nagusiki endosoma/lisosoma sisteman kokatu da, bere aktibitaterako
behar duen pH azidoa aurkitzen duen lekuetan alegia (Kinoshita eta lank., 2003).
Domeinu berdinetan ere beren aktitibitatea egin dezaketen arren, eta horrela denean
gainera haien arteko leia izaten duten arren (Capell eta lank., 2002), sekretasa horiek
leku ezberdinetan gauzatu ohi dute beren proteolisia, eta horren erakusgarri da, gainera,
zelula barneko garraioan APP moztuko duten sekretasek bide ezberdinak jarraitzea; β-
sekretasa axoietarantz modu anterogradoan garraiatzen da, eta α-sekretasak, bestalde,
garraio somatodendritikoa du (Capell eta lank., 2002). Presenilinak, berriz, bi aldeetara
jotzeko joera izango du, nahiz eta, gure emaitzen arabera, eta aurretik ere esan den
bezala, postsinaptikoki maila altuagoan kokatu (Ribaut-Barassin eta lank., 2000).
Hori guztia lotuz, esan da APPren proteolisi amiloidogenikoak lipid raft-ekin erlazioa
duela (Ehehalt eta lank., 2003) eta, horren ondorioz, Ehehalt eta lankideek proposatu
158

Eztabaida
dute, APPren multzo bat lipid raft-etan kokatu eta prozesu amiloidogenikoa jasaten
duela, eta beste multzo bat hortik kanpoko mintzetan kokatzen dela eta prozesu ez-
amiloidogenikoa jasaten duela (Ehehalt eta lank., 2003).
Beraz, eta aipatutako guztiarekin, guztiz zilegia da APPren proteolisi amiloidogenikoa
sinapsiaren alde presinaptikoan eta bertako lipid raft-etan kokatuta gerta daitekeela
proposatzea , eta, bide ez-amiloidogenikoa, berriz, sinapsitik kanpo gertatuko dela
pentsatzea.
5.5. Laburbilduz
Azken urteetan plazaratu diren emaitzek gure emaitzekin batera, APP-ren proteolisia
sinapsian gerta daitekeela iradokitzen dute.
APPren proteolisi bide nagusia bide ez-amiloidogenikoa da (Postina, 2008), eta
nagusiki sinapsia ez den zelularen beste zenbait gunetan gertatzen da proteolisi hori.
ADAM10 sinapsian ere kokatzen den arren, beraren kokagunea ez dago sinapsian
aberastuta; BACE1 eta PS1ren presentzia, berriz bai. Datu horiek sinapsian gertatuko
den proteolisi bide nagusia bide amiloidogenikoa dela adierazten dute.
Bestalde, presinaptikoki kokatzen dira BACE1 eta APP, baita PS1 ere maila baxuagoan
bada ere. Bide amiloidogenikoa lipid raft-etan gertatzen dela frogatu da; lipid raft-etatik
kanpo, berriz, bide ez-amiloidogenikoa. Hori dela eta, uste dugu sinapsiko lipid-raft-
etan mozketa amiloidogenikoa gerta daitekeela, eta bertan sortu daitekeela β-amiloide
peptidoa.
APPren proteolisia modu konstitutiboan eta erregulatuan gertatzen da. Bai bide ez-
amiloidogenikoa, eta baita amiloidogenikoa ere (Hooper eta Turner, 2002) Sinapsian
gertatuko den proteolisi amiloidogenikoa modu erregulatuan gertatzen dela pentsa
dezakegu. Modu horretan, βA peptidoa eratuko da sinapsiaren aktibitatearen arabera.
Sortutako peptidoak bere funtzio fisiologikoak beteko ditu, zenbait hartzaile aktibatuz,
eta baita sinapsiaren gehiegizko aktibitatea ekidinaz ere. Horrela, gaixotasun-egoeran,
erregulazio horren desoreka bat gerta liteke. Horrek βA gehiegi pilatzea ekar dezake.
APPren proteolisi amiloidogenikoa zelularen hainbat gunetan gerta daitekeela esan den
arren, garrantzi handikoa da sinapsian proteolisi lokala gerta daitekeelako hipotesi hau.
Gure emaitzen arabera, presinapsian gertatuko litzateke βA peptidoa ekoizteko mozketa.
Emaitza horiek Alzheimer gaixotasunaren testuinguruan garrantzizkoak izan daitezke,
159

Eztabaida
APPren sinapsiko metabolismoaren deserregularizazioaren ondorioz βΑen metaketak
gerta baitaitezke. Alzheimer gaixotasunean, neuroendekapenezko beste zenbait
sintomaren aurretik, sinapsietan arazoak gertatzen hasten dira (Selkoe, 2002). Arazo
horiek, sinapsian bertan gertatzen ari den APPren proteolisiaren alterazioen ondorioz
gerta daitezke. Hau da, baldintza fisiologikoetan, mintz presinaptikoan, βA peptidoa
sortzen da, eta bere ekoizpen-garbiketa prozesua orekatua dago. Gainera, prozesu ez-
amiloidogenikoa ere gertatuko da aldi berean zelularen beste gune batzuetan, eta horren
ondorioz sortutako zatikiek funtzio fisiologikoak beteko dituzte. Bide ez-
amiloidogenikotik sortutako sAPPα zatia ere xixkuetan garraiatua eta exozitatua izaten
da (Muresan eta lank., 2009). Gaixotasun-egoeran, berriz, axoietan zehar gertatzen den
proteinen garraioan arazoak daudenean, βA peptidoaren ekoizpen maila handitzen dela
ikusi da (Stokin eta lank., 2005). Maila presinaptikoan gertatzen ari zen APPren
proteolisi erregulatu horrek akatsak izan ditzake, eta horren ondorioz Aβ peptidoaren
metaketak gerta daitezke eta horrek sinapsiaren funtzio fisiologikoan eta plastizitate
sinaptikoan arazoak eragin ditzake.
160

6. ONDORIOAK


Ondorioak
6. ONDORIOAK
1. APP arratoi helduen hipokanpoko CA1 guneko neuronen mintzetan kokatzen da,
bai mintz sinaptikoetan, eta baita mintz estrasinaptikoetan ere. Sinapsian modu
aberastuan kokatzen da mintz estrasinaptikoekin alderatuz.
2. PS1 arratoi helduen hipokanpoko CA1 guneko neuronen mintzetan kokatzen da,
bai mintz sinaptikoetan, eta baita mintz estrasinaptikoetan ere. Sinapsian modu
aberastuan kokatzen da mintz estrasinaptikoekin alderatuz.
3. APP sinapsian kokatzen denean mintz presinaptikoan kokatzen da nagusiki.
4. PS1 sinapsian kokatzen denean mintz postsinaptikoan kokatzen da nagusiki.
5. APP molekula osoa presinaptikoki kokatzen da.
6. APPren proteolisiaren ondorioz sortzen diren CTFα eta CTFβ zatiak, mintz
estrasinaptikoan kokatzen dira nagusiki. Sinapsian daudenean mintz
presinaptikoan kokatzen dira.
7. CTFγ zatikia, mintz presinaptikoan kokatzen da nagusiki.
8. APPren proteolisiaren ondorioz sortzen diren sAPP zatiak ehunaren
homogeneizatuaren zati disolbagarrian kokatzen dira.
163


7. BIBLIOGRAFIA


Bibliografia
Alfandari D., Cousin H., Gaultier A., Smith K., White J.M., Darribère T., DeSimone D.W. 2001. Curr Biol. 11(12): 918-930 Amaral D.G. 1978. J Comp Neurol. 182(4 Pt 2):851-914 Amaral D.G., Dolorfo C., Alvarez-Royo B.1991. Hippocampus 1(4):415-436 Amaral D.G., Witter M.P. 1989. Neuroscience. 31(3):571-91. Amaral D.G., Witter M.P. 1995. The Rat Nervous System (21). (2.Edizioa. Ed. Paxinos G.) Academic Press. Anderson J.P., Esch F.S., Keim P.S., Sambamurti K., Lieberburg I., Robakis N.K. 1991. Neurosci Lett 128(1):126-128 Angeletti B., Waldron K.J., Freeman K.B., Bawagan H., Hussain I., Miller C.C., Lau K.F., Tennant M.E., Dennison C., Robinson N.J., Dingwall C. 2005. J.Biol.Chem. 280(18): 17930-17937 Annaert W.G., Levesque L., Craessaerts K., Dierinck I., Snellings G., Westaway D., George-Hyslop P.S., Cordell B., Fraser P., De Strooper B. 1999. J.Cell Biol. 147 (2), 277-294 Apelt J., Schliebs R., Beck M., Roβner S., Bigl V. 1997. Int. J. Devl. Neurosci. 15, 95-112 Asai M., Hattori C., Szabo B., Sasagawa N., Maruyama K., Tanuma S., Ishiura S. 2003. Biochem.Biophys.Res.Commun. 301, 231-235 Barger S.W., Mattson M.P. 1995. Biochem J. 311, 45-47 Baulac S., LaVoie M.J., Kimberly T, Strahle J., Wolfe M.S., Selkoe D.J., Xia W. 2003. Neurobiology of Disease 14, 194-204 Beel A.J., Sanders C.R. 2008. Cell Mol Life Sci. 65(9):1311-34. Beher D., Elle C., Underwood J., Davis J.B., Ward R., Karran E., Masters C.L., Beyreuther K., Multhaup G. 1999. J Neurochem. 72(4):1564-73. Benjannet S., Elagoz A., Wickham L, Mamarbachi M., Munzer J.S., Basak A., Lazure C., Cromlish J.A., Sisodia S., Checler F., Chrétien M., Seidah N. 2001. J. Biol. Chem 276(14) 10879-10887 Bennett B.D., Babu-Khan S., Loeloff R., Louis J.C., Curran E., Citron M., Vassar R. 2000a. J. Biol. Chem 275(27) 20647-20651 Bennett B.D., Denis P., Haniu M., Teplow D.B., Kahn S., Louis J.C., Citron M., Vassar R. 2000b. J. Biol. Chem 275(48) 37712-37717
167

Bibliografia
Berry M.M., Standring S.M., Bannister L.H. 1998. Anatomía de Gray. 38 Edizioa. Harcourt Brace. Besshoh S., Bawa D., Teves L., Wallace M.C., Gurd J.W. 2005. J Neurochem. 93(1):186-94. Billings L.M., Oddo S., Green K.N., McGaugh J.L., LaFerla F.M. 2005. Neuron. 45, 675-688 Blobel C.P. 2000. Curr Opin Cell Biol. 12(5):606-12 . Blobel C.P. 2005. Nat Rev Mol Cell Biol. 6(1):32-43. Bradford M.M.1976.Anal Biochem.72:248-54 Brown M.S., Goldstein J.L.1997.Cell..89, 331-340 Brown M.S., Ye J., Rawson R.B., Goldstein J.L. 2000. Cell 100, 391-398 Bunsey M., Eichenbaum H. 1993. Behav Neurosci. 107(5):740-7 Busciglio J., Gabuzda D.H., Matsudaira P., Yanker B.A. 1993. Procl.Natl.Acad.Sci.U.S.A.90, 2092-2096 Busciglio J., Hartmann H., Lorenzo A., Wong C., Baumann K., Sommer B., Staufenbiel M., Yankner B.A.1997. J Neurosci. 17(13):5101-7. Buxbaum J.D., Liu K.N., Luo Y., Slack J.L., Stocking K.L., Peschon J.J., Johnson R.S., Castner B.J., Cerretti D.P., Black R.A. 1998a. J.Biol.Chem. 273(43): 27765-27767 Buxbaum J.D., Thinakaran G., Koliatsos V., O´Callahan J., Slunt H.H., Price D.L., Sisodia S.S. 1998b. J. Neurosci 18(23), 9629-9637 Caillé I., Allinquant B., Dupont E., Bouillot C., Langer A., Müller U., Prochiantz A. 2004. Development and Disease 131, 2173-2181 Capell A., Beher D., Prokop S., Steiner H., Kaether C., Shearman M.S., Haass C. 2005. J. Biol.Chem, 280, 6471-6478 Capell A., Grünberg J., Pesold B., Diehlmann A., Citron M., Nixon R., Beyreuther K., Selkoe D.J., Haass C. 1998. J. Biol. Chem. 273(6): 3205-3211 Capell A., Meyn L., Fluhrer R., Teplow D.B., Walter J., Haass C. 2002. J. Biol. Chem 277(7), 5637-5643 Capell A., Saffrich R., Olivo J.C., Meyn L., Walter J., Grünberg J., Mathews P., Nixon R., Dotti C., Haass C. 1997. J Neurochem. 69(6):2432-40. Capell A., Steiner H., Willem M., Kaiser H., Meyer C., Walter J., Lammich S., Multhaup G., Haass C. 2000. J. Biol. Chem 275(40), 30849-30854
168

Bibliografia
Caporaso G.L., Takei K., Gandy S.E., Matteoli M., Mundigl O., Greengard P., De Camilli P.1994. J. Neurosci. 14(5), 3122-3138 Card J.P., Meade R.P., Davis L.G. 1988. Neuron. 1(9):835-846 Chan S.L., Mattson M.P. 1999. J Neurosci Res. 58(1):167-90. Chávez-Gutiérrez L., Tolia A., Maes E., Li T., Wong P.C., De Strooper B. 2008. J. Biol. Chem. 283, 20096-20105 Chin J.H., Ma L., MacTavish D., Jhamandas J.H. 2007. J. Neurosci. 27(35): 9262-9269 Chyung J.H., Raper D.M., Selkoe D.J. 2005. J. Biol. Chem 280(6), 4383-4392 Cirrito J.R., Kang J.E., Lee J., Stewart F.R., Verges D.K., Silverio L.M., Bu G., Mennerick S., Holtzman D.M. 2008. Neuron. 58(1):42-51. Cirrito J.R., Yamada K.A., Finn M.B., Sloviter R.S., Bales K.R., May P.C., Schoepp D.D., Paul S.M., Mennerick S., Holtzman D.M. 2005. Neuron. 48(6):913-22. Citron M., Dile T.S., Capell A., Haass C., Teplow D.B., Selkoe D.J. 1996. Neuron. 17, 171-179 Cohen R.S., Blomberg F., Berzins K., Siekevitz P. 1977. J.Cell.Biol. 74:181-203 Cole S.L., Vassar R. 2007. Mol Degeneration. 2 (22) Cole S.L., Vassar R. 2008. J. Biol. Chem 283(44) 29621-29625 Cook D.G., Sung J.C., Golde T.E., Felsenstein K.M., Wojczyk B.S., Tanzi R.E., Trojanowski J.Q., Lee V.M., Doms R.W. 1996. Proc Natl Acad Sci U S A. 93(17):9223-8. Creemers J.W., Ines Dominguez D., Plets E., Serneels L., Taylor N.A., Multhaup G., Craessaerts K., Annaert W., De Strooper B. 2001. J Biol Chem. 276(6):4211-7. Cupers P., Bentahir M., Craessaerts K., Orlans I., Vanderstichele H., Saftig P., De Strooper B., Annaert W. 2001. J.Cell.Biol. 154(4), 731-740 Dawson GR, Seabrook GR, Zheng H, Smith DW, Graham S, O'Dowd G, Bowery BJ, Boyce S, Trumbauer ME, Chen HY, Van der Ploeg LH, Sirinathsinghji DJ. 1999. Neuroscience. 90(1):1-13. De Camilli P., Haucke V., Takei K., Mugnaini E. 2001. Synapses. (Ed. Cowan M., Südhof T.C., Stevens C.F.). De Strooper B. 2003. Neuron 38, 9-12
169

Bibliografia
De Strooper B., Beullens M., Contreras B., Levesque L., Craessaerts K., Cordell B., Moechars D., Bollen M., Fraser P., George-Hyslop P.S., Van Leuven F. 1997 J Biol Chem. 272(6):3590-8. De Strooper B., Saftig P., Craessaerts K., Vandersticheles H., Guhde G., Annaert W., Von Figura K., Van Leuven F. 1998. Nature 391, 387-390 Delint-Ramírez I., Salcedo-Tello P., Bermudez-Rattoni F. 2008. J Neurochem. 106(4):1658-68. Dewachter I., Ris L., Croes S., Borghgraef P., Devijver H., Voets T., Nilius B., Godaux E., Van Leuven F. 2008. Neurobiol Aging. 29(5):639-52. Dewji N.N. 2006. J Alzheimers Dis.10(2-3):277-90. Dewji N.N., Singer S.J. 1996. Proc Natl Acad Sci U S A. 93(22):12575-80 Dewji N.N., Singer S.J. 1998. Proc Natl Acad Sci U S A. 95(25):15055-60. Diana RA, Yonelinas AP, Ranganath C.2007. Trends Cogn Sci.11(9):379-86. Dineley K.T., Westerman M., Bui D., Bell K., Ashe K.H., Sweatt J.D. 2001. J. Neurosci. 21(12): 4125-4133 Doan A., Thinakaran G., Borchelt D.R., Slunt H.H., Ratovitsky T., Podlisny M., Selkoe D.J., Seeger M., Gandy S.E., Price D.L., Sisodia S.S. 1996. Neuron. 17, 1023-1030 Dolleman-Van der Weel M.J., Witter M.P. 1992. Eur.J.Neurosci. Suppl.5:69 Dries D.R., Yu G. 2008. Curr Alzheimer Res. Duan W., Rangnekar V.M., Mattson M.P. 1999. J.Neurochem. 72(6):2312-2322 Edbauer D., Winkler E., Haass C., Steiner H. 2002. Proc Natl Acad Sci U S A. 99(13):8666-71. Ehehalt R., Keller P., Haass C., Thiele C., Simons K. 2003. J Cell Biol. 160(1):113-23. Elder G.A., Tezapsidis N., Carter J., Shioi J., Bouras C., Li H.C., Johnston J.M., Efthimiopoulos S., Friedrich V.L. Jr, Robakis N.K. 1996. J Neurosci Res. 45(3):308-20. Esch F.S., Keim P.S., Beattie E.C., Blacher R.W., Culwell A.R., Oltersdorf T., McClure D., Ward P.J. 1990. Science. 248(4959):1122-1124 Esler W.P., Kimberly W.T., Ostaszewski B.L., Diehl T.S., Moore C.L., Tsai J.Y., Rahmati T., Xia W., Selkoe D.J., Wolfe M.S.2000.Nature Cell Biol. 2, 428-434 Eugenin E.A., King J.E., Nath A., Calderon T.M., Zukin R.S., Bennett M.V., Berman J.W. 2007. Proc Natl Acad Sci U S A. 104(9):3438-43.
170

Bibliografia
Francis R., McGrath G., Zhang J, Ruddy D.A., Sym M., Apfeld J., Nicoll M., Maxwell M., Hai B., Ellis M.C., Parks A.L., Xu W., Li J., Gurney M., Myers R.L., Himes C.S., Hiebsch R., Ruble C., Nye J.S., Curtis D. 2002. Dev.Cell 3, 85-97 Freund T.F., Gulyás A.I., Acsady L., Görcs T., Tóth K. 1990. Proc.Natl.Acad.Sci. USA. 87:8501-8505 Friesen L. eta Woolridge N. CMAJ/JAMC, 2008. (Patterson C., Feightner J.W., Garcia A., Hsiung G.Y.R., MacKnight C., Sadovnick A.D. 2008. CMAJ 178(5): 548-556) Furukawa K., Barger S.W., Blalock E.M., Mattson M.P. 1996a. Nature 379, 74-78 Furukawa K., Mattson M.P. 1998. Neurosci. 83(2): 429-438 Georgakopoulos A., Marambaud P., Efthimiopoulos S., Shioi J., Cui W., Li H.C., Schütte M., Gordon R., Holstein G.R., Martinelli G., Mehta P., Friedrich V.L. Jr, Robakis N.K. 1999. Mol Cell 4(6):893-902 Giliberto L., Borghi R., Piccini A., Mangerini R., Sorbi S., Cirmena G., Garuti A., Ghetti B., Tagliavini F., Mughal M.R., Mattson M.P., Zhu X., Wang X., Guglielmotto M., Tamagno E., Tabaton M. 2009. J Biol Chem. 2009. 284(14):9027-38. Glenner G.G., Wong C.W. 1984. Biochem. Biophys. Res. Commun. 120, 885-890 Goutte C., Tsunozaki M., Hale V.A.,Priess J.R. 2002. Procl.Natl.Acad.Sci.USA. 99(2), 775-779 Gralle M., Ferreira S.T. 2007. Prog Neurobiol. 82(1):11-32. Greenfield J.P., Tsai J., Gouras G.K., Hai B., Thinakaran G., Checler F., Sisodia S.S., Greengard P., Xu H. 1999. Proc Natl Acad Sci U S A. 96(2):742-7. Gu, Y., Chen, F., Sanjo, N., Kawarai, T., Hasegawa H., Duthie M., Li W., Ruan X., Luthra A., Mount H.T.J., Tandon A., Rraser P.E., George-Hyslop P. 2003. J.Biol.Chem. 278. 7374-7380 Haapasalo A., Kim D.Y., Carey B.W., Turunen M.K., Pettingell W.H., Kovacs D.M. 2007. J Biol Chem. 282(12):9063-72. Haass C., Koo E.H., Mellon A., Hung A.Y., Selkoe D.J. 1992. Nature 357, 500-503 Haass C., Lemere C.A., Capell A., Citron M., Seubert P., Schenk D., Lannfelt L., Selkoe D.J. 1995. Nat Med.1(12):1291-6. Haass C., Schlossmacher M.G., Hung A.Y., Vigo-Pelfrey C., Mellon A., Ostaszewski B.L., Lieberburg I., Koo E.H., Schenk D., Teplow D.B., Selkoe D.J. 1992. Nature 359, 322-325 Haass C., Selkoe D.J. 1998. Nature. 391(6665):339-40.
171

Bibliografia
Haniu M., Denis P., Young Y., Mendiaz E.A., Fuller J., Hui J.O., Bennet B.D., Kahn S., Ross S., Burgess T., Katta V., Rogers G., Vassar R., Citron M. 2000. J. Biol. Chem 275(28) 21099-21106 Hartmann D., De Strooper B., Serneels L, Craessaerts K., Herreman A., Annaert W., Umans L., Lϋbke T., Illert A. L., von Figura K., Saftig P. 2002. Hum. Mol. Genet 11 (21): 2615-2624 He W., Lu Y., Qahwash I., Hu X.Y., Chang A., Yan R. 2004. Nat. Med. 10(9): 959-965 Heber S., Herms J., Gajic V., Hainfellner J., Aguzzi A., Rülicke T., Kretzschmar H., von Koch C., Sisodia S., Tremml P., Lipp H.P., Wolfer D.P., Müller U. 2000. J. Neurosci 20(21), 7951-7963 Hébert SS, Serneels L, Dejaegere T, Horré K, Dabrowski M, Baert V, Annaert W, Hartmann D, De Strooper B.2004.Neurobiol Dis.17(2):260-72 Herl L., Thomas A.V., Lill C.M., Banks M., Deng A., Jones P.B., Spoelgen R., Hyman B.T., Berezovska O. 2009. Mol Cell Neurosci. Herms J., Anliker B., Heber S., Ring S., Fuhrmann M., Kretzschmar H., Sisodia S., Müller U. 2004. EMBO J. 23, 4106-4115 Hoe H.S., Fu Z., Makarova A., Lee J.Y., Lu C., Feng L., Pajoohesh-Ganji A., Matsuoka Y., Hyman B.T., Ehlers M.D., Vicini S., Pak D.T., Rebeck G.W. 2009. J Biol Chem. 284(13):8495-506. Epub 2009 Jan 21. Hoffman J., Pietrzik U., Kummer M.P., Twiesselmann C., Bauer C., Herzog V. 1999. J. Histochem. Cytochem. 47(3):373-382 Hooper N.M., Turner A.J. 2002. Curr Med Chem.9(11):1107-19. Hsu S.M., Raine L. 1981. J Histochem Cytochem. 29(11):1349-53. Hung A.Y., Haass C., Nitsch R.M., Qiu W.Q., Citron M., Wurtman R.J., Growdon J.H., Selkoe D.J. 1993. J.Biol.Chem. 268(31):22959-22962 Huovilla A.P.J., Turner A.J., Pelto-Huikko M., Kärkkäinen L., Ortiz R.M. 2005. Trends Biochem. Sci. 30:413-4122 Hur J.Y., Welander H., Behbahani H., Aoki M., Franberg J., Winblad B., Frykman S., Tjernberg L.O. 2008. FEBS J. 2008. 275: 1174-1187 Huse J.T., Pijak D.S, Leslie G.J., Lee V.M., Doms R.W. 2000. J Biol Chem. 27;275(43):33729-37. Hussain I., Powell D., Howlett D.R., Tew D.G., Meek T.D., Chapman C., Gloger I.S., Murphy K.E., Southan C.D., Ryan D.M., Smith T.S., Simmons D.L., Walsh F.S., Dingwall C. , Christie G. 1999. Mol.Cell.Neurosci. 14, 419-427
172

Bibliografia
Ikin A.F., Annaert W.G., Takei K., De Camilli P., Jahn R., Greengard P., Buxbaum J.D. 1996. J Biol Chem. 271(50):31783-6. Ishida A., Furukawa K., Keller J.N., Mattson M.P. 1997 Neuroreport 8, 2133-2137 Jacobsen L., Madsen P., Nielsen M.S., Geraerts W.P., Gliemann J., Smit A.B., Petersen C.M. 2002. FEBS Lett. 511(1-3):155-8. Janes P.W., Saha N., Barton W.A., Kolev M.V., Wimmer-Kleikamp S.H., Nievergall E., Blobel C.P., Himanen J.P., Lackmann M., Nikolov D.B. 2005. Cell 123(2):291-304 Jay T.M., Glowinski J., Thierry A.M. 1989. Brain Res. 505:337-340 Jay T.M., Witter M.P. 1991. J.Comp.Neurol. 313:574-586 Jozwiak K., Krzysko K.A., Bojarski L., Gacia M., Filipek S. 2008. ChemMedChem. 3(4):627-34. Jutras I., Laplante A., Boulais J., Brunet S., Thinakaran G., Desjardins M. 2005. J.Biol.Chem 280 (43), 36310-36317 Kaether C., Lammich S., Edbauer D., Ertl M., Rietdorf J., Capell A., Steiner H., Haass C. 2002. J.Cell.Biol. 158 (3), 551-561 Kaether C., Scheuermann J., Fassler M., Zilow S., Shirotani K., Valkova C., Novak B., Kacmar S., Steiner H., Haass C. 2007. EMBO reports 8 (8) 743-748 Kaether C., Schmitt S., Willem M. Haass C. 2006. Traffic 7, 408-415 Kaether C., Skehel P., Dotti C.G. 2000. Mol Biol Cell. 11(4):1213-24 Kahle W., Frotscher M. 2008. Atlas de Anatomía. 3.Tomoa. 9.edizioa. Editorial Médica Panamericana. Kalvodova L., Kahya N., Schwille P., Ehehalt R., Verkade P., Drechsel D., Simons K. 2005. J.Biol.Chem. 280(44): 36815-36823 Kamal A., Almenar-Queralt A., LeBlanc J.F., Roberts E.A., Goldstein L.S. 2001. Nature. 414(6864):643-8. Kamal A., Stokin G.B., Yang Z., Xia C.H., Goldstein L.S. 2000. Neuron. 28(2):449-59. Kamenetz F., Tomita T., Hsieh H., Seabrook G., Bochelt D., Iwatsubo T., Sisodia S., Malinow R. 2003. Neuron. 37, 925-937 Kandel E.R. 2001. Principios de Neurociencia. IX.zatia (63). (4.edizioa. Ed: Kandel E.R., Schwartz J.H., Jessell T.M.) Mcgraw-Hill-Interamericana. Kandel E.R., Kupfermann I., Iversen S. 2001. Principios de Neurociencia. IX.zatia (62). (4.edizioa. Ed: Kandel E.R., Schwartz J.H., Jessell T.M.) Mcgraw-Hill-Interamericana.
173

Bibliografia
Kandel E.R., Siegelbaum S.A. 2001. Principios de Neurociencia. III zatia (10). (4.edizioa. Ed: Kandel E.R., Schwartz J.H., Jessell T.M.) Mcgraw-Hill-Interamericana. Kang J., Lemaire H.G., Unterbeck A., Salbaum J.M., Masters C.L., Grzeschik K.H., Multhaup G., Beyreuther K., Müller-Hill B. 1987. Nature 325, 733-736 Kasa P., Papp H., Pakaski M. 2001. Brain Res. 909(1-2):159-69. Katona I., Urbán G.M., Wallace M., Ledent C., Jung K.M., Piomelli D., Mackie K., Freund T.F. 2006. J Neurosci. 26(21):5628-37. Kim D.Y., Ingano L.A., Kovacs D.M. 2002. J Biol Chem. 277(51):49976-81. Kim J., Schekman R.. 2004. Proc.Natl.Acad.Sci. USA. 101, 905-906. Kim S.H., Yin Y.I., Li Y.M., Sisodia S.S. 2004. J. Biol. Chem. 279, 48615-48619 Kim T.W., Wu K., Xu J.L., McAuliffe G., Tanzi R.E., Wasco W., Black I.B. 1995. Mol. Brain Res. 32, 36-44 Kimberly W.T. eta Wolfe M.S. 2003. J.Neurosci. Res. 74:353-360 Kinoshita A., Fukumoto H., Shah T., Whelan C.M., Irizarry M.C., Hyman B.T. 2003. J Cell Sci. 116(Pt 16):3339-46. Kins S., Lauther N., Szodorai A., Beyreuther K. 2006. Neurodegener Dis. 3(4-5):218-26. Review. Kirazov E., Kirazov L., Bigl V., Schliebs R. 2001. Int J Dev Neurosci. 19(3):287-96. Kitaguchi N., Takahashi Y., Tokushima Y., Shiojiri S., Ito H. 1988. Nature 331, 530-532 Knight D., Iliadi K., Charlton M.P., Atwood H.L., Boulianne G.L. 2007. Dev Neurobiol. 67(12):1598-613. Koike H., Tomioka S., Sorimachi H., Saido T.C., Maruyama K., Okuyama A., Fujisawa-Sehara A., Ohno S., Suzuki K., Ishiura S. 1999. Biochem. J. 343, 371-375 Kojro E., Gimpl G., Lammich S., Marz W., Fahrenholz F. 2001. Proc Natl Acad Sci U S A. 98(10):5815-20. Kojro E., Postina R., Buro C., Meiringer C., Gehrig-Burger K., Fahrenholz F. 2006. FASEB J. 20(3):512-514 Koo E.H., Sisodia S.S., Archer D.R., Martin L.J., Weidemann A., Beyreuther K., Fischer P., Masters C.L., Price D.L. 1990. Proc Natl Acad Sci U S A. 87(4):1561-5. Koo E.H., Squazzo S.L. 1994. J Biol Chem. 269(26):17386-9.
174

Bibliografia
Kovacs D.M., Fausett H.J., Page K.J., Kim T.W., Moir R.D., Merriam D.E., Hollister R.D., Hallmark O.G., Mancini R., Felsenstein K.M., Hyman B.T., Tanzi R.E., Wasco W. 1996. Nat Med. 2(2):224-9. Krettek J.E., Price J.L. 1977. J.Comp.Neurol. 172:723-752 Krivosheya D., Tapia L., Levinson J.N., Huang K., Kang Y., Hines R., Ting A.K., Craig A.M., Mei L., Bamji S.X., El-Husseini A. 2008. J Biol Chem. 283(47):32944-56 LaFerla F.M. 2002. Nature. 3, 862-872 LaFerla F.M., Green K.N., Oddo S. 2007. Nature. 8, 499-509 Lah J.J., Heilman C.J., Nash N.R., Rees H.D., Yi H., Counts S.E., Levey A.I. 1997. J Neurosci. 17(6):1971-80. Lah J.J., Levey A.I. 2000. Mol Cell Neurosci. 16(2):111-26. Lammich S., Kojro E., Postina R., Gilbert S., Pfeiffer R., Jasionowski M. Haass C., Fahrenholz F. 1999. Proc.Natl.Acad.Sci.USA 96(7):3922-3927 Laudon H., Hansson E.M., Melen K., Bergman A., Farmery M.R., Winblad B., Lendahl U., von Heijne G., Naslund J. 2005. J.Biol. Chem. 280, 35352-35360 LaVoie M.J., Fraering P.C., Ostaszewski B.L, Ye W., Kimberly W.T., Wolfe M.S., Selkoe D.J.2003. J. Biol.Chem 278(39), 37213-37222 Lazarov O., Lee M., Peterson D.A., Sisodia S.S. 2002. J. Neurosci. 22(22):9785-9793 Lazarov O., Morfini G.A., Lee E.B., Farah M.H., Szodorai A., DeBoer S.R., Koliatsos V.E., Kins S., Lee V.M., Wong P.C., Price D.L., Brady S.T., Sisodia S.S. 2005. J Neurosci. 25(9):2386-95. Lee R.K., Wurtman R.J., Cox A.J., Nitsch R.M. 1995. Proc Natl Acad Sci. USA. 92(17):8083-8087 Lemberg M.K., Martoglio B. 2002. Mol.Cell 10, 735-744 Lemberg M.K., Martoglio B. 2004. FEBS Letters 564, 213-218 Lesné S., Ali C., Gabriel C., Croci N., MacKenzie E.T., Glabe C.G., Plotkine M., Marchand-Verrecchia C., Vivien D., Buisson A. 2005. J. Neurosci. 25(41), 9367-9377 Li X., Greenwald I. 1996. Neuron. 17, 1015-1021 Li X., Greenwald I. 1998. Proc.Natl.Acad.Sci. USA .1998. 95, 7109-7114
175

Bibliografia
Li Y.M., Xu M., Lai M.T., Huang Q., Castro J.L., DiMuzio-Mower J., Harrison T., Lellis C., Nadin A., Neduvelil J.G., Register R.B., Sardana M.K., Shearman M.S., Smith A.L., Shi X.P., Yin K.C., Shafer J.A., Gardell S. 2000. Nature 405, 689-694 Li Z.W., Stark G., Götz J., Rülicke T., Gschwind M., Huber G., Müller U., Weissmann C. 1996. Proc Natl Acad Sci USA 93:6158–6162. Lin X., Koelsch G., Wu S., Downs D., Dashti A., Tang J. 2000. Procl. Natl. Acad. Sci. 97(4), 1456-1460 Liu Q., Wu J. 2006. Acta Pharmacol. Sin 27(10): 1277-1286 Liu Y., Zhang Y.W., Wang X., Zhang H., You X., Liao F.F., Xu H. 2009. J. Biol. Chem. Loechel F., Gilpin BJ, Engvall E., Albrechtsen R., Wewer U.M. 1998. J. Biol. Chem. 273, 16993-16997Behav Brain Res 95:65–76. Cell Death Diff 5:858–866. Löffler J., Huber G. 1992. J Neurochem. 59(4):1316-24. López de Jesús M., Sallés J., Meana J.J., Callado L.F. 2006. Neurosci. 140:635-643 Lorente de Nó R. 1934. J.Physiol. Neurol. 46 :113-177 Luján R., Roberts J.D.B., Shigemoto R., Ohishi H., Somogyi P. 1997. J.Chem. Neuroanat. 13:219-241 Luo W.J., Wang H., Li H., Kim B.S., Shah S., Lee H.J., Thinakaran G., Kim T.W., Yu G., Xu H. 2003. J.Biol.Chem 278 (10), 7850-7854 Lyckman A.W., Confaloni A.M., Thinakaran G., Sisodia S.S., Moya K.L. 1998. J.Biol.Chem. 273 (18) 11100-11106 Marcello E., Epis R., Di Luca M. 2008. Eur J Pharmacology 585, 109-118 Marcello E., Gardoni F., Mauceri D., Romorini S., Jeromin A., Epis R., Borroni B., Cattabeni F., Sala C., Padovani A., Di Luca M. 2007. J Neurosci. 27(7):1682-91. Marquez-Sterling N.R., Lo A.C.Y., Sisodia S.S., Koo E.H. 1997. J. Neurosci, 17(1), 140-151 Massone S., Argellati F., Passalacqua M., Armirotti A., Melone L., d'Abramo C., Marinari U.M., Domenicotti C., Pronzato M.A., Ricciarelli R. 2007. Biochem Biophys Res Commun. 362(3):633-8. Masters C.L., Simms G., Weinman N.A., Multhaup G., McDonald B.L., Beyreuther K. 1985. Procl.Natl. Acad. Sci. USA 82, 4245-4249 Matsuura D., Taguchi K., Yagisawa H., Maekawa S. 2007. Neurosci Lett. 16;423(2):158-61. Epub 2007 Jul 28
176

Bibliografia
Mattson M.P. 1997. Physiol Rev. 77(4):1081-132. Mattson M.P., Cheng B., Culwell A.R., Esch F.S., Lieberburg I. Rydel R.E. 1993. Neuron. 10(2), 243-54 May P., Rohlmann A., Bock H.H., Zurhove K., Marth J.D., Schomburg E.D., Noebels J.L., Beffert U., Sweatt J.D., Weeber E.J., Herz J. 2004. Mol Cell Biol. 24(20):8872-83. Mills J., Laurent Charest D., Lam F., Beyreuther K., Ida N., Pelech S.L., Reiner P.B. 1997. J. Neurosci. 17(24):9415-9422 Milward E.A., Papadopoulos R., Fuller S.J., Moir R.D., Small D. Beyreuther K., Masters C.L. 1992. Neuron. 9(1), 129-137 Mizoguchi A., Nakanishi H., Kimura K., Matsubara K., Ozaki-Kuroda K., Katata T., Honda T., Kiyohara Y., Heo K., Higashi M., Tsutsumi T., Sonoda S., Ide C., Takai Y. 2002. J Cell Biol. eb 4;156(3):555-65. Morin P.J., Abraham C.R., Amaratunga A., Johnson R.J., Huber G., Sandell J.H., Fine R.E. 1993. J Neurochem. 61(2):464-73. Moussaoui S., Czech C., Pradier L., Blanchard V., Bonici B., Gohin M., Imperato A., Revah F. 1996. FEBS Lett. 383(3):219-22. Mucke L., Abraham C.R., Masliah E. 1996. Ann.NY Acad. Sci. 777, 82-88 Müller T., Meyer H.E., Egensperger R., Marcus K. 2008. Progress in Neurobiology 85, 393-406 Müller U., Cristina N., Li Z.W., Wolfer D.P., Lipp H.P., Rülicke T., Brandner S., Aguzzi A., Weissmann C. 1994. Cell 79:755–765. Muresan V., Varvel N.H., Lamb B.T., Muresan Z. 2009. J.Neurosci. 29(11): 3565-3578 Ni Y., Zhao X., Bao G, Zou L, Teng L, Wang Z., Song M., Xiong J., Bai Y., Pei G. 2006. Nature Medicine. 12(12) 1390-1396 Nitsch R.M., Deng M., Growdon J.H., Wurtman R.J. 1996. J.Biol.Chem 271(8):4188-4194 Nitsch R.M., Slack B.E., Farber S.A., Borghesani P.R., Schulz J.G., Kim C., Felder C.C., Growdon J.H., Wurtman R.J. 1993. Ann. N.Y. Acad. Sci. 695: 122-127 Nitsch R.M., Slack B.E., Wurtman R.J., Growdon J.H. 1992. Science 258(5080):304-307 Nunan J., Small D.H. 2000 FEBS Lett. 483, 6-10 Oh, Y.S., Turner R.J. 2005. Biochemistry. 44, 11821-11828
177

Bibliografia
Ohsawa I., Takamura C., Kohsaka S. 2001. J.Neurochem 76, 1411-1420 Osenkowski P., Ye W., Wang R., Wolfe M. Selkoe D.J. 2008. J. Biol. Chem. 283 (33) 22529-22540 Ouimet C.C., Baerwald K.D., Gandy S.E., Greengard P. 1994. J. Comp. Neurol. 348(2):244-260 Papp H., Pakaski M., Kasa P. 2002. Neurochem Int. 41(6):429-35. Parameshwaran K., Dhanasekaran M., Suppiramaniam V. 2008. Exp. Neurol. 210, 7-13 Pardossi-Piquard R., Petit A., Kawarai T., Sunyach C., Alves da Costa C., Vincent B., Ring S., D'Adamio L., Shen J., Müller U., St George Hyslop P., Checler F. 2005. Neuron. 46(4):541-54. Parent A.T., Barnes N.Y., Taniguchi Y., Thinakaran G., Sisodia S.S. 2005. J Neurosci. 2005. 25(6):1540-9. Parks A.L., Curtis D. 2007. TRENDS in Genetics 23(3), 140-150 Parvathy S, Hussain I, Karran EH, Turner AJ, Hooper NM. 1999. Biochemistry. 38(30):9728-34. Pasternak S.H., Bagshaw R.D., Guiral M., Zhang S., Ackerley C.A., Pak B.J., Callahan J.W., Mahuran D.J. 2003. J.Biol.Chem. 278(29), 26687-26694 Pastorino L., Ikin A.F., Nairn A.C., Pursnani A., Buxbaum J.D. 2002. Mol Cell Neurosci. 19(2):175-85. Paxinos G., Kus L., Ashwell K.W.S., Watson C. 1999. Chemoarchitectonic Atlas of The Rat Forebrain. Academic Press. Pearson H.A., Peers C. 2006. J.Physiol 575 (1) 5-10 Peschon J.J., Slack J., Reddy P., Stocking K.L., Sunnarborg S.W., Lee D.C., Russell W.E., Castner B.J., Johnson R.S., Fitzner J.N., Boyce R.W., Nelson N., Kozlosky C.J., Wofson M.F., Rauch C.T., Cerretti D.P., Paxton R.J., March C.J., Black R.A. 1998. Science 282, 1281-1284 Pettit D.L., Shao Z., Yakel J.L. 2001. J. Neurosci. 21(1): 120 Pfrieger F.W. 2003. Biochim Biophys Acta. 1610(2):271-80. Phillips G.R., Huang J.K., Wang Y., Tanaka H., Shapiro L., Zhang W., Shan W.S., Arndt K., Frank M., Gordon R.E., Gawinowicz M.A., Zhao Y., Colman D.r. 2001. Neuron. 32: 63-77 Pietrzik C.U., Hoffmann J., Stöber K., Chen C.Y., Bauer C., Otero D.A.C., Roch J.M., Herzog V. 1998. Proc. Natl. Acad. Sci. USA 95, 1770-1775
178

Bibliografia
Plant L.D., Boyle J.P., Smith I.F., Peers C., Pearson H.A. 2003. J. Neurosci. 23(13), 5531-5535 Ponte P., Gonzalez De-Whitt P., Schilling J., Miller J., Hsu D., Greenberg B., Davis K., Wallace W., Lieberburg I., Fuller F., Cordell B. 1988. Nature 331 525-527 Postina R. 2008 Curr Alzheimer Res.; 5(2): 179-186 Postina R., Schroeder A., Dewachter I., Bohl J., Schmitt U., Kojro E., Prinzen C., Endres K., Hiemke C., Blessing M., Flamez P., Dequenne A., Godaux E., van Leuven F., Fahrenholz F. 2004. J. Clin. Invest. 113, 1456-1464 Priller C., Bauer T., Mitteregger G., Krebs B., Kretzschmar H.A., Herms J. 2006. J. Neurosci. 26(27), 7212-7221 Primakoff P., Myles D.G. 2000. Trends Genet. 16(2):83-87 Prokop S., Shirotani K., Edbauer D., Haass C., Steiner H. 2004. J. Biol. Chem 279(22), 23255-23261 Purves D., Augustine G.J., Fitzpatrick D., Hall W.C., Lamantia A.S., Mcnamara J.O., Williams S.M. 2008a. Neurociencia. 3. edizioa (24). Editorial Médica Panamericana. Purves D., Augustine G.J., Fitzpatrick D., Hall W.C., Lamantia A.S., Mcnamara J.O., Williams S.M. 2008b. Neurociencia. 3. edizioa (5). Editorial Médica Panamericana. Purves D., Augustine G.J., Fitzpatrick D., Hall W.C., Lamantia A.S., Mcnamara J.O., Williams S.M. 2008c. Neurociencia. 3. edizioa (6). Editorial Médica Panamericana. Puzzo D., Privitera L., Leznik E., Fà M., Staniszewski A., Palmeri A., Arancio O.2008. J. Neurosci. 28(53): 14537-14545 Ratovitsky T., Slunt H.H., Thinakaran G., Price D.L., Sisodia S.S., Borchelt D.R. 1997. J.Biol.Chem. 272, 24536-24541 Ray W.J., Yao M., Mumm J., Schroeter E.H., Saftig P., Wolfe M., Selkoe D.J., Kopan R., Goate A.M. 1999. J.Biol.Chem 274(51), 36801-36807 Reinhard C., Hébert S.S., De Strooper B. 2005. EMBO J. 24(23) 3996-4006 Reiss K., Maretzky T., Ludwig A., Tousseyn T., de Strooper B., Hartmann D., Saftig P. 2005. EMBO J. 24(4):742-52. Ribaut-Barassin C., Moussaoui S., Brugg B., Haeberlé A.M., Huber G., Imperato A., Delhaye-Bouchaud N., Mariani J., Bailly Y.J. 2000 Synapse, 35(2):96-110. Riddell D.R., Christie G., Hussain I., Dingwall C. 2001. Curr Biol. 11(16):1288-93
179

Bibliografia
Robert S.J., Zugaza J.L., Fischmeister R., Gardier A.M., Lezoualc'h F. 2001. J.Biol.Chem 276(48):44881-44888 Roberts S.B., Ripellino J.A., Ingalls K.M., Robakis N.K. eta Felsenstein K.M. 1994; J.Biol.Chem., 269; 3111-3116 Rolls E.T., Kesner R.P. 2006. Prog Neurobiol. 79(1):1-48. Rossjohn J., Cappai R., Feil S.C., Henry A., McKinstry W.J., Galatis D., Hesse L., Multhaup G., Beyreuther K., Masters C.L., Parker M.W. 1999. Nat Struct Biol. 6(4):327-31. Runz H., Rietdorf J., Tomic I.,de Bernard M., Beyreuther K., Pepperkok R., Hartmann T. 2002. J. Neurosci. 22(5): 1679-1689 Sabo S.L., Ikin A.F., Buxbaum J.D., Greengard P. 2003. 23(13), 5407-5415 Sannerud R. eta Annaert W. 2008. Semin Cell Dev Biol Sato T., Diehl T.S., Narayanan S., Funamoto S., Ihara Y., De Strooper B., Steiner H., Haass C., Wolfe M.S. 2007. J.Biol.Chem. 282 (47), 33985-33993 Scheuermann S., Hambsch B., Hesse L., Stumm J., Schmidt C., Beher D., Bayer T.A., Beyreuther K., Multhaup G. 2001. J.Biol.Chem. 276(36):33923-9 Scholefield Z., Yates E.A., Wayne G., Amour A., McDowell W., Turnbull J.E. 2003. J.Cell.Biol. 163(1): 97-107 Schrenk-Siemens K., Perez-Alcala S., Richter J., Lacroix E., Rahuel J., Korte M., Müller U., Barde Y.A., Bibel M. 2008. Stem Cells 26, 2153-2163 Schubert W., Prior R., Weidemann A., Dircksen H., Multhaup G., Masters C.L., Beyreuther K. 1991.Brain Res. 563(1-2), 184-194 Scoville W.B., Milner B. 1957. J Neurol Neurosurg Psychiatry. 20(1):11-21. Seabrook G.R., Smith D.W., Bowery B.J., Easter A., Reynolds T., Fitzjohn S.M., Morton R.A., Zheng H., Dawson G.R., Sirinathsinghji D.J., Davies C.H., Collingridge G.L., Hill R.G. 1999. Neuropharmacology. 38(3):349-59. Seabrook GR, Rosahl TW. 1999. Neuropharmacology. 38(1):1-17 Selkoe D.J. 2002. Science. 298: 789-791 Selkoe D.J. 2006. Nature Medicine. 12(7) 758-759 Selkoe D.J., Podlisny M.B., Joachim C.L., Vickers E.A., Lee G., Fritz L.C., Oltersdorf T. 1988. Procl. Natl. Acad. Sci. USA 85, 7341-7345
180

Bibliografia
Shah S., Lee S.F., Tabuchi K., Hao Y.H., Yu C., LaPlant Q., Ball H., Ill C.E.D., Südhof T., Yu G. 2005. Cell 122, 435-447 Shah S., Yu G. 2006. Mol Interventions 6 (2) 74-76 Sheng B., Song B., Zheng Z., Zhou F., Lu G., Zhao N., Zhang X., Gong Y. 2008. Neurosci Lett. 400(3): 327-331 Sheng J.G., Price D.L., Koliatsos V.E. 2003. Exp Neurol. 184(2):1053-7. Shi W., Chen H., Sun J., Buckley S., Zhao J., Anderson K.D., Williams R.G., Warburton D. 2003. Dev.Biol. 15; 261 (2): 371-380 Shigematsu K., McGeer P.L., McGeer E.G. 1992. Brain Res. 592(1-2), 353-357 Shimizu H., Tosaki A., Kaneko K., Hisano T., Sakurai T., Nukina N. 2008. Mol Cell Biol 28(11) 3663-3671 Shoji M., Golde T.E., Ghiso J., Cheung T.T., Estus S., Shaffer L.M., Cai X.D., McKay D.M., Tintner R., Frangione B., Younkin S.G. 1992. Science 258, 126-129 Siman R., Salidas S. 2004. Neurosci. 129: 615-628 Simons M., Ikonen E., Tienari P.J., Cid-Arregui A., Mönning U., Beyreuther K., Dotti C.G. 1995. J Neurosci Res.41(1):121-8. Sinha S., Anderson J.P., Barbour R., Basi G.S., Caccavello R., Davis D., Doan M., Dovey H.F., Frigon N., Hong J., Jacobson-Croak K., Jewett N., Keim P., Knops J., Lieberburg I., Power M., Tan H., Tatsuno G., Tung J., Schenk D., Seuber P., Suomensaari S.M., Wang S., Walker D., Zhao J., McConlogue L, John V. 1999. Lett Nature 402: 537-540 Sisodia S.S. 1992; Proc.Natl.Sci.USA., 89: 6075-6079 Sisodia S.S., Koo E.H., Hoffman P.N., Perry G., Price D.L. 1993. J Neurosci. 13(7):3136-42. Smith D.M., Mizumori S.J. 2006. J Neurosci. 26(12):3154-63. Soba P., Eggert S., Wagner K., Zentgraf H., Siehl K., Kreger S., Löwer A., Langer A., Merdes G., Paro R., Masters C.L., Müller U., Kins S., Beyreuther K. 2005. EMBO J.24(20):3624-34. Spasic D., Annaert W. 2007. J. Cell Science 121, 413-420 Spasic D., Annaert W. 2008. J. Cell Sci. 121, 413-420 Spasic D., Raemaekers T., Dillen K., Declerck I., Baert V., Serneels L., Füllekrug J., Annaert W. (2007) J. Cell. Biol. 176 (5), 629-640
181

Bibliografia
Spoelgen R., von Arnim C.A., Thomas A.V., Peltan I.D., Koker M., Deng A., Irizarry M.C., Andersen O.M., Willnow T.E., Hyman B.T. 2006. J.Neurosci. 26(2): 418-428 Steinbach J.P., Müller U., Leist M., Li Z.-W., Nicotera P., Aguzzi A. 1998 Steiner H., Fluhrer R., Haass C. 2008. J. Biol. Chem. 283 (44): 29627-29631 Stokin G.B., Lillo C., Falzone T.L., Brusch R.G., Rockenstein E., Mount S.L., Raman R., Davies P., Masliah E., Williams D.S., Goldstein L.S.B. 2005. Science. 307:1282-1288 Struhl G., Adachi A. 2000. Mol Cell 6, 625-636 Sun X., He G., Song W. 2006. FASEB J. 20(9):1369-76. Suzuki S., Kiyosue K., Hazama S., Ogura A., Kashihara M., Hara T., Koshimizu H., Kojima M. 2007. J. Neurosci. 27(24):6417-6427 Swanson L.W., Köhler C., Björklund A. 1987. Handbook of Chemical Neuroanatomy 5. Integrated Systems of the CNS. PartI (Ed.Björklund A., Hökfelt T., Swanson L.) Elsevier, Amsterdan. 125-227 Sweatt JD. 2001. J Neurochem. 76(1):1-10. Takasugi N., Tomita T., hayashi I., Tsuruoka M., Niimura M., Takahashi Y., Thinakaran G., Iwatsubo T. 2003. Nature 422, 438-441 Tanzi R.E., McClatchey A.I., Lamperti E.D., Villa-Komaroff L., Gusella J.F., Neve R.L. 1988. Nature 331 528-530 Tarassishin L., Yin Y.I., Bassit B., Li Y.M. 2004. Proc. Natl. Acad. Sci. USA. 101(49): 17050-17055 Taylor C.J. Ireland D.R., Ballagh I., Bourne K., Marechal N.M., Turner P.R., Bilkey D.K., Tate W.P., Abraham W.C. 2008. Neurobiol. Dis. 31(2): 250-260 Thinakaran G., Borchelt D.R., Lee M.K., Slunt H.H., Spitzer L, Kim G., Ratovitsky T., Davenport F., Nordstedt C., Seeger M., Hardy J., Levey A.I., Gandy S.E., Jenkins N.A., Copeland N.G., Price D.L., Sisodia S.S. 1996a. Neuron. 17, 181-190 Thinakaran G., Koo E.H. 2008. J. Biol. Chem. 283(44) 29615-29619 Thinakaran G., Teplow D.B., Siman R., Greenberg B., Sisodia S.S. 1996b. J Biol Chem. 271(16):9390-7. Thinakaran, G., Harris C.L., Ratovitsky T., Davenport F., Slunt H.H., Price D.L., Borchelt D.R., Sisodia S.S. 1997. J.Biol.Chem. 272, 28415-28422 Tomimoto H., Akiguchi I. Wakita H., Nakamura S., Kimura J. 1995. Brain Res 672, 187-195
182

Bibliografia
Tominaga-Yoshino K., Uetsuki T., Yoshikawa K., Ogura A. 2001. Brain Res. 918, 121-130 Tremml P., Lipp H.P., Müller U., Ricceri L., Wolfer D.P. 1998. Tsai J.Y., Wolfe M.S., Xia W. 2002. Curr.Med. Chem. 9(11): 1087-1106 Turner A.J., Fisk L., Nalivaeva N.N. 2004. Ann. N.Y. Acad. Sci. 1035: 1-20 Turner P.R., O´Connor K., Tate W.P, Abraham W.C. 2003. Prog. Neurobiol. 70, 1-32 Tyszkiewicz J.P., Yan Z. 2005. J. Neurophysiol. 93: 3102-3111 Uemura K., Kuzuya A., Shimozono Y., Aoyagi N., Ando K., Shimohama S., Kinoshita A. 2007. J Biol Chem. 282(21):15823-32. Uemura K., Lill C.M., Banks M., Asada M., Aoyagi N., Ando K., Kubota M., Kihara T., Nishimoto T., Sugimoto H., Takahashi R., Hyman B.T., Shimohama S., Berezovska O., Kinoshita A. 2009 J. Neurochem.108(2):350-60. Uemura T. 1998. Cell. 93(7):1095-8. Ulus IH, Wurtman RJ. 1997. J Pharmacol Exp Ther. 281(1):149-54. Urano Y., Hayashi I., Isoo N., Reid P.C., Shibasaki Y., Noguchi N., Tomita T., Iwatsubo T., Hamakubo T., Kodama T. 2005. J. Lipid Res. 46, 904-912 Van Gassen G., Annaert W., Van Broeckhoven C. 2000. Neurobiol. Dis. 7, 135-151 Van Groen T., Wyss J.M. 1990. J.Comp.Neurol. 302:515-528 Vassar R., Bennett B.D., Babu-Khan S., Kahn S., Mendiaz E.A., Denis P., Teplow D.B., Ross S., Amarante P., Loeloff R., Luo Y., Fisher S., Fuller J., Edenson S., Lile J., Jarosinski M.A., Biere A.L., Curran E., Burgess T., Louis J.C., Collins F., Treanor J., Rogers G., Citron M. 1999. Science 286, 735-741 Vassar R., Citron M. 2000. Neuron. 27(3):419-22. Verdier Y., Penke B. 2004. Curr. Protein Pept. Sci. 5, 19-21 Vetrivel K.S., Meckler X., Chen Y., Nguyen P.D., Seidah N.G., Vassar R., Wong P.C., Fukata M., Kounnas M.Z., Thinakaran G. 2008. J. Biol.Chem. Vetrivel K.S., Zhang Y.W., Xu H., Thinakaran G. 2006. Mol. Neurodeg. 1:4 von Koch C.S., Zheng H., Chen H., Trumbauer M., Thinakaran G., van der Ploeg L.H., Price D.L., Sisodia S.S. 1997. Neurobiol Aging 18:661– 669
183

Bibliografia
Wahle T. Thal D.R., Sastre M., Rentmeister A., Bogdanovic N., Famulok M., Heneka M.T., Walter J. 2006. J. Neurosci. 26(49) 12838-12846 Wahrle S., Das P., Nyborg A.C., McLendon C., Shoji M., Kawarabayashi T., Younkin L.H., Younkin S.G., Golde T.E. 2002. Neurobiol Dis. 9(1):11-23. Walsh D.M., Selkoe D.J.2007. J. Neurochem 101, 1172-1184 Walter J., Capell A., Grünberg J., Pesold B., Schindzielorz A., Prior R., Podlisny M.B., Fraser P., Hyslop P.S., Selkoe D.J., Haass C. 1996 Mol Med. 2(6):673-91. Walter J., Fluhrer R., Hartung B., Willem M., Kaether C., Capell A., Lammich S., Multhaup G., Haass C. 2001b. J Biol Chem. 276(18):14634-41 Walter J., Kaether C., Steiner H., Haass C. 2001a. Curr Opin Neurobiol. 11(5):585-90. Wang P., Yang G., Mosier D.R., Chang P., Zaidi T., Gong Y.D., Zhao N.M., Dominguez B., Lee K.F., Gan W.B., Zheng H. 2005. J. Neurosci. 25(5), 1219-1225 Wang R., Meschia J.F., Cotter R.J., Sisodia S.S. 1991. J.Biol.Chem. 266, 25; 16960-16964 Wang Y., Greig N.H., Yu Q.S., Mattson M.P. 2007. Neurobiol Aging. Watanabe N., Tomita T., Sato C., Kitamura T., Morohashi Y., Iwatsubo T. 2005. J. Biol. Chem. 280 (51), 41967-41975 Wouterlood F.G., Saldana E, Witter M.P. 1990. J.Comp.Neurol. 296:179-203 Weidemann A., Eggert S., Reinhard F.B., Vogel M., Paliga K., Baier G., Masters C.L., Beyreuther K., Evin G. 2002. Biochemistry. 41(8):2825-35. Weskamp G., Cai H., Brodie T.A., Higashyama S., Manova K., Ludwig T., Blobel C.P. 2002. Mol.Cell.Biol. 22, 1537-1544 Wickham L., Benjannet S., Marcinkiewicz E., Chretien M., Seidah N.G., 2005. J.Neurochem. 92(1): 93-102 Willem M., Garratt A.N., Novak B., Citron M., Kaufmann S., Rittger A, De Strooper B., Saftig P., Birchmeier C., Haass C. 2006. Science 314, 664-666 Wolfe M.S. Curr Top Med Chem. 2008. 8(1):2-8 Review Wolfe M.S., De Los Angeles J., Miller D.D., Xia W., Selkoe D.J. 1999c. Biochemistry 38(35):11223-11230. Wolfe M.S., Kopan R. 2004. Science 305, 1119-1123 Wolfe M.S., Xia W., Moore C.L., Leatherwood D.D., Ostaszewski B., Rahmati T., Donkor I.O., Selkoe D.J. 1999a. Biochemistry. 38(15):4720-7.
184

Bibliografia
Wolfe M.S., Xia W., Ostaszewski B.L., Diehl T.S, Kimberly W.T., Selkoe D.J. 1999b. Nature 398, 513-517 Wolfsberg T.G., Primakoff P., Myles D.G., White J.M. 1995a. J.Cell. Biol. 131; 275-278 Wolfsberg T.G., Straight P.D., Gerena R.L., Huovila A.P., Primakoff P., Myles D.G., White J.M. 1995b. Dev.Biol. 169, 378-383 Wong H.K., Sakurai T., Oyama F., Kaneko K., Wada K., Miyazaki H., Kurosawa M., De Strooper B., Saftig P., Nukina N. 2005. J. Biol. Chem 280(24) 23009-23017 Xie J., Guo Q. 2005. J. Biol. Chem. 280(14): 13824-13832 Xu H., Sweeney D., Wang R., Thinakaran G., Lo A., Sisodia S.S., Greengard P., Gandy S. 1997. Proc. Natl. Acad. Sci. USA. 94(8): 3748-3752 Yamaguchi Y. 2001. Semin.Cell Dev. Biol. 12(2): 99-106 Yamazaki T., Selkoe D.J., Koo E.H. 1995. J. Cell Biol. 129(2), 431-442 Yan R., Bienkowski M.J., Shuck M.E., Miao H., Tory M.C., Pauley A.M., Brashler J.R., Stratman N.C., Mathews W.R., Buhl A.E., Carter D.B., Tomasselli A.G., Parodi L.A., Heinrikson R.L., Gurney M.E. 1999. Lett.Nature 402: 533-537 Yan R., Munzner J.B., Shuck M.E., Bienkowski M.J. 2001 J. Biol. Chem 276(36) 34019-34027 Yang G., Gong Y.D., Gong K., Jiang W.L., Kwon E., Wang P., Zheng H., Zhang X.F., Gan W.B., Zhao N.M. 2005a. Neurosci. Lett. 384: 66-71 Yang P., Baker A., Hagg T. 2006. Progress in Neurobiology 79; 73-94 Yang P., Baker K.A., Hagg T. 2005b. J. Comp. Neurol. 490, 163-179 Ye J., Davé U.P., Grishin N.V., Goldstein J.L., Brown M. 2000. Procl.Natl.Acad.Sci.U.S.A.97, 5123-5128 Yong V.W., Power C., Forsyth P., Edwards D.R. 2001. Nat Rev Neurosci. 2(7):502-511 Yu G., Nishimura M., Arawaka S., Levitan d., Zhang L., Tandon A., Song Y.Q., Rogaeva E., Chen F., Kawarai T., Supala A., Levesque L., Yu H., Yang D.S., Holmes E., Milman P., Liang Y., Zhang D.M., Xu D.H., Sato C., Rogaev E., Smith M., Janus C., Zhang Y., Aebersold R., Farrer L., Sandro S., Brunl A., Geroge-Hyslop P.S. 2000. Nature 407, 48-54 Yu S.P. 2003. Prog Neurobiol 70, 363-386
185

Bibliografia
Yu W.H., Cuervo A.M., Kumar A., Peterhoff C., Schmidt S.D., Lee J.H., Mohan P.S., Mercken M., Farmery M.R., Tjernberg L.O., Jiang Y., Duff K., Uchiyama Y., Näslund J., Mathews P.M., Cataldo A.M., Nixon R.A. 2005. J.Cell.Biol 171, 87-98 Yu W.H., Kumar A., Peterhoff C., Kulnane L.S., Uchiyama Y., Lamb B.T., Cuervo A.M., Nixon R.A. 2004. Int. J. Biochem. Cell Biol 36, 2531-2540 Zhang Y. eta Xu H. 2007. Curr. Molecular Medicine, 7, 687-696 Zhao G., Cui M.Z., Mao G., Dong Y., Tan J., Sun L., Xu X. 2005. J. Biol. Chem. 280(45):37689-97 Zhao J., Chen H., Peschon J.J., Shi W., Zhang Y., Frank S.J., Warburton D. 2001. Dev.Biol. 1; 232 (1): 204-218 Zheng H., Jiang M., Trumbauer M.E., Sirinathsinghji D.J.S., Hopkins R., Smith D.W., Heavens R.P., Dawson G.R., Boyce S., Conner M.W., Stevens K.A., Slunt H.H., Sisodia S.S., Chen H.Y., Van der Ploeg L.H.T. 1995. Cell 81, 525-531
186











![[hasiera] - euskadi.eus filemoda bat?", entzuten da horrelakoetan kalean. I kus ie r laz on t Ikusi erlazionaturiko materiala Gizartearen aldetik ere gero eta hedatuago dago, itxuraz](https://static.fdocuments.co/doc/165x107/5e1feb55cb27dd6d20588fc7/hasiera-bat-entzuten-da-horrelakoetan-kalean-i-kus-ie-r-laz-on-t-ikusi.jpg)

