
FORMAS ESPECIALES DE CÁNCER DE MAMA
60
FORMAS ESPECIALES DE CÁNCER DE MAMA MÁSTER INTERNACIONAL DE ESPECIALIZACIÓN EN MASTOLOGÍA-SENOLOGÍA 2021 CARMEN MORIYÓN HOSPITAL DE CABUEÑES. GIJÓN
Transcript of FORMAS ESPECIALES DE CÁNCER DE MAMA

FORMAS ESPECIALES DE CÁNCER DE MAMA
MÁSTER INTERNACIONAL DE ESPECIALIZACIÓN EN MASTOLOGÍA-SENOLOGÍA
2021
CARMEN MORIYÓN
HOSPITAL DE CABUEÑES. GIJÓN



Hamartoma
Masa bien delimitada, generalmente encapsulada, formada por componentes de tejido mamario normal.
Terminología aceptable: adenolipoma, condrolipoma, hamartoma mioide.
Clínica.
• Masa palpable, blanda o asintomática detectada por mamografía. <5% de tumores de mama.
• Hamartomas múltiples y bilaterales pueden asociarse al sind de Cowden (mutPTEN. Mama, piel, tiroides, endometrio).
Macro.
• Masas redondas u ovales que a la sección pareen tejido mamario normal, lipoma o fibroadenoma.
Micro.
• Generalmente encapsulados.
• Mezcla de ductos, lobulillo y tejido adiposo en variada proporción.
• Fibrosis prominente y lobulillos atróficos en general.
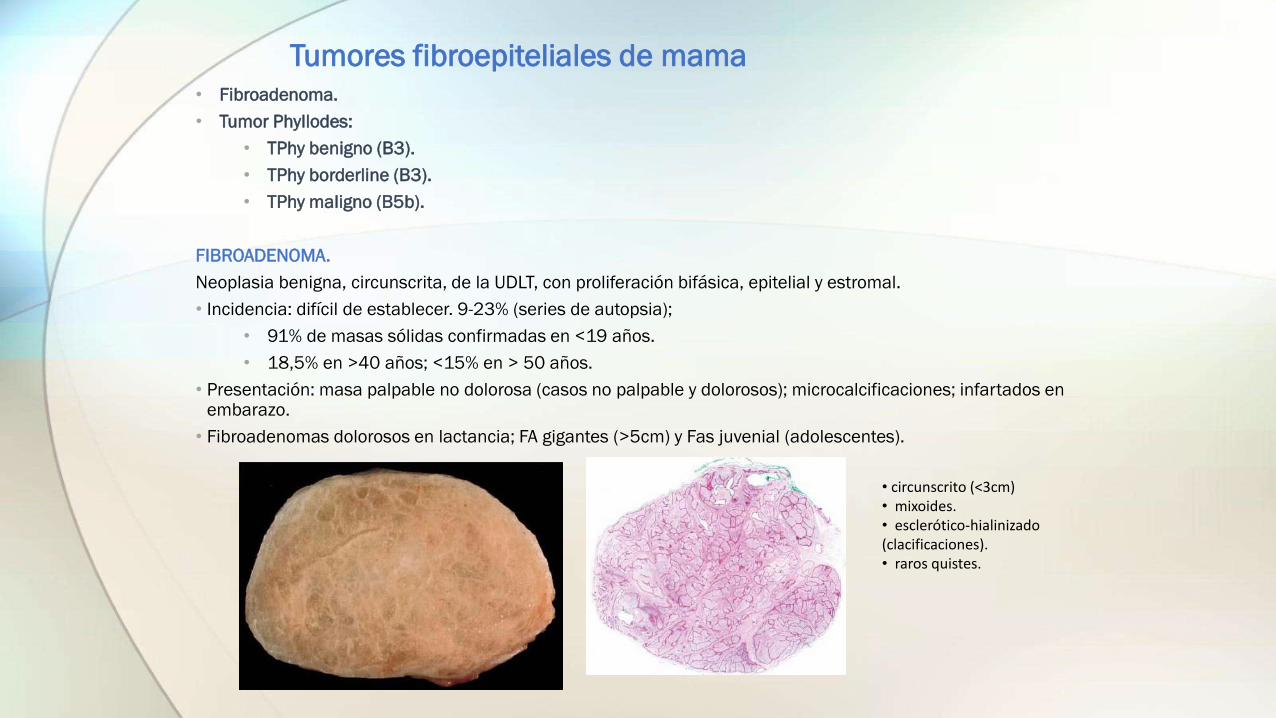
Tumores fibroepiteliales de mama• Fibroadenoma.
• Tumor Phyllodes:
• TPhy benigno (B3).
• TPhy borderline (B3).
• TPhy maligno (B5b).
FIBROADENOMA.
Neoplasia benigna, circunscrita, de la UDLT, con proliferación bifásica, epitelial y estromal.
• Incidencia: difícil de establecer. 9-23% (series de autopsia);
• 91% de masas sólidas confirmadas en <19 años.
• 18,5% en >40 años; <15% en > 50 años.
• Presentación: masa palpable no dolorosa (casos no palpable y dolorosos); microcalcificaciones; infartados en embarazo.
• Fibroadenomas dolorosos en lactancia; FA gigantes (>5cm) y Fas juvenial (adolescentes).
• circunscrito (<3cm)• mixoides.• esclerótico-hialinizado(clacificaciones).• raros quistes.
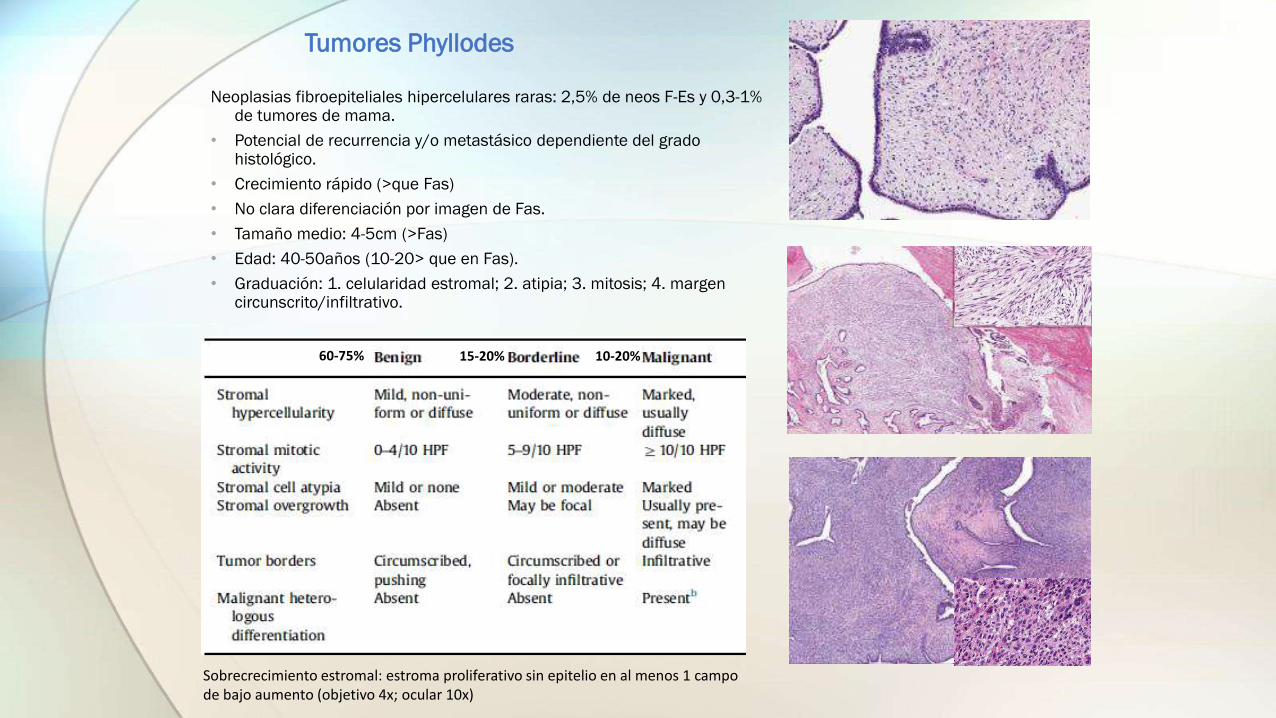
Tumores Phyllodes
Neoplasias fibroepiteliales hipercelulares raras: 2,5% de neos F-Es y 0,3-1% de tumores de mama.
• Potencial de recurrencia y/o metastásico dependiente del grado histológico.
• Crecimiento rápido (>que Fas)
• No clara diferenciación por imagen de Fas.
• Tamaño medio: 4-5cm (>Fas)
• Edad: 40-50años (10-20> que en Fas).
• Graduación: 1. celularidad estromal; 2. atipia; 3. mitosis; 4. margen circunscrito/infiltrativo.
Sobrecrecimiento estromal: estroma proliferativo sin epitelio en al menos 1 campo de bajo aumento (objetivo 4x; ocular 10x)
60-75% 15-20% 10-20%
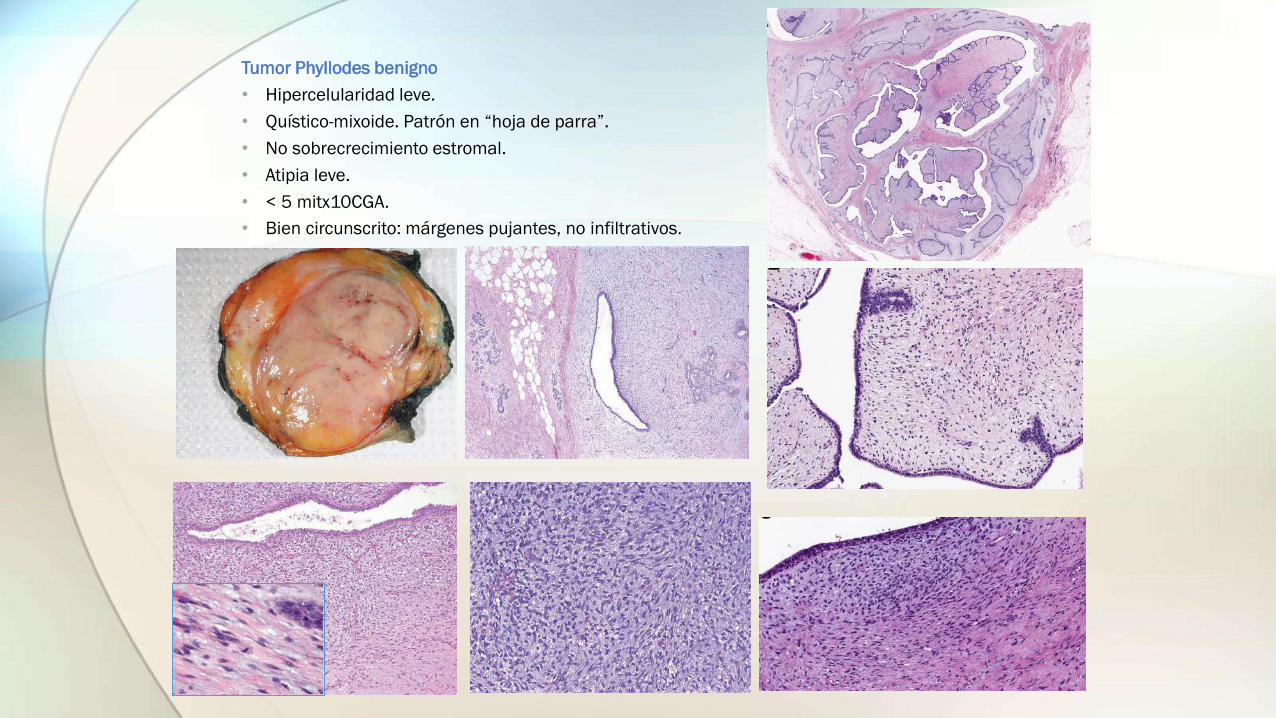
Tumor Phyllodes benigno
• Hipercelularidad leve.
• Quístico-mixoide. Patrón en “hoja de parra”.
• No sobrecrecimiento estromal.
• Atipia leve.
• < 5 mitx10CGA.
• Bien circunscrito: márgenes pujantes, no infiltrativos.

Tumor Phyllodes benigno
• Hipercelularidad leve. Celularidad estromal usualmente mayor que en FA
• Acentuación estromal subepitelial.
• Heterogeneidad estromal.
• Pueden verse células gigantes estromales (no confundir con maligno)
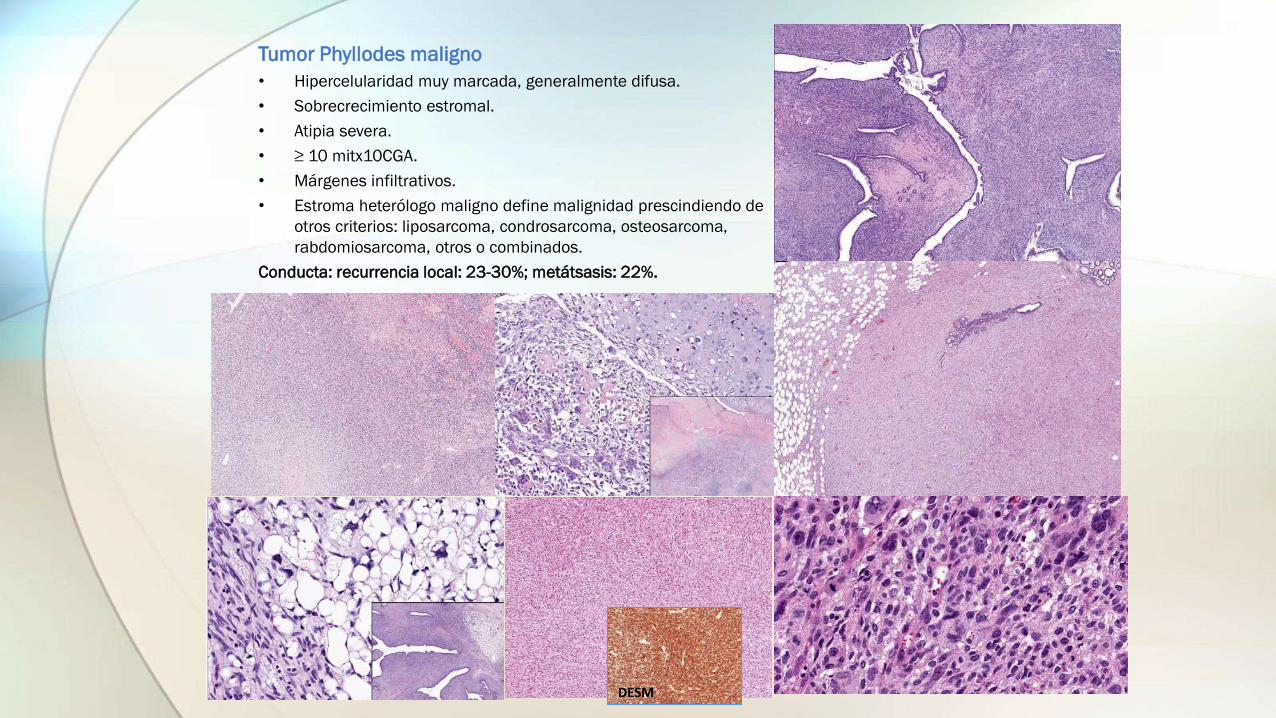
Tumor Phyllodes maligno
• Hipercelularidad muy marcada, generalmente difusa.
• Sobrecrecimiento estromal.
• Atipia severa.
• ≥ 10 mitx10CGA.
• Márgenes infiltrativos.
• Estroma heterólogo maligno define malignidad prescindiendo de
otros criterios: liposarcoma, condrosarcoma, osteosarcoma,
rabdomiosarcoma, otros o combinados.
Conducta: recurrencia local: 23-30%; metátsasis: 22%.
DESM

Ching TW. J Clin Pathol. 2018. 240 casos de TPhys
Recurrencia global
• La mayoría de TPhys se comportan de manera benigna, sin recurrencias o metástasis tras escisión.
• Ratio de recurrencia global: 5-13,2%, 85% locales, benignas.
• Tiempo: 2 años de media (1-20 años), menor en malignos.
• 65% 1 recurrencia; 28%, 5% y 1,2% experimentan 2, 3 y 6 recurrencias respectivamente.
• Rasgos patológicos relacionados con recurrencia: alto grado, mitosis, incremento de celularidad estromal,
atipia/pleomorfismo estromal, sobrecrecimiento estromal, necrosis, margen infiltrativo.
Recurrencia dependiente de escisión
• Escisión local:
• Benigno: 21%; Borderline: 46%; Maligno: 65%.
• Escisión amplia:
▪ Benigno: 8%; Borderline: 29%; Maligno: 36%.
Recurrencia según margen quirúrgico
• El estatus del margen es uno de los más importantes
Predictores de recurrencia
Metástasis:
• <10% global, la mayoría en malignos (25%)
• ESTÁ CLARO QUE TPHYS PUEDEN RECURRIR
INDEPENDIENTEMENTE DEL GRADO, AUNQUE EXISTA UNA
RELACIÓN Y QUE EL MARGEN ES UN FACTOR IMPORTANTE EN
PREDECIR RECURRENCIAS.
• LAS RECURRENCIAS SON MÁS FRECUENTEMENTE DEL MISMO
GRADO, AUNQUE ENTRE 8-10% PUEDEN SER DE MÁS ALTO
GRADO.


Neoplasias mesenquimales de la mama. OMS 2019
Tumores vasculares.
• Hemangioma
• Angiomatosis
• Lesiones vasculares atípicas
• Angiosarcoma post-radiación de la mama
• Angiosarcoma primario de la mama
Tumores fibroblásticos y miofibroblásticos
• Fascitis nodular
• Miofibroblastoma
• Fibromatosis desmoide
• Tumor miofibroblástico inflamatorio
Tumores de vaina de nervio periférico
• Schwannoma
• Neurofibroma
• Tumor de células granulares
Tumores de músculo liso
• Leiomioma
• Leiomiosarcoma
Tumores adipocíticos
• Lipoma
• Angiolipoma
LiposarcomaTumores vasculares.
• Hemangioma
• Angiomatosis
• Lesiones vasculares atípicas
• Angiosarcoma post-radiación de la mama
• Angiosarcoma primario de la mama
Tumores fibroblásticos y miofibroblásticos
• Fascitis nodular
• Miofibroblastoma
• Fibromatosis desmoide
• Tumor miofibroblástico inflamatorio
Tumores de vaina de nervio periférico
• Schwannoma
• Neurofibroma
• Tumor de células granulares
Otros tumores y lesiones tumor-like de la mama
• Hiperplasia estromal pseudoangiomatosa de la mama
Incidencia.
• Sarcomas: 30-50 casos/millón/año.
• Neoplasias benignas: 100 veces superior.
Etiología y patogénesis.
• Desconocida para la mayoría
• Otras etiologías: Virus de Epstein Barr (tumores de músculo liso en inmunodeprimidos), HHV8 (sarcoma de Kaposi)
• Linfedema: asociado a angiosarcoma (síndrome de Stewart-Treves); angiosarcoma post-rediación.
• Asociados a síndromes hereditarios: síndrome de Maffucci (tumores vasculares+condroides); esclerosis tuberosa (angiomiolipomas renal-hepático); síndrome de Cowden (lipomas+hemangiomas).
Clínica.
• Típicamente se presentan como masas indoloras con ratios de crecimiento variables.
• La mayoría <5 cm.
Características histopatológicas generales.
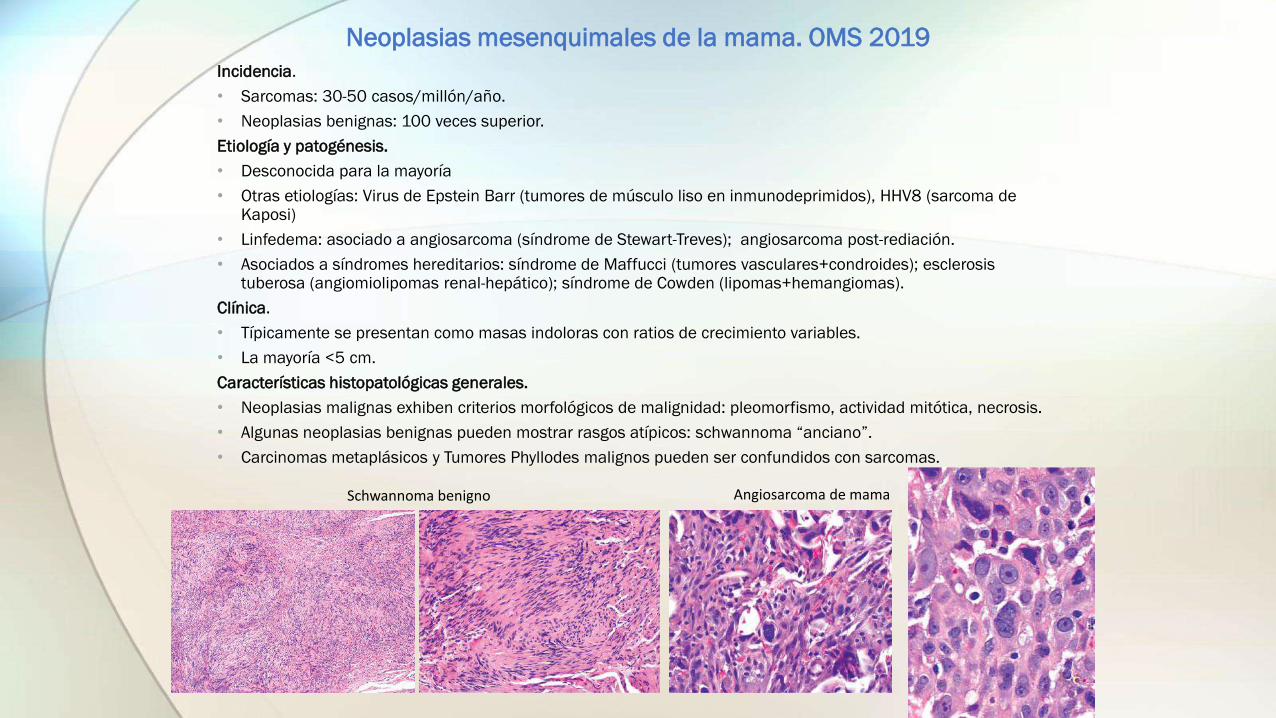
• Neoplasias malignas exhiben criterios morfológicos de malignidad: pleomorfismo, actividad mitótica, necrosis.
• Algunas neoplasias benignas pueden mostrar rasgos atípicos: schwannoma “anciano”.
• Carcinomas metaplásicos y Tumores Phyllodes malignos pueden ser confundidos con sarcomas.
Neoplasias mesenquimales de la mama. OMS 2019
Schwannoma benigno Angiosarcoma de mama
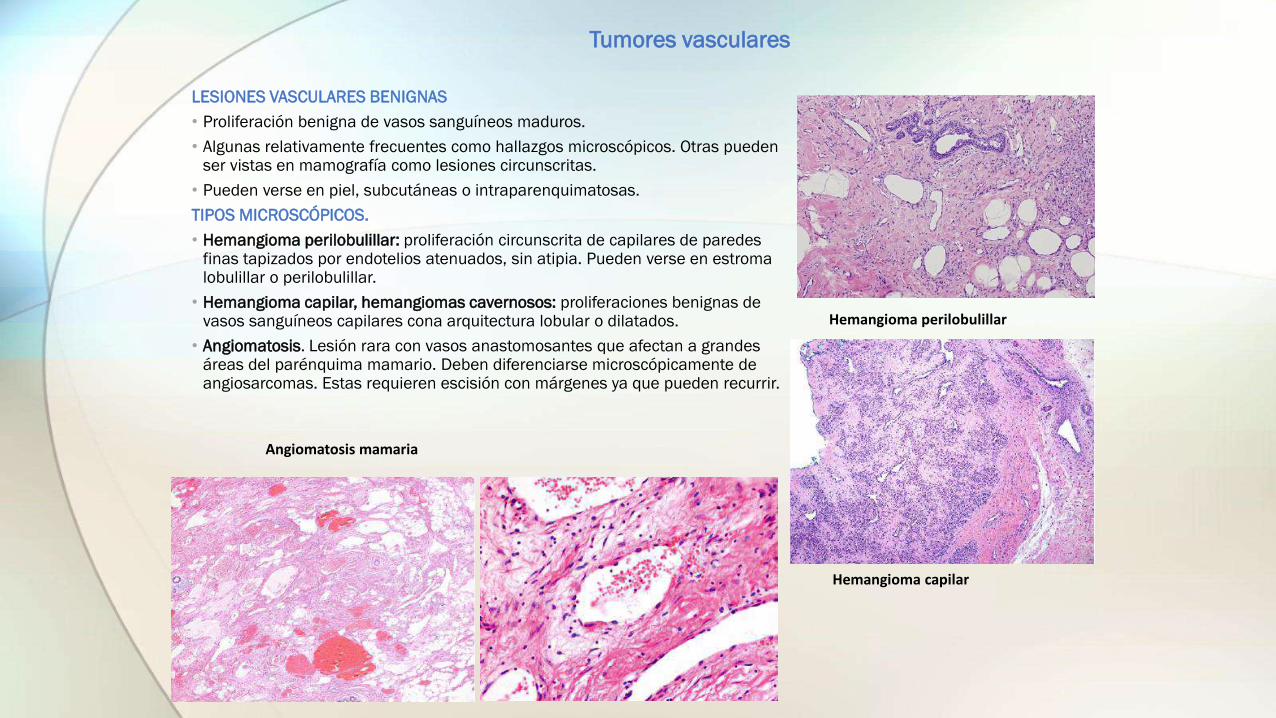
LESIONES VASCULARES BENIGNAS
• Proliferación benigna de vasos sanguíneos maduros.
• Algunas relativamente frecuentes como hallazgos microscópicos. Otras pueden ser vistas en mamografía como lesiones circunscritas.
• Pueden verse en piel, subcutáneas o intraparenquimatosas.
TIPOS MICROSCÓPICOS.
• Hemangioma perilobulillar: proliferación circunscrita de capilares de paredes finas tapizados por endotelios atenuados, sin atipia. Pueden verse en estroma lobulillar o perilobulillar.
• Hemangioma capilar, hemangiomas cavernosos: proliferaciones benignas de vasos sanguíneos capilares cona arquitectura lobular o dilatados.
• Angiomatosis. Lesión rara con vasos anastomosantes que afectan a grandes áreas del parénquima mamario. Deben diferenciarse microscópicamente de angiosarcomas. Estas requieren escisión con márgenes ya que pueden recurrir.
Tumores vasculares
Hemangioma perilobulillar
Hemangioma capilar
Angiomatosis mamaria

LESIONES VASCULARES ATÍPICAS.
• Proliferaciones vasculares atípicas en piel irradiada, con frecuencia múltiples y pequeñas.
Clínica.
• Pápulas eritematosas o marrones, o placas pequeñas (0,5 cm).
• Pacientes más jóvenes que angiosarcomas y período de latencia post-radiación más corto (3-4 a).
Patología.
• Lesiones relativamente bien circunscritas, típicamente centradas en dermis superficial o media.
• Canales finos de tipo linfático o vascular, tapizados por endotelios con núcleos prominentes hipercromáticos, autentica atipia y estratificación celular como visto en angiosarcoma no se ve aquí.
• Subtipos linfático y vascular.
• Expresan marcadores vasculares (CD34, CD31) o linfático (podoplanina).
• Ausencia de sobreexpresión o amplificación MYC típica de angiosarcomas.
Pronóstico.
• Puede mostrar recurrencias. Progresión a angiosarcoma muy rara y controvertida.
Tumores vasculares

ANGIOSARCOMA POST-RADIACIÓN DE LA MAMA
Neoplasia vascular maligna mamaria, secundaria a radiación en piel o parénquima mamario. (0,04% de neoplasias malignas de la mama)
Localización:
• Piel de pared torácica o cicatriz residual en el campo de radiación.
Clínica.
• Período de latencia post-RTx de 5-6 a.
• Parches eritematosos o violáceos cutáneos múltiples. Pápulas o nódulos.
• Puede aparecer en el contexto de linfedema (síndrome de Stewart-Treves)
• Más frecuentemente visto en el contexto de cirugía conservadora de mama.
Patología.
• Neoplasia más frecuentemente de localización dérmica, con variable infiltración de tejido subcutáneo.
• Morfologías variables, con canales vasculares disecantes, tapizados por endotelios atípicos o apariencias más sólidas con crecimiento intraluminal.
Tumores vasculares

Tumores vasculares
ANGIOSARCOMA POST-RADIACIÓN DE LA MAMA
Criterios diagnósticos:
• Esenciales: Antecedente de RTx, intervalo >3 a. Predominio de afectación dérmica-subcutánea. Crecimiento vasoformativo con atipia.
• Deseable: sobreexpresión c-MYC por IHQ; amplificación c-myc por FISH.
Factores pronóstico-predictivos.
• Alta ratio de recurrencia loco-regional (50%).
• Metástasis en pulmón, mama contralateral, piel, hígado, hueso.
• Metástasis axilares raras.
• Supervivencia libre de enfermedad: <3 a. supervivencia media: <5 a.
Estudio de oncogén c-myc por técnica de Hibridación In Situ con Fluorescencia (FISH)
Izquierda: lesión vascular atípica sin amplificación (una señal roja-gen c-myc por célula)
Centro y derecha: amplificación c-myc. Múltiples señales rojas gen c-myc por célula.
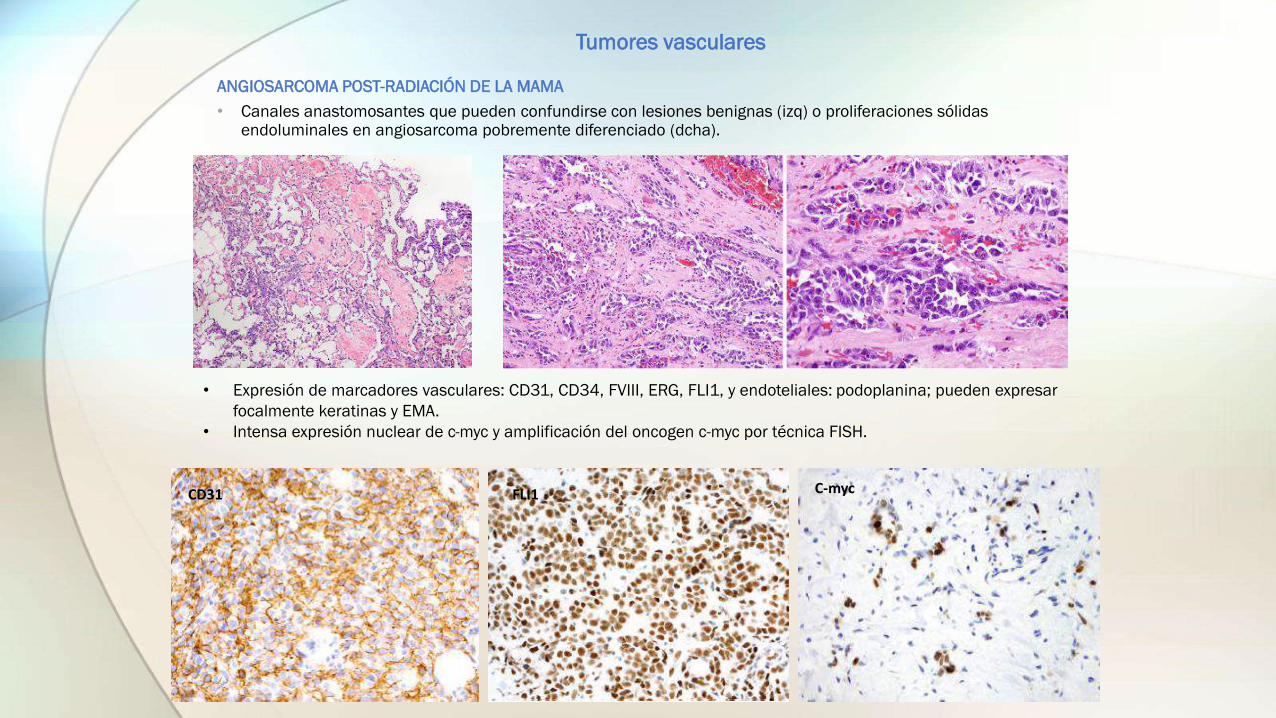
ANGIOSARCOMA POST-RADIACIÓN DE LA MAMA
• Canales anastomosantes que pueden confundirse con lesiones benignas (izq) o proliferaciones sólidas endoluminales en angiosarcoma pobremente diferenciado (dcha).
Tumores vasculares
• Expresión de marcadores vasculares: CD31, CD34, FVIII, ERG, FLI1, y endoteliales: podoplanina; pueden expresar
focalmente keratinas y EMA.
• Intensa expresión nuclear de c-myc y amplificación del oncogen c-myc por técnica FISH.
CD31 FLI1 C-myc
Tumores vasculares
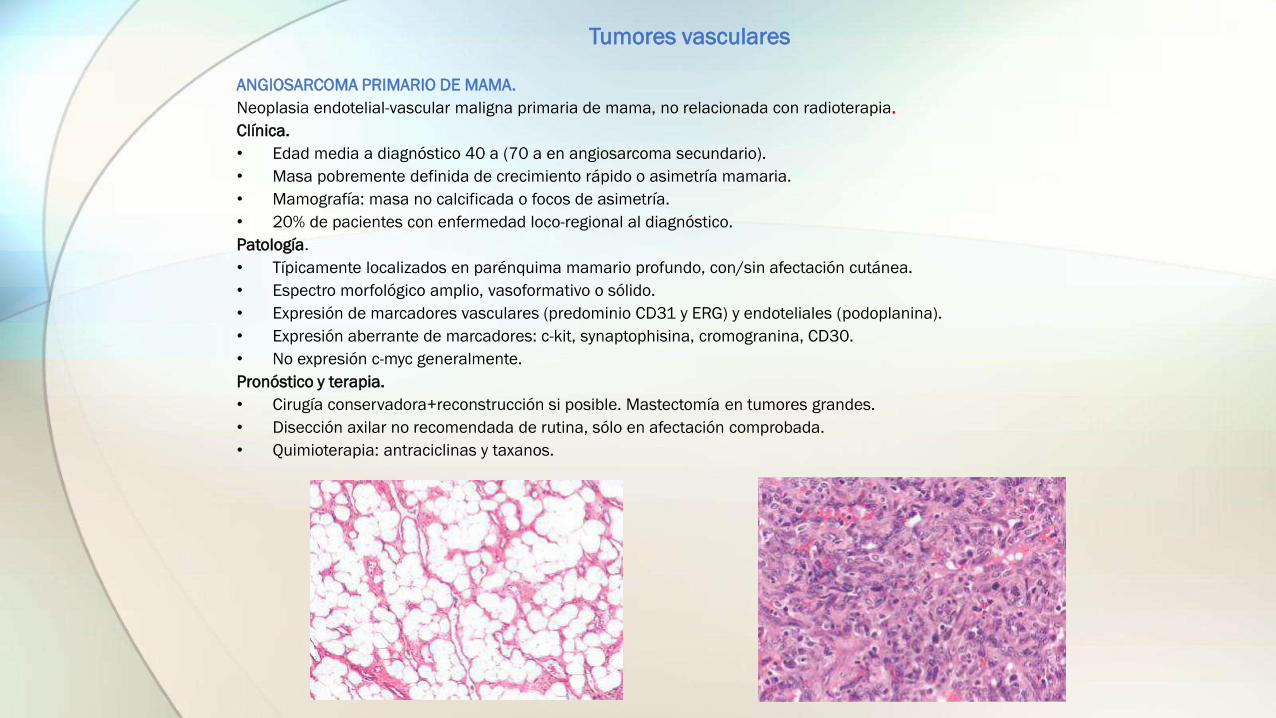
ANGIOSARCOMA PRIMARIO DE MAMA.
Neoplasia endotelial-vascular maligna primaria de mama, no relacionada con radioterapia.
Clínica.
• Edad media a diagnóstico 40 a (70 a en angiosarcoma secundario).
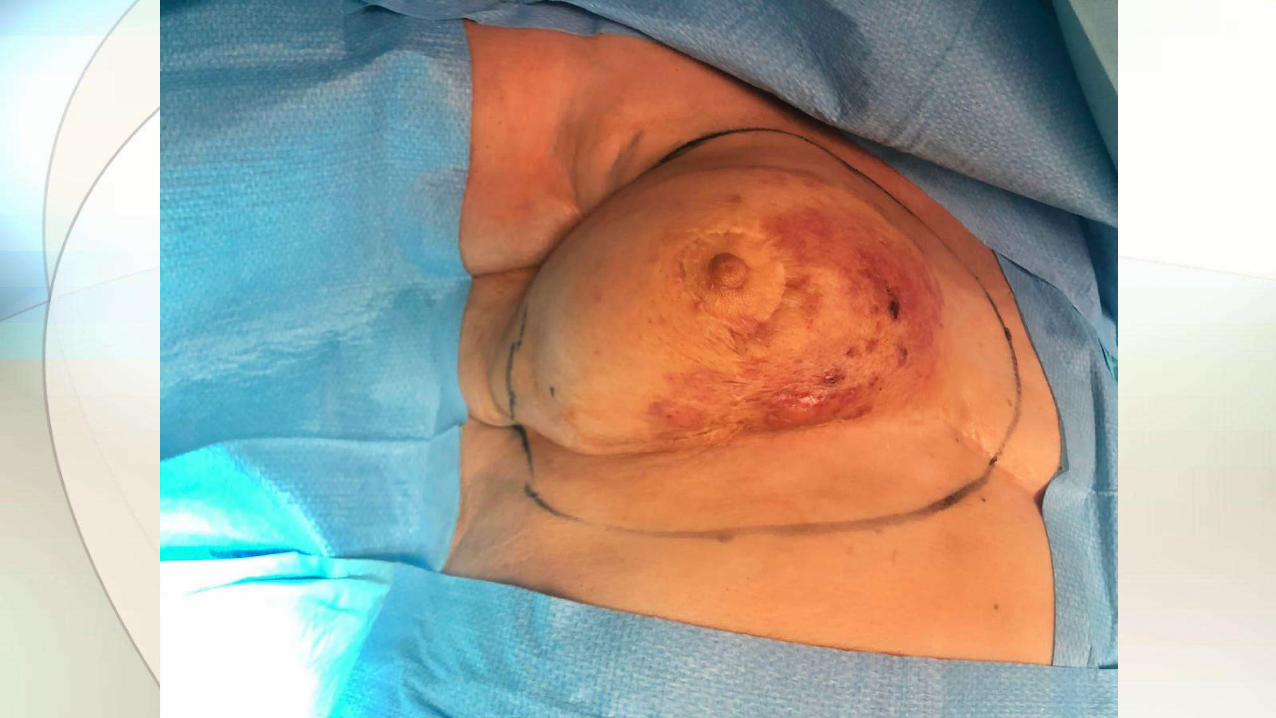
• Masa pobremente definida de crecimiento rápido o asimetría mamaria.
• Mamografía: masa no calcificada o focos de asimetría.
• 20% de pacientes con enfermedad loco-regional al diagnóstico.
Patología.
• Típicamente localizados en parénquima mamario profundo, con/sin afectación cutánea.
• Espectro morfológico amplio, vasoformativo o sólido.
• Expresión de marcadores vasculares (predominio CD31 y ERG) y endoteliales (podoplanina).
• Expresión aberrante de marcadores: c-kit, synaptophisina, cromogranina, CD30.
• No expresión c-myc generalmente.
Pronóstico y terapia.
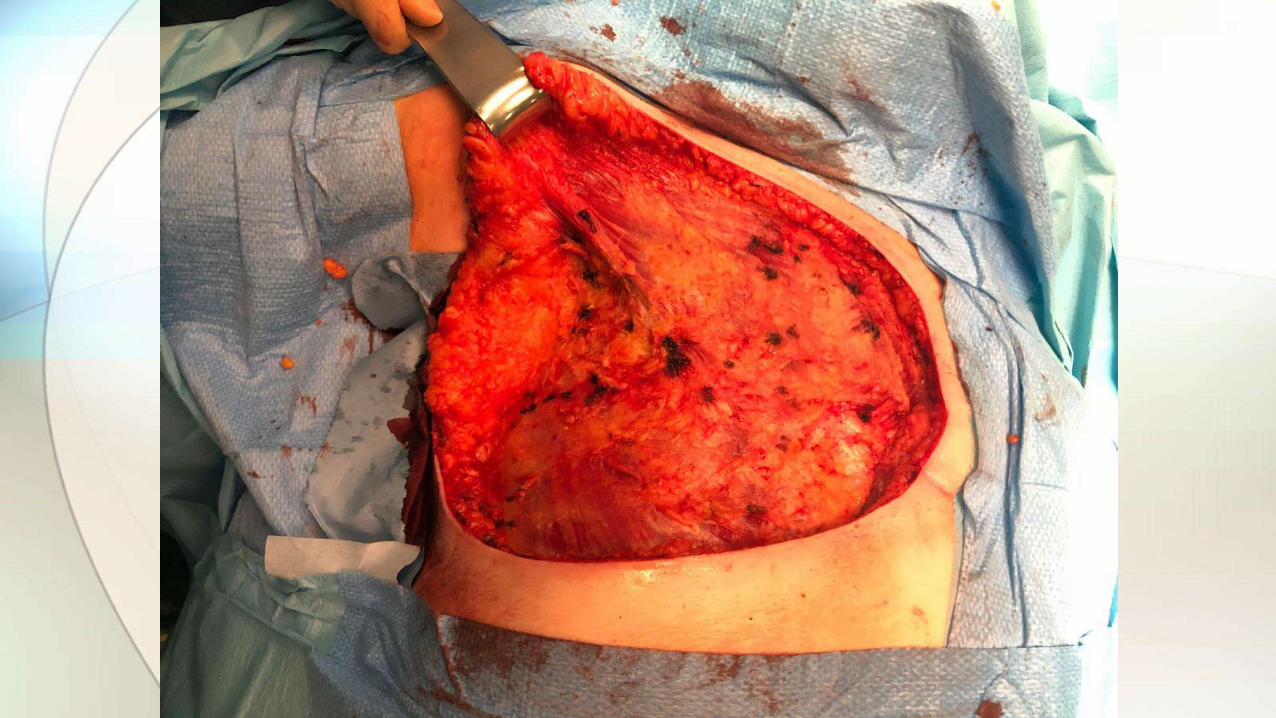
• Cirugía conservadora+reconstrucción si posible. Mastectomía en tumores grandes.
• Disección axilar no recomendada de rutina, sólo en afectación comprobada.
• Quimioterapia: antraciclinas y taxanos.



Tumores fibroblásticos y miofibroblásticos de la mama
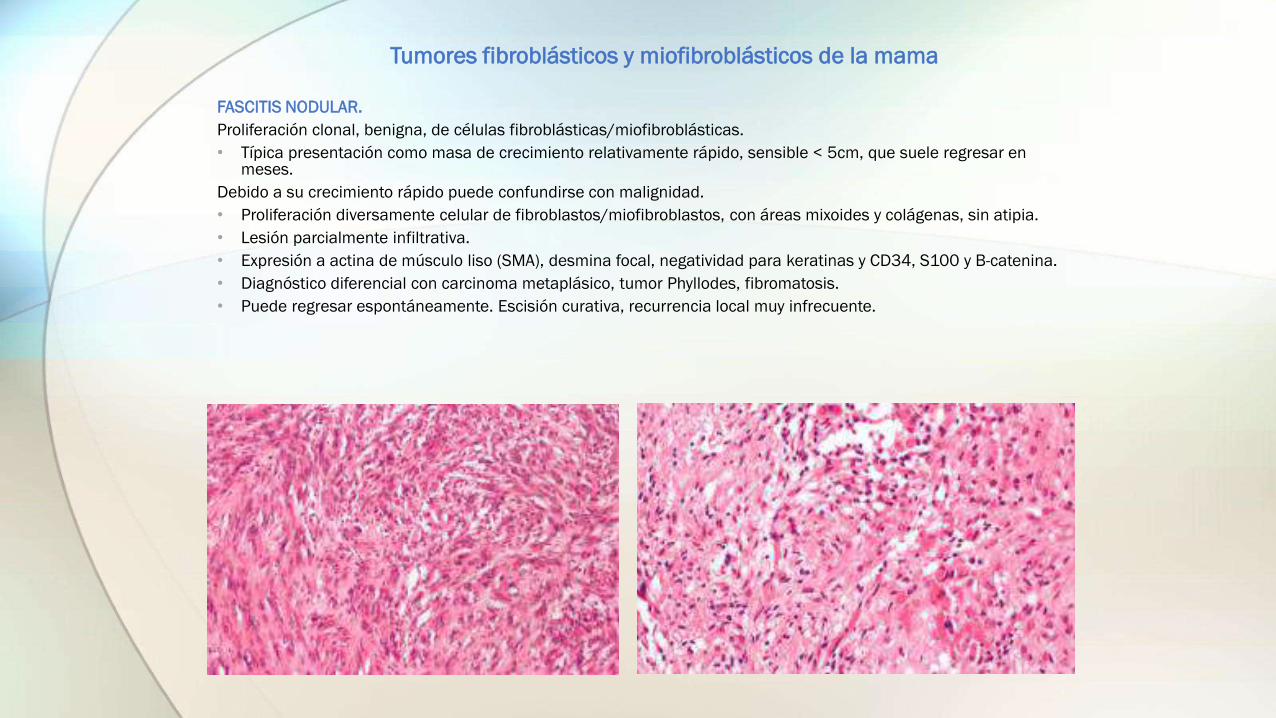
FASCITIS NODULAR.
Proliferación clonal, benigna, de células fibroblásticas/miofibroblásticas.
• Típica presentación como masa de crecimiento relativamente rápido, sensible < 5cm, que suele regresar en meses.
Debido a su crecimiento rápido puede confundirse con malignidad.
• Proliferación diversamente celular de fibroblastos/miofibroblastos, con áreas mixoides y colágenas, sin atipia.
• Lesión parcialmente infiltrativa.
• Expresión a actina de músculo liso (SMA), desmina focal, negatividad para keratinas y CD34, S100 y B-catenina.
• Diagnóstico diferencial con carcinoma metaplásico, tumor Phyllodes, fibromatosis.
• Puede regresar espontáneamente. Escisión curativa, recurrencia local muy infrecuente.
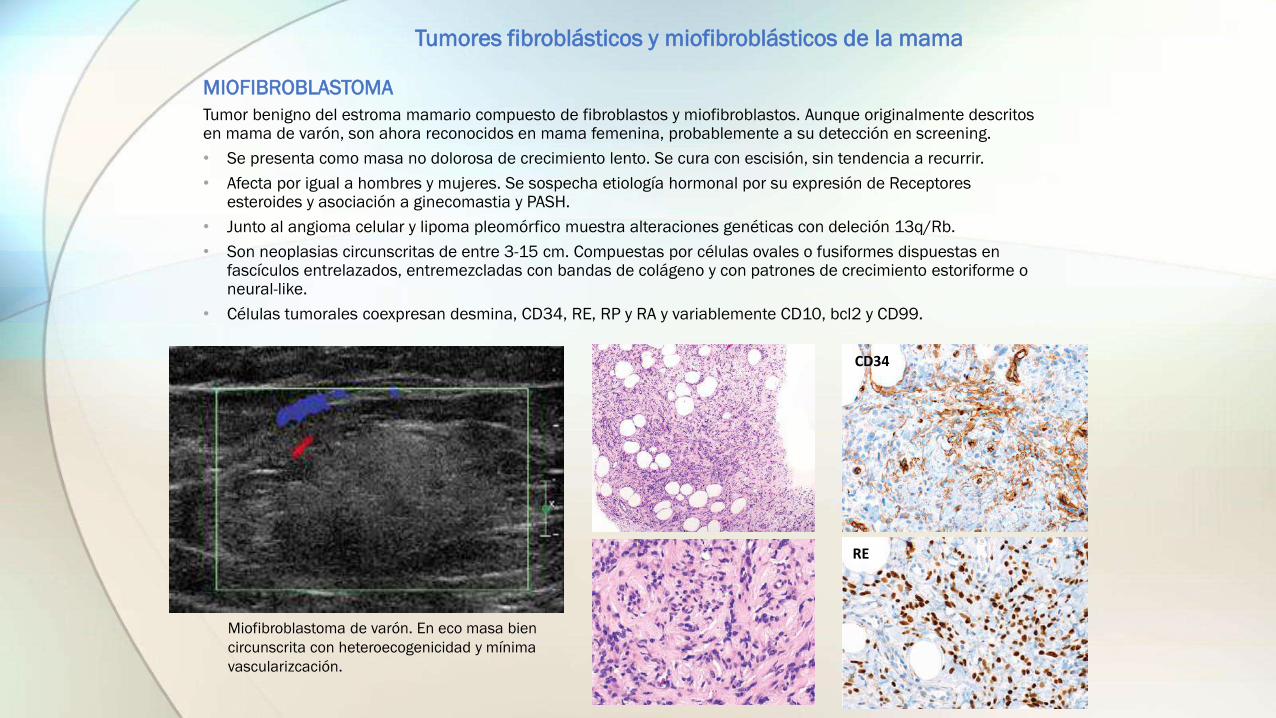
MIOFIBROBLASTOMA
Tumor benigno del estroma mamario compuesto de fibroblastos y miofibroblastos. Aunque originalmente descritos en mama de varón, son ahora reconocidos en mama femenina, probablemente a su detección en screening.
• Se presenta como masa no dolorosa de crecimiento lento. Se cura con escisión, sin tendencia a recurrir.
• Afecta por igual a hombres y mujeres. Se sospecha etiología hormonal por su expresión de Receptores esteroides y asociación a ginecomastia y PASH.
• Junto al angioma celular y lipoma pleomórfico muestra alteraciones genéticas con deleción 13q/Rb.
• Son neoplasias circunscritas de entre 3-15 cm. Compuestas por células ovales o fusiformes dispuestas en fascículos entrelazados, entremezcladas con bandas de colágeno y con patrones de crecimiento estoriforme o neural-like.
• Células tumorales coexpresan desmina, CD34, RE, RP y RA y variablemente CD10, bcl2 y CD99.
Tumores fibroblásticos y miofibroblásticos de la mama
Miofibroblastoma de varón. En eco masa bien
circunscrita con heteroecogenicidad y mínima
vascularizcación.
CD34
RE
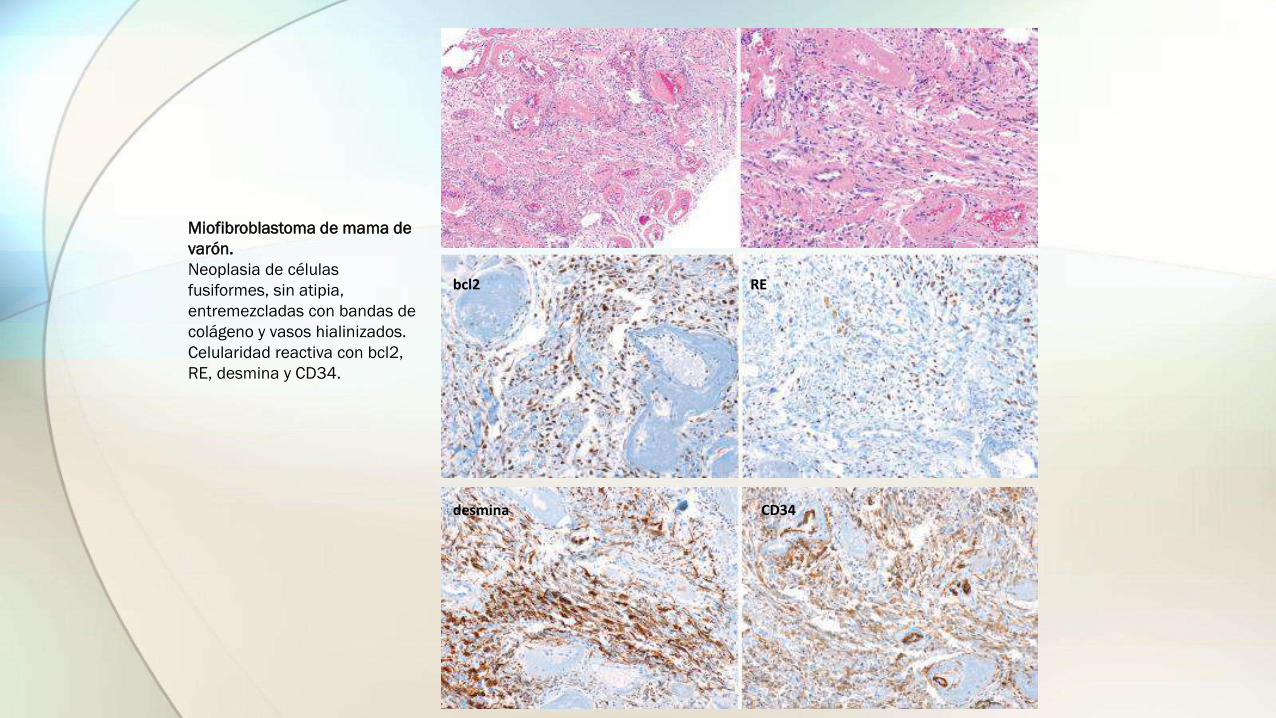
Miofibroblastoma de mama de
varón.
Neoplasia de células
fusiformes, sin atipia,
entremezcladas con bandas de
colágeno y vasos hialinizados.
Celularidad reactiva con bcl2,
RE, desmina y CD34.
bcl2 RE
desmina CD34
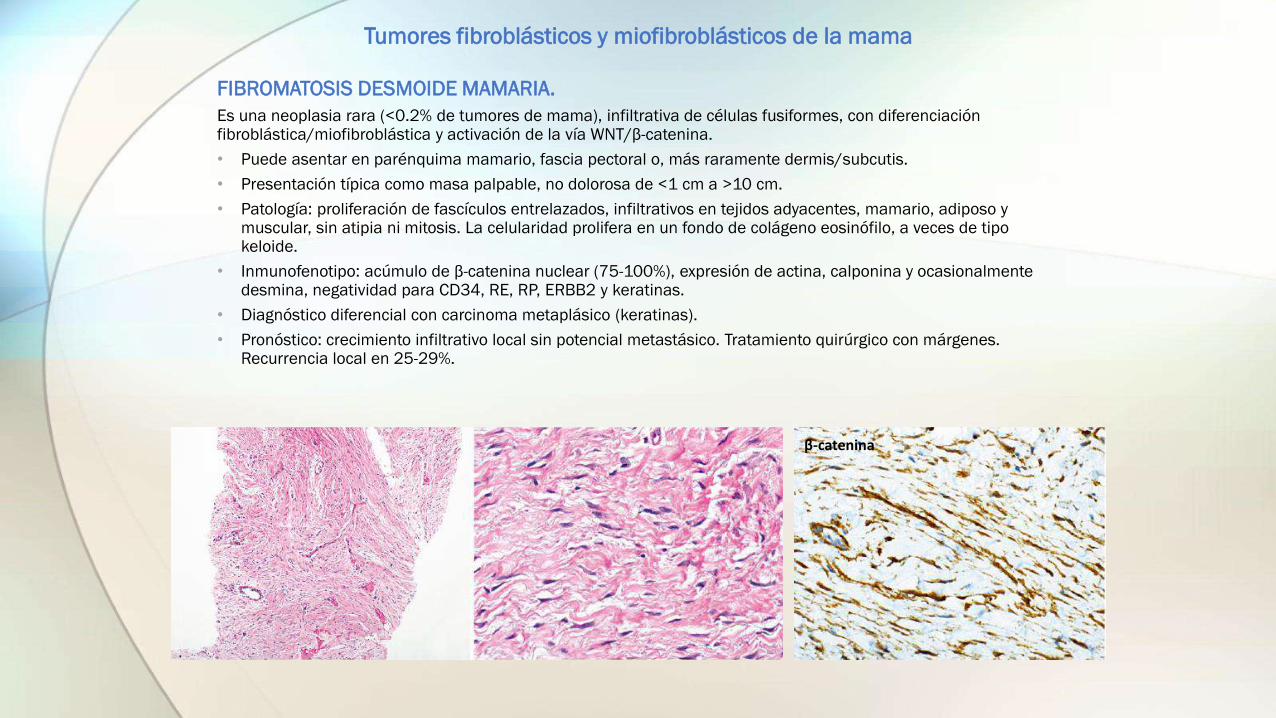
FIBROMATOSIS DESMOIDE MAMARIA.
Es una neoplasia rara (<0.2% de tumores de mama), infiltrativa de células fusiformes, con diferenciación fibroblástica/miofibroblástica y activación de la vía WNT/β-catenina.
• Puede asentar en parénquima mamario, fascia pectoral o, más raramente dermis/subcutis.
• Presentación típica como masa palpable, no dolorosa de <1 cm a >10 cm.
• Patología: proliferación de fascículos entrelazados, infiltrativos en tejidos adyacentes, mamario, adiposo y muscular, sin atipia ni mitosis. La celularidad prolifera en un fondo de colágeno eosinófilo, a veces de tipo keloide.
• Inmunofenotipo: acúmulo de β-catenina nuclear (75-100%), expresión de actina, calponina y ocasionalmente desmina, negatividad para CD34, RE, RP, ERBB2 y keratinas.
• Diagnóstico diferencial con carcinoma metaplásico (keratinas).
• Pronóstico: crecimiento infiltrativo local sin potencial metastásico. Tratamiento quirúrgico con márgenes. Recurrencia local en 25-29%.
Tumores fibroblásticos y miofibroblásticos de la mama
β-catenina
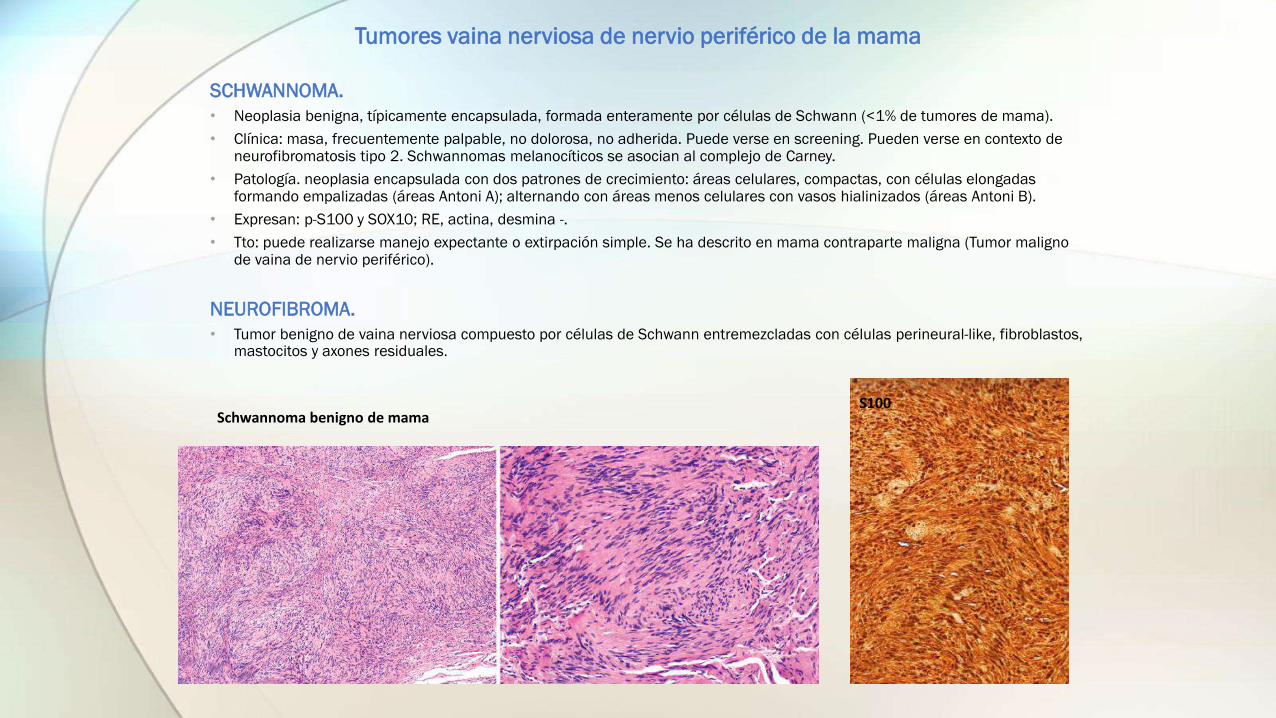
SCHWANNOMA.
• Neoplasia benigna, típicamente encapsulada, formada enteramente por células de Schwann (<1% de tumores de mama).
• Clínica: masa, frecuentemente palpable, no dolorosa, no adherida. Puede verse en screening. Pueden verse en contexto de neurofibromatosis tipo 2. Schwannomas melanocíticos se asocian al complejo de Carney.
• Patología. neoplasia encapsulada con dos patrones de crecimiento: áreas celulares, compactas, con células elongadas formando empalizadas (áreas Antoni A); alternando con áreas menos celulares con vasos hialinizados (áreas Antoni B).
• Expresan: p-S100 y SOX10; RE, actina, desmina -.
• Tto: puede realizarse manejo expectante o extirpación simple. Se ha descrito en mama contraparte maligna (Tumor maligno de vaina de nervio periférico).
NEUROFIBROMA.
• Tumor benigno de vaina nerviosa compuesto por células de Schwann entremezcladas con células perineural-like, fibroblastos, mastocitos y axones residuales.
Tumores vaina nerviosa de nervio periférico de la mama
Schwannoma benigno de mamaS100
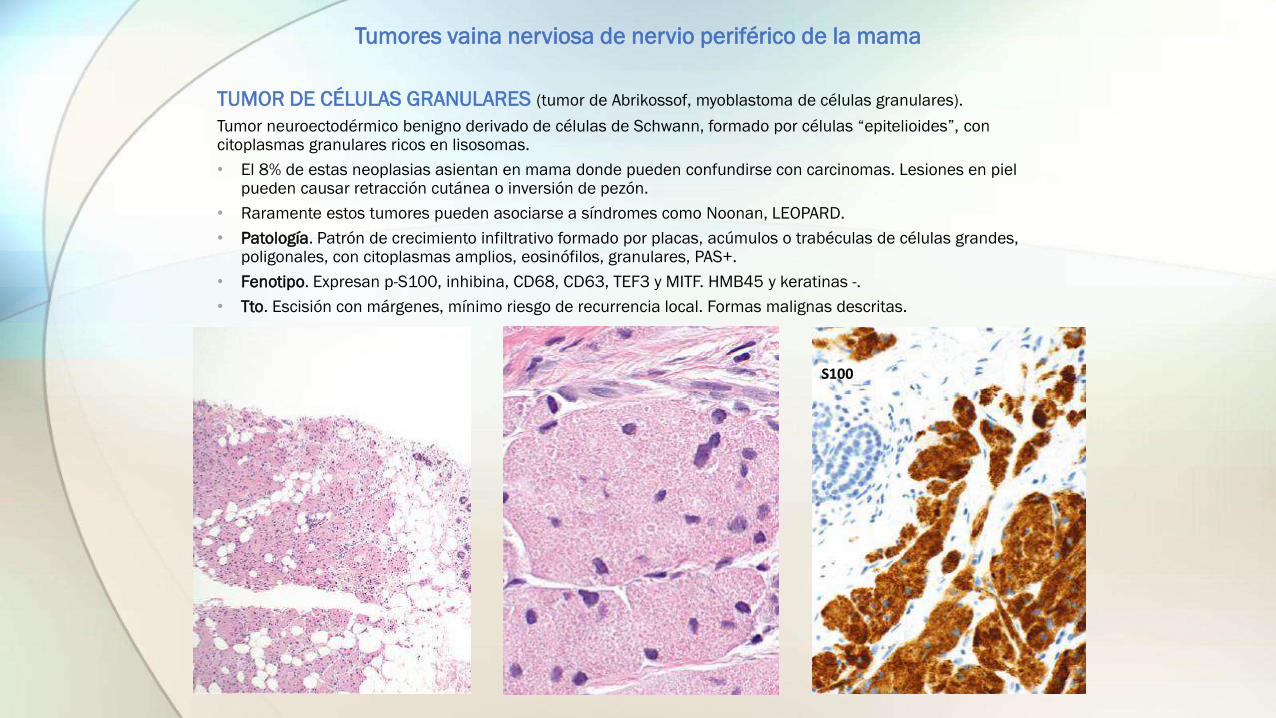
TUMOR DE CÉLULAS GRANULARES (tumor de Abrikossof, myoblastoma de células granulares).
Tumor neuroectodérmico benigno derivado de células de Schwann, formado por células “epitelioides”, con citoplasmas granulares ricos en lisosomas.
• El 8% de estas neoplasias asientan en mama donde pueden confundirse con carcinomas. Lesiones en piel pueden causar retracción cutánea o inversión de pezón.
• Raramente estos tumores pueden asociarse a síndromes como Noonan, LEOPARD.
• Patología. Patrón de crecimiento infiltrativo formado por placas, acúmulos o trabéculas de células grandes, poligonales, con citoplasmas amplios, eosinófilos, granulares, PAS+.
• Fenotipo. Expresan p-S100, inhibina, CD68, CD63, TEF3 y MITF. HMB45 y keratinas -.
• Tto. Escisión con márgenes, mínimo riesgo de recurrencia local. Formas malignas descritas.
Tumores vaina nerviosa de nervio periférico de la mama
S100
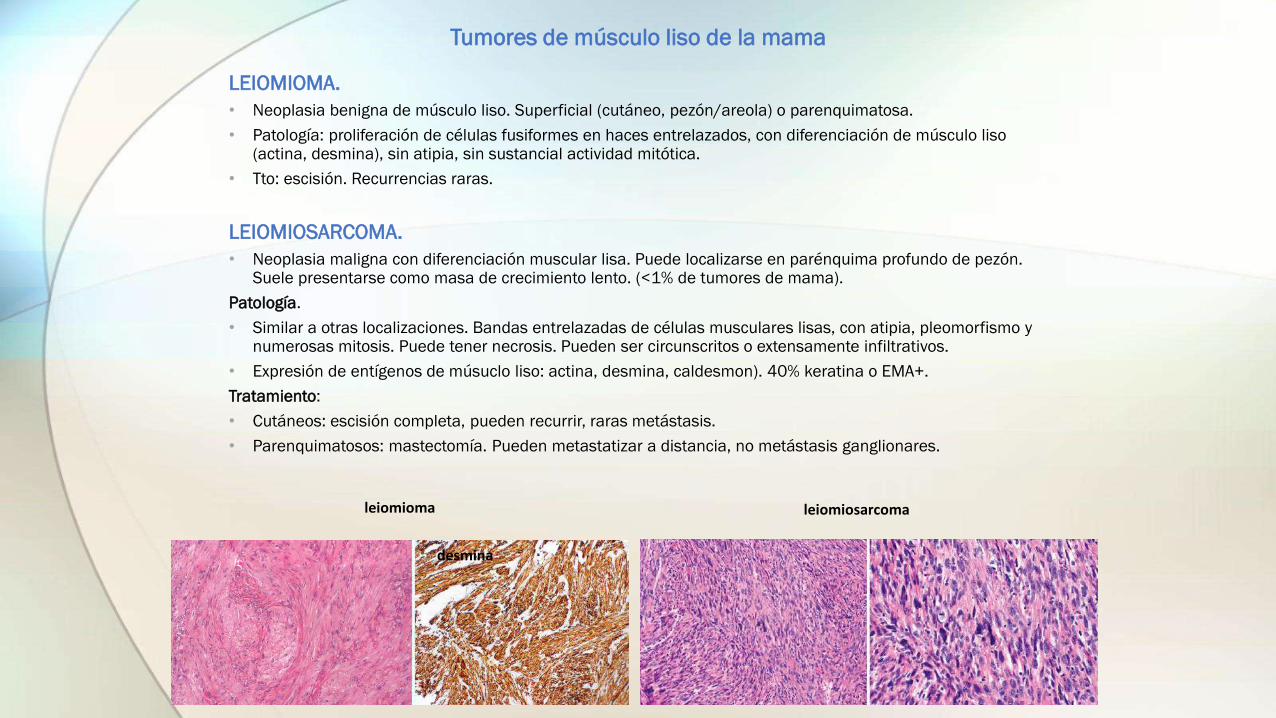
Tumores de músculo liso de la mama
LEIOMIOMA.
• Neoplasia benigna de músculo liso. Superficial (cutáneo, pezón/areola) o parenquimatosa.
• Patología: proliferación de células fusiformes en haces entrelazados, con diferenciación de músculo liso (actina, desmina), sin atipia, sin sustancial actividad mitótica.
• Tto: escisión. Recurrencias raras.
LEIOMIOSARCOMA.
• Neoplasia maligna con diferenciación muscular lisa. Puede localizarse en parénquima profundo de pezón. Suele presentarse como masa de crecimiento lento. (<1% de tumores de mama).
Patología.
• Similar a otras localizaciones. Bandas entrelazadas de células musculares lisas, con atipia, pleomorfismo y numerosas mitosis. Puede tener necrosis. Pueden ser circunscritos o extensamente infiltrativos.
• Expresión de entígenos de músuclo liso: actina, desmina, caldesmon). 40% keratina o EMA+.
Tratamiento:
• Cutáneos: escisión completa, pueden recurrir, raras metástasis.
• Parenquimatosos: mastectomía. Pueden metastatizar a distancia, no metástasis ganglionares.
leiomioma leiomiosarcoma
desmina
Tumores adipocíticos de la mama
LIPOMA.
• Neoplasia benigna de adipocitos maduros, sin atipia. Generalmente son masas asintomáticas.
ANGIOLIPOMA.
• Neoplasia benigna de grasa madura con grupos de vasos, que pueden contener microtrombos hialinos.
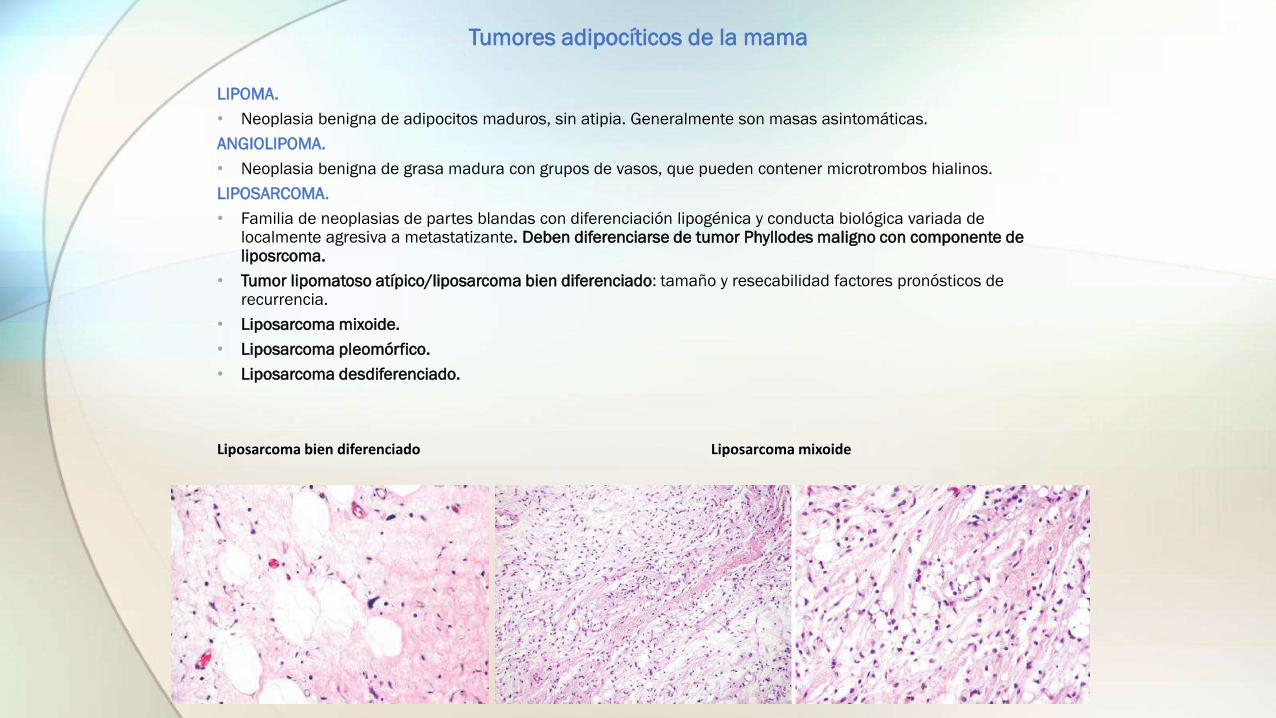
LIPOSARCOMA.
• Familia de neoplasias de partes blandas con diferenciación lipogénica y conducta biológica variada de localmente agresiva a metastatizante. Deben diferenciarse de tumor Phyllodes maligno con componente de liposrcoma.
• Tumor lipomatoso atípico/liposarcoma bien diferenciado: tamaño y resecabilidad factores pronósticos de recurrencia.
• Liposarcoma mixoide.
• Liposarcoma pleomórfico.
• Liposarcoma desdiferenciado.
Liposarcoma bien diferenciado Liposarcoma mixoide

Hiperplasia estromal seudoangiomatosa del estroma mamario (PASH)
Hiperplasia estromal seudoangiomatosa (PASH) es una proliferación miofibroblástica estroma, lineando espacios seudovasculares. Parece ser una lesión hormonal-inducida. Se puede ver en varones asociado a ginecomastia.
• Puede verse como hallazgo incidental en biopsias de mama por otras patologías (hasta en 23% en biopsias de rutina).
• Como masa palpable o visible en imagen. PASH nodular. Similar a un fibroadenoma macroscópicamente.
• Asociado a otras lesiones: hamartoma, fibroadenoma, Y. Phyllodes.
Patología.
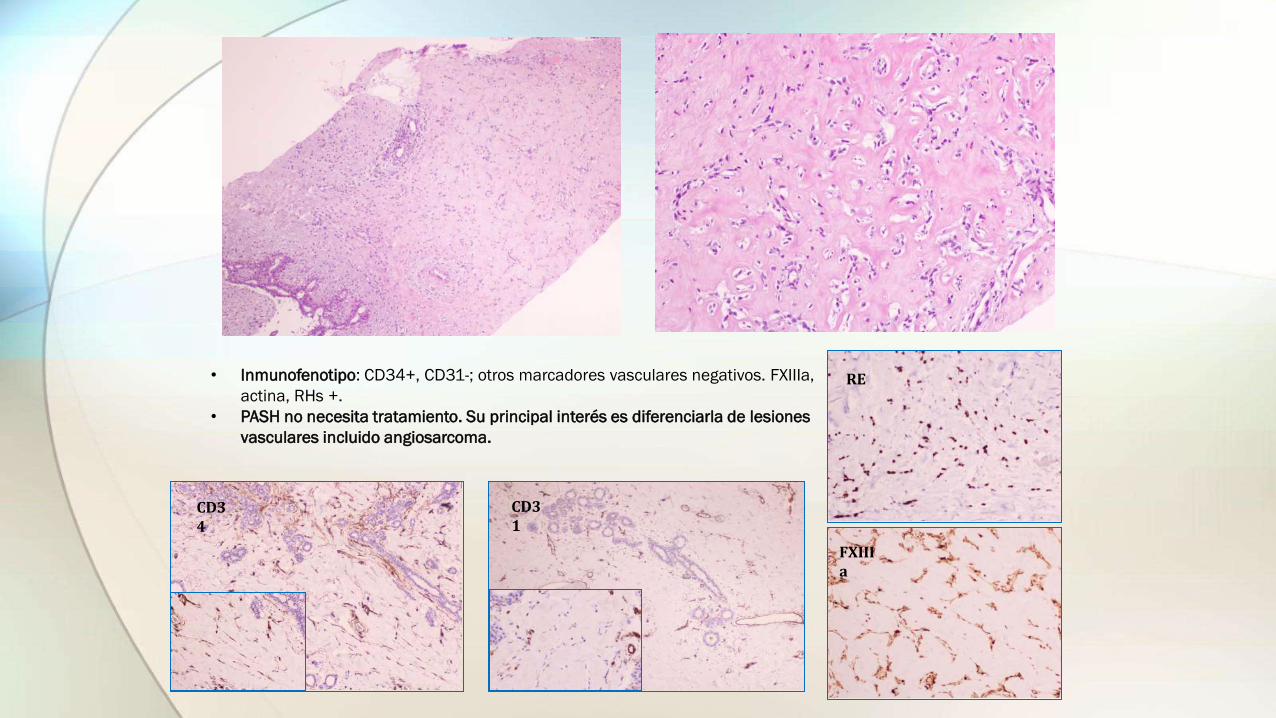
• Estroma colágeno con canales en “hendidura”, sin células sanguíneas, lineados por células fusiformes que pueden parecer células endoteliales de hendiduras vasculares.
• Las células fusiformes (miofibroblastos), no tienen atipia, ni mitosis. Celularidad variable.
CD34
CD31
RE
FXIIIa
• Inmunofenotipo: CD34+, CD31-; otros marcadores vasculares negativos. FXIIIa,
actina, RHs +.
• PASH no necesita tratamiento. Su principal interés es diferenciarla de lesiones
vasculares incluido angiosarcoma.


Las neoplasias hematopoyéticas y linfoides de la mama son muy raras. Linfomas no-Hodgkin (LNH) constituyen el subtipo más frecuente y suponen aproximadamente el 2% de LNH extranodales y <1% de todos los LNH.
El diagnóstico actual de proliferaciones hematolinfoides se basa en un análisis multiparamétrico de alteraciones:
• Clínica: estatus inmune del paciente, distribución y apariencia de las alteraciones clínicas.
• Morfocitología: arquitectura del infiltrado, formación de masa, infiltración destructiva y tipo celular.
• Análisis fenotípico: expresión de moléculas de diferenciación hematopoyética y otras que permiten delinear el tipo celular proliferante y cuyo estudio es fundamental para el diagnóstico y tipaje específico de la proliferación.
• Estudios moleculares-genéticos: permiten establecer la clonalidad del infiltrado maligno, delimitar el tipo histológico de la neoplasia y estudiar la expresión o anomalías de genes fundamentales para el diagnóstico, pronóstico o respuesta a terapias dirigidas.
Neoplasias hematolinfoides de la mama
Linfoma no hodgkin marginal extranodal (MALT) de mama,
diagnóstico en BAG
La aproximación multiparamétrica al diagnóstico de
neoplasias hematolinfoides permite este diagnóstico
en muestras de Biopsia con Aguja Gruesa (BAG), en
la mayoría de casos. El material permite estudios de:
• Morfología.
• Inmunocitoquímica.
• Citometría de flujo.
• Estudios moleculares y citogenéticos.

Definición: linfoma confinado a una o ambas mamas y/o ganglios regionales, en ausencia de historia previa de linfoma. Una definición menos rigurosa incluiría casos con masa dominante o síntomas a la presentación en mama, en paciente sin linfoma conocido.
• Raro. <0,5% de todas las neoplasias malignas de mama. 2% de linfomas extranodales.
• La mayoría en pacientes de mediana edad. Jóvenes y lactantes pueden también ser afectadas. 11% bilaterales.
Clínica: masas mamarias no dolorosas, generalmente sin síntomas constitucionales. Afectación cutánea rara.
Tipos histológicos más frecuentes corresponden a LNH-B.
• Linfoma difuso de célula grande B (LDCG-B) 56-85% de casos (60% aprox.).
• Linfoma de zona marginal extranodal (linfoma MALT) 10-30%.
• Linfoma folicular (LF) 10-20%.
Neoplasias hematolinfoides de la mamaLinfoma primario de mama.
Linfoma difuso de célula grande B. diagnóstico en BAG de
mama
Grupos de neoplasias hematolinfoides (OMS 2016).
• Neoplasias de precursores linfoides:
• Linfoma/leucemia linfoblástica B
• Linfoma/leucemia linfoblástico T y NK.
• Neoplasias de células B maduras.
• Neoplasias de células T y Natural Killer (NK).
• Linfoma de Hodgkin.
• Trastornos linfoproliferativos asociados a
inmunodeficiencia.
• Neoplasias histiocíticas y de células dendríticas.
• Leucemias mieloides/neoplasias mieloides y sarcoma
mieloide.

Linfoma de zona marginal extranodal de tejido linfoide asociado a mucosas (linfoma MALT).
LNH-B extranodal de “bajo grado”, que recapitula rasgos morfofenotípicos del tejido linfoide asociado a mucosas. Compuesto de células linfoides pequeñas, incluyendo células de zona marginal.
• 7-8% de linfomas B, 2% asentando en mama.
Clínica: masa detectada en examen físico o mamografía. No síntomas constitucionales.
Patología.
• Similar a linfomas MALT de otras localizaciones. Recapitula formaciones linfoides de mucosas como placas de Peyer intestinales: folículos linfoides con zonas marginales expandidas y proliferación de linfocitos pequeños B.
• Fenómenos asociados: colonización folicular, lesión linfoepitelial, diferenciación plasmacítica con/sin Igsclonales, deposición de material amiloide.
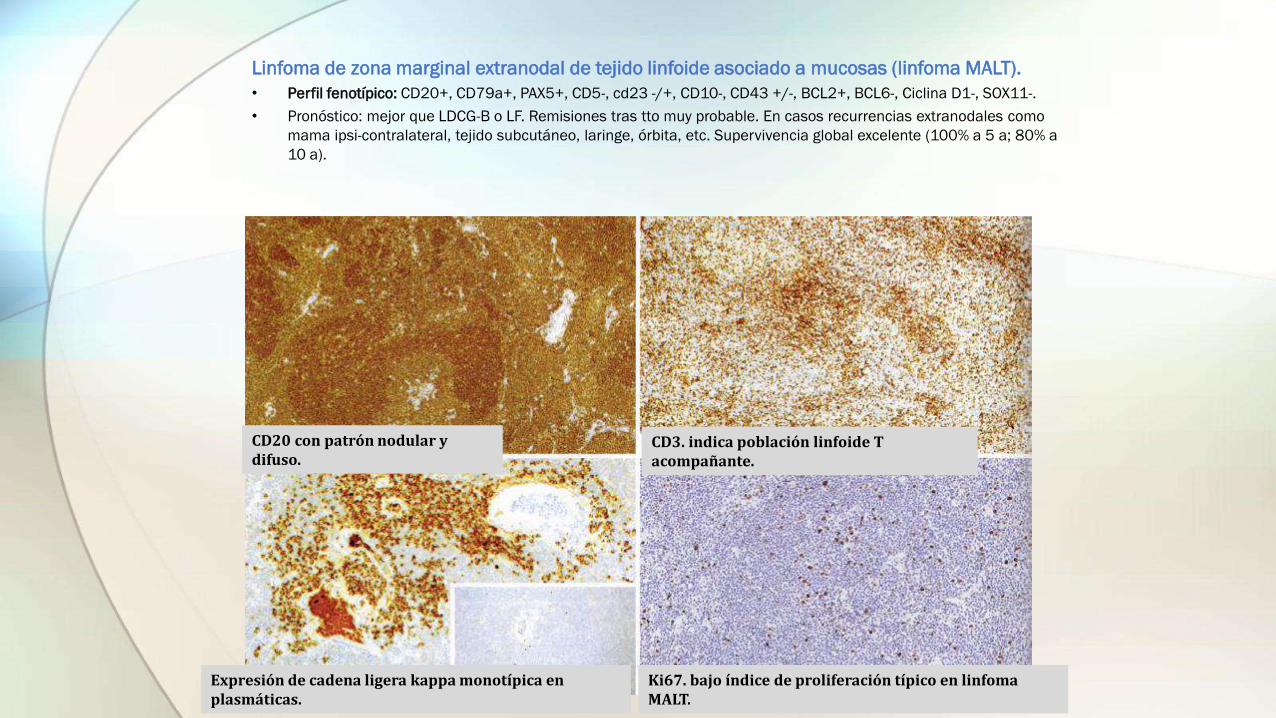
• Perfil fenotípico: CD20+, CD79a+, PAX5+, CD5-, cd23 -/+, CD10-, CD43 +/-, BCL2+, BCL6-, Ciclina D1-, SOX11-.
Neoplasias hematolinfoides de la mama
Linfoma de zona marginal extranodal de tejido linfoide asociado a mucosas (linfoma MALT).
• Perfil fenotípico: CD20+, CD79a+, PAX5+, CD5-, cd23 -/+, CD10-, CD43 +/-, BCL2+, BCL6-, Ciclina D1-, SOX11-.
• Pronóstico: mejor que LDCG-B o LF. Remisiones tras tto muy probable. En casos recurrencias extranodales como
mama ipsi-contralateral, tejido subcutáneo, laringe, órbita, etc. Supervivencia global excelente (100% a 5 a; 80% a
10 a).
CD20 con patrón nodular y difuso.
CD3. indica población linfoide T acompañante.
Expresión de cadena ligera kappa monotípica en plasmáticas.
Ki67. bajo índice de proliferación típico en linfoma MALT.
Neoplasias hematolinfoides de la mama
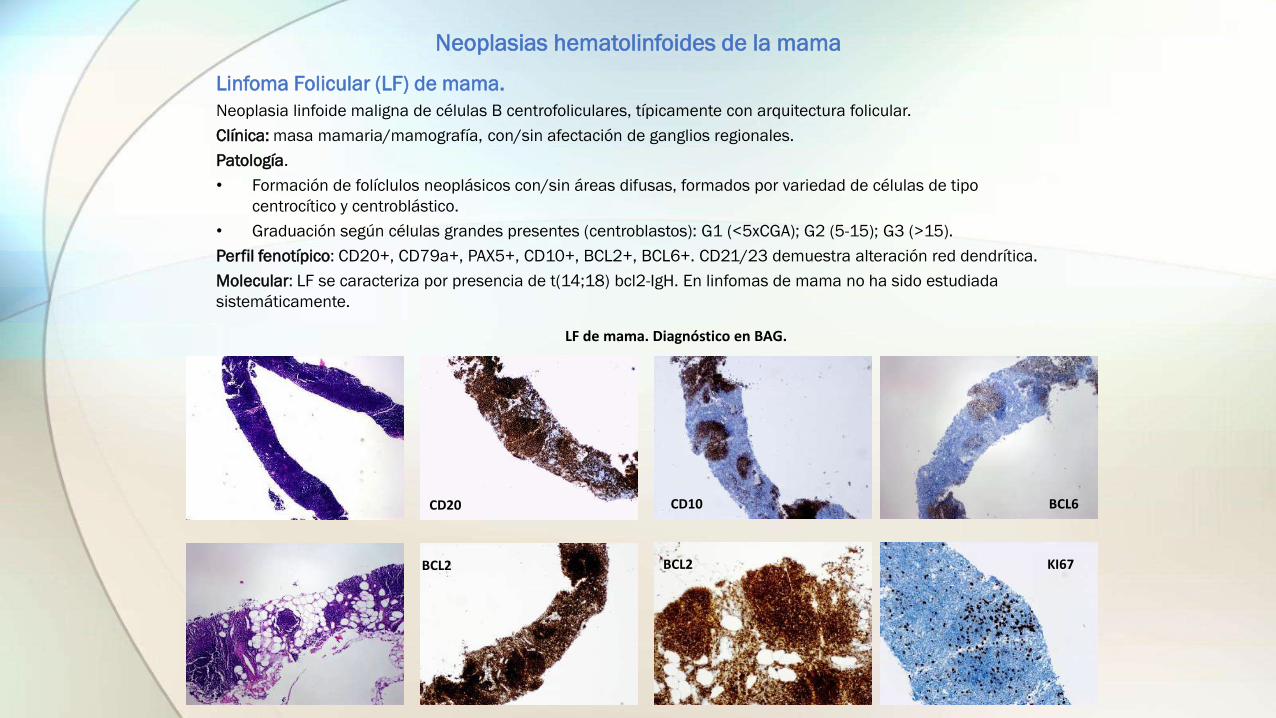
Linfoma Folicular (LF) de mama.
Neoplasia linfoide maligna de células B centrofoliculares, típicamente con arquitectura folicular.
Clínica: masa mamaria/mamografía, con/sin afectación de ganglios regionales.
Patología.
• Formación de folíclulos neoplásicos con/sin áreas difusas, formados por variedad de células de tipo
centrocítico y centroblástico.
• Graduación según células grandes presentes (centroblastos): G1 (<5xCGA); G2 (5-15); G3 (>15).
Perfil fenotípico: CD20+, CD79a+, PAX5+, CD10+, BCL2+, BCL6+. CD21/23 demuestra alteración red dendrítica.
Molecular: LF se caracteriza por presencia de t(14;18) bcl2-IgH. En linfomas de mama no ha sido estudiada
sistemáticamente.
CD20 CD10
BCL2 BCL2 KI67
BCL6
LF de mama. Diagnóstico en BAG.

Neoplasias hematolinfoides de la mama
Linfoma Difuso de célula grande B (LDCG-B) de la mama.
LDCG-B es una proliferación neoplásica difusa de linfocitos grandes B. Es el tipo más frecuente de linfoma afectando a
mama. La afectación puede ser primaria o secundaria a proceso sistémico, con ratio 1:1.
Clínica: masa mamaria/mamografía de screening (10%), con/sin afectación de ganglios regionales. Puede dar eritema
cutáneo, edema o retracción.
Patología.
• Proliferación de linfocitos B, grandes (centroblastos/inmunoblastos) con patrón de crecimiento difuso. Puede afectar
lobulillos dando impresión de nódulos.
Perfil fenotípico: CD19+, CD20+, CD79a+, PAX5+, BCL2+/-, CD10+/-, BCL6+/-, MUM1+/-. Alto índice de proliferación.
• Subclasificación tiene importancia pronóstica y terapéutica: se realiza en función de expresión de CD10/bcl6 (Centro
germinal-like) o MUM1 (células activada-ABC).
CD20
KI67

Neoplasias hematolinfoides de la mama
Linfoma de Burkitt.Linfoma de células B maduras, altamente agresivo, caracterizado por frecuente presentación extranodal o como leucemia aguda, con muy alta actividad proliferativa y, generalmente traslocación MYC.
Clínica. Presentación con masa voluminosa, de muy rápido crecimiento, frecuentemente bilateral, puede relacionarse con la pubertad, embarazo o lactancia.
Epidemiología: 3 tipos epidemiológicos. 1. endémico; 2. esporádico. 3. asociado a inmunodeficiencia-HIV.
• En >90% de casos se demuestra integración del virus de Epstein-Barr (EBV).
Patología.
• Proliferación monótona, difusa, de linfocitos de tamaño mediano, con muy alta actividad mitótica y apoptosis.
• Macrófagos fagocitando restos celulares entremezclados dando apariencia “en cielo estrellado”.
Inmunofenotipo: marcadores B + (CD20, CD19, CD79a, PAX5), marcadores centro-germinal positivos (CD10+, BCL6+), CD43+. CD5-, CD23-, BCL2-, TdT-. Ki67 100%.
Molecular: t(8;14)MYC-IgH. Demostración de EBV.
CD20 bcl6
CD10 bcl2 Ki67
Neoplasias hematolinfoides de la mama
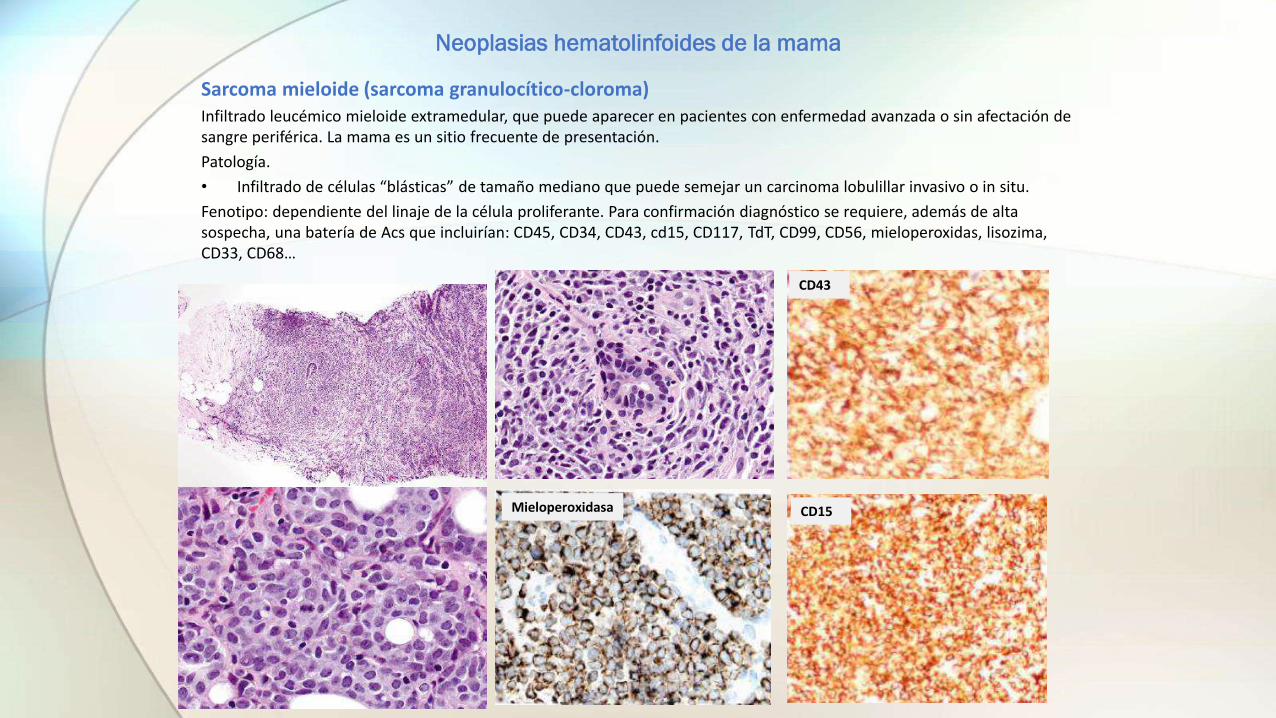
Sarcoma mieloide (sarcoma granulocítico-cloroma)Infiltrado leucémico mieloide extramedular, que puede aparecer en pacientes con enfermedad avanzada o sin afectación de sangre periférica. La mama es un sitio frecuente de presentación.
Patología.
• Infiltrado de células “blásticas” de tamaño mediano que puede semejar un carcinoma lobulillar invasivo o in situ.
Fenotipo: dependiente del linaje de la célula proliferante. Para confirmación diagnóstico se requiere, además de alta sospecha, una batería de Acs que incluirían: CD45, CD34, CD43, cd15, CD117, TdT, CD99, CD56, mieloperoxidas, lisozima, CD33, CD68…
CD43
CD15Mieloperoxidasa

Linfoma anaplásico de célula grande asociado a implante mamario.
Linfoma anaplásico de célula grande asociado a implante (prótesis) mamario (Breast-Implant-Asociated; BIA-ALCL) es un linfoma T con morfología e inmunofenotipo similar a linfomas anaplásicos CD30+, ALK-negativos que tiene como localización única, especial, periimplante mamario, generalmente confinado a la cápsula fibrosa y un pronóstico excelente.
Localización.
• Este único y especial linfoma asienta en mamas con implantes. La mayoría de pacientes presentan efusión periimplante. Aproximadamente el 5% son bilaterales.
• 1/3 de casos se presentan con masa. 20% muestran adenopatías regionales, axila o supraclavicular.
Mujer. 29 años.
• Implantes mamarios bilaterales (2015).
• NAGOTEX® textured surface (referencia IMP-EHR 360).
• Implantes glúteos bilaterales.
Motivo de consulta
• Aumento de volumen mama izquierda.
• Dolor.
• Fiebre (Tª38ºC).
Exploración.
• Mama izquierda: aumento de tamaño, congestiva, blanda, dolor difuso.
• Mama derecha normal.
• Axilas: sin hallazgos.
Analítica: normal.

Ecografía mamaria.
• Implantes retropectorales.
• Líquido periprotésico izquierdo. Extracción de 250cc.
RNM.
• Colección periprotésica izq importante; dcha escasa.
• Cápsula periprotésica izquierda difusamente engrosada, irregular.
• No masas captantes ni adenopatías.
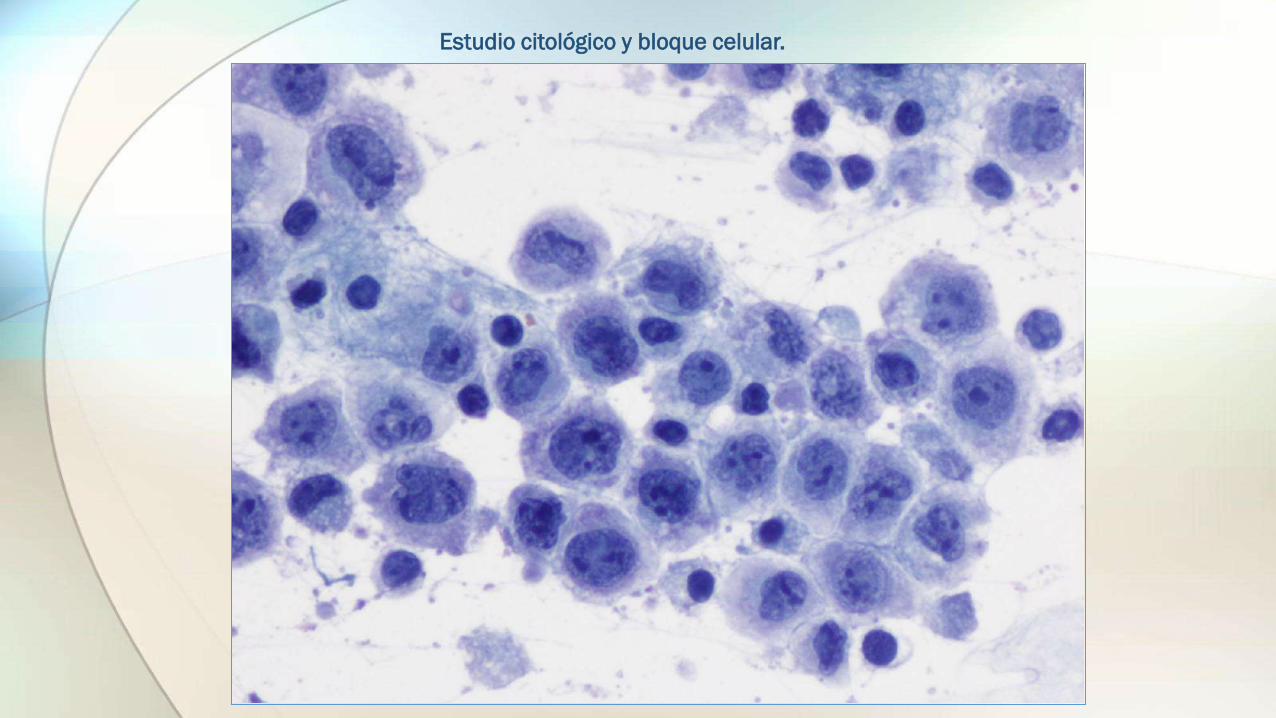
Estudio citológico y bloque celular.
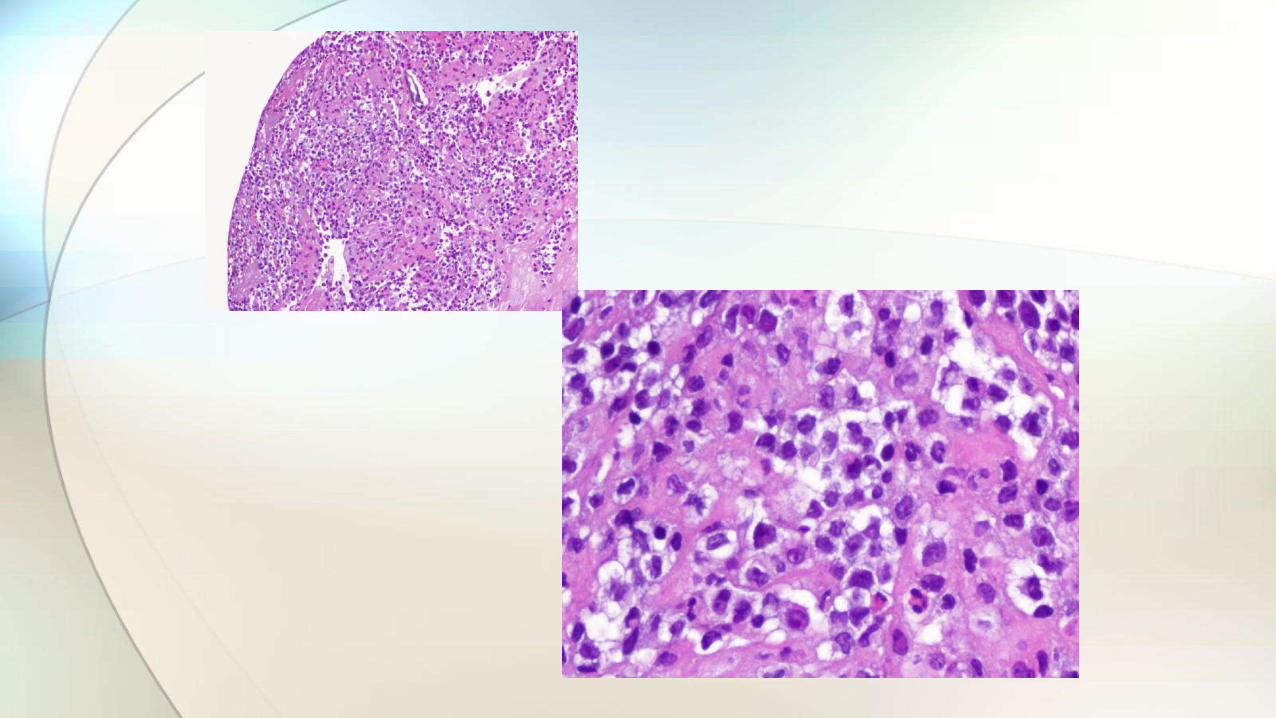
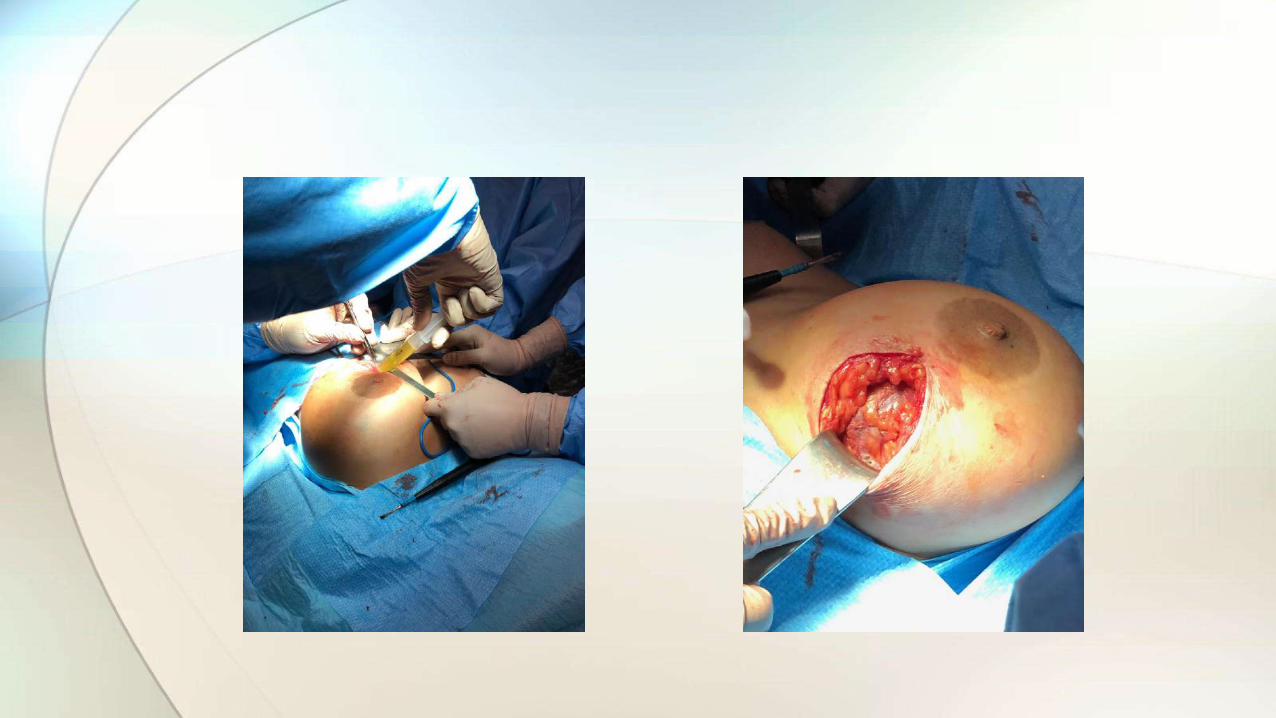
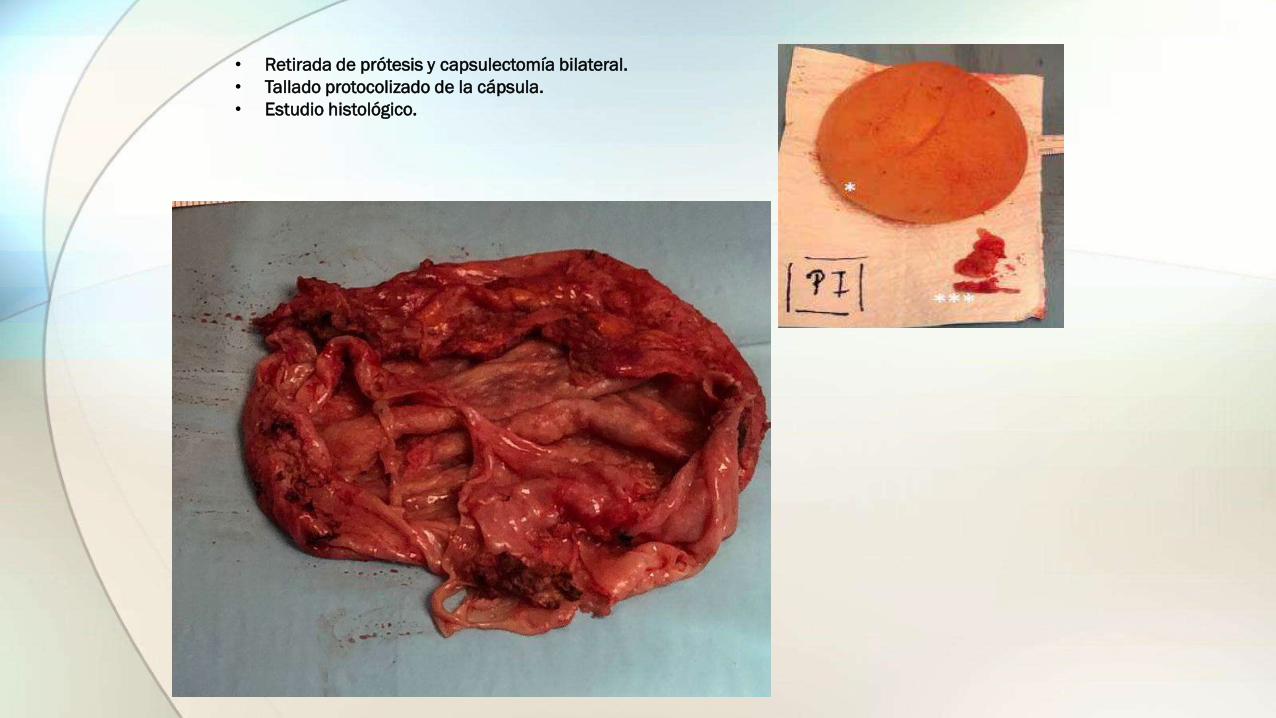
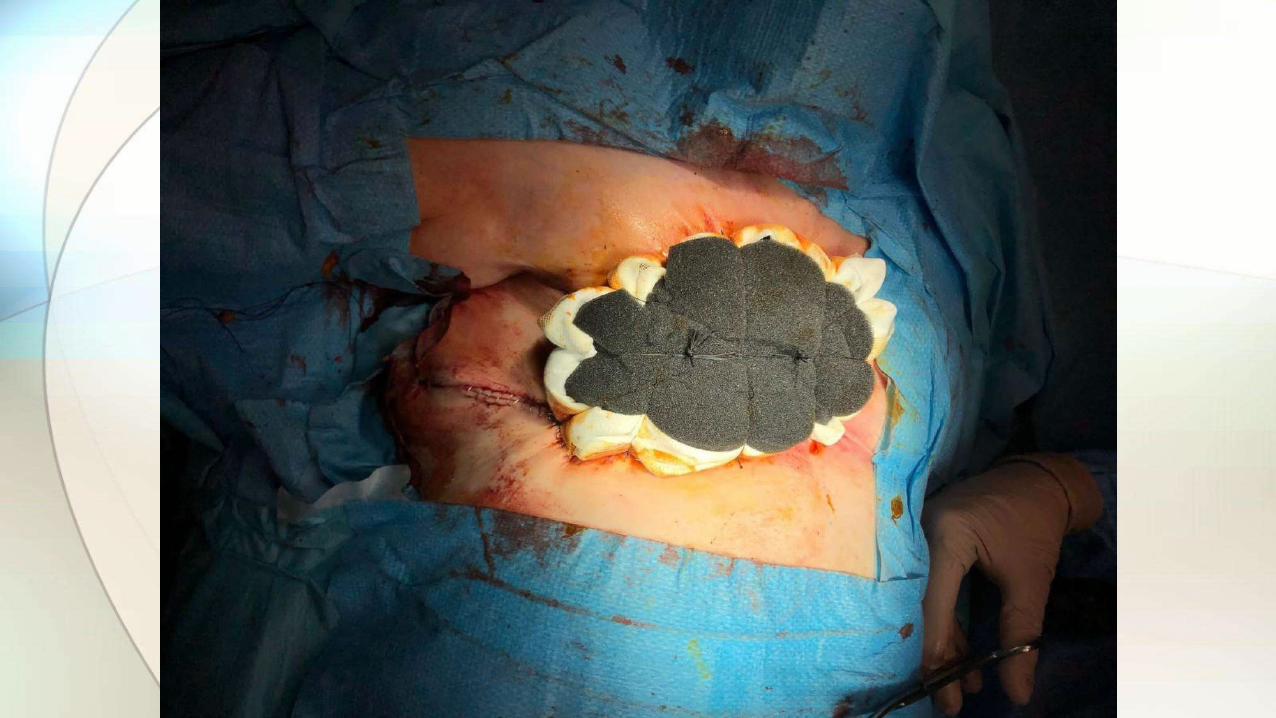
• Retirada de prótesis y capsulectomía bilateral.
• Tallado protocolizado de la cápsula.
• Estudio histológico.

BIA-ALCL es un tipo poco frecuente de linfoma T, asociado a cirugía de prótesis mamaria, con expresión característica de CD30, ALK- (Entidad provisional OMS 2016)
Incidencia estimada.
• 1-3 x millón de mujeres con implantes?.
• Actualmente > 700 casos comunicados en el mundo.
• Casi 400.000 nuevos implantes en USA por año.
2011: FDA declaración: “ no es posible confirmar con certeza estadística que implantes mamarios causen ALCL”.
2016. OMS IAB-ALCL reconocida como entidad provisional.
2017. FDA declaración: existe una relación entre implantes mamarios y ALCL. 414 casos reconocidos y 9 muertes atribuibles.
• Riesgo actualmente predicho por organizaciones de salud entre 1:3817 y 1:30000 implantes, 70x en implantes texturizados (Doren et al. 2017; FDA. 2018).
ESPAÑA
• Sistema de Vigilancia de Productos Sanitarios - AEMPS (última actualización: abril 2019)
• 36 sospechas de LACG.
• 26 casos confirmados.
• Notificación obligatoria.
• El número estimado de mujeres implantadas en España en los últimos 10 de años es de 500.000.
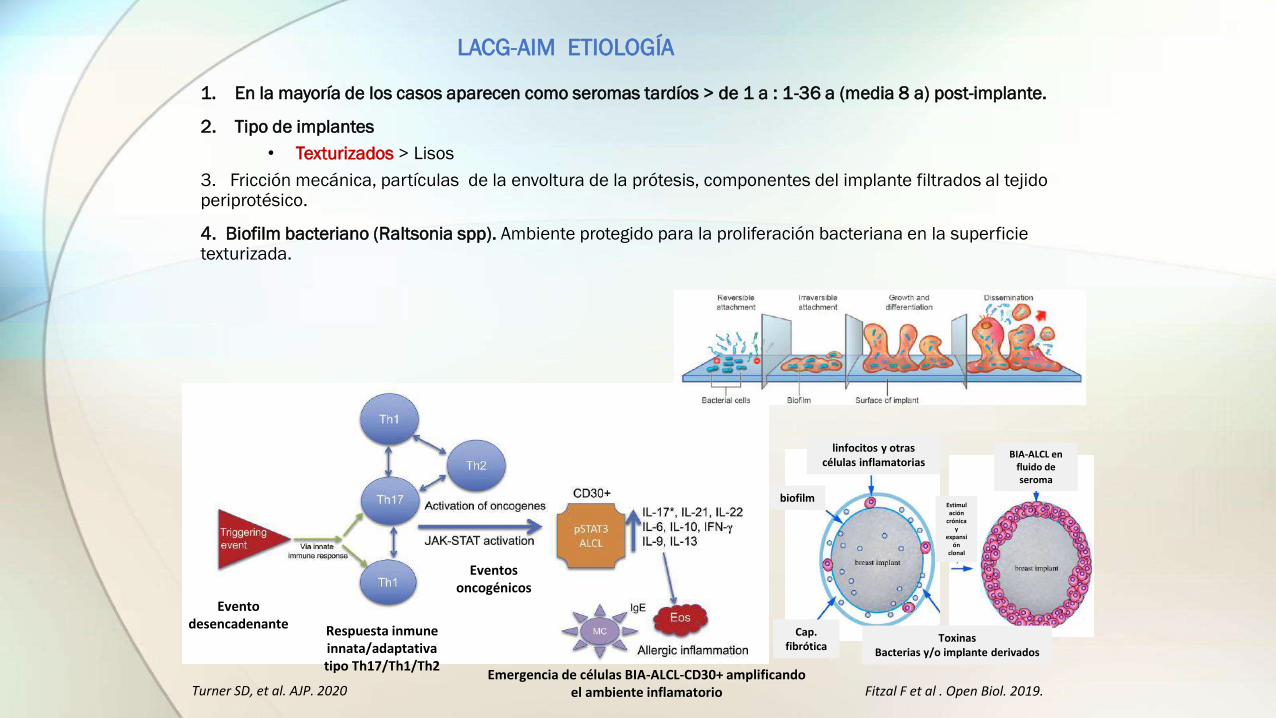
LACG-AIM ETIOLOGÍA
1. En la mayoría de los casos aparecen como seromas tardíos > de 1 a : 1-36 a (media 8 a) post-implante.
2. Tipo de implantes
• Texturizados > Lisos
3. Fricción mecánica, partículas de la envoltura de la prótesis, componentes del implante filtrados al tejido periprotésico.
4. Biofilm bacteriano (Raltsonia spp). Ambiente protegido para la proliferación bacteriana en la superficie texturizada.
Evento desencadenante Respuesta inmune
innata/adaptativa tipo Th17/Th1/Th2
Eventos oncogénicos
Emergencia de células BIA-ALCL-CD30+ amplificando el ambiente inflamatorioTurner SD, et al. AJP. 2020 Fitzal F et al . Open Biol. 2019.
linfocitos y otras células inflamatorias
biofilm
Cap. fibrótica
ToxinasBacterias y/o implante derivados
Estimulación
crónica y
expansión
clonal
BIA-ALCL en fluido de seroma

LACG-AIM Estadiaje
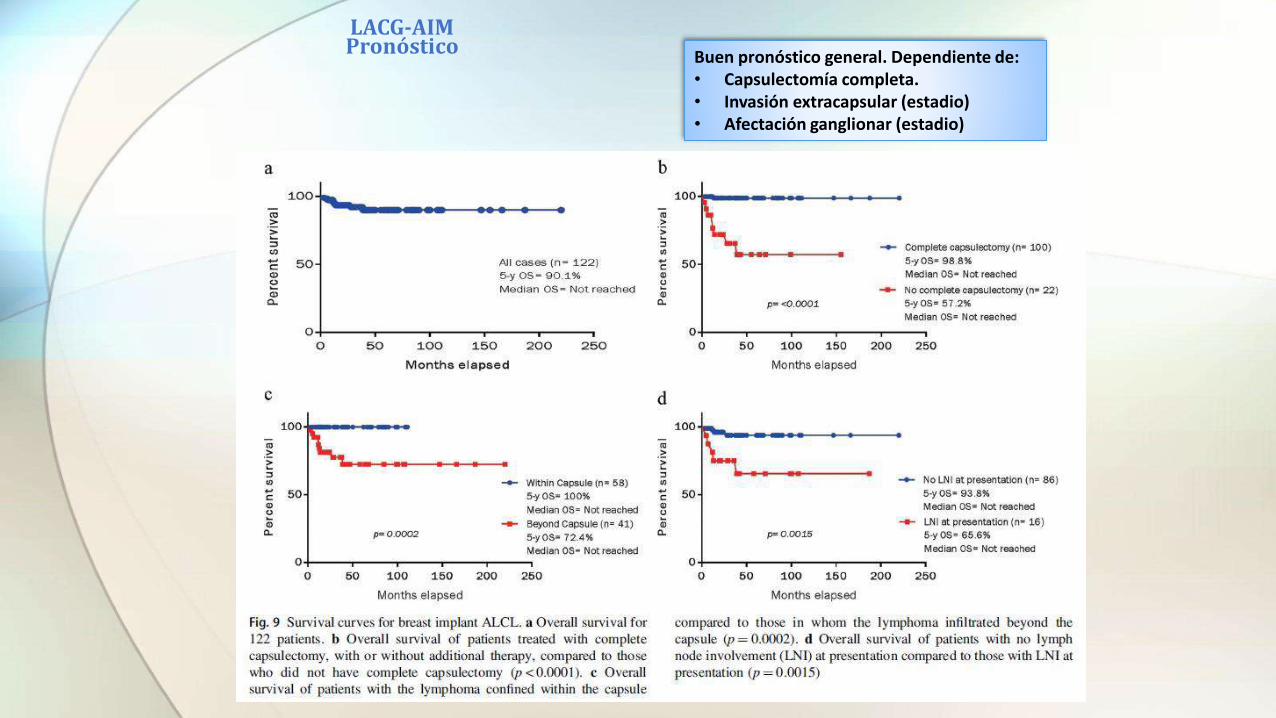
LACG-AIM Pronóstico Buen pronóstico general. Dependiente de:
• Capsulectomía completa.• Invasión extracapsular (estadio)• Afectación ganglionar (estadio)

LACG-AIM
TRATAMIENTO
CAPSULECTOMÍA TOTAL CON RESECCIÓN DE PRÓTESIS COMPLETA.• Valoración de resección total de prótesis contralateral.
+
OPCIÓN 1: ENFERMEDAD LOCALIZADA-ESTADIOS IA-B-C• Si capsulaectomía parcial o resección incompleta de prótesis, con enfermedad
residual: tto adyuvante (valorar radioterapia).
OPCIÓN 2: ESTADIOS IIA-B• Si capsulectomía parcial o resección incompleta, con enfermedad residual: tto
adyuvante con RT y/o QT sistémica (si afectación contralateral o contraindicación de RTx).
OPCIÓN 3: ESTADIOS III-IV.• Tto sistémico con inmuno/quimioterapia.• RTx como paliación (masas ulceradas, sangrantes o dolorosas).

NUESTRO CASO
EVOLUCIÓN
• Reordenamiento TCR+; reordenamientos IRF4/DUSP22 y TP63 -.
• T3N0, estadio IC.
• PET-TC al mes de la cirugía → Sin alteraciones (M0)
• No precisó tratamiento adicional.
• Asintomática, buena evolución (11 meses).
• Las guías clínicas de la NCCN recomiendan un seguimiento clínico y examen físico cada 3 a 6 meses durante 2 años.
• PET-TC cada 6 meses durante 2 años.







