
ANEXO I LISTA DE LOS NOMBRES, FORMA...
22
1 ANEXO I LISTA DE LOS NOMBRES, FORMA FARMACÉUTICA, CONCENTRACIONES DEL MEDICAMENTO, FORMA DE ADMINISTRACIÓN, SOLICITANTE / TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN, ENVASADO Y TAMAÑO DEL ENVASE EN LOS ESTADOS MIEMBROS
Transcript of ANEXO I LISTA DE LOS NOMBRES, FORMA...

1
ANEXO I
LISTA DE LOS NOMBRES, FORMA FARMACÉUTICA, CONCENTRACIONES DELMEDICAMENTO, FORMA DE ADMINISTRACIÓN, SOLICITANTE / TITULAR DE LA
AUTORIZACIÓN DE COMERCIALIZACIÓN, ENVASADO Y TAMAÑO DEL ENVASE ENLOS ESTADOS MIEMBROS

Est
ado
Mie
mbr
oTi
tula
r de
laA
utor
izac
ión
deC
omer
cial
izac
ión
Nom
bre
arbi
trar
ioD
osis
Form
aFa
rmac
éutic
aV
ía d
ead
min
istra
ción
Env
ase
Con
teni
do(c
once
ntra
ción
)Ta
mañ
o de
l env
ase
Aus
tria
Boe
hrin
ger
Inge
lhei
mA
ustri
a G
mbH
Act
ilyse
20, 5
0 m
gPo
lvo
y di
solv
ente
para
solu
ción
inye
ctab
le y
par
ape
rfus
ión
Vía
intra
veno
saPo
lvo:
vial
(vid
rio)
Dis
olve
nte:
vial
(vid
rio)
1 m
g/m
ló 2
mg/
ml
1 vi
al c
on p
olvo
+1
vial
con
dis
olve
nte
Bél
gica
n.v.
Boe
hrin
ger
Inge
lhei
m s.
a.A
ctily
se10
, 20,
50 m
gPo
lvo
y di
solv
ente
para
solu
ción
inye
ctab
le y
par
ape
rfus
ión
Vía
intra
veno
saPo
lvo:
vial
(vid
rio)
Dis
olve
nte:
vial
(vid
rio)
1 m
g/m
ló 2
mg/
ml
1 vi
al c
on p
olvo
+1
vial
con
dis
olve
nte
100
mg
1 vi
al c
on p
olvo
+2
vial
es c
on d
isol
vent
eD
inam
arca
Boe
hrin
ger
Inge
lhei
mIn
tern
atio
nal G
mbH
Act
ilyse
10, 2
0,50
mg
Polv
o y
diso
lven
tepa
ra so
luci
ónin
yect
able
y p
ara
perf
usió
n
Vía
intra
veno
saPo
lvo:
vial
(vid
rio)
Dis
olve
nte:
vial
(vid
rio)
1 m
g/m
ló 2
mg/
ml
1 vi
al c
on p
olvo
+1
vial
con
dis
olve
nte
100
mg
1 vi
al c
on p
olvo
+2
vial
es c
on d
isol
vent
eFi
nlan
dia
Boe
hrin
ger
Inge
lhei
mIn
tern
atio
nal G
mbH
Act
ilyse
10, 2
0,50
mg
Polv
o y
diso
lven
tepa
ra so
luci
ónin
yect
able
y p
ara
perf
usió
n
Vía
intra
veno
saPo
lvo:
vial
(vid
rio)
Dis
olve
nte:
vial
(vid
rio)
1 m
g/m
ló 2
mg/
ml
1 vi
al c
on p
olvo
+1
vial
con
dis
olve
nte
100
mg
1 vi
al c
on p
olvo
+2
vial
es c
on d
isol
vent
eFr
anci
aB
oehr
inge
rIn
gelh
eim
Fran
ce
Act
ilyse
10, 2
0,50
mg
Polv
o y
diso
lven
tepa
ra so
luci
ónin
yect
able
y p
ara
perf
usió
n
Vía
intra
veno
saPo
lvo:
vial
(vid
rio)
Dis
olve
nte:
vial
(vid
rio)
1 m
g/m
ló 2
mg/
ml
1 vi
al c
on p
olvo
+1
vial
con
dis
olve
nte
100
mg
1 vi
al c
on p
olvo
+2
vial
es c
on d
isol
vent
eA
lem
ania
Boe
hrin
ger
Inge
lhei
m P
harm
aK
G
Act
ilyse
10, 2
0,50
mg
Polv
o y
diso
lven
tepa
ra so
luci
ónin
yect
able
y p
ara
perf
usió
n
Vía
intra
veno
saPo
lvo:
vial
(vid
rio)
Dis
olve
nte:
vial
(vid
rio)
1 m
g/m
ló 2
mg/
ml
1 vi
al c
on p
olvo
+1
vial
con
dis
olve
nte
100
mg
1 vi
al c
on p
olvo
+2
vial
es c
on d
isol
vent
e

3
Est
ado
Mie
mbr
oTi
tula
r de
laA
utor
izac
ión
deC
omer
cial
izac
ión
Nom
bre
arbi
trar
ioD
osis
Form
aFa
rmac
éutic
aV
ía d
ead
min
istra
ción
Env
ase
Con
teni
do(c
once
ntra
ción
)Ta
mañ
o de
l env
ase
Gre
cia
Boe
hrin
ger
Inge
lhei
mIn
tern
atio
nal G
mbH
Act
ilyse
10, 2
0,50
mg
Polv
o y
diso
lven
tepa
ra so
luci
ónin
yect
able
y p
ara
perf
usió
n
Vía
intra
veno
saPo
lvo:
vial
(vid
rio)
Dis
olve
nte:
vial
(vid
rio)
1 m
g/m
ló 2
mg/
ml
1 vi
al c
on p
olvo
+1
vial
con
dis
olve
nte
Irla
nda
Boe
hrin
ger
Inge
lhei
m L
td,
UK
Act
ilyse
10, 2
0,50
mg
Polv
o y
diso
lven
tepa
ra so
luci
ónin
yect
able
y p
ara
perf
usió
n
Vía
intra
veno
saPo
lvo:
vial
(vid
rio)
Dis
olve
nte:
vial
(vid
rio)
1 m
g/m
ló 2
mg/
ml
1 vi
al c
on p
olvo
+1
vial
con
dis
olve
nte
100
mg
1 vi
al c
on p
olvo
+2
vial
es c
on d
isol
vent
eIta
liaB
oehr
inge
rIn
gelh
eim
Ital
ia sp
aA
ctily
se20
, 50
mg
Polv
o y
diso
lven
tepa
ra so
luci
ónin
yect
able
y p
ara
perf
usió
n
Vía
intra
veno
saPo
lvo:
vial
(vid
rio)
Dis
olve
nte:
vial
(vid
rio)
1 m
g/m
ló 2
mg/
ml
1 vi
al c
on p
olvo
+1
vial
con
dis
olve
nte
Luxe
mbu
rgo
n.v.
Boe
hrin
ger
Inge
lhei
m s.
a.,
Bel
gium
Act
ilyse
10, 2
0,50
mg
Polv
o y
diso
lven
tepa
ra so
luci
ónin
yect
able
y p
ara
perf
usió
n
Vía
intra
veno
saPo
lvo:
vial
(vid
rio)
Dis
olve
nte:
vial
(vid
rio)
1 m
g/m
ló 2
mg/
ml
1 vi
al c
on p
olvo
+1
vial
con
dis
olve
nte
100
mg
1 vi
al c
on p
olvo
+2
vial
es c
on d
isol
vent
ePa
íses
Baj
osB
oehr
inge
rIn
gelh
eim
b.v
.A
ctily
se10
, 20,
50 m
gPo
lvo
y di
solv
ente
para
solu
ción
inye
ctab
le y
par
ape
rfus
ión
Vía
intra
veno
saPo
lvo:
vial
(vid
rio)
Dis
olve
nte:
vial
(vid
rio)
1 m
g/m
ló 2
mg/
ml
1 vi
al c
on p
olvo
+1
vial
con
dis
olve
nte
100
mg
1 vi
al c
on p
olvo
+2
vial
es c
on d
isol
vent
ePo
rtuga
lB
oehr
inge
rIn
gelh
eim
, Lda
Act
ilyse
10, 2
0,50
mg
Polv
o y
diso
lven
tepa
ra so
luci
ónin
yect
able
y p
ara
perf
usió
n
Vía
intra
veno
saPo
lvo:
vial
(vid
rio)
Dis
olve
nte:
vial
(vid
rio)
1 m
g/m
ló 2
mg/
ml
1 vi
al c
on p
olvo
+1
vial
con
dis
olve
nte
100
mg
1 vi
al c
on p
olvo
+2
vial
es c
on d
isol
vent
e

4
Est
ado
Mie
mbr
oTi
tula
r de
laA
utor
izac
ión
deC
omer
cial
izac
ión
Nom
bre
arbi
trar
ioD
osis
Form
aFa
rmac
éutic
aV
ía d
ead
min
istra
ción
Env
ase
Con
teni
do(c
once
ntra
ción
)Ta
mañ
o de
l env
ase
Espa
ñaB
oehr
inge
rIn
gelh
eim
Inte
rnat
iona
l Gm
bHA
ctily
se10
, 20,
50 m
gPo
lvo
y di
solv
ente
para
solu
ción
inye
ctab
le y
par
ape
rfus
ión
Vía
intra
veno
saPo
lvo:
vial
(vid
rio)
Dis
olve
nte:
vial
(vid
rio)
1 m
g/m
ló 2
mg/
ml
1 vi
al c
on p
olvo
+1
vial
con
dis
olve
nte
100
mg
1 vi
al c
on p
olvo
+2
vial
es c
on d
isol
vent
e
Suec
iaB
oehr
inge
rIn
gelh
eim
Inte
rnat
iona
l Gm
bHA
ctily
se10
, 20,
50 m
gPo
lvo
y di
solv
ente
para
solu
ción
inye
ctab
le y
par
ape
rfus
ión
Vía
intra
veno
saPo
lvo:
vial
(vid
rio)
Dis
olve
nte:
vial
(vid
rio)
1 m
g/m
ló 2
mg/
ml
1 vi
al c
on p
olvo
+1
vial
con
dis
olve
nte
Rei
no U
nido
Boe
hrin
ger
Inge
lhei
m L
tdA
ctily
se10
, 20,
50 m
gPo
lvo
y di
solv
ente
para
solu
ción
inye
ctab
le y
par
ape
rfus
ión
Vía
intra
veno
saPo
lvo:
vial
(vid
rio)
Dis
olve
nte:
vial
(vid
rio)
1 m
g/m
ló 2
mg/
ml
1 vi
al c
on p
olvo
+1
vial
con
dis
olve
nte
100
mg
1 vi
al c
on p
olvo
+2
vial
es c
on d
isol
vent
e

5
ANEXO II
CONCLUSIONES CIENTÍFICAS Y MOTIVOS DE LA MODIFICACIÓN DEL RESUMENDE LAS CARACTERÍSTICAS DEL PRODUCTO PRESENTADAS POR LA EMEA

6
CONCLUSIONES CIENTÍFICAS
RESUMEN GENERAL DE LA EVALUACIÓN CIENTÍFICA DEL ACTILYSE (véase el AnexoI) Base para el procedimiento de arbitraje:La base para el procedimiento de arbitraje consistía en establecer si existen suficientes datos clínicoscon respecto a la eficacia así como también a la seguridad, por ejemplo, el riesgo de hemorragiaintracraneal, para conceder una autorización de comercialización para la nueva indicación "Para eltratamiento fibrinolítico del ataque isquémico agudo", sin poner en peligro la salud pública. Enparticular, la principal preocupación de la mayoría de los Estados miembros afectados es que no sehayan repetido los resultados favorables del ensayo pivotal estadounidense (el Estudio NINDS B) nien los estudios europeos (ECASS-I y ECASS-II) ni en otro ensayo realizado en Estados Unidos(Atlantis).Asimismo, se estima que es necesario añadir requisitos en el Resumen de las Características delProducto para garantizar la seguridad en su uso.Formularon preguntas los Países Bajos, España, Grecia y Reino Unido.
El procedimiento de arbitraje finaliza con las siguientes conclusiones:
- los resultados alcanzados en los ensayos realizados en Estados Unidos se pueden extrapolar, encierta medida, al entorno europeo. El hecho de que los estudios europeos den resultados menosestables se explica por las diferencias en el diseño del ensayo y en los criterios de inclusión, yaque sólo algunos de los pacientes fueron tratados en las tres horas siguientes al ataque.Asimismo, los resultados del metaanálisis publicado manifiestan una tendencia a seguir lamisma dirección que los ensayos estadounidenses, aunque la cantidad de pacientes esdemasiado reducida para que los criterios de evaluación de la eficacia alcancen importanciaestadística,
- en la práctica clínica se puede tratar el medicamento con arreglo a las restricciones del resumenmodificado de las características del producto a fin de aprovechar al máximo los beneficios deltratamiento.
- es muy probable que hubiera dificultades para encontrar pacientes para realizar un estudiocontrolado con placebo en un entorno europeo, con una población de pacientes como la que sedefine en el Resumen de las Características del Producto modificado, y administrarles alteplasaen un plazo de tres horas, y tal vez no sea factible. No obstante, para los investigadoreseuropeos podría ser aceptable un estudio similar realizado con la misma población depacientes, pero en un plazo de tres a cuatro horas, ya que en general los datos de eficacia seconsideran aquí insuficientes y se sabe que el riesgo de hemorragia aumenta a medida que pasael tiempo desde que se presentan los síntomas,
- unos ensayos clínicos aleatorios controlados con placebo, realizados en grupos especiales depacientes de alto riesgo, tendrían un interés general para el conjunto de la sociedad médica, y lademostración de eficacia y seguridad en las poblaciones de alto riesgo, fuera de lasrestricciones que se contemplan en el Resumen de las Características del Producto, seconsidera tanto ética como factible. Sin embargo, sería difícil extrapolar los resultados que seobtuvieran de dichos estudios a toda la población que ha sufrido un infarto, ya que, para hacerla extrapolación, sería crucial contar con una población de pacientes similar a la base de datosoriginal de los estudios realizados en Estados Unidos.
Por consiguiente, se considera aceptable autorizar la comercialización de la alteplasa para eltratamiento fibrinolítico del infarto isquémico agudo cuando se inicia el tratamiento en las tres horassiguientes a la aparición de los síntomas del ataque y tras descartar previamente una hemorragiaintracraneal mediante las técnicas adecuadas de obtención de imágenes, con la condición de que selleve a cabo un estudio europeo que lo confirme y de que se asuman los compromisos necesarios.
El artículo 32.4 de la D 2001/83/CE (antiguo artículo 13.4 de la Directiva 75/319) proporciona la basejurídica para exigir condiciones que se consideran fundamentales para el uso seguro y eficaz del

7
medicamento, incluida la farmacovigilancia. Partiendo de esta base, el Comité de EspecialidadesFarmacéuticas exige a la empresa que asuma los siguientes compromisos y que dichos compromisossean evaluados anualmente por el Comité:
- La empresa llevará a cabo un estudio aleatorio, controlado con placebo, para confirmar laeficacia y la seguridad del medicamento, que se realizará en un margen de tiempo de tres acuatro horas, con una población de pacientes idéntica a la que figura en el criterio de inclusióndel Resumen de las Características del Producto / los ensayos realizados por NINDS enEstados Unidos. Asimismo, la empresa se compromete a presentar semestralmente informesdetallados sobre el avance de la aplicación de este estudio, que incluyan información sobre lainclusión y el seguimiento de nuevos pacientes, a partir de la fecha en que la Comisión adoptela Decisión sobre la indicación del infarto, que deberán ser examinados por el Comité deEspecialidades Farmacéuticas y de forma paralela a la presentación de los Informes Periódicosde Seguridad. El estudio comenzará (incorporación del primer paciente) en un plazo de seismeses a partir de la fecha en que la Comisión adopte la Decisión, y el informe definitivo sobreel estudio habrá de ser presentado en un plazo de seis meses a partir de que se finalice elestudio con el último paciente.
- La empresa realizará un estudio de supervisión en Europa con posterioridad a lacomercialización: "un estudio de control de seguridad SITS-MOST" Asimismo, la empresa secompromete a presentar semestralmente informes detallados sobre el avance de la aplicacióndel estudio, que incluyan información sobre la inclusión y el seguimiento de nuevos pacientes,a partir de la fecha en que la Comisión adopte la Decisión sobre la indicación del infarto, quedeberán ser revisados por el Comité de Especialidades Farmacéuticas y de forma paralela a lapresentación de los Informes Periódicos de Seguridad. El estudio comenzará (incorporación delprimer paciente) en un plazo de seis meses a partir de la fecha en que la Comisión adopte laDecisión, y el informe definitivo sobre el estudio habrá de ser presentado en un plazo de seismeses a partir de que se finalice el estudio con el último paciente.
- La empresa presentará Informes Periódicos de Seguridad a los seis, doce, dieciocho yveinticuatro meses, a los tres años, a los cuatro años y como parte de la solicitud de renovación,a partir de la Decisión de la Comisión con respecto a la indicación sobre el infarto.
- Los protocolos de estudio definitivos para los estudios ECASS III y SITS-MOST sepresentarán al Comité de Especialidades Farmacéuticas, para su examen, a más tardar el 17 dejulio de 2002.
La nueva evaluación del perfil de beneficio/riesgo de Actilyse, realizada por el Comité deEspecialidades Farmacéuticas a partir de dichas condiciones, definirá el curso de los acontecimientosque habrá que seguir con respecto a la autorización de comercialización en cuestión.
Con posterioridad a la reunión entre los expertos y el grupo de asesores científicos del Comité deEspecialidades Farmacéuticas y tras las alegaciones verbales presentadas por la empresa, se introducennumerosas modificaciones en el Resumen de las Características del Producto, como se detalla en elAnexo III del Dictamen de remisión del Comité. Se considera que el Resumen de las Característicasdel Producto propuesto brinda ahora la información adecuada.
El Comité de Especialidades Farmacéuticas, tras considerar:- el informe de evaluación del Procedimiento de Reconocimiento Mutuo del Estado miembro de
referencia,- las cuestiones sometidas a arbitraje,- las respuestas escritas proporcionadas por la empresa,- el informe de evaluación del ponente y del ponente adjunto sobre estas respuestas, su examen
conjunto y el apéndice al examen conjunto definitivo,- los comentarios de los miembros del Comité de Especialidades Farmacéuticas,- las conclusiones y recomendaciones de la reunión entre los expertos y el grupo de asesores
científicos del Comité de Especialidades Farmacéuticas,

8
- las alegaciones verbales de la empresa,- y los compromisos asumidos por la empresa,
llega a la conclusión de que se han resuelto las cuestiones planteadas por Países Bajos, España, Greciay Reino Unido. Dado que la proporción de riesgo / beneficio de la alteplasa, teniendo en cuenta lascondiciones que menciona el Comité y que acuerda la empresa, se considera positiva para laindicación y la posología declaradas, hay que conceder la autorización de comercialización. Noobstante, habría que modificar el Resumen de las Características del Producto que consta en el AnexoIII del Dictamen del Comité de Especialidades Farmacéuticas.

9
ANEXO III
RESUMEN MODIFICADO DE LAS CARACTERÍSTICAS DEL PRODUCTO DEL ESTADO MIEMBRO DE REFERENCIA

10
1. DENOMINACIÓN DEL MEDICAMENTO
Actilyse
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Principio activo:
La solución reconstituida contiene 1mg de alteplasa/1ml.
1 vial con 467 mg de polvo contiene: 10 mg de alteplasa o1 vial con 933 mg de polvo contiene: 20 mg de alteplasa o1 vial con 2.333 mg de polvo contiene: 50 mg de alteplasa o1 vial con 4.666 mg de polvo contiene: 100 mg de alteplasa
La alteplasa es producida mediante la técnica de ADN recombinante, utilizando una línea celularovárica de hamster chino. La actividad específica de la alteplasa, patrón de referencia interno, es de580.000 UI/mg. Esto ha sido confirmado por comparación con el segundo patrón internacional de laO.M.S. para t-PA. La especificación para la actividad específica de la alteplasa es de 522.000 a696.000 UI/mg.
El pH de la solución reconstituida es de 7,3 � 0,5.
Lista de excipientes, en 6.1.
3. FORMA FARMACÉUTICA
Polvo y disolvente para solución inyectable y para perfusión.
4. DATOS CLÍNICOS
4.1. Indicaciones terapéuticas
4.1.1. Tratamiento trombolítico en el infarto agudo de miocardio
- Régimen de dosificación de 90 minutos (acelerado) (ver posología y forma de administración):para pacientes en los cuales el tratamiento puede iniciarse dentro de las 6 h después de lapresentación de los síntomas.
- Régimen de dosificación de 3 h (ver posología y forma de administración): para pacientes en loscuales el tratamiento puede iniciarse entre las 6 y 12 h después de la presentación de lossíntomas, siempre que se haya comprobado la indicación.
Actilyse ha demostrado reducir la mortalidad al cabo de 30 días en pacientes con infarto de miocardioagudo.
4.1.2. Tratamiento trombolítico en embolia pulmonar masiva aguda con inestabilidad hemodinámica.
El diagnóstico deberá ser confirmado, siempre que sea posible, mediante medios objetivos como p.ej.angiografía pulmonar o procedimientos no invasivos como la gammagrafía isotópica pulmonar. Nohay evidencia de efectos positivos sobre la mortalidad y morbilidad tardía relacionadas con la emboliapulmonar.
11
4.1.3. Tratamiento fibrinolítico del ictus isquémico agudo
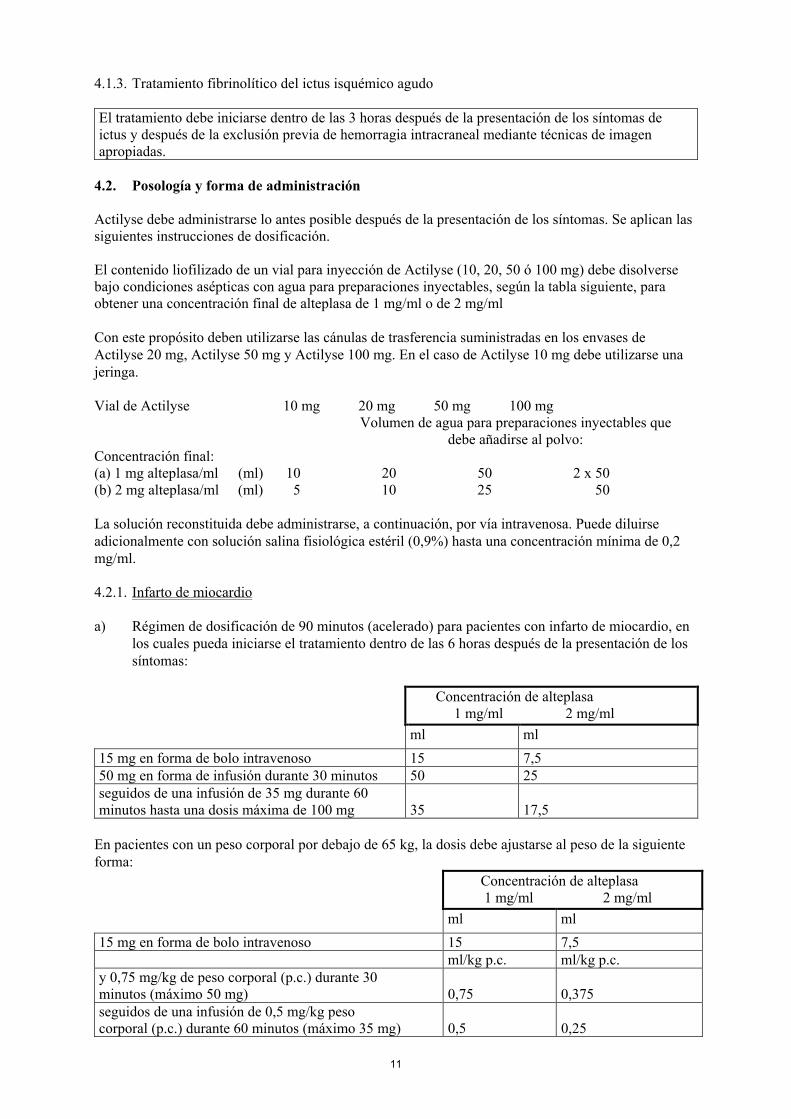
El tratamiento debe iniciarse dentro de las 3 horas después de la presentación de los síntomas deictus y después de la exclusión previa de hemorragia intracraneal mediante técnicas de imagenapropiadas.
4.2. Posología y forma de administración
Actilyse debe administrarse lo antes posible después de la presentación de los síntomas. Se aplican lassiguientes instrucciones de dosificación.
El contenido liofilizado de un vial para inyección de Actilyse (10, 20, 50 ó 100 mg) debe disolversebajo condiciones asépticas con agua para preparaciones inyectables, según la tabla siguiente, paraobtener una concentración final de alteplasa de 1 mg/ml o de 2 mg/ml
Con este propósito deben utilizarse las cánulas de trasferencia suministradas en los envases deActilyse 20 mg, Actilyse 50 mg y Actilyse 100 mg. En el caso de Actilyse 10 mg debe utilizarse unajeringa.
Vial de Actilyse 10 mg 20 mg 50 mg 100 mgVolumen de agua para preparaciones inyectables que
debe añadirse al polvo:Concentración final:(a) 1 mg alteplasa/ml (ml) 10 20 50 2 x 50(b) 2 mg alteplasa/ml (ml) 5 10 25 50
La solución reconstituida debe administrarse, a continuación, por vía intravenosa. Puede diluirseadicionalmente con solución salina fisiológica estéril (0,9%) hasta una concentración mínima de 0,2mg/ml.
4.2.1. Infarto de miocardio
a) Régimen de dosificación de 90 minutos (acelerado) para pacientes con infarto de miocardio, enlos cuales pueda iniciarse el tratamiento dentro de las 6 horas después de la presentación de lossíntomas:
Concentración de alteplasa 1 mg/ml 2 mg/mlml ml
15 mg en forma de bolo intravenoso 15 7,550 mg en forma de infusión durante 30 minutos 50 25seguidos de una infusión de 35 mg durante 60minutos hasta una dosis máxima de 100 mg 35 17,5
En pacientes con un peso corporal por debajo de 65 kg, la dosis debe ajustarse al peso de la siguienteforma:
Concentración de alteplasa 1 mg/ml 2 mg/mlml ml
15 mg en forma de bolo intravenoso 15 7,5ml/kg p.c. ml/kg p.c.
y 0,75 mg/kg de peso corporal (p.c.) durante 30minutos (máximo 50 mg) 0,75 0,375seguidos de una infusión de 0,5 mg/kg pesocorporal (p.c.) durante 60 minutos (máximo 35 mg) 0,5 0,25

12
b) Régimen de dosificación de 3 horas para pacientes en los cuales pueda iniciarse el tratamientoentre las 6 y 12 horas después de la presentación de los síntomas:
Concentración de alteplasa 1 mg/ml 2 mg/mlml ml
10 mg en forma de bolo intravenoso 10 550 mg en forma de infusión durante la primerahora 50 25
ml/30 min ml/30 minseguidos por infusiones de 10 mg durante 30minutos, hasta una dosis máxima de 100 mgdurante 3 horas
10 5
En pacientes con un peso corporal por debajo de 65 kg, la dosis total no debería ser superior a 1,5mg/kg.
La dosis máxima aceptada de alteplasa es de 100 mg.
Tratamiento coadyuvante:Debe iniciarse el tratamiento con ácido acetilsalicílico lo antes posible, tras la presentación de lossíntomas, y continuarse durante los primeros meses después del infarto de miocardio. La dosisrecomendada es de 160-300 mg/día.
Debe administrarse heparina de forma concomitante al menos durante 24 horas o más (al menosdurante 48 h con el régimen de dosificación acelerado). Se recomienda comenzar con un bolointravenoso inicial de 5.000 UI, previo al tratamiento trombolítico, y continuar con una infusión de1.000 UI/hora. La dosis de heparina debe ajustarse de acuerdo con mediciones repetidas de valores deaPTT (tiempo parcial de tromboplastina activada), de 1,5 a 2,5 veces el valor basal.
4.2.2. Embolia pulmonar
Debe administrarse una dosis total de 100 mg de alteplasa en 2 horas. El régimen de dosificación conel que se tiene mayor experiencia es el siguiente:
Concentración de alteplasa 1 mg/ml 2 mg/mlml ml
10 mg en forma de bolo intravenoso durante 1-2minutos
10 5
seguido de 90 mg como infusión intravenosadurante 2 horas
90 45
En pacientes con un peso corporal por debajo de 65 kg, la dosis total no debería ser superior a 1,5mg/kg.
Tratamiento coadyuvante:Después del tratamiento con Actilyse debe iniciarse (o reanudarse) un tratamiento con heparina si losvalores aPTT son inferiores al doble del límite superior normal. La infusión debe ajustarse de acuerdocon valores aPTT, de 1,5 a 2,5 veces el valor basal.

13
4.2.3. Ictus isquémico agudo
El tratamiento debe ser realizado por un médico experto en cuidados neurológicos. (verContraindicaciones y Advertencias y precauciones especiales de empleo)
La dosis recomendada es de 0,9 mg de principio activo/kg de peso (hasta un máximo de 90 mg)infundidos por vía intravenosa durante 60 minutos con un 10 % de la dosis total administrado comobolo intravenoso inicial.
El tratamiento con Actilyse debe iniciarse dentro de las 3 horas después de la presentación de lossíntomas.
Tratamiento coadyuvante:La seguridad y eficacia de este régimen con la administración concomitante de heparina y ácidoacetilsalicílico durante las primeras 24 horas después de la presentación de los síntomas no han sidosuficientemente evaluadas. La administración de ácido acetilsalicílico o heparina intravenosa debeevitarse en las primeras 24 horas después del tratamiento con Actilyse. Si se requiere heparina paraotra indicación (p.ej. prevención de la trombosis venosa profunda) la dosis no debe exceder las 10.000UI por día, administrada por vía subcutánea.
4.3 Contraindicaciones
Como todos los agentes trombolíticos, Actilyse no debe administrarse en casos en los que existe unalto riesgo de hemorragia como p.ej.:- diátesis hemorrágica conocida- pacientes que reciben anticoagulantes orales, p.ej. warfarina sódica- hemorragia grave o peligrosa manifiesta o reciente- sospecha o historia conocida de hemorragia intracraneal- sospecha de hemorragia subaracnoidea o situación después de una hemorragia subaracnoidea
por aneurisma- cualquier historia de lesión del sistema nervioso central (es decir, neoplasia, aneurisma, cirugía
intracraneal o espinal)- retinopatía hemorrágica, p.ej. en diabetes (trastornos de la visión pueden indicar una retinopatía
hemorrágica)- masaje cardíaco externo traumático reciente (menos de 10 días), parto obstétrico reciente,
punción reciente de un vaso sanguíneo no comprimible (p.ej. punción de la vena yugular osubclavia)
- hipertensión arterial grave no controlada- endocarditis bacteriana, pericarditis- pancreatitis aguda- enfermedad gastrointestinal ulcerativa documentada durante los últimos 3 meses, varices
esofágicas, aneurismas arteriales, malformaciones venosas/arteriales- neoplasia con riesgo de hemorragia aumentado- enfermedad hepática grave, incluyendo insuficiencia hepática, cirrosis, hipertensión portal
(varices esofágicas) y hepatitis activa- cirugía mayor o traumatismo importante en los últimos 3 meses
4.3.1. Contraindicaciones adicionales en el infarto agudo de miocardio:cualquier historia de ictus
4.3.2. Contraindicaciones adicionales en la embolia pulmonar aguda:cualquier historia de ictus
4.3.3. Contraindicaciones adicionales en el ictus isquémico agudo:- cuando los síntomas de accidente isquémico empiezan más de 3 horas antes del inicio de la
infusión o cuando se desconoce la hora del inicio de los síntomas,- déficit neurológico leve o síntomas de rápida mejora antes del inicio de la infusión,

14
- ictus grave evaluado clínicamente (p.ej. NIHSS>25) y/o por técnicas de imagen apropiadas,- convulsiones al inicio del ictus,- evidencia de una hemorragia intracraneal en la TC- síntomas que sugieran hemorragia subaracnoidea, incluso con TC normal- administración de heparina dentro de las 48 horas previas y un tiempo de tromboplastina que
exceda el límite superior normal,- pacientes con historia previa de ictus y diabetes concomitante,- ictus previo en los últimos 3 meses,- recuento plaquetar inferior a 100.000/mm3,- presión sanguínea sistólica > 185 o presión sanguínea diastólica > 110 mm Hg, o controles
agresivos (medicación por vía intravenosa) necesarios para reducir la presión sanguínea a estoslímites,
- niveles de glucosa en sangre < 50 ó > 400 mg/dl.
Uso en niños y pacientes ancianosActilyse no está indicado en el tratamiento del ictus agudo en niños menores de 18 años o adultosmayores de 80 años.
4.4 Advertencias y precauciones especiales de empleo
El tratamiento trombolítico/fibrinolítico requiere una monitorización adecuada. Actilyse debe serutilizado sólo por médicos entrenados y con experiencia en la administración de tratamientotrombolítico y con los medios para monitorizar esta administración.
El riesgo de hemorragia intracraneal es mayor en pacientes de edad avanzada, por lo tanto en estospacientes debe valorarse cuidadosamente la relación beneficio/riesgo.
Hasta el momento sólo existe experiencia limitada en la administración de Actilyse a niños.
Como con todos los agentes trombolíticos, el beneficio terapéutico esperado debe ponderarsecuidadosamente frente al posible riesgo, especialmente en pacientes con:- traumatismos menores recientes, como biopsias, punciones de vasos mayores, inyecciones
intramusculares, masaje cardíaco para resucitación- condiciones con un mayor riesgo de hemorragia no mencionadas en el apartado 4.3.Debe evitarse la utilización de catéteres rígidos.
4.4.1. Advertencias y precauciones especiales adicionales en el infarto agudo de miocardio:
No debe administrarse una dosis superior a 100 mg de alteplasa debido a que ha sido asociada con unincremento adicional de hemorragias intracraneales.
Debe procederse con especial cuidado, para asegurar que la dosis de alteplasa que se infundecorresponda a la descrita en el apartado 4.2. Posología y forma de Administración.
Existe experiencia limitada con la administración repetida de Actilyse. No se sospecha que Actilysepueda provocar reacciones anafilácticas. Si se presenta alguna reacción anafilactoide, debesuspenderse la infusión e iniciarse un tratamiento adecuado.
Como con todos los agentes trombolíticos, el beneficio terapéutico esperado debe ser ponderadocuidadosmente frente al posible riesgo, especialmente en pacientes con la presión sanguínea sistólica >160 mm Hg.
4.4.2. Advertencias y precauciones especiales adicionales en la embolia pulmonar aguda:
Las mismas que para el infarto agudo de miocardio (4.4.1.)

15
4.4.3. Advertencias y precauciones especiales adicionales en el ictus isquémico agudo:
Precauciones especiales de empleoEl tratamiento debe ser realizado sólo por un médico entrenado y con experiencia en cuidadosneurológicos.
Advertencias especiales / condiciones con una relación beneficio/riesgo reducidaComparado con otras indicaciones, los pacientes con ictus isquémico agudo tratados con Actilysetienen un riesgo marcadamente aumentado de hemorragia intracraneal, ya que la hemorragia tienelugar principalmente en el área del infarto. Esto es aplicable en particular a los casos siguientes:- todas las situaciones enumeradas en el apartado 4.3 y en general todas las situaciones que
conlleven alto riesgo de hemorragia- aneurisma pequeño asintomático de los vasos cerebrales.- pacientes pre-tratados con ácido acetilsalicílico (AAS) pueden tener un mayor riesgo de
hemorragia intracerebral, sobre todo si se retrasa el tratamiento con Actilyse. No debeadministrarse más de 0,9 mg de alteplasa/kg de peso corporal (90 mg como máx.) debido alriesgo aumentado de hemorragia cerebral.
El tratamiento de los pacientes no debe iniciarse más tarde de 3 horas después de la presentación delos síntomas (ver 4.3 Contraindicaciones) debido a una relación beneficio/riesgo no favorable, basadoprincipalmente en lo siguiente:- disminución de los efectos positivos del tratamiento con el tiempo- aumento de la tasa de mortalidad, especialmente en pacientes con un tratamiento previo con
AAS- aumento del riesgo con respecto a hemorragias sintomáticas
La monitorización de la presión sanguínea durante la administración del tratamiento y hasta 24 horasdespués parece justificado; también se recomienda un tratamiento antihipertensivo por vía intravenosasi la presión sanguínea sistólica es > 180 mm Hg o la presión sanguínea diastólica es > 105 mm Hg.
El beneficio terapéutico se reduce en pacientes con un ictus previo o en los cuales se conoce unadiabetes no controlada; de este modo la relación beneficio/riesgo en estos pacientes se consideramenos favorable pero todavía positiva.
En pacientes con ictus muy leve, el riesgo supera el beneficio esperado (ver 4.3 Contraindicaciones).
Los pacientes con ictus muy grave presentan un riesgo mayor de hemorragia intracerebral y muerte, yno deben ser tratados (ver 4.3 Contraindicaciones).
Los pacientes con infartos extensos presentan un mayor riesgo de resultados insatisfactorios,incluyendo hemorragia grave y muerte. En estos pacientes, debe considerarse cuidadosamente larelación beneficio/riesgo.
En los pacientes con ictus la probabilidad de un buen desenlace disminuye al aumentar la edad, alaumentar la severidad del ictus y con los niveles de glucosa en sangre altos en el momento del ingreso,mientras que la probabilidad de discapacidad grave y muerte o hemorragias intracraneales destacablesaumenta, independientemente del tratamiento. Los pacientes de más de 80 años, pacientes con ictusgrave (evaluado clínicamente y/o mediante técnicas de imagen apropiadas) y pacientes con nivelesbasales de glucosa en sangre < 50 mg/dl ó > 400 mg/dl, no deben ser tratados con Actilyse (ver 4.3Contraindicaciones).
Otras advertencias especialesLa reperfusión del área isquémica puede inducir a edema cerebral en la zona del infarto. Debido a unriesgo aumentado de hemorragia, el tratamiento con inhibidores de la agregación plaquetaria no debeiniciarse dentro de las primeras 24 horas después de la trombolisis con alteplasa.

16
4.5 Interacción con otros medicamentos y otras formas de interacción
El riesgo de hemorragia aumenta si se administran derivados cumarínicos, anticoagulantes orales,inhibidores de la agregación plaquetaria, heparina no fraccionada o heparina de bajo peso molecular(LMWH) u otros agentes que inhiban la coagulación (antes, durante o dentro de la primeras 24 horasdespués del tratamiento con Actilyse) (ver 4.3 Contraindicaciones).
4.6 Embarazo y lactancia
Existe experiencia muy limitada en la utilización de Actilyse durante el embarazo y la lactancia. Encaso de una enfermedad aguda con peligro para la vida de la paciente, debe evaluarse el beneficiofrente al riesgo potencial.En animales gestantes, no se han observado efectos teratogénicos tras la infusión i.v. de dosisfarmacológicamente efectivas. En conejos, dosis superiores a 3 mg/kg/día dieron lugar a efectosembriotóxicos (embrioletalidad, retrasos en el crecimiento). No se observaron efectos en el desarrolloperi-postnatal o sobre parámetros de fertilidad, con dosis de hasta 10 mg/kg/día, en ratas.
4.7 Efectos sobre la capacidad para conducir y utilizar máquinas
No procede.
4.8 Reacciones adversas
La reacción adversa más frecuente asociada a Actilyse es la hemorragia con un descenso en los valoresdel hematocrito y/o hemoglobina. El tipo de hemorragias asociadas al tratamiento trombolítico puededividirse en dos grandes categorías:- hemorragias superficiales, normalmente a partir de punciones o vasos sanguíneos lesionados,- hemorragias internas en el tracto gastrointestinal o urogenital, retroperitoneo o SNC, o
hemorragia en órganos parenquimatosos.La hemorragia intracerebral sintomática es el acontecimiento adverso principal de Actilyse en eltratamiento del ictus isquémico agudo (hasta un 10 % de los pacientes).
En estudios clínicos con Actilyse se observó ocasionalmente una pérdida de sangre significativadebida a una hemorragia gastrointestinal, urogenital o retroperitoneal. Se observan con bastantefrecuencia equimosis, epistaxis y hemorragia gingival, pero por regla general no requieren untratamiento específico. En estudios donde los pacientes fueron tratados conforme a la rutina clínica, esdecir, sin cateterización aguda del corazón izquierdo, sólo fue necesaria ocasionalmente unatransfusión sanguínea. En el tratamiento del infarto agudo de miocardio y la embolia pulmonar agudase describieron raramente (menos del 1 %) casos de hemorragia intracraneal.
Si se produce una hemorragia potencialmente peligrosa, en particular hemorragia cerebral, debeinterrumpirse el tratamiento fibrinolítico. Sin embargo, por regla general, no es necesario sustituir losfactores de coagulación debido a la corta vida media de la alteplasa y al efecto mínimo sobre losfactores de la coagulación sistémicos. La mayoría de los pacientes que presentan hemorragia puedencontrolarse mediante interrupción del tratamiento trombolítico y anticoagulantes, sustitutivos delplasma y aplicación de presión manual a un vaso comprimible. Debe considerarse la administración deprotamina si se ha administrado heparina dentro de las 4 horas después de la presentación de lahemorragia. Puede indicarse el uso racional de productos de transfusión en pacientes que no respondana estas medidas conservadoras. Después de cada administración debe realizarse una reevaluaciónmediante análisis clínicos y de laboratorio, para considerar la necesidad de transfundir crioprecipitado,plasma congelado reciente y plaquetas. Con infusión de crioprecipitado es deseable alcanzar un nivelde fibrinógeno de 1 g/l. Como última alternativa se dispone de agentes antifibrinolíticos.
El tratamiento con Actilyse puede producir, raramente, embolización de cristales de colesterol oembolización trombótica. En los órganos afectados, esto puede conducir a las correspondientesconsecuencias (p.ej. insuficiencia renal en el caso de afectar al riñón).

17
En pacientes que reciben Actilyse para el infarto de miocardio, una reperfusión efectiva se vefrecuentemente acompañada de arritmias. Estas pueden requerir la aplicación de tratamientosantiarrítmicos convencionales.
Los pacientes con infarto de miocardio o embolia pulmonar pueden experimentar acontecimientosrelacionados con la enfermedad, tales como insuficiencia cardíaca, isquemia recurrente, angina, parocardíaco, shock cardiogénico, reinfarto, trastornos valvulares (p. ej. rotura de válvula aórtica) yembolia pulmonar. Estos acontecimientos han sido comunicados también después del tratamientotrombolítico y pueden ser una amenaza para la vida del paciente, llegando a producir la muerte.
Raramente se ha informado sobre náuseas, vómitos, caída de la presión arterial y elevación de latemperatura. Estas reacciones también pueden tener lugar como síntomas acompañantes del infarto demiocardio.
Como ocurre con otros agentes trombolíticos, se ha informado en casos aislados de acontecimientosrelacionados con el sistema nervioso central (p. ej. convulsiones), a menudo asociados aacontecimientos de isquemia concurrentes o hemorragia cerebrovascular.
Raramente se ha informado de reacciones anafilactoides. Estas son normalmente leves, pero en casosaislados pueden suponer un riesgo para la vida. Pueden aparecer en forma de exantema, urticaria,broncoespasmo, angioedema, hipotensión, shock o cualquier otro síntoma asociado con reaccionesalérgicas. Si se producen debe iniciarse el tratamiento antialérgico convencional. Raramente y a dosisbajas se ha observado formación transitoria de anticuerpos contra Actilyse, pero no pudo establecersela relevancia clínica de este hallazgo.
4.9 Sobredosis
A pesar de la especificidad relativa por la fibrina, puede producirse una reducción clínica significativadel fibrinógeno y otros componentes de la coagulación sanguínea después de una sobredosificación.En la mayoría de los casos, es suficiente esperar la regeneración fisiológica de estos factores despuésde haber finalizado el tratamiento con Actilyse. Sin embargo, si se presentan hemorragias graves, serecomienda la infusión de plasma congelado reciente o sangre fresca y, si fuese necesario, puedenadministrarse antifibrinolíticos sintéticos.
5. PROPIEDADES FARMACOLÓGICAS
5.1 Propiedades farmacodinámicas
Grupo fármaco-terapéutico: agente trombolítico, código ATC: B01AD02
El componente activo de Actilyse es la alteplasa, una glucoproteína que activa directamente elplasminógeno a plasmina. Cuando se administra por vía intravenosa, la alteplasa permanecerelativamente inactiva en el sistema circulatorio. Una vez se conjuga con la fibrina, es activada,induciendo la conversión del plasminógeno en plasmina, lo cual produce la disolución del coágulo defibrina.
En un estudio que incluía más de 40.000 pacientes con infarto de miocardio agudo (GUSTO), laadministración de 100 mg de alteplasa durante 90 minutos, con infusión concomitante de heparina i.v.,redujo la mortalidad a los 30 días (6,3%), en comparación con la administración de estreptoquinasa,1,5 millones U durante 60 minutos, con heparina s.c. o i.v. (7,3%). Los pacientes tratados con Actilysemostraron una mayor permeabilidad de los vasos relacionados con el infarto a los 60 y 90 minutos dela trombolisis que los pacientes tratados con estreptoquinasa. No se encontraron diferencias en lapermeabilidad a los 180 minutos, ni más tarde.
La mortalidad al cabo de 30 días se reduce, en comparación con pacientes que no reciben terapiatrombolítica.

18
La liberación de �-hidroxibutirato-deshidrogenasa (HBDH) se reduce. La función ventricular globalasí como la motilidad de la pared regional resulta menos afectada, en comparación con pacientes queno reciben terapia trombolítica.
Estudios en el infarto de miocardioUn estudio controlado con placebo realizado con 100 mg de alteplasa durante 3 horas (LATE),demostró una reducción de la mortalidad al cabo de 30 días, en comparación con el placebo, enpacientes tratados dentro de las 6 - 12 horas después de la presentación de los síntomas. El tratamientopuede ser beneficioso en los casos en que todavía se observan signos claros de infarto de miocardio, siel tratamiento se inicia hasta 24 horas después de la presentación de los síntomas.
Estudios en la embolia pulmonarEn pacientes con embolia pulmonar masiva aguda e inestabilidad hemodinámica, el tratamientotrombolítico con Actilyse conduce a una rápida reducción del tamaño del trombo y a una disminuciónde la presión arterial pulmonar. No se dispone de datos sobre mortalidad.
Estudios en el ictus agudoEn dos estudios de Estados Unidos (NINDS A/B) una proporción de pacientes significativamentemayor, cuando se compara con el placebo, presentó un desenlace favorable (sin o con mínimadiscapacidad). Estos hallazgos no se confirmaron en dos estudios europeos ni en un estudio adicionalde Estados Unidos. Sin embargo, en estos estudios la mayoría de pacientes no fueron tratados dentrode las 3 horas después de la presentación del ictus. En un meta-análisis de todos los pacientes tratadosdentro de las 3 horas después de la presentación del ictus, se confirmó el efecto beneficioso de laalteplasa. La diferencia de riesgo en frente a placebo para una buena recuperación fue del 14,9% (CI95% 8,1% a 21,7%), a pesar de un riesgo aumentado de hemorragia intracraneal grave y mortal. Losdatos no permiten sacar una conclusión definitiva sobre el efecto del tratamiento sobre la mortalidad.No obstante, en conjunto el beneficio/riesgo de la alteplasa, administrada dentro de las 3 horas despuésde la presentación del ictus y considerando las precauciones que constan en la ficha técnica, seconsidera favorable.
Un meta-análisis de todos los datos clínicos demuestra que el agente es menos eficaz en pacientestratados más tarde de 3 horas después de la presentación de los síntomas (de 3 a 6 horas) encomparación con aquellos tratados dentro de las 3 horas después de la presentación de los síntomas,mientras que los riesgos fueron superiores, lo cual hace la relación beneficio/riesgo de alteplasa nofavorable fuera del intervalo de 0-3 horas.
Debido a su especificidad relativa por la fibrina, la alteplasa, a dosis de 100 mg, produce una modestadisminución de los niveles de fibrinógeno circulante hasta el 60% aproximadamente a las 4 horas, lacual, generalmente, es revertida a más del 80% después de 24 horas. El plasminógeno y la �-2-antiplasmina disminuyen hasta aproximadamente el 20% y el 35%, respectivamente, después de 4horas y aumentan de nuevo a más del 80% a las 24 horas. Sólo en unos pocos pacientes se observa unamarcada y prolongada disminución del nivel de fibrinógeno circulante.
5.2 Propiedades farmacocinéticas
La alteplasa se elimina rápidamente de la sangre circulante y se metaboliza principalmente a través delhígado (aclaramiento plasmático 550 - 680 ml/min). La vida media plasmática relevante, T1/2 alfa es de4 - 5 minutos. Esto significa que, al cabo de 20 minutos, menos de un 10% del valor inicial estápresente en el plasma. Para la cantidad residual que permanece en un compartimento profundo, sedeterminó una vida media beta de aprox. 40 minutos.
5.3 Datos preclínicos sobre seguridad
En estudios de toxicidad subcrónica, en ratas y monos titís, no se han observado efectos secundariosinesperados.

19
En estudios de mutagenicidad, no se observaron indicios de potencial mutagénico.
6. DATOS FARMACÉUTICOS
6.1 Lista de excipientes
Polvo para solución:L-ArgininaAcido fosfórico 10%Polisorbato 80
Disolvente:Agua para preparaciones inyectables
6.2 Incompatibilidades
La solución reconstituida puede diluirse adicionalmente con solución salina fisiológica estéril (0,9%)hasta 1:5.
Sin embargo, no puede diluirse adicionalmente con agua para preparaciones inyectables o solucionesde carbohidratos para infusión, p.ej. dextrosa.
Actilyse no debe mezclarse con otros fármacos, ni en el mismo vial de infusión ni en el mismo catéter(ni con heparina).
6.3 Período de validez
36 meses
Se ha demostrado una estabilidad física y química de la solución reconstituida de 24 horas a 2-8ºC yde 8 horas a 25ºC. Desde el punto de vista microbiológico, el preparado debe administrarse de manerainmediata.
6.4 Precauciones especiales de conservación
No conservar a temperatura superior a 25ºC.Conservar en el envase original para preservarlo de la luz.
6.5 Naturaleza y contenido del recipiente
Polvo para solución:Viales de vidrio esterilizados de 10, 20, 50 ó 100 ml, que se cierran con tapones de liofilización degoma butílica, de color gris, siliconados y estériles, y se precintan con cápsulas "flip-off" dealuminio/plástico.
Disolvente:El agua para preparaciones inyectables se envasa en viales de 10, 20, 50 ó 2 x 50 ml, según el tamañode los viales de rt-PA. Los viales de agua para preparaciones inyectables se cierran con tapones degoma adecuados y cápsulas "flip-off" de aluminio/plástico.
Cánula de transferencia (incluida únicamente en los envases de 20 mg, 50 mg y 100 mg)
Tamaños de envase:10 mg1 vial con 467 mg de polvo para solución para perfusión

20
1 vial con 10 ml de agua para preparaciones inyectables20 mg1 vial con 933 mg de polvo para solución para perfusión1 vial con 20 ml de agua para preparaciones inyectables1 cánula de transferencia50 mg1 vial con 2.333 mg de polvo para solución para perfusión1 vial con 50 ml de agua para preparaciones inyectables1 cánula de transferencia100 mg1 vial con 4.666 mg de polvo para solución para perfusión2 viales con 50 ml de agua para preparaciones inyectables2 cánulas de transferencia
6.6 Instrucciones de uso, manipulación y eliminación (en su caso)
Ninguna especial
7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Boehringer Ingelheim Pharma KGBinger Straße 17355216 Ingelheim
8. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
9. FECHA DE LA PRIMERA AUTORIZACIÓN/RENOVACIÓN DE LAAUTORIZACIÓN
10. FECHA DE LA REVISIÓN DEL TEXTO

21
ANEXO IV
CONDICIONES PARA EL USO SEGURO Y EFECTIVO DEL MEDICAMENTO

22
CONDICIONES PARA EL USO SEGURO Y EFECTIVO DEL MEDICAMENTO
- La empresa llevará a cabo un estudio aleatorio, controlado con placebo (ECASS III) paraconfirmar la eficacia y la seguridad del medicamento, que se realizará en un margen de tiempo detres a cuatro horas, con una población de pacientes idéntica a la que figura en el criterio deinclusión del Resumen de las Características del Producto / los ensayos realizados por NINDS enEstados Unidos. Asimismo, la empresa se compromete a presentar semestralmente informesdetallados sobre el avance de la aplicación de este estudio, que incluyan información sobre lainclusión y el seguimiento de nuevos pacientes, a partir de la fecha en que la Comisión adopte laDecisión sobre la indicación del infarto, que deberán ser examinados por el Comité deEspecialidades Farmacéuticas y de forma paralela a la presentación de los Informes Periódicos deSeguridad. El estudio comenzará (incorporación del primer paciente) en un plazo de seis meses apartir de la fecha en que la Comisión adopte la Decisión, y el informe definitivo sobre el estudiohabrá de ser presentado en un plazo de seis meses a partir de que se finalice el estudio con elúltimo paciente.
- La empresa realizará un estudio de supervisión en Europa con posterioridad a lacomercialización: "un estudio de control de seguridad SITS-MOST" Asimismo, la empresa secompromete a presentar semestralmente informes detallados sobre el avance de la aplicación delestudio, que incluyan información sobre la inclusión y el seguimiento de nuevos pacientes, apartir de la fecha en que la Comisión adopte la Decisión sobre la indicación del infarto, quedeberán ser examinados por el Comité de Especialidades Farmacéuticas y de forma paralela a lapresentación de los Informes Periódicos de Seguridad. El estudio comenzará (incorporación delprimer paciente) en un plazo de tres meses a partir de la fecha en que la Comisión adopte laDecisión, y el informe definitivo sobre el estudio habrá de ser presentado en un plazo de seismeses a partir de que se finalice el estudio con el último paciente.
- La empresa presentará Informes Periódicos de Seguridad a los seis, doce, dieciocho yveinticuatro meses, a los tres años, a los cuatro años y como parte de la solicitud de renovación, apartir de la Decisión de la Comisión con respecto a la indicación sobre el infarto.
- Los protocolos de estudio definitivos para los estudios ECASS III y SITS-MOST se presentaránal Comité de Especialidades Farmacéuticas, para su examen, a más tardar el 17 de julio de 2002.


















