Búsqueda automática de genes -...
46
Búsqueda automática de genes Manuel Calaza Departamento de Matemática Aplicada Universidad de Santiago de Compostela
Transcript of Búsqueda automática de genes -...

Búsqueda automática de genes
Manuel CalazaDepartamento de Matemática AplicadaUniversidad de Santiago de Compostela

Manuel CalazaDepto Matemática Aplicada
Introducción
La predicción y anotación de genes por métodos computacionales es el primer paso hacia la comprensión del contenido funcional de los genomas.Como la mayoría de los genes humanos tienen intrones, la clave está en detectar las señales de los sitios de splice y localizar las regiones codificantes.En los años 80 se desarrollaron programas usando sólo señales de splice y codones. Más tarde, en los 90, se añadieron técnicas estadísticas, lingüísticas y de aprendizaje automático. Últimamente, se han añadido técnicas de comparación entre genomas.
Manuel CalazaDepto Matemática Aplicada
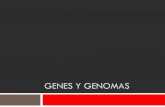
Predicción de genes ab initio
Predicción de señales.Predicción de exones.Ensamblaje de exones.
Manuel CalazaDepto Matemática Aplicada
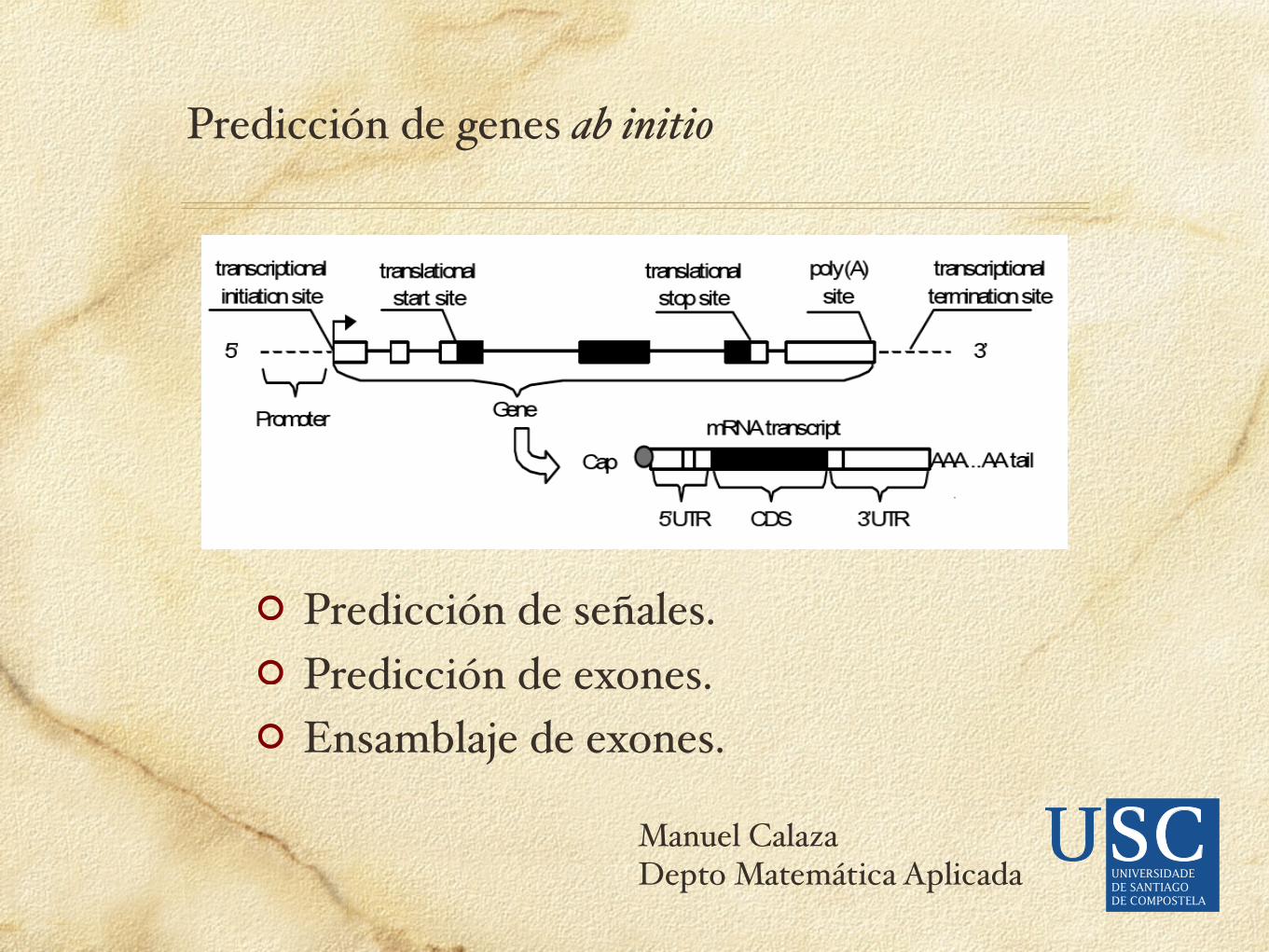
Señales
START GCCGCCRCCATGG
DONOR CAG|GTRAGT
ACCEPTOR (Y)nNCAG|G
BRANCH CTRAY
STOP TAA, TAG, TGA

Manuel CalazaDepto Matemática Aplicada
Señales
Estas secuencias consenso se construyen a partir de las frecuencias de aparición de los nucleótidos en cada una de las posiciones de la señal, sobre un conjunto de datos de entrenamiento. Se organizan de forma matricial:
Donor A G G T R A G T

Manuel CalazaDepto Matemática Aplicada
Señales
Branch
Acceptor
Senapathy et al, 1990.
Y Y Y Y Y Y Y Y Y Y N C A G G

Manuel CalazaDepto Matemática Aplicada
Señales
Nos permite puntuar las señales como sumas de las puntuaciones Sij de cada una de las bases según su posición en la señal.
€
Mij
€
fij =Mij
100
€
fi
€
Sij = log fijf i
WEIGHT MATRIX MODEL (WMM)

Manuel CalazaDepto Matemática Aplicada
Señales
El WMM presenta varios problemas:
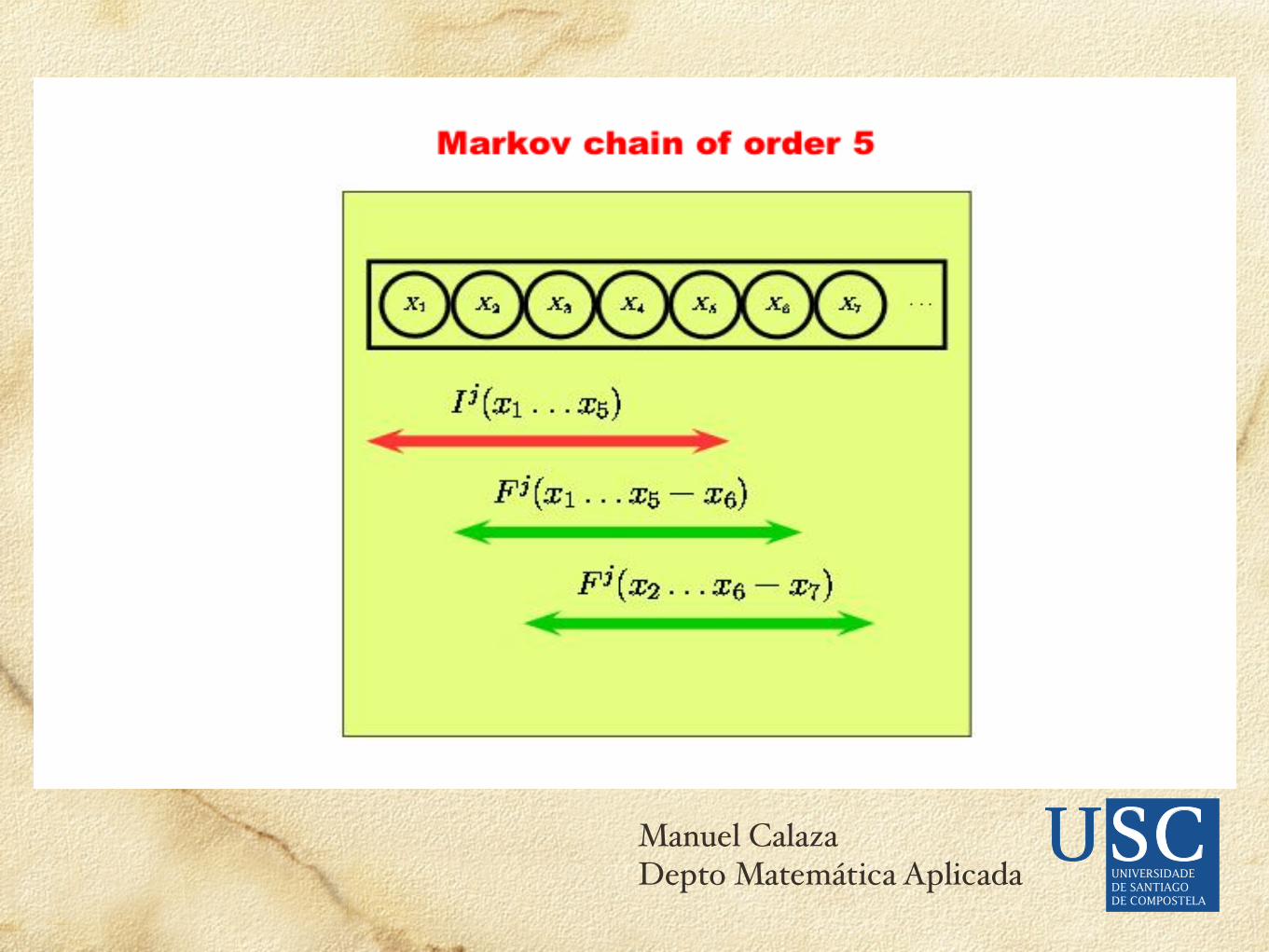
Asume independencia en posiciones diferentes de las señales. (Dependencia de Markov: equivale a extender las filas de la matriz para considerar duplas, tripletas,...pero necesitaríamos más datos de entrenamiento (WAM); Dividir (con un arbol de decisión) los datos de entrenamiento en subgrupos de secuencias donde se pueda suponer esa independencia y adoptar distintos WMMs).
Ignora el contenido de GC de la secuencia. (Adoptar dos WMMs distintos según el contenido alto o bajo de GC en la secuencia).

Manuel CalazaDepto Matemática Aplicada
A pesar de estos refinamientos, el mejor error medio (falsos positivos + falsos negativos) que se puede obtener (Pertea et al. 2001) para acceptor o donor es del orden del 5%.En regiones grandes con 40 señales verdaderas y 4000 falsas, por cada señal verdadera que perdamos, ganaremos 100 falsas!!!!El reconocimiento efectivo de las señales de splice sólo puede hacerse en combinación con el reconocimiento de exones.
Señales

Manuel CalazaDepto Matemática Aplicada
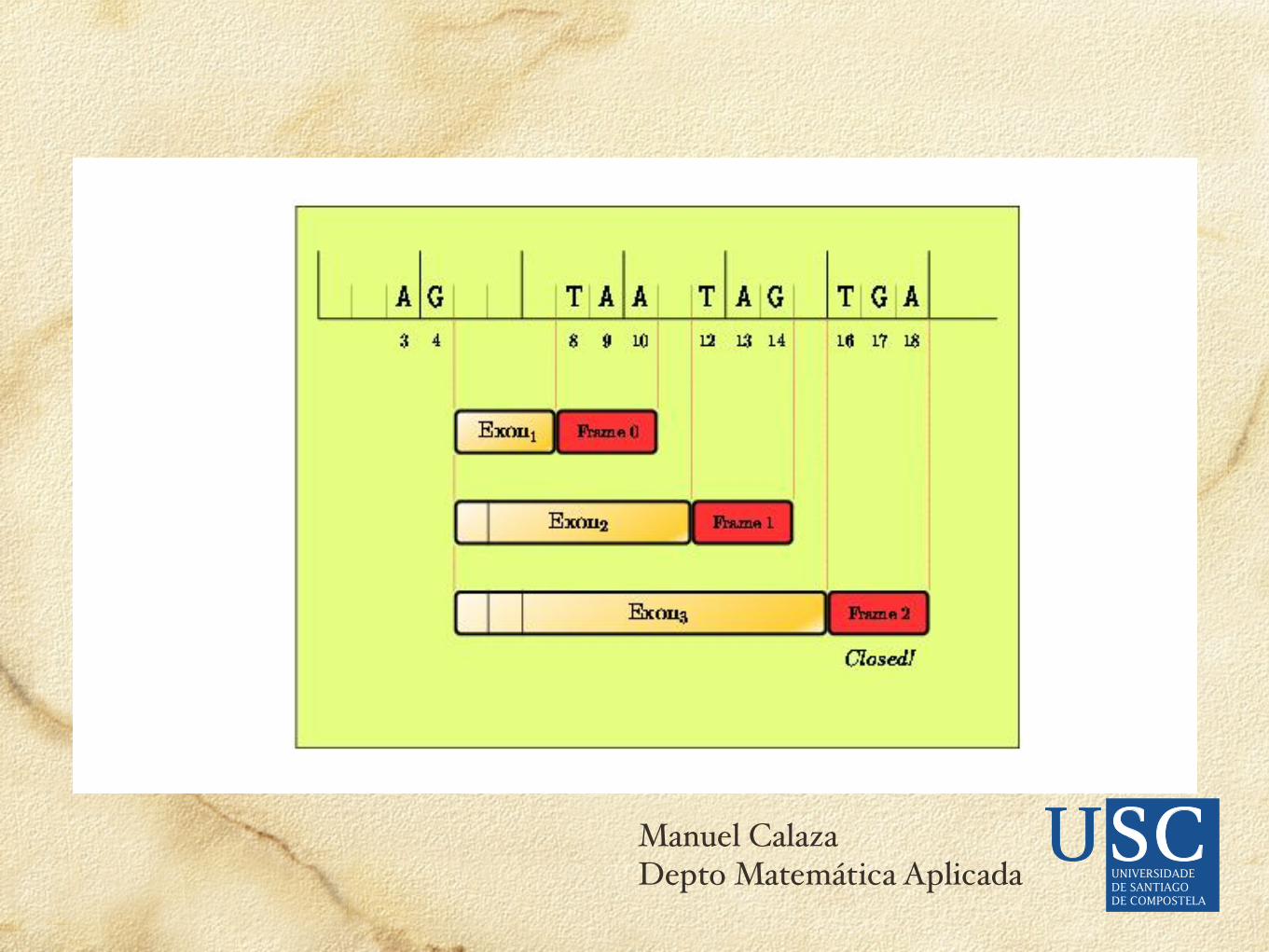
Una vez detectadas las señales, si construimos todos los exones formales con todas las señales, a partir de un error del 5% en señales podemos cometer errores en torno al 10% en exones.La puntuación de los posibles exones que formalmente definen las señales se hace valorando su potencialidad codificante.
Exones
score de frecuencias de aparición de hexámeros en un marco de lectura

Manuel CalazaDepto Matemática Aplicada
score de preferencia
score log-odd
frecuencia de aparición del hexámero w en el marco i calculado a partir de secuencias de entrenamiento.frecuencia de aparición del hexámero w en en los intrones que flanquean exones calculado a partir de las mismas secuencias de entrenamiento.
Exones
fE(w, i)
fI(w)
LE(w, i) = log
(fE(w, i)
fI(w)
)
PE(w, i) =fE(w, i)
fE(w, i) − fI(w)

Manuel CalazaDepto Matemática Aplicada
Podemos definir medidas de este tipo para las regiones intrónicas que flanquean exones:
Son inútiles en regiones exónicas no CDS (UTRs).
El tamaño del exón interno codificante sigue una distribución log-normal centrada en 125 pb.
PI(w) =fI(w)
fE(w) + fI(w)LI(w) = log
(fI(w)
fE(w)
)
fE(w) = (fE(w, 0) + fE(w, 1) + fE(w, 2))/3
Exones

Manuel CalazaDepto Matemática Aplicada
Consideremos secuencias con un exón auténtico o bien un pseudo-exón (AG-algo aleatorio-GT).Sobre este conjunto definimos una serie de variables x=(x1, x2, ...,xn) cuantificadoras del potencial codificante:
x1= media de los LI(w) para todos los hexámeros a lo largo de la región intrónica que flanquea al extremo 5’.x2= score del acceptor.x3= la media de los LE(w,i) sobre todos los hexámeros y maximizando luego en i.x4= el tamaño del exón.
.............xn= media de los LI(w) para todos los hexámeros a lo largo de la región intrónica que flanquea al extremo 3’.
Exones

Manuel CalazaDepto Matemática Aplicada
De este modo, cada secuencia del conjunto de entrenamiento se representa por un punto en el espacio n-dimensional.
Existen varios métodos estadísticos y de aprendizaje automático para construir una función de discriminación óptima (minimización de falsos positivos y falsos negativos).
Esta función (el predictor de exones) es una “hypersuperficie” de decisión en el espacio n-dimensional que separa los exones auténticos de los pseudo-exones aleatorios.
Los algoritmos más empleados para esta tarea han sido LDA-(algoritmo de discriminación lineal) y QDA-(algoritmo de discriminación cuadrática)
Exones

Manuel CalazaDepto Matemática Aplicada
El problema de ensamblar exones en un gen es que las posibilidades de combinación crecen exponencialmente respecto al número de candidatos a exones.La búsqueda de un óptimo sin evaluar todas las posibilidades se puede hacer desde la programación dinámica.
(FGENEH >>>> LDA en exones + DP en ensamblaje)
Ensamblaje de exones

Manuel CalazaDepto Matemática Aplicada
La otra alternativa actualmente son los modelos de Markov ocultos generalizados (HMMs).
Los diferentes tipos de componentes estructurales (como exones e intrones) están caracterizados por un estado y el modelo del gen está generado por una máquina de estados.
Empezando de 5’ a 3’, cada par de bases está generada por una probabilidad de emisión condicionada al estado actual, y la transición entre estados está gobernada por una probabilidad de transición.
Ensamblaje de exones

Manuel CalazaDepto Matemática Aplicada
Todos los parámetros de las probabilidades de emisión y transición se aprenden de un conjunto de datos de entrenamiento.
¿Cuál sería el conjunto de estados consecutivos que hacen máxima la probabilidad de observar la secuencia a examen, condicionada a unas probabilidades de emisión y transición fijadas?
Para responder a esta pregunta, se puede utilizar el algoritmo de Viterbi.
Ensamblaje de exones

Manuel CalazaDepto Matemática Aplicada
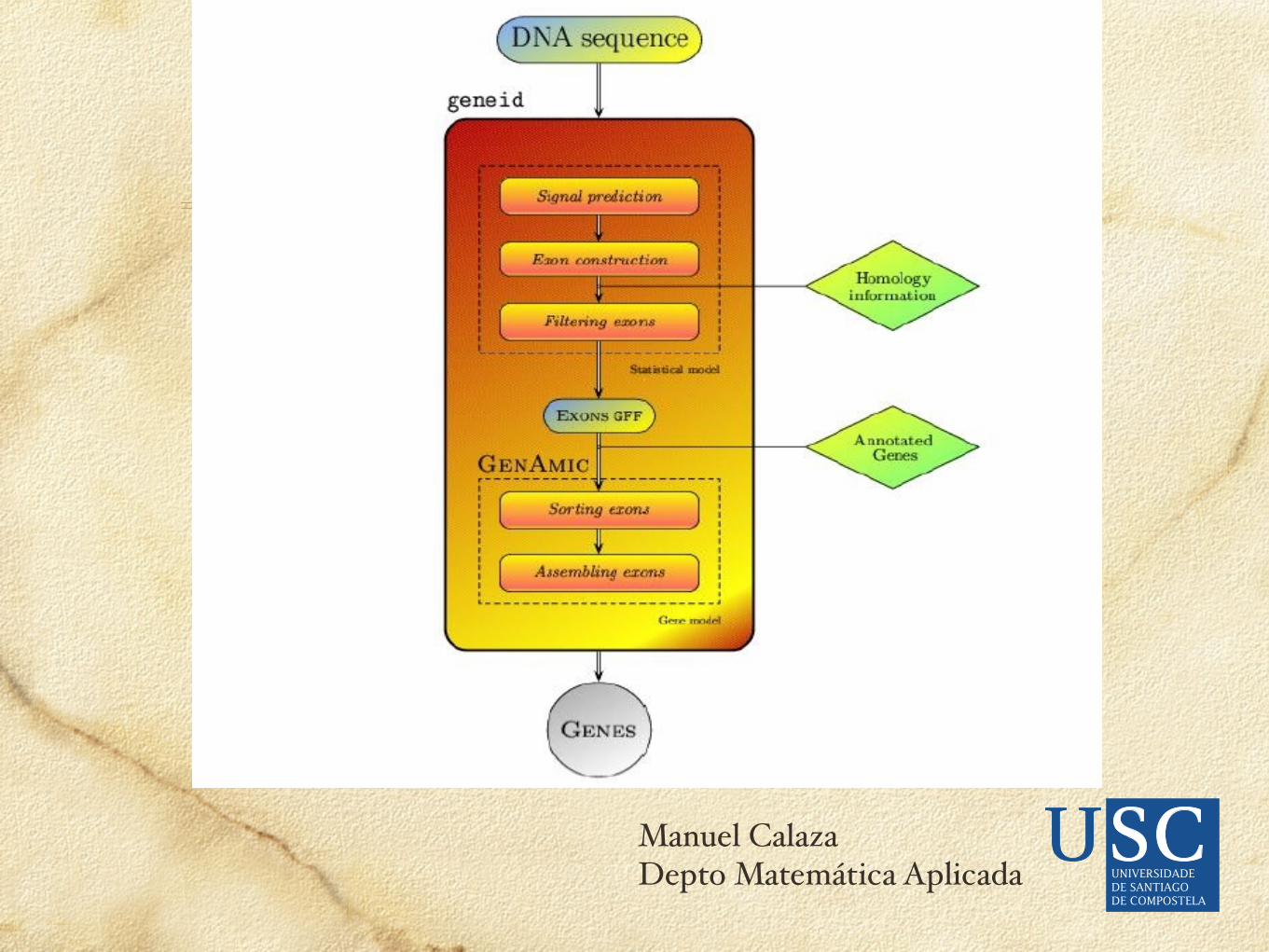
Un ejemplo: GeneID
Manuel CalazaDepto Matemática Aplicada

Manuel CalazaDepto Matemática Aplicada

Manuel CalazaDepto Matemática Aplicada
Manuel CalazaDepto Matemática Aplicada

Manuel CalazaDepto Matemática Aplicada
Manuel CalazaDepto Matemática Aplicada
Manuel CalazaDepto Matemática Aplicada
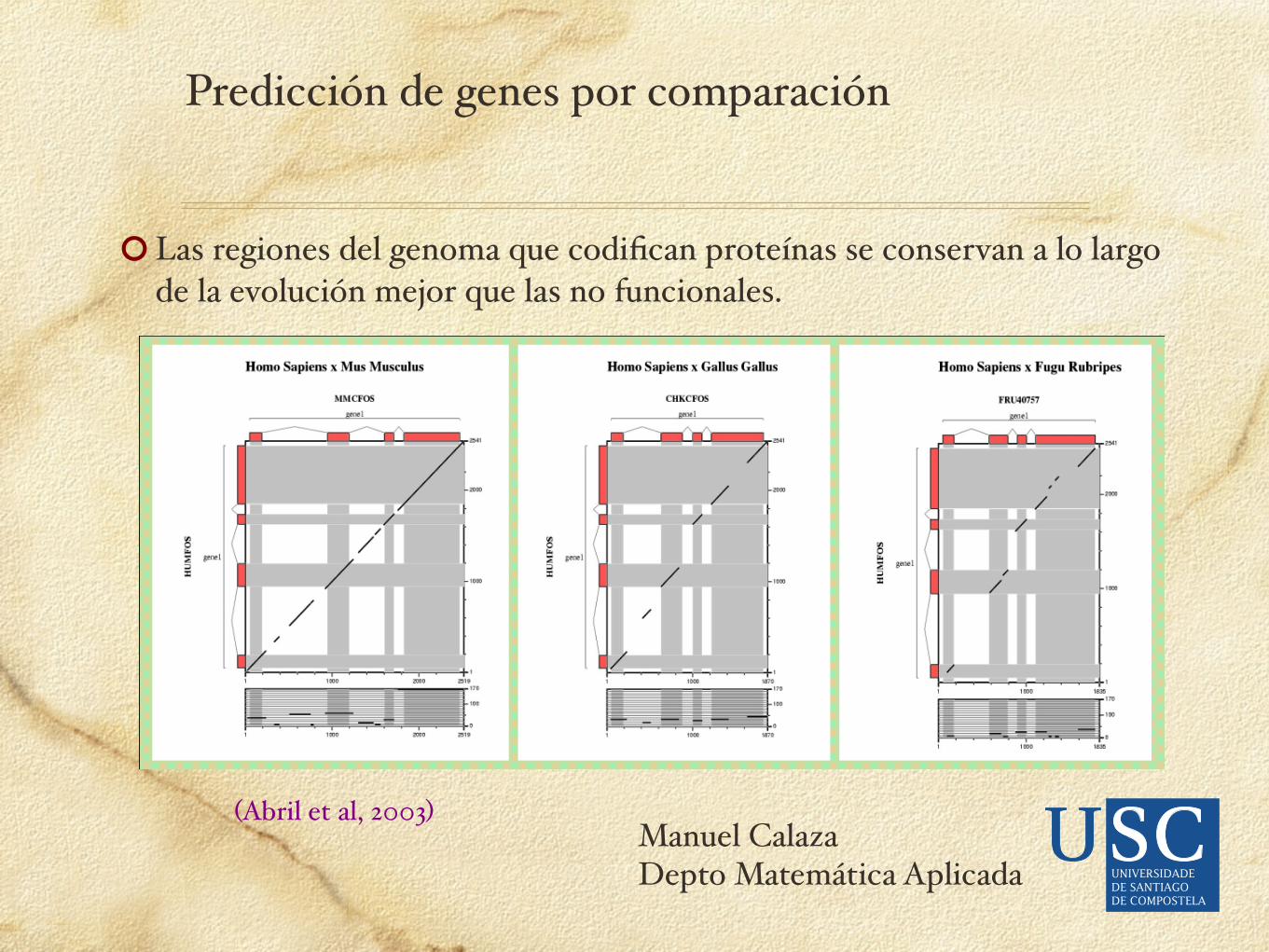
Las regiones del genoma que codifican proteínas se conservan a lo largo de la evolución mejor que las no funcionales.
(Abril et al, 2003)
Predicción de genes por comparación

Manuel CalazaDepto Matemática Aplicada
Con Blastx una query genómica es traducida (6 frames) en un conjunto de secuencias de aminoácidos y comparadas con una base de datos de secuencias de aminoácidos.
Complementariamente, se necesita información sobre codones START y STOP, etc que Blastx no puede proporcionar.
Query genómica contra target proteica

Manuel CalazaDepto Matemática Aplicada
A partir de las secuencia target con un buen score repecto de nuestra secuencia query, podemos realizar un alineamiento “splice” entre ellas.Este tipo de alineamientos permiten grandes gaps, que deberían corresponderse con las regiones intrónicas de nuestra secuencia query, y regiones de similitud casi completa, que deberían corresponderse con exones altamente probables de nuestra secuencia query.Finalmente, queda la cuestión de encontrar la mejor estructura exónica posible para lo que se emplea generalmente DP.
(PROCRUSTES, GENEWISE-ENSEMBL)
Query genómica contra target proteica

Manuel CalazaDepto Matemática Aplicada
Alternativamente, los resultados de realizar comparaciones con bases de datos pueden integrarse en los programas ab initio.El score de un candidato a exón, además de estar definido por los scores de las señales que lo delimitan y su potencial codificante, se incrementa en función de su similitud con secuencias codificantes conocidas.Se potencia la aparición en la predicción final del gen de exones con fuerte similitud con secuencias codificantes conocidas (de la misma u otra especie).
(GENOMESCAN extiende GENSCAN)
Query genómica contra target proteica

Manuel CalazaDepto Matemática Aplicada
Con la disponibilidad de más y más genomas completos, y asumiendo que las regiones conservadas por la evolución coresponden a exones codificantes de genes homólogos, la comparación de secuencias de genomas completos están ganando popularidad.EXOFISH predice exones humanos basándose en la utilización de comparaciones (usando TBLASTX) con una base de datos de secuencias de Tetraodon nigroviridis.
Query genómica contra target genómica

Manuel CalazaDepto Matemática Aplicada
Desde otro punto de vista (Pedersen et al y Blayo et al.), se puede plantear el problema como una extensión del problema del alineamiento de secuencias: dadas dos secuencias genómicas codificantes de genes homólogos, predecir la estructura exónica de cada una de e*as maximizando el score del alineamiento de las dos secuencias.Algunos programas como SLAM y DOUBLESCAN combinan dos modelos de Markov ocultos independientes, uno para realizar el alineamiento de las secuencias y otro para realizar la predicción génica, realizándose las dos tareas simultáneamente, sin que una se derive completamente de la otra.
Query genómica contra target genómica

Manuel CalazaDepto Matemática Aplicada
Otro tipo de programas separan claramente la predicción de genes del alineamiento de secuencias (ROSETTA, SGP1). Comienzan por realizar un alineamiento entre las dos secuencias y luego prediccen la estructura de genes de forma que los exones sean compatibles con los alineamientos.Todos estos enfoques comparten un problema evidente que es la necesidad de secuencias homólogas (e incluso siendo secuencias homólogas, si ha habido reordenaciones de genes o pérdida de regiones sinténicas en la evolución, puede haber genes en una secuencia sin homólogo en la otra)
Query genómica contra target genómica

Manuel CalazaDepto Matemática Aplicada
Para superar esta limitación, los programas TWINSCAN y SGP2 utilizan un enfoque similar al empleado por GENOMESCAN para introducir la similitud a proteínas conocidas al sistema de score de GENSCAN.Esencialmente, la secuencia query es comparada con una colección de secuencias del genoma informante y el resultado de la comparación es utilizado para modificar los scores de los exones de algún programa de predicción ab initio.
GENSCAN + BLASTN >>>> TWINSCAN
GENEID + TBLASTX >>>> SGP2
Query genómica contra target genómica

Manuel CalazaDepto Matemática Aplicada
Medidas:Para evaluar los programas de predicción génica sobre una secuencia test, se compara la estructura predicha con la real.Las mediciones se hacen a distintos niveles: de nucleótido, de exón y de gen.En cada nivel hay dos medidas básicas: la sensitividad y la especificidad.
Exactitud en la predicción de genes

Manuel CalazaDepto Matemática Aplicada
La sensitividad es la proporción de elementos reales que han sido correctamente predichos. La especificidad es la proporción de elementos predichos que son correctos.
TP, nº de elementos codificantes correctamente predichos,TN, nº de elementos no codificantes correctamente predichos,FP, nº de elementos no codificantes predichos como codificantes, yFN, nº de elementos codificantes predichos como no codificantes.
Medidas.
Sen =TP
TP + FNEsp =
TP
TP + FP

Manuel CalazaDepto Matemática Aplicada
A nivel de nucleótidos la medida más utilizada es el coeficiente de correlación, CC:
Medidas.
CC =TP · TN − FN · FP
√(TP + FN) · (TN + FP ) · (TP + FP ) · (TN + FN)

Manuel CalazaDepto Matemática Aplicada
A nivel de exón, se consideran correctamente predichos los exones que coinciden totalmente con los reales (incluidas las fronteras 5’ y 3’), un exón predicho se considera incorrecto si no tiene superposición con ningún exón real, y un exón real se considera perdido si no tiene superposición con ningún exón predicho.
Una medida considerada efectiva a este nivel es la media entre la especifidad y la sensitividad.
Medidas.

Manuel CalazaDepto Matemática Aplicada
A nivel de gen, se considera que un gen está correctamente predicho si todas las regiones codificantes han sido identificadas, todas las fronteras entre exón e intrón son correctas, y todos los exones están incluídos en el gen real.
Medidas.

Manuel CalazaDepto Matemática Aplicada
En 1996, Burset y Guigó, realizaron una comparativa.
Se evaluaron programas sobre 570 secuencias genómicas de vertebrados que contenían un único gen, depositadas en GenBank después de 1993 (para evitar entrenamiento de los programas sobre esas secuencias).
A nivel de nucleótidos, CC varió entre 0’65 y 0’78.
A nivel de exones, la media entre sensitividad y especificidad varió entre 0’37 y 0’60.
Mediciones.

Manuel CalazaDepto Matemática Aplicada
En 2001, Rogic et al, realizaron otra nueva comparativa.Se evaluaron 7 programas sobre secuencias genómicas que contenían un único gen, depositadas en GenBank después de la fecha de aparición de los programas.Los resultados han mejorado respecto de la comparativa anterior.A nivel de nucleótidos, CC varió entre 0’66 y 0’91.A nivel de exones, la media entre sensitividad y especificidad varió entre 0’43 y 0’76.
Mediciones.

Manuel CalazaDepto Matemática Aplicada
Estas comparativas pueden reflejar una situación mejor de la real. Fundamentalmente porque las secuencias eran cortas y con un único gen de estructura simple. Estas secuencias no son representativas del contexto actual: secuencias largas de baja densidad codificante, codificando varios genes y/o genes incompletos, con estructuras génicas complejas.
Mediciones.

Manuel CalazaDepto Matemática Aplicada
Sobre la anotación génica actual del cromosoma 22 se ha realizado una nueva comparativa con los resultados que se sospechaban: la precisión desciende considerablemente.El CC de GENSCAN baja desde el 0’91 en la comparativa de Rogic et al. hasta el 0’64 en el cromosoma 22.Pero incluso programas más sofisticados que utilizan comparación con secuencias (como GENEWISE-ENSEMBL) se quedan en valores de CC en torno a 0’75.
¡Prudencia!
Mediciones.

Manuel CalazaDepto Matemática Aplicada
Actualmente hay tres bases de datos públicas de anotaciones de genes humanos:
EBI&Sanger Institute ENSEMBL,
http://www.ensembl.org
UCSC Genome Browser,y
http://genome.ucsc.edu
NCBI LocusLink.
http://www.ncbi.nim.nih.gov/LocusLink/
Sistemas de anotación génica.

Manuel CalazaDepto Matemática Aplicada
Los genes anotados en ENSEMBL han sido generados automáticamente por herramientas propias de ENSEMBL. El motor básico de anotación es GENEWISE.Para un número determinado de cromosomas (6, 13, 14, 20, 22) también están disponibles las anotaciones manuales del sistema VEGA (http://vega.sanger.ac.uk/Homo_sapiens/).Hay tres tipos de genes anotados en ENSEMBL:
Los que codifican completamente una proteína,
Los que presentan gran homología con proteínas de otros organismos,
Los predichos por GENESCAN que presentan gran homología con proteínas o mRNA de vertebrados.
Conservadoramente, predice en total de unos 24.500 genes.
Sistemas de anotación génica.

Manuel CalazaDepto Matemática Aplicada
El UCSC Genome Browser proporciona un acceso rápido y muy informativo a las anotaciones.Parte de las anotaciones e informaciones son realizadas en la UCSC a partir de datos públicos y el resto son proporcionados por colaboradores.El motor básico de anotación es BLAT que predice genes por comparación usando alineamientos muy rápidos (en comparación con BLAST) entre proteínas de primates y otros vertebrados con el genoma humano.En sus predicciones incluyen, además de los 24.500 genes de ENSEMBL, 25.600 genes de TWINSCAN, 32.400 genes de GENEID, 39.800 genes de FGENESH++ y 45.000 genes de GENSCAN.
Sistemas de anotación génica.

Manuel CalazaDepto Matemática Aplicada
El NCBI LocusLink predice genes por comparación usando alineamientos con MegaBLAST de secuencias de genes de RefSeq y mRNA con el genoma humano.Además, los genes predichos por GENOMESCAN son anotados sólo si no se solapan con los predichos por los alineaminetos con mRNA.GENOMESCAN ha predicho 38.600 genes.
Sistemas de anotación génica.

Manuel CalazaDepto Matemática Aplicada
Las anotaciones proporcionadas por estos sistemas hay que considerarlas altamente hipotéticas dada la precisión de los programas actuales de predicción de genes.Falta un largo camino para que los sistemas automáticos puedan predecir todos los genes dentro de una secuencia genómica.Se necesita conocer mejor lo que realmente es un gen, y los procesos biológicos involucrados en la especificación génica (especialmente lo referente a los sitios de splice y codón de START).
Sistemas de anotación génica.