
Medicamentos hu érfanos - INICIO · Osteogénesis imperfecta(huesos de cristal) Amèlie ......
47
Medicamentos Medicamentos hu hu é é rfanos rfanos M M ó ó nica Salda nica Salda ñ ñ a y Javier Galiana a y Javier Galiana UGC UGC Intercentros Intercentros de Farmacolog de Farmacolog í í a Cl a Cl í í nica nica HUPM C HUPM C á á diz y HU Puerto Real diz y HU Puerto Real mayo, 2013 mayo, 2013
Transcript of Medicamentos hu érfanos - INICIO · Osteogénesis imperfecta(huesos de cristal) Amèlie ......

Medicamentos Medicamentos huhuéérfanosrfanos
MMóónica Saldanica Saldañña y Javier Galianaa y Javier GalianaUGC UGC IntercentrosIntercentros de Farmacologde Farmacologíía Cla ClíínicanicaHUPM CHUPM Cáádiz y HU Puerto Realdiz y HU Puerto Realmayo, 2013mayo, 2013


Osteogénesis imperfecta(huesos de cristal) Amèlie (2001) Leontiasis ósea la máscara ( 1985) retinitsis pigmentaria bailando en la oscuridad CADASIL mar adentro Acondroplasia Vías cruzadas(2003) Cosas que diría con sólo mirarla El inolvidable Simon Birch Sídrome de MorquioPorfiria intermitente aguda La locura del rey Jorge Adrenoleucodistrofia El aceite de la vida Xeroderma pigmentoso Los otros Síndrome de Proteo El hombre elefante Esclerosis múltiple Hilary y Jackie (1998) ELA(esclerosis lateral amiotrófica) El orgullo de los Yanquis Espina bífida Amor ciego Síndrome de Tourette El código Tic(1999) Fibrosis quística Álex: the life of a child




•DESIGNACIÓN DE MEDICAMENTO HUÉRFANO
•Criterios•Debe ser dirigido para el tratamiento, prevención o diagnóstico de una enfermedad que sea amenazante para la vida o que produzca un deterioro crónico•La prevalencia no debe ser superior a 5 entre 10.000 y debe ser improbable que el marketing del producto pueda generar suficiente retorno como para justificar los costos de su desarrollo•No existen métodos autorizados que sean satisfactorios para el diagnóstico, prevención o tratamiento de la patología concernida. Pero si tales métodos existen se espera que el producto aporte un beneficio relevante a las personas afectadas
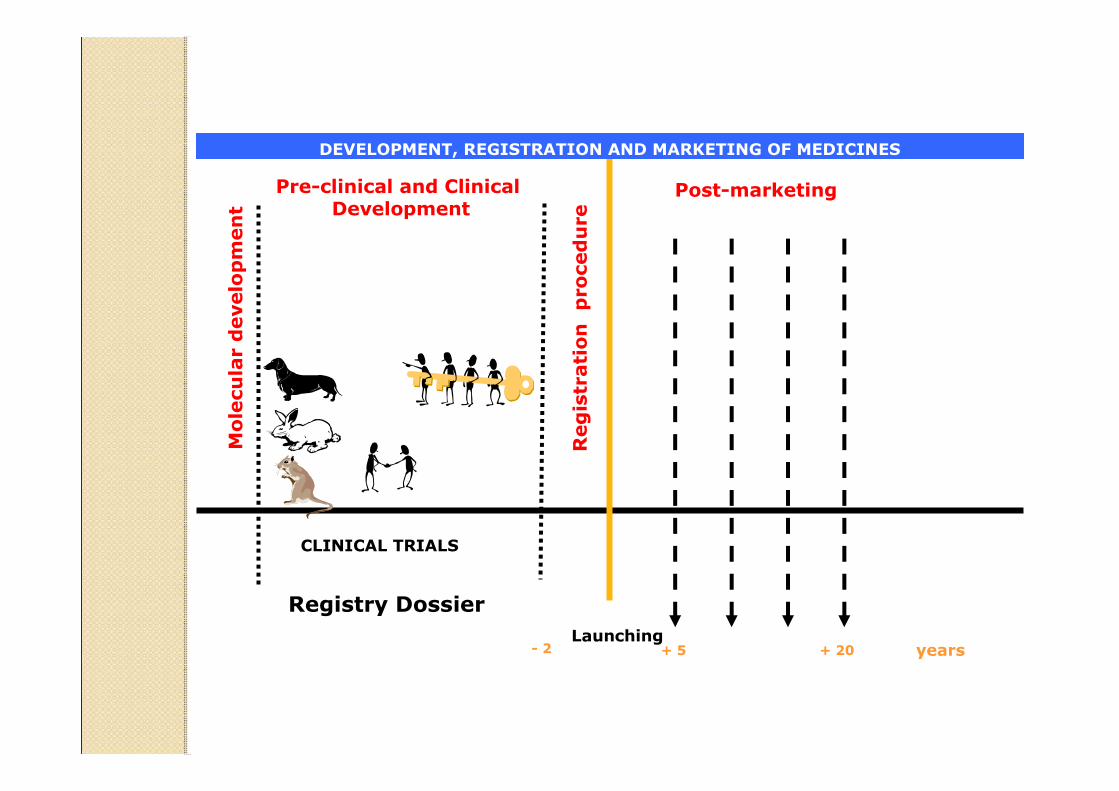
Pre-clinical and Clinical Development
Molecular development
Launching+ 5- 2 years
Registration procedure
+ 20
DEVELOPMENT, REGISTRATION AND MARKETING OF MEDICINES
Post-marketing
CLINICAL TRIALS
Registry Dossier


http://http://www.emea.europa.euwww.emea.europa.eu//htmshtms/human//human/orphorph
ansans//opinions.htmopinions.htm
Comité para Medicamentos Huérfanos (COMP)
Examinar las aplicaciones para la designación del Medicamento Huérfano.
Aconseja a la Comisión Europea sobre el desarrollo de la políticas y las guías sobre los medicamentos huérfanos en Europa
Ayuda la Comisión de la UE enlazando internacionalmente asuntos relacionados con los medicamentos huérfanos y haciendo de enlace entre los grupos de apoyo de pacientes.

ComitComitéé de medicamentos hude medicamentos huéérfanosrfanos
El Comité estará compuesto por:
� Un miembro nombrado por cada uno de los Estados miembros,
� tres miembros nombrados por la Comisión para representar a las asociaciones de pacientes y
� tres miembros nombrados por la Comisión previa recomendación de la Agencia.
� Los miembros del Comité serán nombrados por un período de tres años, renovable.
� Podrán hacerse acompañar por expertos


CommissionCommission GuidelineGuideline (ENTR/6283/00) (ENTR/6283/00) ““Medical Medical PlausibilityPlausibility””
1.Bases para el uso del medicamento en la indicación “huérfana” propuesta
2.Cuando la indicación afecta a una faceta particular de la enfermedad se debe aportar una justificación para el uso restringido en esa “sub”indicación

ObjetivosObjetivos
1. Destacar el nivel de evidencia requerido para apoyar el respaldo médico (credibilidad o verosimilitud) para el empleo del producto en la patología en cuestión
2. Nivel de evidencia requerido para apoyar la aceptación de un beneficio clínicamente significativo

Balance beneficio / riesgoBalance beneficio / riesgo
El tratamiento necesariamente se puede asociar con ciertos riesgos.
Tales riesgos se deben balancear frente a los beneficios esperados en orden a conceder o no la autorización para la venta, de acuerdo con los criterios de seguridad, calidad y eficacia (Directiva 2001/83/EC)

GuidelineGuideline (ENTR/6283/00)(ENTR/6283/00)
En el momento de la designación “los beneficios significativos se deben basar sobre suposiciones bien justificadas. Tales suposiciones deben ser verosímiles y asentadas sobre fundamentos farmacológicos sólidos”.
También se establece que para apoyar la noción de beneficio relevante se deben aportar datos sobre una mayor eficacia potencial, mejor perfil de seguridad y/o propiedades farmacocinéticas favorables

Aporte de datos y referenciasAporte de datos y referencias
1. Documentación científica apropiada
2. “También se pueden aportar otras formas de referencias o informes no publicados, así como consideraciones de expertos”

Con frecuencia, en el momento de la designación no se dispone de experiencia clínica o es muy escasa. Por ello es de gran importancia que se discuta la relevancia de los modelos in vitro e in vivo en el contexto de la patología interesada

El espíritu de la legislación de huérfanos prevéque la aplicación se pueda llevar a cabo en cualquier momento del desarrollo del medicamento.
El beneficio significativo o relevante se debe basar en la evidencia disponible en ese momento

• En ausencia de datos previos, el hecho de que el nuevo producto pueda tener un mecanismo de acción diferente no se considera por sí mismo argumento suficiente para justificar la suposición de beneficio relevante.
• Basado en la evidencia disponible, el sponsor debe justificar que ese mecanismo de acción puede trasladarse a una mejor eficacia. Una nueva diana farmacológica no necesariamente da lugar a una mejoría en la eficacia o en la seguridad

• Los datos sobre seguridad casi nunca son totalmente disponibles ni fiables hasta que no se ha hecho un uso amplio de un producto
• Por ello, es una argumentación válida suponer mayor beneficio potencial para un producto que ya se viene utilizando ampliamente para otra indicación y que por lo tanto, se le conoce su perfil indeseable.

• El riesgo teórico de un producto autorizado no se puede comparar con la falta de riesgo teórico de un producto en desarrollo.
• Por ejemplo: un producto transgénico nuevo tiene un potencial de antigenicidad asociado mayor que el potencial de transmisión viral asociado a un derivado hematológico que lleve en el mercado 2 o 3 décadas

Beneficios significativos previos a la Beneficios significativos previos a la concesiconcesióón de la autorizacin de la autorizacióón de ventan de venta
•El beneficio se debe basar en datos y revisión crítica de las ventajas o contribución relevante para el “cuidado” del paciente en el contexto de los métodos ya autorizados para la indicación huérfana.
•Tales datos se han debido generar durante el desarrollo clínico del producto

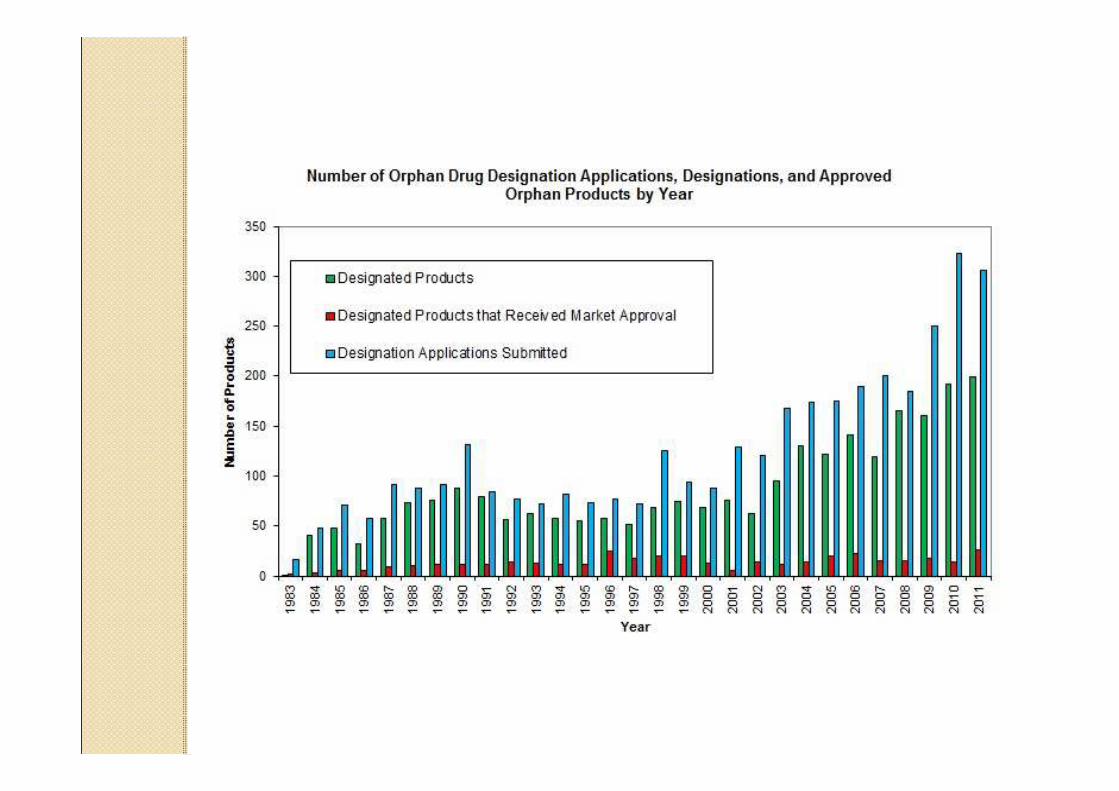
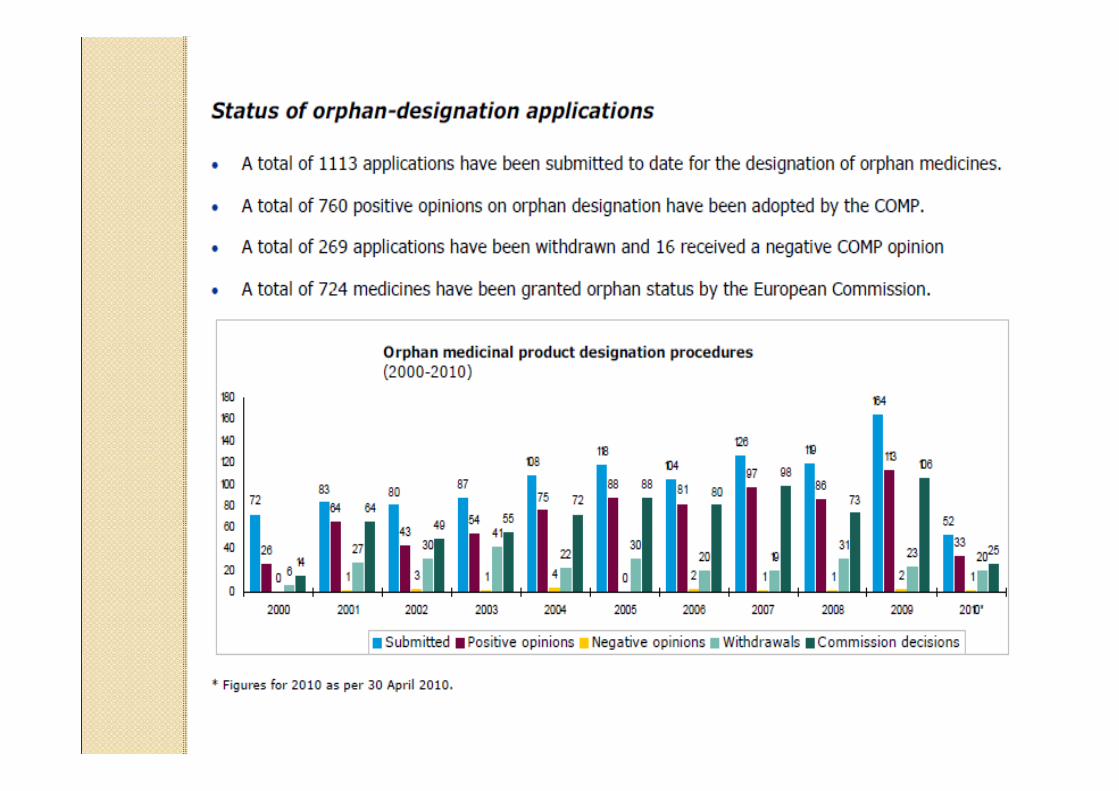
¿¿QuQuéé conlleva la designaciconlleva la designacióón de n de medicamento humedicamento huéérfano? rfano?
Puede obtenerse en cualquier etapa del desarrollo de un producto, siempre que sea presentada una adecuada argumentación científica acerca del uso específico que se pretende. El procedimiento de designación no lleva tasas de ningún tipo.La designación de un medicamento como huérfano, no garantiza su uso en la condición designada, y no implica necesariamente que el producto satisfaga los criterios de eficacia, seguridad y calidad, necesarios para la concesión de la Autorización de comercio.

En la UE,En la UE, la legislacila legislacióón proporciona n proporciona incentivos para los promotores o para incentivos para los promotores o para la industria farmacla industria farmacééutica para el utica para el desarrollo de MHdesarrollo de MH
Los medicamentos que puedan beneficiarse de los incentivos, son designados mediante el procedimiento comunitario de designación de medicamento huérfano

1.1. Exclusividad comercial: Exclusividad comercial:
En la Comunidad Europea 10 años siguientes a la autorización de venta.
Los productos similares, que puedan competir directamente, no podrán ser comercializados.
2. Acceso al procedimiento centralizado: 2. Acceso al procedimiento centralizado:
Los MH acceden de forma directa

3. Exenci3. Exencióón de tasas:n de tasas:
La EMEA recurre a una contribución especial concedida por la Comisión Europea, previa aprobación anual del Parlamento Europeo, para eximir de tasas a los medicamentos huérfanos.
Reducción valedera para todas las actividades centralizadas, incluyendo tasa de solicitud de autorización de comercialización, inspecciones, variaciones, y asistencia a la elaboración de protocolos.

4.4. Asistencia en la elaboraciAsistencia en la elaboracióón de n de protocolos:protocolos:
La EMEA podrá prestar asesoramiento científico a fin de optimizar el desarrollo del medicamento.
Orientar en la preparación del expediente para que cumpla todos los requisitos reglamentarios (mayores garantías de éxito para la autorización de comercialización).

5.Investigaci5.Investigacióón subvencionada por la n subvencionada por la UniUnióón Europea:n Europea:
Las organizaciones que desarrollan medicamentos huérfanos pueden optar a subvenciones de programas e iniciativas de la comunidad o de los Estados miembros destinados a apoyar la investigación y el desarrollo, incluyendo el Programa Marco Comunitario.

La Agencia estimula a las compañias que desarrollan medicamentos huérfanos para que consideren si se pueden clasificar como empresas micro, pequeñas o intermedias (SME).
Estas compañías se benefician de incentivos adicionales que incluyen apoyo administrativo y procedimental desde la propia Agencia, asícomo reducción de tasas

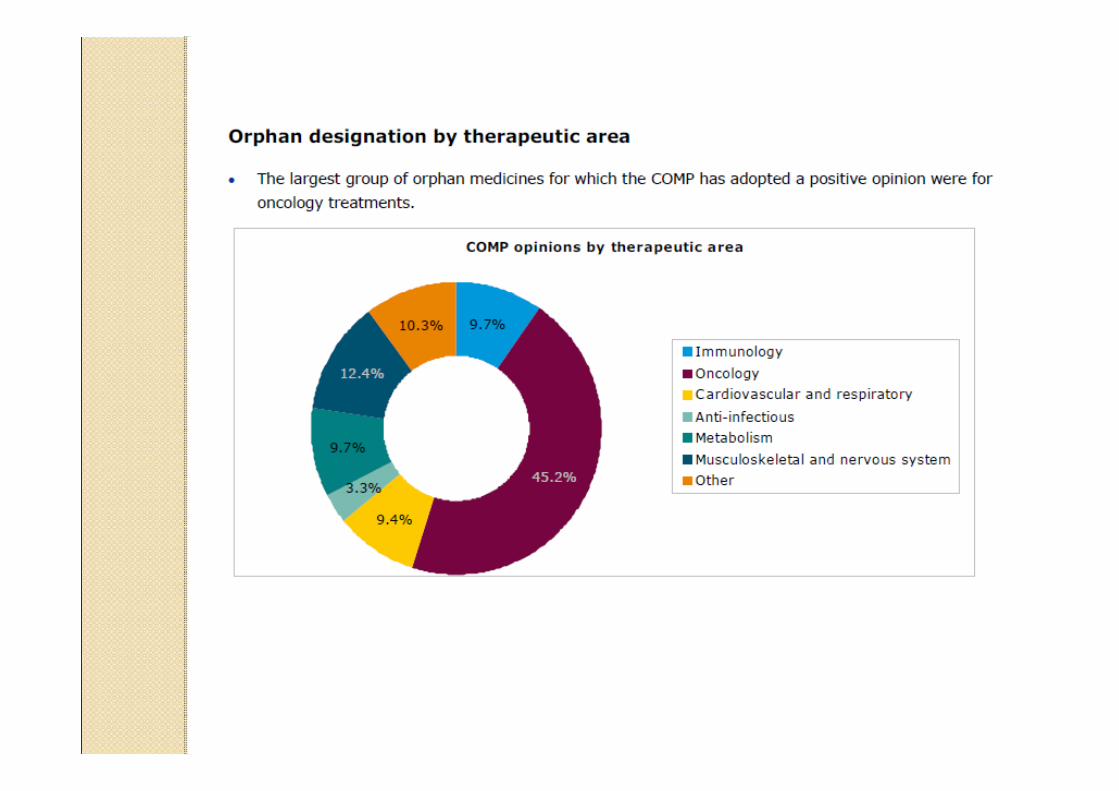
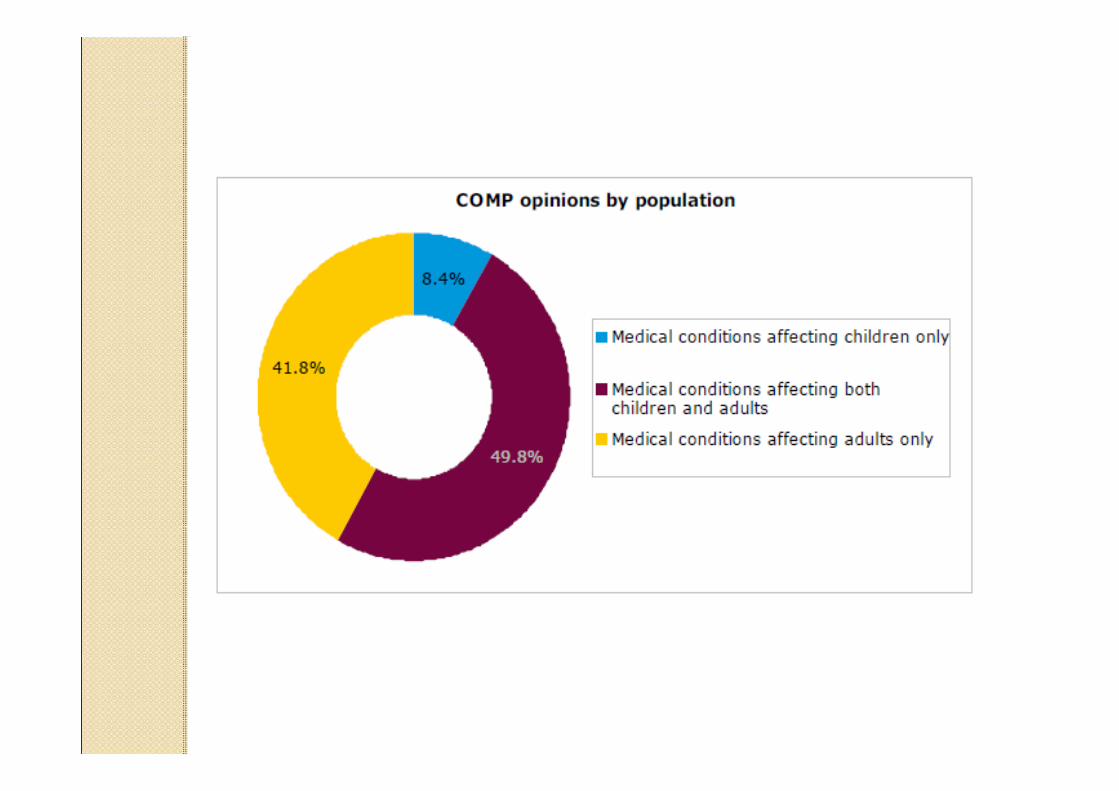
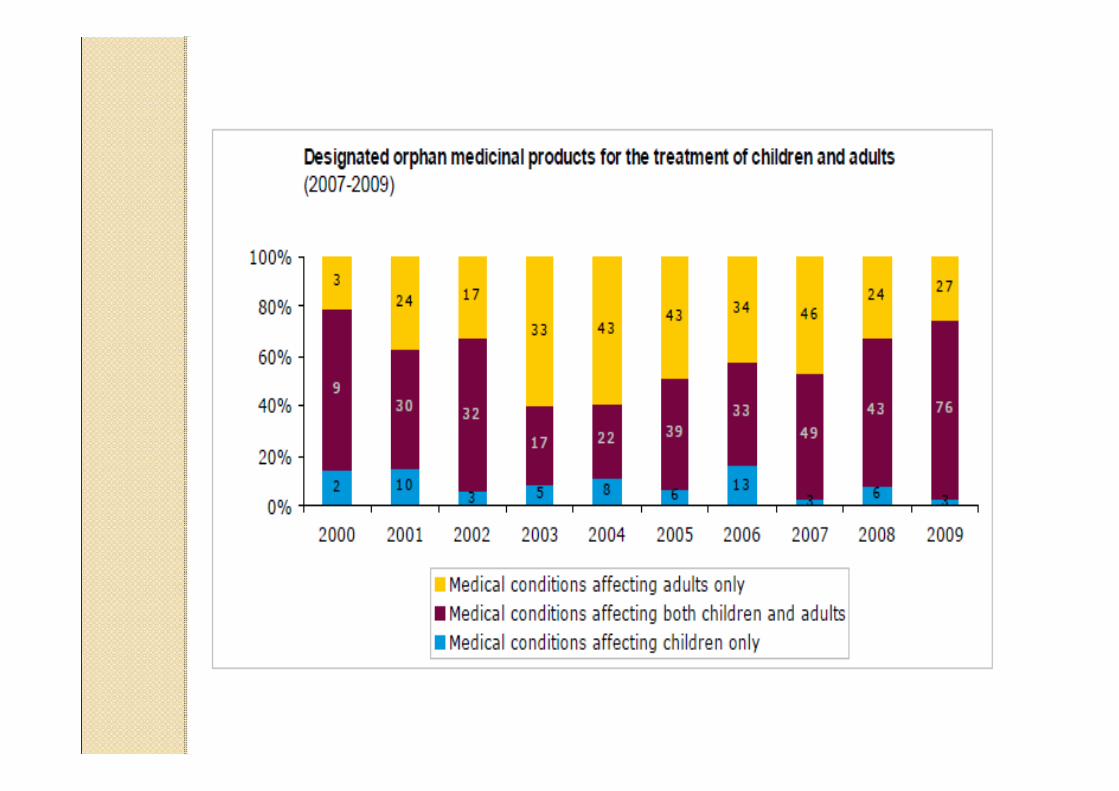

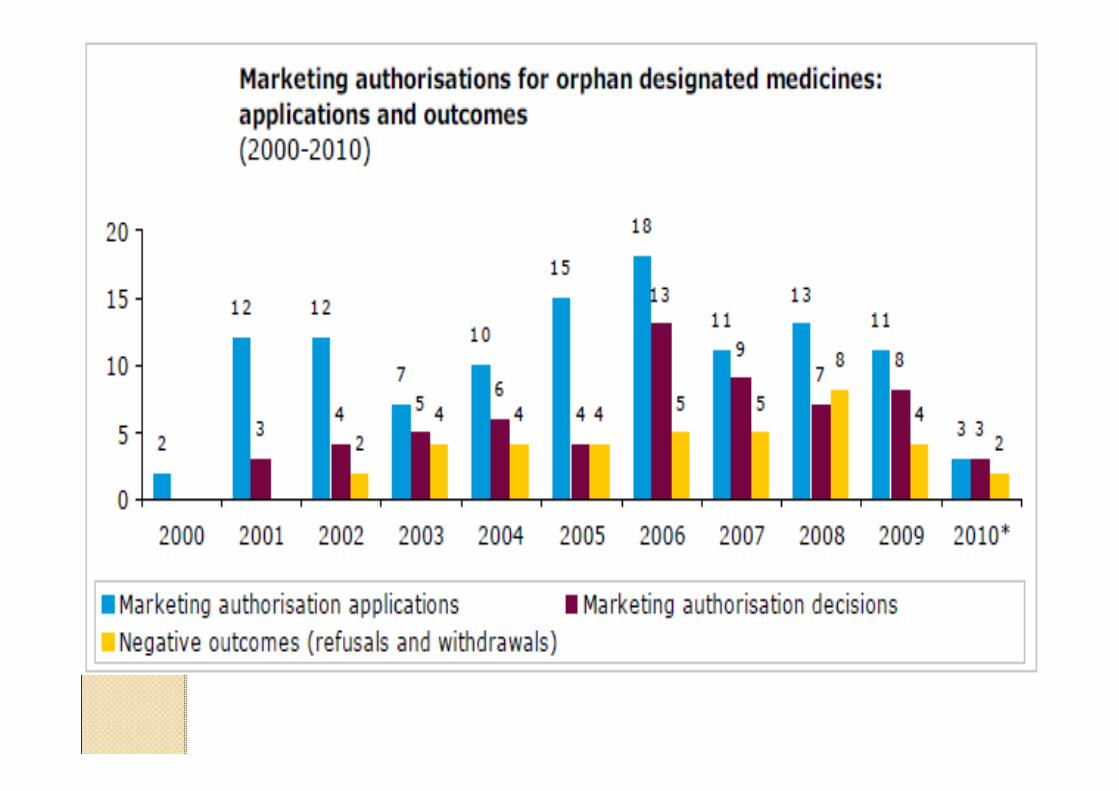


Data providers and sources to identify existingauthorised medicinal products in the European Union and European Economic Area
Spain: www.portalfarma.com; www.agemed.es

Enzyme replacement therapy iscurrently available for some lysosomaldiseases:
Gaucher disease, Fabry disease, MPS I, [MPS II],MPS VIGlycogen storage disease type II.

MEDICAMENTOS HUMEDICAMENTOS HUÉÉRFANOSRFANOS
� No constituyen ningún grupo farmacológico ni terapéutico delimitado
� Forman una relación tan amplia como heterogénea y abierta
� Han obligado a reconsideraciones metodológicas en el desarrollo de los estudios clínicos (“n” muy bajas)
� Aumentan la necesidad de estudios colaborativos� Obligan a una consideración de los criterios de
evidencia desde una óptica mas “permisiva”� Obligan a una evaluación rigurosa del cociente
beneficio/riesgo

One of Many Rare Disease One of Many Rare Disease HeroesHeroes……
EMA/EURORDIS/IRDIRC/ICORD
representingCollaborative international rare disease
efforts

NeuropediatraFarmacia
Industria
NiñoFamilia
Asociaciones
SAS MEDICAMENTOMEDICAMENTO
HUHUÉÉRFANORFANO

Muchas gracias


















