
Maestría en Ciencias en Nanociencias · ii Resumen de la tesis que presenta José Luis Zamora Cruz...
88
Centro de Investigación Científica y de Educación Superior de Ensenada, Baja California Maestría en Ciencias en Nanociencias Evaluación de nanocompuestos para la conversión de glucosa mediante fotocatálisis Tesis para cubrir parcialmente los requisitos necesarios para obtener el grado de Maestro en Ciencias Presenta: José Luis Zamora Cruz Ensenada, Baja California, México 2017
Transcript of Maestría en Ciencias en Nanociencias · ii Resumen de la tesis que presenta José Luis Zamora Cruz...

Centro de Investigación Científica y de Educación Superior de Ensenada, Baja California
Maestría en Ciencias
en Nanociencias
Evaluación de nanocompuestos para la conversión de glucosa mediante fotocatálisis
Tesis para cubrir parcialmente los requisitos necesarios para obtener el grado de
Maestro en Ciencias
Presenta:
José Luis Zamora Cruz
Ensenada, Baja California, México 2017

Tesis defendida por
José Luis Zamora Cruz
y aprobada por el siguiente Comité
José Luis Zamora Cruz © 2017 Queda prohibida la reproducción parcial o total de esta obra sin el permiso formal y explícito del autor y director de la tesis.
Dr. Oscar Raymond Herrera Director de tesis
Miembros del comité Dra. Alma Georgina Navarrete Alcalá
Dr. Manuel Herrera Zaldívar
Dr. Oscar Eugenio Jaime Acuña
Dr. Sergio Fuentes Moyado Coordinador del Posgrado en nanociencias
Dra. Rufina Hernández Martínez Directora de Estudios de Posgrado

ii
Resumen de la tesis que presenta José Luis Zamora Cruz como requisito parcial para la obtención del grado de Maestro en Ciencias en Nanociencias.
Evaluación de nanocompuestos para la conversión de glucosa mediante fotocatálisis
Resumen aprobado por:
_______________________ Dr. Oscar Raymond Herrera
Director de tesis La fotocatálisis plasmónica ha recibido atención recientemente como una tecnología de alto rendimiento en la producción de compuestos de alto valor agregado como sorbitol y acido glucurónico, al usar metales nobles como oro, plata y cobre soportados en nanocompuestos. Estos compuestos juegan un papel importante en la industria farmacéutica y la industria de los hidrocarburos. El presente estudio reporta los resultados obtenidos en la conversión de la glucosa a productos de valor agregado, empleando nanocompuestos basados en nanopartículas de metales y/o semiconductoras contenidas en el interior y en la superficie de matrices zeolíticas tipo mordenita. Se desarrollaron nanocompuestos metal-mordenita como 30Ag-MOR, 30Au-MOR, semiconductor-mordenita como 20CdS-MOR y metal/semiconductor-mordenita como 30Au/20CdS-MOR y 10Ag/20CdS-MOR. Los nanocompuestos fueron caracterizados detalladamente en su morfologia, estructura cristalina, composición superficial, área superficial, porosidad y composición química. La difracción de rayos X mostró el crecimiento de la zeolita tipo mordenita de alta cristalinidad. Las microscopias electrónicas de transmisión y de barrido presentaron los aspectos morfológicos de los polvos caracterizados por granos de empaquetamientos de cristalitos en forma de agujas. Las nanopartículas metálicas y/o semiconductoras se observan distribuidas homogéneamente dispersas en la superficie de los cristalitos. La espectroscopia fotoelectrónica de rayos X confirmó el contenido de los valores predichos para los nanocompuestos. El análisis de adsorción de N2 junto con la técnica BET y las curvas-t proporcionaron los valores del área superficial y el volumen de los mesoporos de cada uno de los nanocompuestos. Por otra parte, las propiedades ópticas se caracterizaron por espectroscopia de absorción UV-Visible, mostrando las propiedades de absorción (fotoactivas) de cada uno de los nanocompuestos empleados. Las propiedades fotocatalíticas plasmónicas de los nanocompuestos fueron evaluadas al aplicar radiación ultravioleta en un fotorreactor con una longitud de onda de 254 nm y con agitación de 200 rpm. Los nanocompuestos se sometieron a prueba en diferentes tiempos de radiación de 15 minutos, 2 horas y 4 horas en soluciones de glucosa en agua y acetonitrilo mostrando su alta capacidad fotocatalítica en la conversión de la glucosa. La determinación de los productos de conversión de glucosa al usar los nanocompuestos y radiación UV, se efectuó por cromatografía liquida para determinar la concentración de sorbitol y de ácido glucurónico. Los resultados muestran la conversión de glucosa en ácido glucurónico. Esto demuestra la posibilidad de usar estos nanocompuestos en la conversión de glucosa para obtener productos de alto valor agregado para usos industriales y farmacéuticos. Palabras clave: nanocompuestos, fotocatálisis plasmónica, fotoreactor, cromatografía liquida.

iii
Abstract of the thesis presented by José Luis Zamora Cruz as a partial requirement to obtain the Master of Science degree in Nanoscience.
Evaluation of nanocomposites for the conversion of glucose by photocatalysis
Abstract approved by: ____________________________________
Ph. D. Oscar Raymond Herrera Thesis Director
The plasmonic photocatalysis has recently received attention as a high performance technology in the production of high added value compounds such as sorbitol and glucuronic acid, using noble metals such as gold, silver and copper supported in nanocomposites. These compounds play an important role in the pharmaceutical and hydrocarbons industry. The present study reports the results obtained in the conversion of glucose to value-added products, using nanocomposites based in nanoparticles of metal and /or semiconductor supported on and in the structure of zeolites type mordenite. Metal-mordenite nanocomposites were developed as 30Ag-MOR, 30Au-MOR, semiconductor-mordenite as 20CdS-MOR and metal/semiconductor-mordenite as 30Au/20CdS-MOR, 10Ag /20CdS-MOR. All nanocomposites were characterized in detail in their morphology, crystalline structure, surface composition, surface area and mesoporous volume and chemical composition. X-ray diffraction showed the growth of the high crystallinity mordenite zeolite. The transmission and scanning electron microscopes presented the morphological aspects of the powders characterized by grains of packing of crystallites in the form of needles. The metallic and / or semiconducting nanoparticles are observed distributed homogeneously dispersed on the surface of the crystallites. The X-ray photoelectron spectroscopy confirmed the content of nanoparticles embedded in the surface of nanocomposites. The analysis of N2 adsorption and the BET technique together with the t-plot report showed that the nanocomposites have a surface area and mesoporous volume. On the other hand, the optical properties were characterized by UV-Visible absorption spectroscopy showing the absorption (photoactive) properties of each of the nanocomposites. The plasmonic photocatalytic properties of all nanocomposites were evaluated, using ultraviolet radiation in a photoreactor with a wavelength of 254 nm and with agitation of 200 rpm. Nanocomposites were tested for different time of radiation of 15 minutes, 2 hours and 4 hours in glucose solutions of water and acetonitrile, showing its high photocatalytic ability in the conversion of glucose. The determination of glucose conversion products using nanocomposites and UV radiation was carried out by liquid chromatography to determine the concentration of sorbitol and glucuronic acid. The results show the conversion of glucose into glucuronic acid. This demonstrates the possibility to use these nanocomposites in the conversion of glucose to obtain products of high added value for industrial and pharmaceutical uses. Keywords: nanocomposites, plasmonic photocatalysis, photoreactor, liquid chromatography.

iv
Dedicatoria
Donde se encuentren dedico la presente a
todos los estudiantes mexicanos que hacen
estudios de posgrado y que quieren el bienestar
para México.

v
Agradecimientos Un profundo agradecimiento a mi director de tesis Dr. Oscar Raymond Herrera por su apoyo y consejos en este proyecto de investigación. Especial dedicación a todos los integrantes del comité de tesis:
Dra. Alma Georgina Navarrete Alcalá
Dr. Manuel Herrera Zaldívar
Dr. Oscar Eugenio Jaime Acuña
y al siguiente personal del CNYN y del CICESE:
Eduardo Morales por su apoyo en la operación del HPLC
David Domínguez Vargas por su apoyo en el XPS
Francisco Ruiz Medina por su apoyo en el TEM
Israel Gradilla Martínez por su apoyo en el SEM
Eloísa Aparicio Ceja por su apoyo en los rayos X
Miguel Estrada por su apoyo con el análisis BET
Elizabeth Avilés Becerril por su apoyo en Biblioteca
Juan Antonio Peralta Centro del Computo en el CNYN.
Ramon Espinosa Bastida por su apoyo incondicional
Gracias a
CONACYT por la beca durante los dos años de estudio.
DGAPA-DAPIIT, IN110316 “Heteroestructuras Magnetoeléctricas de Materiales Multiferroicos”.

vi
Tabla de contenido
Página Resumen en español……………………………………………………………..……………...……...……………………………
ii Resumen en inglés…………………………………………………………….………………………….…………………….……..
iii Dedicatorias…………………………………………………………………….……………………………….…………………………
iv Agradecimientos……………………………………………………….……………………………………..……………….….......
v Lista de figuras………………………………………………………….………………………………….…..……………....…......
viii Lista de tablas…………………………………………………………….……………………………………….………………………
ix Capítulo 1. Introducción………………………………………...…..………………………………………………………….. 1
Capítulo 2. Hipótesis y objetivos………………………...….……………………………………………………………… 6
2.1 Hipótesis…………….........................................................................…...…............................ 6
2.2 Objetivos generales…………………..…………….………….……..……………………………..………………… 6
2.3 Objetivos específicos……………………………….…………………………………..………………………………. 6
Capítulo 3. Métodos y técnicas experimentales………………..…..………………….………………………….… 7
3.1 Materiales empleados……………………………………………………………………….…………………………. 7
3.1.1 Nanocompuestos fotoactivos……………………………………….…….……….………………………. 7
3.1.2. La glucosa……………………………………………………………………………………………….…………… 10
3.1.2.1 Activación de la glucosa…………………..………………………………………………………. 10
3.1.3 Posibles derivados de la glucosa………………………….……………………………………………….. 12
3.1.3.1 Sorbitol………………………..………………………………………………………………………….. 12
3.1.3.2 Acido glucurónico…………………………………………………………………………………….. 13
3.2 Caracterización estructural, morfológica y de composición química……..……….………...…... 13
3.2.1 Difracción de rayos X..……………………………………………………………….…….………….……….. 14
3.2.2 Microscopia electrónica de transmisión………………………..……………….….……….………… 15
3.2.3 Microscopia electrónica de barrido…………………………………………….….…….………………. 17
3.2.4 Espectroscopia de dispersión de energía………………………………………….….…….………… 18
3.2.5 Espectroscopia fotoelectrónica de rayos X……………………………………….….…….………… 20
3.2.6 Análisis de adsorción física…………………….……………………………….…………..……….………. 24
3.3 Características ópticas…….….………………………………………………………….….……..……….…….…… 28
3.3.1 Espectroscopia de absorción UV-Visible…………………………………….……………….……….. 28

vii
3.4 Evaluación fotocatalítica.……………………………………………………….……………..……..…….…………. 29
3.4.1 Fotoreactor………………………………………………………………………………….………………….….. 29
3.4.2 Cromatografía líquida……………………………………….………………………….……….……….…….. 30
Capítulo 4. Resultados y discusiones…………………………………………….………………………………………… 35
4.1 Características estructurales y morfológicas................................…...…............................. 35
4.1.1 Difracción de rayos X..………………………………………………………………..……….………………. 35
4.1.2 Microscopia electrónica de transmisión y de barrido……………………..…….……………… 36
4.2 Características fisicoquímicas y absorción óptica…………………….……………..……….……………. 37
4.2.1 Análisis químico…………………………………………………………………………………….…………….. 37
4.2.2 Análisis químico de superficie…………………………………………………..………...….…………… 38
4.2.3 Análisis de adsorción física…………………………………………………………………..………………. 39
4.2.4 Espectroscopia de absorción UV-Visible……………………..……………………………………….. 42
4.3 Evaluación fotocatalítica para la degradación de glucosa……………………..………………………. 43
4.3.1 Calibración de las soluciones estándares …….…….……………..…………….………………….. 44
4.3.1.1 Resultados para la glucosa………..….………………………..…..………….……………….. 44
4.3.1.2 Resultados para el sorbitol ………………………………….…………………..……………… 46
4.3.1.3 Resultados para el ácido glucurónico………………………..……………….……..…….. 47
4.3.2 Resultados preliminares de la fotoreacción durante 4 horas……………..………………… 50
4.3.3 Evaluación de los sensibilizadores de la fotoreacción durante 15 minutos…………… 56
4.3.4 Evaluación de la fotodegradación en procesos de 2 horas de radiación…………..…… 64
Capítulo 5. Conclusiones y recomendaciones………………………………………………………………………... 70
Literatura citada………………………………………………………………………..……………………………………………... 72

viii
Lista de figuras
Figura
Página
1 a) Respuesta del plasmón de superficie bajo un campo eléctrico oscilante y b) Sinergia entre un metal y un semiconductor en el proceso de fotoreducción y fotoxidación………………………….…………………………………………………………………………………
2
2 Características de la fotocatálisis plasmónica………………..………………………..….…….……. 2
3 Publicaciones de la fotocatálisis plasmónica en los últimos 10 años……………………….. 3
4 Volumen del mercado de productos fotocatalíticos….………………..……..……..……………. 3
5 Sustancias con alto valor agregado obtenidos a partir de la foto-oxidación selectiva de glucosa con TiO2 …………………………………………………………………………………………………
4
6 Simulación del crecimiento de nanopartículas de metal y/o semiconductor soportados en la matriz de la mordenita………………………………………………………………….
8
7 Estructura de la mordenita. Sistema de coordenadas y sus dimensiones………………… 9
8 Formación de glucosa por fotosíntesis……………………………………………………………………. 10
9 Proyección de la estructura química de la glucosa: a) Fischer y b) Harworth………….. 11
10 Mutarrotación en la estructura de la glucosa…………………………………………………………. 11
11 Proyección de Fischer para el sorbitol…………………………………………………………………….. 12
12 Proyección de Fischer para el ácido glucurónico…………………………………………………….. 13
13 Ley de Bragg. Relación entre la dispersión interplanar y el ángulo de incidencia……. 14
14 Esquema de un difractómetro de rayos X tipo Bragg-Bretano…………………………………. 15
15 Difractograma de rayos X de la mordenita natural. Se ilustran algunos índices de Miller………………………………….………………………………..……………………..…………………….……
15
16 a) Esquema de la óptica y b) de la sección transversal de un MET …………….…………… 16
17 Micrografía de MET de polvos del nanocompuesto 20CdS-MOR……………………………. 17
18 Esquema de la óptica de un MEB y una imagen representativa de la muestra 20CdS-MOR ……………………………………………………………………………………..………………………………..
18
19 Interacción de un haz de electrones con la materia …………………..……………………………. 19
20 a) Esquema de la emisión de fotoelectrones bajo la radiación de rayos X; b) comparación del espesor de superficie con el tamaño de material…………………………..
21
21 Proceso de foto-ionización y energías del fotoelectrón expulsado..………….……………… 22
22 Espectro general de EFX del óxido de bario……………………………………………………………… 23

ix
23 Esquema general de un aparato para el análisis de adsorción …………………………..…... 24
24 Isotermas de adsorción y desorción a) tipo I y b) tipo IV …………………………………….…….. 25
25 Curvas-t para la adsorción de N2 a 76K en zeolitas naturales………………………………….. 27
26 Esquema de la óptica de un espectrómetro UV-Visible…………………………………………… 29
27 Fotoreactor de 254 nm de radiación UV…………………………………………………………………. 30
28 Esquema de un HPLC………………………………………………………………………………………………. 32
29 Cromatogramas con detectores: a) DIR y b) DLM……………………………………………………. 33
30 Comparativo de los patrones de difracción de los nanocompuestos con el patrón teórico de la mordenita……………………………………………………………………………………………
35
31 Imágenes representativas de MET de las muestras a) 30Au/20CdS-MOR, b) 10Ag/20CdS-MOR y c) 30Au-MOR….………………………………………………………………………..
36
32 Imágenes representativas de MEB de polvos de los nanocompuestos…..................... 37
33 Espectro general de XPS del nanocompuesto 30Ag-MOR…………………..……………………. 39
34 Isoterma de adsorción y desorción del nanocompuesto 10Ag/20CdS-MOR……..……… 40
35 Curva de calibración para el nanocompuesto 10Ag/20CdS-MOR…………..………………… 41
36 Zonas de la curva-t para el nanocompuesto 10Ag/20CdS-MOR …….…..…..………………. 42
37 Espectros de absorción óptica de los nanocompuestos: a) 30Ag-MOR y 30Au-MOR y b) 20CdS-MOR, 30Au/20CdS-MOR y 10Ag/20CdS-MOR…………………………………………..
43
38 Cromatogramas de los detectores: a) DIR y b) DLM para las soluciones estándares de glucosa sin fotocatalizador………………………………………………………………………………….
45
39 Curva de calibración obtenida con el detector DIR para la solución estándar de glucosa……………………………………………………………………………………………………………………
45
40 Cromatogramas de los detectores: a) DIR y b) DLM para las soluciones estándares de sorbitol sin fotocatalizador………………………………………………………………………………….
46
41
Curva de calibración obtenida con el detector DIR para la solución estándar de sorbitol…………………………………………………………………………………………………………………….
47
42 Cromatogramas de los detectores: a) DIR y b) DLM para las soluciones estándares de ácido glucurónico sin fotocatalizador…………………………………………………………………
48
43 Curva de calibración obtenida con el detector DLM para la solución estándar de ácido glucurónico…………………………………………………………………………………………………….
49

x
44
Cromatogramas obtenidos con los detectores: a) DIR y b) DLM para la solución de glucosa, fotocatalizador 30Ag-MOR y 4 horas de radiación………………………………………
51
45 Cromatogramas obtenidos con los detectores: a) DIR y b) DLM para la solución de glucosa, fotocatalizador 30Au-MOR y 4 horas de radiación……………………………………..
52
46 Cromatogramas obtenidos con los detectores: a) DIR y b) DLM para la solución de glucosa, fotocatalizador 20CdS-MOR y 4 horas de radiación……………………………………
53
47 Cromatogramas obtenidos con los detectores: a) DIR y b) DLM para la solución de glucosa, fotocatalizador 30Au/20CdS-MOR y 4 horas de radiación………………………….
54
48 Cromatogramas obtenidos con los detectores: a) DIR y b) DLM para la solución de glucosa, fotocatalizador 10Ag/20CdS-MOR y 4 horas de radiación…………………………..
55
49 Cromatogramas de los detectores: a) DIR y b) DLM para la solución con acetonitrilo sin fotocatalizador y 15 minutos de radiación UV…………………………………………………….
57
50 Cromatogramas de los detectores: a) DIR y b) DLM para la solución de NaOH sin fotocatalizador y 15 minutos de radiación UV………………………………………………………….
58
51 Cromatogramas de los detectores: a) DIR y b) DLM para las soluciones de ACN, NaOH, glucosa, fotocatalizador 10Ag/20CdS-MOR y 15 minutos de radiación UV…..
59
52 Cromatogramas de los detectores: a) DIR y b) DLM para las soluciones con ACN, o NaOH, glucosa, fotocatalizador 30Au-MOR y 15 minutos de radiación UV……………….
60
53 Cromatogramas de los detectores: a) DIR y b) DLM para las soluciones de ACN, NaOH, glucosa, fotocatalizador 30Ag-MOR y 15 minutos de radiación UV……………….
61
54 Cromatogramas de los detectores: a) DIR y b) DLM para las soluciones de ACN, NaOH, glucosa, fotocatalizador 20CdS-MOR y 15 minutos de radiación UV……………..
62
55 Cromatogramas de los detectores: a) DIR y b) DLM para las soluciones de ACN, NaOH, glucosa, fotocatalizador 30Au/20CdS-MOR y 15 minutos de radiación UV……
63
56 Cromatogramas de los detectores: a) DIR y b) DLM para la solución de acetonitrilo sin fotocatalizador y bajo 2 horas de radiación UV…………………………………………………..
65
57 Cromatogramas de los detectores: a) DIR y b) DLM para la solución de acetonitrilo, glucosa, fotocatalizador 10Ag/20CdS-MOR y 2 horas de radiación UV…………………….
66
58 Cromatogramas de los detectores: a) DIR y b) DLM para la solución de acetonitrilo, glucosa, fotocatalizador 30Ag-MOR y 2 horas de radiación UV………………………………..
67
59 Cromatogramas de los detectores: a) DIR y b) DLM para la solución de acetonitrilo, glucosa, fotocatalizador 30Au-MOR y 2 horas de radiación UV…………………………………
68

xi
60 Cromatogramas de los detectores: a) DIR y b) DLM para la solución de acetonitrilo, glucosa, fotocatalizador 20CdS-MOR y 2 horas de radiación UV……………….……………..
69

xii
Lista de tablas
Tabla Página
1 Aplicaciones de la fotocatálisis…….……….….…………….….……….….…………….……….……….. 4
2 Análisis químico obtenido por EDS de algunas zeolitas sintéticas tipo mordenita………………………..……………………………………………………………………………………… 20
3 Tiempos de retención de algunos carbohidratos……………………………………………………… 34
4 Análisis químico de los nanocompuestos tipo mordenita………………………..………………. 38
5 Datos obtenidos del analisis BET para el nanocompuesto 10Ag/20CdS-MOR………….. 40
6 Parámetros texturales para el nanocompuesto 10Ag/20CdS-MOR………………………….. 42
7 Comparativo entre las concentraciones estándares y estimadas para la glucosa…….. 46
8 Comparativo entre las concentraciones estándares y estimadas del sorbitol……………... 47
9 Comparativo entre las concentraciones estándares y estimadas del ácido glucurónico………………………………………………………………………………………………………………. 49
10 Tiempos de retención promedio por detector y por reactivo ……………………….…………. 49
11 Porcentaje de conversión de glucosa con el fotocatalizador 30Ag-MOR y 4 horas de radiación………………………………………………………………………………………………………………….. 51
12 Porcentaje de conversión de glucosa con el fotocatalizador 30Au-MOR y 4 horas de radiación………………………………………………………………………………………………………………….. 52
13 Porcentaje de conversión de glucosa con el fotocatalizador 20CdS-MOR y 4 horas de radiación……………………………………………………………………………………………………………..…… 53
14 Porcentaje de conversión de glucosa con el fotocatalizador 30Au/20CdS-MOR y 4 horas de radiación………………………………………………………………………………………………….… 54
15 Porcentaje de conversión de glucosa con el fotocatalizador 10Ag/20CdS-MOR y 4 horas de radiación………………………………………………………………………………………………….… 55
16 Conversión de glucosa por fotocatalizador y por hora de radiación…………………….. 56
17 Conversión de glucosa con solución de acetonitrilo y 15 minutos de radiación…….….. 57
18 Conversión de glucosa con solución de NaOH y 15 minutos de radiación……..………….. 58
19 Conversión de glucosa en solución con acetonitrilo o NaOH, el fotocatalizador 10Ag/20CdS-MOR y 15 minutos de radiación…………………………………………………………… 59
20 Conversión de glucosa en solución con acetonitrilo o NaOH usando el fotocatalizador 30Au-MOR bajo 15 minutos de radiación………………………………………………………………….. 60

xiii
21 Conversión de glucosa en solución con acetonitrilo o NaOH y el fotocatalizador 30Ag-MOR bajo 15 minutos de radiación………………………………………………………………………….. 61
22 Conversión de glucosa en solución con acetonitrilo o NaOH y el fotocatalizador 20CdS-MOR bajo 15 minutos de radiación……………………………………………………………….. 62
23 Conversión de la glucosa en solución con acetonitrilo o NaOH y el fotocatalizador 30Au/20CdS-MOR bajo 15 minutos de radiación……………………………………………………… 63
24 Conversión de la glucosa en solución con acetonitrilo sin fotocatalizador y 2 horas de radiación……………………………………………………………………………………………………………… 65
25 Conversión de glucosa con solución de acetonitrilo, el fotocatalizador 10Ag/20CdS-MOR y 2 horas de radiación……………………………………………………………………………………… 66
26 Conversión de glucosa con solución de acetonitrilo, el fotocatalizador 30Ag-MOR y 2 horas de radiación……………………………………………………………………………………………………. 67
27 Conversión de glucosa con solución de acetonitrilo, el fotocatalizador 30Au-MOR y 2 horas de radiación……………………………………………………………………………………………………. 68
28 Conversión de glucosa con solución de acetonitrilo, el fotocatalizador 20CdS-MOR y 2 horas de radiación……………………..………………………………………………………………………….. 69

1
Capítulo 1. Introducción
El desarrollo de la catálisis se ha efectuado en diferentes periodos, iniciando con Jöns Jacob Berzelius quien
sistemáticamente investigó la fermentación del alcohol en 1835 (Lindström et al., 2017). A continuación,
se definen algunos conceptos importantes con el propósito de introducir el presente proyecto de
investigación.
La catálisis contempla el uso de catalizadores, homogéneos y/o heterogéneos, con lo cual se disminuye la
energía de activación y la reacción sea más rápida y con menos consumo de energía. El catalizador ayuda
a la reacción, no sufre cambio químico y no se destruye en el proceso. La catálisis homogénea es cuando
el catalizador se encuentra en la misma fase que las sustancias reaccionantes. La catálisis heterogénea es
cuando un catalizador se encuentra en fase diferente a las sustancias reaccionantes. La energía de
activación es la energía necesaria para que los reactivos reaccionen entre sí y produzcan nuevos productos
(Atkins et al., 2010). Después del desarrollo de la catálisis y con la explicación del efecto fotoeléctrico por
Albert Einstein, se llevaron a cabo un gran número de aplicaciones, entre otros, la fotocatálisis.
La fotocatálisis en química es la aceleración de una reacción mediante la luz (fotoreacción) en presencia
de un catalizador. La fotocatálisis, en principio, usa semiconductores para absorber fotones para crear
electrones y huecos activos, los cuales pueden iniciar la reducción u oxidación de sustancias químicas
(James et al., 2009).
En particular, la fotocatálisis plasmónica se refiere a la fotocatálisis donde el fotocatalizador posee en su
conformación elementos metálicos nanoestructurados. Asi, la fotocatálisis plasmónica tiene como
característica más sobresaliente, el plasmón de resonancia de superficie (SPR: Surface Plasmon
Resonance), que se genera en la superficie del metal, cuando se hace incidir un haz de luz de longitud de
onda característica y el campo electromagnético de la luz provoca que los electrones libres en las
nanopartículas de metal oscilen provocando que los electrones se polaricen a un lado u otro de la
superficie. De este modo, los electrones libres en la banda de conducción oscilan colectiva y
coherentemente en resonancia con la frecuencia del campo electromagnético de la luz incidente, lo cual
causa un oscilador dipolar como se ilustra en la Figura 1 a). La Figura 1 b) (Zhang, 2014) muestra el
esquema de bandas de un semiconductor acoplado a un plasmón de superficie. De modo que, al hacer
incidir luz se generan electrones en la banda de conducción del semiconductor, los cuales se sincronizan

2
con los electrones del plasmón incrementando la actividad de los procesos de oxidación y reducción,
reforzada por la generación de huecos en la banda de valencia.
Figura 1. a) Respuesta del plasmón de superficie bajo un campo eléctrico oscilante y b) Sinergia entre un metal y un semiconductor en el proceso de fotoreducción y fotoxidación. Tomado de Zhang, 2014.
La fotocatálisis plasmónica (Figura 2) (Zhang et al., 2013) presenta las características con respecto a un
fotocatalizador tipo metal/semiconductor con nanopartículas de metales nobles y las propiedades del
plasmón de resonancia de superficie localizada. La Figura 3 presenta el tren anual de publicaciones en los
últimos 10 años con respecto al tema de la “fotocatálisis plasmónica” (Web of Science, Citation Report).
Figura 2. Características de la fotocatálisis plasmónica. Tomado de Zhang et al., 2013.

3
Figura 3. Publicaciones de la fotocatálisis plasmónica en los últimos 10 años. Modificado de Web of Science, 2016.
Por otro lado, la Figura 4 (Observatory briefing, 2016) muestra el mercado global en el sector de la
fotocatálisis en general. Los fotocatalizadores a nanoescala son incluidos en estos valores, pero no se
especifican en detalle. De cualquier modo, los productos a nanoescala se han incrementado en los últimos
años y esta tendencia es muy probable que continúe. El volumen del mercado global en 2014 se esperaba
que alcanzara 1.7 mil millones de dólares. Se estima un crecimiento anual de 14.3 %, esto, significa que
para el 2017 se estima un volumen de 1.9 mil millones de dólares.
Figura 4. Volumen del mercado de productos fotocatáliticos (Observatory briefing, 2016).

4
Los principales campos de aplicación de la fotocatálisis en base a las propiedades oxidativas y a las
propiedades superhidrofílicas se muestran en la Tabla 1.
Tabla 1. Aplicaciones de la fotocatálisis (Murakami, 2010).
La oxidación fotocatalítica de la glucosa y su selectividad en la producción de compuestos orgánicos con
alto valor agregado como el ácido glucárico, el ácido glucónico y el arabitol en solución liquida se pueden
obtener en la presencia de fotocatalizadores nanoestructurados de TiO2, como se ilustra en la Figura 5, en
el reporte de Colmenares et al., 2011 se encontraron posibles rutas de producción, se detectó CO2 y trazas
de hidrocarburos en fase líquida. En este proceso la glucosa es oxidada bajo la presencia de TiO2 en polvo
usado como fotocatalizador, el TiO2 presenta una selectividad a los productos antes mencionados
(Colmenares et al., 2011).
Figura 5. Sustancias con alto valor agregado obtenidos a partir de la foto-oxidación selectiva de glucosa con TiO2 (Colmenares et al., 2011).

5
El presente proyecto de investigación tiene como propósito implementar la fotocatálisis plasmónica en la
conversión de glucosa para producir productos de valor agregado. Para lo cual, se probarán cinco
nanocompuestos fotocatalizados y sintetizados previamente y donde la presencia de nanopartículas
metálicas y semiconductoras pueden generar productos nuevos.

6
Capítulo 2. Hipótesis y Objetivos
2.1. Hipótesis
Es posible la conversión de glucosa a productos de alto valor agregado como sorbitol y ácido glucurónico
mediante el empleo de fotocatalizadores plasmónicos conformados por nanopartículas metálicas y/o
semiconductoras soportados en matrices zeolíticas tipo mordenita.
2.2. Objetivos generales
Evaluar mediante procesos de fotocatálisis el uso de nanocompuestos plasmónicos conformados por
nanopartículas metálicas y/o semiconductoras soportados en matrices zeolíticas tipo mordenita para la
conversión de glucosa en productos de alto valor agregado.
2.3. Objetivos específicos
1. Realizar la caracterización estructural, morfológica, así como de la composición química de los
nanocompuestos fotocatalizadores sintetizados y seleccionados previamente empleando las
técnicas de rayos X, microscopia electrónica de barrido y transmisión, espectroscopia de
dispersión de rayos X y fotoelectrónica de rayos X, así como evaluar las propiedades físico-
químicas mediante espectroscopia de absorción UV-visible y adsorción física de gases.
2. Realizar la evaluación fotocatalítica de cada uno de los nanocompuestos seleccionados en la
degradación de glucosa y establecer las condiciones óptimas para obtener subproductos o
derivados como sorbitol y ácido glucurónico.

7
Capítulo 3. Métodos y técnicas experimentales
3.1. Materiales empleados
3.1.1 Nanocompuestos fotoactivos
Las zeolitas tipo mordenita son aluminosilicatos cristalinos hidratados que tienen una estructura porosa
regular. El área superficial que poseen las zeolitas y su gran capacidad de atraer e incorporar a su
estructura iones positivos, hacen que se les conozca como tamices moleculares. Existen varios tipos de
zeolitas y una de las más utilizadas es la mordenita (MOR). Los nanocompuestos empleados en este trabajo
son compuestos nanoestructurados tipo metal-mordenita, metal/semiconductor-mordenita y
semiconductor-mordenita sintetizados en procesos de un solo paso a partir de residuos sólidos derivados
de la conversión de energía geotérmica, mediante soluciones acuosas libres de solventes orgánicos y de
plantillas orgánicas, donde se incluye al ion o la combinación de iones que conforman la parte metálica
y/o semiconductora de un modo controlado, dosificado y repetible (Jaime-Acuña et al. 2016a) .
Los fotocatalizadores utilizados en este proyecto son:
A. Metal-mordenita
1. 30Ag-MOR
2. 30Au-MOR
B. Metal/semiconductor-mordenita
3. 30Au/20CdS-MOR
4. 10Ag/20CdS-MOR
C. Semiconductor-mordenita
5. 20CdS-MOR
La estructura descriptiva de las nanopartículas metal, metal/semiconductor o semiconductor crecidas
dentro y fuera de la matriz de mordenita se muestra en la figura 6. Los números presentes en cada nombre
de los nanocompuestos corresponde con la concentración del metal y/o el semiconductor empleados en
el proceso de síntesis de cada compuesto como se describe en Jaime-Acuña 2014.

8
Figura 6. Simulación del crecimiento de nanopartículas de metal y/o semiconductor soportados en la matriz de la mordenita. Tomado de Jaime-Acuña, et al., 2016.
La mordenita es uno de los silicatos que se consideran tamices moleculares debido a su estructura
ortorrómbica de mesoporos y canales conformados con el enrejado de átomos de Si y Al, coordinados
tetraédricamente con el oxígeno y expresado en tetraedros de SiO4 o AlO-4 (Weitkamp, 2000). La
estructura (Figura 7) de la mordenita se caracteriza por la presencia de dos tipos de canales en la dirección
[001]: uno de 12 miembros con un acceso libre de 0.65 x 0.7 nm y otro de 8 miembros con una ventana
de 0.26 x 0.57 nm. Las zeolitas naturales, contrario a las zeolitas sintéticas, están formadas de dos tipos
de porosidad: (i) primaria, debido a la presencia de microporos y (ii) secundaria que son los mesoporos
(Gregg et al., 1982). En los mesoporos de la matriz zeolítica se efectúa la difusión y la catálisis heterogénea
(Liu et al., 2004). La porosidad primaria está caracterizada por el volumen de microporos y la porosidad
secundaria por la distribución de mesoporos y por su área externa entre otros. Estos parámetros se
determinarán a partir de mediciones de adsorción de N2 a 76 K y mediante el análisis BET.

9
Figura 7. Estructura de la mordenita. Sistema de coordenadas y sus dimensiones (Asociación internacional de zeolitas, 2016).
La fórmula química de la mordenita es |Na+8 (H2O)24| [Al8Si40 O96]-MOR y la estructura cristalina es
ortorrómbica con parámetros de celda a = 18.2560 Å, b = 20.5340 Å, c = 7.5420 Å y α = β = γ = 900, con un
volumen de 2827.3 Å3.
Basándose en lo reportado por Jha et al., 2016 las propiedades catalíticas de las zeolitas son:
1. Capacidad de intercambio catiónico (CEC)
2. Volumen y diámetro de los poros y canales
3. Gravedad específica
4. Composición química y características morfológicas
5. Presencia de cargas microscópicas negativas sobre superficies pequeñas y/o los poros internos que aumentan su intercambio iónico o hidrofilicidad.

10
3.1.2. La glucosa
Los carbohidratos son los compuestos más abundantes en la naturaleza. La mayoría de las plantas y
animales sintetizan y metabolizan los carbohidratos, usándolos para almacenar energía y llevarla a las
células. Las plantas sintetizan los carbohidratos a través de la fotosíntesis en una serie de reacciones
complejas que utilizan luz solar como fuente de energía para convertir el bióxido de carbono (CO2) y agua
(H2O) en D-glucosa (C6H12O6) y oxigeno (O2) (Figura 8). Se usa el prefijo D cuando la glucosa es de origen
natural y no sintetizada. La mayoría de las células oxidan la glucosa en CO2 y H2O para proporcionar la
energía necesaria a sus células. Los animales también pueden almacenar energía al unir varias moléculas
de glucosa y formar glucógeno (Wade, 2006). En las plantas una gran cantidad de moléculas de glucosa
se pueden unir y formar almidón para almacenar energía y/o para formar celulosa para soporte de la
planta. La glucosa es la fuente de energía primaria para organismos vivos y se usa terapéuticamente en el
reemplazo de líquidos y nutrientes (PubChem, 2017a).
Figura 8. Formación de glucosa por fotosíntesis.
3.1.2.1. Activación de la glucosa
La fórmula química de la glucosa se puede representar en forma lineal (proyección de Fischer) o en forma
cíclica (proyección de Harworth) (Figura 9a y b). La estructura de anillo de la glucosa se genera cuando el
grupo OH (hidroxilo) del quinto átomo de carbono reacciona con el grupo aldehído (H-C=O) en la primera
posición de la proyección de Fischer y el grupo carbonilo (C=O) en la misma posición anterior. La estructura
de la Figura 10a con el grupo OH dirigido hacia abajo y en la posición del primer carbono representa lo que
se conoce como la forma alfa (α) y cuando el grupo OH está dirigido hacia arriba es la forma beta (β) de la
D-glucosa y se conocen como anómeros debido a la orientación del grupo OH en su estructura cíclica.

11
Figura 9. Proyección de la estructura química de la glucosa: a) Fischer y b) Harworth. Tomada de Smith et al., 2007.
En solución acuosa, la glucosa o D-glucosa existe como una mezcla de tres anómeros en equilibrio (Figura
10a, b y c). La Interconversión entre ellas se conoce como mutarrotación (del latín mutare, que significa
"cambiar"). En equilibrio, la mezcla consiste en aproximadamente 36% de α-D-glucosa, 64% de β-D-
glucosa y menos de 0.02% en la forma de cadena abierta de aldehído (proyección de Fischer).
Figura 10. Mutarrotación en la estructura de la glucosa. Tomada de Smith et al., 2007.
Para el presente proyecto es importante tener la glucosa en su forma más reactiva, la cual es en solución
acuosa para usar los tres anómeros. Se usarán soluciones de acetonitrilo para sensibilizar la estructura en
forma de anillo de la glucosa y poder hacerla más activa. Los grupos en las sustancias que activan las
reacciones se llaman activadores o sensibilizadores y los grupos que disminuyen la activación de una
reacción se llaman desactivadores (Smith et al., 2007).

12
3.1.3. Posibles derivados de la glucosa
Los posibles derivados a considerar en esta investigación son el sorbitol y el ácido glucurónico que son
derivados de reducción y oxidación de la glucosa.
3.1.3.1. Sorbitol
El D-sorbitol (Figura 11) se produce por la reducción del grupo carbonilo en la D-glucosa. Industrialmente,
el sorbitol se produce por la hidrogenación catalítica de la glucosa con níquel. El sorbitol es un alcohol de
azúcar con aproximadamente la mitad del dulzor de la sacarosa. El sorbitol se produce naturalmente y
también se produce sintéticamente a partir de la glucosa. Anteriormente se usaba como diurético y
todavía se puede usar como laxante y en soluciones de irrigación para algunos procedimientos quirúrgicos.
También se usa en forma de solución para el acondicionamiento de la humedad de cremas y las lociones
cosméticas, en las pastas de dientes, en el tabaco, en la gelatina y como agente de envoltura para papel.
El sorbitol se presenta ampliamente en los frutos de la familia de las rosáceas, que incluyen las manzanas,
peras, cerezas y albaricoques los que contienen cantidades apreciables. Las fuentes ricas son los frutos de
las especies sorbus y crataegus. El sorbitol se utiliza para la fabricación de sorbosa, propilenglicol, ácido
ascórbico, resinas, plastificantes y como mezclas anticongelantes con glicerol o glicol (PubChem, 2017b).
Figura 11. Proyección de Fischer para el sorbitol.

13
3.1.3.2. Ácido glucurónico
El ácido D-glucurónico es un ácido de azúcar formado por la oxidación del carbono 6 de la glucosa (Figura
12). Además de ser un metabolito (que son más fáciles de excretar por vía renal o hepática (en la bilis))
soluble en agua y que se excreta fácilmente, el ácido glucurónico también desempeña un papel en la
desintoxicación de ciertos fármacos y toxinas al conjugarse con ellos para formar glucurónicos. La
formación de glucurónidos es la reacción que consiste en la conjugación con sustancias endógenas (ácido
glucurónico); estas reacciones son de carácter sintético (PubChem, 2017c).
Figura 12. Proyección de Fischer para el ácido glucurónico.
3.2. Caracterización estructural, morfológica y de composición química
Para el presente proyecto es importante conocer las características estructurales, morfológicas y de la
composición química. Para ello se emplearon las técnicas de difracción de rayos X, la microscopia
electrónica de transmisión (TEM del inglés Transmission Electron Microscopy), la microscopia electrónica
de barrido (SEM del inglés Scanning Electron Microscopy), la espectroscopia de dispersión de energía
(EDX/EDS del inglés Energy Dispersive X-ray/Energy Dispersive Spectroscopy) , la espectroscopia
fotoelectrónica de rayos X (XPS del inglés X-ray Photoelectron Spectroscopy) y por adsorción de gases
(técnica BET de los autores Brunauer-Emmett-Teller).

14
3.2.1. Difracción de rayos-X
Para conocer la estructura cristalina de los fotocatalizadores se emplea la ley de Bragg donde se hace
incidir un haz de luz de longitud de onda, λ, que será dispersado por los planos cristalinos característicos
(Figura 13). El ángulo entre el haz de luz incidente y los planos de la red cristalina se llama teta (θ). El
ángulo entre el haz incidente y el haz dispersado 2θ. Los planos paralelos de la red cristalina están
separados un espacio dhkl. La diferencia entre los planos adyacentes de la red del cristal es igual a 2d sen
θ. La interferencia constructiva entre los planos adyacentes ocurre cuando esta es un numero entero de
la longitud de onda λ (Kittel, 2005).
Figura 13. Ley de Bragg. Relación entre la dispersión interplanar y el ángulo de incidencia. Tomado de
Speakman, 2016.
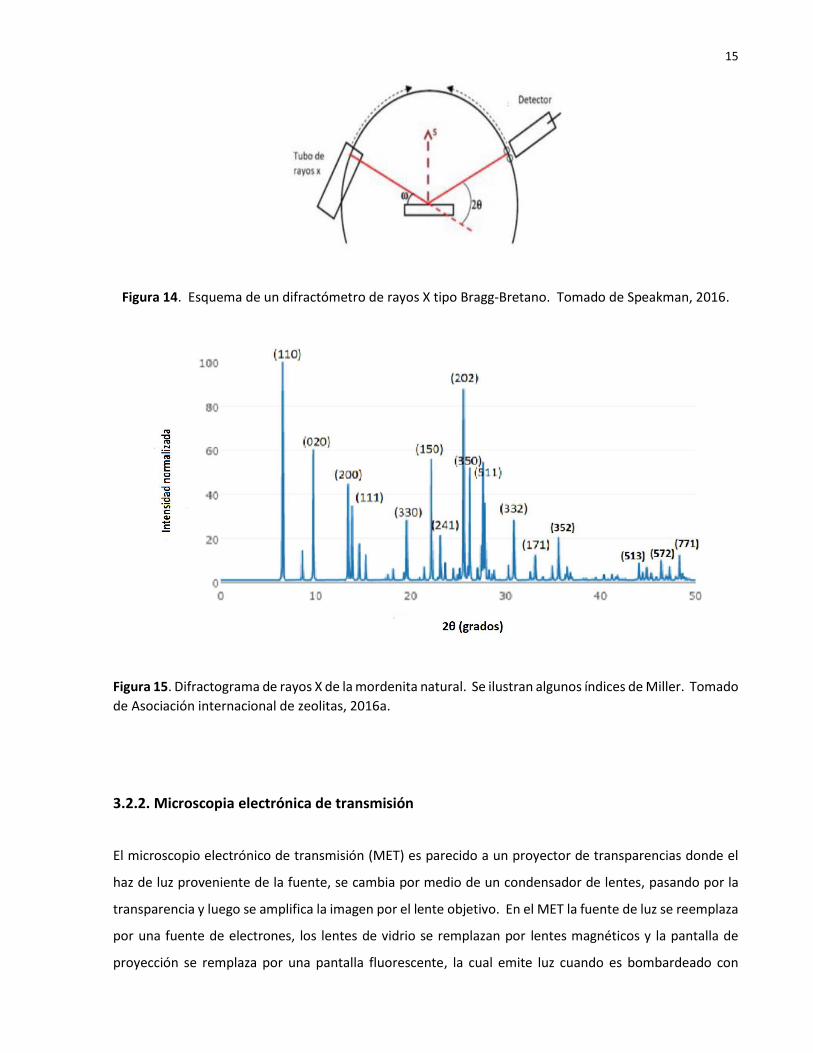
La caracterización estructural de los fotocatalizadores tipo mordenita se efectuará en un instrumento de
geometría Bragg-Bretano como se muestra en la Figura 14 (Speakman, 2016). Ésta proporciona que los
ángulos de difracción estén bien definidos. El detector y la fuente de rayos X se colocan a igual distancia
y diferentes ángulos de la superficie de la muestra. Al aplicar la ley de Bragg y usar el difractómetro Bragg-
Bretano, se genera el difractograma. En la Figura 15 se muestra un difractograma representativo de los
polvos de zeolita tipo mordenita, el cual ha sido indexado y se ilustran algunos de los planos cristalinos
con sus índices de Miller (Asociación internacional de zeolitas, 2016).
15
Figura 14. Esquema de un difractómetro de rayos X tipo Bragg-Bretano. Tomado de Speakman, 2016.
Figura 15. Difractograma de rayos X de la mordenita natural. Se ilustran algunos índices de Miller. Tomado
de Asociación internacional de zeolitas, 2016a.
3.2.2. Microscopia electrónica de transmisión
El microscopio electrónico de transmisión (MET) es parecido a un proyector de transparencias donde el
haz de luz proveniente de la fuente, se cambia por medio de un condensador de lentes, pasando por la
transparencia y luego se amplifica la imagen por el lente objetivo. En el MET la fuente de luz se reemplaza
por una fuente de electrones, los lentes de vidrio se remplazan por lentes magnéticos y la pantalla de
proyección se remplaza por una pantalla fluorescente, la cual emite luz cuando es bombardeado con

16
electrones. En la actualidad se emplea un dispositivo electrónico de imágenes como una cámara de CCD
(charge-couple device). La muestra debe ser muy delgada para que permita a los electrones pasar a través
de ella y obtener su micrografía. La figura 16a presenta el esquema del MET similar a un proyector de
transparencias; mientras la Figura 16b indica las partes de un MET moderno.
Figura 16. a) Esquema de la óptica y b) de la sección transversal de un MET. Tomado de FEI™, 2017.
El Centro de Nanociencias y Nanotecnología (CNYN) cuenta con un MET moderno (JEOL LEM-2100). Este
es del tipo de microscopio electrónico de transmisión por barrido (METB), el cual combina las técnicas de
transmisión y barrido. El METB cuenta con un cañón de electrones tipo emisión de campo-Schottky, modo
barrido, adquisición digital de imágenes y análisis químico por medio de espectroscopia de dispersión de
energía (EDS). La Figura 17 ilustra los cristales en forma de agujas que son característicos de la mordenita
sintetizada y utilizada en este trabajo.

17
Figura 17. Micrografía de MET de polvos del nanocompuesto 20CdS-MOR.
3.2.3. Microscopia electrónica de barrido
El microscopio electrónico de barrido (MEB), es parecido al MET, y consiste en una columna óptica de
electrones, un sistema de vacío, componentes electrónicos y el software. La columna es más corta que la
del MET, ya que los únicos lentes son para enfocar a los electrones sobre la superficie de la muestra, ver
figura 18 (Jaime-Acuña, 2010). No hay lentes debajo de la muestra y la cámara para la muestra es más
grande que la del MET debido a que en el MEB la única restricción en el tamaño de la muestra es el tamaño
de la cámara. La técnica de MEB consiste en hacer incidir un haz de electrones sobre una muestra, los
cuales generan señales que son capturadas en el detector. Los electrones que bombardean la muestra
generan electrones secundarios(ES) de baja energía que provienen de los átomos de la muestra cercanos
a la superficie y la intensidad de las emisiones varía en función del ángulo que forma el haz incidente con
la superficie del material y dependiendo del relieve de ésta (Jaime-Acuña et al., 2016). Otra característica
es, si la muestra es lo suficientemente delgada, el MEB puede trabajar en el modo de MET, si cuenta con
un detector localizado por debajo de la superficie del material ya que la muestra emite electrones
(catoluminiscencia) y emite rayos X. Los rayos X se usan ampliamente para hacer análisis químico en el
MEB.

18
Figura 18. Esquema de la óptica de un MEB y una imagen representativa de la muestra 20CdS-MOR. Tomado de Jaime-Acuña, 2010.
El MEB a utilizar es el JEO JSM-5300 con detector para hacer análisis químico por dispersión de energía y
la adquisición de imágenes. Lo que será de utilidad para analizar las características superficiales, los
mesoporos y los microporos en los nanocompuestos.
3.2.4. Espectroscopia de dispersión de energía
En el MEB un haz de electrones barre la superficie de la muestra. Cuando los electrones golpean la
muestra, una variedad de señales se generan y es la detección de una señal especifica es la que indica la
composición química elemental de ésta. Las tres señales que proporcionan la mayor información en el
MEB/MET son los electrones secundarios, los electrones retro-dispersados y los rayos X (Figura 19) (Zinin,
2016).
Los electrones secundarios son emitidos de la superficie externa de la muestra produciendo una imagen
de ésta. Los contrastes de la imagen son determinados por la morfología de la muestra. Una imagen de
alta resolución se puede obtener debido al diámetro pequeño del haz de electrones primarios.

19
Los contrastes producidos en la imagen están determinados por el numero atómico de los elementos en
la muestra. Por lo tanto, la imagen mostrará la distribución de las diferentes fases químicas en el
nanocompuesto, ya que estos electrones son emitidos desde lo profundo del material, pero la resolución
de la imagen de estos electrones no es tan buena como la de los electrones secundarios.
La interacción del haz de electrones primario con los átomos de la muestra provoca la transición de
electrones entre los orbitales, lo que resulta en la emisión de rayos X. Los rayos X tienen una energía
característica del elemento que los emite. La medición y detección de la energía permite hacer análisis
químico elemental (EDX: Energy Dispersive X-ray Spectroscopy y/o EDS: Energy Dispersive Spectroscopy)
o espectroscopia de dispersión de energía (EDE), proporcionando rápidamente un análisis cualitativo y/o
cuantitativo a una profundidad en la muestra de 1 a 2 micras. Los rayos X también se emplean para
representar la distribución de los elementos en la muestra (Zinin, 2016).
Figura 19. Interacción de un haz de electrones con la materia (Zinin, 2016).
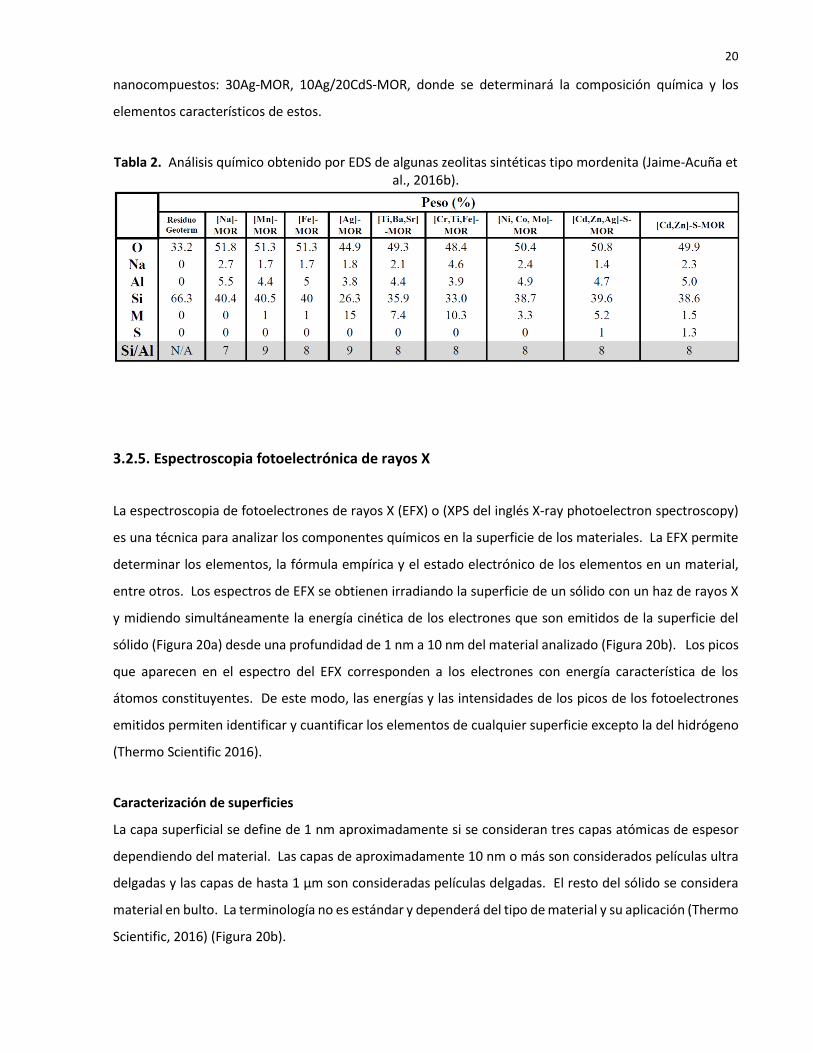
La Tabla 2 (Jaime-Acuña et al., 2016), proporciona los resultados del análisis químico de nanocompuestos
fotocatlizadores, algunos de los cuales seran usados en el presente proyecto de investigación, estos son:
Ag-MOR, 10Ag/20CdS-MOR, CdS-MOR. En este trabajo, se hará el análisis químico para dos
20
nanocompuestos: 30Ag-MOR, 10Ag/20CdS-MOR, donde se determinará la composición química y los
elementos característicos de estos.
Tabla 2. Análisis químico obtenido por EDS de algunas zeolitas sintéticas tipo mordenita (Jaime-Acuña et al., 2016b).
3.2.5. Espectroscopia fotoelectrónica de rayos X
La espectroscopia de fotoelectrones de rayos X (EFX) o (XPS del inglés X-ray photoelectron spectroscopy)
es una técnica para analizar los componentes químicos en la superficie de los materiales. La EFX permite
determinar los elementos, la fórmula empírica y el estado electrónico de los elementos en un material,
entre otros. Los espectros de EFX se obtienen irradiando la superficie de un sólido con un haz de rayos X
y midiendo simultáneamente la energía cinética de los electrones que son emitidos de la superficie del
sólido (Figura 20a) desde una profundidad de 1 nm a 10 nm del material analizado (Figura 20b). Los picos
que aparecen en el espectro del EFX corresponden a los electrones con energía característica de los
átomos constituyentes. De este modo, las energías y las intensidades de los picos de los fotoelectrones
emitidos permiten identificar y cuantificar los elementos de cualquier superficie excepto la del hidrógeno
(Thermo Scientific 2016).
Caracterización de superficies
La capa superficial se define de 1 nm aproximadamente si se consideran tres capas atómicas de espesor
dependiendo del material. Las capas de aproximadamente 10 nm o más son considerados películas ultra
delgadas y las capas de hasta 1 µm son consideradas películas delgadas. El resto del sólido se considera
material en bulto. La terminología no es estándar y dependerá del tipo de material y su aplicación (Thermo
Scientific, 2016) (Figura 20b).

21
Figura 20. a) Esquema de la emisión de fotoelectrones bajo la radiación de rayos X; b) comparación del espesor de superficie con el tamaño de material. Tomado de Thermo Scientific, 2016.
Propiedades de superficies
El entendimiento del análisis de superficie ayuda en el entendimiento de las siguientes áreas: catálisis,
semiconductores, microcircuitos, corrosión, oxidación, lubricación, etc.
Principios básicos de EFX
La EFX se utiliza para foto-ionizar y analizar la distribución de energía cinética de los fotoelectrones
emitidos de la superficie de una muestra. Para analizar la superficie se usa una fuente de excitación de
radiación de 200 – 2000 eV, la cual se considera radiación de rayos X suaves; ver la Figura 21 de Thermo
Scientific, 2016.
El proceso de foto-ionización se expresa químicamente como:
X0 + hv X+ + e- (2)
Donde, X0 es la superficie del material en estado sólido, hv es la energía de la radiación electromagnética
del fotón ionizante (rayos X), según la relación de Einstein E = hv. X+ es la superficie del material ionizado
y e- es el electrón expulsado (de uno de los niveles de energía K, L, M, etc.). Aplicando la ley de la
conservación de la energía, considerando únicamente la energía cinética de cada uno de los componentes
tenemos,
E(X0) + hv = E(X+) + E(e-) (3)

22
donde,
E(e-) = EC = energía cinética del electrón expulsado
e implica que E(X0) + hv = E(X+) + EC (4)
y EC = hv – [E(X+) – E(X0)] (5)
donde, la diferencia [E(X+) - E(X0)] = BE es conocida como, la energía de unión o energía de enlace de un
electron (BE del inglés Binding energy). La diferencia de energías, E(X+) - E(X0), representa la energía entre
el átomo ionizado y el átomo neutro o metálico.
Figura 21. Proceso de fotoionización y energías del fotoelectrón expulsado. Tomado de Thermo Scientific, 2016).
Espectro general
Para cada elemento habrá una energía de enlace asociada a cada orbital atómico y así, cada elemento
químico dará un grupo de picos característicos en el espectro fotoelectrónico EFX a determinadas energías
de enlace como se observa en la Figura 22 (Thermo Scientific, 2016). La presencia de picos a determinada
energía de enlace indicará la presencia de un elemento en particular en la muestra de estudio. Además,
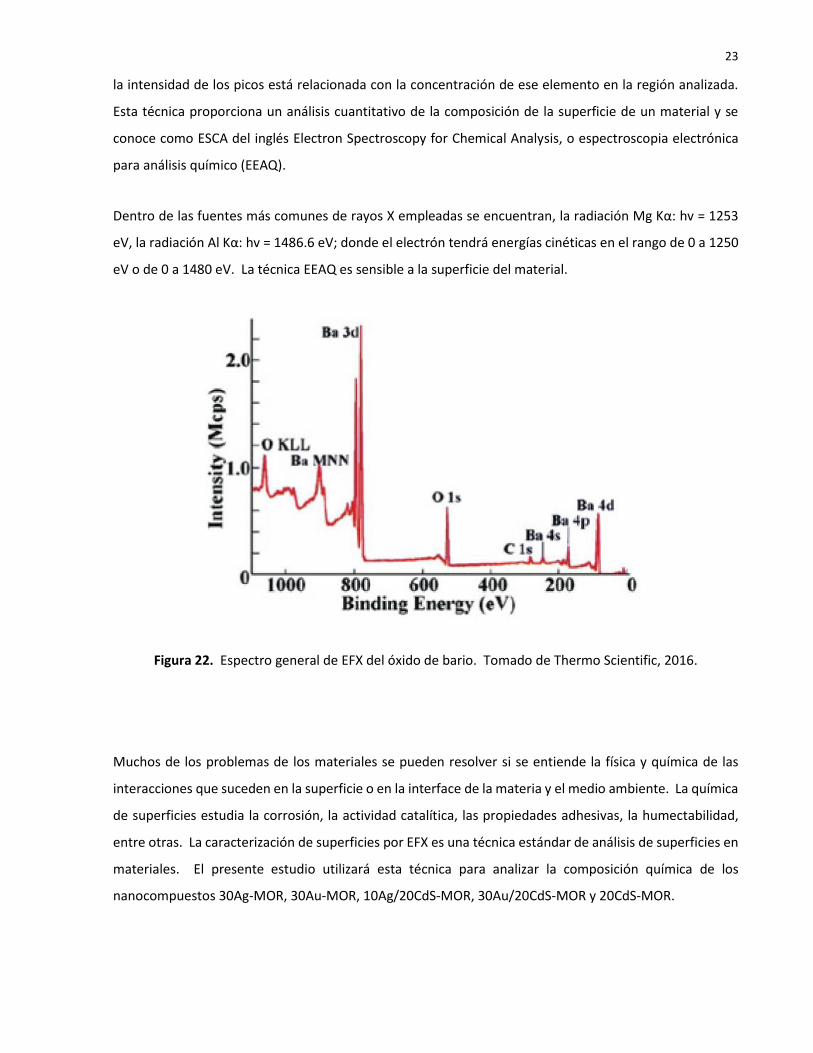
23
la intensidad de los picos está relacionada con la concentración de ese elemento en la región analizada.
Esta técnica proporciona un análisis cuantitativo de la composición de la superficie de un material y se
conoce como ESCA del inglés Electron Spectroscopy for Chemical Analysis, o espectroscopia electrónica
para análisis químico (EEAQ).
Dentro de las fuentes más comunes de rayos X empleadas se encuentran, la radiación Mg Kα: hv = 1253
eV, la radiación Al Kα: hv = 1486.6 eV; donde el electrón tendrá energías cinéticas en el rango de 0 a 1250
eV o de 0 a 1480 eV. La técnica EEAQ es sensible a la superficie del material.
Figura 22. Espectro general de EFX del óxido de bario. Tomado de Thermo Scientific, 2016.
Muchos de los problemas de los materiales se pueden resolver si se entiende la física y química de las
interacciones que suceden en la superficie o en la interface de la materia y el medio ambiente. La química
de superficies estudia la corrosión, la actividad catalítica, las propiedades adhesivas, la humectabilidad,
entre otras. La caracterización de superficies por EFX es una técnica estándar de análisis de superficies en
materiales. El presente estudio utilizará esta técnica para analizar la composición química de los
nanocompuestos 30Ag-MOR, 30Au-MOR, 10Ag/20CdS-MOR, 30Au/20CdS-MOR y 20CdS-MOR.

24
3.2.6. Análisis de adsorción física
La adsorción fisica, similar a la tensión superficial, es una consecuencia de la energía en la superficie del
sólido. La mayoría de los átomos que forman un sólido están ligados a otros átomos. Los átomos junto a
la superficie están ligados parcialmente a los átomos internos, debido a la interacción de las fuerzas de
Van der Waals. Los átomos en la superficie son más reactivos y atraen a los gases, los vapores y a los
líquidos para balancear las fuerzas atómicas superficiales. El conocimiento del área superficial ayuda a
entender cómo los sólidos reaccionan con otros materiales.
Equipo de adsorción de N2
Para determinar el área superficial de un sólido, la muestra es pre-tratada con calor, vacío y/o con una
corriente de gas para remover los contaminantes adsorbidos por contacto con la atmosfera como agua y
bióxido de carbono. El aparato de adsorción y desorción de gases que se emplea para las medidas de
adsorción físicas y para el cálculo de las isotermas de adsorción y desorción es el de la Figura 23 de
Micromeritic, 2017. El esquema muestra los componentes principales. 1) colector de análisis, 2) Sistema
de vacío con válvula a colector, 3) fuente del gas de adsorción, 4) Sensor de temperatura y transductor de
presión, 5) equipo para registro de señales del sensor de temperatura y del transductor de presión, 6) tubo
de muestra de conocido espacio libre o vacío, 7) Tubo de muestra conectado a colector de análisis, 8)
medio para reducir la temperatura de la muestra con nitrógeno líquido (NL).
Figura 23. Esquema general de un aparato para el análisis de adsorción. Tomado de Micromeritics, 2017.

25
Mediciones de las isotermas de adsorción y desorción BET
Las mediciones se efectúan con la ecuación correspondiente a las isotermas de adsorción y desorción BET
(Figura 24):
1
[Va(Po
P − 1)]
= 𝐶−1
𝑉𝑚 𝐶 x
𝑃
𝑃𝑜 +
1
𝑉𝑚 𝐶 (6)
donde, P es la presión parcial del gas adsorbido en equilibrio con la superficie a 77. 4 K (Punto de ebullición
del nitrógeno líquido), en pascales (Pa). Po es la presión de saturación del gas adsorbido en pascales. Va
es el volumen del gas adsorbido a presión y temperatura estándar (273.15 K y presión atmosférica 1.013
x 1015 Pa) en mililitros de la columna de mercurio. Vm es el volumen del gas adsorbido a condiciones de
temperatura y presión estándar (ETP) para producir una monocapa en la superficie de la muestra, en
mililitros. C es la constante relacionada a la entalpia de adsorción del gas adsorbido en la muestra. Los
valores de Va se obtienen de tres o más valores de P/Po. Luego, los valores de 1
[Va(Po
P − 1)]
en la ecuación
6 se grafican en función de P/P0 (Figura 24). La operación del aparato de la Figura 23 proporcionará los
parámetros necesarios para graficar las isotermas de adsorción y desorción BET.
Figura 24. Isotermas de adsorción y desorción: a) tipo I y b) tipo IV (Micromeritics. 2016).

26
Curvas de calibración BET
La curva de calibración BET es una línea recta ajustada de la ecuación 6. Las presiones relativas de
operación del aparato de la Figura 23 deben ser de aproximadamente 0.05 a 0.3. Los datos se consideran
adecuados, si el coeficiente de correlación, r, de la curva de calibración no es menor de 0.9975, lo cual
significa que r2 > 0.995. Posteriormente se obtienen los valores de la pendiente, S = (C – 1)/VmC, y la
intersección con el eje Y, Yint = 1/ VmC de la ecuacion 6, para asi, obtener los valores de Vm y C. Con
estos valores podemos determinar el area superficial, SABET en m2/g con la ecuación (Micromeritics®
2017):
SABET = 4.3532 x Vm (7)
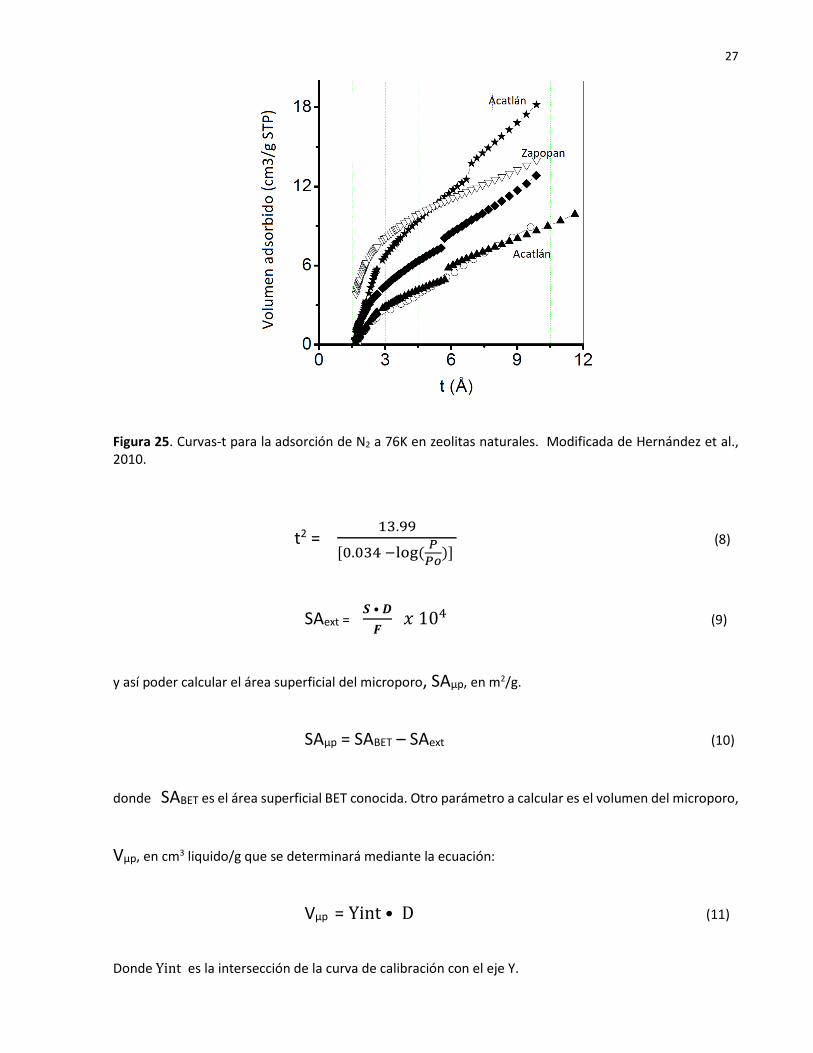
Las Curvas-t
La curvas-t (Figura 25) proporcionan información asociada con la formación de las capas de adsorción
sobre las paredes de materiales porosos como son las zeolitas al expresar la dependencia del volumen
adsorbido con el espesor según la ecuación de Harkins y Jura. Empleando las ecuaciones 6 y 7 se podrá
obtener primeramente el área superficial y la operación del equipo de la Figura 23 proporciona los
siguientes parámetros: el factor de corrección del área superficial, F, para generar el reporte de la curva-t
y el valor del factor de conversión de densidad para las propiedades de adsorción, D. Los parámetros F y
D se emplean en la ecuación 9 para determinar el área superficial externa, SAext, m2/g.
27
Figura 25. Curvas-t para la adsorción de N2 a 76K en zeolitas naturales. Modificada de Hernández et al., 2010.
t2 = 13.99
[0.034 −log(𝑃
𝑃𝑜)]
(8)
SAext = 𝑺 • 𝑫
𝑭 𝑥 104 (9)
y así poder calcular el área superficial del microporo, SAµp, en m2/g.
SAµp = SABET – SAext (10)
donde SABET es el área superficial BET conocida. Otro parámetro a calcular es el volumen del microporo,
Vµp, en cm3 liquido/g que se determinará mediante la ecuación:
Vµp = Yint • D (11)
Donde Yint es la intersección de la curva de calibración con el eje Y.

28
3.3. Caracterización óptica
Las nanopartículas tienen propiedades ópticas que son sensibles a su tamaño, a su estado de aglomeración
y al índice de refracción en la superficie de estas, lo que hace a la espectroscopia UV-Visible una técnica
invaluable en la caracterización de estos materiales. Las nanopartículas hechas de materiales como la
plata y el oro interactúan fuertemente con la luz a ciertas longitudes de onda y las propiedades ópticas de
estos materiales son la base para el campo de la plasmónica (nanoComposix, 2017).
3.3.1. Espectroscopia de absorción UV-Visible
La espectroscopia UV-Visible es una técnica usada para cuantificar la luz que es absorbida, transmitida y
reflejada por una muestra. En la forma más simple se coloca una muestra entre la fuente de luz y el
fotodetector, se mide la intensidad de la luz antes y después de que pasa por la muestra. Las medidas son
comparadas a cada longitud de onda para cuantificar la absorbancia o transmitancia de la muestra. Los
datos se grafican como absorbancia o transmitancia en función de la longitud de onda.
La transmitancia (T) se define como la fracción de fotones que pasan a través de una muestra sobre el
número de fotones incidentes, T = IT/ I0, donde IT es la intensidad de la luz que se detecta en el Detector,
I0 es la intensidad de la fuente de luz. En una solución diluida el valor de la absorbancia (A) se mide a
valores de longitud de onda específicos siendo proporcionales a la concentración. La absorbancia se puede
determinar mediante la ley de Beer A = ɛbC, donde ɛ es el factor de absorción del material, b es el espesor
del porta muestras o cubeta y C es la concentración de la muestra. Si analizamos un sólido como una
película delgada, se utiliza la ley de Beer para sólidos (Figura 26), donde, α es el coeficiente de absorción
del solido o película delgada, y d es el espesor del sólido o de la película delgada.
La Figura 26 representa el esquema de un espectrómetro de luz ultravioleta y/o luz visible, si este cuenta
con una lámpara de UV y una del visible. La pre-óptica se emplea para dejar pasar longitudes de onda
determinadas, se tiene la cubeta o porta muestras, se corrige la luz transmitida que se detecta en el
detector.

29
Figura 26. Esquema de la óptica de un espectrómetro UV-Visible. Tomado de Agilent, 2016.
3.4. Evaluación fotocatalítica
3.4.1. Fotoreactor
El proceso de conversión de glucosa se realizará empleando la caja de luz CROSS LINKER® CL-508 de UVITEC
CAMBRIDEGE para proporcionar la radiación UV de 254 nm en la cual se adoptó un contenedor para las
soluciones y se agregó el agitador magnético como se muestra en la Figura 27 (Jaime-Acuña, et al., 2016).
Las muestras se agitarán a 300 rpm y se programará el fotoreactor para aplicar radiación ultravioleta por
dos horas o más. Se cargarán 30 ml de cada una de las muestras de análisis.
La caja de luz posee varios ajustes manuales como son: energía de radiación en Joules, tiempo de
radiación. En este proyecto se utilizará el tiempo de radiación en minutos.

30
Figura 27. Fotoreactor de 254 nm de radiación UV. Modificado de Jaime-Acuña, et al., 2016.
3.4.2. Cromatografía Líquida
La cromatografía líquida (LC del inglés Liquid Chromatography) es una técnica de separación molecular
que involucra:
• La inyección de un pequeño volumen de muestra
• Pasar la muestra por un tubo empaquetado con partículas porosas (la fase liquida)
• Transportar por gravedad los componentes de la muestra con un líquido a un tubo empaquetado (la columna).
Los componentes de la muestra son separados unos de otros por la columna empacada la cual involucra
interacciones físicas y/o químicas entre las moléculas de la muestra y las partículas del empaque. Las
componentes separadas de la muestra se recogen a la salida de la columna de empaque y se identifican
por alguna técnica de medición, como la espectrofotometría la cual mide la intensidad de los colores, o
por algún otro dispositivo que puede medir la cantidad de muestra separada. Comparando esta técnica
con la Cromatografía Liquida de Alto Rendimiento (HPLC del inglés High Performance Liquid
Chromatography), esta última es una técnica de separación que involucra:

31
• La inyección de pequeñas cantidades de una muestra liquidad en un tubo empacado con pequeñas partículas de 3 a 5 micrómetros (µm) en diámetro, llamada fase estacionaria.
• Las componentes de la muestra son movidos por un líquido (la fase móvil) a una columna empacada (la columna) por una bomba de alta presión.
Las componentes son separadas por la columna y por la interacción de las componentes físico y/o químicos
de las moléculas de la muestra y las partículas del empaque de la columna. Los componentes separados
son detectados a la salida de la columna por un detector y con una computadora se genera un
cromatograma.
En principio la LC y la HPLC funcionan del mismo modo excepto por la velocidad, la eficiencia, la
sensibilidad y la fácil operación del equipo de HPLC haciendo esta última técnica superior que la LC.
El análisis por cromatografía liquida se realizará en un equipo HPLC 1100 de Agilent, ver la figura 28. El
método de operación del HPLC depende de los componentes que serán analizados. Por ejemplo, para
carbohidratos que difieren del que se usa para proteínas. El método de operación contempla el tipo de
columna de separación, el volumen de inyección de la muestra, la temperatura de operación, el flujo que
se requiere para operar la bomba y el tipo de detector de especies en una muestra. Existen varios tipos
de detectores como son el detector de índice de refracción (DIR), el detector de longitud de ondas múltiple
(DLM), el detector de fluorescencia, entre otros. Para nuestro proyecto se usarán los dos primeros, el DIR
y el DLM. En este estudio se usará el siguiente método de operación del HPLC 1100, Agilent:
Columna: ZORBAX, Análisis de Carbohidratos, 4.6 mm x 150 mm (5µm)
Fase Móvil: 75/25 de Acetonitrilo/Agua de grado HPLC
Velocidad de Flujo: 0.8 mL/min
Temperatura: 30 0C
Detectores: HP1100 DIR, DLM (230 nm de longitud de onda)
Volumen de Muestra Inyectada: 20 µL
Componentes del HPLC
Las principales componentes del cromatógrafo de alto desempeño se muestran en la figura 28. El HPLC
empleado es un cromatógrafo de alta presión. La función de la bomba (1) es empujar el líquido (la fase
móvil o solventes) por el cromatógrafo a una velocidad de flujo específico, que se expresa en mililitros por
minuto (mL/min). El flujo normalmente es de 1 a 2 mL/min y la presión de la bomba puede ser de 400 a

32
600 bar. En un experimento de cromatografía, la bomba puede suministrar una composición constante
de la fase móvil (flujo isocrático), o una composición de la fase móvil creciente (flujo de gradiente). El
inyector (2) sirve para introducir la muestra líquida dentro del flujo de corriente de la fase móvil y los
volúmenes de muestra que se inyectan son de 5 a 20 microlitros (µL) y el inyector debe de poder resistir
la alta presión del líquido del sistema. Una automuestra es una versión automática para cuando se tienen
varias muestras para analizar o cuando la inyección manual no es muy práctica. La columna (3) se
considera el “corazón del cromatógrafo”; la fase estacionaria de la columna separa las componentes de
interés en la muestra, usando varios parámetros físicos y químicos. Las pequeñas partículas dentro de la
columna son las que causan la despresurización en un flujo normal. La bomba debe empujar fuerte para
mover la fase móvil por la columna, lo cual causa la alta presión en el cromatógrafo. El detector (4) puede
detectar las moléculas que van saliendo de la columna, estos sirven para medir la cantidad de los
componentes de una muestra y genera los datos que llegan a la computadora, la cual genera los
cromatogramas relativos a los componentes en una muestra.
El HPLC se usa óptimamente en la separación de compuestos químicos y biológicos que son no-volátiles
como son:
• Sales como cloruro de sodio; fosfato de potasio
• Productos naturales como extractos de plantas, hierbas medicinales, etc.
• Compuestos orgánicos como polímeros, polietileno, etc.
• Farmacéutico como tylenol, aspirina, etc.
Figura 28. Esquema de un HPLC. Modificado de Agilent, 2015.

33
Cromatogramas
Los cromatogramas en la Figura 29 muestran los picos de los frentes, que son los solventes de la fase móvil
que arrastra los compuestos de análisis a través del HPLC. La identificación de un compuesto individual se
efectúa con el parámetro más común, el tiempo de retención en minutos que corresponde al tiempo
después de la inyección de una muestra. Dependiendo del detector que se use, la identificación de un
compuesto se basa por su estructura química, peso molecular o algún otro parámetro molecular. En un
análisis cuantitativo la cantidad de un compuesto se determina por su concentración; en un cromatograma
se determina la altura de los picos medidos desde la línea base y se determina el área de los picos en cada
cromatograma. Para hacer un análisis cuantitativo de un compuesto determinado, se inyecta el
compuesto de interés con una composición conocida y se mide la altura del pico o el área del pico. En este
estudio se usará el área de los picos.
Para analizar residuos o trazas de compuestos que son difíciles de separar o de detectar, se requiere una
alta resolución de separaciones y detectores muy sensibles. Estos compuestos por lo general se presentan
en un cromatograma con picos muy pequeños y/o área de los picos muy pequeñas, con concentraciones
en peso del 1% o en partes por millón (ppm) o en concentraciones más bajas.
Figura 29. Cromatogramas con detectores: a) DIR y b) DLM.
34
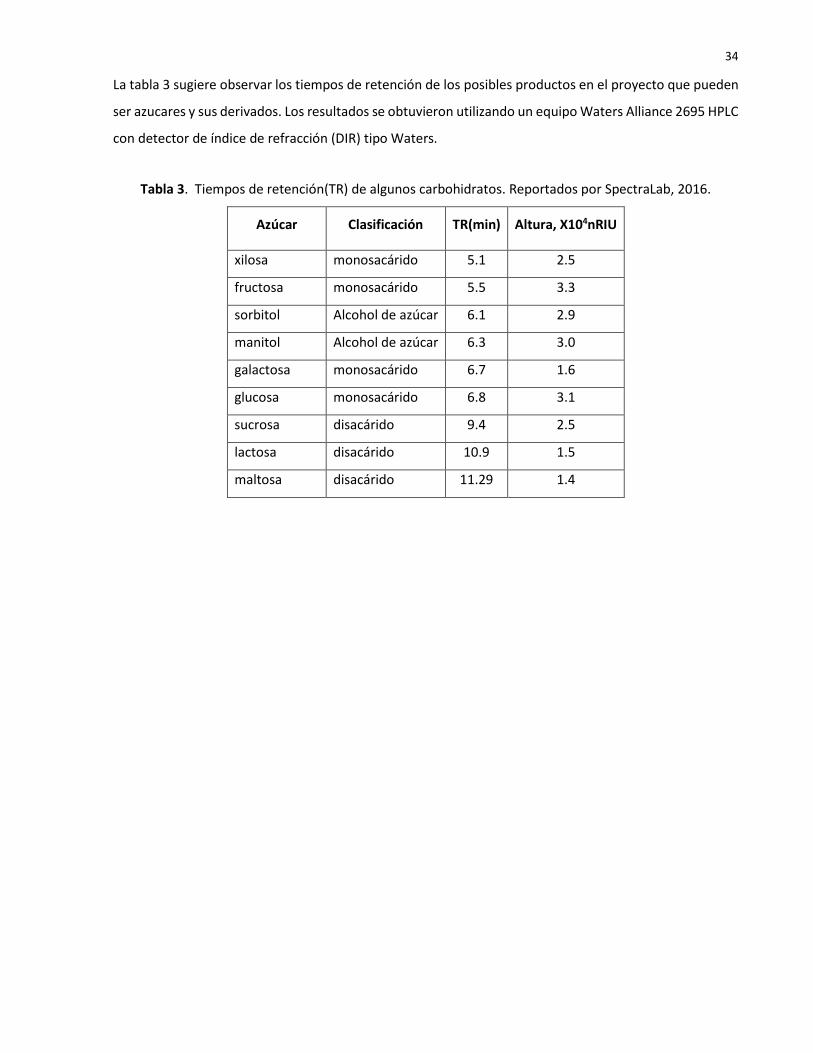
La tabla 3 sugiere observar los tiempos de retención de los posibles productos en el proyecto que pueden
ser azucares y sus derivados. Los resultados se obtuvieron utilizando un equipo Waters Alliance 2695 HPLC
con detector de índice de refracción (DIR) tipo Waters.
Tabla 3. Tiempos de retención(TR) de algunos carbohidratos. Reportados por SpectraLab, 2016.
Azúcar Clasificación TR(min) Altura, X104nRIU
xilosa monosacárido 5.1 2.5
fructosa monosacárido 5.5 3.3
sorbitol Alcohol de azúcar 6.1 2.9
manitol Alcohol de azúcar 6.3 3.0
galactosa monosacárido 6.7 1.6
glucosa monosacárido 6.8 3.1
sucrosa disacárido 9.4 2.5
lactosa disacárido 10.9 1.5
maltosa disacárido 11.29 1.4

35
Capítulo 4. Resultados y Discusiones
4.1. Características estructurales y morfológicas
4.1.1. Difracción de rayos X
Los patrones de difracción de las muestras analizadas (Figura 30) son típicos de zeolitas tipo mordenita,
según la tarjeta ICSD 68445 reportada para la mordenita sódica (Na-MOR) y para algunas muestras de
Metal-MOR. La Figura 30 muestra la comparación entre los patrones de difracción de los nanocompuestos
y el reportado para Na-MOR, obtenidos en el difractómetro Phillips X’s Pert con radiación CuKα. Todos los
nanocompuestos exhiben los picos característicos de una estructura cristalina tipo mordenita típica de las
zeolitas naturales.
10 15 20 25 30
ICSD 68445 202
350 511150
241330
200111
021 310
221 131 420
2 teta grados
Inte
nsi
dad
10Ag/20CdS-MOR
30Au/20CdS-MOR
30Ag-MOR
30Au-MOR
20CdS-MOR
Figura 30. Comparativo de los patrones de difracción de los nanocompuestos con el patrón teórico de la mordenita.

36
La estructura cristalina de los nancompuestos sintetizados (Jaime-Acuna at al 2016) corresponde en su
mayoría a los picos bien definidos de la mordenita sódica, esto significa que presentan modificaciones,
pero mantiene la estructura básica de la mordenita como lo muestran los difractogramas de rayos X de
la Figura 30. La aparente amorfización de la estructura cristalina de los fotocatalizadores es debido a las
incrustaciones de metales y/o semiconductores incrustados en la superficie y/o en los poros y/o canales
de la estructura misma de la mordenita.
4.1.2. Microscopia electrónica de transmisión y de barrido
La microscopia electrónica de transmisión (MET) proporciona diferentes imágenes de algunos de los
nanocompuestos utilizados en este estudio. Las imágenes (Figura 31a y b) nos muestran que las
nanopartículas metálicas de Au, Ag y el semiconductor CdS están bien soportadas por la matriz zeolítica.
La Figura 31b nos muestra la característica morfológica de la mordenita, el empaquetamiento de cristalitos
en forma de agujas.
Figura 31. Imágenes representativas de MET de las muestras a) 30Au/20CdS-MOR, b) 10Ag/20CdS-MOR y c) 30Au-MOR.
Las imágenes MEB de la Figura 32 son representativas de los polvos de los diferentes nanocompuestos a
utilizar como fotocatalizadores en esta investigación. En general las imágenes de los polvos nos muestran
la microestructura característica donde los granos están constituidos por empaquetamientos de cristalitos
en forma de agujas en cuya superficie crecen las nanopartículas metálicas y/o semiconductoras.

37
Figura 32. Imágenes representativas de MEB de polvos de los nanocompuestos.
4.2. Características fisicoquímicas y absorción óptica
4.2.1. Análisis químico
El análisis químico para los nanocompuestos 30Ag-MOR, 10Ag/20CdS-MOR, se efectuó usando el MEB
(SEM, JEOL- JIB-4500) con el cual se hace análisis químico de EDS. La tabla 4 resume los porcentajes en
pesos y porcentajes atomicos. La diferencia de la relacion de Si/Al por lo reportado y lo obtenido es de
una unida menor, Jaime-Acuña, 2016 reportó una oscilacion de 7 y 9 para muestras de Na-MOR y Ag-MOR
y el presente analisis presenta un relacion de Si/Al de 6 y 8 para los nanocompuestos analizados. Estos
valores indican que los nanocompuestos tienen varios sitios acidos para sitios de inserción de las
nanopartículas; las partículas ionicas en los sitios acidos determinan el tamaño de partiícula. Por otra
parte, la disminucion en sitios acidos se debe a la contaminacion de Ca.

38
Tabla 4. Análisis químico de los nanocompuestos tipo mordenita obtenido por EDS
4.2.2. Análisis químico de superficie
El espectrograma general del nanocompuesto de plata (Figura 33), se obtuvo con una radiación de Al Kα:
hν = 1486.6 eV, en el equipo XPS ESPEC de alta resolución. Se realizó el análisis cualitativo de 1 nm de
espesor (tres monocapas) aproximadamente. Los datos de las energías de enlace para cada elemento en
la superficie del fotocatalizador se obtuvieron del manual del equipo utilizado y del NIST Standard
Reference Database 20. Los datos del espectro general concuerdan con los reportados en la literatura. La
composición de la mordenita sintética incluye Al, Si, H y O. El espectro nos muestra el contenido de Ag en
la superficie y también nos muestra que la plata se encuentra en la superficie en estado metálico.

39
1400 1200 1000 800 600 400 200 0
0
10000
20000
Ag MNN
Si 2s
Ag 3p3/2
Ag 3d3/2C
PS
Energia de Enlace
O 1s
Ag 3d3/2
Si 2p1/2
Al 2p3/2
C 1s
Figura 33. Espectro general de XPS del nanocompuesto 30Ag-MOR.
4.2.3 Análisis de adsorción física
Isotermas de adsorción de N2
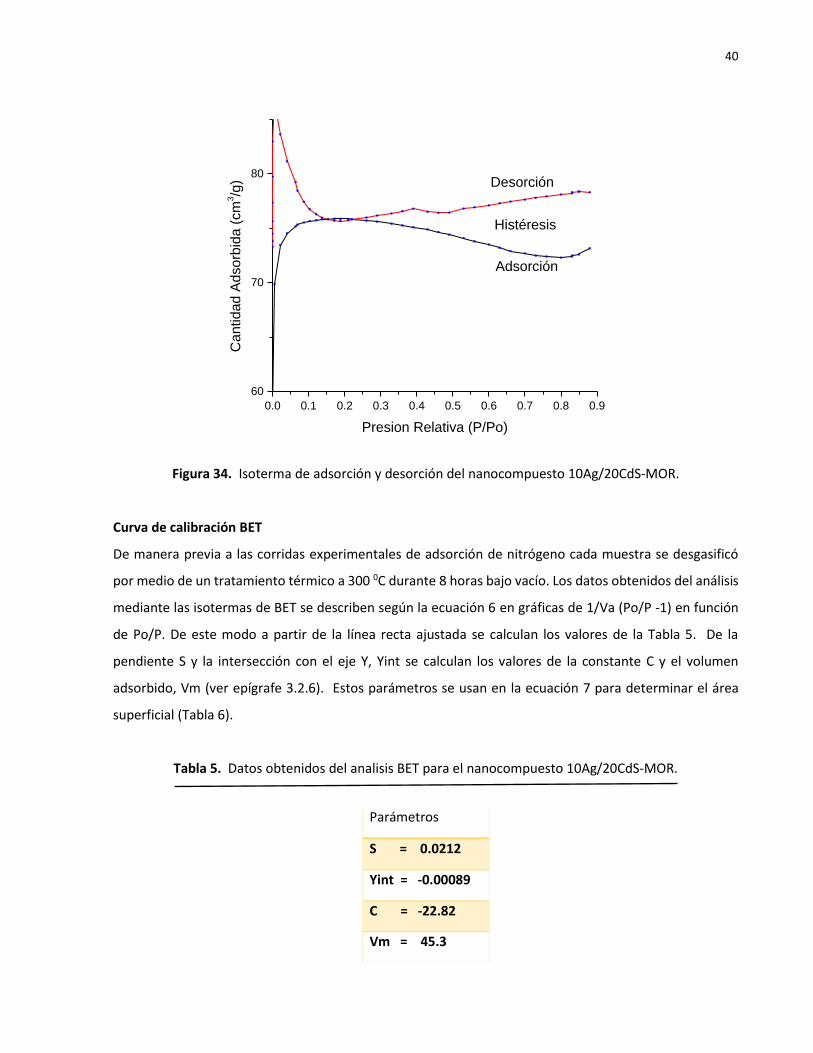
La isoterma de la Figura 34 correspondiente al nanocompuesto 10Ag/20CdS-MOR muestra algunas
anomalías. La histéresis no se cerró en su extremo derecho y la abertura puede significar que la muestra
no se desgasificó completamente. La Figura 34 indica que el nanocompuesto posee una alta adsorción a
presiones relativamente bajas de 0.1 y decae a un mínimo a presiones relativas de 0.8. A partir de 0.8 la
isoterma de adsorción crece sin llegar a cerrar con la curva de desorción.
40
0.0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9
60
70
80
Ca
ntid
ad
Adso
rbid
a (
cm
3/g
)
Presion Relativa (P/Po)
Desorción
Adsorción
Histéresis
Figura 34. Isoterma de adsorción y desorción del nanocompuesto 10Ag/20CdS-MOR.
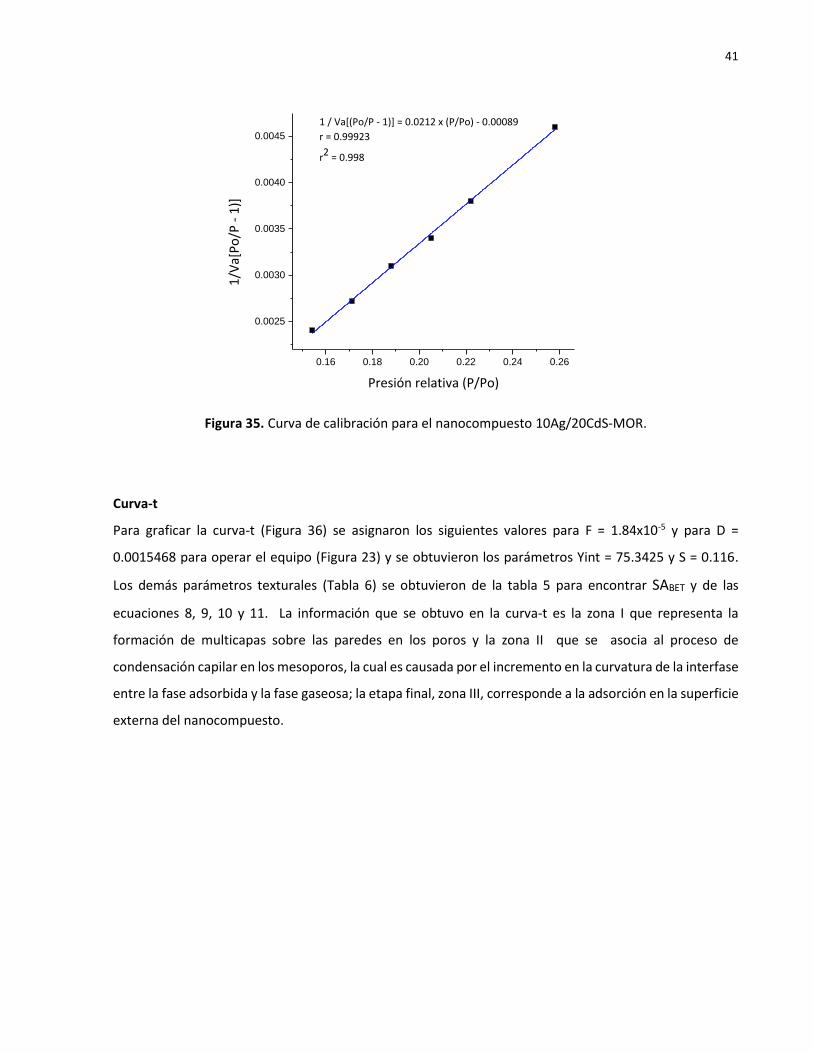
Curva de calibración BET
De manera previa a las corridas experimentales de adsorción de nitrógeno cada muestra se desgasificó
por medio de un tratamiento térmico a 300 0C durante 8 horas bajo vacío. Los datos obtenidos del análisis
mediante las isotermas de BET se describen según la ecuación 6 en gráficas de 1/Va (Po/P -1) en función
de Po/P. De este modo a partir de la línea recta ajustada se calculan los valores de la Tabla 5. De la
pendiente S y la intersección con el eje Y, Yint se calculan los valores de la constante C y el volumen
adsorbido, Vm (ver epígrafe 3.2.6). Estos parámetros se usan en la ecuación 7 para determinar el área
superficial (Tabla 6).
Tabla 5. Datos obtenidos del analisis BET para el nanocompuesto 10Ag/20CdS-MOR.
Parámetros
S = 0.0212
Yint = -0.00089
C = -22.82
Vm = 45.3
41
Figura 35. Curva de calibración para el nanocompuesto 10Ag/20CdS-MOR.
Curva-t
Para graficar la curva-t (Figura 36) se asignaron los siguientes valores para F = 1.84x10-5 y para D =
0.0015468 para operar el equipo (Figura 23) y se obtuvieron los parámetros Yint = 75.3425 y S = 0.116.
Los demás parámetros texturales (Tabla 6) se obtuvieron de la tabla 5 para encontrar SABET y de las
ecuaciones 8, 9, 10 y 11. La información que se obtuvo en la curva-t es la zona I que representa la
formación de multicapas sobre las paredes en los poros y la zona II que se asocia al proceso de
condensación capilar en los mesoporos, la cual es causada por el incremento en la curvatura de la interfase
entre la fase adsorbida y la fase gaseosa; la etapa final, zona III, corresponde a la adsorción en la superficie
externa del nanocompuesto.
0.16 0.18 0.20 0.22 0.24 0.26
0.0025
0.0030
0.0035
0.0040
0.0045
1/V
a[P
o/P
- 1
)]
Presión relativa (P/Po)
1 / Va[(Po/P - 1)] = 0.0212 x (P/Po) - 0.00089
r = 0.99923
r2 = 0.998

42
3.8 4.0 4.2 4.4
75.75
75.80
75.85
III
Ad
sorc
ion
(cm
3/g
STP
)
Espesor (t (A))
I
II
Figura 36. Zonas de la curva-t para el nanocompuesto 10Ag/20CdS-MOR.
Tabla 6. Parámetros texturales para el nanocompuesto 10Ag/20CdS-MOR.
Comparando el área superficial BET (SABET) del nanocompuesto (Tabla 7) y las zeolitas naturales
tipo mordenita, notamos que hay una gran diferencia con respecto al área superficial de
adsorción. Por ejemplo, las zeolitas de Acatlán, Mx., presentan una SABET = 30.8 m2/g y la zeolita
de Zapopan, Jalisco, Mx., muestran una SABET = 31.8 m2/g (Hernández et al., 2010).
4.2.4. Espectroscopia de absorción UV-Visible
Las soluciones fueron preparadas con 1.4 mg de cada nanocompuesto (30Ag-MOR, 30Au-MOR, 20CdS-
MOR, 30Au/20CdS-MOR, 10Ag/20CdS-MOR) por 3 mL de agua desionizada. Se vertieron 20 µL en la cubeta
Nanocompuesto SABET(m2g-1) SAEXT (m2g-1) SAµp (m2g-1) Vµp (cm3 g-1)
10Ag/20CdS-MOR 197.1 9.752 187.35 0.1165
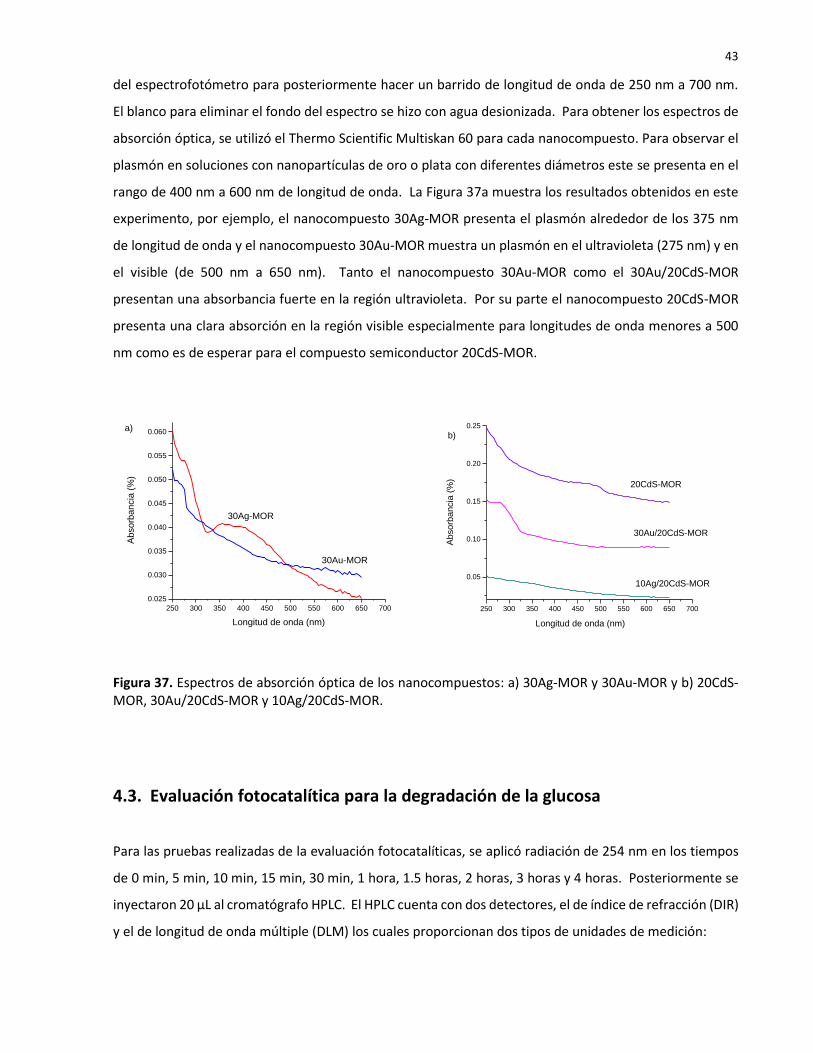
43
del espectrofotómetro para posteriormente hacer un barrido de longitud de onda de 250 nm a 700 nm.
El blanco para eliminar el fondo del espectro se hizo con agua desionizada. Para obtener los espectros de
absorción óptica, se utilizó el Thermo Scientific Multiskan 60 para cada nanocompuesto. Para observar el
plasmón en soluciones con nanopartículas de oro o plata con diferentes diámetros este se presenta en el
rango de 400 nm a 600 nm de longitud de onda. La Figura 37a muestra los resultados obtenidos en este
experimento, por ejemplo, el nanocompuesto 30Ag-MOR presenta el plasmón alrededor de los 375 nm
de longitud de onda y el nanocompuesto 30Au-MOR muestra un plasmón en el ultravioleta (275 nm) y en
el visible (de 500 nm a 650 nm). Tanto el nanocompuesto 30Au-MOR como el 30Au/20CdS-MOR
presentan una absorbancia fuerte en la región ultravioleta. Por su parte el nanocompuesto 20CdS-MOR
presenta una clara absorción en la región visible especialmente para longitudes de onda menores a 500
nm como es de esperar para el compuesto semiconductor 20CdS-MOR.
250 300 350 400 450 500 550 600 650 700
0.025
0.030
0.035
0.040
0.045
0.050
0.055
0.060
30Ag-MOR
Absorb
ancia
(%
)
Longitud de onda (nm)
30Au-MOR
a)
250 300 350 400 450 500 550 600 650 700
0.05
0.10
0.15
0.20
0.25
20CdS-MOR
30Au/20CdS-MOR
Absorb
ancia
(%
)
Longitud de onda (nm)
10Ag/20CdS-MOR
b)
Figura 37. Espectros de absorción óptica de los nanocompuestos: a) 30Ag-MOR y 30Au-MOR y b) 20CdS-MOR, 30Au/20CdS-MOR y 10Ag/20CdS-MOR.
4.3. Evaluación fotocatalítica para la degradación de la glucosa
Para las pruebas realizadas de la evaluación fotocatalíticas, se aplicó radiación de 254 nm en los tiempos
de 0 min, 5 min, 10 min, 15 min, 30 min, 1 hora, 1.5 horas, 2 horas, 3 horas y 4 horas. Posteriormente se
inyectaron 20 µL al cromatógrafo HPLC. El HPLC cuenta con dos detectores, el de índice de refracción (DIR)
y el de longitud de onda múltiple (DLM) los cuales proporcionan dos tipos de unidades de medición:

44
1) Para la señal del DIR:
a) la altura de los picos se expresa en nRIU del inglés nanorefractive index units.
b) el área bajo la curva se expresa en nRIU* s del inglés nRIU per second
2) Para la señal DLM:
1) la altura se expresa en mAU del inglés milli absorbance unit
2) el área se expresa en mAU*s del inglés milli absorbance unit per Second
4.3.1. Calibración de las soluciones estándares
Se prepararon tres soluciones estándares por cada compuesto de glucosa, de sorbitol y de ácido
glucurónico con concentraciones iniciales de 5 mg/mL, 2.5 mg/mL y 1.25 mg/mL (Colmenares et al., 2011).
Los resultados del HPLC se presentan en los cromatogramas para cada solución estándar, los cuales
proporcionarán los tiempos de retención y las áreas de cada pico. Se graficarán las curvas de calibración
estándares empleando las concentraciones anteriores en función de las áreas de los picos (señales) que
se obtenga por cada cromatograma. Los cromatogramas presentan un pico o varios picos al inicio de estos.
Estos picos representan “el frente” asociado a los solventes (agua y acetonitrilo) empleados como fase
móvil para mover la muestra a través de la columna de separación del HPLC.
4.3.1.1 Resultados para la glucosa
Los cromatogramas obtenidos (Figura 38) presentan las señales de los detectores DIR y DLM para cada
una de las soluciones estándares de la glucosa (1.5 mg/mL, 2.5 mg/mL y 5 mg/mL) sin fotocatalizador. El
pico para los tiempos de retención entre 2 y 3 minutos representa el frente. Los cromatogramas del
detector DIR (Figura 38a) sugieren que la glucosa tiene un tiempo de retención entre 6 y 6.5 minutos por
cada solución estándar. Los cromatogramas del detector DLM (Figura 38b) muestran que el tiempo de
retención de las soluciones estándares están entre 6 y 6.5 minutos coincidiendo con los resultados del
detector DIR; sin embargo, presentan una pequeña señal perceptible para la concentración de glucosa
más de 2.5 mg/mL. El reporte generado por los cromatogramas de la Figura 38a proporcionó las áreas
de la Tabla 7, las cuales se grafican en función de las concentraciones estándares (1.5 mg/mL, 2.5 mg/mL
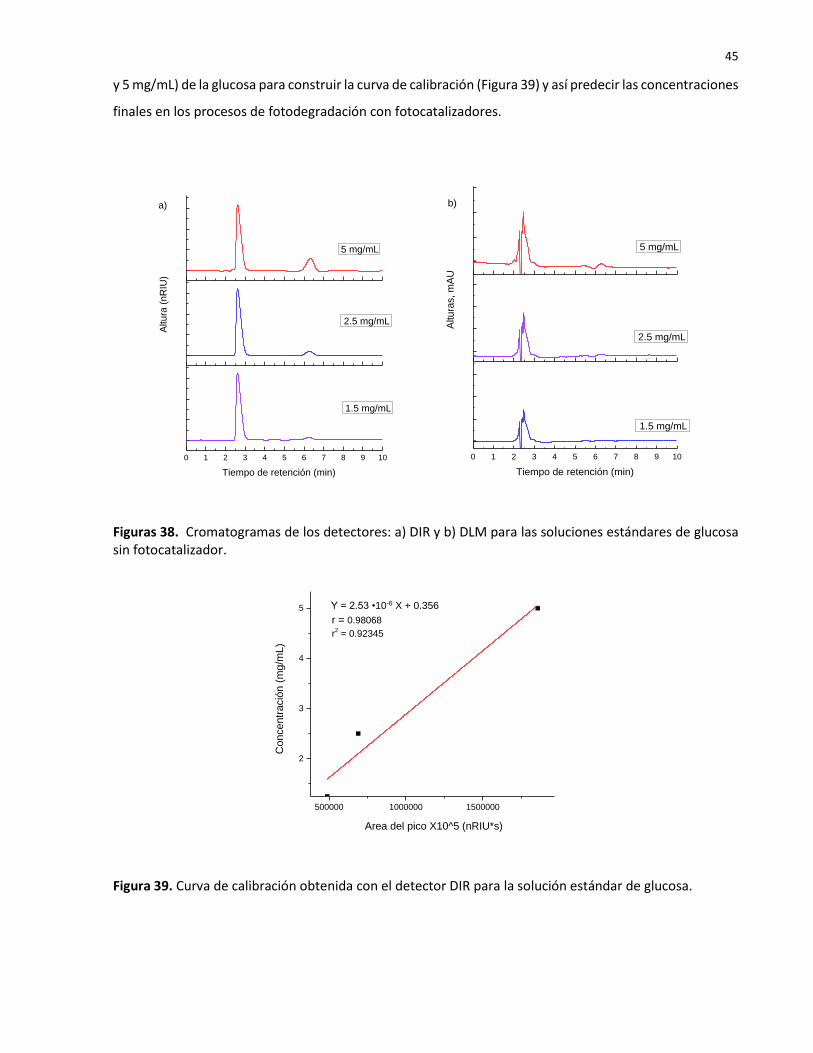
45
y 5 mg/mL) de la glucosa para construir la curva de calibración (Figura 39) y así predecir las concentraciones
finales en los procesos de fotodegradación con fotocatalizadores.
0 1 2 3 4 5 6 7 8 9 10
1.5 mg/mL
2.5 mg/mL
a)
Tiempo de retención (min)
Altu
ra (
nR
IU)
5 mg/mL
0 1 2 3 4 5 6 7 8 9 10
Tiempo de retención (min)
1.5 mg/mL
Altura
s, m
AU
2.5 mg/mL
5 mg/mL
b)
Figuras 38. Cromatogramas de los detectores: a) DIR y b) DLM para las soluciones estándares de glucosa sin fotocatalizador.
Figura 39. Curva de calibración obtenida con el detector DIR para la solución estándar de glucosa.
500000 1000000 1500000
2
3
4
5
Co
nce
ntr
ació
n (
mg
/mL
)
Area del pico X10^5 (nRIU*s)
Y = 2.53 •10-6 X + 0.356
r = 0.98068
r2 = 0.92345
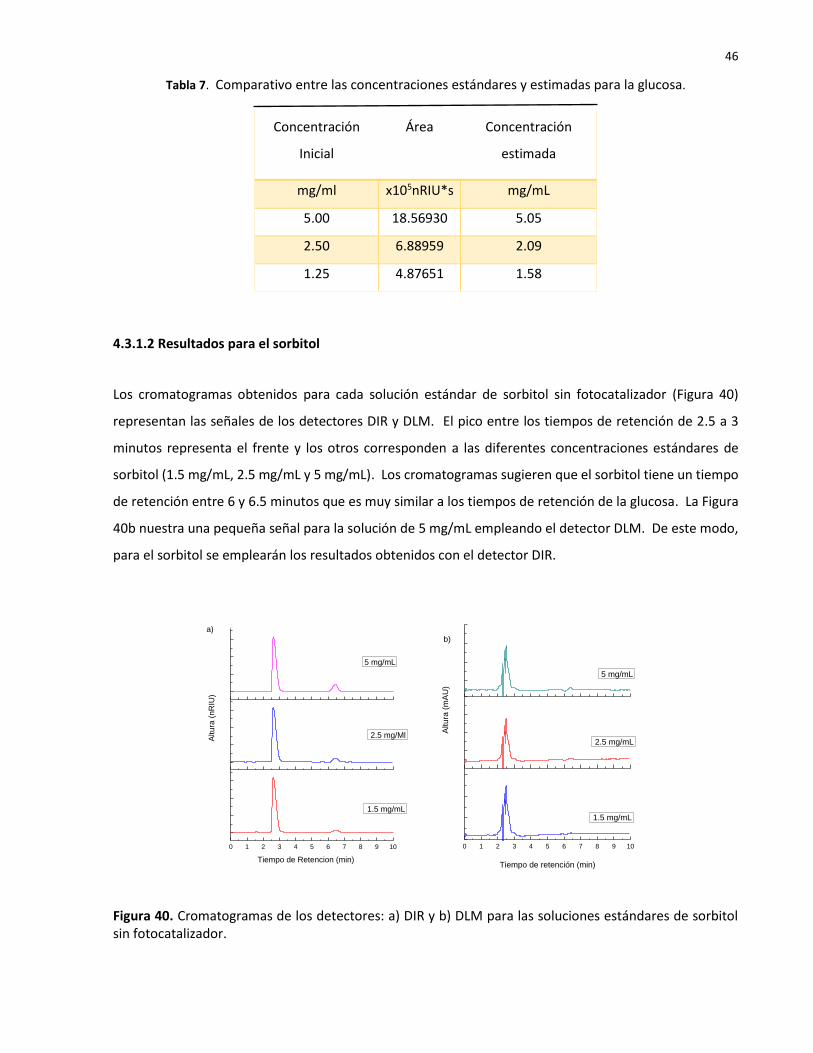
46
Tabla 7. Comparativo entre las concentraciones estándares y estimadas para la glucosa.
Concentración
Inicial
Área Concentración
estimada
mg/ml x105nRIU*s mg/mL
5.00 18.56930 5.05
2.50 6.88959 2.09
1.25 4.87651 1.58
4.3.1.2 Resultados para el sorbitol
Los cromatogramas obtenidos para cada solución estándar de sorbitol sin fotocatalizador (Figura 40)
representan las señales de los detectores DIR y DLM. El pico entre los tiempos de retención de 2.5 a 3
minutos representa el frente y los otros corresponden a las diferentes concentraciones estándares de
sorbitol (1.5 mg/mL, 2.5 mg/mL y 5 mg/mL). Los cromatogramas sugieren que el sorbitol tiene un tiempo
de retención entre 6 y 6.5 minutos que es muy similar a los tiempos de retención de la glucosa. La Figura
40b nuestra una pequeña señal para la solución de 5 mg/mL empleando el detector DLM. De este modo,
para el sorbitol se emplearán los resultados obtenidos con el detector DIR.
0 1 2 3 4 5 6 7 8 9 10
1.5 mg/mL
Tiempo de Retencion (min)
2.5 mg/MlAltu
ra (
nR
IU)
a)
5 mg/mL
0 1 2 3 4 5 6 7 8 9 10
Altu
ra (
mA
U)
Tiempo de retención (min)
1.5 mg/mL
b)
2.5 mg/mL
5 mg/mL
Figura 40. Cromatogramas de los detectores: a) DIR y b) DLM para las soluciones estándares de sorbitol sin fotocatalizador.

47
El reporte generado por los cromatogramas de la Figura 40a proporcionó las áreas de la Tabla 8, estas se
grafican en función de las concentraciones estándares (1.5 mg/mL, 2.5 mg/mL y 5 mg/mL) del sorbitol para
construir la curva de calibración (Figura 41) y así predecir las concentraciones finales.
Figura 41. Curva de calibración obtenida con el detector DIR para la solución estándar de sorbitol.
Tabla 8. Comparativo entre las concentraciones estándares y estimadas del sorbitol.
Concentración Inicial Área Concentración estimada
mg/ml X105nRIU*s mg/mL
5.00 11.58360 4.97
2.50 6.47782 2.6
1.25 4.01709 1.44
4.3.1.3 Resultados para el ácido glucurónico
Los cromatogramas obtenidos para cada solución estándar de ácido glucurónico sin fotocatalizador (Figura
42) son las señales de los detectores DIR y DLM. Los cromatogramas de la Figura 42a son del detector DIR,
donde el pico entre los tiempos de retención de 2.5 a 3 minutos representa el frente y se observa que el
400000 600000 800000 1000000 1200000
2
3
4
5C
once
ntr
ació
n (
mg
/mL
)
Area (nRIU*sec)
Y = 4.666 •10-6 X – 0.4344
r = 0.99906
r2 = 0. 99623

48
ácido glucurónico no fue detectado con el detector DIR. Los cromatogramas de la figura 42b representa
el detector DLM, donde se muestra el frente entre 2 y 3 minutos y también indica la señal del ácido
glucurónico entre 7 y 8 minutos. Para el ácido glucurónico se emplearán los resultados obtenidos con el
detector DLM.
0 1 2 3 4 5 6 7 8 9 10
Tiempo de retención (min)
1.5 mg/mL
Altu
ra (
mR
IU) 5 mg/mL
a)
0 1 2 3 4 5 6 7 8 9 10
0.0
1.2
2.4
3.6
0.0
1.4
2.8
4.2
0.0
1.4
2.8
4.2
Tiempo de retención (min)
1.5 mg/mL
2.5 mg/mL
Altura
(m
AU
)
5 mg/mL
b)
Figura 42. Cromatogramas de los detectores: a) DIR y b) DLM para las soluciones estándares de ácido glucurónico sin fotocatalizador.
Los resultados obtenidos en los cromatogramas de la Figura 42b (Tabla 9) se emplearán para construir la
curva de calibración (Figura 43) y para predecir las concentraciones estimadas (Tabla 9).
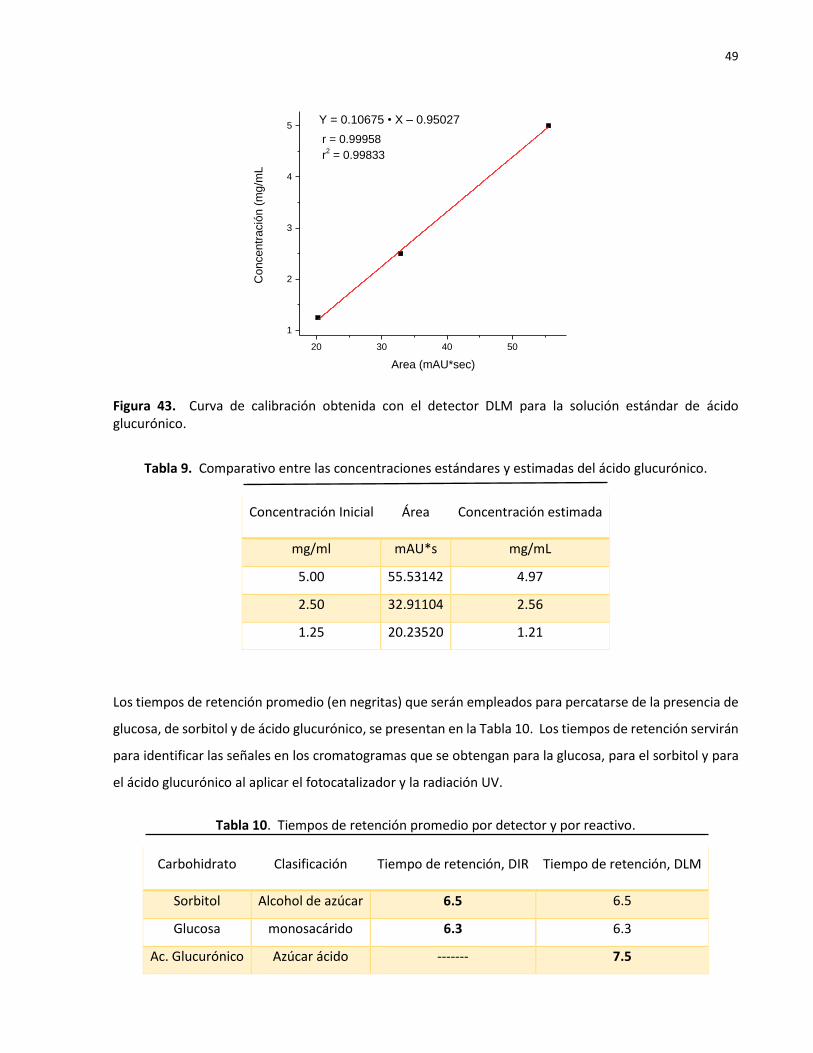
49
20 30 40 50
1
2
3
4
5
Concentr
ació
n (
mg/m
L
Area (mAU*sec)
Y = 0.10675 • X – 0.95027
r = 0.99958
r2 = 0.99833
Figura 43. Curva de calibración obtenida con el detector DLM para la solución estándar de ácido glucurónico.
Tabla 9. Comparativo entre las concentraciones estándares y estimadas del ácido glucurónico.
Concentración Inicial Área Concentración estimada
mg/ml mAU*s mg/mL
5.00 55.53142 4.97
2.50 32.91104 2.56
1.25 20.23520 1.21
Los tiempos de retención promedio (en negritas) que serán empleados para percatarse de la presencia de
glucosa, de sorbitol y de ácido glucurónico, se presentan en la Tabla 10. Los tiempos de retención servirán
para identificar las señales en los cromatogramas que se obtengan para la glucosa, para el sorbitol y para
el ácido glucurónico al aplicar el fotocatalizador y la radiación UV.
Tabla 10. Tiempos de retención promedio por detector y por reactivo.
Carbohidrato Clasificación Tiempo de retención, DIR Tiempo de retención, DLM
Sorbitol Alcohol de azúcar 6.5 6.5
Glucosa monosacárido 6.3 6.3
Ac. Glucurónico Azúcar ácido ------- 7.5

50
4.3.2. Resultados preliminares de la fotoreacción durante 4 horas Las primeras pruebas se hicieron de cuatro horas y se usaron los fotocatalizadores 30Ag-MOR, 30Au-MOR,
20CdS-MOR, 30Au/20CdS-MOR, 10Ag/20CdS-MOR. Se preparó la solución de glucosa (Colmenares et al.,
2011) agregando 2 g de glucosa y aforando con 100 mL de agua desionizada obteniendo una concentración
inicial de 20 mg/mL y se tomaron 30 mL de esta solución para posteriormente agregar 0.02 gramos de
fotocatalizador. Se aplicó radiación UV de 254 nm de longitud de onda en el fotoreactor. En cada hora se
tomaron 20 µL del fotoreactor los cuales fueron inyectados en la columna del HPLC para obtener los
cromatogramas. En cada ocasión se tomó el pH y la temperatura. El HPLC se calibró para el detector DIR
y el detector DLM (con longitud de onda de 230 nm) y se corrió el HPLC con un tiempo de retención de 10
minutos.
La conversión de glucosa se determina como sigue:
% Conversión = 100 × [Ci – Cf]/ Ci (12)
donde
Ci = concentración inicio para el tiempo cero.
Cf = concentración final para un tiempo t.
Resultados para el fotocatalizador 30Ag-MOR
Los cromatogramas para el detector DIR (Figura 44a) indican los frentes de la fase móvil (la solución de
agua y acetonitrilo) entre 2.5 y 3 minutos y las señales de la glucosa usando el fotocatalizador 30Ag-MOR
por cada hora bajo la radiación UV en el fotoreactor. Estos cromatogramas proporcionaron los tiempos
de retención y las áreas de cada pico (Tabla 11). Los cromatogramas para el detector DLM (Figura 44b)
presentan las señales de un producto nuevo entre 5 y 5.5 minutos, pero diferente a los buscados en este
proyecto (el sorbitol y el ácido glucurónico). Las otras señales corresponden a la glucosa y a una posible
traza de ácido glucurónico.
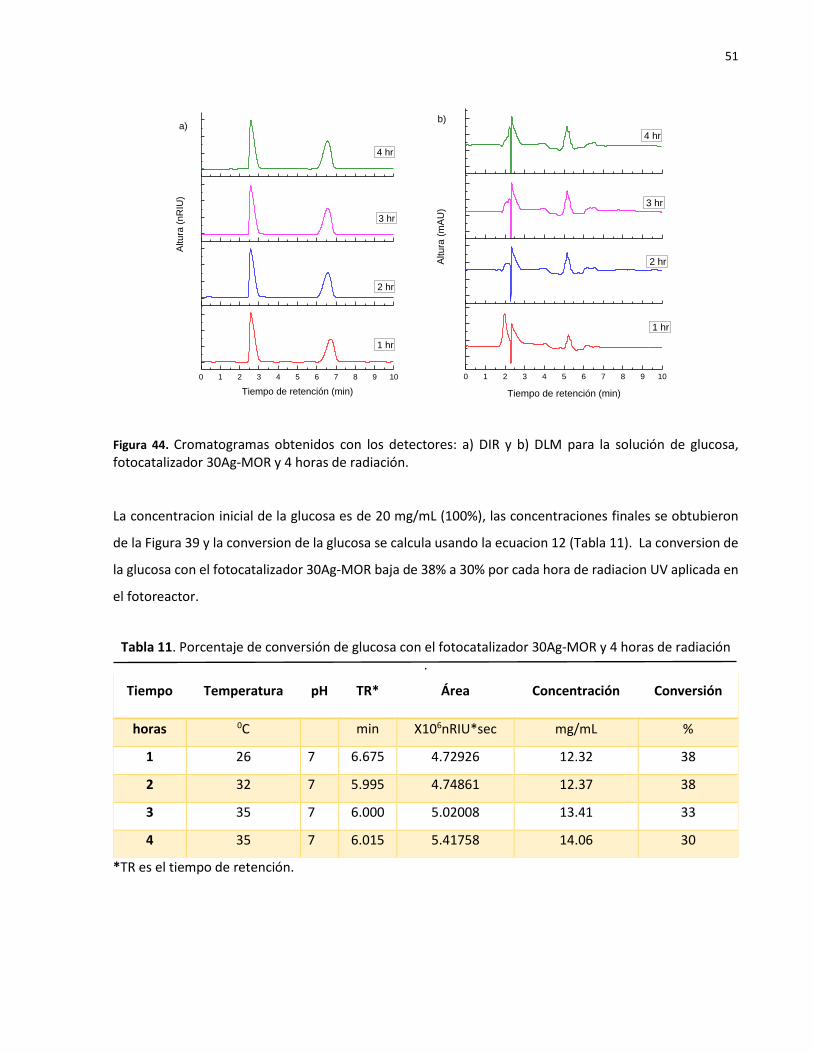
51
0 1 2 3 4 5 6 7 8 9 10
Altu
ra (
nR
IU)
Tiempo de retención (min)
1 hr
2 hr
3 hr
4 hr
a)
0 1 2 3 4 5 6 7 8 9 10
Altura
(m
AU
)
Tiempo de retención (min)
1 hr
b)
2 hr
3 hr
4 hr
Figura 44. Cromatogramas obtenidos con los detectores: a) DIR y b) DLM para la solución de glucosa, fotocatalizador 30Ag-MOR y 4 horas de radiación. La concentracion inicial de la glucosa es de 20 mg/mL (100%), las concentraciones finales se obtubieron
de la Figura 39 y la conversion de la glucosa se calcula usando la ecuacion 12 (Tabla 11). La conversion de
la glucosa con el fotocatalizador 30Ag-MOR baja de 38% a 30% por cada hora de radiacion UV aplicada en
el fotoreactor.
Tabla 11. Porcentaje de conversión de glucosa con el fotocatalizador 30Ag-MOR y 4 horas de radiación .
Tiempo Temperatura pH TR* Área Concentración Conversión
horas 0C min X106nRIU*sec mg/mL %
1 26 7 6.675 4.72926 12.32 38
2 32 7 5.995 4.74861 12.37 38
3 35 7 6.000 5.02008 13.41 33
4 35 7 6.015 5.41758 14.06 30
*TR es el tiempo de retención.
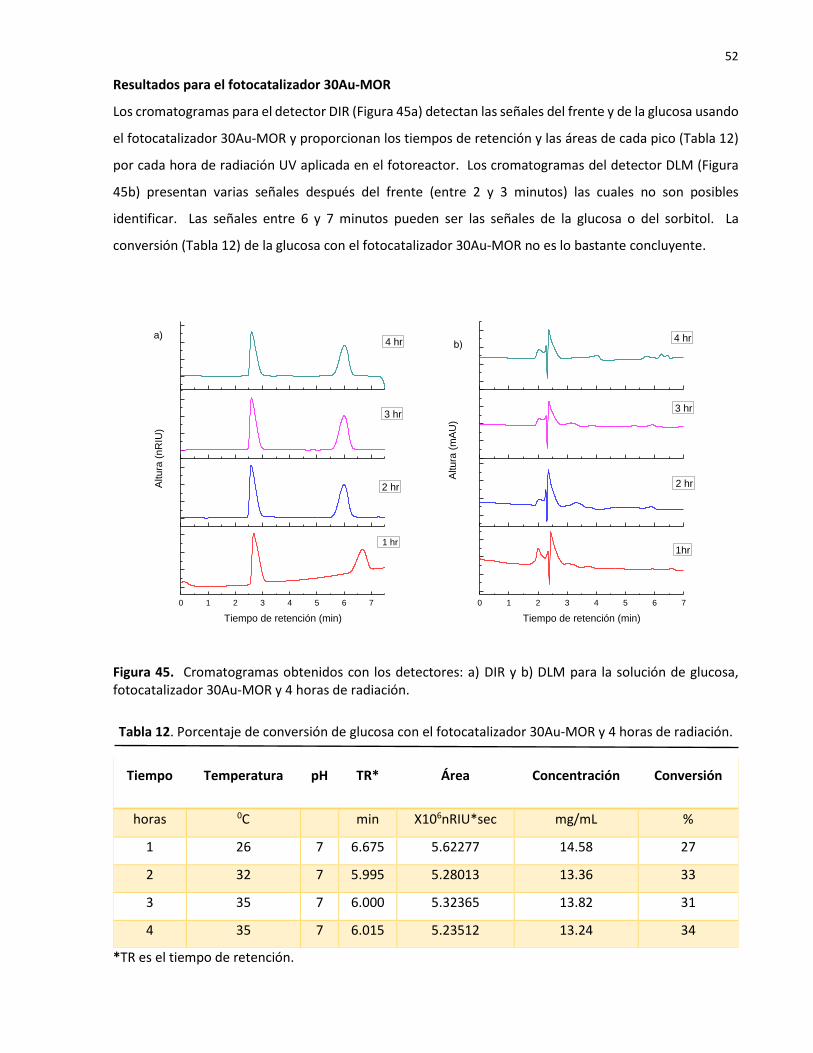
52
Resultados para el fotocatalizador 30Au-MOR
Los cromatogramas para el detector DIR (Figura 45a) detectan las señales del frente y de la glucosa usando
el fotocatalizador 30Au-MOR y proporcionan los tiempos de retención y las áreas de cada pico (Tabla 12)
por cada hora de radiación UV aplicada en el fotoreactor. Los cromatogramas del detector DLM (Figura
45b) presentan varias señales después del frente (entre 2 y 3 minutos) las cuales no son posibles
identificar. Las señales entre 6 y 7 minutos pueden ser las señales de la glucosa o del sorbitol. La
conversión (Tabla 12) de la glucosa con el fotocatalizador 30Au-MOR no es lo bastante concluyente.
0 1 2 3 4 5 6 7
Tiempo de retención (min)
1 hr
Altu
ra (
nR
IU)
2 hr
3 hr
4 hra)
0 1 2 3 4 5 6 7
Altu
ra (
mA
U)
Tiempo de retención (min)
1hr
b)
2 hr
3 hr
4 hr
Figura 45. Cromatogramas obtenidos con los detectores: a) DIR y b) DLM para la solución de glucosa, fotocatalizador 30Au-MOR y 4 horas de radiación.
Tabla 12. Porcentaje de conversión de glucosa con el fotocatalizador 30Au-MOR y 4 horas de radiación.
Tiempo Temperatura pH TR* Área Concentración Conversión
horas 0C min X106nRIU*sec mg/mL %
1 26 7 6.675 5.62277 14.58 27
2 32 7 5.995 5.28013 13.36 33
3 35 7 6.000 5.32365 13.82 31
4 35 7 6.015 5.23512 13.24 34
*TR es el tiempo de retención.

53
Resultados para el fotocatalizador 20CdS-MOR
Los cromatogramas de la Figura 46a con el detector DIR muestran los picos del frente y de la glucosa con
los cuales se obtuvieron los tiempos de retención y las áreas de cada pico (Tabla 13). Los cromatogramas
de la Figura 46b obtenidas con el detector DLM presentan una traza de un producto nuevo entre 5 y 5.5
minutos similar al observado en la Figura 44b. Las señales entre 6 y 7 minutos (Figura 46b) pueden ser
asociadas a la glucosa y/o al sorbitol. La conversión (Tabla 13) de la glucosa con el fotocatalizador 20CdS-
MOR baja de 35 % y 33%.
0 1 2 3 4 5 6 7 8 9 10
Altura
(nR
IU)
Tiempo de retención (min)
1 hr
2 hr
3 hr
4 hr
a)
0 1 2 3 4 5 6 7 8 9 10
Altu
ra (
mA
U)
Tiempo de retención (min)
1 hr
b)
2 hr
3 hr
4 hr
Figura 46. Cromatogramas obtenidos con los detectores: a) DIR y b) DLM para la solución de glucosa, fotocatalizador 20CdS-MOR y 4 horas de radiación.
Tabla 13. Porcentaje de conversión de glucosa con el fotocatalizador 20CdS-MOR y 4 horas de radiación.
Tiempo Temperatura pH TR* Área Concentración Conversión
horas 0C min X106nRIU*sec mg/mL %
1 26 7 6.728 4.97055 12.93 35
2 32 7 6.574 4.94162 12.86 36
3 35 7 6.530 5.09100 13.24 34
4 35 7 6.516 5.18470 13.47 33
*TR es el tiempo de retención.
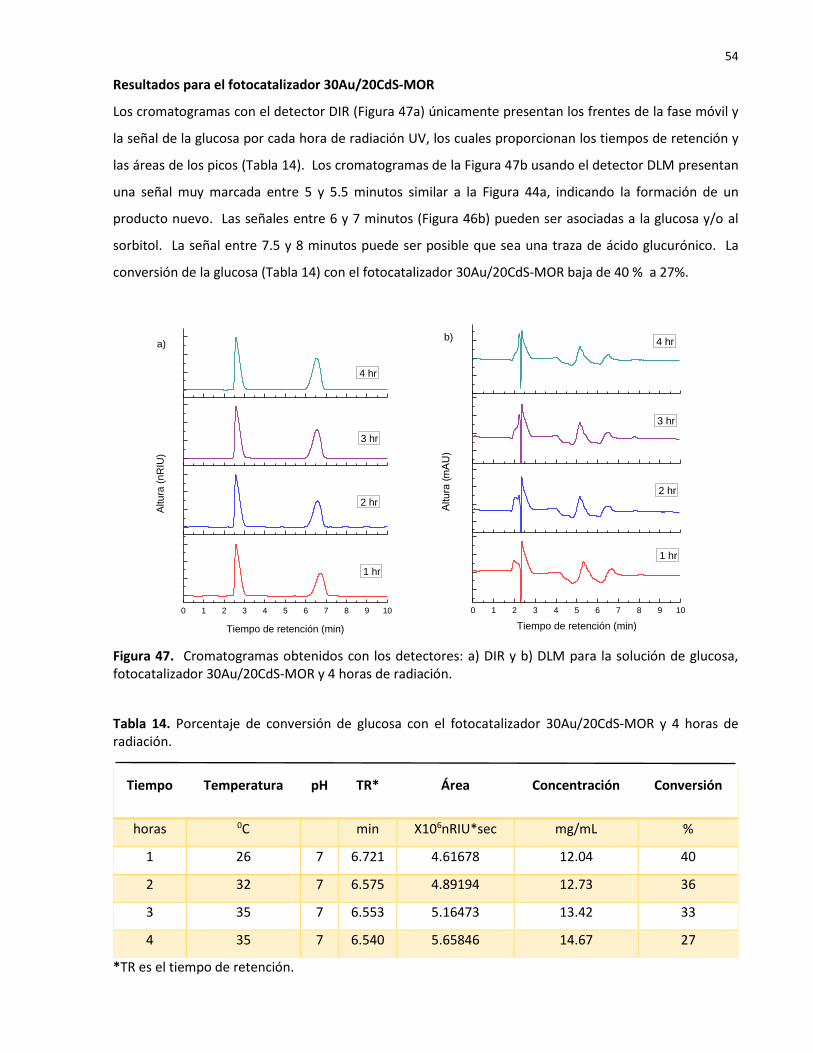
54
Resultados para el fotocatalizador 30Au/20CdS-MOR
Los cromatogramas con el detector DIR (Figura 47a) únicamente presentan los frentes de la fase móvil y
la señal de la glucosa por cada hora de radiación UV, los cuales proporcionan los tiempos de retención y
las áreas de los picos (Tabla 14). Los cromatogramas de la Figura 47b usando el detector DLM presentan
una señal muy marcada entre 5 y 5.5 minutos similar a la Figura 44a, indicando la formación de un
producto nuevo. Las señales entre 6 y 7 minutos (Figura 46b) pueden ser asociadas a la glucosa y/o al
sorbitol. La señal entre 7.5 y 8 minutos puede ser posible que sea una traza de ácido glucurónico. La
conversión de la glucosa (Tabla 14) con el fotocatalizador 30Au/20CdS-MOR baja de 40 % a 27%.
0 1 2 3 4 5 6 7 8 9 10
Altu
ra (
nR
IU)
Tiempo de retención (min)
1 hr
a)
2 hr
3 hr
4 hr
0 1 2 3 4 5 6 7 8 9 10
Altura
(m
AU
)
Tiempo de retención (min)
1 hr
2 hr
3 hr
4 hrb)
Figura 47. Cromatogramas obtenidos con los detectores: a) DIR y b) DLM para la solución de glucosa, fotocatalizador 30Au/20CdS-MOR y 4 horas de radiación.
Tabla 14. Porcentaje de conversión de glucosa con el fotocatalizador 30Au/20CdS-MOR y 4 horas de radiación.
Tiempo Temperatura pH TR* Área Concentración Conversión
horas 0C min X106nRIU*sec mg/mL %
1 26 7 6.721 4.61678 12.04 40
2 32 7 6.575 4.89194 12.73 36
3 35 7 6.553 5.16473 13.42 33
4 35 7 6.540 5.65846 14.67 27
*TR es el tiempo de retención.
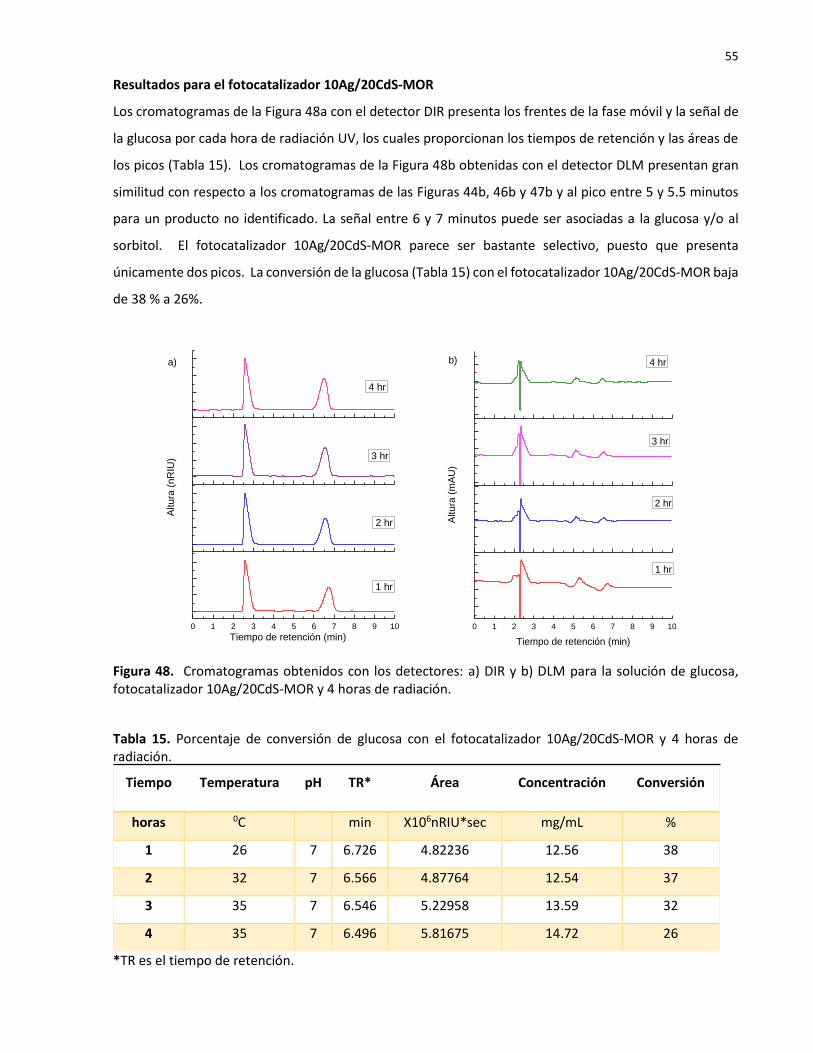
55
Resultados para el fotocatalizador 10Ag/20CdS-MOR
Los cromatogramas de la Figura 48a con el detector DIR presenta los frentes de la fase móvil y la señal de
la glucosa por cada hora de radiación UV, los cuales proporcionan los tiempos de retención y las áreas de
los picos (Tabla 15). Los cromatogramas de la Figura 48b obtenidas con el detector DLM presentan gran
similitud con respecto a los cromatogramas de las Figuras 44b, 46b y 47b y al pico entre 5 y 5.5 minutos
para un producto no identificado. La señal entre 6 y 7 minutos puede ser asociadas a la glucosa y/o al
sorbitol. El fotocatalizador 10Ag/20CdS-MOR parece ser bastante selectivo, puesto que presenta
únicamente dos picos. La conversión de la glucosa (Tabla 15) con el fotocatalizador 10Ag/20CdS-MOR baja
de 38 % a 26%.
0 1 2 3 4 5 6 7 8 9 10
Altu
ra (
nR
IU)
Tiempo de retención (min)
1 hr
a)
2 hr
3 hr
4 hr
0 1 2 3 4 5 6 7 8 9 10
Altura
(m
AU
)
Tiempo de retención (min)
1 hr
b)
2 hr
3 hr
4 hr
Figura 48. Cromatogramas obtenidos con los detectores: a) DIR y b) DLM para la solución de glucosa, fotocatalizador 10Ag/20CdS-MOR y 4 horas de radiación.
Tabla 15. Porcentaje de conversión de glucosa con el fotocatalizador 10Ag/20CdS-MOR y 4 horas de radiación.
Tiempo Temperatura pH TR* Área Concentración Conversión
horas 0C min X106nRIU*sec mg/mL %
1 26 7 6.726 4.82236 12.56 38
2 32 7 6.566 4.87764 12.54 37
3 35 7 6.546 5.22958 13.59 32
4 35 7 6.496 5.81675 14.72 26
*TR es el tiempo de retención.
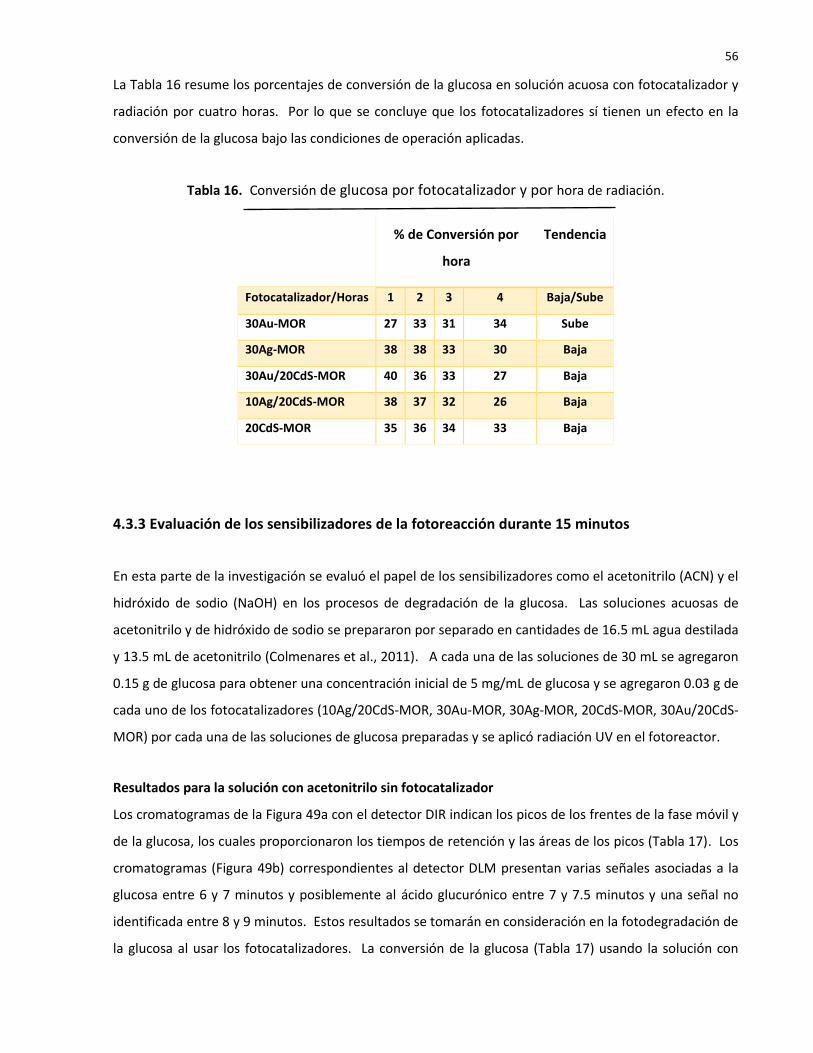
56
La Tabla 16 resume los porcentajes de conversión de la glucosa en solución acuosa con fotocatalizador y
radiación por cuatro horas. Por lo que se concluye que los fotocatalizadores sí tienen un efecto en la
conversión de la glucosa bajo las condiciones de operación aplicadas.
Tabla 16. Conversión de glucosa por fotocatalizador y por hora de radiación.
% de Conversión por
hora
Tendencia
Fotocatalizador/Horas 1 2 3 4 Baja/Sube
30Au-MOR 27 33 31 34 Sube
30Ag-MOR 38 38 33 30 Baja
30Au/20CdS-MOR 40 36 33 27 Baja
10Ag/20CdS-MOR 38 37 32 26 Baja
20CdS-MOR 35 36 34 33 Baja
4.3.3 Evaluación de los sensibilizadores de la fotoreacción durante 15 minutos
En esta parte de la investigación se evaluó el papel de los sensibilizadores como el acetonitrilo (ACN) y el
hidróxido de sodio (NaOH) en los procesos de degradación de la glucosa. Las soluciones acuosas de
acetonitrilo y de hidróxido de sodio se prepararon por separado en cantidades de 16.5 mL agua destilada
y 13.5 mL de acetonitrilo (Colmenares et al., 2011). A cada una de las soluciones de 30 mL se agregaron
0.15 g de glucosa para obtener una concentración inicial de 5 mg/mL de glucosa y se agregaron 0.03 g de
cada uno de los fotocatalizadores (10Ag/20CdS-MOR, 30Au-MOR, 30Ag-MOR, 20CdS-MOR, 30Au/20CdS-
MOR) por cada una de las soluciones de glucosa preparadas y se aplicó radiación UV en el fotoreactor.
Resultados para la solución con acetonitrilo sin fotocatalizador Los cromatogramas de la Figura 49a con el detector DIR indican los picos de los frentes de la fase móvil y
de la glucosa, los cuales proporcionaron los tiempos de retención y las áreas de los picos (Tabla 17). Los
cromatogramas (Figura 49b) correspondientes al detector DLM presentan varias señales asociadas a la
glucosa entre 6 y 7 minutos y posiblemente al ácido glucurónico entre 7 y 7.5 minutos y una señal no
identificada entre 8 y 9 minutos. Estos resultados se tomarán en consideración en la fotodegradación de
la glucosa al usar los fotocatalizadores. La conversión de la glucosa (Tabla 17) usando la solución con

57
acetonitrilo fue de hasta el 40% indicando que el acetonitrilo sensibiliza la estructura de la glucosa
posiblemente en enómeros.
0 1 2 3 4 5 6 7 8 9 10
Altura
(m
RIU
)
Tiempo de retención (min)
0 min
a)
5 min
10 min
15 min
0 1 2 3 4 5 6 7 8 9 10
Altura
(m
AU
)
Tiempo de retención (min)
0 min
b)
5 min
10 min
15 min
Figura 49. Cromatogramas de los detectores: a) DIR y b) DLM para la solución con acetonitrilo sin fotocatalizador y 15 minutos de radiación UV.
Tabla 17. Conversión de glucosa con solución de acetonitrilo y 15 minutos de radiación.
Tiempo de radiación 0 min 5 min 10 min 15 min
Tiempo Retención(min) 7.181 7.096 6.278 6.261
Área bajo la Curva(X106nRIU*s) 1.35219 0.910370 1.24297 1.46543
Concentración mg/mL 4 3 4 4
% de Conversión 20 40 20 20
Resultados para la solución con hidróxido de sodio sin fotocatalizador
Los cromatogramas de la Figura 50a con el detector DIR indican los frentes de la fase móvil y de la glucosa,
los cuales proporcionaron los tiempos de retención y las áreas de los picos (Tabla 18). Los cromatogramas
(Figura 50b) con el detector DLM presentan una señal entre 7 y 8 minutos la cual puede ser asociada con
el ácido glucurónico. Esto es algo a considerar cuando se usen los fotocatalizadores. Las concentraciones
finales y el porcentaje de conversión para las soluciones con acetonitrilo (Tabla 18) se calcularon utilizando
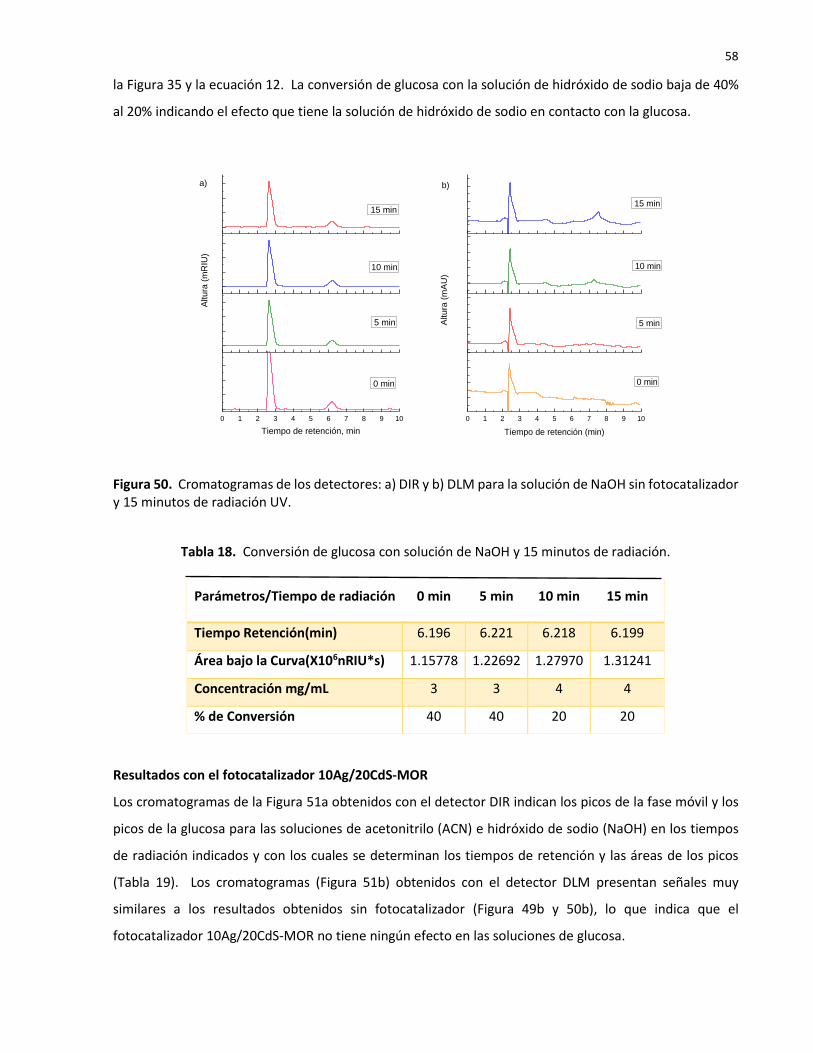
58
la Figura 35 y la ecuación 12. La conversión de glucosa con la solución de hidróxido de sodio baja de 40%
al 20% indicando el efecto que tiene la solución de hidróxido de sodio en contacto con la glucosa.
0 1 2 3 4 5 6 7 8 9 10
0 min
Tiempo de retención, min
5 min
10 min
15 minA
ltu
ra (
mR
IU)
a)
0 1 2 3 4 5 6 7 8 9 10
0 min
Tiempo de retención (min)
Altu
ra (
mA
U)
b)
5 min
10 min
15 min
Figura 50. Cromatogramas de los detectores: a) DIR y b) DLM para la solución de NaOH sin fotocatalizador y 15 minutos de radiación UV.
Tabla 18. Conversión de glucosa con solución de NaOH y 15 minutos de radiación.
Resultados con el fotocatalizador 10Ag/20CdS-MOR
Los cromatogramas de la Figura 51a obtenidos con el detector DIR indican los picos de la fase móvil y los
picos de la glucosa para las soluciones de acetonitrilo (ACN) e hidróxido de sodio (NaOH) en los tiempos
de radiación indicados y con los cuales se determinan los tiempos de retención y las áreas de los picos
(Tabla 19). Los cromatogramas (Figura 51b) obtenidos con el detector DLM presentan señales muy
similares a los resultados obtenidos sin fotocatalizador (Figura 49b y 50b), lo que indica que el
fotocatalizador 10Ag/20CdS-MOR no tiene ningún efecto en las soluciones de glucosa.
Parámetros/Tiempo de radiación 0 min 5 min 10 min 15 min
Tiempo Retención(min) 6.196 6.221 6.218 6.199
Área bajo la Curva(X106nRIU*s) 1.15778 1.22692 1.27970 1.31241
Concentración mg/mL 3 3 4 4
% de Conversión 40 40 20 20

59
Las concentraciones finales y los porcentajes de conversión se obtuvieron de la curva de calibración (Figura
39) y la ecuación 12 para los resultados de la Tabla 19, la cual muestra los valores de la conversión de
glucosa con acetonitrilo o hidróxido de sodio y el fotocatalizador 10Ag/20CdS-MOR. A los 10 minutos de
radiación existe un error de calibración del HPLC.
0 1 2 3 4 5 6 7 8 9 10
5 min;ACN
Tiempo de retención (min)
Altura
(m
RIU
)
a)
10 min;ACN
15 min;ACN
5 min;NaOH
10 min;NaOH
15 min;NaOH
0 1 2 3 4 5 6 7 8 9 10
5 min; ACNA
ltura
(m
AU
)
Tiempo de retención (min)
b)
10 min;ACN
15 min; ACN
5 min;NaOH
10 min; NaOH
15 min; NaOH
Figura 51. Cromatogramas de los detectores: a) DIR y b) DLM para las soluciones de ACN, NaOH, glucosa, fotocatalizador 10Ag/20CdS-MOR y 15 minutos de radiación UV.
Tabla 19. Conversión de glucosa en solución con acetonitrilo o NaOH, el fotocatalizador 10Ag/20CdS-MOR y 15 minutos de radiación.
Acetonitrilo Hidróxido de sodio
Tiempo de radiación 5 min 10 min 15 min 5 min 10 min 15min
Tiempo Retención(min) 6.277 6.279 6.276 6.435 6.401 6.291
Área bajo la Curva(106nRIU*s) 1.28162 1.39778 1.29435 1.33192 2.99792 1.47743
Concentración de glucosa mg/mL 4 4 4 4 error 4
% de conversión 20 20 20 20 error 20
Resultados con el fotocatalizador 30Au-MOR
Los cromatogramas de la Figura 52a obtenidos con el detector DIR indican los picos de la fase móvil y los
de la glucosa los cuales proporcionan los tiempos de retención y las áreas de los picos (Tabla 20). Los
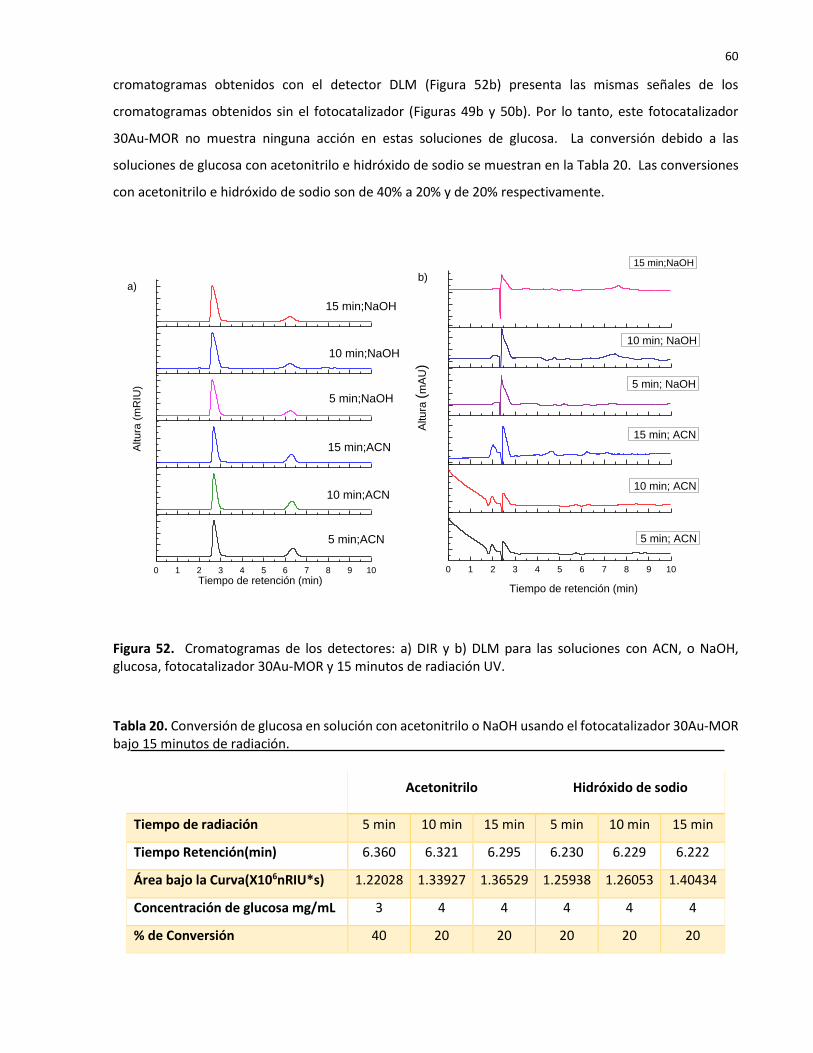
60
cromatogramas obtenidos con el detector DLM (Figura 52b) presenta las mismas señales de los
cromatogramas obtenidos sin el fotocatalizador (Figuras 49b y 50b). Por lo tanto, este fotocatalizador
30Au-MOR no muestra ninguna acción en estas soluciones de glucosa. La conversión debido a las
soluciones de glucosa con acetonitrilo e hidróxido de sodio se muestran en la Tabla 20. Las conversiones
con acetonitrilo e hidróxido de sodio son de 40% a 20% y de 20% respectivamente.
0 1 2 3 4 5 6 7 8 9 10
5 min;ACN
Tiempo de retención (min)
Altu
ra (
mR
IU)
10 min;ACN
15 min;ACN
5 min;NaOH
10 min;NaOH
15 min;NaOH
a)
0 1 2 3 4 5 6 7 8 9 10
5 min; ACN
Tiempo de retención (min)
Altura
(m
AU
)
10 min; ACN
15 min; ACN
5 min; NaOH
b)
10 min; NaOH
15 min;NaOH
Figura 52. Cromatogramas de los detectores: a) DIR y b) DLM para las soluciones con ACN, o NaOH, glucosa, fotocatalizador 30Au-MOR y 15 minutos de radiación UV.
Tabla 20. Conversión de glucosa en solución con acetonitrilo o NaOH usando el fotocatalizador 30Au-MOR bajo 15 minutos de radiación.
Acetonitrilo Hidróxido de sodio
Tiempo de radiación 5 min 10 min 15 min 5 min 10 min 15 min
Tiempo Retención(min) 6.360 6.321 6.295 6.230 6.229 6.222
Área bajo la Curva(X106nRIU*s) 1.22028 1.33927 1.36529 1.25938 1.26053 1.40434
Concentración de glucosa mg/mL 3 4 4 4 4 4
% de Conversión 40 20 20 20 20 20
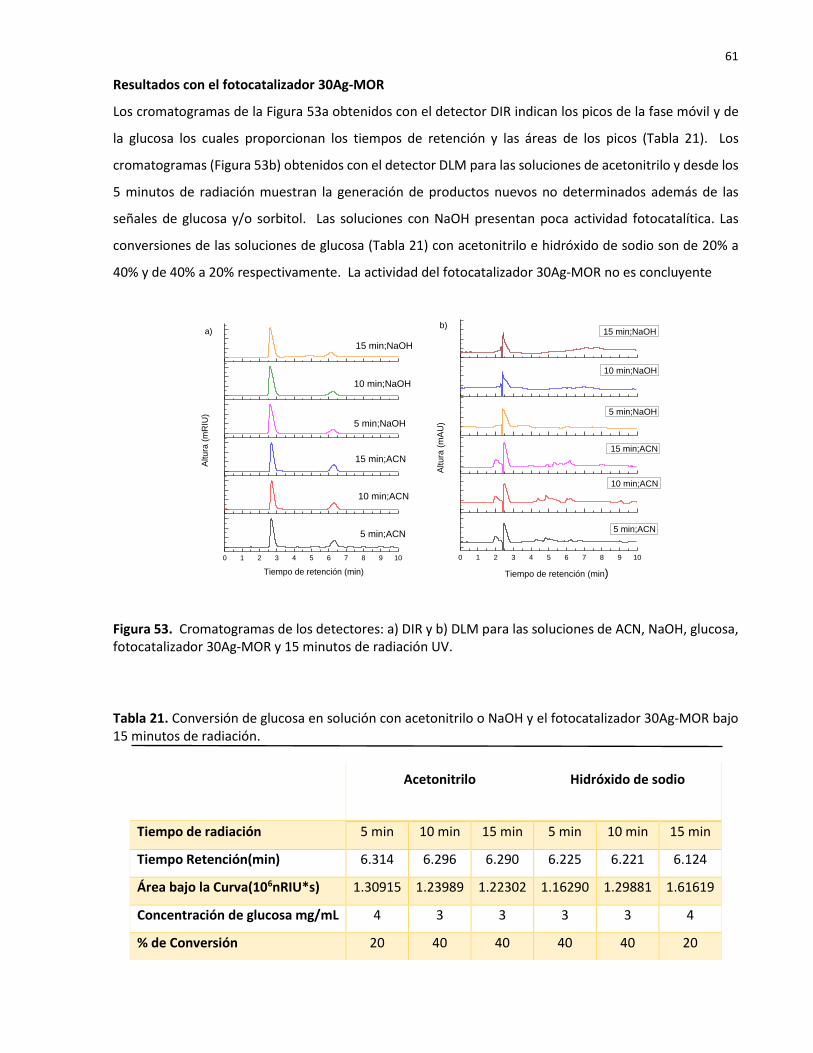
61
Resultados con el fotocatalizador 30Ag-MOR
Los cromatogramas de la Figura 53a obtenidos con el detector DIR indican los picos de la fase móvil y de
la glucosa los cuales proporcionan los tiempos de retención y las áreas de los picos (Tabla 21). Los
cromatogramas (Figura 53b) obtenidos con el detector DLM para las soluciones de acetonitrilo y desde los
5 minutos de radiación muestran la generación de productos nuevos no determinados además de las
señales de glucosa y/o sorbitol. Las soluciones con NaOH presentan poca actividad fotocatalítica. Las
conversiones de las soluciones de glucosa (Tabla 21) con acetonitrilo e hidróxido de sodio son de 20% a
40% y de 40% a 20% respectivamente. La actividad del fotocatalizador 30Ag-MOR no es concluyente
0 1 2 3 4 5 6 7 8 9 10
5 min;ACN
Tiempo de retención (min)
Altura
(m
RIU
)
10 min;ACN
15 min;ACN
5 min;NaOH
10 min;NaOH
15 min;NaOH
a)
0 1 2 3 4 5 6 7 8 9 10
5 min;ACN
Tiempo de retención (min)
Altu
ra (
mA
U)
b)
10 min;ACN
15 min;ACN
5 min;NaOH
10 min;NaOH
15 min;NaOH
Figura 53. Cromatogramas de los detectores: a) DIR y b) DLM para las soluciones de ACN, NaOH, glucosa, fotocatalizador 30Ag-MOR y 15 minutos de radiación UV.
Tabla 21. Conversión de glucosa en solución con acetonitrilo o NaOH y el fotocatalizador 30Ag-MOR bajo 15 minutos de radiación.
Acetonitrilo Hidróxido de sodio
Tiempo de radiación 5 min 10 min 15 min 5 min 10 min 15 min
Tiempo Retención(min) 6.314 6.296 6.290 6.225 6.221 6.124
Área bajo la Curva(106nRIU*s) 1.30915 1.23989 1.22302 1.16290 1.29881 1.61619
Concentración de glucosa mg/mL 4 3 3 3 3 4
% de Conversión 20 40 40 40 40 20

62
Resultados con el fotocatalizador 20CdS-MOR
Los cromatogramas de la Figura 54a obtenidos con el detector DIR indican los picos de la fase móvil y de
la glucosa los cuales proporcionan los tiempos de retención y las áreas de los picos (Tabla 22). Los
cromatogramas (Figura 54b) obtenidos con el detector DLM son similares a los observados en la Figura
49b con respecto a los tiempos entre 6 y 7 minutos en la solución con acetonitrilo. Se muestra poca
actividad a los 15 minutos de radiación para las soluciones con NaOH. La conversión de la glucosa (Tabla
22) con acetonitrilo sube de 20 % y 40 % y con NaOH bajan de 40 % y 20 %.
0 1 2 3 4 5 6 7 8 9 10
5 min;ACN
Tiempo de retención (min)
Altura
(m
RIU
)
a)
10min;ACN
15 min;ACN
5 min;NaOH
10min;NaOH
15 min;NaOH
0 1 2 3 4 5 6 7 8 9 10
5 min;ACN
b)
10min;ACN
15 min;ACN
5 min;NaOH
10min;NaOH
15 min;NaOH
Tiempo de retención (min)
Altura
(m
AU
)
Figura 54. Cromatogramas de los detectores: a) DIR y b) DLM para las soluciones de ACN, NaOH, glucosa, fotocatalizador 20CdS-MOR y 15 minutos de radiación UV.
Tabla 22. Conversión de glucosa en solución con acetonitrilo o NaOH y el fotocatalizador 20CdS-MOR bajo 15 minutos de radiación.
Acetonitrilo Hidróxido de sodio
Tiempo de radiación 5 min 10 min 15 min 5 min 10 min 15 min
Tiempo Retención(min) 6.377 6.349 6.326 6.749 6.552 6.308
Área bajo la Curva(106nRIU*s) 1.28543 1.18968 1.14656 1.15341 0.996436 1.4316
Concentración de glucosa mg/mL 4 3 3 3 3 4
% de Conversión 20 40 40 40 40 20
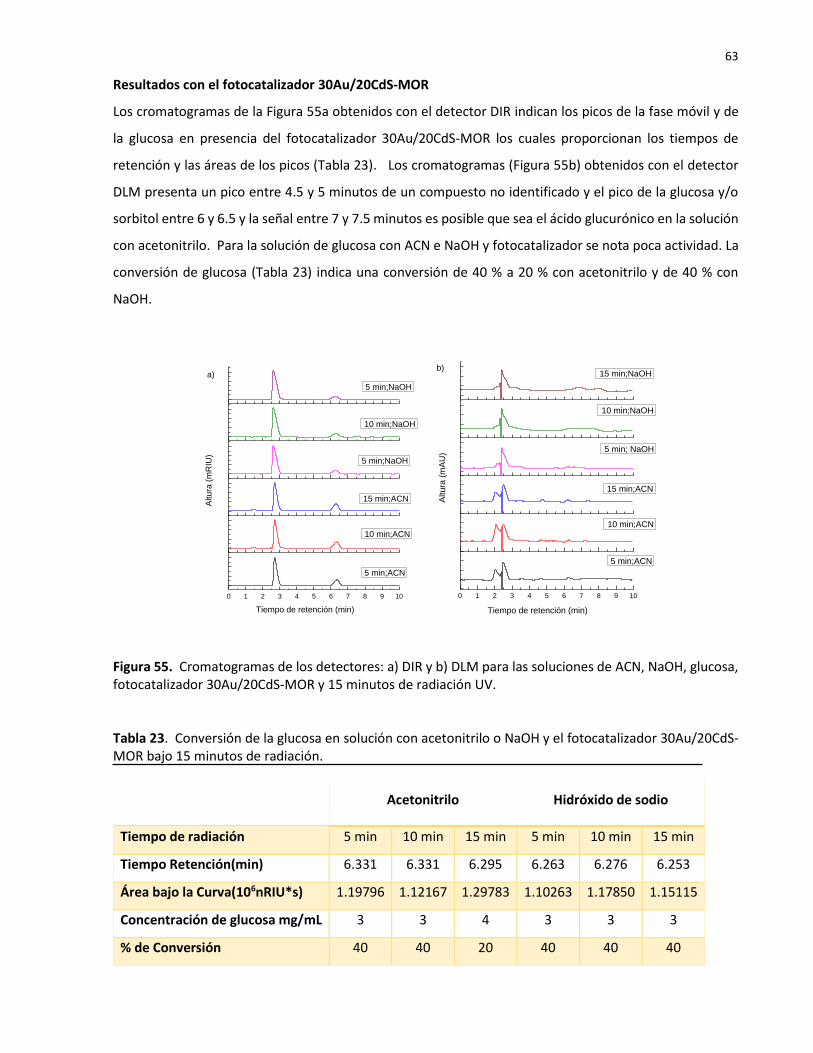
63
Resultados con el fotocatalizador 30Au/20CdS-MOR Los cromatogramas de la Figura 55a obtenidos con el detector DIR indican los picos de la fase móvil y de
la glucosa en presencia del fotocatalizador 30Au/20CdS-MOR los cuales proporcionan los tiempos de
retención y las áreas de los picos (Tabla 23). Los cromatogramas (Figura 55b) obtenidos con el detector
DLM presenta un pico entre 4.5 y 5 minutos de un compuesto no identificado y el pico de la glucosa y/o
sorbitol entre 6 y 6.5 y la señal entre 7 y 7.5 minutos es posible que sea el ácido glucurónico en la solución
con acetonitrilo. Para la solución de glucosa con ACN e NaOH y fotocatalizador se nota poca actividad. La
conversión de glucosa (Tabla 23) indica una conversión de 40 % a 20 % con acetonitrilo y de 40 % con
NaOH.
0 1 2 3 4 5 6 7 8 9 10
5 min;ACN
Tiempo de retención (min)
Altu
ra (
mR
IU)
10 min;ACN
15 min;ACN
5 min;NaOH
10 min;NaOH
5 min;NaOH
a)
0 1 2 3 4 5 6 7 8 9 10
5 min;ACN
Tiempo de retención (min)
Altura
(m
AU
)
b)
10 min;ACN
15 min;ACN
5 min; NaOH
10 min;NaOH
15 min;NaOH
Figura 55. Cromatogramas de los detectores: a) DIR y b) DLM para las soluciones de ACN, NaOH, glucosa, fotocatalizador 30Au/20CdS-MOR y 15 minutos de radiación UV. Tabla 23. Conversión de la glucosa en solución con acetonitrilo o NaOH y el fotocatalizador 30Au/20CdS-MOR bajo 15 minutos de radiación.
Acetonitrilo Hidróxido de sodio
Tiempo de radiación 5 min 10 min 15 min 5 min 10 min 15 min
Tiempo Retención(min) 6.331 6.331 6.295 6.263 6.276 6.253
Área bajo la Curva(106nRIU*s) 1.19796 1.12167 1.29783 1.10263 1.17850 1.15115
Concentración de glucosa mg/mL 3 3 4 3 3 3
% de Conversión 40 40 20 40 40 40

64
4.3.4 Evaluación de la fotodegradación en procesos de 2 horas de radiación
En esta parte del proyecto, la evaluación de la fotodegradación de la glucosa considerará los resultados
previos. Para ello se prepararon soluciones de 30 mL con 16.5 mL de agua destilada, 13.5 mL de acetonitrilo
y 0.15 g de glucosa obteniendo una concentración inicial de 5 mg/mL de solución con glucosa (Colmenares
et al., 2011). Se agregaron 0.03 g de cada uno de los fotocatalizadores (10Ag/20CdS-MOR, 30Ag-MOR,
30Au-MOR, 20CdS-MOR) a cada 30 mL de solución de glucosa y se utilizó una radiación de 254 nm durante
2 horas en el fotoreactor. A diferencia de los análisis anteriores el tiempo de radiación se extiende hasta
las 2 horas.
Resultados para la solución sin fotocatalizador
Los cromatogramas de la Figura 56a y b son los resultados obtenidos con los detectores DIR y DLM
respectivamente a diferentes tiempos de radiación entre 0 y 2 horas. Los cromatogramas de la Figura 56a
muestra los picos de la fase móvil y de la glucosa permitiendo obtener los tiempos de retención y el área
de cada pico (Tabla 24). Los cromatogramas (Figura 56b) indican que la glucosa es activa en esta solución;
se formaron picos nuevos antes y después de los tiempos correspondientes a las señales de la glucosa y el
sorbitol; sin presentar señal para el ácido glucurónico. El pico entre 11 y 12 minutos corresponde a un
compuesto nuevo no identificado. Los picos generados se considerarán para los subsiguientes resultados
al aplicar los fotocatalizadores. Las concentraciones finales y los porcentajes de conversión se obtendrán
de la Figura 39 y la ecuación 12 para los resultados de las tablas subsiguientes. La conversión de la glucosa
(Tabla 24) en solución con acetonitrilo baja de 40 % a 0 % en dos horas de radiación. Esto es un indicador
de la fotosensibilización de la glucosa sin fotocatalizador.

65
0 2 4 6 8 10 12
Altura
(nR
IU)
Tiempo de retención (min)
0 min
a)
5 min
10 min
15 min
30 min
1 hr
1.5 hr
2 hr
0 1 2 3 4 5 6 7 8 9 10 11 12 13
0 min
Altura
(m
AU
)
Tiempo de retención (min)
b)
5 min
10 min
15 min
30 min
1 hr
1.5 hr
2 hr
Figura 56. Cromatogramas de los detectores: a) DIR y b) DLM para la solución de acetonitrilo sin fotocatalizador y 2 horas de radiación UV.
Tabla 24. Conversión de la glucosa en solución con acetonitrilo sin fotocatalizador y 2 horas de radiación.
Tiempo de radiación 0 min 5 min 10 min 15 min 30 min 1 hr 1.5hr 2 hr
Tiempo Retención(min) 6.224 6.217 6.231 6.209 6.215 6.191 6.190 6.176
Área bajo la Curva(106nRIU*s) 1.221 1.308 1.489 1.284 1.318 1.377 1.469 1.692
Concentración de glucosa mg/mL 3 4 4 4 4 4 4 5
% de Conversión 40 20 20 20 20 20 20 0
Resultados con el fotocatalizador 10Ag/20CdS-MOR
Los cromatogramas de la Figura 57a obtenidos con el detector DIR muestran los picos de la fase móvil y
de la glucosa con la presencia del fotocatalizador 10Ag/20CdS-MOR y 2 horas de radiación UV. Con los
cuales se obtuvieron el tiempo de retención y las áreas de los picos (Tabla 25). Los cromatogramas (Figura
57b) obtenidos con el detector DLM presentan el pico de la glucosa y/o el sorbitol entre 6 y 6.5 minutos,
la señal entre 8 y 9 minutos es un producto nuevo no identificado y la señal entre 10 y 12 minutos es
similar a la de la Figura 56b. La conversión de la glucosa con el fotocatalizador 10Ag/20CdS-MOR se
mantiene en 20% durante las 2 horas de radiación.
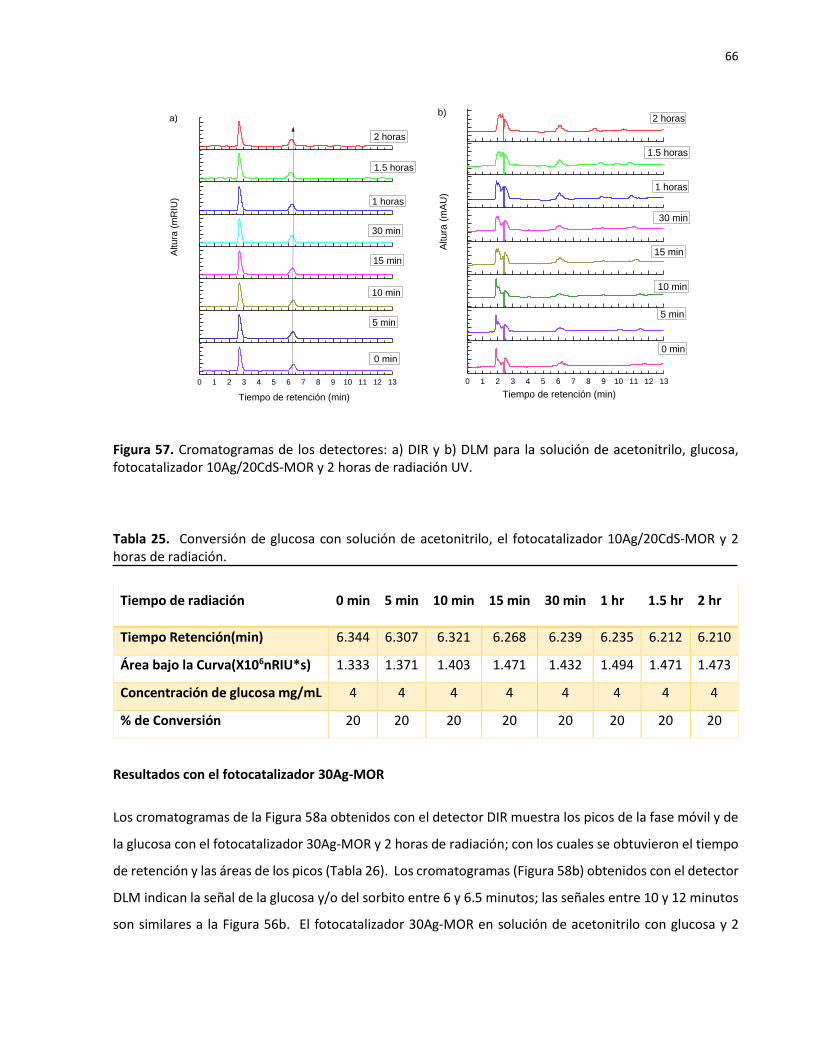
66
0 1 2 3 4 5 6 7 8 9 10 11 12 13
a)
Tiempo de retención (min)
Altura
(m
RIU
)
0 min
5 min
10 min
15 min
30 min
1 horas
1.5 horas
2 horas
0 1 2 3 4 5 6 7 8 9 10 11 12 13
Tiempo de retención (min)
Altura
(m
AU
)
b)
0 min
2 horas
5 min
10 min
15 min
30 min
1 horas
1.5 horas
Figura 57. Cromatogramas de los detectores: a) DIR y b) DLM para la solución de acetonitrilo, glucosa, fotocatalizador 10Ag/20CdS-MOR y 2 horas de radiación UV.
Tabla 25. Conversión de glucosa con solución de acetonitrilo, el fotocatalizador 10Ag/20CdS-MOR y 2 horas de radiación.
Tiempo de radiación 0 min 5 min 10 min 15 min 30 min 1 hr 1.5 hr 2 hr
Tiempo Retención(min) 6.344 6.307 6.321 6.268 6.239 6.235 6.212 6.210
Área bajo la Curva(X106nRIU*s) 1.333 1.371 1.403 1.471 1.432 1.494 1.471 1.473
Concentración de glucosa mg/mL 4 4 4 4 4 4 4 4
% de Conversión 20 20 20 20 20 20 20 20
Resultados con el fotocatalizador 30Ag-MOR
Los cromatogramas de la Figura 58a obtenidos con el detector DIR muestra los picos de la fase móvil y de
la glucosa con el fotocatalizador 30Ag-MOR y 2 horas de radiación; con los cuales se obtuvieron el tiempo
de retención y las áreas de los picos (Tabla 26). Los cromatogramas (Figura 58b) obtenidos con el detector
DLM indican la señal de la glucosa y/o del sorbito entre 6 y 6.5 minutos; las señales entre 10 y 12 minutos
son similares a la Figura 56b. El fotocatalizador 30Ag-MOR en solución de acetonitrilo con glucosa y 2

67
horas de radiación no presenta actividad fotocatalítica. La conversión de la glucosa baja de 20 % a 0 % a
partir de los 5 minutos de radiación (Tabla 26) debido a la actividad de la solución y no del fotocatalizador.
0 1 2 3 4 5 6 7 8 9 10 11 12 13
0 min
a)
5 min
10 min
15 min
30 min
1 horas
1.5 horas
2 horas
Tiempo de retención (min)
Altura
(m
RIU
)
0 1 2 3 4 5 6 7 8 9 10 11 12 13
0 min
5 min
10 min
Tiempo de retención (min)
Altura
(m
AU
)
15 min
30 min
1 hora
1.5 horas
2 horasb)
Figura 58. Cromatogramas de los detectores: a) DIR y b) DLM para la solución de acetonitrilo, glucosa, fotocatalizador 30Ag-MOR y 2 horas de radiación UV. Tabla 26. Conversión de glucosa con solución de acetonitrilo, el fotocatalizador 30Ag-MOR y 2 horas de radiación.
Tiempo de radiación 0 min 5 min 10 min 15 min 30 min 1 hr 1.5hr 2 hr
Tiempo Retención(min) 6.207 6.183 6.187 6.190 6.181 6.182 6.158 6.144
Área bajo la Curva(X106nRIU*s) 1.418 1.279 1.329 1.199 1.338 1.464 1.477 1.783
Concentración de glucosa mg/mL 4 4 4 3 4 4 4 5
% de Conversión 20 20 20 40 20 20 20 0
Resultados con el fotocatalizador 30Au-MOR
Los cromatogramas de la Figura 59a obtenidos con el detector DIR muestran los picos de la fase móvil y
de la glucosa con el fotocatalizador 30Au-MOR y 2 horas de radiación; se obtuvieron los tiempos de
retención y las áreas de los picos (Tabla 27). Los cromatogramas (Figura 59b) obtenidos con el detector
DLM muestran que el primer cromatograma con 0 minutos de radiación se corrió hasta un tiempo de

68
retención de 10 minutos, el cual muestra la señal de la glucosa únicamente. A partir de los 5 minutos de
radiación los cromatogramas muestran un pico entre 5 y 5.5 minutos similar a la Figura 56b. Los picos
entre 6 y 7 minutos es posible que sea los de la glucosa y/o el sorbitol. El pico entre 8.5 y 9 minutos y 1.5
horas de radiación es posible que sea un producto nuevo no identificado. Los picos entre 10 y 12 minutos
son similares a los de la Figura 56b. La conversión de glucosa baja a partir de los 5 minutos de radiación
de 40 % a 20% (Tabla 27). Parece existir actividad catalítica hasta la 1.5 horas de radiación.
0 1 2 3 4 5 6 7 8 9 10
Altura
(nR
IU)
0 min
Tiempo de retención (min)
a)
5 min
10 min
15 min
30 min
1 horas
1.5 horas
2 horas
0 1 2 3 4 5 6 7 8 9 10 11 12 13
0 min
Altura
(m
AU
)
Tiempo de retención (min)
b)
5 min
10 min
15 min
30 min
1 hora
1.5 horas
2 horas
Figura 59. Cromatogramas de los detectores: a) DIR y b) DLM para la solución de acetonitrilo, glucosa, fotocatalizador 30Au-MOR y 2 horas de radiación UV.
Tabla 27. Conversión de glucosa con solución de acetonitrilo, el fotocatalizador 30Au-MOR y 2 horas de radiación.
Tiempo de radiación 0 min 5 min 10 min 15 min 30 min 1 hr 1.5hr 2 hr
Tiempo Retención(min) 6.228 6.198 6.183 6.195 6.139 6.132 6.155 6.118
Área bajo la Curva(106nRIU*s) 1.292 1.200 1.289 1.294 1.272 1.366 1.405 1.337
Concentración de glucosa mg/mL 4 3 4 4 4 4 4 4
% de Conversión 20 40 20 20 20 20 20 20
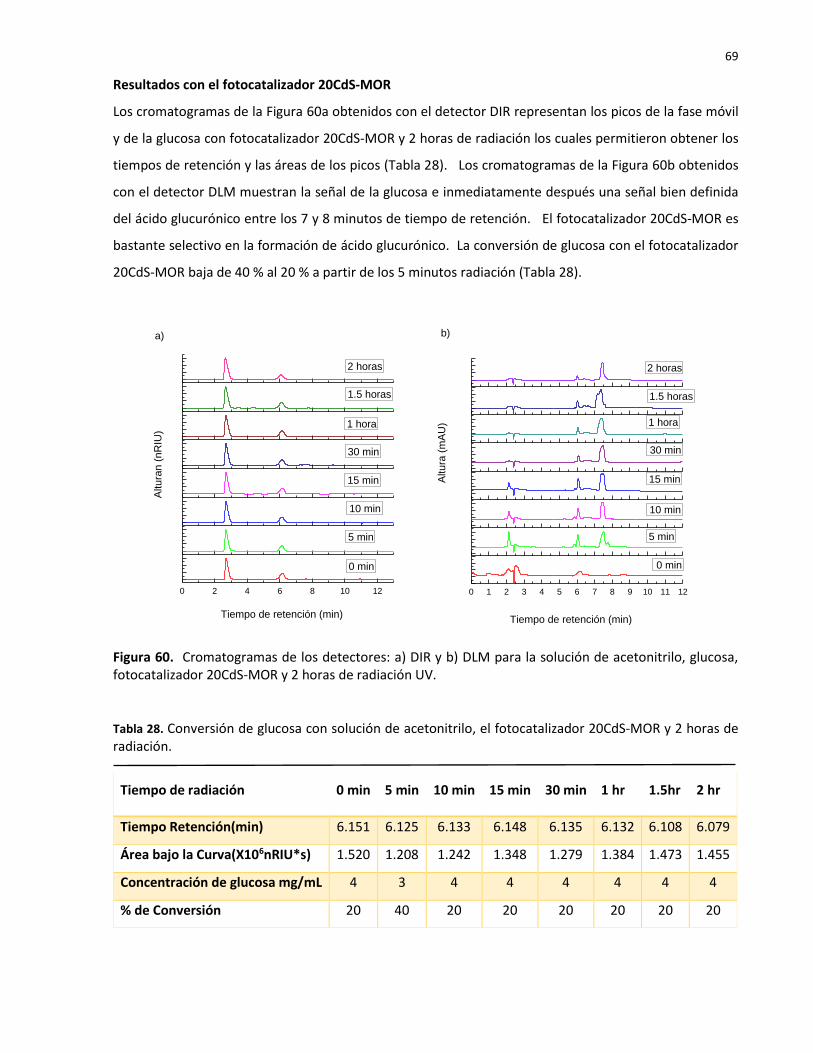
69
Resultados con el fotocatalizador 20CdS-MOR
Los cromatogramas de la Figura 60a obtenidos con el detector DIR representan los picos de la fase móvil
y de la glucosa con fotocatalizador 20CdS-MOR y 2 horas de radiación los cuales permitieron obtener los
tiempos de retención y las áreas de los picos (Tabla 28). Los cromatogramas de la Figura 60b obtenidos
con el detector DLM muestran la señal de la glucosa e inmediatamente después una señal bien definida
del ácido glucurónico entre los 7 y 8 minutos de tiempo de retención. El fotocatalizador 20CdS-MOR es
bastante selectivo en la formación de ácido glucurónico. La conversión de glucosa con el fotocatalizador
20CdS-MOR baja de 40 % al 20 % a partir de los 5 minutos radiación (Tabla 28).
0 2 4 6 8 10 12
0 min
Tiempo de retención (min)
Altura
n (
nR
IU)
a)
5 min
10 min
15 min
30 min
1 hora
1.5 horas
2 horas
0 1 2 3 4 5 6 7 8 9 10 11 12
0 min
Altura
(m
AU
)
Tiempo de retención (min)
b)
5 min
10 min
15 min
30 min
1 hora
1.5 horas
2 horas
Figura 60. Cromatogramas de los detectores: a) DIR y b) DLM para la solución de acetonitrilo, glucosa, fotocatalizador 20CdS-MOR y 2 horas de radiación UV. Tabla 28. Conversión de glucosa con solución de acetonitrilo, el fotocatalizador 20CdS-MOR y 2 horas de radiación.
Tiempo de radiación 0 min 5 min 10 min 15 min 30 min 1 hr 1.5hr 2 hr
Tiempo Retención(min) 6.151 6.125 6.133 6.148 6.135 6.132 6.108 6.079
Área bajo la Curva(X106nRIU*s) 1.520 1.208 1.242 1.348 1.279 1.384 1.473 1.455
Concentración de glucosa mg/mL 4 3 4 4 4 4 4 4
% de Conversión 20 40 20 20 20 20 20 20

70
Capítulo 5. Conclusiones y recomendaciones
1. Los fotocatalizadores sintéticos utilizados en el presente proyecto presentan las
características de la mordenita en cuanto a la estructura cristalina con sus microporos y
mesoporos y su presencia como matriz sobre la que crecen las nanopartículas metálicas y/o
semiconductoras.
2. El análisis de adsorción de N2 con la técnica de BET y la curva-t para determinar el área
superficial de las muestras y el volumen de los microporos, demostró que se tienen áreas
superficiales y tamaño de mesoporos superiores a los de la mordenita natural. Esta es una
característica importante en la catálisis heterogénea y en particular en la fotocatálisis.
3. De los estudios realizados se encuentra que los fotocatalizadores tienen potencial de
aplicación en la conversión de glucosa a productos de valor agregado.
4. Se encontró una mayor acción sensibilizadora del acetonitrilo respecto al hidróxido de sodio
en la degradación de la glucosa.
5. En los procesos de evaluación fotocatalítica se detectó la posible presencia de algún otro
derivado de la glucosa no previsto al inicio de la investigación.
6. Los nanocompuestos 30Au/20CdS-MOR, 30Ag-MOR, 10Ag/20CdS-MOR, 20CdS-MOR
mostraron gran actividad fotocatalítica en la degradación de la glucosa para las soluciones sin
sensibilizador.
7. El fotocatalizador 20CdS-MOR en solución de acetonitrilo demostró gran selectividad en la
generación de nuevos productos, especialmente en la producción de ácido glucurónico.

71
Recomendaciones
1. Los nanocompuestos sintéticos se pueden modificar o funcionalizar, para hacerlos más selectivos
como fotocatalizadores para la degradación de glucosa a productos de valor agregado, con
técnicas como la desaluminización del enrejado estructural de la matriz zeolítica, oxidar las
nanopartículas metálicas, entre otras.
2. Para identificar plenamente los productos de conversión de glucosa es necesario realizar o utilizar
otras técnicas de caracterización como la espectroscopía por infrarrojo y la espectroscopía UV-
Visible en fase líquida antes y después de la fotoreacción.

72
Literatura citada
Agilent. 2015. Agilent ZORBAX Carbohydrate Analysis Column. Data Sheet. Recuperado el 20 de junio de 2015 de: http://www.agilent.com/cs/library/datasheets/public/820629-008D.pdf
Agilent. 2016. UV-Vis spectroscopy-benefits of photo diode array. Recuperado el 23 de junio de 2016 de: http://www.agilent.com/en/products/uv-vis-uv-vis-nir/uv-vis-uv-vis-nir-accessories/photodiode-array
Asociacion internacional de zeolitas, 2016a, Database of zeolites, Recuperado el 14 de abril de 2016 de: http://america.iza-structure.org/IZA-SC/pow_plot.php
Asociación internacional de zeolitas, 2016b. Database of zeolite structures. Recuperado el 14 de abril de 2016 de: http://america.iza-structure.org/IZA-SC/framework.php?STC=MOR
Atkins, P., de Paula, J. 2010. Physical chemistry. (9th ed). WH. Freeman and Company. New York
Colmenares, J. C., Magdziarz, A., Bielejewska, A. 2011. High-value chemicals obtained from selective photo-oxidation of glucose in the presence of nanostructured titanium photocatalysts. Bioresource technology, 102(24), 11254-11257.
Costa, A., Lourenço, C., Marrucho, I., & Branco, L. C. 2013. Alternative methods for preparation of gluconate salts using heterogeneous non-metal catalysts. Journal of Materials Science and Engineering. B, 3(6B).
FEI™. 2017. An introduction to electron microscopy booklet. Recuperado el 10 de agosto de 2016 de: https://www.fei.com/documents/introduction-to-microscopy-document/
Gregg, S. J., Sing, K. S. W.,Adsorption, 1982. Surface area and porosity. (2nd ed). Academic Press, London.
Hernández, M. A., Rojas, F., Portillo, R., Castelán, R., Perez, G., 2010. Estructura porosa y propiedades estructurales de mordenita y clinoptilolita. Superficies y Vacio 23 (S)51-56.
Jaime-Acuña, O.E., Villavicencio-García, H., Vázquez-González, R., Petranovskiib, V., Raymond-Herrera, O., 2016. Synthesis of nanostructured metal–, semiconductor–, and metal/semiconductor–mordenite composites from geothermal waste. Journal of Applied Research and Technology 14 (4) 232–238.
Jaime-Acuña, O.E., Villavicencio-García, H., Vázquez-González, R., Petranovskiib, V., Raymond-Herrera, O., 2016. Síntesis de materiales multifuncionales nano estructurados a partir de salmueras de la geotérmica de Cerro Prieto. Revista Geotermia Vol. 29, Núm. Enero- junio.Recuperado en 12 de octubre de 2017 de: http://geotermia.org.mx/geotermia/revistageotermia/Geotermia-Vol29-1.pdf
Jaime-Acuña, O.E., 2010. Obtención y caracterización de nanopartículas semiconductoras y/o catiónicas en zeolitas tipo MOR para aplicaciones fotocataliticas, Tesis de Maestría en Ciencia. CICESE. Ensenada, B. C. México.
Jaime-Acuña, O.E., 2014. Nanocompuestos fotocatalíticos a partir de zeolitas sintéticas, Tesis de Doctorado en Ciencias. CICESE. Ensenada, B. C. México.

73
James, B. D., Baum, G. N., Perez, J., & Baum, K. N. (2009). Technoeconomic analysis of photoelectrochemical (PEC) hydrogen production. Directed Technologies, Inc., Arlington, VA (United States). Kittel, C., 2005. Introduction to solid state physics, John Wiley.
Jha, B., Singh, D. 2016. Basics of Zeolites. 5-31. 10.1007/978-981-10-1404-8_2.
Lindstrom, B., Pettersson, L. J. 2017. A brief history of catalysis. Recuperado el 15 de julio de 2007 de: http://image.sciencenet.cn/olddata/kexue.com.cn/upload/blog/file/2008/10/200810102212944318.pdf
Liu, Z., Ottaviani, M. F., Abrams, L., Lei, X., & Turro, N. J. (2004). Characterization of the External Surface of Silicalites Employing Electron Paramagnetic Resonance. The Journal of Physical Chemistry A, 108(39), 8040-8047.
microReport. 2017. Automatic section of data points and optimization of BET surface area. Recuperado el 21 de septiembre de 2017 de: http://micro-report.com/automatic-bet-surface-area/#more-183
Micromeritics. 2016. Gas adsorption theory. Recuperado el 23 de septiembre de 2016 de: http://www.micromeritics.com/Repository/Files/Gas_Adsorption_Theory_poster.pdf
Micromeritics. 2017. Micromeritic BET Surface area and porosity analyzer. Recuperado el 23 de septiembre de 2017 de: https://borch.agsci.colostate.edu/files/2016/02/BET-Analyzer-Generalized-SOP.pdf
Micromeritics. 2017. Volumetric gas adsorption apparatus. Part Number 200-42902-00. Recuperado el 23 de septiembre de 2017 de: http://www.micromeritics.com/Library/Scientific-Posters.aspx
Murakami, T., Fujishima, A. 2010. Expanding industrialization of photocatalysis. Recuperado el 24 de marzo de 2010 de: https://sangakukan.jp/journal/journal_contents/2010/06/articles/1006-03-2/1006-03-2_earticle.html
nanoComposix, 2017. Gold Nanoparticles: Optical Properties. Recuperado el 10 de enero de 2017 de: https://nanocomposix.com/pages/gold-nanoparticles-optical-properties
PubChem, 2017a. D-Glucose. Recuperado el 13 de noviembre de 2017 de: https://pubchem.ncbi.nlm.nih.gov/compound/22814120
PubChem, 2017b. D-Sorbitol. Recuperado el 13 de noviembre de 2017 de: https://pubchem.ncbi.nlm.nih.gov/compound/D-Sorbitol#section=Top
PubChem, 2017c. D-Glucuronic Acid. Recuperado el 13 de noviembre de 2017 de: https://pubchem.ncbi.nlm.nih.gov/compound/65041#section=Top
ObservatoryNano briefing. 2011. Applications of photocatalysis. Recuperado en 2015 de: http://nanopinion.archiv.zsi.at/sites/default/files/observatorynano_briefing_no.10_applications_for_photocatalysts.pdf
Smith, B. M., March, J., 2007. March’s advanced organic chemistry: reactions, mechanisms, and structure, (6th. Ed). John Wiley and Sons.

74
Speakman, S. A., 2016. Basics of X-Ray Diffraction. Recuperado el 5 de julio de 2016 de: http://prism.mit.edu/xray/oldsite/1%20Basics%20of%20X-Ray%20Powder%20Diffraction.pdf
SpectraLab. 2016. Sugar Analysis by HPLC-RI. Recuperado el 12 de enero de 2016 de: http://www.spectralabsci.com/PDFFiles/Sugars%20Analysis%20by%20HPLC-RI.pdf
Thermo Scientific. 2016. Thermo scientific XPS: what is XPS? Recuperado el 25 de mayo de 2016 de: https://xpssimplified.com/whatisxps.php
Uvitec. 2015. CrossLinker 509. Recuperado de: http://www.cleaverscientific.com/wp-content/uploads/2015/06/CL-508-Cross-Linker.pdf
Wade, L.G., 2006. Organic Chemistry. (6th ed). Pearson Prentice Hall.
Web of Science. 2016. Busqueda básica. Recuperada el 14 de marzo de 2016 de: http://oreon.dgbiblio.unam.mx/F/2MDM7NNB8UI78H7J3AVT4HJHQBNHH5JI5DK7N4QEPBM37THCMP-07576?func=find-&request=web+of+science&find_code=WTI&adjacent=N&x=61&y=16
Weitkamp, J. 2000. Zeolites and catalysis. Solid State Ionics, 131(1), 175-188
Zhang, X., 2014. Plasmonic photocatalysts of supported gold nanoparticles for organic conversions. Doctoral dissertation, Queensland University of Technology, Brisbane QLD. Australia.
Zhang, X., Chen, Y. L., Liu, R. S., & Tsai, D. P., 2013. Plasmonic photocatalysis. Reports on Progress in Physics, 76(4), 046401.
Zinin, P. 2017. Microanalysis in electron microscopy (EDS and WDS). University of Hawaii, Honolulu.USA. Recuperado el 21 de Agosto de 2017 de: http://www.soest.hawaii.edu/HIGP/Faculty/sksharma/GG711/GG711Lec14EDS.pdf


















