
Informes Periódicos de Seguridad: procedimiento...
29
Informes Periódicos de Seguridad: procedimiento, elaboración y envío Laboratorios Maymó, S.A. Francisco Jaramillo 1-9-2015
Transcript of Informes Periódicos de Seguridad: procedimiento...

Informes Periódicos de Seguridad: procedimiento, elaboración y envío Laboratorios Maymó, S.A.
Francisco Jaramillo 1-9-2015

Informes Periódicos de Seguridad: procedimiento, elaboración y envío
pág. 1
Universitat Autònoma de Barcelona
Facultad de Medicina
Máster en Farmacología
Trabajo Fin de Máster
Título: Informes Periódicos de Seguridad: procedimiento, elaboración
y envío Descripción de las prácticas correspondiente al Trabajo de Fin de Máster en
Farmacología.
El trabajo se realizó en la empresa farmacéutica Laboratorios Maymó,
S.A., bajo la dirección de Doña Mª Dolores Cainzos.
Francisco Jaramillo María Dolores Cainzos Caridad Pontes
Alumno Tutor Empresa Tutor Académico
Barcelona, 1 de septiembre del 2015

Informes Periódicos de Seguridad: procedimiento, elaboración y envío
pág. 2
Contenido RESUMEN .......................................................................................................................................... 3
1. INTRODUCCIÓN .......................................................................................................................... 3
2. TECNICAS ...................................................................................................................................... 6
2.1 INFORMES PERIODICOS DE SEGURIDAD (IPS) ........................................................... 6
2.1.1 Definición ............................................................................................................................ 6
2.1.2 Contenido ............................................................................................................................ 7
2.1.3 Calendario .......................................................................................................................... 9
2.2 PROCEDIMIENTO NORMALIZADO DE TRABAJO (PNT) ......................................... 13
2.2.1 Definición .......................................................................................................................... 13
2.2.2 PNT de uso frecuente en Farmacovigilancia Veterinaria ............................................. 13
2.2.3 Descripción Detallada del Sistema de Farmacovigilancia (DDPS por sus siglas en
inglés) ......................................................................................................................................... 14
2.3 ESTUDIOS POST AUTORIZACIÓN .................................................................................. 15
2.4 BASE DE DATOS ................................................................................................................... 15
3. CONOCIMIENTOS Y HABLILIDADES ADQUIRIDAS ....................................................... 16
3.1 INFORMES PERIÓDICOS DE SEGURIDAD (IPS) ......................................................... 16
3.1.1 Elaboración del calendario de IPS ................................................................................. 16
3.1.2 Archivo de la documentación virtual y real relacionada con los IPS .......................... 17
3.1.3 Pago de tasas ..................................................................................................................... 17
3.1.4 Consideraciones a futuro sobre los IPS .......................................................................... 18
3.2 PROCEDIMIENTO NORMALIZADO DE TRABAJO (PNT) ......................................... 19
3.2.1 Actualización de PNT ...................................................................................................... 19
3.2.2 Descripción Detallada del Sistema de Farmacovigilancia (DDPS por sus siglas en
inglés) ......................................................................................................................................... 21
4. BIBLIOGRAFÍA .......................................................................................................................... 22
ANEXOS ............................................................................................................................................ 23
ANEXO 1: MODELO DE UN INFORME PERIÓDICO DE SEGURIDAD PARA UN
PRODUCTO Y TITULAR FICTICIO ....................................................................................... 23
ANEXO 2: TARJETA VERDE PARA NOTIFICACIÓN, POR PROFESIONALES
SANITARIOS, DE SOSPECHAS DE REACCIONES ADVERSAS A MEDICAMENTOS
SANITARIOS................................................................................................................................ 27

Informes Periódicos de Seguridad: procedimiento, elaboración y envío
pág. 3
RESUMEN
Durante las prácticas como parte de mi formación en el Master Oficial en Farmacología
de la Universitat Autónoma de Barcelona se me fue designado los Laboratorios Maymó.
Mi función ha sido colaborar con el departamento de Farmacovigilancia realizando
prototipos y modelos que simulen las situaciones que se pueden presentar en las
condiciones reales:
He colaborado en la redacción de algunos apartados que forman parte de los
modelos que simulan a los IPSs (Informe Periódico de Seguridad) de productos
registrados en España ante la Agencia Española de Medicamentos y Productos
Sanitarios (AEMPS).
He realizado prototipos y modelos simulados de los PNTs (Procedimiento
Normalizado de Operación).
En tal virtud, esta memoria describirá parte de los procesos de reporte de IPSs para la
Autoridad Nacional Competente, así como los PNTs usados para la gestión del sistema
de Farmacovigilancia.
De la misma manera en la introducción se describirá las funciones generales de un
técnico en Farmacovigilancia, sus habilidades y competencias.
1. INTRODUCCIÓN
La farmacovigilancia ha sido definida por la Organización Mundial de la Salud (OMS)
de la siguiente manera: "La farmacovigilancia se ocupa de la detección, la evaluación y
la prevención de los riesgos asociados a los medicamentos una vez comercializados."
(Uppsala Monitoring Centre 2001), “este principio también se aplica para los
productos de uso veterinario” (Comisión Europea 2011).

Informes Periódicos de Seguridad: procedimiento, elaboración y envío
pág. 4
“La Farmacovigilancia Veterinaria (FV) es la actividad de salud pública, sanidad
animal y de protección del medio ambiente que tiene por objetivo la identificación,
cuantificación, evaluación, prevención y minimización de los riesgos derivados del uso
de los medicamentos veterinarios (MVs) una vez comercializados. Por lo tanto, está
orientada inevitablemente a la toma de decisiones que permitan mantener en el
mercado MVs con una relación beneficio-riesgo adecuada, o bien suspender su uso
cuando esto no sea posible.” (Agencia Española de Medicamentos y Productos
Sanitarios 2013)
Los productos medicinales veterinarios ya han sido evaluados respecto a sus
características físico-químicas, actividad in vitro e in vivo, toxicidad, farmacocinética y
actividad farmacológica en las fases previas a su autorización, pero siempre en un
número limitado de animales; al momento de ser comercializados podría presentarse
reacciones adversas desconocidas que solo se manifestarán al exponer a un número
elevado de animales a dicho fármaco. “Los estudios posteriores a la comercialización
(fase IV) tienden más a centrar sus objetivos en los efectos indeseables de los
medicamentos” (Laporte y Tognoni 2007).
Es una sección esencial en el control y regulación de la industria farmacéutica; por tanto
los profesionales de la salud como la industria farmacéutica veterinaria deben mantener
al tanto sobre eventos adversos a la autoridad nacional competente (ANC).
El titular de un producto farmacéutico veterinario está obligado a emitir informes
regulares a la ANC posterior a la aprobación de comercialización conforme a la
legislación europea vigente.
La pronta toma de decisiones de la Autoridad Nacional Competente frente a un evento
adverso puede ser determinante en la prevención de un daño a la población o al medio
ambiente, estas medidas pueden ir desde una modificación en el Resumen de
características del Producto, (SPC) por sus siglas en inglés, hasta una suspensión de la
autorización de comercialización. Para esto la ANC se vale de un sistema de
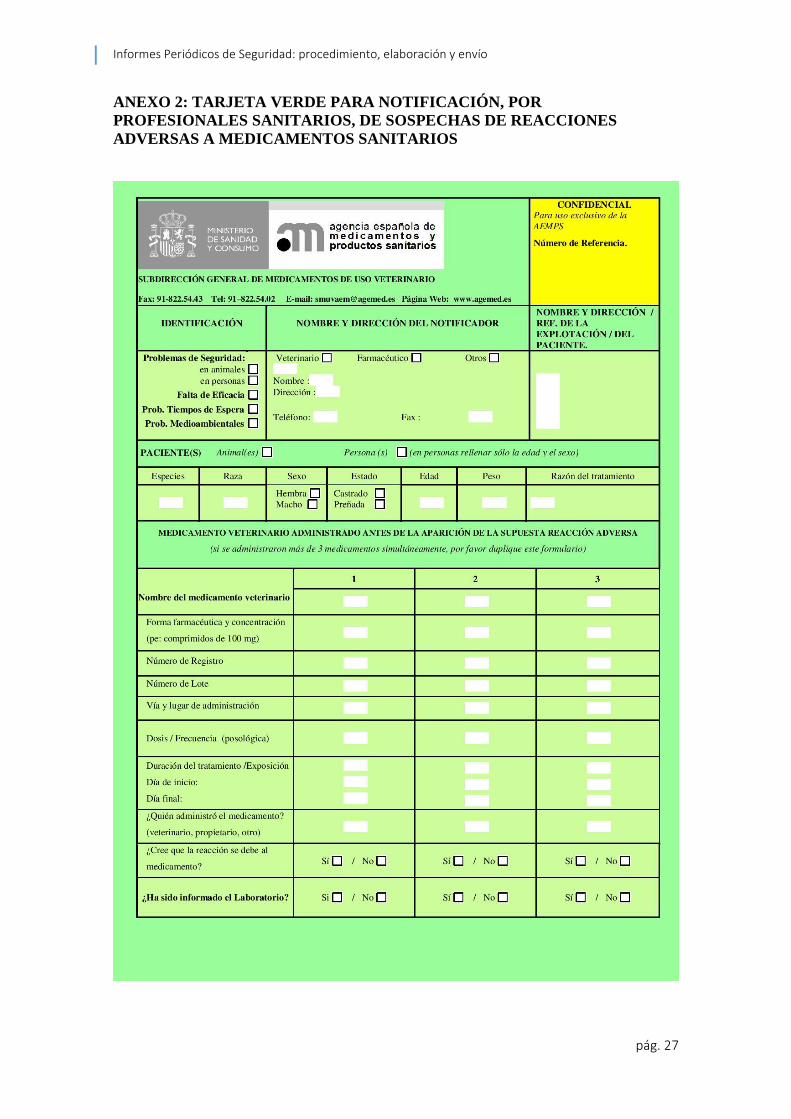
notificaciones por parte de los profesionales de la salud veterinaria llamado tarjeta verde
“Es el formulario de uso exclusivo por los profesionales sanitarios para notificar las
SAEs. Recoge la información armonizada a nivel de la UE para la notificación de SAEs
por los profesionales sanitarios. Editada en color verde, lleva impresa la dirección de

Informes Periódicos de Seguridad: procedimiento, elaboración y envío
pág. 5
la AEMPS y su franqueo es en destino, para facilitar su envío.” (Agencia Española de
Medicamentos y Productos Sanitarios 2013) (ver anexo 2)
Es por esto que, el técnico en farmacovigilancia es el profesional cuya tarea consiste en
recopilar los posibles eventos adversos, tanto en países miembros del Espacio
Económico Europeo (EEE), o en terceros países, valorarlos y, de ser necesario,
reportarlos inmediatamente a la Autoridad Nacional Competente, en caso del estado
español a la Agencia Española de Medicamentos y Productos Sanitarios (AEMPS).
A nivel interno, el responsable de Farmacovigilancia (RFV) (QPPV por sus siglas en
inglés) debe organizar las acciones del titular de la autorización de comercialización de
un medicamento (TAC) para que la gestión de Farmacovigilancia sea íntegra y tenga
una trazabilidad total, los esfuerzos no deben estar duplicados y las responsabilidades
deben ser claras. “El TAC debe disponer de PNT aprobados por la dirección del TAC y
el RFV que describan de manera adecuada las funciones y actividades que se lleven a
cabo en materia de farmacovigilancia. Si la compañía tiene PNT globales para dichas
actividades, el RFV debe conocerlos para asegurar su consistencia y cumplimiento. El
RFV debe asegurar la implementación de los mismos.” (Agencia Española de
Medicamentos y Productos Sanitarios 2011).
Con estos antecedentes podemos destacar las siguientes funciones del técnico en
Farmacovigilancia:
Conocer la legislación concerniente a la farmacovigilancia de productos
veterinarios
Estar en la capacidad de evaluar el grado de causalidad entre la droga
suministrada y el evento adverso
Diseñar calendarios que le permitan tener conocimiento cabal de las fechas en
que tienen que presentarse los IPSs de los productos registrados por el titular
Conocer las cifras de ventas para la preparación y elaboración de los IPSs
Recolectar datos científicos y técnicos así como informes de terceros ya sean
profesionales de la salud o propietarios para realizar un oportuno informe a la
ANC
Notificar a la ANC dentro de los plazos establecidos una reacción adversa seria
Comunicar al interior de la empresa la aparición de una reacción adversa seria

Informes Periódicos de Seguridad: procedimiento, elaboración y envío
pág. 6
Conocer a fondo el sistema de farmacovigilancia de la empresa y colaborar en su
constante desarrollo
Estar en constante contacto con las autoridades para poder armonizar criterios
Elaborar Procedimientos Normalizados de Trabajo (PNT) que brinden
transparencia a las acciones tomadas por el titular
Conocer las normas de las Buenas Prácticas en Farmacovigilancia
Capacitar a los empleados y distribuidores que podrían recopilar notificaciones
de eventos adversos
Sistematizar las bases de datos para poder tener a mano toda la documentación
virtual del departamento
Archivar toda la documentación física de manera cuidadosa y ordenada
realizando su previa digitalización
El técnico de Farmacovigilancia debe tener las siguientes competencias:
Poseer al menos una licenciatura relacionada con las ciencias de la salud
Mantener una continua capacitación
Tener criterios suficientes para establecer causalidades
Capacidad de comprensión de lenguaje legal
Capacidad de comunicación en español e inglés
2. TECNICAS
2.1 INFORMES PERIODICOS DE SEGURIDAD (IPS)
2.1.1 Definición
“Los IPSs son documentos importantes de farmacovigilancia. Proporcionan una
oportunidad para el TAC de revisar el perfil de seguridad de sus productos y asegurar
que el Resumen de las Características del Producto (SPC) y otra información del
producto están al día” (Comisión Europea 2011).

Informes Periódicos de Seguridad: procedimiento, elaboración y envío
pág. 7
2.1.2 Contenido
El IPS debe tener información de todas las presentaciones autorizadas así como de todas
sus formas farmacológicas y especies de destino. “El informe de actualización
periódica de la seguridad deberá incluir una evaluación científica de los beneficios y
riesgos asociados al medicamento veterinario” (The European Parliament and the
Council of The European Union 2001). Los términos deben estar acorde a la
terminología VeDDRA.
La EMA recomienda que los puntos que debería tener un IPS al menos son los
siguientes:
Nombre del titular
Nombre del producto
Número de autorización de mercado
Número de procedimiento, si es aplicable
Fecha de inicio del ciclo de presentación
Periodo cubierto por el IPS
Fecha inicial de puesta en el mercado del espacio económico europeo (EEE)
Orden cronológico del IPS
La última versión de la ficha técnica debe ser incluida como anexo y, si se han
producido cambios significativos, estos deben ser explicados debidamente en el IPS. En
el caso de productos de registro antiguo que no cuentan con una versión actualizada de
ficha técnica, como estipula el Volumen 9 B en el punto 6.3.1.3 Parte I, se ha de
adjuntar el prospecto del producto.
Cabe señalar que el Volumen 9 B contiene las guías para la Farmacovigilancia
veterinaria. Estas pautas son delineadas por la Comisión Europea en consulta con la
EMA, los Estados Miembro y las partes interesadas. Puede ser consultado en el enlace
de internet http://ec.europa.eu/health/files/eudralex/vol-9/vol_9b_2011-10.pdf
Un punto importante que exige la Autoridad Nacional Competente en el IPS es el
cálculo de la dosis de exposición al producto. Esta debe ser calculada tomando en
cuenta factores como:

Informes Periódicos de Seguridad: procedimiento, elaboración y envío
pág. 8
Volumen de ventas: se informará las cantidades del producto comercializadas
durante el periodo cubierto por el IPS tanto en España como en los países donde
se exportan productos; durante la preparación de los IPSs se solicita al
responsable que informe la cantidad, presentaciones y especies de destino.
Número de animales tratados: en la preparación del IPS y, con la información ya
recibida, se calcula el número de animales tratados, infiriendo la cantidad de
venta con las dosis que deben recibir los animales; su cálculo en la mayoría de
los caso se dará con el peso del animal multiplicado por la dosis diaria y por los
días de tratamiento tomando en cuenta siempre el máximo de exposición
posible. Respecto a los pesos, el Volumen 9 B en el punto 6.3.1.4 Parte I
dispone de una detallada tabla de pesos normalizados para el cálculo (ver tabla
1). Cabe señalar que en ocasiones, debido a la forma farmacéutica, no es posible
el cálculo de una dosis individual; de ser ese el caso, en el apartado
correspondiente del IPS se procede a justificar ante la Autoridad Nacional
Competente y se informa solamente la cantidad de producto vendido.
Especies y sub poblaciones Peso estándar (Kg)
caballo 550
perro 20
gato 5
vaca 550
ternera de engorde 150
ternera recién nacida 50
cerda / verraco 160
cerdo de finalización 60
cerdo de destete 25
oveja 60
cordero 10
aves, broiler 1
aves, gallina de postura 2

Informes Periódicos de Seguridad: procedimiento, elaboración y envío
pág. 9
aves, pavo 10
conejo 1.5
Se debe informar la existencia de eventos adversos y expresarlos en relación con las
ventas a manera de incidencias; se debe hacer una evaluación siguiendo el modelo del
line listings. De ser necesario evaluar la causalidad se la hará con el código ABON así:
Categoría A: Probable
Categoría B: Posible
Categoría O: Inclasificable/no-evaluable (eventos dónde la información
es insuficiente para sacar una conclusión)
Categoría O1: Cuestionable (eventos donde otros factores impiden sacar
una conclusión, pero la asociación al producto no puede ser descartada)
Categoría N: Improbable
En el apartado de reportes no espontáneos se consulta fuentes varias, como
publicaciones científicas, estudios post-autorización; también se consulta las bases de
datos médicas y las páginas web de EMA y FDA en las que pudiese estar detallada
alguna información de interés, usamos como palabras claves de la búsqueda el o los
principios activos y las especies de destino. Toda información relevante será incluida en
el IPS.
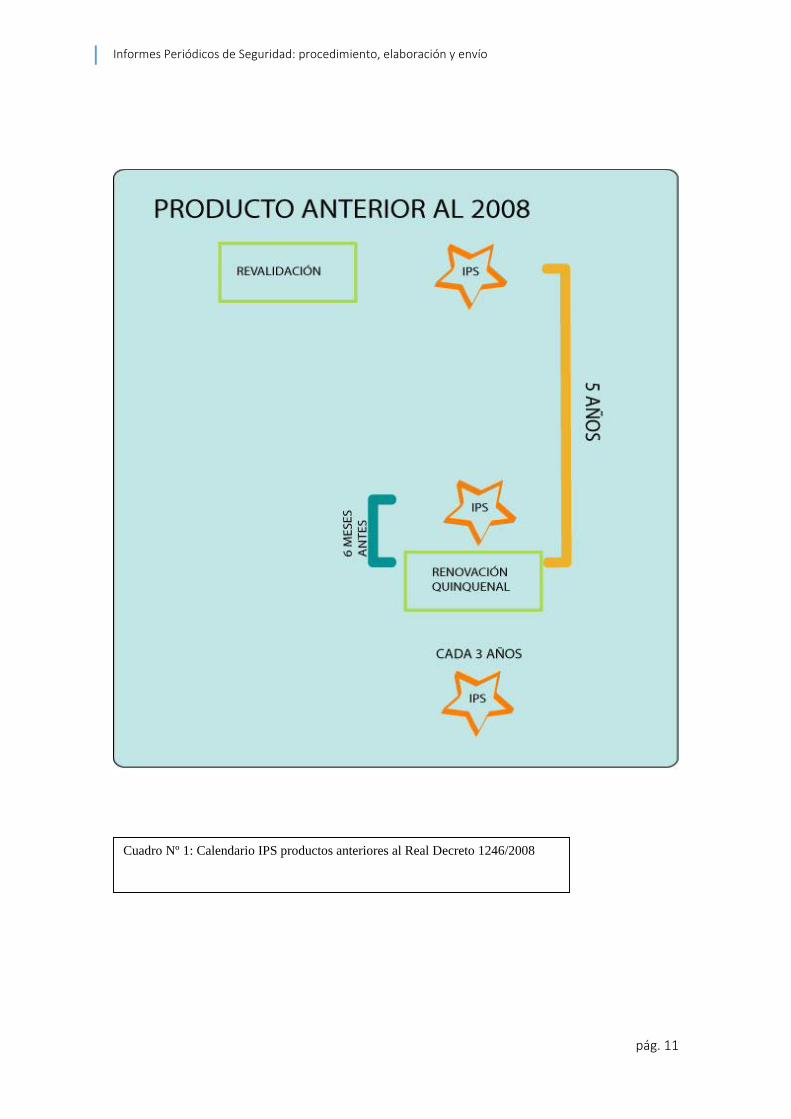
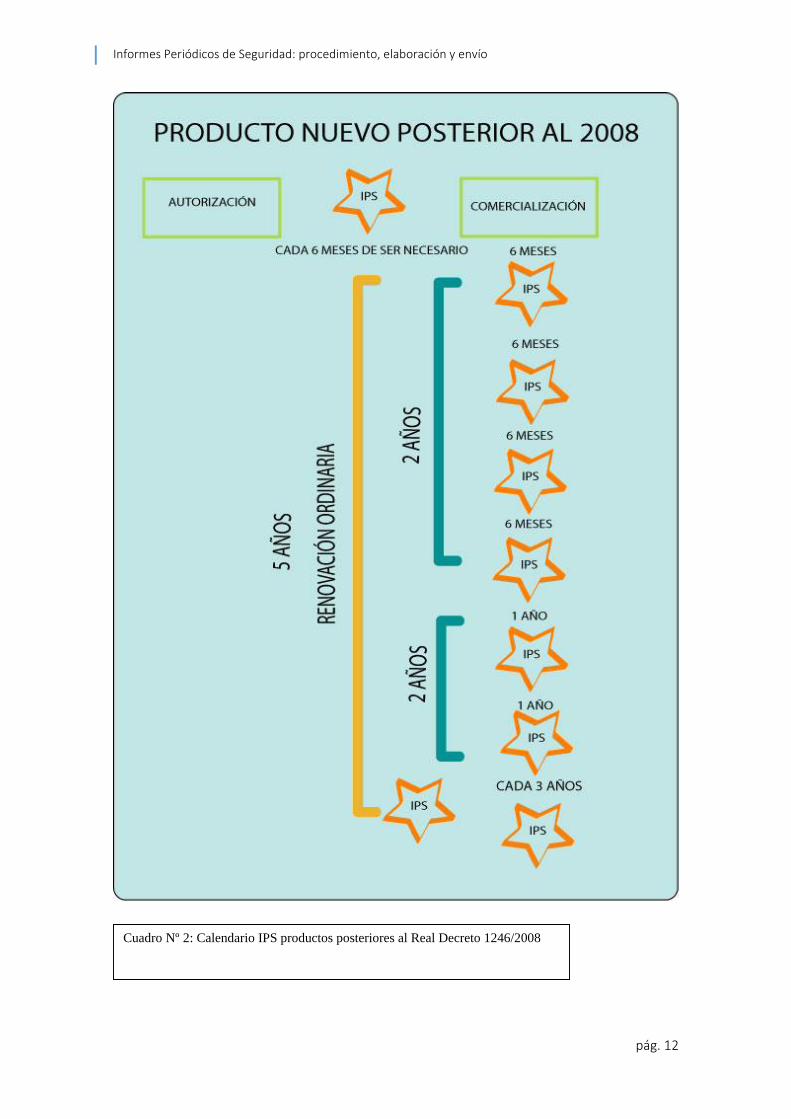
2.1.3 Calendario
Los IPS se elaboran post autorización en periodos fijados por la EMA en la Directiva
2001/82/EC artículo 75 (5) así:
Semestralmente durante los primeros dos años
Anualmente durante los siguientes dos años
A partir de entonces en intervalos de tres años
Tabla Nº 1: Tabla de pesos estándar
Fuente: Volumen 9B

Informes Periódicos de Seguridad: procedimiento, elaboración y envío
pág. 10
Si el producto ya ha sido autorizado pero no se está comercializando se deberá elaborar
IPSs semestralmente hasta la puesta en el mercado del mismo.
El IPS puede ser presentado a la ANC sesenta días después de su fecha de fin de
cobertura en idioma inglés para fármacos de reconocimiento mutuo o en español si se
trata de un informe para la AEMPS de un producto con registro nacional.
Los IPS se remiten a la AEMPS conforme a su calendario previamente establecido por
el Volumen 9B de la Comisión Europea.
Para el caso concreto de España se requiere que a los medicamentos veterinarios
autorizados con anterioridad a la fecha de entrada en vigor del Real Decreto 1246 del
año 2008 “…para otorgarles la autorización indefinida y que les sea de aplicación el
sistema de renovación de las autorizaciones de comercialización previsto en este real
decreto, deberán renovar dicha autorización, de acuerdo con las instrucciones de
ordenación del proceso que dicte la Agencia Española de Medicamentos y Productos
Sanitarios.” (Boletin Oficial del Estado 2008).
Todos los productos, tanto los autorizados previo al Real Decreto 1246 / 2008 como los
posteriores deberán someterse a la Renovación quinquenal “La renovación quinquenal
de una autorización consiste en una revisión administrativa y técnica de toda la
documentación del medicamento.” (Agencia Española de Medicamentos y Productos
Sanitarios 2012).
Para esta renovación se puede presentar un addendum como lo estipula el Volumen 9B
en su inciso 6.4.1.2 de haber un periodo sin informar ya que bajo ninguna circunstancia
se puede dejar un periodo sin cobertura de información de IPS; de esta manera, a lo
establecido por el calendario del volumen 9B, se complementa por los IPS enviados en
los procesos de Revalidación Extraordinaria o Renovación Ordinaria, de ser este el caso
del producto en cuestión ya que la Renovación es un proceso independiente al
calendario de IPSs.
Anteriormente se enviaba la documentación física por correo certificado, actualmente
se envía por email. Para el pago de las tasas se utiliza la sede electrónica de la página
web de AEMPS que genera tres documentos de pago para la agencia y para el titular.
Informes Periódicos de Seguridad: procedimiento, elaboración y envío
pág. 11
Cuadro Nº 1: Calendario IPS productos anteriores al Real Decreto 1246/2008
Informes Periódicos de Seguridad: procedimiento, elaboración y envío
pág. 12
Cuadro Nº 2: Calendario IPS productos posteriores al Real Decreto 1246/2008

Informes Periódicos de Seguridad: procedimiento, elaboración y envío
pág. 13
2.2 PROCEDIMIENTO NORMALIZADO DE TRABAJO (PNT)
2.2.1 Definición
“Un elemento esencial en cualquier sistema de farmacovigilancia es que existan
procedimientos claros escritos para el lugar de trabajo” (Comisión Europea 2011). Los
PNT deben estar redactados de forma clara y ser fáciles de seguir por el personal de la
empresa, deben permitir documentar las actividades y darles trazabilidad.
2.2.2 PNT de uso frecuente en Farmacovigilancia Veterinaria
La EMA sugiere que los titulares tengan Procedimientos Normalizados de Trabajo
claros y documentados de al menos estas áreas (ver tabla 2) aunque es potestad del
titular redactar más PNT o englobar varias tareas en uno solo.
Nombre del PNT Actividad Actividades del RFV (QPPV) y procedimiento de
reemplazo a aplicarse en su ausencia
Describe las actividades del RFV (QPPV) y el
proceso que se aplica en su ausencia
La recolección, procesamiento, control de
calidad, la codificación, clasificación, revisión
veterinaria y la notificación de los Eventos
Adversos Sospechosos (SAE)
Describe el procesamiento, control, codificación,
clasificación, revisión y notificación de los SAE
Seguimiento de los reportes, por información
perdida y por información de la evolución, y
resultados de los caso (s)
Describe el seguimiento que se da a los reportes,
sea el caso de información perdida o de evolución
de los casos
Detección de reportes duplicados Describe el sistema de detección de reportes
duplicados
Informes expeditos Describe el proceso de reporte expedito
Reporte electrónico Describe el reporte electrónico
Informe Periódico de Seguridad Describe los IPS, preparación, proceso, control,
revisión y reporte
Actividades de farmacovigilancia globales que se
aplican a todos los productos
Se describen las actividades globales de
farmacovigilancia que se aplican a todos los
productos: detección y revisión de la señal,
riesgo/beneficio, evaluación y presentación de
informes y comunicación notificantes a las
autoridades competentes y profesionales de la
salud animal
Interacciones entre problemas de seguridad y
defectos del producto
Describe las interacciones entre los problemas de
seguridad y los defectos del producto
Respuesta a las solicitudes de información de las
autoridades competentes
Describe la respuesta a las solicitudes de
información por parte de las autoridades
competentes
Manejo de las restricciones de seguridad urgentes
y variaciones de seguridad
Describe el manejo de las restricciones de
seguridad urgentes y variaciones de seguridad
Cumplimiento de los compromisos con las
autoridades competentes en relación con las
autorizaciones de comercialización
Describe el cumplimiento de los compromisos
con las autoridades en relación con las
autorizaciones de comercialización
Gestión y uso de bases de datos u otros sistemas
de registro
Describe la gestión y uso de las bases de datos y
de otros sistemas

Informes Periódicos de Seguridad: procedimiento, elaboración y envío
pág. 14
Auditoría interna del sistema de
farmacovigilancia
Describe la auditoría interna del sistema de
farmacovigilancia
Capacitación Describe las labores de capacitación ofrecida al
personal de la empresa
Archivo Describe el archivo de toda la documentación de
farmacovigilancia
2.2.3 Descripción Detallada del Sistema de Farmacovigilancia (DDPS por sus siglas
en inglés)
El DDPS, como se señala en el apartado 2.3 del Volumen 9B, es un requisito que el
titular debe entregar a la Autoridad Competente al registrar un producto, en el cual
prueba que cuenta con los servicios de un RFV (QPPV por sus siglas en inglés) y que
cuenta con las medidas necesarias para la recogida y notificación de cualquier evento
adverso.
“El DDPS debe incluir una visión general del sistema de farmacovigilancia
suministrando información sobre los elementos clave de dicho sistema” (Comisión
Europea 2011).
El DDPS contendrá información sobre:
QPPV datos, formación y descripción de tareas
Procedimiento en caso de ausencia del QPPV
Información sobre la empresa y de sus actividades de farmacovigilancia
Diagramas de flujo sobre las tareas de farmacovigilancia
Listado de los PNTs
Descripción de la base de datos
Acuerdos contractuales con terceros en torno a labores de farmacovigilancia
Programa de capacitación
Gestión de documentación
Gestión de calidad
Tabla Nº 2: listado de PNTs recomendados
Fuente: Volumen 9B

Informes Periódicos de Seguridad: procedimiento, elaboración y envío
pág. 15
2.3 ESTUDIOS POST AUTORIZACIÓN
“Las condiciones de realización de estos estudios se ajustarán a las condiciones que se
establezcan en la autorización de comercialización.” (Agencia Española de
Medicamentos y Productos Sanitarios 2014).
Los estudios post autorización son importantes para detectar reacciones como:
Efectos a largo plazo
Efectos de baja frecuencia
Relevancia clínica
Eficacia en la práctica clínica
Reacciones adversas
Seguridad del usuario
Validez del tiempo de espera
Se complementan con el programa de reporte espontáneo y, pueden ser requeridos por
las autoridades o por iniciativa del mismo titular.
2.4 BASE DE DATOS
Un punto de importancia crítica en farmacovigilancia es la base de datos. Esta base,
como se indica en el volumen 9B punto 2.3.3 inciso d, debe ser capaz de:
Compilar los informes de seguridad
Gestionar los informes expeditos electrónicos
Detectar señales
Intercambio y acceso a la información mundial
La EMA pide que estas bases y sus sistemas de validación sean descriptos en el DDPS,
también se debe indicar las responsabilidades de la operación de la base y su ubicación.
Debe estar conforme a los sistemas acordados internacionalmente para la presentación
electrónica de informes sobre reacciones adversas.

Informes Periódicos de Seguridad: procedimiento, elaboración y envío
pág. 16
Se deben tomar medidas para cuidar la confidencialidad y la seguridad de los datos
incluyendo un control severo para que solo las personas autorizadas tengan acceso a los
documentos y a la base de datos. Se sugiere incorporar un software para la detección de
reportes duplicados.
3. CONOCIMIENTOS Y HABLILIDADES ADQUIRIDAS
3.1 INFORMES PERIÓDICOS DE SEGURIDAD (IPS)
3.1.1 Elaboración del calendario de IPS
Para tener un completo conocimiento de la realización de los Informes Periódicos de
Seguridad (IPS) se redactó modelos basados en los requerimientos dados en el volumen
9 B.
En el presente Trabajo de Fin de Máster se muestra un modelo de IPS para un producto
ficticio (Pentax 500 inyectable de Laboratorio UAB, ver anexo 1) con los puntos que
exige el Volumen 9B en su inciso 6.3.1
Algunos puntos que son importantes resaltar son:
El punto 1 está dado por los datos del producto y los periodos que se está
informando en el IPS. En este ejemplo sería el cuarto informe semestral de un
producto de autorización nacional
El punto 2 informa de la existencias de medidas tomadas por razones de
seguridad indicando alcance, estatus y fecha
El punto 3 incluirá la ficha técnica (en este ejemplo no está incluido ya que se
trata de un producto ficticio)
En el punto 4 se reporta las ventas totales en el periodo y las especies a las que
fue destinada
También se da el cálculo de animales teóricamente tratados; se debe considerar
que: algunos productos tienen presentaciones individuales y el número de
unidades del producto será igual al número de animales tratados; en la mayoría

Informes Periódicos de Seguridad: procedimiento, elaboración y envío
pág. 17
de productos el número de animales tratados se calcula con la dosis y tiempo de
administración autorizados (usando la dosis y duración máximas)
En el punto 5 se establece una relación entre el volumen de ventas del producto
y el número de eventos adversos reportados
El punto 6 se incluye una revisión de datos de seguridad durante el periodo de
reporte
Punto 7, una revisión de datos sobre eventos adversos de otras fuentes como
estudios post autorización, publicaciones de eventos adversos o experiencias de
usuarios
El punto 8 describe si se han dado eventos adversos por diversos errores
Punto 9, es un análisis científico de los datos y una evaluación crítica del
beneficio-riesgo del producto con la nueva información disponible
El punto 10 informa si se ha recibido información de importancia a partir del
cierre de datos para el reporte
En el punto 11 los reportes individuales deben ser presentados como line listings
siguiendo un modelo dado en el volumen 9 B anexo 2.4
3.1.2 Archivo de la documentación virtual y real relacionada con los IPS
El Volumen 9B establece entre las responsabilidades del Responsable de
Farmacovigilancia (RFV) (QPPV por sus siglas en inglés) está mantener el correcto
archivo de los documentos reales y de las bases de datos de la empresa. Durante la
práctica se digitalizó documentos, se creó archivos virtuales; clasificando y archivando
de manera adecuada dichos documentos.
3.1.3 Pago de tasas
Para remitir los IPS se deben cancelar las respectivas tasas (ver tabla 3)
Descripción Euros
Tasa por evaluación de informe periódico de seguridad semestral
de un medicamento veterinario, esté o no registrado el
medicamento en España
382.71
Tasa por evaluación de informe periódico de seguridad anual de
un medicamento veterinario, esté o no registrado el medicamento
en España
754.84

Informes Periódicos de Seguridad: procedimiento, elaboración y envío
pág. 18
Tasa por evaluación de informe periódico de seguridad trienal o
superior a tres años de un medicamento veterinario, esté o no
registrado el medicamento en España
2273.52
3.1.4 Consideraciones a futuro sobre los IPS
Desde el año pasado se está discutiendo en el seno de la Comisión cómo mejorar el
sistema de Informes Periódicos de Seguridad, “La presente propuesta introduce un
enfoque de la farmacovigilancia basado en los riesgos, conforme al cual se relajan
determinados requisitos que no contribuyen eficazmente a la salud pública, la sanidad
animal o la protección del medio ambiente (por ejemplo, la presentación de informes de
seguridad actualizados periódicamente)” (Comisión Europea 2014).
Tanto el sector público como privado han visto la necesidad de mejorar aspectos como
la carga normativa, la falta de disponibilidad de medicamentos veterinarios
especialmente en mercados pequeños como los de las abejas y mejorar el
funcionamiento del mercado interior.
Se pide que la regulación sea congruente con las particularidades del mercado de
productos farmacéuticos veterinarios, ya que el hecho de haber varias especies lo
fragmenta; además la lógica de precios es distinta, llevando a que estos sean mucho más
bajos que los de humana.
Los objetivos de la Comisión Europea al revisar la normativa serán seguir precautelando
por la salud pública, la sanidad animal, la seguridad alimentaria y el medio ambiente
pero al mismo tiempo:
Aumentar la disponibilidad de medicamentos veterinarios
Reducir las cargas administrativas
Estimular la competitividad y la innovación
Mejorar el funcionamiento del mercado interior
Hacer frente al riesgo de farmacorresistencias
Tabla Nº 3: Tabla de valores de tasas para la remisión de IPS
Fuente: AEMPS

Informes Periódicos de Seguridad: procedimiento, elaboración y envío
pág. 19
Respecto a la farmacovigilancia, lo que busca la propuesta es un cambio de enfoque a
una farmacovigilancia basada en riesgos, retirando requisitos que no contribuyen a la
salud pública, sanidad animal o protección del medio ambiente, por ejemplo la
presentación de IPS actualizados periódicamente. Una de las acciones de la Agencia es
crear una base de datos centralizada para gestionar los efectos adversos de las medicinas
autorizadas en toda la Unión Europea.
El resultado de esta propuesta se verá en la nueva legislación en el año 2017 o 2018.
3.2 PROCEDIMIENTO NORMALIZADO DE TRABAJO (PNT)
3.2.1 Actualización de PNT
3.2.1.1 PNT de Auditoría Interna
A manera de ejemplo, y para comprender lo que requiere la legislación en la práctica, se
realizó como ejercicio de comprensión un prototipo de una check list para registrar el
cumplimiento de lo estipulado en el Volumen 9 B respecto a:
RFV (QPPV).- existencia, procedimiento en ausencia, facultades
Personal: responsabilidades y capacitación
Documentación: existencia, actualización y aprobación de PNTs
Notificaciones: calendarios de IPS y notificaciones de eventos adversos
Base de datos: sistemas de seguridad y respaldos
Gestión de datos: gestión de datos electrónicos, gestión de archivos físicos
Calidad: plan, manual de calidad
Auditorías: programa, verificación de externalidad, feedback

Informes Periódicos de Seguridad: procedimiento, elaboración y envío
pág. 20
3.2.1.2 PNT de Reporte Electrónico
Continuando con los prototipos se redactó un modelo de addendum al PNT de Reporte
Electrónico con los tutoriales de la EMA a cerca de:
cómo realizar un reporte electrónico
pasos a seguir si es la primera vez que se realiza un reporte electrónico
preguntas y errores más frecuentes
3.2.1.3 PNT de Recolección de Eventos Sospechosos Adversos (SAE por sus siglas en
inglés)
Se elaboró una simulación de guía de trabajo que articula las acciones del PNT de
Recolección de SAE con los PNT de Entrenamiento y de Actividades de
Farmacovigilancia Globales. Con este prototipo se detallan las acciones a tomar para
recopilar información de posibles eventos adversos (SAE) cuando:
Se inicia una relación con un nuevo distribuidor
Con los distribuidores existentes
Puesta en mercado de un nuevo producto
Con el personal de la empresa
3.2.1.4 PNT de Digitalización de Documentos
Se redactó un documento prototipo para la gestión, escaneo, registro y archivo de los
documentos relacionados a las tareas de Farmacovigilancia donde se establecen los
procedimientos y se delegan las responsabilidades. Como anexo se redactó una hoja de
control del personal que manipula dichos documentos para, de esta manera poder dar el
seguimiento debido.

Informes Periódicos de Seguridad: procedimiento, elaboración y envío
pág. 21
3.2.1.5 PNT de Recogida de Datos de Diferentes Orígenes
Esta simulación de PNT establece el procedimiento a tomarse cuando se dan reportes de
SAE originados en fuentes externas a la empresa. Establece responsabilidades al interior
de la empresa y da el procedimiento a realizarse por parte del personal. Lo articula con
el PNT de Recolección de SAE.
Como parte de esta simulación se redactó una hoja de recogida de SAE de diferentes
orígenes a ser usada por el personal señalado en este PNT.
3.2.2 Descripción Detallada del Sistema de Farmacovigilancia (DDPS por sus siglas
en inglés)
Se observó los cambios que tiene un DDPS conforme se dan sus diferentes versiones
debidamente documentadas.

Informes Periódicos de Seguridad: procedimiento, elaboración y envío
pág. 22
4. BIBLIOGRAFÍA
Agencia Española de Medicamentos y Productos Sanitarios. Editado por Comité Técnico de
Inspección. 2011. Consultado en: http://www.aemps.gob.es/industria/Inspeccion-
BPFV/docs/CTI.BPFV.127.00.11.pdf (último acceso: 19 de 06 de 2015).
Agencia Española de Medicamentos y Productos Sanitarios. 07 de 11 de 2014. Consultado en:
http://www.aemps.gob.es/investigacionClinica/medicamentos-vet/EPAs/home.htm
(último acceso: 25 de 06 de 2015).
Agencia Española de Medicamentos y Productos Sanitarios. «Buenas Prácticas de
Farmacovigilancia» Editado por Comité Técnico del Sistema Español de Farmacovigilancia
de Medicamentos Veterinarios. 08 de 08 de 2013. Consultado en:
http://www.aemps.gob.es/vigilancia/medicamentosVeterinarios/docs/bp_fv_vet_agosto2
013.pdf (último acceso: 16 de 07 de 2015).
Agencia Española de Medicamentos y Productos Sanitarios. «Renovación Quinquenal de
Medicamentos Veterinarios». 08 de 10 de 2012. Consultado en:
http://www.aemps.gob.es/informa/circulares/medicamentosVeterinarios/2012/docs/Circ
ular_4-2012.pdf (último acceso: 09 de 07 de 2015).
Boletin Oficial del Estado. «Real Decreto 1246/2008 del 18 de julio de 2008.» Por el que se regula el
procedimiento de autorización, registro y farmacovigilancia de los medicamentos
veterinarios fabricados industrialmente. Vol. 193. BOE-A-2008-13682, 18 de 07 de 2008.
Comisión Europea. «Propuesta de reglamento del Parlamento Europeo y del Consejo sobre los
medicamentos veterinarios.» Vol. 2014/0257 (COD). Bruselas, 10 de 09 de 2014.
Comisión Europea . Volume 9B of The Rules Governing Medicinal Products in the European Union.
2011. Consultado en: http://ec.europa.eu/health/files/eudralex/vol-9/vol_9b_2011-
10.pdf (último acceso: 06 de 2015).
Laporte, J. R., y G. Tognoni. Principios de epidemiología del medicamento. Segunda edición.
Barcelona: Masson - Salvat Medicina, 2007.
The European Parliament and the Council of The European Union. «Directive 2001/82/EC of the
European Parliament.» Official Journal of the European Communities, 2001.
Uppsala Monitoring Centre. «Vigilancia de la Seguridad de los Medicamentos.» Guía, the Uppsala
Monitoring Center, Uppsala, 2001.

Informes Periódicos de Seguridad: procedimiento, elaboración y envío
pág. 23
ANEXOS
ANEXO 1: MODELO DE UN INFORME PERIÓDICO DE SEGURIDAD PARA
UN PRODUCTO Y TITULAR FICTICIO
1.- Detalles del titular y del producto
i) Nombre del titular: Laboratorio UAB
ii) Nombre del Producto: Pentax 500 inyectable
iii) Número de autorización: 123 ESP
iv) Número de procedimiento, si es aplicable: procedimiento nacional
v) Fecha de inicio para el ciclo de presentación de IPS: 01 de agosto 2013
vi) Periodo cubierto por el IPS: 01 de febrero 2015 al 01 de agosto 2015
vii) Fecha de puesta en el mercado del EEE, entendido como la fecha
cuando la primera presentación del producto fue puesta en mercado por
primera vez en cualquier estado miembro en caso de autorizaciones
nacionales, o en algún país dentro del EEE en caso de productos de
autorización centralizada: 01 de agosto 2013
viii) Orden cronológico del IPS: cuarto informe semestral
2.- Actualización sobre medidas regulatorias o acciones del titular
tomadas por razones de seguridad
No se ha tomado ninguna medida de seguridad con el producto en ningún lugar del
mundo durante el periodo cubierto por este Informe Periódico de Seguridad.
3.- Ficha técnica (Resumen de las características del producto)

Informes Periódicos de Seguridad: procedimiento, elaboración y envío
pág. 24
Ver anexos (en este ejemplo no se incluye ya que es un producto ficticio).
4.- Estimaciones de exposición
i) Volumen de ventas: el detalle de las ventas es el siguiente
País Total ventas
España 250 ml x 1000 + 500 ml x 500
Total 500,000 ml = 500 L
No ha habido ventas en otros países, del total de ventas la distribución por especie fue
bovino (50%) y equino (50%).
ii) Número de animales tratados
BOVINO:
Volumen de ventas: 250 L = 250,000 ml
Dosis de Pentax 500 inyectable: 10 ml/animal por día por 5 días = 50 ml
250,000 ml / 50 ml = 5,000 dosis teóricas
EQUINO:
Volumen de ventas: 250 L = 250,000 ml
Dosis de Pentax 500 inyectable: 10 ml/animal por día por 5 días = 50 ml
250,000 ml / 50 ml = 5,000 dosis teóricas
5.- Incidencia de eventos adversos

Informes Periódicos de Seguridad: procedimiento, elaboración y envío
pág. 25
No se han reportado eventos adversos durante el periodo cubierto por el IPS y por lo
tanto no puede ser calculada la incidencia.
6.- Revisión de datos
No se han reportado sospechas de eventos adversos durante el periodo de este IPS.
7.- Reportes no espontáneos
No existen datos a disposición de otras fuentes. Tampoco se ha encontrado información
de utilidad en las fuentes bibliográficas ni publicaciones científicas.
8.- Otra información
No se han dado eventos adversos por errores de prescripción o errores de nombre.
9.- Evaluación global de seguridad
La evaluación de seguridad de este medicamento no ha cambiado porque no ha sido
reportado ningún evento adverso.
10.- Información importante recibida después del punto de cierre de
datos

Informes Periódicos de Seguridad: procedimiento, elaboración y envío
pág. 26
No se ha recibido ninguna información después de la fecha de cierre de datos que pueda
afectar el beneficio-riesgo.
11.- IPS “line listings”
No es aplicable porque no han ocurrido eventos adversos durante el periodo
concerniente a este IPS.
Informes Periódicos de Seguridad: procedimiento, elaboración y envío
pág. 27
ANEXO 2: TARJETA VERDE PARA NOTIFICACIÓN, POR
PROFESIONALES SANITARIOS, DE SOSPECHAS DE REACCIONES
ADVERSAS A MEDICAMENTOS SANITARIOS

Informes Periódicos de Seguridad: procedimiento, elaboración y envío
pág. 28


















