
INSTITUTO POLITÉCNICO NACIONAL Aldo.pdf · sistema de doble híbrido en levadura y la...
120
INSTITUTO POLITÉCNICO NACIONAL “PREDICCIÓN Y ANÁLISIS COMPUTACIONAL DE NUEVAS INTERACCIONES ENTRE PROTEÍNAS QUE PARTICIPAN EN LA PATOGÉNESIS DE Helicobacter pylori” T E S I S Que para obtener el grado de: MAESTRO EN CIENCIAS EN BIOTECNOLOGÍA GENÓMICA Presenta: ALDO SEGURA CABRERA Reynosa, Tamaulipas, México, Junio del 2009
Transcript of INSTITUTO POLITÉCNICO NACIONAL Aldo.pdf · sistema de doble híbrido en levadura y la...

INSTITUTO POLITÉCNICO NACIONAL
“PREDICCIÓN Y ANÁLISIS COMPUTACIONAL DE
NUEVAS INTERACCIONES ENTRE PROTEÍNAS QUE
PARTICIPAN EN LA PATOGÉNESIS DE Helicobacter pylori”
T E S I S
Que para obtener el grado de:
MAESTRO EN CIENCIAS EN BIOTECNOLOGÍA GENÓMICA
Presenta:
ALDO SEGURA CABRERA
Reynosa, Tamaulipas, México, Junio del 2009



ÍNDICE
CONTENIDO ................................................................................................................................ PÁGINA
ABREVIATURAS ...................................................................................................................................... i
RELACIÓN DE CUADROS Y FIGURAS ............................................................................................... ii
RESUMEN ............................................................................................................................................... iv
ABSTRACT .............................................................................................................................................. vi
1 INTRODUCCIÓN .................................................................................................................................. 1
2 ANTECEDENTES ................................................................................................................................. 3
2.1 Historia y características generales de Helicobacter pylori ................................................................. 3
2.2 La infección por Helicobacter pylori ................................................................................................... 3
2.2.1 Enfermedades producidas por Helicobacter pylori .......................................................................... 3
2.3 Aspectos moleculares de Helicobacter pylori ..................................................................................... 5
2.3.1 Genómica ................................................................................................................................... 5
2.3.2 Transcriptómica y proteómica ................................................................................................... 7
2.3.3 Interactómica ............................................................................................................................. 9
2.4 Métodos para la determinación de interacciones entre proteínas ...................................................... 11
2.4.1 Métodos experimentales .......................................................................................................... 12
2.4.1.1 Sistema de dos híbridos en levadura ............................................................................. 12
2.4.1.2 Purificación por afinidad en tándem acoplada a espectrometría de masas ............................. 14
2.4.1.3 Interacciones entre proteínas a nivel atómico ................................................................ 16
2.4.2 Métodos computacionales ....................................................................................................... 16
2.5 Bases de datos de interacciones entre proteínas ................................................................................ 20
2.5.1 BIND (Biomolecular Interaction Network Database) ............................................................. 21
2.5.2 DIP (Database of Interacting Proteins) ................................................................................... 21
2.5.3 MINT (Molecular INTeraction database) .............................................................................. 21
2.5.4 3did (3D interacting domains) ................................................................................................. 22
2.5.5 IntAct (IntAct database) .......................................................................................................... 22
2.5.6 HPRD (The Human Protein Reference Database) .................................................................. 22
2.6 Calidad de los métodos de determinación de interacciones entre proteínas ...................................... 23
3 PLANTEAMIENTO DEL PROBLEMA ............................................................................................. 25
4 JUSTIFICACIÓN ................................................................................................................................. 25

5 OBJETIVOS ......................................................................................................................................... 26
5.1 Objetivo general ................................................................................................................................. 26
5.2 Objetivos específicos ......................................................................................................................... 26
6 HIPÓTESIS .......................................................................................................................................... 26
7 METODOLOGÍA ................................................................................................................................. 27
7.1 Obtención de las proteínas de participación conocida en la patogénesis (proteínas raíz) ................. 27
7.2 Construcción de la red de interacciones entre proteínas de H. pylori ................................................ 27
7.3 Predicción de nuevas interacciones y proteínas involucradas en la patogénesis de H. pylori ........................................................................................................................................................ 28
7.4 Análisis del papel de las predicciones en patogénesis ....................................................................... 29
8 RESULTADOS .................................................................................................................................... 30
8.1 Proteínas asociadas a motilidad y quimiotaxis .................................................................................. 34
8.2 Proteínas asociadas a la adaptación al medio ácido ........................................................................... 39
8.3 Proteínas asociadas al metabolismo del nitrógeno ............................................................................ 41
8.4 Proteínas asociadas con el sistema de secreción tipo IV ................................................................... 43
8.5 Proteínas asociadas con la respuesta a estrés ..................................................................................... 45
8.6 Proteínas asociadas a la síntesis de lipopolisacáridos ........................................................................ 45
9 DISCUSIÓN ......................................................................................................................................... 50
9.1 Proteínas asociadas a motilidad y quimiotaxis .................................................................................. 54
9.2 Proteínas asociadas a la adaptación al medio ácido ........................................................................... 57
9.3 Proteínas asociadas al metabolismo del nitrógeno ............................................................................ 60
9.4 Proteínas asociadas con el sistema de secreción tipo IV ................................................................... 61
9.5 Proteínas asociadas con la respuesta a estrés ..................................................................................... 63
9.6 Proteínas asociadas a la síntesis de lipopolisacáridos ........................................................................ 66
9.7 Interconexión entre agrupamientos de proteínas ............................................................................... 68
9.8 Limitaciones del estudio ................................................................................................................... 69
10 CONCLUSIONES .............................................................................................................................. 71
11 RECOMENDACIONES ..................................................................................................................... 72
12 REFERENCIAS .................................................................................................................................. 73
13 GLOSARIO ...................................................................................................................................... 102
14 APÉNDICE ....................................................................................................................................... 106

i
ABREVIATURAS
3D: Tridimensional. ADN: Ácido desoxirribonucleico. ARN: Ácido ribonucleico. ATP: Adenosín trifosfato. BLAST : de las siglas en inglés, herramienta para la búsqueda del alineamiento local básico. COX: Ciclo oxigenasa. MALDI-MS : Espectrometría de masas, variante desorción/ionización láser asistida por matriz. MALT : Linfoma asociado a mucosa. NCBI : Centro Nacional para la Información Biotecnológica de EE.UU., por sus siglas en inglés. ORF: Marco de lectura abierto, por sus siglas en inglés. PIANA : Análisis de redes e interacciones entre proteínas, por sus siglas en inglés. RMN : Resonancia magnética nuclear. ROS: Especies reactivas de oxígeno. STRING: Herramienta de búsqueda para la recuperación de interacciones de genes/proteínas, por sus siglas en inglés. TAP-MS: Purificación por afinidad en tándem acoplada a espectrometría de masas, por sus siglas en inglés. Y2H: Sistema de doble híbrido en levadura, por sus siglas en inglés.

ii
RELACIÓN DE CUADROS Y FIGURAS
CONTENIDO ............................................................................................................................. PÁGINA
Cuadro1. Asociación entre el contexto funcional de proteínas (GUP) y su conocida función
en patogénesis .......................................................................................................................................... 31
Cuadro 2. Valores de GUP para cada una de la proteínas que integraron la red de
interacción y su probable asociación en patogénesis de H. pylori ........................................................... 32
Cuadro 3. Lista de proteínas candidato asociadas a quimiotaxis y motilidad ........................................ 35
Cuadro 4. Lista de proteínas candidato asociadas a adaptación al medio ácido .................................... 40
Cuadro 5. Lista de proteínas candidato asociadas al metabolismo del nitrógeno .................................. 42
Cuadro 6. Lista de proteínas candidato asociadas al sistema de secreción tipo IV ................................ 43
Cuadro 7. Lista de proteínas candidato asociadas a proteínas de respuesta a estrés .............................. 46
Cuadro 8. Lista de proteínas candidato asociadas a síntesis de lipopolisacáridos ................................. 48
Figura 1. Método de doble hibrido en levadura ...................................................................................... 14
Figura 2. Método de purificación por afinidad en tándem acoplado a espectrometría de
masas ........................................................................................................................................................ 15
Figura 3. Métodos computacionales de predicción de interacciones entre proteínas basados
en el contexto genómico .......................................................................................................................... 18
Figura 4. Construcción de la red de interacción a partir de las proteínas raíz obtenidas de la
base de datos MvirDB .............................................................................................................................. 27
Figura 5. Cálculo del GUP de una proteína, del valor predictivo positivo y la sensibilidad a
diferentes umbrales de GUP .................................................................................................................... 28
Figura 6. Red de interacción entre proteínas asociadas a patogénesis de H. pylori ............................... 33
Figura 7. Agrupamiento de proteínas asociadas a la quimiotaxis y motilidad ....................................... 34
Figura 8. Estructura cristalográfica donde se observa la interacción entre las proteínas
CheA y CheY de E. coli ........................................................................................................................... 36

iii
Figura 9. Organización a nivel genómico de los genes fliF, fliG y fliH , respecto a los genes
candidato dxs y lepA ................................................................................................................................ 38
Figura 10. Agrupamiento de proteínas asociadas a la adaptación al medio ácido ................................. 39
Figura 11. Estructura cristalográfica donde se observa la interacción entre la proteína NikR
y el ADN de E. coli .................................................................................................................................. 40
Figura 12. Agrupamiento de proteínas asociadas a el metabolismo del nitrógeno ................................ 41
Figura 13. Agrupamiento de proteínas asociadas al sistema de secreción tipo IV ................................. 43
Figura 14. Agrupamiento de proteínas asociadas a la respuesta a estrés ................................................ 45
Figura 15. Estructura cristalográfica donde se observa la interacción entre la subunidad
alfa de la ADN polimerasa con TrxA, durante el proceso de reparación del ADN del
bacteriófago T7 ........................................................................................................................................ 47
Figura 16. Agrupamiento de proteínas asociadas a síntesis de lipopolisacáridos .................................. 48

iv
Predicción y análisis computacional de nuevas interacciones entre proteínas que
participan en la patogénesis de Helicobacter pylori
Resumen
La información de interacciones entre proteínas producto de experimentos a gran
escala en humano y otros organismos eucariotes ha sido de gran valor para la comprensión
a nivel de sistemas de diversos procesos biológicos. Sin embargo, en organismos
procariotes se dispone de limitada información sobre las interacciones entre proteínas;
siendo Helicobacter pylori (H. pylori) el primer organismo de este tipo estudiado a nivel de
las interacciones entre proteinas. Para contribuir al entendimiento de la participación de las
proteínas en la patogénesis de H. pylori, se construyó una red de interacción para 127
proteínas asociadas a patogénesis, combinado datos de interactomas obtenidos a nivel
experimental, predicciones de alta confiabilidad y análisis in silico. La red resultante
presentó un total de 330 proteínas con 1,271 interacciones. La predicción de nuevas
interacciones entre proteínas se realizó mediante los métodos de búsqueda de interólogos y
basados en el contexto genómico, en combinación con información derivada de
experimentos de los interactomas disponibles y minería de datos. Todas las proteínas
presentes en la red se agruparon de acuerdo a procesos celulares específicos, esto indica
que el análisis realizado esta en concordancia con la evidencia experimental disponible de
interacciones proteína-proteína. Para predecir nuevas proteínas patogénicas se realizó un
análisis del contexto funcional para cada una de las proteínas que integraron la red.
Cincuenta proteínas candidato se obtuvieron con valor predictivo positivo y sensibilidad
superior al 60%. El análisis in silico predice la asociación de las proteínas candidato con los
procesos de quimiotaxis y motilidad, adaptación al medio ácido, respuesta al estrés, sistema
de secreción tipo IV, síntesis de lipopolisacáridos y formación de biopelículas. Se demostró
la importancia del análisis computacional de redes de interacción para caracterizar la
posible función de proteínas hipotéticas o para explorar nuevas funciones de proteínas
conocidas. Así, se proponen funciones para 13 proteínas hipotéticas, para la proteína CheA,
para el regulador transcripcional Nik.R y para la superóxido dismutasa SodB. CheA es una
proteína de conocida función en quimiotaxis; en el presente estudio se determinó su posible
asociación con la exportación de proteínas flagelares durante el ensamblaje del flagelo y

v
con la formación de biopelículas, el cual es un proceso poco comprendido en el contexto de
la patogénesis de H. pylori. NikR fue asociada con la adaptación al medio acido y SodB
con la inhibición de la apoptosis de las células del hospedero. Finalmente, estas nuevas
interacciones proporcionan una base teórica para descubrir nuevos factores asociados a
patogénesis mediante su validación experimental y para el desarrollo de estrategias
terapéuticas novedosas.

vi
Computational prediction and analysis of novel interactions between proteins
involved in Helicobacter pylori pathogenesis
Abstract
Information on protein interactions produced by large-scale experiments in human
and other eukaryotic organisms has contributed to the understanding of biological processes
at system level. However, only a limited amount of interaction data is available for
prokaryotes, the main information was produced from Helicobacter pylori (H. pylori). To
better understand the participation of proteins in H. pylori pathogenesis, we built a protein
interaction network for 127 known proteins involved in H. pylori pathogenesis and
expanded the network by combining experimental interactome data, high confidence
predictions and in silico analysis. The resulting interaction network includes 1,271
interactions between 330 proteins. To predict protein–protein interactions, the interolog and
genomic context methods were used in combination with the information derived from
existing high-throughput interactome assays and data mining. All the proteins present in the
network were clearly located in different clusters, which resulted to have a correspondence
with specific cellular processes. This indicates that our analysis is reliable. To predict novel
pathogenic proteins an analysis of functional context for each protein was carried out. In
this network, 50 pathogenesis candidate proteins were identified with more than 60%
positively predictive value and sensitivity. The in silico analysis predicts the involvement of
candidate proteins in chemotaxis and motility, acid adaptation, response to stress, secretion
system type IV, lipopolysaccharides synthesis and biofilm formation. The importance of
computational analysis in protein interaction networks to characterize the possible function
of unknown proteins or to explore new function of known proteins was demonstrated.
These include 13 hypothetical proteins, the CheA protein, the transcriptional regulator
NikR and superoxide dismutase SodB. CheA, a known chemotactic protein, was shown to
be associated with protein exportation during flagellar assembly, and appears to be involved
in early biofilm formation; such process is still poor understood in the H. pylori
pathogenesis context. NikR was associated to acid adaptation and SodB has been involved
in the inhibition of host cell apoptosis. Finally, these new interactions provide a theoretical
basis for finding new factors in pathogenesis by further experimental validation and for the
development of novel therapeutic strategies.

1
1. Introducción
Las interacciones entre proteínas desempeñan un papel fundamental en varios
aspectos de la organización estructural y funcional de la célula, y van desde la formación de
estructuras macromoleculares y complejos enzimáticos, hasta la regulación y transducción
de señales (Pawson et al., 2000, 2002).
Actualmente, se cuenta con bases de datos que contienen información sobre
interacciones proteína-proteína obtenida mediante métodos experimentales, siendo el
sistema de doble híbrido en levadura y la purificación por afinidad acoplada a
espectrometría de masas los que han sido aplicados a escala genómica. Así mismo, estos
métodos sólo han logrado cubrir una pequeña fracción del proteoma de los organismos en
estudio. La compresión global de la interacción entre proteínas, como en el caso de
eucariotes, no ha alcanzado el mismo nivel de desarrollo para el caso de las bacterias.
Helicobacter pylori (H. pylori) es uno de los organismos mas estudiados a este nivel. Esta
bacteria, infecta a más del 50% de la población mundial y en México se estima que más del
66% de la población adulta está infectada con esta bacteria (Torres et al., 1998). La
infección por H. pylori es la principal causa de gastritis crónica, ulceras pépticas, y es
considerado un factor de riesgo para el desarrollo de adenocarcinoma gástrico y linfomas
gástricos (Montecucco et al., 2001; Peek y Blaser, 2002). A pesar de la importancia de H.
pylori como patógeno humano, queda mucho por aprender de su biología y los mecanismos
por los que causa enfermedad. Los resultados del mapa de interacción de las proteínas
determinado experimentalmente en H. pylori sólo cubren el 46% de su proteoma (Rain et
al., 2001), así mismo se estima que aproximadamente el 50% de estas interacciones son
falsos positivos y negativos, dadas las limitaciones actuales de las técnicas para su estudio
(Cusick et al., 2005). De esta manera, existe una gran cantidad y diversidad de
interacciones entre proteínas asociadas a patogénesis que no han sido determinadas en esta
bacteria. En el presente trabajo, mediante métodos computacionales, se integró información
derivada de las diferentes bases de datos de carácter público, la cual permitió predecir y
analizar interacciones entre proteínas asociadas a patogénesis de H. pylori. El método parte
de la construcción de una red de interacciones entre proteínas a partir de una base de datos
de proteínas de asociación conocida a patogénesis. Analizando el contexto funcional de
cada una de las proteínas que integraron la red se identificaron nuevas interacciones y

2
proteínas asociadas a patogénesis. También se demuestra que este tipo de análisis puede ser
utilizado para visualizar en contexto diferentes procesos biológicos y para generar nuevas
hipótesis de acerca de la función y evolución de las proteínas; principalmente, las
hipotéticas y proteínas no caracterizadas funcionalmente. Finalmente, se proporciona una
lista de 50 proteínas candidato, la cual es evaluada mediante el uso de anotaciones
funcionales disponibles en bases de datos públicas y de la búsqueda en la literatura. Se
concluye discutiendo el papel de las predicciones en la patogénesis de H. pylori y las
limitaciones del presente estudio.

3
2. Antecedentes
2.1. Historia y características generales de Helicobacter pylori
En 1975, Colin y Jones observaron bacilos Gram negativos en el 80% de los
pacientes con úlcera gástrica. En 1983 Marshall y Warren hallaron bacilos con forma de
“S” en biopsias gástricas y las cultivaron usando técnicas para Campylobacter por lo que
los denominaron Campylobacter pyloridis. En 1987 Marshall y Godwin realizaron la
primera revisión de la nomenclatura denominándolo Campylobacter pylori; posteriormente,
mediante estudios filogenéticos se demostró que el microorganismo no pertenece al género
Campylobacter y se sugirió un género diferente; Helicobacter (Goodwin et al., 1989).
2.2. La infección por H. pylori
La infección por H. pylori es la infección bacteriana más común en el ser humano y
por lo menos la mitad de la población mundial está infectada. Su reservorio primario es el
estómago humano y su vía de transmisión no está completamente clara pero parece ser por
la ruta oral-oral y fecal-oral. La magnitud de la prevalencia esta relacionada con las
condiciones sanitarias y económicas, siendo más alta en los países subdesarrollados (Cave,
1997).
2.2.1. Enfermedades producidas por H. pylori
Todos los infectados con H. pylori desarrollan gastritis crónica, la cual es
asintomática. Solo el 20% tendrán una entidad clínica que incluye úlceras pépticas (15%),
cáncer gástrico (1-2%), linfoma gástrico (menos del 1%); por lo tanto la mayoría de los
infectados permanecen asintomáticos (Parkin et al., 1988; Nomura et al., 1994; Shimiyama
et al., 1997; Strobel et al., 1998; Weel et al., 1998; Ye et al., 1999). La aparición de estas
entidades clínicas depende de la interacción entre las características genéticas del huésped,
del medio ambiente y del genotipo infectante; actualmente no hay medios clínicos o de
laboratorio que identifiquen pacientes que podrían tener secuelas por la infección o
pacientes portadores asintomáticos.
En varias zonas a nivel mundial se ha demostrado que H. pylori es la principal causa
de ulcera péptica, siendo el 90% duodenales y hasta 80% gástricas; en otros países la

4
prevalencia puede disminuir como por ejemplo en EE.UU. debido a mayor consumo de
anti-inflamatorios no esteroideos (AINES); la evidencia demuestra que la erradicación del
agente causal reduce la recurrencia de ulceras pépticas (Cover et al., 1995; Everhart et al.,
2000). La proporción de adenocarcinomas gástricos atribuibles a H. pylori va desde el 60 a
90%, diversos estudios han documentado que H. pylori es la principal causa de este tumor
(Guarner et al., 1993; Kobayashi et al., 1993).
El cáncer gástrico es la segunda de las neoplasias más frecuentes del tubo digestivo
en todo el mundo y en México es la cuarta y quinta para el sexo femenino y masculino,
respectivamente (Parkin, 2004). Hasta el momento no se ha identificado que H. pylori
produzca mutaciones o carcinogénesis de manera directa, por lo tanto se considera que este
efecto lo realiza de manera indirecta estando relacionado con una fuerte respuesta
inflamatoria que se desencadena en el estómago infectado la cual se sigue por una serie de
cambios morfológicos y moleculares que pueden darse en el orden: gastritis crónica,
gastritis crónica atrófica, metaplasia intestinal, displasia y cáncer (Correa y Piazuelo, 2008).
Esta secuencia de cambios, sugiere que la inflamación crónica es un pre-requisito para el
desarrollo de tumores. La modulación de este proceso inflamatorio, puede determinar en
gran parte que se produzca o no un desenlace neoplásico. La diferencia entre estas dos
posibilidades es la secreción de ácido: los pacientes con úlcera duodenal presentan
secreción normal o aumentada de ácido, y los pacientes con úlcera gástrica, generalmente
presentan hipocloridia (Correa y Piazuelo, 2008).
La mayoría de las cepas de H. pylori se pueden agrupar en dos fenotipos distintos
con base en la presencia o no de la isla de patogenicidad cag (cag-PAI, por sus siglas en
inglés), la cual es una región del ADN que contiene de 30 a 40 genes, que codifican
factores de virulencia, incluyendo el gen cagA, que codifica la proteína citotóxica CagA
(Cecini et al., 1996; Cover et al., 1995; Evans et al., 1998). Las cepas que expresan esta
proteína son altamente virulentas e inducen mayor producción de citoquinas inflamatorias
como la interleucina 8 (IL–8), e inducen simultáneamente proliferación y apoptosis celular
significativa (Pan et al., 1997; Rudi et al., 1998). Estas proteínas Cag son introducidas por
H. pylori en el interior de la célula epitelial, por un mecanismo secretor mediante el cual la
proteína es inyectada. En el citoplasma la proteína CagA es fosforilada, en grado variable, a
mayor fosforilación, mayor potencial oncogénico de la cepa de H. pylori (Odenbreit et al.,

5
2000; Stein et al., 2000). La proteína CagA fosforilada interactúa con diversas moléculas y
activan proteínas cinasas (Backert et al., 2001; Yamazaki et al., 2003). Cuando una persona
está infectada por H. pylori el riesgo de cáncer gástrico es de 2 a 3 veces mayor, pero si
tiene anticuerpos anti-Cag A el riesgo aumenta hasta 11 veces pudiendo aumentar hasta 20
veces dependiendo del grado de fosforilación. Más aún, si se combina con la alteración
genética del gen que codifica la síntesis de la interleucina 1-β (IL-1β), el riesgo puede
aumentar hasta 87 veces (Covacci et al., 1993; Manetti et al., 1995; Cover et al., 1995). La
IL-1β, es una citocina proinflamatoria y un muy potente inhibidor de la secreción de ácido
(100 veces más potente que el omeprazol) (Susuki et al., 2002; Graham et al., 2003).
2.3. Genómica funcional de H. pylori
2.3.1. Genómica
En la actualidad se ha determinado la secuencia del genoma completo de seis cepas de
H. pylori (acceso a Entrez Genome marzo 2009): 1) la cepa 26695 que fue aislada en el
Reino Unido de un paciente con gastritis (Tomb et al., 1997); 2) la cepa J99 aislada en
Estados Unidos de un paciente con úlcera duodenal (Alm et al., 1999); 3) la cepa HPAG1
aislada en Suecia de un paciente con gastritis atrófica crónica (Oh et al., 2006); 4) la cepa
Shi470, esta proviene de un aislado del antro gástrico de un paciente amerindio de Perú; 5)
la cepa P12, proviene de un aislado clínico de un paciente con úlcera duodenal; y 6) la cepa
G27, fue aislada de una endoscopia practicada a un paciente en Toscana, Italia (Baltrus et
al., 2009).
El cromosoma circular de la cepa 26695 contiene 1,667,867-pb, el de la J99
1,643,831-pb, el de la HPAG1 1, 596,366-pb, el de la Shi470 1,608,548-pb, el de la P12
1,673,813-pb y el de la G27 1,652,983-pb. El promedio en el contenido de G + C es del
39% en todas las cepas secuenciadas. La cepa HPAG1 presenta 43 genes que no son
detectables o están representados de manera incompleta en los genomas de J99 y 26695. De
estos 43 genes, tres tienen relación con sistemas de restricción-modificación de otras
especies bacterianas.
Los productos predichos de 15 de los 43 genes antes mencionados presentan alta
similitud con otras proteínas reportadas en las otras cepas de H. pylori; dos de estos ORFs
codifican para genes asociados con citotoxina cag localizados en una cag-PAI, otros cuatro

6
están relacionados con componentes de sistemas de restricción-modificación, finalmente
uno más está relacionado también con proteínas Cag (CagY), sin embargo se encuentra
localizado fuera de la isla de patogenicidad (Oh et al., 2006). CagY forma parte de una
estructura en forma de pili que es el núcleo del sistema de secreción tipo IV requerido para
la translocación de la proteína CagA en las células del epitelio gástrico; la introducción de
CagA lleva a la activación en cascada de una serie de cinasas que trae consigo cambios
morfológicos en las células del huésped e induce la producción de interleucina-8. Así
mismo, HPAG1 carece de 29 genes presentes en las otras dos cepas; cinco de éstos son
miembros de los sistemas de restricción-modificación, dos son miembros de la cag-PAI,
una proteína periférica de membrana, una integrasa/recombinasa putativa y se desconoce la
función de los otros 20. La mayoría de los genes de J99 y 26695 que están perdidos en
HPAG1 se localizan en las llamadas zonas de plasticidad. Los tres genomas contienen dos
copias de los genes 16S y dos juegos de los genes 5S-23S del ARN ribosomal;
adicionalmente la cepa 26695 contiene un gen extra del 5S ARNr. Del mismo modo, la
cepa G27 posee 58 genes que no están presentes en las cepas HPAG1, J99 y 26695. La
mayoría de estos genes específicos de G27 codifican para proteínas hipotéticas (Baltrus et
al., 2009).
Un análisis de sintenia de las cepas HPAG1, 26695 y J99 muestran que en contraste
a otras bacterias los genes ARNr de H. pylori no están situados continuamente en el
cromosoma, sugiriendo que su regulación es más compleja que la de otros procariotes. Los
tres genomas codifican para 36 especies de ARNs de transferencia, aparentemente
localizados en las mismas regiones del mapa cromosomal en las dos cepas. Otras
características que comparten las cepas son: ausencia de un origen de replicación
identificable, longitud promedio de secuencias codificables, de donde se han detectado
1536 ORFs para la cepa HPAG1, 1576 para 26695 y 1489 para J99 representando el 91%,
el 90% y el 90% del genoma respectivamente (Boneca et al., 2003).

7
2.3.2. Transcriptómica y proteómica
En la patogénesis de H. pylori, diversos factores externos influyen, entre los que
destaca el pH de las secreciones gástricas, el cual es esencial para el establecimiento y
colonización de la mucosa estomacal. Breves exposiciones de la bacteria a pH de 3.5
genera un incremento en la expresión de proteínas de choque térmico y de la colonización
estomacal; estos cambios en la fisiología bacteriana se consideran una respuesta a señales
fisicoquímicas a través de regulación génica (Tomb et al., 1997).
Allan et al., (2001) identificó genes diferencialmente expresados cuando la cepa
26695 fue expuesta a medios con pH 4.0 o pH 7.0. Entre los genes que incrementaron su
expresión a pH bajo fueron: cagA, HP0681 y HP1289 (únicos en H. pylori) los cuales
participan en el sistema de secreción tipo IV; así como también, genes que codifican para
enzimas involucradas en la síntesis de fosfolípidos y lipopolisacáridos. Por otra parte,
existen genes cuya expresión génica disminuye a pH bajo, este es el caso de genes que
codifican para proteínas involucradas en la síntesis de nucleótidos, proteínas flagelares y
proteínas periféricas de membrana. En otro estudio Ang et al., (2001) diseñaron un
dispositivo de microarreglos donde productos de PCR fueron fijados a una membrana;
representando 1543 ORFs de la cepa 26695. Este método fue utilizado para evaluar la
respuesta transcripcional global a las 48 hrs. de crecimiento a pH 5.5 o pH 7.2. Alrededor
de 53 ORFs fueron altamente expresados en ambas condiciones; sugiriendo que bajo
condiciones estresantes estos genes son esenciales en la fisiología bacteriana. Así mismo,
445 ORFs fueron expresados de manera estable en ambas condiciones sin variación
significativa. Esto implica que sus productos probablemente son necesarios para mantener
las funciones básicas en la célula bacteriana. A pH bajo, un total de 80 ORFs incrementaron
significativamente sus niveles de expresión, mientras que en otros 4 ORFs su expresión fue
suprimida. Los restantes 952 ORFs no fueron detectables bajo cualquiera de las
condiciones de pH.
Scott et al., (2007) sometió a H. pylori a una exposición crónica (10 días) en medio
ambiente gástrico y determinó el perfil de expresión génica. La comparación del
transcriptoma “in vitro” e “in vivo” a pH 7.4 reveló dos grupos de genes de función
conocida que incrementan al doble su expresión; tres de estos grupos responden tanto al
medio ácido “in vitro” como a la infección gástrica.

8
Otros investigadores han demostrado que la infección por H. pylori afecta el
equilibrio entre proliferación y apoptosis de las células epiteliales gástricas. En particular
H. pylori ha mostrado tener la capacidad de inducir la transcripción de la ciclina D1, uno de
los reguladores del ciclo celular de las células antes citadas (Hirata et al., 2001). Del mismo
modo, la proteína-1 de membrana de H. pylori ha mostrado capacidad de inducir la sobre
expresión del factor de necrosis tumoral alfa y producir tumores en ratones desnudos;
sugiriendo que este podría ser un mecanismo celular involucrado en el desarrollo de cáncer
gástrico (Suganuma et al., 2001). Otro sistema molecular que es inducido por H. pylori y
que ha mostrado tener relación con el cáncer gástrico es el sistema de la ciclo oxigenasa 2
(COX 2). El análisis de biopsias de 160 pacientes, 97 con dispepsia no ulcerosa (47
negativos y 50 positivos para H. pylori) y 63 con cáncer gástrico, fue realizado para evaluar
tanto la expresión de COX 2 como la proliferación celular y la apoptosis (Wambura et al.,
2002). En pacientes con dispepsia no ulcerosa y positivos para H. pylori, la expresión de
COX 2 fue significativamente más alta en los que los negativos. Por su parte las biopsias de
pacientes con cáncer gástrico presentaron una mayor expresión de COX 2, en las regiones
anatomicas que corresponden al cuerpo y antro gastrico, que los pacientes con dispepsia no
ulcerosa. Estos resultados sugieren que la expresión de COX 2 está asociada con la pérdida
de la integridad en el epitelio gástrico y por lo tanto participa en la carcinogénesis gástrica.
Por otra parte, después de la secuenciación de las cepas J99 y 26695, Jungblut et al., (2000)
realizaron un análisis comparativo del proteoma de H. pylori en las dos cepas antes citadas
y una más, cuyo genoma no ha sido secuenciado (cepa SS1). El análisis del perfil proteico
por 2D-PAGE reveló 1863 bandas para la cepa 26695, 1662 para la J99 y 1448 para SS1;
estos perfiles revelan alta variabilidad en los productos de expresión de cada cepa.
Posteriormente utilizando espectrometría de masas en la variante de desorción/ionización
láser asistida por matriz (MALDI-MS), mediante la cual se obtiene la huella digital de las
proteínas, se identificaron 152 proteínas, incluidos nueve factores de virulencia y 28
antígenos. Se confirmo la expresión de 27 ORFs hipotéticos y 6 ORFs desconocidos y se
observo que sólo un pequeño número de proteínas eran comunes para las cepas estudiadas.
Una aproximación similar fue realizada para caracterizar los subproteomas con el
fin de identificar proteínas importantes en la interacción huésped-patógeno (Backert et al.,
2005). Para ello se realizó el análisis de la composición de dos fracciones proteicas de H.

9
pylori; que corresponden a las proteínas solubles y la otra con las proteínas de la estructura
celular (incluidas las de membrana). Las 50 proteínas más abundantes en el análisis de 2D-
PAGE fueron identificadas mediante su huella digital por MALDI-MS. Numerosas
proteínas periféricas de membrana, 4 proteínas Cag, la citotoxina vacuolizante (VacA),
otros potenciales factores de virulencia y pocas proteínas ribosomales fueron identificadas
en la fracción de proteínas de la estructura celular. En contraste, la enzima catalasa (Kat A),
la gamma-glutamiltranspeptidasa (Ggt), y la proteína activadora de neutrófilos (NapA) se
encontraron exclusivamente en la fracción de proteínas solubles. La identificación de estos
subproteomas es de suma importancia para identificar nuevos factores de virulencia y
antígenos de potencial valor terapéutico o diagnóstico contra las infecciones causadas por
este patógeno.
2.3.3. Interactómica
Con la disponibilidad de la secuencia completa de genomas de varios organismos se
han desarrollado aproximaciones globales para un mejor entendimiento de las funciones de
las proteínas. Así, se han determinado varios mapas de interacción (interactomas) tanto para
eucariotes como en procariotes. El primer mapa de interacciones entre proteínas
determinado para un procarionte fue precisamente el de H. pylori (Rain et al., 2001). La
estrategia experimental fue el sistema de dos híbridos en levadura; se analizaron 261
proteínas contra una biblioteca compleja de fragmentos genómicos de la cepa 26695,
utilizando aproximadamente 10 millones de clonas. De manera general se encontraron más
de 1200 interacciones entre proteínas de H. pylori, cubriendo alrededor del 46.6% del
proteoma. Explorando el mapa de interacciones proteína-proteína se visualizaron diferentes
rutas celulares y se predijo la función de varias proteínas, éste es el caso de sistemas de
quimiotaxis. El genoma de H. pylori presenta tres homólogos de proteínas de E. coli que
están involucradas en rutas quimiotacticas (CheA, CheW and CheY) y proteínas tales como
TlpA similares a receptores de quimiotaxis (MCPs).
Los resultados mostraron que la proteína CheA interactúa con CheY y CheW; y los
distintos dominios interactuantes fueron identificados de manera precisa. El dominio de
CheA que se une a CheY precisamente traslapa con su dominio interactuante; esta
asignación se realizó mediante un estudio estructural del homólogo en E. coli (Welch et al.,

10
1998). El sitio de unión de TlpA para CheW fue localizado en un dominio susceptible de
metilación de una proteína conocida en E. coli, por lo tanto fue implicado en la
transducción de señales quimiotácticas.
El complejo ureasa también fue examinado. La actividad ureasa es esencial para la
patogenicidad de H. pylori y su síntesis requiere dos subunidades estructurales UreA y
UreB, y el producto de cuatro genes accesorios: ureE, ureF, ureG, ureH14; los respectivos
homólogos en E. coli juegan un papel importante en la incorporación de níquel (Cussac et
al., 1992). El mapa de interacción revela la conexión entre UreA y UreB, y una de las dos
interacciones homo-oligoméricas de las subunidades estructurales (UreA); el homodímero
UreB no fue detectado. Otra conexión fue observada entre proteínas accesorias UreH y
UreA mediante sus subunidades estructurales; esto es consistente con el presunto papel de
chaperona de UreH (Mobley et al., 1995). Un nuevo vínculo estructural fue encontrado
entre UreG y UreE. Adicionalmente a la proteínas accesorias, el operón ureasa codifica
para una proteína interna de membrana (UreI); esencial para la adaptación a pH ácido
(Skouloubris et al., 1998) y funcionan como un canal para urea dependiente de pH (Weeks
et al., 2000). El tercer dominio citoplasmático de esta proteína revela una interacción
potencial con la proteína ExbD involucrada en la transmisión de energía mediante fuerza
motora de protones a receptores fuera de la membrana celular.
La combinación de datos genómicos y proteómicos también ha permitido predecir la
función de las proteínas en el contexto de las redes de interacción. El proteoma de H. pylori
contiene un homólogo de la proteína HolB en E. coli. En E. coli esta proteína interactúa con
HolA para formar parte del núcleo de la ADN polimerasa (Dong et al., 1993). Los
resultados en el mapa de interacción de H. pylori muestra un alto puntaje de interacción
entre HolB y un polipéptido no caracterizado (HP1247). Un alineamiento entre HolA de E.
coli y HP1247 remarca similitud a nivel estructural. Así, tomando como referencia a HolA
le fue asignada una función similar a la proteína HP1247 en H. pylori. Varias de las
interacciones también fueron analizadas en el contexto de la estructura tridimensional de las
proteínas. La ARN polimerasa procariota está compuesta de un núcleo (α2ββ’) asociado
con un factor sigma. En H. pylori, las subunidades beta están fusionadas en un polipéptido
sencillo (RpoB). Uno de los dos factores sigma alternativos presentes en H. pylori
(HP1032) es similar a la proteína FliA de E. coli, la cual es necesaria para la transcripción

11
de genes involucrados en la biosíntesis del flagelo (Liu y Matsumura, 1995). De esta
manera se identificó de manera precisa una región de RpoB que interacciona con FliA de H.
pylori. Este trabajo es el único que se ha realizado a gran escala para H. pylori y ha
permitido la identificación de complejos que están presentes en otros organismos, por
ejemplo en E. coli y C. jejuni. Así mismo, se ha complementado el análisis a nivel de
secuencia para inferir homología y estudios de predicción de operones basados en la
localización de genes en el cromosoma. Finalmente, los dominios interactuantes
identificados fueron analizados en el contexto de la estructura tridimensional de las
proteínas. Precisamente, este tipo de análisis contextuales permitieron la asignación de
funciones para proteínas no caracterizadas, proporcionando materia prima para futuros
ensayos biológicos (Rain et al., 2001).
2.4. Métodos para la determinación de interacciones entre proteínas
Las interacciones entre proteínas desempeñan un papel fundamental en varios
aspectos de la organización estructural y funcional de la célula (Pawson et al., 2000; 2002).
Las interacciones entre proteínas básicamente pueden ocurrir en los siguientes niveles: i)
las que ocurren entre dominios de una misma cadena polipeptídica, ii) las que ocurren entre
dominios de diferentes cadenas polipeptídicas en proteínas multiméricas, y iii) las que
ocurren de manera transitoria en complejos formados entre proteínas independientes
(Teichmann et al., 2001).
Hasta la actualización correspondiente al 23 de marzo del 2009 en el Centro
Nacional para la Información Biotecnológica de EE.UU. (NCBI, por sus siglas en ingles) se
dispone de las secuencias de aminoácidos codificadas por 867 genomas secuenciados
completamente. Además, se dispone de un gran número de datos obtenidos a través de
microarreglos de ADN; e información sobre interacciones proteína-proteína obtenida
mediante los métodos de dos híbridos en levadura, la purificación por afinidad y los
microarreglos de proteínas acoplados a espectrometría de masas, cristalografía de rayos X,
co-inmunoprecipitación, resonancia magnética nuclear (RMN) y resonancia de plasmones
de superficie (BIAcore). No obstante la gran cantidad de información proporcionada por
estos métodos, debido a dificultades para su escalamiento sólo dos de ellos han sido
aplicados a escala genómica: el sistema de dos híbridos en levadura y la purificación por

12
afinidad acoplada a espectrometría de masas. Ejemplos de aplicación de estos métodos son
los mapas de interacción entre proteínas determinados para Saccharomyces cerevisiae (Uetz
et al., 2000; Ito et al., 2001; Gavin et al., 2002; Ho et al., 2002), Caenorhabditis elegans
(Walhout et al., 2000), Helicobacter pylori (Rain et al., 2001), Drosophila melanogaster
(Giot et al., 2003) y Campilobacter jejuni (Parish et al., 2007). Los resultados
proporcionados por estas aproximaciones de análisis global junto con la de experimentos a
pequeña escala han dado lugar a la creación de una amplia variedad de bases de datos, al
tiempo que la visión sobre la función de las proteínas ha evolucionado. Desde el punto de
vista clásico, la función de una proteína, está determinada por su acción local; mientras que
en un contexto sistémico la función de una proteína se define como el entramado de
interacciones con otras moléculas. Esta nueva forma de comprender a los sistemas
biológicos desde un punto de vista contextual es el objeto de estudio de la biología de
sistemas.
Análisis de genómica funcional han mostrado que la complejidad de los organismos
no depende del número de proteínas codificadas en sus genomas, sino del número de
interacciones entre ellas (Claverie, 2001). Así, la planta Arabidopsis thaliana, el nemátodo
Caenorhabditis elegans, la mosca Drosophila melanogaster y el ser humano poseen un
número similar de genes (25,498; 19,099; 14,100 y 30,000-35,000; respectivamente). Por
ello, la optimización y el desarrollo de métodos para predecir y analizar los detalles acerca
de las interacciones entre proteínas tanto a nivel atómico como a escala genómica sigue
siendo una de las áreas en constate desarrollo (Edwards et al., 2002; Jansen et al., 2002).
Una vez dilucidadas las redes de interacciones o “interactoma” en sistemas celulares
modelos, será posible el diseño de fármacos más específicos y la ingeniería de procesos
celulares (Serrano, 2007).
2.4.1. Métodos experimentales
2.4.1.1 Sistema de doble híbrido en levadura (Y2H)
El desarrollo de la técnica de Y2H se fundamenta en que activadores
transcripcionales eucariotas tienen al menos dos tipos de dominios, uno que se une
directamente a la región promotora (dominio de unión al ADN) y otro que activa la

13
transcripción (dominio de activación). Se ha demostrado que la separación del dominio de
unión (BD) y del dominio de activación (AD) da como resultado una inactividad
transcripcional, la cual es restablecida si el BD se asocia físicamente con un AD (Fields y
Song, 1989). Así, un segmento de ADN que codifica para una proteína de interés es
fusionado al respectivo que codifica para un BD (construcción cebo o carnada). Un
procedimiento similar es utilizado para crear una quimera que incluye un segmento
codificante para otra proteína de interés fusionado a un AD (construcción presa). Ambas
construcciones son clonadas por separado en vectores de expresión e introducidas en una
célula de levadura. Una vez expresadas las proteínas fusionadas y si éstas interaccionan el
factor transcripcional podrá ensamblarse, activando la transcripción de un gen reportero
(Figura 1). Los sistemas Y2H más ampliamente utilizados son los basados en GAL4/LexA,
donde GAL4 controla la expresión del gen LacZ que codifica para la beta-galactosidasa.
Para realizar un escaneo a nivel genómico dos variantes del sistema Y2H han sido
desarrolladas: escaneo basado en una matriz y escaneo sobre una biblioteca. En la primer
variante de las antes mencionadas, se crea una matriz de clonas “presa”, donde cada clona
expresa una proteína “presa” particular en un pocillo de una placa de reacción. Entonces,
cada proteína “cebo” es acoplada sobre un arreglo de clonas “presa” y aquellas donde dos
proteínas interaccionan son seleccionadas en base a la expresión de un gen reportero y a su
posición en la placa de reacción. En la segunda variante, cada “cebo” es escaneado contra
una biblioteca que contiene segmentos al azar de cADN u ORFs. Así, aquellas proteínas
que interaccionan serán seleccionadas por su capacidad de crecer sobre sustratos
específicos y caracterizadas mediante secuenciación. Las principales ventajas del método
Y2H son: rapidez y sencillez, detección de interacciones transitorias y que es realizado in
vivo.

14
Figura 1. Método de doble híbrido en levadura.
Sin embargo el método también presenta desventajas, entre ellas: interferencia entre
los dominios fusionados, detección de interacciones uno a uno dejando de lado los efectos
cooperativos, su capacidad de detectar interacciones transitorias la hace susceptible de
uniones inespecíficas, los procesos de modificación post-traduccional y de plegamiento
pueden variar entre la levadura y otros organismos. Esto hace difícil escanear proteínas de
mamíferos y de procariontes mediante Y2H, así como también proteínas de membrana. Se
pueden utilizar diferentes técnicas de análisis in vitro para validar los resultados de Y2H
pueden (Shoemaker y Panchenko, 2007).
2.4.1.2. Purificación por afinidad en tándem acoplada a espectrometría de masas
(TAP-MS)
Este método consiste en la generación de una construcción que incluye un segmento
génico que codifica para una proteína de interés con un segmento que codifica para la
etiqueta TAP. Una etiqueta TAP está formada por la proteína A de Staphylococcus con
dominios de unión para inmunoglobulina G (IgG), y un péptido de unión a calmodulina
separado por un sitio de rompimiento para la proteasa del virus del tabaco (Rigaut et al.,
1999; Puig et al., 2001). Posteriormente, la construcción se expresa en una célula de

15
levadura donde esta puede formar complejos con otras proteínas. En el primer paso de la
purificación, la proteína A se une a IgG que se encuentra fija a una matriz; después se
procede a realizar un lavado para eliminar los contaminantes, se adiciona la proteasa y se
rompe la unión entre la proteína A y la matriz de IgG. El producto del primer paso es
posteriormente eluido mediante su incubación con perlas cubiertas de calmodulina en
presencia de calcio; nuevamente se realiza un lavado y los complejos formados con la
proteína de interés, son purificados bajo condiciones nativas, por lo tanto, manteniendo la
integridad del complejo. Los componentes del complejo formado son resueltos por 2D-
PAGE, digeridos enzimáticamente por proteasas y los fragmentos son identificados
mediante espectrometría de masas (Figura 2). La principal ventaja de este método es su
capacidad para determinar complejos multiproteicos, a diferencia de Y2H donde sólo
resuelve interacciones uno contra uno, dejando de lado los efectos cooperativos. Entre las
desventajas del método se encuentran: sólo se detectan interacciones estables, por lo cual
las transitorias no se detectan, la proteína no se expresa en el momento de la lisis y
finalmente, la interferencia del TAP-cassette en la interacción (Shoemaker y Panchenko,
2007).
Figura 2. Método de purificación por afinidad en tándem acoplado a espectrometría de masas.

16
2.4.1.3. Interacciones entre proteínas a nivel atómico
La información a nivel atómico más detallada de las interacciones entre proteínas
puede ser proporcionada por la cristalografía de rayos X y espectroscopía de resonancia
magnética nuclear, pero el número de complejos determinados por estas técnicas sigue
siendo bajo (Tong et al., 2001). Al mismo tiempo, la caracterización en tiempo real de
interacciones entre proteínas in vivo puede ser alcanzada mediante técnicas
espectroscópicas, las cuales requieren la adición de una marca fluorescente a la proteína de
interés (Lippincott-Schwartz y Patterson, 2003; Piehler, 2005). Otro método efectivo para
determinar interacciones entre ligandos solubles y receptores inmovilizados sobre una placa
de oro es la llamada resonancia de plasmones de superficie (Cooper, 2003; Karlsson, 2004).
Recientemente, se han desarrollado nuevos métodos para analizar la interacción entre
proteínas a nivel atómico. Éste es el caso de la microscopía de fuerza atómica (Yang et al.,
2003) y de técnicas fluorescentes, que son capaces de caracterizar cambios
conformacionales en las proteínas durante la interacción (Margittai et al., 2003).
2.4.2. Métodos computacionales
En el análisis funcional de un genoma, un paso crítico es la comprensión de las
interacciones entre las proteínas que codifica (Eisenberg et al., 2000). Por ello se ha
desarrollado un conjunto de métodos teóricos para abordar el problema de la predicción de
interacciones entre proteínas a partir del análisis de sus secuencias, de información de los
genomas y de datos cristalográficos de complejos macromoleculares. De manera general,
los métodos utilizan como hipótesis que si se conoce el modo de interacción entre dos
proteínas o dos dominios de proteínas, cualquier interacción que pueda ocurrir entre
proteínas homólogas involucra contactos del mismo tipo (Teichmann et al., 2001). Sin
embargo, existen algunos casos en los que proteínas homólogas poseen diferentes modos de
interacción. Por ejemplo, la enzima nucleótido difosfato quinasa se presenta en la
naturaleza en forma de hexámero o tetrámero dependiendo de la especie (Morera et al.,
1994).
El reconocimiento proteína-proteína está determinado por las propiedades físicas y
químicas de la superficie interfacial, la cual ha sido caracterizada en términos de su
geometría (tamaño, forma y complementariedad) y de su naturaleza química (tipo de grupo

17
químico y aminoácido, hidrofobicidad, interacciones electrostáticas y enlaces por puente de
hidrógeno). El análisis de las superficies de contacto entre proteínas o región interfacial
tuvo sus inicios con el trabajo de Chothia y Janin (1975). A partir de ese año y hasta la
fecha, siguen apareciendo diferentes trabajos sobre el tema (Janin y Chothia, 1990;
Lawrence y Colman, 1993; Jones y Thornton, 1997; Chakrabarti y Janin, 2002; Headd et
al., 2007; Yan et al., 2008).
Se han desarrollado métodos que utilizan la información contenida en los genomas
o también llamados métodos basados en el contexto genómico, entre los principales están:
fusión génica, perfiles filogenéticos y vecindad génica. Por ejemplo, un evento de fusión
génica representa la fusión física de dos genes relacionados a nivel funcional y separados
físicamente, para producir un solo gen multifuncional (Figura 3). Dandekar et al., (1998)
utilizaron la información asociada a la conservación del orden de genes ortólogos y la
arquitectura de operones en 6 genomas de procariotas y 3 de arqueas. Con este método, se
identificaron para cada genoma alrededor de 100 proteínas involucradas en interacciones
físicas directas previamente descritas a nivel experimental. El método desarrollado por
Pellegrini et al. (1996), denominado perfiles filogenéticos, se basó en la co-evolución de
proteínas que interactúan, dicho de otra manera presencia-ausencia de genes ortólogos en
genomas diferentes. Si el patrón de proteínas ortólogas (presencia-ausencia) se conserva en
organismos de la misma especie, se debe probablemente a que una de las proteínas no
puede ejercer su función sin la otra (Figura 3). Huynen et al. (2000) aplicaron este método
al genoma de Mycoplasma genitalium, prediciendo 34% de los pares como interactuantes y
un 29% adicional pertenecientes a la misma vía metabólica o proceso funcional. Así
mismo, métodos basados en la fusión de dominios se fundamentan en la hipótesis de que
cuando dos proteínas A y B contienen dominios homólogos a dominios diferentes de una
tercera proteína en otro organismo, pero A y B no son homólogas entre sí, entonces A y B
interaccionan (Marcotte et al., 1999; Enright et al., 1999).

18
Figura 3. Métodos computacionales de predicción de interacciones entre proteínas basados en el
contexto genómico. (A) Representación del método de vecindad génica para 8 genomas completos, mostrando
un par de genes (marcados con color rojo y azul) que están físicamente próximos en los 8 genomas. Además,
se muestra en un par de genomas un evento de fusión génica entre dos genes (marcados en color amarillo y
azul claro). (B) Representación de perfiles filogenéticos de los genes de la representación previa. Los tres
pares de genes tienen los mismos patrones de co-ocurrencia en los 8 genomas, por tanto, sus productos
podrían interaccionar físicamente. (C) Representación de dos alineamientos de secuencias para un par de
familias de proteínas, se muestran las regiones conservadas marcadas en color rojo y azul, respectivamente.
Para cada familia de proteínas, mutaciones co-relacionadas (marcadas en color verde) están presentes en sub-
arboles idénticos, indicando que estos sitios podrían estar involucrados en interacciones entre proteínas de
cada una de estas familias.
La limitación de los métodos basados en el contexto genómico radica en que sólo
son aplicables a genomas secuenciados completamente, para tener la certeza de la ausencia
de genes específicos; por otro lado son aplicables solamente en bacterias, donde el orden de
los genes es una característica relevante; y por último dependen de la calidad del
alineamiento múltiple de las proteínas en estudio (Valencia y Pazos, 2002).

19
En 2001, se desarrolló un método de predicción de patrones de secuencia
característicos, observados con regularidad, en pares de proteínas cuya interacción ha sido
comprobada de forma experimental (Sprinzak y Margalit, 2001). Una de las proteínas posee
un patrón de secuencia característico, definido en la clasificación InterPro (Apweiler et al.,
2001), y la otra proteína que interacciona un segundo patrón característico. A este conjunto
de pares de patrones característicos se les denominaron patrones de secuencia
correlacionados.
Wojcik y Schächter (2001) combinaron métodos de búsqueda de similitud de
secuencia y agrupamiento de secuencias (“clustering”) con la información contenida en las
bases de datos acerca de dominios en interacción proteína-proteína. Este método se ha
empleado para predecir el mapa de interacción de proteínas en la bacteria Escherichia coli,
utilizando la información previa del mapa de interacción de H. pylori. Pazos y Valencia
(2001, 2002) combinaron información a nivel de secuencia y genómica para calcular las
distancias evolutivas entre proteínas que pertenecen a familias relacionadas y para calcular
la correlación de mutaciones entre posiciones de un alineamiento múltiple de secuencias.
Esta idea se basa en observaciones previas que indican una correspondencia entre los
árboles filogenéticos de familias de proteínas interactuantes no homólogas (ejemplo: la
insulina y receptor para insulina). Este método se ha aplicado a gran escala, utilizando
67,000 pares de proteínas de E. coli, de los cuales 2,742 fueron predichos correctamente. Se
ha desarrollado un método similar al anterior; en éste se alinean árboles filogenéticos,
representados por matrices, para definir proteínas específicas involucradas en interacciones.
Este método se ha aplicado a más de 18 familias de proteínas con resultados satisfactorios
(Ramani y Marcotte, 2003).
Lu et al. (2002) desarrollaron un método denominado MULTIPROSPECTOR que
se basa en la metodología conocida con el nombre de modelaje por hilvanado (del inglés
“threading”). Esta metodología es utilizada para la predicción de estructura cuaternaria, y
consta de dos fases. Una primera fase ejecuta el método de “threading” PROSPECTOR,
para generar un conjunto de estructuras 3D potenciales de la proteína en estudio. En una
segunda fase, re-evalúan la compatibilidad de las estructuras 3D generadas con las
estructuras molde que forman parte de complejos cristalográficos determinadas
experimentalmente. El método fue evaluado utilizando un conjunto de 40 homodímeros, 15

20
heterodímeros y 69 monómeros, explorados contra una selección de 2,478 estructuras
representativas del banco de datos de proteinas (PDB, por sus siglas en inglés). El método
predijo correctamente 36 homodímeros, 15 heterodímeros y 65 monómeros. Además, se
evaluaron todas las posibles interacciones para proteínas de levadura, de un total de 2,865
predicciones utilizando MULTIPROSPECTOR (Lu et al., 2002), 1,138 están documentadas
en la base de datos de interacciones entre proteínas (DIP, por sus siglas en inglés).
Sprinzak et al., (2003) propusieron un método para evaluar la exactitud de los datos
experimentales sobre interacciones proteína-proteína obtenidos por el método de doble
híbrido. Estos investigadores sugieren que el 50% de las interacciones proteína-proteína
identificadas en la levadura S. cerevisiae corresponden a casos positivos y que el número
total de interacciones para este organismo oscila entre 10,000 y 16,600 interacciones.
Por último, Goffard et al. (2003) desarrollaron el método IPPRED para predecir
interacciones entre proteínas basado en la similitud de secuencias amino acídicas. Para
inferir las interacciones entre proteínas IPPRED utiliza el programa BLAST (Altschul et
al., 1990), datos experimentales documentados en la base de datos BIND (Bader et al.,
2003) y en la literatura científica. Además, IPPRED está disponible a través de una
interfase en el sitio de internet http://cbi.labri.fr/outils/ippred/.
2.5. Bases de datos de interacciones entre proteínas
Para documentar y describir las interacciones entre proteínas, se ha creado un grupo
de bases de datos que incluyen toda la información obtenida por los diferentes métodos
experimentales descritos anteriormente. A continuación se describe de manera general las
principales bases de datos, concentrando la atención en el tipo de datos que contienen y
cómo accederlas.

21
2.5.1. BIND (Biomolecular Interaction Network Database)
http://bond.unleashedinformatics.com/
Esta base de datos, documenta interacciones moleculares que incluyen proteína-
proteína, proteína-ARN, proteína-ADN, proteína-moléculas pequeñas, complejos
moleculares, y vías metabólicas. Los datos experimentales obtenidos por doble-híbrido,
espectrometría de masas, cristalografía, bibliotecas de fagos se incluyen en esta base de
datos (Bader et al., 2003). Actualmente (marzo 2009), BIND documenta 158,489
interacciones, y 3,705 complejos.
2.5.2. DIP (Database of Interacting Proteins)
http://dip.doe-mbi.ucla.edu
Esta base de datos, documenta interacciones moleculares, proteína-proteína,
determinadas experimentalmente por los métodos de doble-híbrido, inmunoprecipitación,
ensayos de bloqueo con anticuerpos monoclonales y anotaciones extraídas de la literatura
científica, entre otros. DIP está implementada como una base de datos relacional, la cual se
define como aquella en donde todos los datos visibles al usuario están organizados
estrictamente como tablas de valores, y en donde todas las operaciones de la base de datos
operan sobre estas tablas. En DIP la base de datos relacional esta formada por cuatro tablas.
Una primera con información sobre las proteínas, una segunda sobre interacciones proteína-
proteína, una tercera que describe detalles sobre los experimentos, y la cuarta tabla contiene
la lista de todas las referencias en Medline. DIP permite la representación visual de las
interacciones así como navegar a través de la red de interacciones proteicas mediante
internet (Xenarios et al., 2001; 2002). Hasta marzo del 2009 el número de proteínas
contenidas en DIP ascendía a 20,728 y el número de interacciones a 57,684.
2.5.3. MINT (Molecular INTeraction database)
http://cbm.bio.uniroma2.it/mint/index.html
MINT contiene información sobre complejos moleculares, interacción entre
dominios, modificaciones enzimáticas de las proteínas interactuantes, constantes de unión y
otros datos cinéticos. Toda la información contenida en MINT es “curada” de manera

22
manual por expertos, y está organizada en una base de datos relacional (Zanzoni et al.,
2002). Actualmente (marzo 2009), MINT tiene documentadas 110,164 interacciones.
2.5.4. 3did (3D interacting domains)
http://gatealoy.pcb.ub.es/3did/
Esta base de datos permite explorar los detalles de interacciones entre dominios
derivados de estructuras tridimensionales (Stein et al., 2005). Para cada dominio, se
proporciona una vista de todas sus interacciones con otros dominios. En algunos casos se
muestran comparaciones estructurales entre las superficies interfaciales de dominios
interactuantes, revelando la variación de interacciones entre pares de familias de dominios.
InterPreTS es un servicio en línea asociado a 3did que predice interacciones entre dominios
basado en la similitud a nivel de secuencia con proteínas de una base de datos de dominios
interactuantes (Aloy y Russell, 2003). Hasta marzo del 2009, 3did tiene documentadas
115,559 interacciones.
2.5.5. IntAct (IntAct database)
http://www.ebi.ac.uk/intact/site/index.jsf
La principal meta de este proyecto es proporcionar un repositorio público con
información referente a interacciones proteína-proteína derivadas experimentalmente
(Kerrien et al., 2007). La base de datos hace un uso extensivo de vocabularios controlados,
los cuales permiten obtener una descripción consistente con los detalles experimentales que
fueron utilizados para generar la información. Para soportar búsquedas generalizadas, estos
vocabularios tienen una estructura jerárquica y dentro de lo posible, son utilizados sistemas
de referencia tales como la base de datos de taxonomía del NCBI o la base de datos de
Ontologías génicas (GO). Actualmente (marzo 2009), IntAct tiene documentadas 189,082
interacciones.
2.5.6. HPRD (The Human Protein Reference Database)
http://www.hprd.org/
HPRD contiene información que ha sido manualmente curada para la mayoría de las
proteínas del ser humano (Peri et al., 2003). Información concerniente a proteínas

23
involucradas con enfermedades humanas es anotada y enlazada a la base de datos de
herencia mendeliana en humanos (OMIM database). El NCBI proporciona un enlace a
HPRD a través de su bases de datos de proteínas de humano (Entrez gene y RefSeq). La
base de datos despliega información sobre las funciones de las proteínas de humano,
incluyendo: interacciones proteína-proteína, modificaciones post-traduccionales, relaciones
enzima-sustrato y asociaciones con enfermedades. Toda la información es “curada” de
manera manual por expertos a través de análisis bioinformáticos de las secuencias de las
proteínas. Actualmente (marzo 2009), HPRD tiene documentadas 38,167 interacciones.
2.6. Calidad de los métodos de determinación de interacciones entre proteínas
El método estándar para evaluar la calidad de las interacciones detectadas a nivel
experimental se basa en que se debe detectar la misma interacción por diferentes métodos
(Schwikowski et al., 2000). Por ejemplo en S. cerevisiae, se han determinado a nivel
experimental y de manera independiente varios interactomas, lo que ha producido un total
de 80,000 interacciones. Sin embargo, sólo 2,400 interacciones fueron comunes a todos los
interactomas de S. cerevisiae (von Mering et al., 2002). Existen varias explicaciones para
esto; posiblemente los métodos no han alcanzado aún su máximo desarrollo; varios de estos
métodos producen una gran cantidad de falsos positivos y negativos, y algunos otros
presentan problemas para ciertos tipos de interacciones. Cada uno de los métodos para
determinar las interacciones entre proteínas, producen una distribución de interacciones
única con respecto a las categorías funcionales de las mismas. Estas diferencias en la
cobertura sugieren que los métodos presentan debilidades y fortalezas. Así, los métodos de
purificación de complejos resuelven pocas interacciones para proteínas involucradas en
transporte y en recepción de señales, debido posiblemente a que estas fracciones del
proteoma por lo general son ricas en proteínas de membrana, las cuales son difíciles de
purificar. Similarmente, el sistema de Y2H, a diferencia de otros métodos, difícilmente
detecta interacciones entre proteínas involucradas en el proceso de traducción (von Mering
et al., 2002).
Otro enfoque para validar la interacciones combina la información que brindan
métodos experimentales independientes con datos 3D obtenidos por cristalografía de rayos
X (Edwards et al., 2002). Siguiendo esta línea de razonamiento, la integración de diferentes

24
bases de datos o diferentes métodos, contribuirá a validar las anotaciones en las mismas
(Gerstein et al., 2002). Un ejemplo de ello lo constituye el método propuesto por Tong et
al. (2002) que consistió en combinar la metodología de despliegue en fagos (“phage
display”) con Y2H. Primeramente, mediante una biblioteca de fagos, se realizó una
búsqueda de secuencias consenso en proteínas de levadura capaces de unirse a dominios de
reconocimiento de péptidos desplegados en la superficie de cada fago. Como resultado de
esta búsqueda se obtiene un entramado de interacciones que conecta a proteínas que poseen
una secuencia consenso con aquellas que poseen dominios de reconocimiento específico
para esas secuencias, a la vez, define sitios de unión en algunas de las proteínas. Un
segundo entramado de interacciones se obtiene de la búsqueda de proteínas que
interaccionan con los dominios de reconocimiento de las secuencias consenso, utilizando el
método de doble-híbrido. Este método ha sido aplicado en la búsqueda de proteínas que
interactúan con dominios SH3; estos dominios se encuentran principalmente en proteínas
involucradas en vías de señalización.

25
3. Planteamiento del problema
El conocimiento del contexto de las interacciones de una proteína es crucial para
comprender las funciones celulares. Sin embargo, los métodos experimentales para
determinar a gran escala las interacciones entre proteínas sólo cubren una pequeña fracción
del proteoma de una célula bajo condiciones específicas. En el mismo contexto, para el
caso de las bacterias, incluido H. pylori, la compresión del papel biológico de las
interacciones entre proteínas no ha alcanzado el mismo nivel de desarrollo como en el caso
de organismos eucariontes, como el humano y la levadura S. cerevisiae. Más aún, para
microorganismos patógenos de humano poco se sabe acerca del papel que juegan en la
patogénesis las interacciones entre proteínas.
4. Justificación
En muy pocas especies bacterianas, incluyendo a H. pylori, se han analizado las
interacciones entre proteínas a nivel global. H. pylori es la bacteria que más personas
infecta en el mundo. En México el 66% de la población adulta está infectada con esta
bacteria. La infección por H. pylori es la principal causa de gastritis crónica, ulceras
pépticas y adenocarcinoma gástrico, también produce linfomas gástricos asociados
amucosa (MALT), anemia ferropénica, y actualmente es tema de discusión que sea la causa
de por lo menos el 50% de los casos de púrpura trombocitopénica idiopática. Dada la
importancia de la H. pylori como patógeno, es necesario comprender los mecanismos
moleculares por los que causa enfermedad. En este sentido, los resultados del mapa de
interacción entre proteínas de H. pylori sólo cubren el 46% de su proteóma, así mismo se
estima que aproximadamente el 50% de los resultados obtenidos en experimentos de
interactómica son falsos positivos o falsos negativos. De tal manera que existe una amplia
cantidad y diversidad de información por confirmar y explorar, a fin de elucidar algunas de
las interacciones entre proteínas asociadas a patogénesis que no han sido determinadas en
esta bacteria. La construcción de un modelo de la red interacciones entre proteínas
asociadas a patogénesis de H. pylori revelará información detallada acerca de los procesos
celulares activados por este patógeno durante el proceso de infección. Entre las posibles
aplicaciones prácticas de este trabajo están la identificación de potenciales blancos para el
diseño racional de fármacos y vacunas, la ingeniería de procesos celulares y el desarrollo de
nuevas estrategias de tratamiento.

26
5. Objetivos
5.1. General
Identificar nuevas interacciones entre proteínas de participación conocida en patogénesis de
H. pylori.
5.2. Específicos
� Construir in silico una red de interacciones, a partir de proteínas que se conoce que
participan en la patogénesis de H. pylori.
� Identificar nuevas interacciones de proteínas asociadas a patogénesis de H. pylori.
� Predecir nuevas proteínas asociadas a patogénesis de H. pylori.
� Analizar el papel de las interacciones predichas en la patogénesis de H. pylori.
� Proponer una lista de proteínas candidato con probable asociación a patogénesis de
H. pylori.
6. Hipótesis
“El análisis de redes de interacción entre proteínas permite asignar funciones adicionales a
proteínas de función patogénica conocida y, predecir nuevas proteínas con probable
asociación a patogénesis de H. pylori”.

27
7. Metodología
7.1. Obtención de las proteínas con participación conocida en la patogénesis (proteínas
raíz)
De la base de datos MvirDB (http://mvirdb.llnl.gov/) se obtuvieron todas aquellas
proteínas con participación conocida en la patogénesis de H. pylori. Esta base de datos
contiene información sobre proteínas que funcionan como toxinas, factores de virulencia y
resistencia a los antibióticos (Zhou et al., 2006).
7.2. Construcción de la red de interacciones entre proteínas de H. pylori
Mediante el sistema de análisis STRING (von Mering et al., 2009) y a partir de las
proteínas raíz se obtuvo una red de interacciones. Los parámetros para la generación de la
red fueron: Active Prediction Methods (Neighborhood, Gene Fusion, Co-occurrence, Co-
expression, Text mining, Experiments y Databases); confidence score (0.7=high
confidence); network depth (1) y edge scaling factor (80%). Esta configuración implica la
obtención de una red de interacciones con un alto nivel de confiabilidad (Figura 4).
Figura 4: Construcción de la red de interacción a partir de las proteínas raíz obtenidas de la base de datos
MvirDB. Las proteínas raíz son introducidas en STRING, el cual construye la red a partir de las citadas
proteínas mediante la adición de sus compañeros de interacción directos.
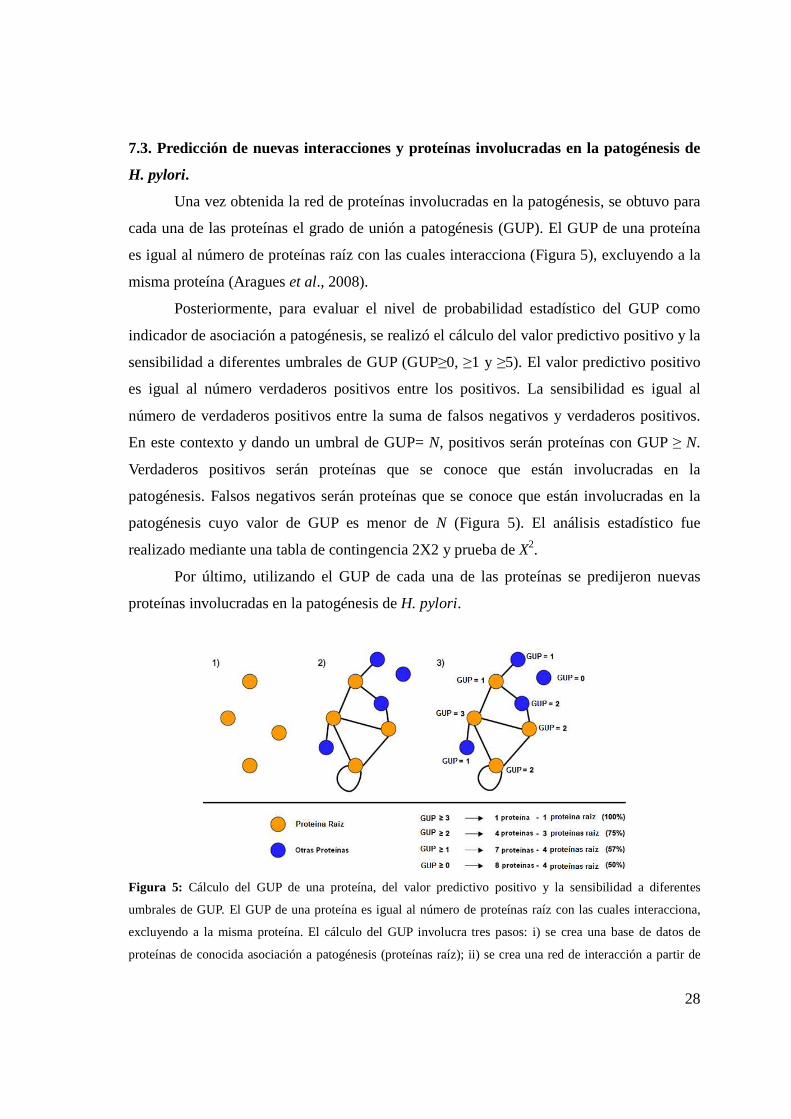
28
7.3. Predicción de nuevas interacciones y proteínas involucradas en la patogénesis de
H. pylori.
Una vez obtenida la red de proteínas involucradas en la patogénesis, se obtuvo para
cada una de las proteínas el grado de unión a patogénesis (GUP). El GUP de una proteína
es igual al número de proteínas raíz con las cuales interacciona (Figura 5), excluyendo a la
misma proteína (Aragues et al., 2008).
Posteriormente, para evaluar el nivel de probabilidad estadístico del GUP como
indicador de asociación a patogénesis, se realizó el cálculo del valor predictivo positivo y la
sensibilidad a diferentes umbrales de GUP (GUP≥0, ≥1 y ≥5). El valor predictivo positivo
es igual al número verdaderos positivos entre los positivos. La sensibilidad es igual al
número de verdaderos positivos entre la suma de falsos negativos y verdaderos positivos.
En este contexto y dando un umbral de GUP= N, positivos serán proteínas con GUP ≥ N.
Verdaderos positivos serán proteínas que se conoce que están involucradas en la
patogénesis. Falsos negativos serán proteínas que se conoce que están involucradas en la
patogénesis cuyo valor de GUP es menor de N (Figura 5). El análisis estadístico fue
realizado mediante una tabla de contingencia 2X2 y prueba de X2.
Por último, utilizando el GUP de cada una de las proteínas se predijeron nuevas
proteínas involucradas en la patogénesis de H. pylori.
Figura 5: Cálculo del GUP de una proteína, del valor predictivo positivo y la sensibilidad a diferentes
umbrales de GUP. El GUP de una proteína es igual al número de proteínas raíz con las cuales interacciona,
excluyendo a la misma proteína. El cálculo del GUP involucra tres pasos: i) se crea una base de datos de
proteínas de conocida asociación a patogénesis (proteínas raíz); ii) se crea una red de interacción a partir de

29
las proteínas raíz; y iii) se calcula el GUP para cada una de las proteínas presentes en la red de interacciones.
En el ejemplo proporcionado, se observa que proteínas con un alto valor de GUP son más probables a estar
asociadas a patogénesis en comparación a aquellas con bajos valores de GUP.
7.4. Análisis del papel de las predicciones en la patogénesis
El análisis funcional de las predicciones se realizó mediante la búsqueda de
anotaciones en las bases de datos Uniprot (The UniProt Consortium, 2008), Pfam
(pfam.sanger.ac.uk) y con el sistema de minería de datos iHop (Hoffmann y Valencia,
2004).
En lo que respecta al análisis de propiedades funcionales a nivel de redes de
interacción, éste fue realizado mediante el software Cytoscape (Shannon et al., 2006) y sus
complementos MCODE (Bader y Hogue, 2006) y BINGO (Maere et al., 2005).

30
8. Resultados
El análisis de redes de interacción entre proteínas es una de las principales
aproximaciones en la caracterización funcional de las proteínas, ya que permite asignar
funciones a proteínas aún no descritas o nuevas funciones a proteínas conocidas. En el
presente trabajo, mediante el análisis de redes de interacción entre proteínas, se realizó la
predicción de nuevas interacciones entre proteínas involucradas en la patogénesis de H.
pylori. Este análisis se sostiene en la hipótesis de que dentro de una red de interacción,
aquellas proteínas que están involucradas en un mismo proceso biológico tienden a
presentar alta conectividad entre ellas. Por lo tanto, proteínas de H. pylori que
interaccionan, de manera frecuente, con aquellas de conocida participación en su
patogénesis son probablemente proteínas que también están involucradas en dicho proceso.
En contraste, con aquellas proteínas que presentan pocas o nulas interacciones con
proteínas de participación conocida involucradas en la patogénesis. El sistema de análisis
STRING 8.0 (Jensen et al., 2009) fue utilizado para construir una red de interacciones a
partir de proteínas de participación conocida en la patogénesis (denominadas proteínas
raíz). STRING ejecuta una serie de algoritmos de predicción basados en contexto genómico
y transfiere interacciones determinadas, a nivel experimental, de organismos modelo hacia
otras especies, mediante la ortología de las respectivas proteínas (interólogos). Los
resultados arrojados por STRING, además de contener interacciones directas, también
incluye interacciones funcionales (indirectas). Es decir, proteínas que comparten algún
sustrato, proteínas que se regulan entre sí a nivel transcripcional y proteínas que participan
en un complejo multi-proteico. Así, en el presente estudio STRING fue capaz de detectar
interacciones entre proteínas asociadas a patogénesis que previamente ya habían sido
descritas mediante experimentos bioquímicos en H. pylori y otras bacterias. Este es el caso
del complejo ureasa, del sistema de transporte ExbD-ExbB-TonB, del sistema de motilidad
y quimiotaxis, y del sistema de secreción tipo IV; todos ellos actúan como complejos multi-
proteicos. Estos ejemplos ilustran las capacidades analíticas del sistema de análisis antes
citado. STRING, en su versión número ocho, incluye información de 2.5 millones de
proteínas pertenecientes a 630 organismos (Jensen et al., 2009).
El conjunto de proteínas de participación conocida en la patogénesis de H. pylori
(proteínas raíz) fue obtenido de la base de datos MvirDB (http://mvirdb.llnl.gov/). Esta base

31
de datos incluye proteínas o genes asociados a toxinas y factores de virulencia. La
información de esta base de datos proviene de revisión de literatura y de información
contenida en otras bases de datos. La lista final del total de proteínas obtenidas de MvirDB
fue de 127 proteínas. Posteriormente, la lista de proteínas raíz fue introducida a STRING.
En 15 proteínas, STRING no encontró interacciones de alta confiabilidad, por lo tanto se
eliminaron. La red resultante cuenta con un total de 330 proteínas y 1,271 interacciones.
Para cada una de las proteínas que integraron la red, se realizó un análisis para determinar
la asociación entre su contexto funcional (GUP) y su conocida función en patogénesis. Este
tipo de análisis ha mostrado ser útil para determinar la asociación de proteínas o genes con
cáncer (Aragues et al., 2008). Los resultados mostraron que el GUP de las proteínas fue un
buen parámetro para predecir si una proteína está asociada a patogénesis de H. pylori. El
nivel de probabilidad estadístico del indicador fue evaluado mediante una prueba de X2
(Cuadro 1). Posteriormente, utilizando el GUP de cada una de las proteínas se predijeron
nuevas proteínas involucradas en la patogénesis de H. pylori con valor predictivo positivo y
sensibilidad por encima del 60%, con respecto al GUP (Cuadro 2). Con estos resultados la
red inicial fue filtrada y se obtuvo una red final de 712 interacciones entre 153 proteínas
involucradas en la patogénesis de H. pylori, de las cuales 50 corresponden a proteínas
candidato (Figura 6). Así, la red de interacciones obtenida está compuesta de proteínas de
participación conocida en la patogénesis de H. pylori y sus compañeros directos de
interacción.
Cuadro 1. Asociación entre el contexto funcional de proteínas (GUP) y su conocida función en patogénesisa.
Proteínas con GUP ≥ 0
Proteínas con
GUP ≥ 1
GUP ≥ 0 vs. GUP ≥ 1
Proteínas con
GUP ≥ 5
GUP ≥ 1 vs. GUP ≥ 5
Proteínas asociadas a patogénesis
127
101
76.04 > 3.84
X2 cal > X2tab (1 gl, 0.05)
54
70.44 > 3.84
X2 cal > X2tab (1 gl, 0.05)
aLa evaluación se realizó comparando proteínas con GUP ≥ 0, GUP ≥ 1 y GUP ≥ 5. El valor de probabilidad (p) de la prueba X2 indica diferencia estadística entre el conjunto de proteínas con GUP ≥0 y aquellas con GUP ≥ 1 y GUP ≥ 5.

32
Cuadro 2. Valores de GUP para cada una de la proteínas que integraron la red de interacción y su probable asociación en la patogénesis de H. pylori. Los valores predictivo positivo y sensibilidad se obtuvieron con relación al GUP.
GUP
No. de proteínas: total /(proteínas raíz)
Valor predictivo positivo (%)
Sensibilidad (%)
≥0 330 (127) 12 100 ≥1 317 (101) 32 80 ≥2 139 (79) 60 61 ≥3 91 (68) 75 53 ≥4 68 (58) 85 45 ≥5 60 (54) 90 42 ≥10 43 (41) 95 32
Mediante los complementos MCODE (Bader y Hogue, 2006) y BiNGO (Maere et
al., 2005), incorporados en el programa Cytoscape (Shannon et al., 2006) y el sistema de
minería de datos iHOP (http://www.ihop-net.org), se realizó el análisis de la red de
interacción entre proteínas para determinar la probable función de proteínas candidato
asociadas con patogénesis en H. pylori. El complemento MCODE permite detectar regiones
dentro de una red de interacción de proteínas muy conectadas ó agrupadas. En general,
estas regiones corresponden a complejos multi-proteicos o vías celulares asociadas con un
proceso biológico en particular (Figura 6). El análisis realizado con MCODE resultó en un
total de seis agrupamientos de proteínas; cada uno de los cuales está asociado con un
proceso de patogénesis en particular. Este resultado permitió ademas, corroborar la
hipótesis de que las proteínas asociadas con un proceso biológico en particular tienden a
presentar alta conectividad entre ellas. Además, los agrupamientos de proteínas fueron
analizados mediante el complemento BiNGO; el cual fue utilizado con el objetivo de
determinar términos y anotaciones de ontología génica (GO terms, por sus siglas en inglés).
Los términos de ontología génica (términos GO) representan un conjunto de vocabularios
controlados (ontologías) para describir un gen o atributos de un producto génico en
términos de su proceso biológico, de sus componentes celulares y su función molecular
(The Gene Ontology Consortium, 2000). Actualmente, para un gen o proteína la gran
mayoría de las bases de datos biológicas incluyen, en algún apartado, la anotación de sus
términos de ontología génica. Esto facilita la búsqueda de información funcional

33
estableciendo un tipo de anotación estándar para un producto génico presente en diferentes
bases de datos.
Figura 6: Red de interacción entre proteínas asociadas a patogénesis de H. pylori. Cada nodo representa una proteína y cada enlace entre dos proteínas representa una interacción entre sí mismas. Las proteínas en color rojo corresponden a las proteínas raíz y las marcadas en verde, a las proteínas candidato. También se muestran los agrupamientos de proteínas producidos por MCODE; cada uno es marcado con un color que corresponde al tipo de proceso biológico asociado a patogénesis.
Como se observa en la Figura 6, los agrupamientos de proteínas obtenidos se
corresponden con procesos celulares especificos asociados a patogénesis de H. pylori. A
continuación se describen las interacciones de las proteínas candidato dentro de cada
agrupamiento.

34
8.1. Motilidad y quimiotaxis
El agrupamiento de proteínas asociado a motilidad y quimiotaxis está constituido
por 13 proteínas candidato de un total de 45 proteínas (Figura 7).
Figura 7: Agrupamiento de proteínas asociadas a la quimiotaxis y motilidad de H. pylori. Las
proteínas en color verde corresponden a proteínas raíz y las marcadas en color rojo, a proteínas candidato. El análisis realizado con BINGO indica que el agrupamiento de proteínas, además
de estar enriquecido con términos GO para los procesos biológicos asociados con en el
ensamblaje del flagelo y quimiotaxis, también lo están aquellos asociados síntesis de
tiamina. La presencia de este último grupo de términos es inesperada ya que aparentemente
no existe alguna relación funcional entre la quimiotaxis, motilidad y síntesis de vitaminas
del complejo B. En el cuadro 3 se enlistan las 13 proteínas candidato correspondiente a este
agrupamiento de proteínas.
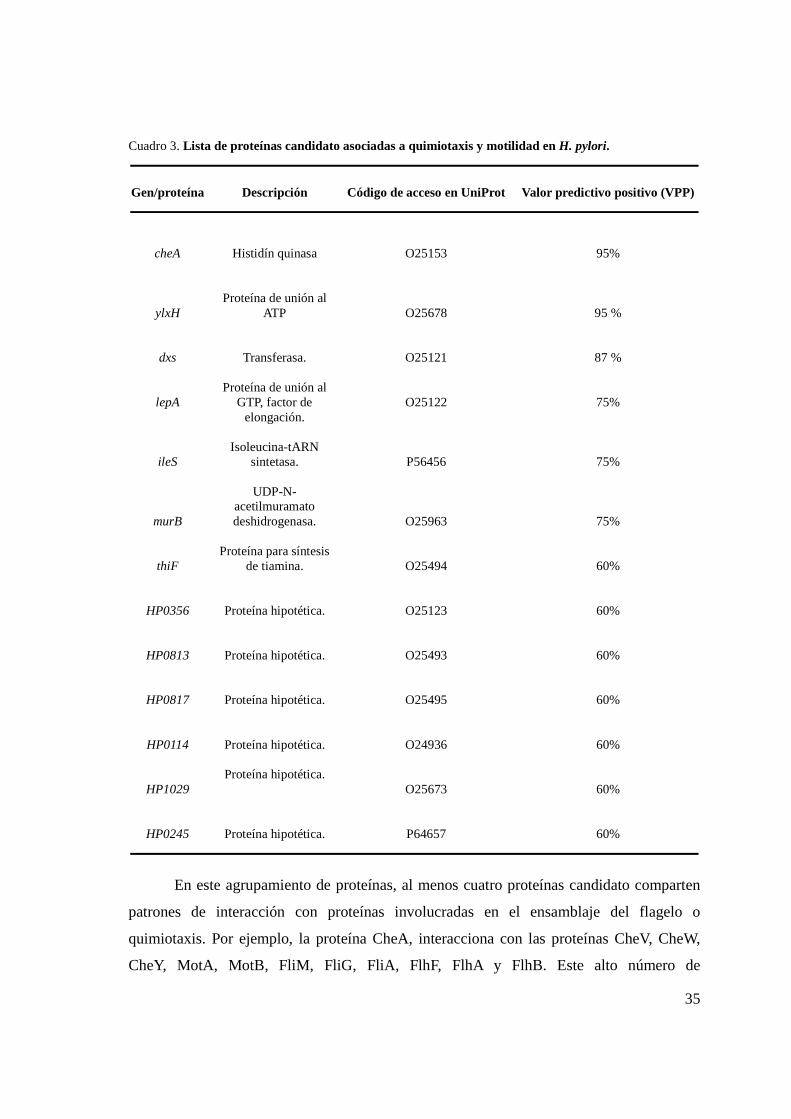
35
Cuadro 3. Lista de proteínas candidato asociadas a quimiotaxis y motilidad en H. pylori.
Gen/proteína
Descripción
Código de acceso en UniProt
Valor predictivo positivo (VPP)
cheA
Histidín quinasa
O25153
95%
ylxH
Proteína de unión al
ATP
O25678
95 %
dxs
Transferasa.
O25121
87 %
lepA
Proteína de unión al
GTP, factor de elongación.
O25122
75%
ileS
Isoleucina-tARN
sintetasa.
P56456
75%
murB
UDP-N-
acetilmuramato deshidrogenasa.
O25963
75%
thiF
Proteína para síntesis
de tiamina.
O25494
60%
HP0356
Proteína hipotética.
O25123
60%
HP0813
Proteína hipotética.
O25493
60%
HP0817
Proteína hipotética.
O25495
60%
HP0114
Proteína hipotética.
O24936
60%
HP1029
Proteína hipotética.
O25673
60%
HP0245
Proteína hipotética.
P64657
60%
En este agrupamiento de proteínas, al menos cuatro proteínas candidato comparten
patrones de interacción con proteínas involucradas en el ensamblaje del flagelo o
quimiotaxis. Por ejemplo, la proteína CheA, interacciona con las proteínas CheV, CheW,
CheY, MotA, MotB, FliM, FliG, FliA, FlhF, FlhA y FlhB. Este alto número de

36
interacciones de CheA se ve reflejado en un alto valor predictivo positivo (Cuadro 3). CheA
es una conocida proteína de los denominados sistemas de dos componentes. Estos sistemas
de proteínas están asociados con la transducción de señales en organismos procariotes y
cumplen funciones en el proceso de quimiotaxis. Así, las interacciones de CheA con CheV,
CheW y CheY, eran esperadas, ya que de las cuatro proteínas se conoce su papel en la
quimiotaxis. Además, algunas de sus interacciones ya han sido determinadas a nivel
experimental en otros procariotes (Figura 8). Sin embargo, las interacciones con las otras
proteínas raíz no eran esperadas, ya que la mayoría están asociadas con el proceso de
exportación de proteínas que conduce al ensamblaje del flagelo.
Figura 8: Estructura cristalográfica donde se observa la interacción entre las proteínas CheA y CheY
de E. coli. Código de acceso a la base de datos PDB: 1EAY. La proteína marcada con el color verde corresponde a CheY y la marcada en color naranja a la proteína CheA (McEvoy et al., 1998).
Las proteínas candidato YlxH, Dxs y ThiF pertenecen a un grupo de proteínas
asociadas con la síntesis de vitaminas del complejo B y las tres presentan interacciones con
algunas de las proteínas raíz citadas. En el caso de la proteína YlxH, ésta interacciona con
las mismas proteínas que CheA. A excepción de CheW, CheV y CheY, las cuales están
asociadas, de manera específica, con quimiotaxis. YlxH es una proteína de unión al ATP y
está asociada con síntesis de Vitamina B12. Existe escasa información acerca de la función
que desempeña esta proteína.
Las proteínas candidato Dxs y ThiF interaccionan con las proteínas raíz FliG, FliH y
FliF y MotA y MotB, respectivamente. El análisis de las anotaciones de Dxs y ThiF en
UniProt revela que son proteínas asociadas a la síntesis de vitaminas del complejo B. las

37
proteínas raíz con las cuales interaccionan Dxs y ThiF participan tanto en la exportación de
proteínas para el ensamblaje del flagelo como en la generación de un gradiente de protones
necesario para la rotación flagelar.
Las proteínas candidato IleS y MurB interaccionan con las proteínas raíz FliQ y
FliL. Las dos últimas proteínas raíz pertenecen a la región estructural del cuerpo basal del
flagelo. Las proteínas de esta región tienen función estructural y en la exportación de
proteínas para el ensamblaje del flagelo. IleS es una proteína que participa en la síntesis de
proteínas, evitando la agregación incorrecta de amino ácidos por el tARN. Por su parte,
MurB es una proteína que participa en síntesis de péptido glicano y está relacionado tanto
en la degradación como en la síntesis de la pared celular (Benson et al., 1996).
Las proteínas LepA y HP0356 interaccionan con FliF, FliG y FliH. Estas últimas
son proteínas de membrana que constituyen parte del sistema de rotación flagelar
participando en la exportación de proteínas para el correcto ensamblaje del flagelo. El
análisis de secuencia de HP0356 mostró que corresponde a una proteína hipotética de la
cual no se dispone de información. En el caso de LepA, no se conoce su función con
certeza en bacterias; el análisis de su secuencia revela la presencia de una familia de
dominios relacionados con la síntesis de proteínas y con la unión a los ribosomas,
dependiente de GTP (accesos de Pfam: PF00679, PF00009, PF03144 y PF06421). A nivel
genómico, LepA tiene una disposición estructural de vecindad con proteínas flagelares y
con la proteína candidato Dxs. Sin embargo, esta disposición estructural sólo se observó en
algunas especies del género Helicobacter, Bacillus, Geobacillus y Oceanobacillus. De tal
manera, que la citada organización a nivel genómico de LepA con proteínas flagelares
podría ser una característica única de pocos géneros bacterianos (Figura 9). Esto es
importante, considerando que aquellos genes que guardan una relación de vecindad en el
genoma, tienden a codificar proteínas que interaccionan o que están asociadas al mismo
proceso biológico (Dandekar et al., 1998).

38
Figura 9: Organización a nivel genómico de los genes fliF, fliG y fliH , respecto a los genes
candidato dxs y lepA. Imagen modificada del sistema de información para patógenos microbianos de EE. UU. (NMPDR, por sus siglas en inglés).
Las proteínas candidato HP0813 y HP0817 son proteínas hipotéticas que
interaccionan con las proteínas raíz MotA y MotB. Estas últimas proteínas están asociadas
con la membrana celular y participan en el proceso de flujo de protones que genera el
torque del flagelo. El análisis de secuencia de estas proteínas hipotéticas sólo reveló
información para la proteína HP0813, la cual presenta un dominio tipo beta-lactamasa
(accesso de PFAM número: PF00753), asociados a la resistencia a los antibióticos.
HP0114 corresponde a una proteína hipotética que interacciona con las proteínas
FlaB y SpsE. FlaB es el componente minoritario del filamento flagelar. Esta región se
encuentra conectada al gancho flagelar, constituido por las proteínas FlgK y FlgL y es la
que realiza el trabajo hidromecánico necesario para la propulsión. Por su parte, SpsE
pertenece a otro agrupamiento de proteínas asociadas con la síntesis de polisacáridos. De
esta manera HP0114 establece un vínculo entre dos agrupamientos de proteínas sin
aparente relación funcional.
De las proteínas hipotéticas HP1029 y HP0245 no se dispone de información. Por lo
tanto, la única probable asociación funcional con este agrupamiento estaría determinada por
sus patrones de interacción. HP1029 interacciona con FliM y FliY, proteínas que
constituyen el llamado “complejo switch” que es el regulador del rotor flagelar. HP0245
interacciona con las proteínas FlgK y FlgL. Ambas proteínas pertenecen a la región
estructural conocida como gancho del flagelo. Se requieren de nuevos estudios para
corroborar la asociación de estas proteínas hipotéticas.
A manera de resumen, en este agrupamiento algunas de las interacciones
encontradas no eran esperadas debido a su aparente carencia de significado biológico. Este

39
el caso de proteínas asociadas a síntesis de vitaminas del complejo B y aquellas
involucradas en el proceso de traducción durante la síntesis de proteínas. Mientras que otras
interacciones eran esperadas, como es el caso de proteínas asociadas específicamente con
quimiotaxis o en el caso de proteínas raíz que dadas sus características estructurales
necesitan estar ancladas a la pared celular.
8.2. Adaptación al medio ácido
El agrupamiento de proteínas asociado a adaptación al medio ácido está constituido
por 4 proteínas candidato de un total de 18 proteínas (Figura 10).
Figura 10: Agrupamiento de proteínas asociadas a la adaptación al medio ácido de H. pylori. Las proteínas marcadas en color rojo corresponden a proteínas raíz y las marcadas en color verde, a proteínas candidato.
El análisis realizado con BINGO sugiere que este agrupamiento de proteínas está
enriquecido con términos GO para los procesos asociados a catálisis de la arginina y
metabolismo de la urea, síntesis de lipopolisacáridos asociados a la pared celular. Del
mismo modo que en el agrupamiento anterior, la presencia de éste último grupo de términos
GO es inesperada, ya que aparentemente no existe una asociación funcional entre el sistema
de adaptación al medio acido y componentes estructurales de la pared celular.

40
Cuadro 4. Lista de proteínas candidato asociadas a adaptación al medio ácido de H. pylori.
Gen/proteína
Descripción
Código de acceso en UniProt Valor predictivo positivo
(VPP)
nikR
Regulador transcripcional
dependiente de níquel.
O25896
87%
glmM
Fosfoglucosamina
mutasa.
P25177
60 %
csrA
Regulador del
almacenamiento de carbono.
O25983
60 %
smpB
Proteína de unión al
tmARN.
O25985
60%
La proteína NikR es un factor de transcripción dependiente de níquel (Figura 11).
NikR interacciona con NixA, UreA, ExbD1 y ExbB1. NixA es una proteína de membrana,
encargada de la importación del níquel al interior de la célula. UreA corresponde a una de
las subunidades de la enzima ureasa; responsable de la adaptación al medio ácido. Por su
parte, ExbD1 y ExbB1 son proteínas que forman un complejo con la proteína TonB. Este
complejo de proteínas se encarga, mediante un gradiente de protones, de la movilización de
complejos que contienen níquel (Schauer et al., 2008).
Figura 11: Estructura cristalográfica donde se observa la interacción entre la proteína NikR y el
ADN de E. coli. Código de acceso a la base de datos PDB: 2HZV. La proteína marcada con el color verde corresponde a NikR y la estructura marcada en color naranja al ADN.

41
Por su parte, la proteína GlmM interacciona con UreA y UreB, ambas forman la
estructura básica del complejo ureasa. GlmM es una enzima de la familia de las
fosfohexosas mutasas. Estas enzimas son importantes en la producción de intermediarios
durante la síntesis de péptido glicano de la pared celular (Jolly et al., 1999).
Las proteínas candidato CsrA y SmpB interaccionan con las proteínas del tipo
ExbB2 y ExbD2 que forman un complejo con TonB. La función de este complejo ya fue
citado anteriormente. CsrA pertenece a una familia de proteínas de unión al ARN que actúa
sobre la traducción de genes y/o sobre la estabilidad de los transcritos. El mecanismo
mediante el cual CsrA activa o suprime la expresión de determinados genes aún no se
conoce. SmpB es una proteína de unión al tmARN. El complejo SmpB-tmARN es
fundamental en un proceso conocido como trans-traducción, el cual es un mecanismo
utilizado por las bacterias para el aseguramiento de la calidad de la síntesis de proteínas
(Keiler et al., 1996; Dulebohn et al., 2007; Moore y Sauer, 2007; Keiler, 2008).
Por último, en este agrupamiento sobresalen interacciones que relacionan al
complejo ureasa con estructuras asociadas a la pared celular, y remarca el papel que podrían
desempeñar una serie de reguladores a nivel transcripcional y traduccional, alterando la
expresión de genes necesarios para la adaptación al medio ácido.
8.3. Metabolismo del nitrógeno
El agrupamiento de proteínas asociado a metabolismo del nitrógeno está constituido
por 7 proteínas candidato de un total de 9 proteínas (Figura 12).
Figura 12: Agrupamiento de proteínas asociadas a el metabolismo del nitrógeno de H. pylori. Las proteínas marcadas en color rojo corresponden a proteínas raíz y las marcadas en color verde, a proteínas candidato.
El análisis con BINGO reveló términos de ontología génica asociados a la síntesis
de glutamina y la síntesis de compuestos nitrogenados. En el cuadro cinco se enlistan las
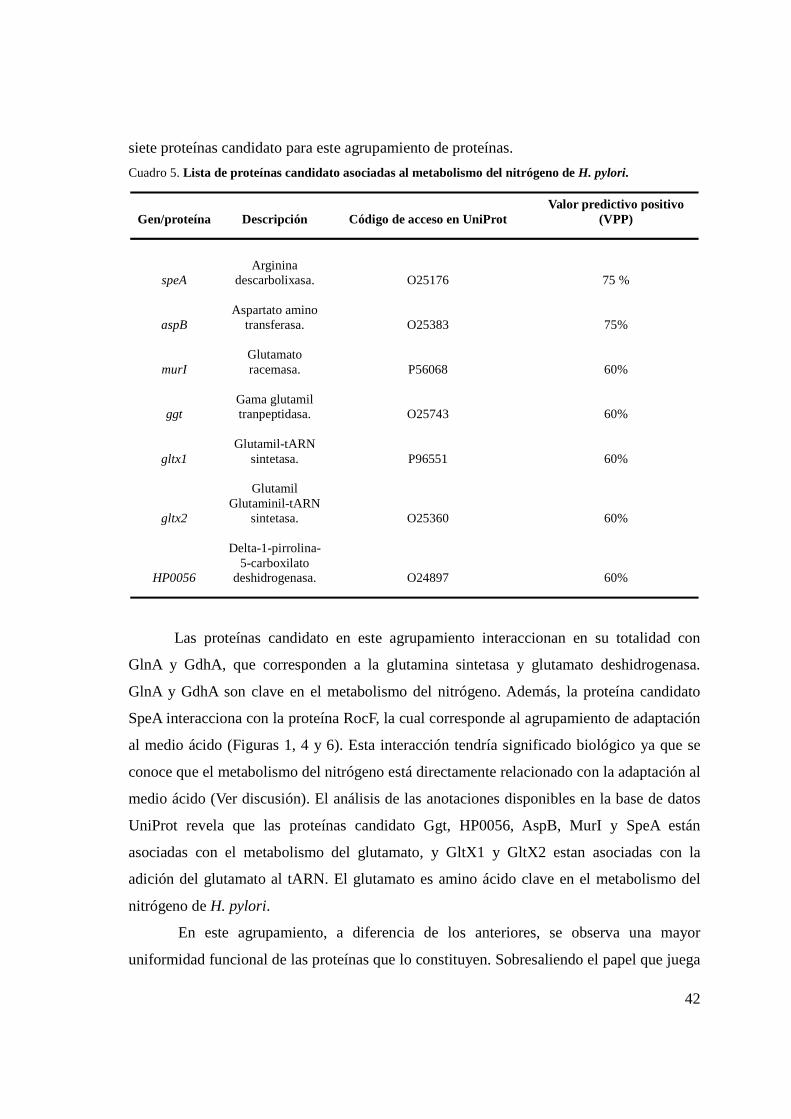
42
siete proteínas candidato para este agrupamiento de proteínas.
Cuadro 5. Lista de proteínas candidato asociadas al metabolismo del nitrógeno de H. pylori.
Gen/proteína
Descripción
Código de acceso en UniProt Valor predictivo positivo
(VPP)
speA
Arginina
descarbolixasa.
O25176
75 %
aspB
Aspartato amino
transferasa.
O25383
75%
murI
Glutamato racemasa.
P56068
60%
ggt
Gama glutamil tranpeptidasa.
O25743
60%
gltx1
Glutamil-tARN
sintetasa.
P96551
60%
gltx2
Glutamil
Glutaminil-tARN sintetasa.
O25360
60%
HP0056
Delta-1-pirrolina-
5-carboxilato deshidrogenasa.
O24897
60%
Las proteínas candidato en este agrupamiento interaccionan en su totalidad con
GlnA y GdhA, que corresponden a la glutamina sintetasa y glutamato deshidrogenasa.
GlnA y GdhA son clave en el metabolismo del nitrógeno. Además, la proteína candidato
SpeA interacciona con la proteína RocF, la cual corresponde al agrupamiento de adaptación
al medio ácido (Figuras 1, 4 y 6). Esta interacción tendría significado biológico ya que se
conoce que el metabolismo del nitrógeno está directamente relacionado con la adaptación al
medio ácido (Ver discusión). El análisis de las anotaciones disponibles en la base de datos
UniProt revela que las proteínas candidato Ggt, HP0056, AspB, MurI y SpeA están
asociadas con el metabolismo del glutamato, y GltX1 y GltX2 estan asociadas con la
adición del glutamato al tARN. El glutamato es amino ácido clave en el metabolismo del
nitrógeno de H. pylori.
En este agrupamiento, a diferencia de los anteriores, se observa una mayor
uniformidad funcional de las proteínas que lo constituyen. Sobresaliendo el papel que juega

43
el glutamato en el metabolismo del nitrógeno y su interconexión con la adaptación al medio
acido.
8.4. Sistema de secreción tipo IV
El agrupamiento de proteínas asociado al sistema de secreción tipo IV está
constituido por 8 proteínas candidato de un total de 42 proteínas (Figura 13).
Figura 13: Agrupamiento de proteínas asociadas al sistema de secreción tipo IV de H. pylori. Las proteínas marcadas en color verde corresponden a proteínas raíz y las marcadas en color rojo, a proteínas candidato.
El análisis con BINGO identificó términos de ontología génica asociados con la
patogénesis, conjugación bacteriana y transporte de moléculas. En el cuadro 6 se enlistan
las 8 proteínas candidato para este agrupamiento de proteínas.
Cuadro 6. Lista de proteínas candidato asociadas al sistema de secreción tipo IV de H. pylori.
Gen/proteína
Descripción
Código de acceso en UniProt Valor predictivo positivo
(VPP)
trbI
Proteína del sistema de
secreción tipo IV, relacionada con VirB10.
O24883
92 %
hopQ
Proteína de membrana
externa.
O25791
87%
HP1342
Proteína de membrana
externa.
O34523
75%
HP0439
Proteína hipotética.
O25187
75%

44
HP0040
Proteína hipotética.
O24881
75%
HP0922
Proteína de membrana
externa.
O25579
60%
HP0120
Proteína hipotética.
O24939
60%
HP0066
Proteína hipotética.
O24906
60%
En primer lugar, las proteínas candidato HopQ y HP1342 corresponden a proteínas
de la membrana externa de H. pylori. Ambas proteínas interaccionan con las proteínas raíz
Cag3, CagS, VacA y CagA, las cuales están asociadas directamente a patogénesis. Por
ejemplo, se conoce que CagA y VacA son potentes cito-toxinas capaces de alterar la vías de
señalización de las células del hospedero (Censini et al., 1996; Akopyants et al., 1998;
Blomstergren et al., 2004).
Del mismo modo, las proteínas candidato HP0922, HP0066 y HP0120 presentan
patrones de interacción similares a HopQ y HP1342. HP0922, HP0066 y HP0120,
presentan interacciones con las proteínas de membrana externa BabA y Omp1, las cuales
son adhesinas conocidas en H. pylori. Además, HP0922, HP0120 y HP0066 interaccionan
por separado con CagA, Cag3 y VacA, respectivamente.
Por otro lado, existe un grupo de proteínas candidato que comparten algunos
patrones de interacción. Tal es el caso de las proteínas HP0040, HP0439 y TrbI. Las tres
proteínas interaccionan con HP0017 y HP0459. Además, TrbI interacciona con Cag5,
CagE, VirB11 y TraG. Estas últimas proteínas raíz, al igual que HP0017 y HP0459,
participan durante el ensamblaje del sistema de transferencia horizontal de genes. HP0040
y HP0439 son proteínas hipotéticas. El análisis de la secuencia de HP0439 revela la
presencia de dominios del tipo VirB8. Este tipo de dominios están presentes en proteínas
trans-membranales que constituyen componentes primarios del sistema de transporte de
ADN (Kumar y Das, 2001).
A manera de resumen, en la figura 7 se observa que este agrupamiento presenta tres
subgrupos. El subgrupo más denso e interconectado corresponde en su totalidad a proteínas
raíz que conforman la estructura básica del sistema de secreción tipo IV encargado de la

45
inyección de cito-toxinas. Otro subgrupo de proteínas que comparte patrones interacción
corresponde a adhesinas y cito-toxinas. El último subgrupo está constituido por proteínas
asociadas a transferencia horizontal de genes. Este agrupamiento de proteínas muestra una
organización estructurada, donde cada subgrupo corresponde a funciones particulares del
sistema de secreción tipo IV.
8.5. Proteínas de respuesta a estrés.
El agrupamiento de proteínas asociado a respuesta a estrés está constituido por 7
proteínas candidato de un total de 16 proteínas (Figura14).
Figura 14: Agrupamiento de proteínas asociadas a la respuesta a estrés de H. pylori. Las proteínas marcadas en verde corresponden a proteínas raíz y las marcadas en color rojo, a proteínas candidato.
El análisis con BINGO sugiere que este agrupamiento de proteínas está enriquecido
con términos GO en procesos biológicos asociados a metabolismo de ácidos nucleicos,
síntesis de macromoléculas, metabolismo de especies reactivas de oxígeno, interconversión
entre nucleótidos y nucleósidos, así como al metabolismo de ribonucleótidos. En el cuadro
7 se enlistan las ocho proteínas candidato.
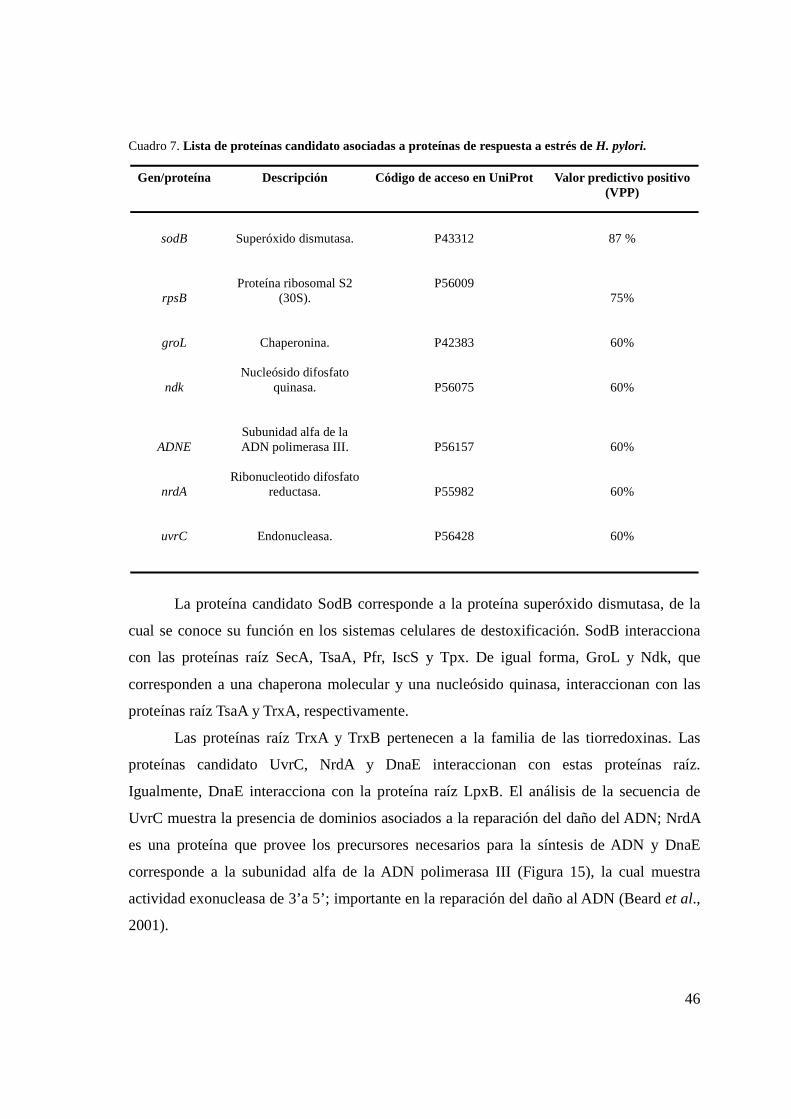
46
Cuadro 7. Lista de proteínas candidato asociadas a proteínas de respuesta a estrés de H. pylori.
Gen/proteína
Descripción Código de acceso en UniProt Valor predictivo positivo (VPP)
sodB
Superóxido dismutasa.
P43312
87 %
rpsB
Proteína ribosomal S2 (30S).
P56009
75%
groL
Chaperonina.
P42383
60%
ndk
Nucleósido difosfato
quinasa.
P56075
60%
ADNE
Subunidad alfa de la ADN polimerasa III.
P56157
60%
nrdA
Ribonucleotido difosfato
reductasa.
P55982
60%
uvrC
Endonucleasa.
P56428
60%
La proteína candidato SodB corresponde a la proteína superóxido dismutasa, de la
cual se conoce su función en los sistemas celulares de destoxificación. SodB interacciona
con las proteínas raíz SecA, TsaA, Pfr, IscS y Tpx. De igual forma, GroL y Ndk, que
corresponden a una chaperona molecular y una nucleósido quinasa, interaccionan con las
proteínas raíz TsaA y TrxA, respectivamente.
Las proteínas raíz TrxA y TrxB pertenecen a la familia de las tiorredoxinas. Las
proteínas candidato UvrC, NrdA y DnaE interaccionan con estas proteínas raíz.
Igualmente, DnaE interacciona con la proteína raíz LpxB. El análisis de la secuencia de
UvrC muestra la presencia de dominios asociados a la reparación del daño del ADN; NrdA
es una proteína que provee los precursores necesarios para la síntesis de ADN y DnaE
corresponde a la subunidad alfa de la ADN polimerasa III (Figura 15), la cual muestra
actividad exonucleasa de 3’a 5’; importante en la reparación del daño al ADN (Beard et al.,
2001).

47
Figura 15: Estructura cristalográfica donde se observa la interacción entre la subunidad alfa de la
ADN polimerasa con TrxA, durante el proceso de reparación del ADN del bacteriófago T7. Código de acceso a la base de datos PDB: 1SKR. La proteína marcada con el color verde corresponde a la ADN polimerasa y la estructura marcada en color naranja a la proteína TrxA.
La proteína candidato RpsB interacciona con las proteínas raíz SecA, FliR y FliA,
las cuales están asociadas a la exportación de proteínas flagelares. RpsB es una proteína
ribosomal encargada de la regulación de la traducción. Aunque sorprende la interacción de
RpsB con las proteínas raíz antes citadas, ya que parece carecer de significado biológico,
diversos estudios en otras bacterias demuestran que RpsB se produce bajo condiciones de
estrés (Jurgen et al., 2001).
Resumiendo, este agrupamiento presenta proteínas candidato con funciones
presuntivas en concordancia con las de las proteínas raíz de este agrupamiento. Entre las
que destacan aquellas asociadas con la reparación del daño al ADN, con la respuesta al
estrés oxidativo y síntesis de proteínas. Mecanismos clave para la sobrevivencia de este
patógeno en ambiente hostil, como lo es el epitelio gástrico.

48
8.6. Síntesis de lipopolisacáridos
El agrupamiento de proteínas asociado a lipopolisacáridos está constituido por 11
proteínas candidato de un total de 20 proteínas (Figura 16).
Figura 16: Agrupamiento de proteínas asociadas a síntesis de lipopolisacáridos de H. pylori. Las proteínas marcadas en color verde corresponden a proteínas raíz y las marcadas en color rojo, a proteínas candidato.
El análisis con BINGO sugiere que este agrupamiento de proteínas esta enriquecido
con términos GO para procesos biológicos asociados a síntesis de lípidos, metabolismo de
carbohidratos, polisacáridos y lipopolisacáridos. En el cuadro 8 se enlistan las nueve
proteínas candidato para este agrupamiento de proteínas.
Cuadro 8. Lista de proteínas candidato asociadas a síntesis de lipopolisacáridos de H. pylori.
Gen/proteína
Descripción
Código de acceso en UniProt Valor predictivo positivo
(VPP)
spsC
Transferasa.
O25130
92%
kdtA
Transferasa.
O25611
75 %
rfaD
Epimerasa.
O25530
75%
HP0102
Proteína hipotética.
O24928
75%
HP0277
Ferredoxina.
O25054
60%
hldE
Transferasa.
O25529
60%
algC
Pirofosforilasa.
O25865
60%
galU
Epimerasa.
O25363
60%

49
flaA1
Epimerasa.
O25511
60%
gmhA
Isomerasa.
O25528
60%
HP1570
Proteína hipotética.
O26090
60%
La proteína candidato SpsC interacciona con las proteínas raíz NeuA, RfbD,
HP0043, WbpB y NolK. Del mismo modo, la proteína candidato HP0102 interacciona con
RfbD, HP0043 y NolK. SpsC corresponde a una proteína con actividad amino transferasa y
HP0102 a una proteína hipotética. Todas las proteínas raíz que interaccionan con SpsC y
HP0102 están asociadas a la síntesis de polisacáridos; en particular con la síntesis de un
componente estructural del antígeno O, la cual es una molécula importante en la virulencia
bacteriana (Stroeher et al., 1995; Rashid et al., 2003).
Las proteínas candidato KdtA y HP0277 interaccionan con las proteínas raíz RfaC,
CoaD. Además, KdtA interacciona con la proteína raíz LpxB. KdtA es una enzima con
actividad transferasa. Las proteínas raíz con las cuales interacciona KdtA están relacionadas
con la síntesis de una estructura llamada núcleo de oligosacárido necesaria para el anclaje
de lipopolisacáridos a la membrana celular. HP0277 corresponde a una ferredoxina. Este
tipo de proteínas confieren resistencia al estrés oxidativo, mediante la reducción de
diferentes tipos de peróxidos a su correspondiente alcohol (Wood et al., 2003).
Por otro lado, un grupo de proteínas candidato comparte algunos patrones de
interacción. Este el caso de la proteínas candidato GmhA, HldE, RfaD, GalU y AlgC.
GmhA, HldE, RfaD interaccionan con las proteínas raíz RfbD y RfaC, las cuales están
relacionadas con el sistema de exportación del antígeno O. Además, RfaD y HldE
interaccionan con HP0043. GalU y AlgC también interaccionan con HP0043 y, además,
con GalE. GmhA, HldE y RfaD presentan funciones de isomerasa, transferasa y epimerasa,
respectivamente. Del mismo modo, GalU y AlgC presentan funciones de epimerasa y
pirofosforilasa.
Las proteínas candidato FlaA1 y HP1570 interaccionan con las proteínas raíz NeuA
y SpsE, las cuales están asociadas con la síntesis de lipopolisacáridos de superficie. FlaA1
corresponde una proteína con actividad epimerasa y HP1570 a una proteína hipotética.
Finalmente, este agrupamiento presenta un conjunto de proteínas que presentan

50
funciones relacionadas con la síntesis de polisacáridos, específicamente con la síntesis del
antígeno O y del núcleo de oligosacárido. Estas últimas son moléculas importantes en la
estructura y función de los lipopolisacáridos de superficie.
9. Discusión
Desde que Warren y Marshall (1983), asociaron por primera vez a H. pylori con la
aparición de ulceras gástricas, pasando por la era genómica y post-genómica; diversos
estudios han proporcionado información respecto a las proteínas y/o genes que son
esenciales durante el proceso de patogénesis de H. pylori. Sin embargo, las funciones
celulares que resultan de la interacción entre las proteínas requieren de ser investigadas
desde una perspectiva holística, es decir el estudio de la biología de sistemas (Aloy y
Russel, 2006; Aloy, 2007). Así, la construcción de redes de interacción entre proteínas es un
punto de partida en el entendimiento de los sistemas celulares. En el caso particular de H.
pylori, las redes de interacción entre proteínas se han investigado mediante métodos
experimentales. Este estudio ha permitido el análisis de aproximadamente el 50% del
proteoma de H. pylori (Rain et al., 2001). En el presente trabajo, se realizó un análisis, a
nivel de sistemas, de la red de interacción entre proteínas asociadas a patogénesis de H.
pylori. Para construir la red de interacción de proteínas se utilizó de una base de datos de
proteínas de función conocida asociada a patogénesis de H. pylori (proteínas raíz).
Inicialmente, a partir de las proteínas raíz se construyó una red de interacción entre
proteínas solamente tomando en cuenta las interacciones reportadas en el interactoma
determinado a nivel experimental por Rain, et al., (2001). El procedimiento anterior se
realizó con el objetivo de determinar la cobertura de la información a nivel experimental de
interacciones entre proteínas disponible para las proteínas raíz. Los resultados obtenidos de
este análisis revelaron una baja cobertura (50%) de interacciones; es decir, a nivel
experimental sólo se dispone de datos de interacciones entre proteínas para el 50% de las
proteínas raíz. En vista de lo anterior se procedió a realizar las predicciones para el restante
50% de proteínas raíz que carecen de datos de interacción a nivel experimental. Las
predicciones fueron realizadas con el programa PIANA, el cual implementa el método de
los interólogos (Aragues et al., 2006); sin embargo, nuevamente los resultados no fueron
satisfactorios; alcanzándose una cobertura del 56%. Esto aún sin descontar que se estima

51
que el 50% de las interacciones reportadas por el método de Y2H son falsos positivos y
negativos; esto es debido a la limitación propia de la metodología (von Mering et al.,
2003). En concordancia a lo reportado por Aragues et al., (2008) con esta baja cobertura no
fue posible aplicar el método de análisis del contexto funcional para cada una de las
proteína que integran una red de interacción (GUP).
Diversos estudios sobre métodos para la determinación de interacciones entre
proteínas a nivel experimental y computacional, sugieren que la mejor manera de
incrementar la cobertura y calidad de un estudio de interactómica es a través de la
combinación de diferentes métodos (Schwikowski et al., 2000; Edwards et al., 2002; von
Mering et al., 2002). Por lo tanto, esta situación remarcó la necesidad de incluir más
métodos de predicción para mejor la cobertura y de encontrar una forma de evaluar la
calidad de las interacciones. El sistema de análisis STRING cumplió con todos los
requerimientos antes citados (Jensen et al., 2009). STRING contiene interacciones entre
proteínas que han sido determinadas a nivel experimental por estudios a gran escala y de
baja escala (interacciones físicas directas). Ejemplos de los primeros, son los resultados
producidos por los ensayos de tipo Y2H y TAP-MS. Los segundos corresponden a los
experimentos clásicos de bioquímica y genómica, reportados de manera individual. Por otro
lado, STRING también predice interacciones indirectas; es decir, proteínas que comparten
vía metabólica o proceso celular específico, proteínas co-reguladas a nivel transcripcional y
proteínas que participan en un complejo multi-proteico. Tomando en cuenta toda esta
información, STRING realiza predicciones mediante el método de los “interólogos”.
Además, utiliza la información contenida en una gran cantidad de genomas secuenciados
completamente y realiza predicciones basadas en el contexto genómico. Los métodos
basados en el contexto genómico son idóneos cuando se requiere predecir interacciones
entre proteínas de organismos procariotes ya que se fundamentan en el orden y contenido
de los genes dentro de un genoma; ambas propiedades son una característica distintiva de
los genomas de estos organismos (Valencia y Pazos, 2002). Por ejemplo, los genes vecinos
en varios genomas (operones potenciales) tienden a codificar proteínas que interaccionan
entre sí. Finalmente, STRING es capaz de determinar con probabilidad estadística la
interacción entre las proteínas mediante la implementación de una función de puntaje. Con
la aplicación de los métodos y filtros citados se logró obtener una red de interacciones entre

52
proteínas con alta cobertura (88%) y confiabilidad de probabilidad estadística (puntaje de
STRING=0.7).
La red de interacción entre proteínas resultado del presente estudio cumple con las
siguientes funciones. Cerca del 40% del genoma de H. pylori aún no está anotado (Boneca
et al., 2003). Entre los principales objetivos de los proyecto sobre los interactomas es
predecir la función de proteínas hipotéticas y proteínas sin función asignada. La red de
interacción entre proteínas resultado del presente estudio cumple con este objetivo ya que
ofrece indicios de probables funciones de proteínas hipotéticas derivadas su interacción con
proteínas de función conocida asociada a patogénesis de H. pylori. Esto es, asociación de
función por “culpabilidad” (Figura 5). Este método fue inicialmente desarrollado para
predecir la posible asociación entre proteínas con cáncer (Aragues et al., 2008). Existen
otros métodos basados en la teoría de grafos que intentan asignar funciones o papeles
biológicos a las proteínas que integran una red de interacciones (Yook et al., 2004). Este
último tipo de métodos han permitido determinar que las redes biológicas adoptan una
organización deacuerdo a la ley de potencias, a este tipo de organización se le conoce como
“libre de escala” (Barabási y Oltvai, 2004). En una red “libre de escala”, existen nodos
(proteínas en este estudio) que concentran una gran cantidad interacciones y otros que
presentan pocas; es por ello que a los primeros se les conoce como “focos de la red”.
Generalmente, para poder caracterizar la organización de una red se realiza el
cálculo de una serie de parámetros, conocidos como parámetros de la topología de la red.
Entre los que destaca el conocido como “grado del nodo”, este parámetro hace referencia al
número total de conexiones que presenta un nodo dentro de una red de interacción, por lo
cual es muy útil para caracterizar a los focos de la red. Esto último es importante debido a
que el análisis del interactoma de S. cerevisiae ha demostrado que los focos de la red de
interacción corresponden a genes esenciales, y que su afectación puede resultar letal para el
organismo (Barabási y Oltvai, 2004). A pesar de la importancia del cálculo de los
parámetros topológicos de una red de interacción, en el presente trabajo éstos no fueron
calculados por las siguientes razones: Primero, uno de los algoritmos de predicción
utilizado por STRING se basa en el conocimiento previo. En este algoritmo, para cada
proteína introducida en STRING, realiza una búsqueda en la literatura, de manera que si
encuentra algún reporte donde se mencione la interacción con otra proteína, STRING la

53
toma como verdadera. Así tendríamos que en una red de interacción construida a base de
datos “curados”, como en el caso del algoritmo citado, se esperaría que aquellas proteínas
sobre las cuales la comunidad científica ha puesto mucho interés sean las que presenten un
mayor número de interacciones. Hoffmann y Valencia, (2003) reportan que en el
interactoma de S. cerevisiae no hay una correlación entre el “grado del nodo” y el numero
de citas en la literatura para 380 genes. De esta manera se justifica la omisión del cálculo de
parámetros topológicos de la red de interacción obtenida en el presente estudio, ya que de
haberlo hecho se podría llegar a conclusiones erróneas; por ejemplo, asignar erróneamente
a una proteína como esencial en la patogénesis de H. pylori solamente basados en su
número total de interacciones. Segundo, Aragues et al., (2008) demuestran
estadísticamente, que el “grado del nodo” no es un buen indicador para predecir la función
de una proteína y que el método basado en el análisis del contexto de una proteína dentro
de una red de interacción no es influenciado por el uso de datos “curados”. Todo este
conjunto de observaciones justifican el uso de la metodología empleada en éste estudio.
A continuación, para cada agrupamiento de proteínas se discuten los resultados
obtenidos en el presente trabajo.

54
9.1. Motilidad y quimiotaxis
Uno de los aspectos fundamentales de la infección de H. pylori es su capacidad de
moverse a través de la capa mucosa del lumen gástrico para así poder acceder a la
superficie del epitelio gástrico. El tránsito hacia el epitelio gástrico está dirigido por la
búsqueda de un nicho que le proporcione los nutrientes y condiciones necesarias para la
proliferación. Este movimiento dirigido requiere de la quimiotaxis y motilidad. Para ello un
conjunto de proteínas actúan de manera concertada ensamblando estructuras o participando
en cascadas de señalización. Este es el caso de la proteína candidato CheA; de la cual es
conocido su papel en los sistemas de transducción de señales en bacterias. Específicamente,
CheA interactúa con CheW y CheV, se auto-fosforila y transfiere su grupo fosfato a CheY.
Esto se traduce en una señal para los motores flagelares, modificando la dirección
rotacional del flagelo. Sin embargo, en el presente estudio se determinó que CheA
interacciona con un grupo de proteínas asociadas a la exportación de proteínas flagelares
para el ensamblaje del flagelo. Estas nuevas interacciones de CheA no han sido estudiadas
previamente, por lo cual podrían indicar una nueva función para esta proteína. Existen en la
literatura científica, dos artículos que soportan algunas de las nuevas interacciones descritas
para CheA. Por ejemplo, en un estudio de proteómica diferencial en C. jenuni sugiere que
CheA, FlhA y FliA (estas dos últimas son proteínas flagelares) son indispensables durante
proceso de formación de biopelículas (Kalmokoff et al., 2006). Un hallazgo similar fue
descrito en Aeromonas spp (Kirov et al., 2004). Ambos trabajos reportan el papel de estas
proteínas durante las primeras etapas de la formación de biopelículas. El aumento de la
expresión de estas proteínas podría relacionarse con la necesidad de exportación de otras
proteínas flagelares necesarias para el ensamblaje del flagelo, con la formación de una
matriz tridimensional que facilitaría el contacto célula-célula o como un componente de
matriz extracelular. Esto es fundamental dada la evidencia de que H. pylori es capaz de
formar biopelículas en la mucosa gástrica del hospedero (Carron et al., 2006; Cellini et al.,
2008). De esta manera, se sugiere una probable función reguladora de CheA en la
exportación de proteínas flagelares durante las primeras etapas de la formación de las
biopelículas en H. pylori.
Por otro lado, en este agrupamiento se encontró que las proteínas candidato YlxH,
ThiF y Dxs, presentan dominios asociados con la síntesis de vitaminas del complejo B. La

55
proteína YlxH presenta interacciones con algunas de la proteínas con las que interacciona
CheA, con excepción de CheW y CheV. Aunque existe poca información de la proteína
YlxH, el análisis de su secuencia con el programa BLASTp resultó con una similitud del
63% (valor E de 3 x 10-63) y un mismo contenido de dominios funcionales con la proteína
FlhG de C. jejuni, de acuerdo al acceso en la base de datos PFAM: PF01656. FlhG
pertenece a una familia de reguladores esenciales para la síntesis proteínas asociadas al
ensamblaje del flagelo. Por ejemplo, existe evidencia de que el homólogo de FlhG (FleN)
en P. aeruginosa regula a nivel transcripcional el número de flagelos (Dasgupta et al., 2000
y 2003). Esto sugiere que YlxH podría ser un homólogo de FlhG, con función similar en H.
pylori. Las proteínas candidato Dxs y ThiF interaccionan con proteínas relacionadas con la
exportación de proteínas flagelares y con proteínas generadoras del torque flagelar.
Posiblemente estas proteínas candidato proporcionen moléculas o intermediarios que sean
útiles en la producción de co-factores en la síntesis de proteínas flagelares. Así, estos
patrones de interacción sugieren un vínculo entre proteínas relacionadas con la síntesis de
vitaminas del complejo B, la quimiotaxis y la exportación de proteínas para el ensamblaje
del flagelo.
Otro hallazgo importante en este agrupamiento de proteínas fue la presencia de
proteínas candidato asociadas con el proceso de traducción en la síntesis de proteínas. Este
fue el caso de las proteínas IleS y LepA. Por ejemplo, en E. coli, LepA se ha asociado a la
síntesis de proteínas de membrana; específicamente, con la fidelidad del proceso de
traducción (Qin et al., 2006; Youngman y Green, 2007). Así mismo, su homólogo en S.
cerevisiae, ha mostrado que es capaz de unirse, de manera GTP dependiente, a los
ribosomas mitocondriales y funcionar como un factor asegurador de la fidelidad de la
síntesis de proteínas mitocondriales, principalmente de aquellas asociadas con la formación
de complejos de la cadena transportadora de electrones (Bauerschmitt, et al., 2008). Esta
última observación es fundamental considerando que uno de los primeros eventos del
ensamblaje del flagelo involucra la inserción de FliF y proteínas relacionadas en la
membrana plasmática para constituir la base del motor flagelar. Posteriormente, FliG, que
corresponde a la principal proteína del rotor, se ensambla sobre la base constituida por FliF
y el proceso prosigue hasta que el ensamblaje del motor se completa. Una vez que se han
constituido las estructuras básicas del flagelo (sistema motor y de exportación), se inicia el

56
proceso de exportación de proteínas para ir ensamblando las estructuras distales del flagelo.
El paso inicial en la exportación de proteínas flagelares consiste en la formación de un
macrocomplejo de proteínas que involucra a las proteínas FliH y FliI. El macrocomplejo,
posteriormente se acopla a las compuertas del sistema de exportación localizado en la base
del flagelo y facilita la entrada de las proteínas para ser exportadas. Diversos estudios
reportan que, en ausencia de FliH, no es posible el acoplamiento a las compuertas del
sistema de exportación; así FliH es esencial en el proceso. Así mismo, mutantes de fliG y
fliF en H. pylori son incapaces de realizar la exportación necesaria para el correcto
ensamble del flagelo (Allan et al., 2000). Estos mecanismos de revisión a nivel traducción
serían fundamentales como un paso previo a la exportación de proteínas que ensamblarían
al flagelo. Este conjunto de evidencias sugiere que IleS y LepA podrían estar asociadas en
la regulación y aseguramiento de la fidelidad de la síntesis de las proteínas FliQ y FliL,
FliF, FliG y FliH, respectivamente; las cuales son clave en las etapas iníciales del proceso,
de ensamblaje y funcionamiento del flagelo.
La proteína candidato MurB interacciona con proteínas asociadas al cuerpo basal
del flagelo. Esta estructura anatómica es la encargada de anclar el flagelo a las envolturas
celulares, da soporte al sistema de exportación de proteínas y al rotor flagelar. MurB es una
proteína que participa en síntesis de péptidoglicanos y está relacionada tanto en la
degradación como en la síntesis de la pared celular. Esto es fundamental considerando que
las proteínas que pertenecen al cuerpo basal del flagelo deben estar ancladas a nivel de la
capa de péptidoglicano. Por lo tanto, dadas las características funcionales de MurB se
sugiere que está asociada con la inserción y anclaje de las proteínas del cuerpo basal del
flagelo.
Para la proteína candidato HP0813 se determinó la presencia un dominio con
actividad beta-lactamasa. La base de datos 3DID, reporta que en varias estructuras
cristalográficas este tipo de dominios, además de participar en la resistencia a los
antibióticos, interacciona con dominios de proteínas relacionadas con el transporte
electrónico a través de la membrana celular y con el reconocimiento de proteínas de la
membrana celular. Acceso a la anotaciones de estos dominios en la base de datos PFAM:
PF00258 y PF01473, respectivamente. Este conjunto de evidencias sugiere que HP0813
podría contribuir, junto con MotA y MotB, en la generación de torque necesario para la

57
rotación flagelar.
La proteína candidato HP0114 parece interaccionar con proteínas del gancho
flagelar y con una proteína asociada a la síntesis de polisacáridos. Estas interacciones que
presenta HP0114 podrían actuar como un vínculo entre proteínas relacionadas con la
síntesis de polisacáridos y proteínas del filamento flagelar. Esto es fundamental
considerando que tanto el ensamblaje del flagelo, como la fuerza rotacional de
desplazamiento implican que las proteínas de dichas estructuras estén glicosiladas para
funcionar de manera óptima. Por ejemplo existe evidencia experimental donde mutantes de
C. jenuni carentes de los genes asociados a glicosilación de las flagelinas presentan
fenotipos no motiles (Guerry et al., 2006; McNally et al., 2006).
Por último, dada la importancia que juega el flagelo en el proceso de colonización y
persistencia de la infección por H. pylori, este agrupamiento de proteínas sugiere una
interconexión entre los sistemas de quimiotaxis, exportación y motilidad. De tal manera
que H. pylori asegura un proceso de síntesis de proteínas fidedigno, principalmente de las
que son indispensables en las primeras etapas del ensamblaje del flagelo.
9.2. Adaptación al medio ácido
La adaptación al medio ácido es la capacidad de H. pylori de mantener el pH peri-
plasmático cercano a la neutralidad; permitiendo que en el citoplasma se mantengan valores
de pH fisiológicos. Varios genes asociados a la adaptación del medio ácido son requeridos
por H. pylori para colonizar modelos animales. Principalmente, genes asociados con la
síntesis de ureasa (Weeks et al., 2000; Mollenhauer-Rektorschek et al., 2002; Marcus et al.,
2005).
Entre los resultados obtenidos para este agrupamiento de proteínas, destaca la
proteína candidato NikR, la cual interacciona con proteínas asociadas con la enzima ureasa
y con el metabolismo del níquel. Es conocido que NikR es un regulador transcripcional
asociado con la homeostasia del níquel. El níquel juega un papel importante en la
colonización y persistencia de H. pylori. De manera específica, el níquel funciona como co-
factor de la enzima ureasa. La enzima se encarga, en gran parte, de la adaptación al medio
ácido. Sin embargo, aunque este metal es esencial para la sobrevivencia de H. pylori, a gran
concentración puede resultar tóxico para la bacteria. Por lo tanto, es necesario un control de

58
su importación. Diversos estudios sugieren que a alta concentración de níquel, NikR es
capaz de unirse a la región promotora de nixA, ureA y del operón exbB-exbD-tonB. Esto
produce dos fenómenos: en primer lugar, se suprime la producción de nixA y de exbB-
exbD-tonB y, simultáneamente, la importación de níquel y se activa la transcripción de ureA
para la utilización de este metal (Abraham et al., 2006; Dosanjh et al., 2007; Zambelli et
al., 2008). Mientras este trabajo estuvo en progreso, un estudio indica que, además de la
función homeostática del níquel, la actividad de NikR está directamente ligada a cambios
en el pH (Li y Zamble, 2009). Así, la inclusión de NikR en este agrupamiento de proteínas
se soporta de manera experimental constituyendo una prueba ciega para dichas
predicciones. La conclusión de las observaciones sugiere que NikR es un regulador
transcripcional con función preponderante en la adaptación al medio ácido y
consecuentemente en patogénesis de H. pylori.
Igualmente, la proteína candidato GlmM interacciona con las subunidades
estructurales del complejo ureasa (UreA-UreB). Aparentemente esta interacción entre
GlmM y UreA-UreB carece de significado biológico, ya que GlmM corresponde a una
enzima asociada a la síntesis de péptido glicano de la pared celular. Además, estudios
realizados en E. coli y S. gordonii sugieren que GlmM participa en el ciclo celular
bacteriano y en la formación de biopelículas (Shimazu et al., 2008). Esta asociación
funcional de GlmM es también soportada por los resultados de este trabajo, ya que GlmM
interacciona con FtsH, una proteína asociada con el citoesqueleto y ciclo celular bacteriano
(resultados no mostrados). Recientemente, se ha reportado a nivel experimental el
interactoma del complejo ureasa en H. pylori. Una de las conclusiones de este trabajo fue
que existe una relación entre el complejo ureasa con proteínas del citoesqueleto y del ciclo
celular bacteriano (Stingl et al., 2008). Estos hallazgos sugieren que tal vínculo esta dado
por la necesidad de crear un compartimento celular que contenga una fracción del complejo
ureasa, facilitando sus actividades en respuesta al pH ácido. A la luz de las observaciones,
se sugiere que GlmM, en conjunto con proteínas que conforman el citoesqueleto
bacteriano, contribuye a la generación un compartimento capaz de contener al complejo
ureasa, facilitando la entrega de sustratos al mismo.
Nuevamente, se presentan proteínas candidato asociadas con el proceso de
traducción en la síntesis de proteínas. Este es el caso de CsrA y SmpB, las cuales

59
interaccionan con proteínas raíz asociadas con el transporte de moléculas a través de la
membrana celular. Por ejemplo, en C. jejuni se demostró que CsrA es esencial para la
sobrevivencia bajo estrés, para la formación de biopelículas y para la invasión de células
del hospedero (Fields y Thompson, 2008). Esta evidencia soporta el papel de CsrA en este
agrupamiento de proteínas, considerando que el medio ambiente ácido por si mismo genera
condiciones de estrés. Adicionalmente, estudios en E. coli, Y. pseudotuberculosis y S.
entérica, demuestran que CsrA actúa como un regulador de genes asociados con virulencia
(Cerca y Jefferson, 2008; Heroven et al., 2008; Kerrinnes et al., 2008).
Por su parte, la proteína candidato SmpB participa en el proceso conocido como
trans-traducción. En bacterias este proceso ha sido estudiado a detalle con el fin de entender
los mecanismos de control de calidad del flujo de la información genética (Keiler et al.,
1996; Dulebohn et al., 2007; Moore y Sauer, 2007; Keiler, 2008). La trans-traducción es un
mecanismo utilizado para rescatar ribosomas secuestrados por mARNs carentes de señales
de terminación apropiadas, por tanto, incapaces de reanudar el proceso de traducción. En
consecuencia, la trans-traducción disminuye el número de cadenas defectuosas de mARN y
además, es utilizada para adicionar a proteínas incompletas, amino ácidos que funcionan
como etiquetas, útiles para la degradación de estas proteínas defectuosas. A pesar de que
este mecanismo ha sido estudiado a detalle, se desconocen muchos aspectos relacionados
con su papel biológico específico. Estudios en cepas de E. coli, Salmonella o Synechocystis
con defectos en la trans-traducción, resultaron ser hipersensibles al efecto de los
antibióticos (Vioque y de la Cruz, 2003; Luidalepp et al., 2005). Deficiencias en la trans-
traducción afecta la habilidad de S. typhimurium de colonizar ratones y de sobrevivir dentro
de los macrófagos (Baumler et al., 1994). Una cepa de Y. pseudotuberculosis con defectos
en la trans-traducción, resultó ser no virulenta en un modelo de infección murino (Okan et
al., 2006).
Del mismo modo que para NikR, durante el desarrollo de este trabajo se publicó un
estudio donde se demuestra que la proteína candidato SmpB es esencial para H. pylori.
Entre los principales resultados de este trabajo se destaca que las afectaciones al gen smpB
producen una disminución de la división celular y que smpB aumenta su expresión en
condiciones de pH bajo, similares a las encontradas a nivel gástrico. Al igual que en NikR,
esta evidencia soporta la validez de la metodología empleada en el presente trabajo.

60
Csra y SmpB, interaccionan con el sistema de transporte constituido por las
proteínas ExbB2, ExbD2 y TonB, su función ya fue citada. Existe evidencia que demuestra
que la estabilidad de TonB es dependiente de la presencia de proteínas del tipo ExbB y
ExbD. Por ejemplo, en E. coli, cuando estas dos últimas proteínas están ausentes, la vida
media de TonB disminuye como producto de su degradación (Lucchetti-Miganeh, et al.,
2008). Esta observación sugiere que CsrA y SmpB, bajos condiciones de pH acidas,
regulan la producción de las proteínas del tipo ExbB y ExbD y, por lo tanto, del transporte
de sustratos por TonB.
En resumen, este agrupamiento de proteínas se evidencia su función en las distintas
redes de regulación, en la modulación de cambios conductuales a nivel celular y en la
expresión de factores de virulencia necesarios para la persistencia y colonización de H.
pylori en su hospedero.
9.3. Metabolismo del nitrógeno
El proceso de asimilación de nitrógeno es esencial para la síntesis de biomoléculas,
tales como amino ácidos, purinas, pirimidinas y amino glucósidos. Las bacterias obtienen el
nitrógeno principalmente de moléculas inorgánicas donde el amoniaco es el compuesto
requerido para el anabolismo del nitrógeno. GlnA y GdhA son las enzimas que participan
en tal proceso. Específicamente, catalizan la condensación del glutamato y amoniaco para
producir glutamina (Eisenberg et al., 1987). Existe evidencia que sugiere que mutaciones
del gen glnA son letales para H. pylori (Garner et al., 1998). En B. subtilis mutaciones de
gltX también resultaron letales para la bacteria (Gagnon et al., 1994). Además, existen
estudios que asocian a GlnA con virulencia de varios patógenos de humanos. Este es caso
de M. tuberculosis, donde GlnA ha mostrado ser esencial para sobrevivencia dentro de los
macrófagos (Tullius et al., 2003) y de S. pneumoniae, que tiene función durante el proceso
de adherencia e invasión (Kloosterman et al., 2006).
Al igual que las proteínas raíz GlnA y GdhA, se sugiere que las proteínas candidato
de este agrupamiento están involucradas con el metabolismo del glutamato. Por ejemplo,
Ggt cataliza la ruptura del glutatión liberando el grupo glutamil que será utilizado por GlnA
y/o GdhA en la síntesis de glutamato. HP0056 oxida a la prolina para producir directamente
glutamato y AspB cataliza la conversión de aspartato a glutamato. MurI está asociada con

61
la síntesis de intermediarios de la pared celular a partir del glutamato. GltX1 y GltX2,
ambas corresponden a glutamil-tARN sintetasas y están asociadas con la síntesis de
proteínas.
Dado que el amoniaco es la principal fuente inorgánica para la obtención de
nitrógeno, existe un vínculo entre el agrupamiento de proteínas asociado a adaptación al
medio ácido y con el agrupamiento de proteínas asociado con el metabolismo del
nitrógeno. Como ya se mencionó, el complejo ureasa degrada la urea para producir dióxido
de carbono y amoniaco. Una parte del amoniaco realiza función de tampón mientras que el
restante es convertido a amonio para ser utilizado en el metabolismo del nitrógeno.
Además, existe otra fuente de amoniaco para la hidrólisis de urea. Esta vía alterna está
asociada con la degradación del amino ácido arginina. Las enzimas responsables de tal
proceso son la arginasa (RocF) y la arginina descarboxilasa (SpeA). Esta última es una de
las proteínas candidato de este agrupamiento de proteínas. Del mismo modo para el
complejo ureasa, la acción de RocF está asociada con la adaptación de H. pylori al medio
ácido, mientras que, SpeA se le ha asociado con el equilibrio del pH intracelular. Las
figuras 6 y 7 resumen el vínculo entre los agrupamientos de proteínas citados y las
proteínas RocF y SpeA. Estas últimas, tienen como función la regulación del pH y
suplementan parte del amoniaco necesario para el óptimo funcionamiento de este
agrupamiento de proteínas.
Por último, este agrupamiento de proteínas da evidencia de la interrelación entre
diferentes sistemas metabólicos, así como también de redundancia biológica que produce
vías alternas para mantener los niveles celulares de glutamato y, por tanto, de la
homeostasia celular que contribuye a la patogénesis de H. pylori.
9.4. Sistema de secreción tipo IV
Muchas cepas de H. pylori contienen la isla de patogenecidad Cag (Cag-PAI, por
sus siglas en inglés), cuyos genes codifican para la mayoría de los componentes del sistema
de secreción tipo IV (Odenbreit, 2005). La presencia de tal isla ha sido asociada con la
virulencia de las cepas de H. pylori (Cover y Blaser, 1999).
En este agrupamiento se identificaron proteínas candidato que interaccionan con
proteínas de función conocida como adhesinas. Este es el caso de las proteínas candidato

62
HopQ, HP1342, HP0922, HP0066 y HP0120. El análisis de las anotaciones disponibles en
la base datos UniProt para estas proteínas candidato sugiere que su localización sub-celular
está asociada con la membrana externa de H. pylori. De manera particular, el análisis de la
secuencia de HP0922 revela la presencia de un grupo de dominios relacionado con la
familia de cito-toxinas de la familia VacA, acceso en PFAM: PF03797, PF02691 y
PF03077. El mismo análisis para la secuencia de HP0066 demuestra la presencia de
dominios de la familia FtsK/SpoIIIE. En E.coli y B. subtilis, las proteínas que contienen
estos dominios participan en la regulación de su ciclo celular y en la transferencia de ADN
inter-cromosómico (Begg et al., 1995; Wu y Errington, 1994). La proteína HP0120
corresponde a una proteína hipotética de la cual sólo se dispone información acerca de su
patrón de interacción, por lo tanto, se requiere de mayores estudios de función para sostener
la predicción.
Las proteínas raíz que interaccionan con las proteínas candidato citadas participan
en tres procesos de la patogénesis de H. pylori. Las proteínas raíz Cag3 y CagS están
asociadas con en el sistema de secreción tipo IV (T4SS), el cual es clave en la secreción e
inyección de cito-toxinas. BabA y Omp1 son proteínas de la membrana externa que
funcionan como adhesinas contribuyendo con el proceso de fijación al epitelio gástrico del
hospedero y VacA es una potente cito-toxina. En el caso de las proteínas raíz CagA y VacA
(cito-toxinas), ambas se introducen en las células del epitelio gástrico. La introducción de
estas cito-toxinas produce cambios en las células gástricas, desde producción de una
respuesta inflamatoria, mediada por la liberación de citoquinas, hasta inducción de
apoptosis celular (Censini et al., 1996; Akopyants et al., 1998; Blomstergren et al., 2004).
Así mismo, se ha documentado que aquellos pacientes que portan cepas de H. pylori con
algunas de las cito-tóxinas y adhesinas citadas anteriormente presentan daño gástrico
severo y son un factor de riesgo para desarrollo de cáncer y ulcera gástrica (Warburton et
al., 1998; Umit et al., 2009).
Estas observaciones sugieren una probable función de HP0922 como cito-toxina,
HP0066 en la transferencia horizontal de genes y de HopQ, HP1342 y HP0120 como
adhesinas. Así mismo, es importante considerar que las proteínas de la membrana externa
son, por lo general, inmunogénicas y, por lo tanto, son un blanco en el desarrollo de
vacunas y de métodos específicos de diagnóstico.

63
Las proteínas candidato HP0040, HP0439 y TrbI comparten algunos patrones de
interacción con proteínas raíz asociadas a la transferencia horizontal de genes. HP0040 y
HP0439 corresponden a proteínas hipotéticas que presentan dominios del tipo VirB9 y
VirB8, respectivamente; ambos tipos de dominio están asociados a estructuras que
participan en la conjugación bacteriana. Por ejemplo, se ha documentado que en A.
tumefaciens, las proteínas que presentan dominios del tipo VirB8 interaccionan con
componentes estructurales del sistema conjugación bacteriana, como son VirB9 y VirB10
(Kumar y Das, 2001). En el caso de TrbI, su homólogo en A. tumefaciens, aunque no es
esencial, incrementa, de manera significativa, la eficiencia del proceso de conjugación
bacteriana (Alt-Morbe et al., 1996). Por lo tanto, se sugiere una función similar de TrbI con
la de su homólogo en A. tumefaciens.
A manera de resumen, considerando la localización sub-celular y el análisis de la
función de las proteínas raíz con las interaccionan las proteínas candidato, se sugiere que
estas últimas podrían estar asociadas al proceso de conjugación bacteriana, de adhesión y
del proceso de inyección de cito-toxinas que alteran la homeostasis de las células del
epitelio gástrico. Además, de acuerdo a la actividad cito-toxica de algunas de estas
proteínas candidato, su presencia en determinadas cepas podría estar asociada a la
intensidad del daño que producen en el epitelio gástrico del hospedero, siendo un factor de
riesgo para desarrollo del cáncer gástrico.
9.5. Proteínas de respuesta a estrés
El estrés oxidativo es un problema experimentado por cualquier ser vivo. La
reducción parcial del oxígeno molecular por la auto-oxidación de flavoproteínas genera una
mezcla de radicales superóxido y de peróxido de hidrógeno (Imlay, 2003). Estos
compuestos son la principal fuente para la producción de especies reactivas de oxígeno, las
cuales son capaces de alterar la función de las proteínas y la estructura del ADN (Imlay y
Linn, 1988; Imlay, 2003). H. pylori es una bacteria microaerofílica vulnerable a los efectos
tóxicos del superóxido, por tanto, debe de contar con mecanismos de destoxificación
eficaces que le permitan colonizar un medio ambiente con condiciones de estrés, tal como
el epitelio gástrico.

64
Entre los resultados obtenidos para este agrupamiento destaca la presencia de las
proteínas candidato SodB, GroL y Ndk. La mayoría de las proteínas raíz con las cuales
interaccionan estas proteínas candidato, a excepción de SecA, presentan actividad
antioxidante. SecA corresponde a una proteína asociada a translocación de macromoléculas
a través de la membrana celular. Así, la interacción entre SodB y SecA es fundamental
considerando que ya se ha demostrado que SodB es secretada por H. pylori. Aunque su
función en la fracción extracelular no está bien documentado, se sugiere que ésta participa
en el proceso de patogénesis (Backert et al., 2005). Por ejemplo, en M. tuberculosis existe
evidencia de que SodB se secreta y es capaz de inhibir la muerte celular programada en las
células infectadas del hospedero (Edwards et al., 2001; Hinchey et al., 2007). Por su parte,
en E. coli y C. jejuni, los homólogos de GroL y Ndk han sido asociadas a proteínas de
repuesta a estrés. En particular, GroL previene alteraciones del plegamiento de las proteínas
y Ndk repara el ADN (Goswami et al., 2006; Klancnik et al., 2006). Además, se ha
demostrado, mediante estudios de proteómica diferencial, que GroL está sobre-producida
en H. pylori cuando infecta las células del epitelio gástrico (Backert et al., 2005). Una
posible explicación a este comportamiento de GroL podría estar relacionado con las etapas
iníciales de la infección. Cuando H. pylori infecta el epitelio gástrico se desencadena una
respuesta inflamatoria de tipo inmunológico. Durante este proceso, puede liberarse al
medio, una gran cantidad de especies reactivas de oxígeno (Bagchi et al., 1996; Nardone et
al., 2004). Por lo tanto, H. pylori debe ser capaz de adaptarse a tales condiciones de estrés.
En estudios proteómicos previos se ha documentado que GroL se secreta por H. pylori y su
presencia en la fracción extracelular se ha asociado con la producción de interleucina 8 (Lin
et al., 2005). Esta citoquina, además de coadyuvar con el funcionamiento del sistema
inmune, se caracteriza por producir inflamación de los tejidos. Esto es fundamental
considerando que uno de los procesos en el establecimiento y persistencia de H. pylori
sobre el epitelio gástrico es la inducción de una respuesta inflamatoria crónica. Así, GroL
sería una proteína fundamental para la persistencia de la infección por H pylori.
Las proteínas candidato UvrC, NrdA y ADNE interaccionan con las proteínas raíz
TrxA y TrxB; las cuales están asociadas a mantener el equilibrio oxido-reducción de la
célula. Existe evidencia que demuestra que la actividad catalítica de NrdA se facilita por la
acción reductora de TrxA y TrxB (Gallardo-Madueño et al., 1998). Por su parte, LpxB es

65
capaz de generar enlaces glucosídicos. Tomando en conjunto las observaciones anteriores,
es posible visualizar una maquinaria completa capaz de producir componentes asociados a
síntesis de ADN; sugiriendo que UvrC, NrdA y DnaE conforman un grupo de proteínas
encargadas de la reparación del daño del ADN.
Por otra parte resalta la presencia de una proteína candidato asociada a regulación
de la traducción. Este es el caso de la proteína RpsB que interacciona con proteínas
asociadas al sistema de exportación de proteínas relacionadas con el ensamblaje del flagelo.
Por ejemplo, en un análisis proteómico de B. subtilis, se ha observado que junto con otras
proteínas de estrés, la presencia de RpsB está aumentada (Jürgen et al., 2001). Del mismo
modo, en Listeria monocytogenes y Bifidobacterium longum, se ha documentado que RpsB
está aumentada bajo condiciones de estrés por alta concentración de NaCl y bilis (Duché et
al., 2002; Sánchez et al., 2005). Esta última observación permite explicar la interacción de
RpsB con las proteínas flagelares FliA y FliR. Durante su tránsito por el tracto
gastrointestinal H. pylori debe tolerar y resistir condiciones de estrés causado por la bilis.
La presencia de bilis es una condición desfavorable para la motilidad de H. pylori. En
consecuencia, el sistema flagelar debería de funcionar aún en respuesta al estrés causado
por la bilis. Posiblemente, RpsB facilita, bajo estas condiciones de estrés, la síntesis de las
proteínas que son importantes para el ensamblaje de las estructuras básicas del flagelo. Por
otra parte, aunque la interacción de RpsB con SecA (esta última proteína ya fue citada),
pareciera carecer de relación funcional, en un estudio proteómico de las proteínas
secretadas por H. pylori, se documentó una gran cantidad de proteínas ribosomales sin
función conocida pero esto sugiere probable función en patogénesis (Backert et al., 2005).
Por lo cual, SecA podría facilitar la exportación RpsB al medio extracelular. Estas
observaciones sugieren que RpsB podría regular bajo condiciones de estrés la síntesis de
proteínas que son útiles para el ensamblaje de estructuras celulares.
Este agrupamiento demuestra que es un grupo de proteínas cuya función permite a
H. pylori sobrevivir bajo una variedad de condiciones de estrés. Es por esto que H. pylori es
hábil para sobrevivir bajo condiciones adversas y persistir en la mucosa gástrica. Por lo
tanto, es interesante conocer cómo funcionan los mecanismos de respuesta a estrés de H.
pylori, lo cual es clave en el entendimiento del mecanismo de persistencia y patogénesis de
dicho patógeno.

66
9.6. Síntesis de lipopolisacáridos
La presencia de lipopolisacárido (LPS) es una de las principales características
distintivas de las bacterias Gram (-) y la serología de estas moléculas ha sido un
procedimiento de rutina para la diferenciación dentro de una especie bacteriana. El LPS es
una molécula abundante en superficie celular y funciona como barrera de protección contra
agentes externos. Una molécula de LPS está constituida por tres elementos básicos: el
lípido A el cual esta embebido en la cara externa, de la membrana externa de la bacteria; el
núcleo de oligosacárido, que funciona como una región de enlace entre las dos caras de la
membrana externa y finalmente el antígeno O que es un determinante serotipo-específico,
presente como una unidad polimérica extendiéndose lejos de la superficie celular. Todas las
proteínas raíz y candidato de este agrupamiento presentan funciones relacionadas con la
síntesis de los componentes básicos del LPS, antes citados.
Por ejemplo, en V. cholera, RfbA, RfbD y RfbE, son proteínas homólogas en H.
pylori de HP0043, RfbD y de la proteína candidato SpsC, respectivamente. RfbA, RfbD y
RfbE participan en la síntesis de perosamina, un componente básico del lipopolisacárido
que constituye al antígeno O y en la síntesis de la matriz de lipopolisacárido que produce
una colonia de bacterias con morfología rugosa específica (Stroeher et al., 1995; Rashid et
al., 2003). La proteína raíz NolK también está asociada con la síntesis del antígeno O en E.
coli (Wang et al., 2006). Mientras que en el complejo Mycobacterium avium se ha
documentado que las proteínas homólogos de RfbD y de la proteína candidato HP0102
están involucrados en la síntesis de lipopolisacáridos de superficie (Miyamoto et al., 2007).
Para el caso de la interacción entre WbpB y la proteína candidato SpsC, existe
evidencia de que en P. aeruginosa y B. pertussis, sus proteínas homólogos, participan en la
síntesis de un raro trisacárido que se incorpora al antígeno O, confiriéndole a estos
patógenos resistencia a la acción lítica del complemento y, por consecuencia, facilita el
proceso de colonización del hospedero (Harvill et al., 2000; Westman et al., 2008).
Las proteínas candidato KdtA y HP0277 comparten patrones de interacción; ambas
interaccionan con las proteínas raíz RfaC y CoaD. Ademas, Kdta interacciona con la
proteínas raíz LpxB. La interacción de KdtA con las proteínas raíz antes citadas, podría
soportarse considerando que en E. coli, K. pneumoniae y S. enterica se ha demostrado que
KdtA, RfaC y CoaD, en conjunto, participan en la síntesis de un oligosacárido que forma

67
parte del núcleo del lipopolisacárido, estructura asociada al anclaje del lipopolisacárido con
la membrana celular (Heinrichs et al., 1998; Izquierdo et al., 2002). LpxB corresponde a
una enzima asociada a síntesis del lípido A. La interacción entre KdtA y LpxB es clave
considerando que el núcleo de oligosacárido, ya citado, está unido al lípido A que permite
el anclaje de la molécula de lipopolisacárido a la membrana celular (Whitfield et al., 1997).
Por su parte, la proteína candidato HP0277 corresponde a una ferredoxina; este tipo de
proteínas están asociadas con la respuesta al estrés oxidativo. La interacción de HP0277
con las proteínas raíz antes citadas podría explicarse, considerando que diversos estudios
sugieren que bajo estrés oxidativo, los diferentes constituyentes estructurales lipídicos
pueden tener serias alteraciones comprometiendo la integridad de la célula bacteria. Este
puede ser caso de los fosfolípidos de la membrana celular que pueden ser oxidados por
radicales libres, llevando a una desestabilización de la bicapa lipídica y, por consiguiente,
de una disfunción de la membrana celular (Hazell y Graham, 1990; Khulusi et al., 1995;
Sun et al., 2003). Considerando lo anterior, podría proponerse que HP0277 contribuye para
evitar la degradación de los diferentes productos lipídicos sintetizados por RfaC y CoaD,
manteniendo así la integridad celular.
Otras proteínas candidato que podrían desempeñar una función similar a la
propuesta para KdtA son GmhA, HldE, RfaD, GalU y AlgC. La evidencia que podría
sostener la predicción de las interacciones para las proteínas candidato antes mencionadas
parte de estudios que demuestran que en E. coli, N. meningitidis y S. typhimurium, las
proteínas homólogos de GmhA, HldE, RfaD, HP0043 y RfaC están asociadas a la síntesis
del núcleo del lipopolisacárido (Sirisena et al., 1992; Shih et al., 2001; Kneidinger et al.,
2002). Del mismo modo, en A. ferrooxidans se ha asociado a los homólogos de GalU y
GalE con la producción de exolipolisacáridado involucrado en la formación de biopelículas
(Barreto et al., 2005). Por su parte, AlgC está implicada en la síntesis del núcleo del
lipopolisacárido en P. aeruginosa (Kooistra et al., 2003).
Las proteínas candidato FlaA1 y HP1570 interaccionan con las proteínas raíz NeuA
y SpsC. Se ha documentado en C. jejuni que PseF y PseC, proteínas homólogas en H.
pylori de NeuA y SpsC, respectivamente, participan en la glicosilación de las flagelinas del
filamento flagelar y que las mutantes carentes de los genes asociados a glicosilación de las
flagelinas presentan fenotipos no motiles (Guerry et al., 2006; McNally et al., 2006).

68
Estudios realizados en A. hydrophila y C. jejuni sugieren que las proteínas homólogos de
FlaA1, NeuA y SpsE participan activamente en la glicosilación de proteínas flagelares
(Guerry et al., 2006; Canals et al., 2007). En C. jejuni, se ha observado que tal glicosilación
está asociada a un fenómeno llamado auto-aglutinación a su vez asociado a la formación de
biopelículas en las células del epitelio intestinal (Guerry, 2007). Así mismo, en C. jejuni se
ha demostrado, a nivel experimental, que existe una interacción física entre las proteínas
ortólogos de HP1570 y NeuA (Parrish et al., 2007). Por tanto, se sugiere que las proteínas
candidato FlaA1 y HP1570 podrían estar asociadas a glicosilación de proteínas flagelares.
Por último, este agrupamiento de proteínas remarca la importancia de las proteínas
involucradas en síntesis del antígeno O y de los lipopolisacáridos de superficie en H. pylori
cuya función está asociada a una respuesta inflamatoria del epitelio gástrico y con la
evasión del sistema inmune. Además, se estable un vínculo entre este agrupamiento de
proteínas y el que corresponde al ensamblaje del flagelo mediante glicosilación de las
flagelinas, proteínas clave para la actividad hidrodinámica del flagelo.
9.7. Interconexión entre agrupamientos de proteínas
La red de interacción entre proteínas del presente estudio, demuestra varios
agrupamientos entre proteínas, cada uno representando un sistema asociado a proceso
especifico durante el proceso de patogénesis de H. pylori. No obstante esta especificidad, la
interconexión entre dichos agrupamientos no ha sido estudiada. En este estudio se
demuestra la interconexión entre el sistema que conforman las proteínas flagelares y el
sistema de síntesis de polisacáridos (Figura 6). Esta relación está dada por la modificación
postraduccional de las flagelinas (glicosilación) lo que les permite ensamblar
metaestructuras (ver discusión para este agrupamiento). Del mismo modo, se determinó una
interconexión entre el sistema de adaptación al medio ácido y el metabolismo del nitrógeno
(Figura 6). Estos dos sistemas forman un bucle que se auto-regula. Por ejemplo, la urea es
producto final del metabolismo de la arginina y otros amino ácidos. Posteriormente, la urea
puede ser desdoblada por la enzima ureasa produciendo CO2 y amoniaco para funcionar
como tampón. Ambos sistemas están asociados a la liberación y asimilación del amoniaco,
por lo tanto contribuyen a la manutención del balance del nitrógeno en H. pylori. Estas
interconexiones de los sistemas dan evidencia de la redundancia inherente a los sistemas

69
biológicos permitiendo vías o caminos alternos para un mismo proceso biológico;
manteniendo la homeostasia celular.
9.8. Limitaciones del estudio
La red de interacción de proteínas del presente estudio tiene limitaciones. Se
combinaron datos de interacción entre proteínas a nivel experimental y predicciones a nivel
computacional. Esta combinación de métodos proporciona una mayor cobertura pero
también se deben considerar sus posibles fallas. Por ejemplo, en el caso de los ensayos de
doble híbrido en levadura no son 100% verificables, ya que un alto número de interacciones
putativas no pueden ser confirmadas por otros métodos (falsos positivos). Además, se ha
observado que interacciones entre proteínas confirmadas por varios métodos difícilmente
podrían ocurrir en la naturaleza puesto que las proteínas que participan en dicha interacción
están confinadas a espacios u organelos celulares diferentes. Este último tipo de
interacciones se denominan falsos positivos biológicos (Cusick et al., 2005). Por otro lado,
los falsos negativos pueden presentarse por varias causas, entre las que destaca la alteración
del plegamiento de la proteína producto de la fusión y por diferentes mecanismos de
modificación postraduccional en la levadura, etc.
La purificación por afinidad en tándem es otro método utilizado para caracterizar
interactomas. Sin embargo, al igual que el sistema de doble híbrido en levaduras las
limitaciones son una pobre cobertura del proteoma del organismo en estudio (Cusick et al.,
2005). Por ejemplo, los interactomas caracterizados de S. cerevisiae (Uetz et al., 2000; Ito
et al., 2001), C. elegans (Li et al., 2004) y H. pylori (Rain et al., 2001) presentan una
cobertura del proteoma de cerca del 15%, 5% y 47%, respectivamente. Esto sin considerar
los probables falsos positivos y negativos. Por tal motivo, para varios organismos modelo
se han caracterizado varios mapas de interacción entre proteínas para validar tales
interacciones.
Por su parte, los métodos in silico también tienen limitaciones ya que la gran
mayoría de los aquí utilizados se basan en la identificación de señales evolutivas. Sin
embargo podrían existir proteínas que carecen, o que no se pueden detectar dichas señales.
Así mismo, la mayoría de los métodos computacionales, basados en el contexto genómico,
no distinguen entre una interacción física o funcional. Otra limitación es que el alto nivel de

70
confiabilidad aplicado durante el filtrado de las interacciones entre proteínas, obtenidas por
STRING, es muy astringente. Esto es, STRING considera que una interacción entre
proteínas es confiable si existen varias fuentes de información o métodos que la soporten.
Por lo cual, las interacciones de proteínas verdaderas poco documentadas o soportadas por
varios métodos (experimental o computacional) son ignoradas. Por lo tanto, la red de
interacción entre proteínas aquí propuesta muy probablemente no representa el total de las
proteínas asociadas a patogénesis de H. pylori. En consecuencia, las predicciones deben de
ser validadas a nivel experimental.

71
10. Conclusiones
En el presente trabajo se construyó una red de interacción entre proteínas a partir de
una base de datos de proteínas con función conocida asociada a patogénesis de H. pylori.
Esto permitió, identificar y priorizar un conjunto de nuevas interacciones y proteínas
asociadas a patogénesis de H. pylori.
El análisis a nivel de sistemas resultó en nuevas funciones para algunas proteínas ya
caracterizadas. Este es el caso de CheA, NikR y SodB. CheA está asociada a exportación de
proteínas flagelares para el ensamblaje del flagelo y participa durante las primeras etapas
del desarrollo de las biopelículas. NikR es una proteína reguladora maestra de un conjunto
de genes clave asociadas a adaptación al medio acido. SodB, además de presentar actividad
antioxidante, se secreta para inhibir la muerte celular programada de las células del
hospedero.
El análisis de la red de interacción entre proteínas del presente predecir nuevas
proteínas con probable asociación a patogénesis y nuevas hipótesis susceptibles de ser
validadas a nivel experimental. Entre las nuevas proteínas se identificó a un grupo de
proteínas candidato asociadas a regulación del proceso de traducción en la síntesis de
proteínas. Estas proteínas están presentes en todos los agrupamientos de proteínas, con la
excepción del sistema de proteínas de secreción tipo IV y el de la síntesis de
lipopolisacáridos. Estas proteínas aparecen justo en aquellos agrupamientos de proteínas
que son esenciales en las primeras etapas de la infección, sugiriendo que son necesarias
para un control de calidad de la síntesis de proteínas durante el proceso de patogénesis de
H. pylori.
Por último, el análisis de la red de interacción entre proteínas da evidencia de las
estrategias evolutivas del patógeno para desarrollo y persistencia del proceso infeccioso.
Así mismo, demuestra que varios de los procesos biológicos asociados a patogénesis de H.
pylori no son independientes. Por el contrario, están interconectados generando un sistema
redundante capaz de modificarse para responder a diferentes estímulos del medio ambiente.

72
11. Recomendaciones
Una vez concluido el presente estudio, se considera interesante investigar sobre
otros aspectos relacionados con las interacciones entre proteínas de H. pylori y, por lo tanto,
se recomienda:
• Extender los análisis expuestos en esta tesis al estudio de otros procesos biológicos
de H. pylori.
• Mejorar el método de predicción basado en el contexto funcional mediante la
inclusión de otros tipos de información experimental, por ejemplo información
estructural de regiones interfaciales derivadas de complejos proteicos depositados
en la base de datos PDB.
• Evaluar si existen diferencias a nivel de interacción entre proteínas de las seis cepas
de H. pylori de las que se dispone su genoma completo.
• Finalmente, seleccionar algunas de las proteínas candidato propuestas y realizar su
validación experimental.

73
12. Referencias
• Abraham, L.O.; Li, Y.; Zamble, D.B. (2006). The metal- and DNA-binding
activities of Helicobacter pylori NikR. J Inorg Biochem. 100(5-6):1005-14.
• Akopyants, N.S.; Clifton, S.W.; Kersulyte, D.; Crabtree, J.E.; Youree, B.E.; Reece,
C.A.; Bukanov, N.O.; Drazek, E.S.; Roe, B.A.; Berg, D.E. (1998). Analyses of the
cag pathogenicity island of Helicobacter pylori. Mol Microbiol. 28(1):37-53.
• Allan, E.; Clayton,C.L.; McLaren, A.; Wallace D.M.; Wren, B.W. (2001).
Characterization of the low pH responses of Helicobacter pylori using genomic
ADN arrays. Microbiology, 147:2285-2292.
• Alm, R.A.; Ling, L.S.; Moir, D.T.; King, B.L.; Brown, E.D.; Doig, P.C.; Smith,
D.R.; Noonan, B.; Guild, B.C.; deJonge, B.L.; Carmel, G.; Tummino, P.J.; Caruso,
A.; Uria-Nickelsen, M.; Mills, D.M.; Ives, C.; Gibson, R.; Merberg, D.; Mills, S.D.;
Jiang, Q.; Taylor, D.E.; Vovis, G.F.; Trust, T.J. (1999). Genomic-sequence
comparison of two unrelated isolates of the human gastric pathogen Helicobacter
pylori. Nature, 14;397(6715):176-80.
• Aloy, P.; Russell, R.B. (2002). Interrogating protein interaction networks through
structural biology. Proc Natl Acad Sci USA., 99: 5896-901.
• Aloy, P.; Russell, R.B. (2003). InterPreTS: protein interaction prediction through
tertiary structure. Bioinformatics, 19:161-2.
• Aloy, P.; Russell, R.B. (2006). Structural systems biology: modelling protein
interactions. Nat Rev Mol Cell Biol. 7(3):188-97.
• Aloy, P. (2007). Shaping the future of interactome networks. Genome Biol.
8(10):316.

74
• Alt-Morbe, J.; Stryker, J.L.; Fuqua, C.; Li, P.L.; Farrand, S.K.; Winans, S.C. (1996).
The conjugal transfer system of Agrobacterium tumefaciens octopine-type Ti
plasmids is closely related to the transfer system of an IncP plasmid and distantly
related to Ti plasmid vir genes. J Bacteriol 178:4248-4257.
• Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. (1990). Basic local
alignment search tool. J Mol Biol., 215:403-10.
• Ang, S.; Lee, C.; Peck, K.; Sindici, M.; Matrubutham, U.; Gleeson, M.A.; Wang,
J.T. (2001). Acid-induces gene expression in Helicobacter pylori: study in genomic
scale by microarray. Infect. Immun., 69:1679-1686.
• Apweiler, R.; Attwood, T.K.; Bairoch, A.; Bateman, A.; Birney, E.; Biswas, M.;
Bucher, P.; Cerutti, L.; Corpet, F.; Croning, M.D.; Durbin, R.; Falquet, L.;
Fleischmann, W.; Gouzy, J.; Hermjakob, H.; Hulo, N.; Jonassen, I.; Kahn, D.;
Kanapin, A.; Karavidopoulou, Y.; Lopez, R.; Marx, B.; Mulder, N.J.; Oinn, T.M.;
Pagni, M.; Servant, F.; Sigrist, C.J.; Zdobnov, E.M. (2001). The InterPro database,
an integrated documentation resource for protein families, domains and functional
sites. Nucleic Acids Res., 29:37-40.
• Ashburner, M., et al. (2000). Gene ontology: tool for the unification of biology. The
Gene Ontology Consortium. Nat. Genet., 25, 25–29.
• Aragues, R.; Jaeggi, D.; Oliva, B. (2006). PIANA: protein interactions and network
analysis. Bioinformatics. 15;22(8):1015-7.
• Aragues, R.; Sander, C.; Oliva, B. (2008). BMC Bioinformatics. 27;9:172.

75
• Bagchi, D.; Bhattacharya, G.; Stohs, S.J. (1996). Production of reactive oxygen
species by gastric cells in association with Helicobacter pylori. Free Radic Res.
24(6):439-50.
• Backert, S.; Moese, S.; Selbach, M.; Brinkmann, V.; Meyer, T.F. (2001).
Phosphorylation of tyrosine 972 of the Helicobacter pylori CagA protein is essential
for induction of a scattering phenotype in gastric epithelial cells. Mol Microbiol.,
42(3):631-44.
• Backert, S.; Kwok, T.; Schmid, M.; Selbach, M.; Moese, S.; Peek, R.M. Jr.; König,
W.; Meyer, T.F.; Jungblut, P.R. (2005). Subproteomes of soluble and structure-
bound Helicobacter pylori proteins analyzed by two-dimensional gel
electrophoresis and mass spectrometry. Proteomics, 5(5):1331-45.
• Bader, G.D.; Betel, D.; Hogue, C.W.V. (2003). BIND: the biomolecular interaction
network database. Nucleic Acids Res., 31:248-50.
• Bader, G.D.; Hogue, C.W. (2003). An automated method for finding molecular
complexes in large protein interaction networks. BMC Bioinformatics. 13;4:2.
• Barabási, A.L.; Oltvai, Z.N. (2004). Network biology: understanding the cell's
functional organization. Nat Rev Genet. 5(2):101-13.
• Bäumler, A.J.; Kusters, G.J.; Stojiljkovic, I.; Heffron, F. (1994). Salmonella
typhimurium loci involved in survival within macrophages. Infect Immun. 62:1623–
1630.
• Bauerschmitt, H.; Funes, S.; Herrmann, J.M. (2008). The membrane-bound GTPase
Guf1 promotes mitochondrial protein synthesis under suboptimal conditions. J Biol
Chem. 283(25):17139-46.

76
• Beard, W.A. Wilson, S.H. (2001). DNA lesion bypass polymerases open up.
Structure. 9(9):759-64.
• Begg, K.J.; Dewar, S.J.; Donachie, W.D. (1995). A new Escherichia coli cell
division gene, ftsK. J Bacteriol 177:6211-6222.
• Benson, T.E.; Walsh, C.T.; Hogle, J.M. (1996). The structure of the substrate-free
form of MurB, an essential enzyme for the synthesis of bacterial cell walls.
Structure 4:47-54.
• Berman, H.M.; Battistuz, T.; Bhat, T.N.; Bluhm, W.F.; Bourne, P.E.; Burkhardt, K.;
FENA, Z.; Gilliland, G.L.; Iype, L.; Jain, S.; Fagan, P.; Marvin, J.; Padilla, D.;
Ravichandran, V.; Schneider, B.; Thanki, N.; Weissig, H.; Westbrook, J.D.;
Zardecki, C. (2002). The Protein Data Bank. Acta Crystallogr D Biol Crystallogr.,
58, 899.
• Blaser, J.M. (1991). Helicobacter pylori. Princ and Pract Inf Dis. Update 9: 3-9.
• Boneca, I.G.; de Reuse, H.; Epinat, J.C.; Pupin, M.; Labigne, A.; Moszer, I. (2003).
A revised annotation and comparative analysis of Helicobacter pylori genomes.
Nucleic Acids Res. 31(6):1704-14.
• Blomstergren, A.; Lundin, A.; Nilsson, C.; Engstrand, L.; Lundeberg, J. (2004).
Comparative analysis of the complete cag pathogenicity island sequence in four
Helicobacter pylori isolates. Gene. 328:85-93.
• Cave, D.R. (1997). Epidemiology and Transmission of Helicobacter pylori
Infection. How Is Helicobacter pylori Transmitted?. Gastroenterology, 113: S9-
S14.

77
• Carron, M.A.; Tran, V.R.; Sugawa, C.; Coticchia, J.M. (2006). Identification of
Helicobacter pylori biofilms in human gastric mucosa. J Gastrointest Surg 10, 712–
717.
• Censini, S.; Lange, C.; Xiang, Z.; Cabtree, J.E.; Giara, P.; Borodovsky, M.;
Rappuoli, R.; Covacci, A. (1996). cag, a Pathogenicity Island of Helicobacter
pylori, Encodes Type I-Specific and Disease-Associated Virulence Factors. Proc
Natl Acad Sci USA., 93: 14648-14653.
• Cellini, L.; Grande, R.; Di Campli, E.; Di Bartolomeo, S.; Di Giulio, M.; Traini, T.;
Trubiani, O. (2008). Characterization of a Helicobacter pylori environmental strain.
J Appl Microbiol. 105(3):761-9.
• Cerca, N.; Jefferson, K.K. (2008). Effect of growth conditions on poly-N-
acetylglucosamine expression and biofilm formation in Escherichia coli. FEMS
Microbiol Lett. 283(1):36-41.
• Chakrabarti, P.; Janin, J. (2002). Dissecting protein-protein recognition sites.
Proteins, 47: 334-43.
• Chothia, C.; Janin, J. (1975). Principles of protein-protein recognition. Nature,
256:705-8.
• Claverie, J.M. (2001). Gene number: What if there are only 30,000 human genes?
Science, 291:1255-7.
• Cooper, M.A. (2003). Label-free screening of bio-molecular interactions. Anal
Bioanal Chem, 377: 834-842.
• Correa, P.; Piazuelo, M.B. (2008). Natural history of Helicobacter pylori infection.
Dig Liver Dis., 2008, 4.

78
• Covacci, A.; Censini, S.; Bugnoli, M. (1993). Molecular Characterization of the
128-kDa Immunodominant Antigen of Helicobacter pylori Associated with
Citotoxicity and Duodenal Ulcer. Proc Natl Acad Sci USA., 90: 5791-5795.
• Cover, T.; Glupczynski, Y.; Lage, A.; Burette, A.; Tummuru, Mr.; Perez-Perez, G.;
Blaser, M. (1995). Serologic Detection of Infection with cagA + Helicobacter
pylori Strains. J Clin Microbiol. 33:1496-1500.
• Cover, T.L.; Blaser, J.M. (1999). Helicobacter pylori factors associated with
disease. Gastroenterology. 117(1):257-61.
• Crennell, S.J.; Garman, E.F.; Laver, W.; Vimr, E.; Taylor, G.L. (1993). Crystal
structure of a bacterial sialidase (from Salmonella typhimurium LT2) shows the
same fold as an influenza virus neuraminidase. Proc Natl Acad Sci USA, 90:9852-6.
• Cussac, V.; Ferrero, R.L.; Labigne, A. (1992). Expression of Helicobacter pylori
urease genes in Escherichia coli grown under nitrogen-limiting conditions. J.
Bacteriol., 174, 2466-2473.
• Cusick, M.E.; Klitgord, N.; Vidal, M.; Hill, D.E. (2005). Interactome: gateway into
systems biology. Hum Mol Genet. 14 Spec No. 2:R171-81.
• Dandekar, T.; Snel, B.; Huynen, M.; Bork, P. (1998). Conservation of gene order: a
fingerprint of proteins that physically interact. Trends Biochem Sci., 23:324-8.
• Dasgupta. N.; Arora, S.K.; Ramphal, R. (2000). fleN, a gene that regulates flagellar
number in Pseudomonas aeruginosa. J Bacteriol. 182(2):357-64.

79
• Dasgupta, N.; Wolfgang, M.C.; Goodman, A.L.; Arora, S.K.; Jyot, J.; Lory, S.;
Ramphal, R. (2003). A four-tiered transcriptional regulatory circuit controls
flagellar biogenesis in Pseudomonas aeruginosa. Mol Microbiol. 50(3):809-24.
• Dong, Z.; Onrust, R.; Skangalis, M.; O'Donnell, M. (1993). ADN polymerase III
accessory proteins. I. holA and holB encoding delta and delta9. J. Biol. Chem., 268,
11758-11765.
• Dosanjh, N.S.; Hammerbacher, N.A.; Michel, S.L. (2007). Characterization of the
Helicobacter pylori NikR-P(ureA) DNA interaction: metal ion requirements and
sequence specificity. Biochemistry. 46(9):2520-9.
• Duché O, Trémoulet F, Namane A, Labadie J; European Listeria Genome
Consortium. (2002). A proteomic analysis of the salt stress response of Listeria
monocytogenes. FEMS Microbiol Lett. 215(2):183-8.
• Dulebohn, D.; Choy, J.; Sundermeier, T.; Okan, N.; Karzai, A.W. (2007). Trans-
translation: the tmRNA-mediated surveillance mechanism for ribosome rescue,
directed protein degradation, and nonstop mRNA decay. Biochemistry.46:4681–
4693.
• Edwards, K.J.; Metherell, L.A.; Yates, M.; Saunders, N.A. (2001). Detection of
rpoB mutations in Mycobacterium tuberculosis by biprobe analysis. J Clin
Microbiol. 39(9):3350-2.
• Edwards, A.M.; Kus, B.; Jansen, R.; Greenbaum, D.; Greenblatt, J.; Gerstein, M.
(2002). Bridging structural biology and genomics: assessing protein interaction data
with known complexes. Trends Genet., 18:529-36.
• Eisenberg, D.; Almassy, R.J.; Janson, C.A.; Chapman, M.S.; Suh, S.W.; Cascio, D.;
Smith, W.W. (1987). Some evolutionary relationships of the primary biological

80
catalysts glutamine synthetase and RuBisCO. Cold Spring Harb Symp Quant Biol.
52:483-90.
• Eisenberg, D.; Marcotte, E.M.; Xenarios, I.; Yeates, T.O. (2000). Protein function
in the postgenomic era. Nature., 405:823-6.
• Enright, A.; Iliopoulos, I.; Kyrpides, N.; Ouzounis, C. (1999). Protein interaction
maps for complete genomes based on gene fusion events. Nature, 402:86-90.
• Evans, D.; Queiroz, D.; Mendes, E.; Evans Jr, D. (1998). Helicobacter pylori CagA
status and s and m alleles of VacA in isolates from individuals with a variety of H.
pylori associated gastric diseases. J Clin Microbiol., 36: 3435-3437.
• Everhart, J.E.; Kruszon-Moran, D.; Perez-Perez, G.I.; Tralka, T.S.; McQuillan, G.
(2000). Seroprevalence and ethnic differences in Helicobacter pylori infection
among adults in the Unites States. J Infect Dis., 181: 1359-1363.
• Fields, S.; Song, O. (1989). A novel genetic system to detect protein-protein
interactions. Nature, 340(6230):245-246.
• Fields, J.A.; Thompson, S.A. (2008). Campylobacter jejuni CsrA mediates
oxidative stress responses, biofilm formation, and host cell invasion. J Bacteriol.
190(9):3411-6.
• Gallardo-Madueño, R.; Leal, J.F.; Dorado, G.; Holmgren, A.; López-Barea, J.;
Pueyo, C. (1998). In vivo transcription of nrdAB operon and of grxA and fpg genes
is triggered in Escherichia coli lacking both thioredoxin and glutaredoxin 1 or
thioredoxin and glutathione, respectively. J Biol Chem. 273(29):18382-8.
• Gallet, X.; Charloteaux, B.; Thomas, A.; Brasseur, R. (2000). A fast method to
predict protein interaction sites from sequences. J Mol Biol., 302:917-26.

81
• Gagnon, Y.; Breton, R.; Putzer, H.; Pelchat, M.; Grunberg-Manago, M.; Lapointe, J.
(1994). Clustering and co-transcription of the Bacillus subtilis genes encoding the
aminoacyl-tRNA synthetases specific for glutamate and for cysteine and the first
enzyme for cysteine biosynthesis. J Biol Chem. 269(10):7473-82.
• Garner, R.M.; Fulkerson, J. Jr.; Mobley, H.L. (1998). Helicobacter pylori glutamine
synthetase lacks features associated with transcriptional and posttranslational
regulation. Infect Immun. 66(5):1839-47.
• Gavin, A.C.; Bösche, M.; Krause, R.; Grandi, P.; Marzioch, M.; Bauer, A.; Schultz,
J.; Rick, J.M.; Michon, A.M.; Cruciat, C.M.; Remor, M.; Höfert, C.; Schelder, M.;
Brajenovic, M.; Ruffner, H.; Merino, A.; Klein, K.; Hudak, M.; Dickson, D.; Rudi,
T.; Gnau, V.; Bauch, A.; Bastuck, S.; Huhse, B.; Leutwein, C.; Heurtier, M.A.;
Copley, R.R.; Edelmann, A.; Querfurth, E.; Rybin, V.; Drewes, G.; Raida, M.;
Bouwmeester, T.; Bork, P.; Seraphin, B.; Kuster, B.; Neubauer, G.; Superti-Furga,
G. (2002). Functional organization of the yeast proteome by systematic analysis of
protein complexes. Nature, 415(6868):141-147.
• Gerstein, M.; Lan, N.; Jansen, R. (2002). Integrating interactomes. Science,
295:284-7.
• Goffard, N.; Garcia, V.; Iragne, F.; Groppi, A.; de Daruvar, A. (2003). IPPRED:
server for proteins interactions inference. Bioinformatics, 19:903-4.
• Goodwin, C.S.; Armstrong, J.A.; Chilvers, T.; Peters, M.; Collins, M.D.; Sly, L.;
McConnell, W.; Harper, W.E.S. (1989). Transfer of Campylobacter pyloridis and
Campylobacter mustelae to Helicobacter gen. nov. as Helicobacter pylori comb.
nov. and Helicobacter mustelae comb. nov., respectively. Int J Syst Bacteriol. 39:
397-405.

82
• Goswami M, Mangoli SH, Jawali N. (2006). Involvement of reactive oxygen
species in the action of ciprofloxacin against Escherichia coli. Antimicrob Agents
Chemother. 50(3):949-54.
• Guerry, P.; Ewing, C.P.; Schirm, M.; Lorenzo, M.; Kelly, J.; Pattarini, D.; Majam,
G.; Thibault, P.; Logan S. (2006). Changes in flagellin glycosylation affect
Campylobacter autoagglutination and virulence. Mol Microbiol. 60(2):299-311.
• Guerry, P. (2007). Campylobacter flagella: not just for motility. Trends Microbiol.
15(10):456-61.
• Graham, D.Y.; Opekun, A.R.; Yamaoka, Y.; Osato, M.S.; el-Zimaity, H.M. (2003).
Early events in protom pump inhibitor associated exacerbation of corpus gastritis.
Aliment Phamacol Ther, 17: 193-200.
• Grigoriev, A. (2001) A relationship between gene expression and protein interaction
on the proteome scale: analysis of the bacteriophage t7 and the yeast
Saccharomyces cerevisiae. Nucleic Acids Res., 29,3513-3519.
• Guarner, J.; Mohar, A.; Parsonnet, J.; Halperin, D. (1993). The association of
Helicobacter pylori with gastric cancer and preneoplastic gastric lesions in Chiapas
Mexico. Cancer, 71: 297-301.
• Harvill, E.T.; Preston, A.; Cotter, P.A.; Allen, A.G.; Maskell, D.J.; Miller, J.F.
(2000). Multiple roles for Bordetella lipopolysaccharide molecules during
respiratory tract infection. Infect Immun. 68(12):6720-8.
• Hazell, S.L.; Graham, D.Y. (1990). Unsaturated fatty acids and viability of
Helicobacter (Campylobacter) pylori. J Clin Microbiol. 28(5):1060-1.

83
• Headd, J.J.; Ban, Y.E.; Brown, P.; Edelsbrunner, H.; Vaidya, M.; Rudolph, J.
(2007). Protein-protein interfaces: properties, preferences, and projections. J
Proteome Res., 6(7):2576-86.
• Hinchey, J.; Lee, S.; Jeon, B.Y.; Basaraba, R.J.; Venkataswamy, M.M.; Chen, B.;
Chan, J.; Braunstein, M.; Orme, I.M.; Derrick, S.C.; Morris, S.L.; Jacobs, W.R. Jr.;
Porcelli, S.A. (2007). Enhanced priming of adaptive immunity by a proapoptotic
mutant of Mycobacterium tuberculosis. J Clin Invest. 117(8):2279-88.
• Hirata, Y.; Maeda, S.; Mitsuno, Y.; Akanuma, M.; Yamaji, Y.; Ogura, K.; Yoshida,
H.; Shiratori, Y.; Omata, M. (2001). Helicobacter pylori activates the cyclin D1
gene through mitogen-activated protein kinase pathway in gastric cancer cells.
Infect. Immun, 69:3965-3971.
• Heinrichs, D.E.; Yethon, J.A.; Amor, P.A.; Whitfield, C. (1998). The assembly
system for the outer core portion of R1- and R4-type lipopolysaccharides of
Escherichia coli. The R1 core-specific beta-glucosyltransferase provides a novel
attachment site for O-polysaccharides. J Biol Chem. 273(45):29497-505.
• Heroven, A.K.; Böhme, K.; Rohde, M.; Dersch, P. (2008). A Csr-type regulatory
system, including small non-coding RNAs, regulates the global virulence regulator
RovA of Yersinia pseudotuberculosis through RovM. Mol Microbiol. 68(5):1179-
95.
• Ho, Y.; Gruhler, A.; Heilbut, A.; Bader, G.D.; Moore, L.; Adams, S.L.; Millar, A.;
Taylor, P.; Bennett, K.; Boutilier, K. (2002). Systematic identification of protein
complexes in Saccharomyces cerevisiae by mass spectrometry. Nature,
415(6868):180-183.
• Hoffmann, R.; Valencia, A. (2003). Life cycles of successful genes. Trends Genet.
19(2):79-81.

84
• Huynen, M.; Snel, B.; Lathe, W.; Bork, P. (2000). Predicting protein function by
genomic context: quantitative evaluation and qualitative. Genome Res ,10:1204-10.
• Imlay, J.A.; Linn, S. (1988). DNA damage and oxygen radical toxicity. Science.
240(4857):1302-9.
• Imlay, J.A. (2003). Pathways of oxidative damage. Annu Rev Microbiol. 57:395-
418.
• Ito, T.; Tashiro, K.; Muta, S.; Ozawa, R.; Chiba, T.; Nishizawa, M.; Yamamoto, K.;
Kuhara, S.; Sakaki, Y. (2000). Toward a protein-protein interaction map of the
budding yeast: A comprehensive system to examine two-hybrid interactions in all
possible combinations between the yeast proteins. Proc Natl Acad Sci USA,
97(3):1143-1147.
• Izquierdo, L.; Merino, S.; Regué, M.; Rodriguez, F.; Tomás, J.M. (2002). Synthesis
of a Klebsiella pneumoniae O-antigen heteropolysaccharide (O12) requires an ABC
2 transporter. J Bacteriol. 185(5):1634-41.
• Janin, J.; Chothia, C. The structure of protein-protein recognition sites. (1990). J
Biol Chem., 265:16027-30.
• Jansen, R.; Greenbaum, D.; Gerstein, M. (2002). Relating whole-genome
expression data with protein-protein interactions. Genome Res., 12:37-46.
• Jensen, L.J.; Kuhn, M.; Stark, M.; Chaffron, S.; Creevey, C.; Muller, J.; Doerks, T.;
Julien, P.; Roth, A.; Simonovic, M.; Bork, P. von Mering, C. (2009). Nucleic Acids
Res. 37(Database issue):D412-6.

85
• Jones, S.; Thornton, J.M. (1997). Prediction of protein-protein interaction sites
using surface patches. J Mol Biol., 272:133-43.
• Jungblut, P.R.; Bumann, D.; Haas, G.; Zimny-Arndt, U.; Holland, P.; Lamer, S.;
Siejak, F.; Aebischer, A.; Meyer, T.F. (2000). Comparative proteome analysis of
Helicobacter pylori. Mol. Microbiol., 36:710-725.
• Jürgen B, Hanschke R, Sarvas M, Hecker M, Schweder T. (2001). Proteome and
transcriptome based analysis of Bacillus subtilis cells overproducing an insoluble
heterologous protein. Appl Microbiol Biotechnol. 55(3):326-32.
• Kalmokoff, M.; Lanthier, P.; Tremblay, T.L.; Foss, M.; Lau, P.C.; Sanders, G.;
Austin, J.; Kelly, J.; Szymanski, C.M. (2006). Proteomic analysis of Campylobacter
jejuni 11168 biofilms reveals a role for the motility complex in biofilm formation. J.
Bacteriol. 188: 4312-4320.
• Karlsson, R. (2004). SPR for molecular interaction analysis: A review of emerging
application areas. J Mol Recognit., 17: 151-161.
• Keiler, K.C.; Waller, P.R.; Sauer, R.T. (1996). Role of a peptide tagging system in
degradation of proteins synthesized from damaged messenger RNA. Science 271:
990–993.
• Keiler, K.C. (2008). Biology of trans-Translation. Ann Rev Microbiol. 62:133–151.
• Kerrien, S.; Alam-Faruque, Y.; Aranda, B.; Bancarz, I.; Bridge, A.; Derow, C.;
Dimmer, E.; Feuermann, M.; Friedrichsen, A.; Huntley, R.; Kohler, C.; Khadake, J.;
Leroy, C.; Liban, A.; Lieftink, C.; Montecchi-Palazzi, L.; Orchard, S.; Risse, J.;
Robbe, K.; Roechert, B.; Thorneycroft, D.; Zhang, Y.; Apweiler, R.; Hermjakob, H.
(2007). IntAct-open source resource for molecular interaction data. Nucleic Acids
Res., 35, 561-5.

86
• Kerrinnes, T.; Zelas, Z.B.; Streckel, W.; Faber, F.; Tietze, E.; Tschäpe, H.; Yaron,
S. (2008). CsrA and CsrB are required for the post-transcriptional control of the
virulence-associated effector protein AvrA of Salmonella enterica. Int J Med
Microbiol.
• Khulusi, S.; Ahmed, H.A.; Patel, P.; Mendall, M.A.; Northfield, T.C. (1995). The
effects of unsaturated fatty acids on Helicobacter pylori in vitro. J Med Microbiol.
42(4):276-82.
• Kirov, S.M.; Castrisios, M.; Shaw, J.G. (2004). Aeromonas flagella (polar and
lateral) are enterocyte adhesins that contribute to biofilm formation on surfaces.
Infect Immun. 72(4):1939-45.
• Klancnik, A.; Botteldoorn, N.; Herman, L.; Mozina, S.S. (2006). Survival and stress
induced expression of groEL and rpoD of Campylobacter jejuni from different
growth phases. Int J Food Microbiol. 112(3):200-7.
• Kloosterman, T.G.; Hendriksen, W.T.; Bijlsma, J.J.; Bootsma, H.J.; van Hijum,
S.A.; Kok, J.; Hermans, P.W.; Kuipers, O.P. (2006). Regulation of glutamine and
glutamate metabolism by GlnR and GlnA in Streptococcus pneumoniae. J Biol
Chem. 281(35):25097-109.
• Kneidinger, B.; Larocque, S.; Brisson, J.R.; Cadotte, N.; Lam, J.S. (2003).
Biosynthesis of 2-acetamido-2,6-dideoxy-L-hexoses in bacteria follows a pattern
distinct from those of the pathways of 6-deoxy-L-hexoses. Biochem J. 371(Pt
3):989-95.
• Kobayashi, K.; Sakamoto, J.; Kito, T.; Yamamura, Y.; Koshikawa, T.; Fujita, M.;
Watanabe, T.; Nakazato, H. (1993). Lewis blood group-related antigen expression
in normal gastric epithelium, intestinal metaplasia, gastric adenoma, and gastric
carcinoma. Am J Gastroenterol., 88: 919-924.

87
• Kooistra, O.; Bedoux, G.; Brecker, L.; Lindner, B.; Sánchez-Carballo, P.; Haras, D.;
Zähringer, U. (2003). Structure of a highly phosphorylated lipopolysaccharide core
in the Delta algC mutants derived from Pseudomonas aeruginosa wild-type strains
PAO1 (serogroup O5) and PAC1R (serogroup O3). Carbohydr Res. 338(23):2667-
77.
• Kumar, R.B.; Das, A. (2001). Functional analysis of the Agrobacterium tumefaciens
T-DNA transport pore protein VirB8. J Bacteriol 183:3636-3641.
• Lawrence, M.C.; Colman, P.M. (1993). Shape complementarity at protein/protein
interfaces. J Mol Biol., 234:946-50.
• Li, S.; Armstrong, C.M.; Bertin, N.; Ge, H.; Milstein, S.; Boxem, M.; Vidalain,
P.O.; Han, J.D.; Chesneau, A.; Hao, T.; Goldberg, D.S.; Li, N.; Martinez, M.; Rual,
J.F.; Lamesch, P.; Xu, L.; Tewari, M.; Wong, S.L.; Zhang, L.V.; Berriz, G.F.;
Jacotot, L.; Vaglio, P.; Reboul, J.; Hirozane-Kishikawa, T.; Li, Q.; Gabel, H.W.;
Elewa, A.; Baumgartner, B.; Rose, D.J.; Yu, H.; Bosak, S.; Sequerra, R.; Fraser, A.;
Mango, S.E.; Saxton, W.M.; Strome, S.; Van Den Heuvel, S.; Piano, F.;
Vandenhaute, J.; Sardet, C.; Gerstein, M.; Doucette-Stamm, L.; Gunsalus, K.C.;
Harper, J.W.; Cusick, M.E.; Roth, F.P.; Hill, D.E.; Vidal, M. (2004). A map of the
interactome network of the metazoan C. elegans. Science. 303(5657):540-3.
• Li, Y.; Zamble, D. (2009). The pH-responsive DNA-binding activity of
Helicobacter pylori NikR. Biochemistry. 48 (11), 2486–2496.
• Lin, S.N.; Ayada, K.; Zhao, Y.; Yokota, K.; Takenaka, R.; Okada, H.; Kan, R.;
Hayashi, S.; Mizuno, M.; Hirai, Y.; Fujinami, Y.; Oguma, K. (2005). Helicobacter
pylori heat-shock protein 60 induces production of the pro-inflammatory cytokine
IL8 in monocytic cells. J Med Microbiol. 54(Pt 3):225-33.

88
• Lippincott-Schwartz, J.; Patterson, G.H. (2003). Development and use of
fluorescent protein markers in living cells. Science, 300: 87-91.
• Liu, X.; Matsumura, P. (1995). An alternative sigma factor controls transcription of
flagellar class-III operons in Escherichia coli: gene sequence, overproduction,
purification and characterization. Gene, 164, 81-84.
• Lu, L.; Lu, H.; Skolnick, J. (2002). MULTIPROSPECTOR: an algorithm for the
prediction of proteinprotein interaction by multimeric threading. Proteins, 49:350-
64.
• Lucchetti-Miganeh, C.; Burrowes, E.; Baysse, C.; Ermel, G. (2008). The post-
transcriptional regulator CsrA plays a central role in the adaptation of bacterial
pathogens to different stages of infection in animal hosts. Microbiology 154: 16-29.
• Luidalepp, H.; Hallier, M.; Felden, B.; Tenson, T. (2005). tmRNA decreases the
bactericidal activity of aminoglycosides and the susceptibility to inhibitors of cell
wall synthesis. RNA Biol.2:70–74.
• Maere, S.; Heymans, K.; Kuiper, M. (2005). BiNGO: a Cytoscape plugin to assess
overrepresentation of gene ontology categories in biological networks.
Bioinformatics. 21(16):3448-9.
• Marcus, E.A.; Moshfegh, A.P.; Sachs, G.; Scott, D.R. (2005). The periplasmic
alpha-carbonic anhydrase activity of Helicobacter pylori is essential for acid
acclimation. J Bacteriol. 187(2):729-38.
• Manetti, R.; Massari, P.; Burroni, D.; De Bernard, M.; Marchini, A.; Olivieri, R.;
Papani, E.; Montecucco, A.; Rappuoli, R.; Telford, J. (1995). Helicobacter pylori
Cytotoxin: Importance of Native Conformation for Induction of Neutralizing
Antibodies. Infect Imm., 63: 4476-4480.

89
• Marcotte, E.M.; Pellegrini, M.; Thompson, M.J.; Yeates, T.O.; Eisenberg, D.
(1999). A combined algorithm for genome-wide prediction of protein function.
Nature, 402:83-6.
• Margittai, M.; Widengren, J.; Schweinberger, E.; Schröder, G.F.; Felekyan, S.;
Haustein, E.; König, M.; Fasshauer, D.; Grubmüller, H.; Jahn, R.; Seidel, C.A.
(2003). Single-molecule fluorescence resonance energy transfer reveals a dynamic
equilibrium between closed and open conformations of syntaxin 1. Proc Natl Acad
Sci USA, 100: 15516-15521.
• McEvoy, M.M.; Hausrath, A.C.; Randolph, G.B.; Remington, S.J.; Dahlquist,
F.W. (1998). Two binding modes reveal flexibility in kinase/response regulator
interactions in the bacterial chemotaxis pathway. Proc.Natl.Acad.Sci.USA 95:
7333-7338.
• McNally, D.J.; Hui, J.P.; Aubry, A.J.; Mui, K.K.; Guerry, P.; Brisson, J.R.; Logan,
S.M.; Soo, E.C. (2006). Functional characterization of the flagellar glycosylation
locus in Campylobacter jejuni 81-176 using a focused metabolomics approach. J
Biol Chem. 281(27):18489-98.
• Mobley, H.L.; Island, M.D.; Hausinger, R. P. (1995). Molecular biology of
microbial ureases. Microbiol. Rev., 59, 451-480.
• Mollenhauer-Rektorschek, M.; Hanauer, G.; Sachs, G.; Melchers, K. (2002).
Expression of UreI is required for intragastric transit and colonization of gerbil
gastric mucosa by Helicobacter pylori. Res Microbiol. 153(10):659-66.
• Morera, S.; Lebras, G.; Lascu, I.; Lacombe, M.L.; Veron, M.; Janin, J. (1994).
Refined X-ray structure of Dictyostelium discoideum nucleoside diphosphate kinase
at 1.8Å resolution. J Mol Biol., 243:873-90.

90
• Moore, S.D.; Sauer, R.T. (2007). The tmRNA system for translational surveillance
and ribosome rescue. Annu Rev Biochem. 76:101–124.
• Murzin, A.G.; Brenner, S.E.; Hubbard, T.; Chothia, C. (1995). SCOP: a structural
classification of proteins database for the investigation of sequences and structures.
J. Mol. Biol,. 247, 536–540.
• Miyamoto, Y.; Mukai, T.; Maeda, Y.; Nakata, N.; Kai, M.; Naka, T.; Yano, I.;
Makino, M. (2007). Characterization of the fucosylation pathway in the
biosynthesis of glycopeptidolipids from Mycobacterium avium complex. J
Bacteriol. 189(15):5515-22.
• Nardone, G.; Rocco, A.; Malfertheiner, P. (2004). Helicobacter pylori and
molecular events in precancerous gastric lesions. Aliment Pharmacol Ther.
20(3):261-70.
• Nomura, A.; Stemmarmann, G.; Chyan, P.; Perez-Perez, G.; Blaser, M. (1994).
Helicobacter pylori infection and risk for duodenal and gastric ulceration. Ann Inter
Med., 120: 977-981.
• Odenbreit, S.; Püls, J.; Sedlmaier, B.; Gerland, E.; Fischer, W.; Haas, R. (2000).
Translocation of Helicobacter pylori CagA into gastric epithelial cells by type IV
secretion. Science, 25;287(5457):1497-500.
• Odenbreit, S. (2005). Adherence properties of Helicobacter pylori: Impact on
pathogenesis and adaptation to the host. Int J Med Microbiol. 295:317–324.
• Oh, J.D.; Kling-Bäckhed, H.; Giannakis, M.; Xu, J.; Fulton, R.S.; Fulton, L.A.;
Cordum, H.S.; Wang, C.; Elliott, G.; Edwards, J.; Mardis, E.R.; Engstrand, L.G.;
Gordon, J.I. (2006). The complete genome sequence of a chronic atrophic gastritis

91
Helicobacter pylori strain: evolution during disease progression. Proc Natl Acad Sci
U S A., 27;103(26):9999-10004.
• Okan, N.A.; Bliska, J.B.; Karzai, A.W. (2006). A Role for the SmpB-SsrA system
in Yersinia pseudotuberculosis pathogenesis. PLoS Pathogens.2:e6.
• Pan, Z.; van der Hulst, R.; Su, W.M.; Feller, M.; Xiao, S.; Tytgat, G.N.; Dankert, J.;
van der Ende, A. (1997). Equally high prevalence of infection with cagA-positive
Helicobacter pylori in chinise patients with pepic ulcer diseases and those with
chronic gastritis-associated dyspepsia. J Clin Microbiol., 35: 1344-1347.
• Parkin, D.; Laara, E.; Muir, C. (1988). Estimates of the world wide frequency of
sixteen major cancer in 1980. Int J Cancer, 41: 184-197.
• Parkin, D.M. (2004). International variation. Oncogene, 23: 6329–40
• Parrish, J.R.; Yu, J.; Liu, G.; Hines, J.A.; Chan, J.E.; Mangiola, B.A.; Zhang, H.;
Pacifico, S.; Fotouhi, F.; DiRita, V.J.; Ideker, T.; Andrews, P.; Finley, R.L. Jr.
(2007). A proteome-wide protein interaction map for Campylobacter jejuni.
Genome Biol. 8(7):R130.
• Pazos, F.; Valencia, A. (2001). Similarity of phylogenetic trees as indicator of
protein-protein interaction. Protein Eng., 14:609-14.
• Pazos, F.; Valencia, A. (2002). In silico two-hybrid system for the selection of
physically interacting proteins pairs. Proteins, 47:219-27.
• Pawson, T.; Nash, P. (2000). Protein-protein interactions define specificity in signal
transduction. Genes Dev., 14:1027-47.

92
• Pawson, T.; Raina, M.; Nash, P. (2002). Interaction domains: from simple binding
events to complex cellular behavior. FEBS Lett., 513:2-10.
• Pellegrini, M.; Marcotte, E.M.; Thompson, M.J.; Eisenberg, D.; Yeates, T.O.
(1999). Assigning protein functions by comparative genome analysis: protein
phylogenetic profiles. Proc Natl Acad Sci USA., 96:4285-8.
• Peri, S.; Navarro, J.D.; Amanchy, R.; Kristiansen, T.Z.; Jonnalagadda, C.K.;
Surendranath, V.; Niranjan, V.; Muthusamy, B.; Gandhi, T.K.; Gronborg, M.;
Ibarrola, N.; Deshpande, N.; Shanker, K.; Shivashankar, H.N.; Rashmi, B.P.;
Ramya, M.A.; Zhao, Z.; Chandrika, K.N.; Padma, N.; Harsha, H.C.; Yatish, A.J.;
Kavitha, M.P.; Menezes, M.; Choudhury, D.R.; Suresh, S.; Ghosh, N.; Saravana, R.;
Chandran, S.; Krishna, S.; Joy, M.; Anand, S.K.; Madavan, V.; Joseph, A.; Wong,
G.W.; Schiemann, W.P.; Constantinescu, S.N.; Huang, L.; Khosravi-Far, R.; Steen,
H.; Tewari, M.; Ghaffari, S.; Blobe, G.C.; Dang, C.V.; Garcia, J.G.; Pevsner, J.;
Jensen, O.N.; Roepstorff, P.; Deshpande, K.S.; Chinnaiyan, A.M.; Hamosh, A.;
Chakravarti, A.; Pandey, A. (2003). Development of human protein reference
database as an initial platform for approaching systems biology in humans. Genome
Res., 13(10):2363-71.
• Piehler, J. (2005). New methodologies for measuring protein interactions in vivo
and in vitro. Curr Opin Struct Biol., 15: 4-14.
• Puig, O.; Caspary, F.; Rigaut, G.; Rutz, B.; Bouveret, E.; Bragado-Nilsson, E.;
Wilm, M.; Séraphin, B. (2001). The tandem affinity purification (TAP) method: A
general procedure of protein complex purification. Methods, 24: 218-229.
• Qin, Y.; Polacek, N.; Vesper, O.; Staub, E.; Einfeldt, E.; Wilson, D.N.; Nierhaus,
K.H. (2006). The highly conserved LepA is a ribosomal elongation factor that back-
translocates the ribosome. Cell 127, 721–733.

93
• Rain, J.C.; Selig, L.; De Reuse, H.; Battaglia, V.; Reverdy, C.; Simon, S.; Lenzen,
G.; Petel, F.; Wojcik, J.; Schächter, V.; Chemama, Y.; Labigne, A.; Legrain, P.
(2001). The protein-protein interaction map of Helicobacter pylori. Nature.
409(6817):211-5.
• Ramani, A.K.; Marcotte, E.M. (2003). Exploiting the co-evolution of interacting
proteins to discover interaction specificity. J Mol Biol ., 327: 273-84.
• Rashid, M.H.; Rajanna, C.; Ali, A.; Karaolis, D.K. (2003). Identification of genes
involved in the switch between the smooth and rugose phenotypes of Vibrio
cholerae. FEMS Microbiol Lett. 227(1):113-9.
• Rigaut, G.; Shevchenko, A.; Rutz, B.; Wilm, M.; Mann, M.; Seraphin, B. (1999). A
generic protein purification method for protein complex characterization and
proteome exploration. Nature, 17:1030-2.
• Rudi, J.; Kolb, C.; Maiwald, M.; Kuck, D.; Sieg, A.; Galle, P.; Stremmel, W.
(1998). Diversity of Helicobacter pylori vacA and cagA Genes and Relationship to
VacA and Protein Expression, Cytotoxin Production, and Associated Diseases. J
Clin Microbiol., 36: 944-948.
• Sánchez, B.; Champomier-Vergès, M.C.; Anglade, P.; Baraige, F.; de Los Reyes-
Gavilán, C.G.; Margolles, A.; Zagorec, M. (2005). Proteomic analysis of global
changes in protein expression during bile salt exposure of Bifidobacterium longum
NCIMB 8809. J Bacteriol. 187(16):5799-808.
• Serrano, L. (2007). Synthetic biology: promises and challenges. Mol Syst Biol.
3:158.
• Schauer, K.; Rodionov, D.A.; de Reuse, H. (2008). New substrates for TonB-
dependent transport: do we only see the 'tip of the iceberg'?. Trends Biochem Sci.
33(7):330-8.

94
• Schwikowski, B.; Uetz, P.; Fields, S. (2000). A network of protein-protein
interactions in yeast. Nature Biotechnol., 18:1257-61.
• Scott, D.R.; Marcus, E.A.; Wen, Y.; Oh, J.; Sachs, G. (2007). Gene expression in
vivo shows that Helicobacter pylori colonizes an acidic niche on the gastric surface.
Proc Natl Acad Sci USA., 104(17):7235-40.
• Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin,
N.; Schwikowski, B.; Ideker, T. (2006). Cytoscape: a software environment for
integrated models of biomolecular interaction networks. Genome Res. 13(11):2498-
504.
• Shih, G.C.; Kahler, C.M.; Carlson, R.W.; Rahman, M.M.; Stephens, D.S. (2001).
gmhX, a novel gene required for the incorporation of L-glycero-D-manno-heptose
into lipooligosaccharide in Neisseria meningitidis. Microbiology. 147(Pt 8):2367-
77.
• Shimazu, K.; Takahashi, Y.; Uchikawa, Y.; Shimazu, Y.; Yajima, A.; Takashima,
E.; Aoba, T.; Konishi, K. (2008). Identification of the Streptococcus gordonii glmM
gene encoding phosphoglucosamine mutase and its role in bacterial cell
morphology, biofilm formation, and sensitivity to antibiotics. FEMS Immunol Med
Microbiol. 53(2):166-77.
• Shimiyama, T.; Fukuda, S.; Tanaka, M.; Mikami, T.; Saito, Y.; Munakata, A.
(1997). High Prevalence of CagA-positive Helicobacter pylori strains in Japanese
asymptomatic patients and gastric cancer patients. Scand J Gastroenterol., 32: 465-
468.

95
• Shoemaker, B.A.; Panchenko, A.R. (2007). Deciphering protein-protein
interactions. Part I. Experimental techniques and databases. PLoS Comput Biol.,
3(3): e42.
• Sirisena, D.M.; Brozek, K.A.; MacLachlan, P.R.; Sanderson, K.E.; Raetz, C.R.
(1992). The rfaC gene of Salmonella typhimurium. Cloning, sequencing, and
enzymatic function in heptose transfer to lipopolysaccharide. J Biol Chem.
267(26):18874-84.
• Skouloubris, S.; Thiberge, J.M.; Labigne, A.; De Reuse, H. (1998). The
Helicobacter pylori UreI protein is not involved in urease activity but is essential for
bacterial survival in vivo. Infect. Immun., 66, 4517-4521.
• Sprinzak, E.; Margalit, H. (2001). Correlated sequencesignatures as markers of
protein-protein interaction. J Mol Biol., 311:681-92.
• Sprinzak, E.; Sattath, S.; Margalit, H. (2003). How reliable are experimental
protein-protein interaction data? J Mol Biol ., 327:919-23.
• Stebbins, C.E. (2005). Structural microbiology at the pathogen-host interface. Cell.
Microbiol., 7: 1227-1236.
• Stein, M.; Rappuoli, R.; Covacci, A. (2000). Tyrosine phosphorylation of the
Helicobacter pylori CagA antigen after cag-driven host cell translocation. Proc Natl
Acad Sci USA., 97(3):1263-8.
• Stein, A.; Russell, R.B.; Aloy, P. (2005). 3did: interacting protein domains of
known three-dimensional structure. Nucleic Acids Res., 33:D413-7.

96
• Stingl, K.; Schauer, K.; Ecobichon, C.; Labigne, A.; Lenormand, P.; Rousselle, J.C.;
Namane, A.; de Reuse, H. (2008). In vivo interactome of Helicobacter pylori urease
revealed by tandem affinity purification. Mol Cell Proteomics. 7(12):2429-41.
• Strobel, S.; Bereswill, S.; Balig, P.; Allgaier, P.; Sonntag, H.G.; Kist, M. (1998).
Identification and analysis of a new vacA genotype variant of Helicobacter pylori in
different patient groups in Germany. J Clin Microbiol., 36: 1285-1289.
• Stroeher, U.H.; Karageorgos, L.E.; Brown, M.H.; Morona, R.; Manning, P.A.
(1995). A putative pathway for perosamine biosynthesis is the first function
encoded within the rfb region of Vibrio cholerae O1. Gene. 166(1):33-42.
• Suganuma, M.; Kurusu, M.; Okabe, S.; Sueoka, N.; Yoshida, M.; Wakatsuki, Y.;
Fujiki, H. (2001). Helicobacter pylori membrane protein 1: a new carcinogenetic
factor of Helicobacter pylori. Cancer Res., 61:6356-6359.
• Suzuki, M.; Suzuki, H.; Kitahora, T.; Miyazawa, M.; Nagahashi, S.; Suzuki, K.;
Ishii, H. (2002). Treatment with a proton pump inhibitor promotes corpus gastritis
in patients with Helicobacter pylori-infected antrum-predominant gastritis. Aliment
Pharmacol Ther., 16: 159-165.
• Tatusov, R.L.; Fedorova, N.D.; Jackson, J.D.; Jacobs, A.R.; Kiryutin, B.; Koonin,
E.V.; Krylov, D.M.; Mazumder, R.; Mekhedov, S.L.; Nikolskaya, A.N.; Rao, B.S.;
Smirnov, S.; Sverdlov, A.V.; Vasudevan, S.; Wolf, Y.I.; Yin, J.J.; Natale, D.A.
(2003). The COG database: an updated version includes eukaryotes. BMC
Bioinformatics, 4, 41.
• Teichmann, S.A.; Murzin, A.G.; Chothia, C. (2001). Determination of protein
function, evolution and interactions by structural genomics. Curr Opin Struct Biol.,
11:354-63.

97
• Tomb, J.F.; White, O.; Kerlavage, A.R.; Clayton, R.A.; Sutton, G.G.; Fleischmann,
R.D.; Ketchum, K.A.; Klenk, H.P.; Gill, S.; Dougherty, B.A.; Nelson, K.;
Quackenbush, J.; Zhou, L.; Kirkness, E.F.; Peterson, S.; Loftus, B.; Richardson, D.;
Dodson, R.; Khalak, H.G.; Glodek, A.; McKenney, K.; Fitzegerald, L.M.; Lee, N.;
Adams, M.D.; Hickey, E.K.; Berg, D.E.; Gocayne, J.D.; Utterback, T.R.; Peterson,
J.D.; Kelley, J.M.; Cotton, M.D.; Weidman, J.M.; Fujii, C.; Bowman, C.; Watthey,
L.; Wallin, E.; Hayes, W.S.; Borodovsky, M.; Karp, P.D.; Smith, H.O.; Fraser,
C.M.; Venter, J.C. (1997). The complete genome sequence of the gastric pathogen
Helicobacter pylori. Nature, 388(6642):539-47.
• Tong AH, Drees B, Nardelli G, Bader GD, Brannetti B, Castagnoli L, Evangelista
M, Ferracuti S, Nelson B, Paoluzi S, Quondam M, Zucconi A, Hogue CW, Fields S,
Boone C, Cesareni G. (2002). A combined experimental and computational strategy
to define protein interaction networks for peptide recognition modules. Science,
295.
• Tullius, M.V.; Harth, G.; Horwitz, M.A. (2003). Glutamine synthetase GlnA1 is
essential for growth of Mycobacterium tuberculosis in human THP-1 macrophages
and guinea pigs. Infect Immun. 71(7):3927-36.
• Umit, H.; Tezel, A.; Bukavaz, S.; Unsal, G.; Otkun, M.; Soylu, A.R.; Tucer, D.;
Otkun, M.; Bilgi, S. (2009). The relationship between virulence factors of
Helicobacter pylori and severity of gastritis in infected patients. Dig Dis Sci.
54(1):103-10.
• Uetz, P.; Giot, L.; Cagney, G.; Mansfield, T.A.; Judson, R.S.; Knight, J.R.;
Lockshon, D.; Narayan, V.; Srinivasan, M.; Pochart, P.; Qureshi-Emili, A.; Li, Y.;
Godwin, B.; Conover, D.; Kalbfleisch, T.; Vijayadamodar, G.; Yang, M.; Johnston,
M.; Fields, S.; Rothberg, J.M. (2000). A comprehensive analysis of protein-protein
interactions in Saccharomyces cerevisiae. Nature, 403(6770):623-627.

98
• Valencia, A.; Pazos, F. (2002). Computational methods for the prediction of protein
interactions. Curr Opin Struct Biol. 12(3):368-73.
• Vioque, A.; de la Cruz, J. (2003). Trans-translation and protein synthesis inhibitors.
FEMS Microbiol Lett.218:9–14.
• von Mering, C.; Krause, R., Snel, B.; Cornell, M.; Oliver, S.G.; Fields, S.; Bork, P.
(2002). Comparative assessment of large-scale data sets of protein-protein
interactions. Nature, 417:399-403.
• Walhout, A.J.; Boulton, S.J.; Vidal, M. (2000). Yeast two-hybrid systems and
protein interaction mapping projects for yeast and worm. Yeast, 17(2):88-94.
• Wanbura, C.; Aoyama, N.; Shirasaka, D.; Sakai, T.; Ikemura, T.; Sakashita, M.;
Maekawa, S.; Kuroda, K.; Inoue, T.; Ebara, S.; Miyamoto, M.; Kasuga, M. (2002).
Effect of Helicobacter pylori-induced cyclooxygenase-2 on gastric epithelial cell
kinetics: implication for gastric carcinogenesis. Helicobacter, 7:129-138.
• Wang, W.; Peng, X.; Wang, Q.; Cheng, J.S.; Wang, L. (2006). Sequence of
Escherichia coli O11 O-antigen gene cluster and identification of molecular markers
specific to O11. Wei Sheng Wu Xue Bao. 46(3):341-6.
• Warburton, V.J.; Everett, S.; Mapstone, N.P.; Axon, A.T.; Hawkey, P.; Dixon, M.F.
(1998). Clinical and histological associations of cagA and vacA genotypes in
Helicobacter pylori gastritis. J Clin Pathol. 51(1):55-61.
• Weeks, D.L.; Eskandari, S.; Scott, D.R.; Sachs, G. (2000). A H+-gated urea
channel: the link between Helicobacter pylori urease and gastric colonization.
Science. 287(5452):482-5.

99
• Weel, J.F.; van der Hulst, R.W.M.; Gerrits, Y.; Roorda, P.; Feller, M.; Dankert, J.;
Tytgat, G.N.; van der Ende, A. (1998). The interrelationship between cytotoxin-
associated gene A, vacuolating cytotoxin and Helicobacter pylori-related diseases. J
Infect Dis., 173: 1171-1175.
• Welch, M.; Chinardet, N.; Mourey, L.; Birck, C.; Samama, J.P. (1998). Structure of
the CheY-binding domain of histidine kinase CheA in complex with CheY. Nature
Struct. Biol., 5, 25-29.
• Westman, E.L.; Preston, A.; Field, R.A.; Lam, J.S. (2008). Biosynthesis of a rare di-
N-acetylated sugar in the lipopolysaccharides of both Pseudomonas aeruginosa and
Bordetella pertussis occurs via an identical scheme despite different gene clusters. J
Bacteriol. 190(18):6060-9.
• Whitfield, C.; Amor, P.A.; Köplin, R. (1997). Modulation of the surface
architecture of gram-negative bacteria by the action of surface polymer:lipid A-core
ligase and by determinants of polymer chain length. Mol Microbiol. 23(4):629-38.
• Wojcik, J.; Schächter, V. (2001). Protein-protein interaction map inference using
interacting domain profile pairs. Bioinformatics, 17 Suppl 1:296-305.
• Wu, L.J.; Errington, J. (1994). Bacillus subtilis spoIIIE protein required for DNA
segregation during asymmetric cell division. Science 264:572-575.
• Yan, C.; Wu, F.; Jernigan, R.L.; Dobbs, D.; Honavar, V. (2008). Characterization of
protein-protein interfaces. Protein J., 27(1):59-70.
• Yang, Y.; Wang, H.; Erie, D.A. (2003). Quantitative characterization of
biomolecular assemblies and interactions using atomic force microscopy. Methods,
29: 175-187.

100
• Yamazaki, S.; Yamakawa, A.; Ito, Y.; Ohtani, M.; Higashi, H.; Hatakeyama, M.;
Azuma, T. (2003). The CagA protein of Helicobacter pylori is translocated into
epithelial cells and binds to SHP-2 in human gastric mucosa. J Infect Dis.,
87(2):334-7. Epub 2002 Dec 19.
• Ye, D.; Willhite, C.; Blanke, S.R. (1999). Identification of the Minimal Intracellular
Vacuolating Domain of the Helicobacter pylori Vacuolating Toxin. J Biol Chemis.,
274: 9277-9282.
• Yook, S.H.; Oltvai, Z.N.; Barabási, A.L. (2004). Functional and topological
characterization of protein interaction networks. Proteomics. 4(4):928-42.
• Youngman, E.M.; Green, R. (2007). Ribosomal translocation: LepA does it
backwards. Curr Biol. 17(4):R136-9.
• Yu, H.; Luscombe, N.M.; Lu, H.X.; Zhu, X.; Xia, Y.; Han, J.D.; Bertin, N.; Chung,
S.; Vidal, M.; Gerstein, M. (2004). Annotation transfer between genomes: protein–
protein interologs and protein–ADN regulogs. Genome Res., 14, 1107–1118.
• Xenarios, I.; Fernández, E.; Salwinski, L.; Joyce-Duan, X.; Thompson, M.J.;
Marcotte, E.M.; Eisenberg, D. (2001). DIP: The database of interacting proteins:
2001 update. Nucleic Acids Res., 29:239-41.
• Xenarios, I.; Salwinski, L.; Duan, X.J.; Higney, P.; Kim, S.M.; Eisenberg, D.
(2002). DIP, the database of interacting proteins: a research tool for studying
cellular networks of protein interactions. Nucleic Acids Res., 30:303-5.
• Zambelli, B.; Danielli, A.; Romagnoli, S.; Neyroz, P.; Ciurli, S.; Scarlato, V.
(2008). High-affinity Ni2+ binding selectively promotes binding of Helicobacter
pylori NikR to its target urease promoter. J Mol Biol. 383(5):1129-43.

101
• Zanzoni, A.; Montecchi-Palazzi, L.; Quondam, M.; Ausiello, G.; Helmer-Citterich,
M.; Cesareni, G. (2002). MINT: a molecular interaction database. FEBS Lett,
513:135-40.

102
13. Glosario
Ácido desoxirribonucleico (ADN): molécula que almacena la información genética.
Amino ácido: unidad monomérica fundamental de las proteínas.
Ácido ribonucleico (ARN): molécula que transmite información genética. Además cumple
con funciones estructurales y de acoplamiento en la maquinaria de traducción de la
informacion.
ARN mensajero: molécula de ARN, copia de un gene, que lleva la información desde el
genoma hasta donde se realiza la traducción.
Bioinformática: disciplina científica emergente que utiliza tecnología de la información
para organizar, analizar y distribuir información biológica con la finalidad de responder
preguntas complejas en biología.
Conformación: arreglo espacial que adopta una molécula, en virtud de los diferentes
ángulos de rotación que pueden adquirir sus enlaces químicos.
Dominio de una proteína: se refiere a una unidad estructural independiente de otras
cadenas en la misma proteína, generalmente es el responsable de las características
funcionales, evolutivas y de plegamiento de las proteinas.
Enzima: proteína con actividad catalítica, es decir; que acelera una reacción química
específica sin destruirse en el proceso. Las moléculas blanco de una enzima se denominan
sustratos.
Eucarionte: organismo con núcleo organizado y cromosomas. Forma parte de un gran
grupo que difiere de los procariontes (las bacterias y organismos similares) que carecen de
núcleo. Existen diferencias fundamentales entre la organización genética de uno y otro

103
grupo.
Genoma: término que denota a todo el material genético de un organismo vivo. En un ser
humano, por ejemplo, se refiere a todas las secuencias de todos los cromosomas de una
célula.
In vitro : se refiere a condiciones experimentales en las que no existen células u organismos
vivos. Condiciones dadas en portaobjetos microscópicos, tubos de ensayo, etcétera
In vivo : se refiere a condiciones experimentales que incorporan células u organismos vivos.
In silico: se referiere a experimentos hechos con el uso de computadoras, por ejemplo
simulaciones o predicciones.
Interactoma: se refiere al conjunto total de interacciones entre las proteínas dentro de un
organismo.
Molécula: conjunto de átomos unidos unos con otros por enlaces fuertes. Es la expresión
mínima de un compuesto o sustancia química. Trabajar con estas entidades primordiales
justifica el uso del término “molecular” para denominar diversas áreas de la investigación
en biología. (Entidad química constituida por la unión de varios átomos. Es la expresión
mínima de un concepto químico. Una macromolécula puede estar constituida.por miles o
hasta millones de átomos, típicamente enlazados en largas cadenas.)
Mutagénesis: proceso por el cual se inducen cambios en el material genético de un
organismo. El proceso puede ser espontáneo o inducido.
Nucleótido: unidad fundamental de los ácidos nucleicos. Constituida por una base
nitrogenada, un azúcar y un fosfato.
Organelo: estructura intracelular especializada en una función determinada, como el

104
núcleo, las mitocondrias, los cloroplastos, los ribosomas, etcétera.
Proteína: polímero constituido por una hebra o cadena lineal de unidades llamadas
aminoácidos. La secuencia de aminoácidos de una proteína determina su estructura y su
función. Las proteínas son los productos primarios codificados por los genes, encargadas de
organizar la actividad bioquímica celular.
Proteoma: el término describe el conjunto total de proteínas expresadas por un genoma
completo, incluyendo las modificadas después de la traducción.
Replicación: proceso por el cual las moléculas de ADN se duplican, generando dos copias
iguales a partir de una sola. El proceso requiere la separación de las dos hebras de la
molécula original.
Ribosoma: organelo encargado de manufacturar proteínas, de acuerdo con las instrucciones
del ADN (presentes en el ARN mensajero). Es un agregado de proteínas y ARN, llamado
ribosomal.
Sintenia: es la localización conservada de genes en posiciones equivalentes en especies
relacionadas.
Sitio activo: zona de una enzima con la que se asocia el sustrato y donde se induce su
transformación química para dar un producto. Los sitios activos frecuentemente son
hendiduras en la superficie de las enzimas.
Sustrato: sustancia o molécula con la que interacciona una enzima, transformándose en
productos.
Traducción: proceso por medio del cual se lee la secuencia de codones del ARN y se
elabora una cadena de proteína, con la secuencia correspondiente, de acuerdo con el código
genético.

105
Transcripción: proceso por el que un gene se expresa, mediante la síntesis de un ARN que
contiene la misma secuencia del gene.
Transcriptoma: el término se refiere al estudio de los perfiles de expressión de todos los
genes presentes en el genoma. Los estudios de expresión sugieren pistas sobre la función de
los genes. Aunque son muchos los métodos para estudiar el transcriptoma el más utilizado
es el microarray de ADN, que permite el análisis simultáneo de la expresión de miles de
genes.
Transducción de señal: frase que se aplica al proceso por el cual una señal molecular (por
ejemplo, la presencia de una hormona en el medio extracelular) es convertida en una
respuesta (por ejemplo, generación de un compuesto en el medio intracelular). Esta
respuesta puede ser después amplificada o atenuada y, a su vez, dar origen a otro evento de
transducción de señal.
Transformación: procedimiento que permite introducir, directamente a células vivas,
moléculas de ADN. Existen varios procedimientos que pueden lograr este objetivo:
tratamiento con calcio, descarga eléctrica, pistolas génicas, etcétera.
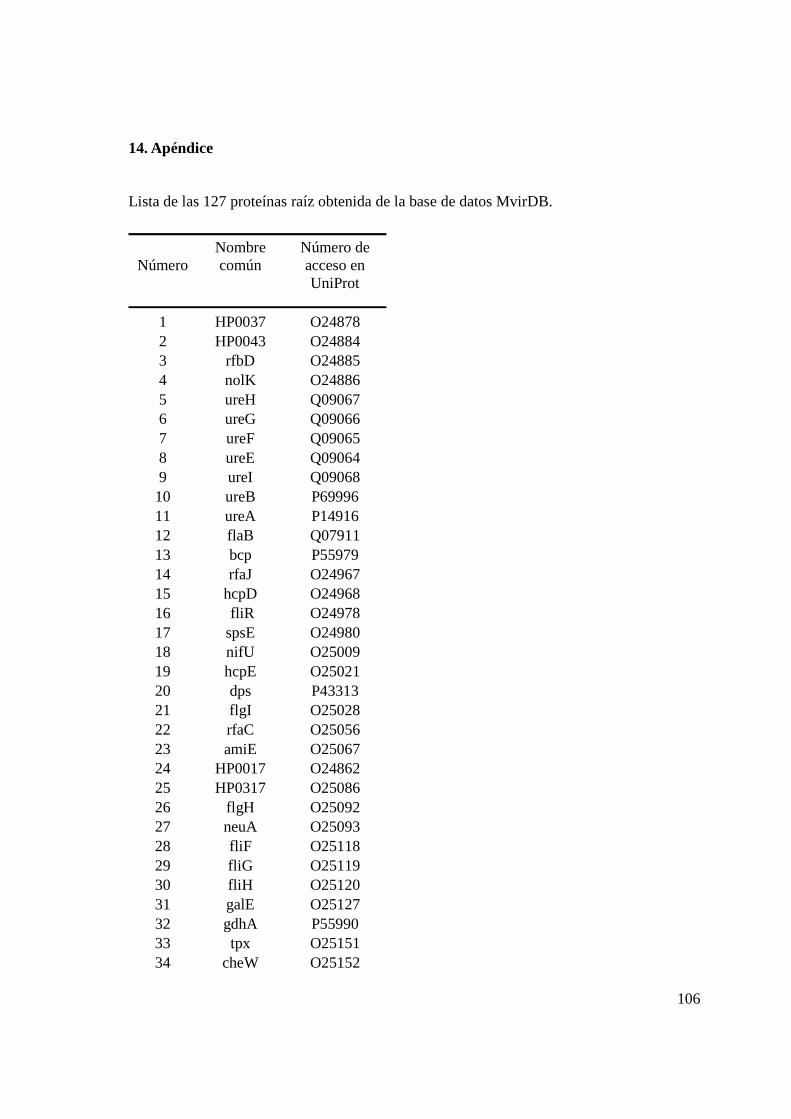
106
14. Apéndice
Lista de las 127 proteínas raíz obtenida de la base de datos MvirDB.
Número
Nombre común
Número de acceso en UniProt
1 HP0037 O24878 2 HP0043 O24884 3 rfbD O24885 4 nolK O24886 5 ureH Q09067 6 ureG Q09066 7 ureF Q09065 8 ureE Q09064 9 ureI Q09068 10 ureB P69996 11 ureA P14916 12 flaB Q07911 13 bcp P55979 14 rfaJ O24967 15 hcpD O24968 16 fliR O24978 17 spsE O24980 18 nifU O25009 19 hcpE O25021 20 dps P43313 21 flgI O25028 22 rfaC O25056 23 amiE O25067 24 HP0017 O24862 25 HP0317 O25086 26 flgH O25092 27 neuA O25093 28 fliF O25118 29 fliG O25119 30 fliH O25120 31 galE O25127 32 gdhA P55990 33 tpx O25151 34 cheW O25152

107
35 HP0019 O24864 36 HP0616 O25337 37 HP0393 O25154 38 nifS O25161 39 HP0441 O25189 40 HP0459 O25206 41 glnA P94845 42 cag1 O25257 43 cag5 O25260 44 virB11 Q7BK04 45 cag-Z O25261 46 cag8 O25263 47 cagT P97245 48 cagS P97227 49 cag16 O25270 50 cag17 O25271 51 cag20 O25274 52 cag22 O25276 53 cagE Q48252 54 cagA P55980 55 fliN O25306 56 flaA P0A0S1 57 pfr P52093 58 wbpB O25390 59 HP0686 O25395 60 feoB O25396 61 fliD P96786 62 fliS O25448 63 flhB P56416 64 secA O25475 65 hpaA P55969 66 HP0807 O25487 67 motA P65410 68 motB P56427 69 trxA P66928 70 trxB P56431 71 lpxB O25537 72 flgE P50610 73 HP0876 O25543 74 vacA P55981 75 flgD O25565 76 fur O25671 77 fliY O25674

108
78 fliM O25675 79 flhF O25679 80 flhA O06758 81 cheY P71403 82 nixA Q48262 83 tly O25718 84 flgK O25744 85 gluP O25788 86 amiF O25836 87 exbB O25897 88 exbD O25898 89 tonB O25899 90 rocF O25949 91 HP1400 O25950 92 fliQ P0A0S3 93 fliI O07025 94 trbB O25965 95 exbB2 O25986 96 exbD2 P64101 97 coaD O26010 98 fliE P67708 99 flgC O26080 100 ceuE O26082 101 tsaA P21762 102 iscS O25008 103 cag7 O25262 104 omp13 O25355 105 alpA O25570 106 HP0913 O25571 107 cag10 O25265 108 cag11 O25266 109 cag14 O25268 110 cag18 O25272 111 cag21 O25275 112 cag24 O25277 113 cag3 O25258 114 cag4 O25259 115 cag9 O25264 116 cagC O25278 117 cagI O25273 118 cagP O25269 119 babA O25840 120 fliP O25394

109
121 fliA O25676 122 flaG O25094 123 flgG O26104 124 omp1 Q1CVE6 125 HP0651 O25366 126 HP0725 O25218 127 traG O25652

















![La cromatografía de gases acoplada a espectrometría de ...nico de los hidrocarburos hallados. La utilidad de estos cocientes se aprecia en el trabajo de Fernández y colaboradores[11],](https://static.fdocuments.co/doc/165x107/5e5fa80159d59e484516464c/la-cromatografa-de-gases-acoplada-a-espectrometra-de-nico-de-los-hidrocarburos.jpg)
