
Metodolog ía Sint ética Aplicada a la S íntesis de F … · Síntesis de Inhibidores Selectivos...
110
Metodolog Metodologí a Sint a Sinté tica Aplicada tica Aplicada a la S a la Sí ntesis de F ntesis de Fá rmacos rmacos Miguel Carda Miguel Carda Má ster en Qu ster en Quí mica Aplicada y Farmacol mica Aplicada y Farmacológica gica Universidad Jaume I Universidad Jaume I NH2 HOOC MeO HN N N N N Ph 3 C (PriO) 2 B Br Br Síntesis de losartan
Transcript of Metodolog ía Sint ética Aplicada a la S íntesis de F … · Síntesis de Inhibidores Selectivos...

MetodologMetodolog íía Sinta Sint éética Aplicada tica Aplicada a la Sa la Sííntesis de Fntesis de F áármacosrmacos
Miguel CardaMiguel CardaMMááster en Quster en Qu íímica Aplicada y Farmacolmica Aplicada y Farmacol óógicagica
Universidad Jaume IUniversidad Jaume I
NH2
HOOCMeO
HN
N
NN
NPh3C
(PriO)2B
Br
Br
Síntesis de losartan


Tema 3
Enfermedades del sistema nervioso central: síntesis de antidepresivos, antiepilépticos y anti-Parkinson
Máster en Química Aplicada y Farmacológica
Universidad Jaume I


Tema 3. Enfermedades del sistema nervioso central
3.1. Neurotransmisores 1
3.1.1. Tipos de neurotransmisores 4
3.2. Inhibidores Selectivos de la Recaptación de Serotonina (ISRS) 6
3.2.1. Liberación de la serotonina 9
3.2.2. Receptores se seronotina 10
3.3. Otros inhibidores selectivos de recaptación de serotonina 12
3.4. Fármacos Inhibidores Selectivos de la Recaptación de Serotonina 12
3.5. Síntesis de Inhibidores Selectivos de la Recaptación de Serotonina 14
3.5.1. Síntesis de escitalopram 14
3.5.1.a. Análisis retrosintético 15
3.5.1.b. Síntesis 16
3.5.1.c. Cuestiones 16
3.5.2. Síntesis de dapoxetina 16
3.5.2.1a. Análisis retrosintético 16
3.5.2.1b. Síntesis 16
3.5.2.2b. Síntesis de dapoxetina empleando (R)-fenilglicina como
material quiral de partida 17
3.5.2.2c. Cuestiones 18
3.5.3. Síntesis de fluoxetina (Prozac) 18
3.5.3.a. Análisis retrosintético 18
3.5.3.b. Síntesis 19
3.5.3.c. Cuestiones 19
3.5.4. Síntesis de sertralina 20
3.5.4.a. Análisis retrosintético 20
3.5.4.b. Síntesis 21 3.5.4.c. Cuestiones 22
3.5.5. Síntesis de paroxetina 25
3.5.5.1a. Análisis retrosintético 25
3.5.5.1b. Síntesis 26
3.5.5.1c. Cuestiones 27
3.5.5.2a. Análisis retrosintético de paroxetina mediante la estrategia de
Adición de Quiralidad (AQ) 27
3.5.5.2b. Síntesis de (+)-paroxetina mediante el empleo de un fragmento
quiral (Adición de Quiralidad) 28
3.5.5.2c. Cuestiones 29
3.5.5.3a. Análisis retrosintético de paroxetina mediante una estrategia
de desimetrización 30

3.5.5.3b. Síntesis de paroxetina mediante desimetrización enantioselectiva
de un diéster simétrico 31
3.5.5.3c. Cuestiones 32
3.6. Síntesis de Inhibidores Selectivos de la Recaptación de Serotonina y
Norepinefrina (ISRSN) 33
3.6.1. Síntesis de venlafaxina 33
3.6.1.a. Análisis retrosintético 34
3.6.1.b. Síntesis 34
3.6.1.c. Cuestiones 35 3.6.2. Síntesis desvenlafaxina 35
3.6.2.a. Análisis retrosintético 35 3.6.2.b. Síntesis 35
3.6.3. Síntesis de milnacipran 35
3.6.3.a. Análisis retrosintético 36
3.6.3.b. Síntesis 36
3.6.3.c. Cuestiones 37
3.6.4. Sïntesis de duloxetina 38
3.6.4.1a. Análisis retrosintético 38
3.6.4.1b. Síntesis 39
3.6.4.1c. Cuestiones 40
3.6.4.2a. Análisis retrosintético de duloxetina mediante reducción
enantioselectiva 40
3.6.4.2b. Síntesis de duloxetina mediante reducción enantioselectiva 41
3.6.4.2c. Cuestiones 42
3.6.4.3a. Análisis retrosintético de duloxetina mediante esterificación
enzimática enantioselectiva 43
3.6.4.3b. Síntesis de duloxetina mediante mediante esterificación
enzimática enantioselectiva 43
3.6.4.3c. Cuestiones 44
3.7. Síntesis de Inhibidores Selectivos de la Recaptación de Noradrenalina (ISRN) 46
3.7.1. Síntesis de atomoxetina 46
3.7.1.1a. Análisis retrosintetico 46
3.7.1.1b. Síntesis 47
3.7.c.1c. Cuestiones 48
3.7.1.2a. Análisis retrosintetico de atomoxetina mediante reacción SNAr 48
3.7.1.2b. Síntesis de atomoxetina mediante reacción SNAr 49
3.7.1.2c. Cuestiones 50
3.8. Síntesis de antidepresivos tricíclicos 51

3.8.1. Síntesis de amitriptilina 51 3.8.1.a. Análisis retrosintético 51 3.8.1.b. Síntesis 52 3.8.1.c. Cuestiones 52
3.8.2. Síntesis de imipramina 52 3.8.2.a. Análisis retrosintético 53 3.8.2.b. Síntesis 53 3.8.2.c. Cuestiones 53
3.9. Epilepsia 54
3.10. Fármacos antiepilépticos 54
3.10.1. GABA: un neurotransmisor inhibitorio cerebral 57
3.10.1.a. Receptores de GABA 59
3.10.2. Fármacos antiepilépticos: gabapentina y pregabalina 63
3.10.3. Modo de acción de la pregabalina 65
3.11. Sintesis de fármacos antiepilépticos 66
3.11.1. Síntesis de gabapentina 67
3.11.1.1a. Análisis retrosintético 67
3.11.1.1b. Síntesis 67
3.11.1.1c. Cuestiones 68
3.11.1.2a. Análisis retrosintético de gabapentina mediante umpolung 68
3.11.1.2b. Síntesis 68
3.11.2. Sintesis de pregabalina 69
3.11.2.1a. Análisis retrosintético 69
3.11.2.1b. Síntesis 69
3.11.2.1c. Cuestiones 70
3.11.2.2a. Análisis retrosintético de (S)-pregabalina mediante el empleo
de un auxiliar quiral 70
3.11.2.2b. Síntesis de (S)-pregabalina mediante el empleo de una
oxazolidinona quiral de Evans 71
3.11.2.2c. Cuestiones 72
3.11.2.3a. Análisis retrosintètico de (S)-pregabalina mediante el empleo
del pool quiral 74
3.11.2.3b. Síntesis de (S)-pregabalina a partir de L-leucina 75
3.11.2.3c. Cuestiones 76
3.11.3. Síntesis de rufinamida 77
3.11.3.a. Análisis retrosintético 78
3.11.3.b. Síntesis 79
3.11.3.c. Cuestiones 79

3.11.4. Síntesis de lacosamida 79
3.11.4.a. Análisis retrosintético 79 3.11.4.b. Síntesis 80
3.11.5. Síntesis de perampanel 81 3.11.5.a. Análisis retrosintético 82 3.11.5.b. Síntesis 83 3.11.5.c. Cuestiones 85
3.12. Enfermedad de Parkinson 87
3.12.1. Causas de la enfermedad de Parkinson 87
3.13. Fármacos antiParkinson 88
3.13.1. Levodopa 89
3.13.2. Agonistas de dopamina 89
3.13.3. Inhibidores de la monoaminooxidasa B: selegilina y rasagilina 91
3.13.4. Liberadores presinápticos de dopamina: amantadina 92
3.13.5. Antagonistas del receptor muscarínico de la acetilcolina: benztropina 93
3.14. Síntesis de fármacos antiParkinson 93
3.14.1. Síntesis de pramiprexol 93
3.14.1.a. Análisis retrosintético 93
3.14.1.b. Síntesis 94
3.14.1.c. Cuestiones 94
3.14.2. Síntesis de ropinirol 95
3.14.2.a. Análisis retrosintético 95
3.14.2.b. Síntesis 96
3.14.2.c. Cuestiones 97
3.14.2.2b. Síntesis a partir de isocromano 97
3.14.2.2c. Cuestiones 98
3.14.3. Síntesis de selegilina 99
3.13.3.a. Análisis retrosintético 99
3.13.3.b. Síntesis 99
3.13.3.c. Cuestiones 101
3.14.4. Síntesis de mesilato de rasagilina 102
3.13.4.a. Análisis retrosintético 102
3.13.4.b. Síntesis 102
Tema 3. Enfermedades del sistema nervioso central 1
3.1. Neurotransmisores
Los neurotransmisores (NT) son los compuestos encargados de transmitir el impulso nervioso entre neuronas. Estos metabolitos son sintetizados por enzimas existentes en el cuerpo neuronal y son almacenados en vesículas de las células presinápticas.
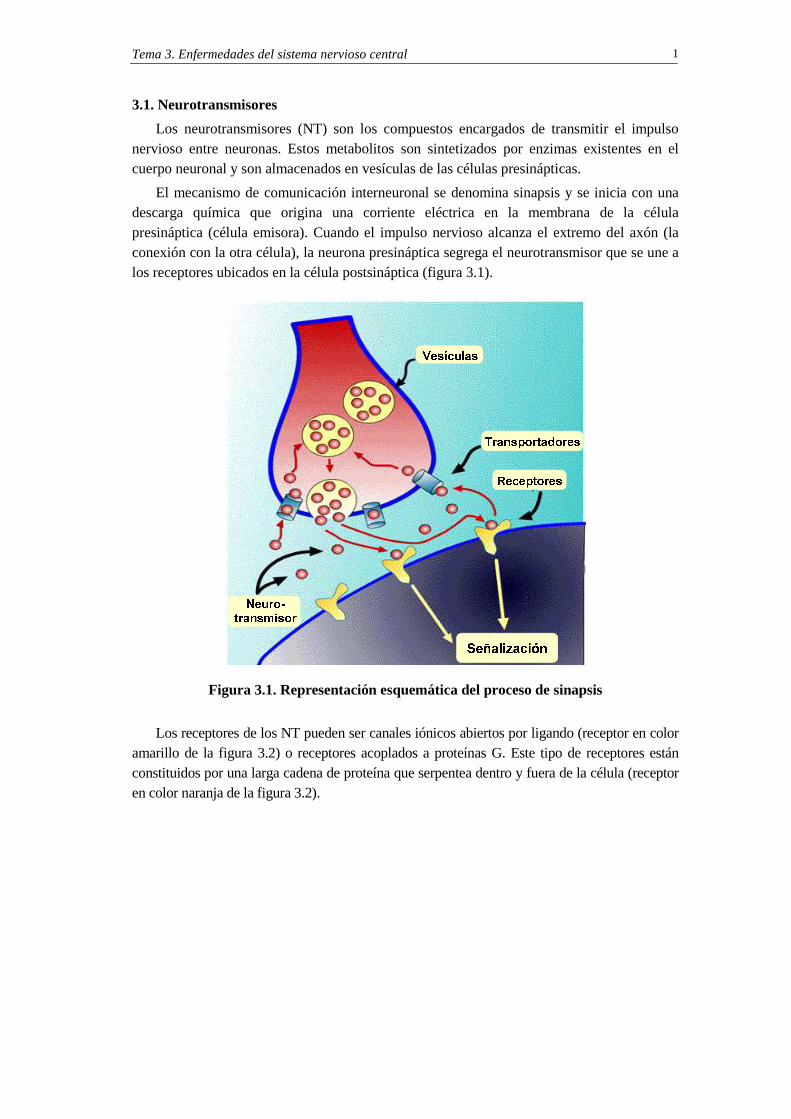
El mecanismo de comunicación interneuronal se denomina sinapsis y se inicia con una descarga química que origina una corriente eléctrica en la membrana de la célula presináptica (célula emisora). Cuando el impulso nervioso alcanza el extremo del axón (la conexión con la otra célula), la neurona presináptica segrega el neurotransmisor que se une a los receptores ubicados en la célula postsináptica (figura 3.1).
Figura 3.1. Representación esquemática del proceso de sinapsis
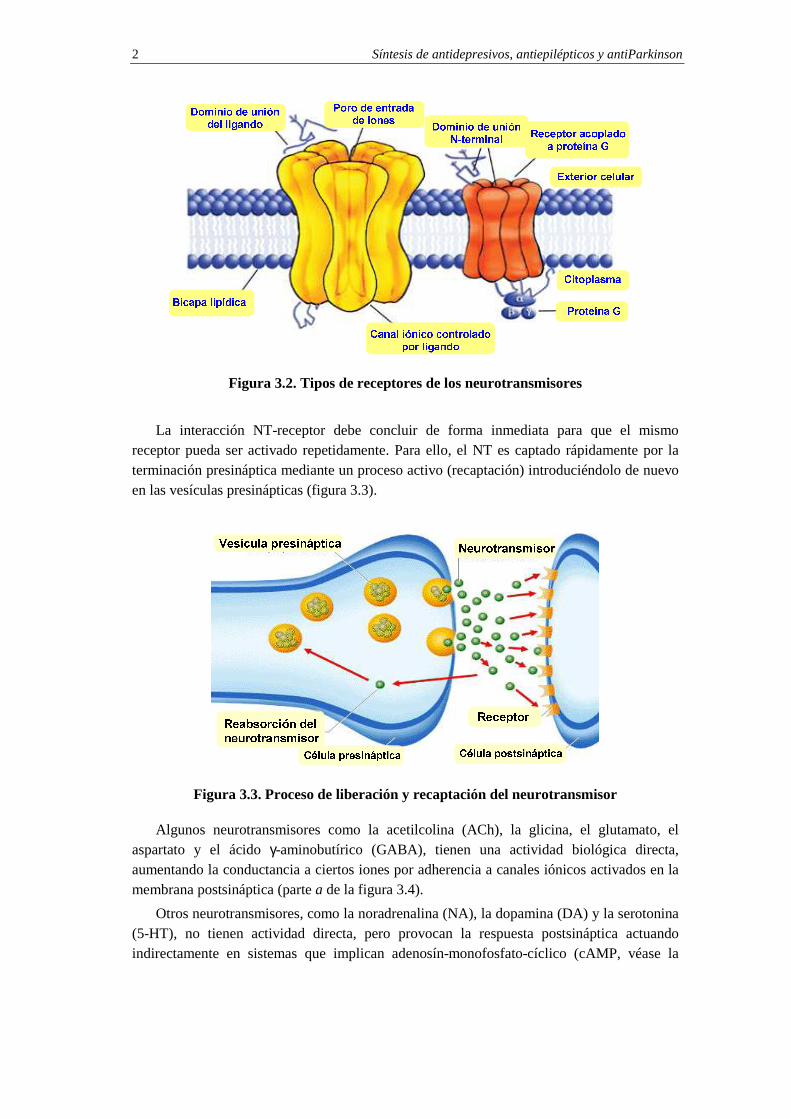
Los receptores de los NT pueden ser canales iónicos abiertos por ligando (receptor en color amarillo de la figura 3.2) o receptores acoplados a proteínas G. Este tipo de receptores están constituidos por una larga cadena de proteína que serpentea dentro y fuera de la célula (receptor en color naranja de la figura 3.2).
Síntesis de antidepresivos, antiepilépticos y antiParkinson 2
Figura 3.2. Tipos de receptores de los neurotransmisores
La interacción NT-receptor debe concluir de forma inmediata para que el mismo receptor pueda ser activado repetidamente. Para ello, el NT es captado rápidamente por la terminación presináptica mediante un proceso activo (recaptación) introduciéndolo de nuevo en las vesículas presinápticas (figura 3.3).
Figura 3.3. Proceso de liberación y recaptación del neurotransmisor
Algunos neurotransmisores como la acetilcolina (ACh), la glicina, el glutamato, el aspartato y el ácido γ-aminobutírico (GABA), tienen una actividad biológica directa, aumentando la conductancia a ciertos iones por adherencia a canales iónicos activados en la membrana postsináptica (parte a de la figura 3.4).
Otros neurotransmisores, como la noradrenalina (NA), la dopamina (DA) y la serotonina (5-HT), no tienen actividad directa, pero provocan la respuesta postsináptica actuando indirectamente en sistemas que implican adenosín-monofosfato-cíclico (cAMP, véase la
Tema 3. Enfermedades del sistema nervioso central 3
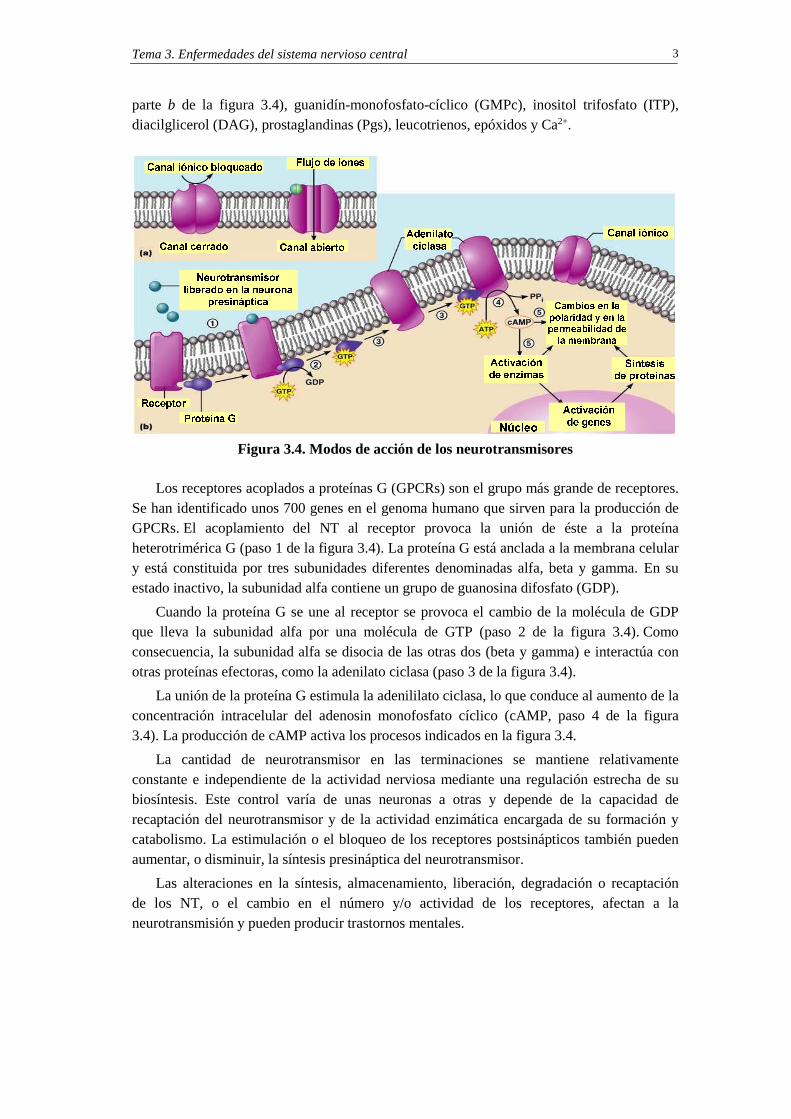
parte b de la figura 3.4), guanidín-monofosfato-cíclico (GMPc), inositol trifosfato (ITP), diacilglicerol (DAG), prostaglandinas (Pgs), leucotrienos, epóxidos y Ca2+.
Figura 3.4. Modos de acción de los neurotransmisores
Los receptores acoplados a proteínas G (GPCRs) son el grupo más grande de receptores. Se han identificado unos 700 genes en el genoma humano que sirven para la producción de GPCRs. El acoplamiento del NT al receptor provoca la unión de éste a la proteína heterotrimérica G (paso 1 de la figura 3.4). La proteína G está anclada a la membrana celular y está constituida por tres subunidades diferentes denominadas alfa, beta y gamma. En su estado inactivo, la subunidad alfa contiene un grupo de guanosina difosfato (GDP).
Cuando la proteína G se une al receptor se provoca el cambio de la molécula de GDP que lleva la subunidad alfa por una molécula de GTP (paso 2 de la figura 3.4). Como consecuencia, la subunidad alfa se disocia de las otras dos (beta y gamma) e interactúa con otras proteínas efectoras, como la adenilato ciclasa (paso 3 de la figura 3.4).
La unión de la proteína G estimula la adenililato ciclasa, lo que conduce al aumento de la concentración intracelular del adenosin monofosfato cíclico (cAMP, paso 4 de la figura 3.4). La producción de cAMP activa los procesos indicados en la figura 3.4.
La cantidad de neurotransmisor en las terminaciones se mantiene relativamente constante e independiente de la actividad nerviosa mediante una regulación estrecha de su biosíntesis. Este control varía de unas neuronas a otras y depende de la capacidad de recaptación del neurotransmisor y de la actividad enzimática encargada de su formación y catabolismo. La estimulación o el bloqueo de los receptores postsinápticos también pueden aumentar, o disminuir, la síntesis presináptica del neurotransmisor.
Las alteraciones en la síntesis, almacenamiento, liberación, degradación o recaptación de los NT, o el cambio en el número y/o actividad de los receptores, afectan a la neurotransmisión y pueden producir trastornos mentales.

Síntesis de antidepresivos, antiepilépticos y antiParkinson 4
3.1.1. Tipos de neurotransmisores
Los principales neurotransmisores pueden clasificarse según su tamaño en:
a) Neurotransmisores de pequeño tamaño de tipo aminoácido: glicina, ácido aspártico, ácido glutámico:
Figura 3.5. Aminoácidos con actividad neurotransmisora
La glicina deriva del metabolismo de la serina y es un NT que actúa en las interneuronas de la médula espinal.
Los aminoácidos glutamato y aspartato son los principales NT excitatorios del sistema nervioso central. Están presentes en la corteza cerebral, el cerebelo y la médula espinal.
b) Neurotransmisores de pequeño tamaño derivados de aminoácidos: GABA, histamina, serotonina, norepinefrina y dopamina:
Figura 3.6. Neurotransmisores derivados de aminoácidos
El ácido γ-aminobutírico (GABA) es el principal NT inhibitorio cerebral. Deriva del ácido glutámico mediante la descarboxilación provocada por la enzima glutamato-descarboxilasa. Tras la interacción con los receptores específicos, el GABA es recaptado y metabolizado.
La histamina es un NT del sistema nervioso central. También interviene decisivamente en las reacciones de hipersensibilidad inmediata y alérgica. La histamina se forma por descarboxilación del aminoácido histidina catalizada por el enzima L-histidina-descarboxilasa.
La serotonina (5-hidroxitriptamina) (5-HT) participa en el control de los estados de sueño y de vigilia. Interviene regulando los estados de ánimo y las emociones y es decisiva en el desencadenamiento de algunos tipos de depresión. También interviene en el control de la temperatura del cuerpo, de la conducta sexual y de ciertos estados alucinatorios inducidos por drogas. La serotonina se origina en el núcleo del rafe (estructuras del encéfalo) y en las neuronas de la línea media de la protuberancia y del meséncefalo.
La norepinefrina (noradrenalina) es el NT de la mayor parte de las fibras simpáticas postganglionares y de muchas neuronas centrales, por ejemplo del locus ceruleus y del hipotálamo. El precursor de la noradrenalina es la tirosina, que se convierte en dopamina,

Tema 3. Enfermedades del sistema nervioso central 5
que a su vez es hidroxilada por la dopamina β-hidroxilasa a noradrenalina. Cuando se libera la noradrenalina interactúa con los receptores adrenérgicos (véase el tema 2), proceso que finaliza con su recaptación por las neuronas presinápticas y su degradación por la monoaminoxidasa (MAO) y por la catecol-O-metiltransferasa (COMT). La tirosina-hidroxilasa y la MAO regulan los niveles intraneuronales de noradrenalina.
La dopamina es el NT de algunas fibras nerviosas y periféricas y de muchas neuronas centrales, por ejemplo en la sustancia negra, el diencéfalo, el área tegmental ventral del tronco cerebral y el hipotálamo. La dopamina se biosintetiza a partir del aminoácido tirosina que es captado por las neuronas dopaminérgicas y convertido en 3,4-dihidroxifenilalanina (dopa) por medio de la tirosina-hidroxilasa. La dopa se descarboxila hasta dopamina por la acción de la descarboxilasa de L-aminoácidos aromáticos. Tras ser liberada, la dopamina interactúa con los receptores dopaminérgicos y el complejo NT-receptor es captado de forma activa por las neuronas presinápticas. La tirosina-hidroxilasa y la MAO regulan las tasas de dopamina en la terminación nerviosa.
c) Neuropéptidos: metabolitos compuestos por más de 3 aminoácidos como la somatostatina, la vasopresina y la oxitocina. Muchos de estos neuropéptidos actúan también como hormonas, denominándose en estos casos neurohormonas.
d) Otros neurotransmisores: acetilcolina
Figura 3.7. Estructura de la acetilcolina
La acetilcolina es el NT fundamental de las neuronas motoras bulbo-espinales, las fibras preganglionares autónomas, las fibras colinérgicas postganglionares (parasimpáticas) y muchos grupos neuronales del SNC, como los de los ganglios basales y de la corteza motora. Se biosintetiza a partir de la colina y de la acetil-coenzima A mitocondrial mediante acción del enzima colinacetiltransferasa. Al ser liberada, la acetilcolina estimula receptores colinérgicos específicos y su interacción finaliza rápidamente por hidrólisis local a colina y acetato mediante la acción de la acetilcolinesterasa. Los niveles de acetilcolina están regulados por la acetilcolintransferasa y por el grado de recaptación de colina.
Los neurotransmisores también se pueden clasificar en función de su estructura química del siguiente modo:

Síntesis de antidepresivos, antiepilépticos y antiParkinson 6
Figura 3.8. Clasificación de los neurotransmisores en función de su estructura química
3.2. Inhibidores Selectivos de la Recaptación de Serotonina (ISRS)
La depresión severa (Trastorno Depresivo Mayor, en inglés Major Depressive Disorder MDD) se manifiesta por una combinación de síntomas como tristeza patológica, apatía, ansiedad, etc, que interfieren en la capacidad para trabajar, estudiar, dormir, comer y disfrutar de actividades que antes eran placenteras.
Se acepta en general que la depresión está relacionada con la reducción de la trasmisión del impulso nervioso en zonas específicas del sistema nervioso central provocada por un déficit de neurotransmisores en la sinapsis. De hecho, todos los antidepresivos actúan aumentando la concentración de aminas neurotransmisoras en la sinapsis.
Una vez producido el impulso nervioso, el 95% de aminas liberadas son vueltas a recaptar por la neurona presináptica en preparación del siguiente impulso. El 5% no recaptado es destruido por la enzima monoaminooxidasa (MAO). Las pérdidas de neurotransmisores son repuestas a partir de precursores metabólicos.
La serotonina se clasifica dentro del grupo de los neurotransmisores adrenérgicos, que son aquéllos que se unen a receptores acoplados a proteína G.

Tema 3. Enfermedades del sistema nervioso central 7
Figura 3.9. Unión de la setononina al receptor postsináptico
La serotonina, también denominada 5-hidroxitriptamina (5-HT) se genera en el organismo mediante hidroxilación del L-triptófano catalizada por el enzima triptófano-hidroxilasa (esquema 3.1). La hidroxilación del triptófano produce el 5-hidroxi-L-triptófano, el cual se transforma en serotonina por descarboxilación catalizada por el enzima 5-hidroxi-L-triptófano descarboxilasa.
Una vez liberada por la neurona presináptica, la serotonina puede ocupar receptores postsinápticos, recaptarse, ocupar autorreceptores o metabolizarse por la MAO mitocondrial y convertirse en ácido 5-hidroxi-indolacético.
Esquema 3.1. Biosíntesis de la serotonina
Los niveles de serotonina están regulados por la disponibilidad de L-triptófano y por la acción de la monoaminooxidasa (MAO). La biodegradación de la serotonina, se lleva a cabo tanto a nivel intracelular como en la hendidura sináptica por la acción enzimática de la MAO, que la convierte en su principal metabolito inactivo: el 5-hidroxi-indolacetaldehído. Este metabolito es oxidado por la enzima aldehído-deshidrogenasa y transformado en ácido 5-hidroxi-indolacético (esquema 3.2).

Síntesis de antidepresivos, antiepilépticos y antiParkinson 8
Esquema 3.2. Degradación enzimática de la serotonina
En humanos existen dos tipos de MAO: MAO-A y MAO-B. La MAO-A es particularmente importante en el catabolismo de monoaminas ingeridas con el alimento. Ambas MAOs juegan un papel clave en la inactivación de los neurotransmisores monoaminérgicos. Así, la serotonina, norepinefrina (noradrenalina), y epinefrina (adrenalina) son degradadas en su mayoría por la MAO-A. La fenetilamina es degradada por la MAO-B, mientras que la dopamina es degradada por ambas MAO.
La unión de la serotonina con el receptor debe concluir de forma inmediata para que el mismo receptor pueda ser activado repetidamente. Una de las vías de eliminación de la serotonina, a parte de su degradación por la MAO, es la recaptación de la misma por parte de la terminación presináptica. En la figura 3.10 se representan de forma esquemática los procesos de formación y recaptación de serotonina.
Neuronapresináptica
Triptófano
5-HTP
Serotonina
Liberación deserotonina
Proteína GReceptor
Neurona postsináptica
Serotonina
Recaptación
Autorreceptores
Destrucción pormonoamino-
oxidasa
Figura 3.10. Liberación y recaptación de serotonina
Los inhibidores selectivos de la recaptación de la serotonina (ISRS) son una clase de antidepresivos utilizados en el tratamiento de la depresión, trastorno por ansiedad y algunos trastornos de la personalidad. Actúan aumentando los niveles extracelulares del neurotransmisor serotonina, inhibiendo su recaptación por la neurona presináptica e

Tema 3. Enfermedades del sistema nervioso central 9
incrementando de esta forma el nivel de serotonina disponible para unirse con el receptor postsináptico.
Aunque existe serotonina en todo el cuerpo ésta es incapaz de atravesar la barrera hematoencefálica, por lo que el cerebro produce su propia serotonina. La biosíntesis de serotonina cerebral depende del aporte del aminoácido L-triptófano. Este es un aminoácido esencial y por tanto el organismo no lo puede biosintetizar. El L-triptófano sólo puede provenir de la dieta, por lo que sus niveles cerebrales dependen, en parte, de los alimentos ingeridos. El L-triptófano abunda en los huevos, la leche, los cereales integrales, el chocolate, la avena, los dátiles, las semillas de sésamo, los garbanzos, las pipas de girasol, las pipas de calabaza y los cacahuetes. Las personas que no ingieren estos alimentos tienen mayor riesgo de deficiencia de triptófano, así como aquellas personas sometidas a altos niveles de estrés. Para un buen metabolismo del triptófano se requieren niveles adecuados de vitamina B6 y de magnesio.
3.2.1. Liberación de la serotonina
La liberación de la serotonina se produce por exocitosis, que es un proceso calcio-dependiente. Así, los iones calcio transitan del exterior al interior de la célula a través de los canales iónicos que atraviesan la membrana de la célula. La apertura de un canal iónico puede lograrse mediante un cambio de voltaje (despolarización o llegada de potencial de acción) o por unión de una sustancia química a un receptor. En la figura 3.11 se indica una representación esquemática de un canal iónico.
Representación esquemática de un canal iónico
1= Dominio del canal iónico. 2=Vestíbulo externo3= Filtro de selección. 4=Diámetro del filtro de selección
5=Sitio de fosforilación. 6=Membrana celular
Figura 3.11. Estructura de un canal inónico
Los canales iónicos de calcio son proteínas oligoméricas constituidos por una subunidad principal α1, que sirve como poro y sensor del cambio de potencial, y diversas subunidades reguladoras o auxiliares tales como la subunidad β, las subunidades α2σ (unidas por puentes disulfuro) y, dependiendo del tejido, una quinta subunidad (véase la figura 3.12).

Síntesis de antidepresivos, antiepilépticos y antiParkinson 10
Figura 3.12. Vista superior de un canal de calcio
Cuando llega un impulso nervioso a la neurona presináptica ésta abre los canales de Ca2+
y los iones entran en la neurona lo que activa el proceso de exocitosis provocándose el traslado de las vesículas a los lugares de su liberación con la ayuda de proteínas de membrana plasmática y de la membrana vesicular. Este proceso desemboca en la liberación del neurotransmisor en el espacio sináptico.
3.2.2. Receptores de serotonina
Los principales receptores de serotonina son el 5-HT1, el 5-HT2 y el 5-HT3. Éstos, a su vez, se subdividen en cuatro subtipos del 5-HT1 (de la A a la D), dos del 5-HT2 (A y B) y uno del 5-HT3. De todos ellos, la mayoría son postsinápticos, pero al menos dos de ellos (el 5-HT1B y el 5-HT1D) pueden ser autorreceptores, modulando la liberación del neurotransmisor. Los receptores de serotonina se localizan en la membrana celular de las células nerviosas. Con la excepción del receptor 5-HT3, que es un canal iónico asociado a ligando, los demás receptores son receptores acoplados a proteínas G, que son también conocidos como receptores 7TM (transmembrana) o "en serpentina", debido a la región incluida en la membrana, que asoma siete veces. En la figura 3.13 se representa esquemáticamente la estructura de un receptor GPCR de serotonina (5-HT).
Figura 3.13. Representación esquemática de un receptor GPCR de serotonina (5-HT)

Tema 3. Enfermedades del sistema nervioso central 11
En la figura inferior 3.14 se ilustra una representación del receptor visto desde la cara extracelular. La flecha señala la zona de interacción con el neurotransmisor.
NT
Figura 3.14. Vista superior extracelular de un receptor GPCR
Los receptores 5-HT1A están asociados a la apertura de canales de K+, presumiblemente de forma directa a través de una proteína G. En las áreas del campo terminal como el hipocampo, los receptores 5-HT1A están también asociados, mediante proteína G, a la inhibición de la actividad de la adenilciclasa.
Los receptores 5-HT1B y 5-HT1D también están asociados a la inhibición de adenilciclasa a través de la proteína G.
Los receptores 5-HT1C y 5-HT2 están asociados a través de la proteína G a la estimulación de la hidrólisis de fosfoinositol (PI).
El receptor 5-HT3 es de tipo canal iónico, por lo que su activación no es mediada por segundo mensajero o a través de proteínas G.
El receptor 5-HT4 está asociado a la estimulación de la actividad de la adenilciclasa y a la inhibición de canales de K+. Se ha demostrado que la inhibición de canales de K+ en neuronas del colículo implica la producción de adenosin monofosfato cíclico (AMPc) y la activación de proteína-quinasa A dependiente de AMPc.
Figura 3.15. Acción de los receptores en el proceso de liberación y recaptación de serotonina

Síntesis de antidepresivos, antiepilépticos y antiParkinson 12
3.3. Otros inhibidores selectivos de recaptación de serotonina Los ISRS pertenecen a una subclase de inhibidores de la recaptación de serotonina que
incluye también a otros inhibidores no selectivos como:
a) Los inhibidores de la recaptación de serotonina-noradrenalina-dopamina.
b) Los inhibidores de la recaptación de serotonina-norepinefrina.
c) Los estimulantes selectivos de la recaptación de serotonina. 3.4. Fármacos inhibidores selectivos de la recaptación de la serotonina
Las primeras moléculas empleadas en el tratamiento del Trastorno Depresivo Mayor fueron los denominados antidepresivos tricíclicos, como la imipramina, que se introdujeron en el mercado en la década de 1950. En la década de l960 se introdujeron los inhibidores de monoaminooxidasa (IMAO), de entre los cuales cabe destacar a la isocarboxazida.
N
Imipramina
ON
NH
HN
O
Isocarboxazida
NMe2
Figura 3.16. Estructuras de fármacos empleados originalmente contra la depresión
Los inhibidores de MAO (IMAO) ejercen su acción antidepresiva aumentando los niveles de monaminas, como la serotonina, la norepinefrina o la dopamina. Desafortunadamente los inhibidores de MAO tienen importantes efectos secundarios, entre los que destaca la supresión de la reabsorción de tiramina, por lo que se debe evitar la administración de inhibidores de MAO con la ingesta de alimentos que contengan una alta concentración de tiramina, tales como alimentos fermentados, arenques, o hígado de pollo, ya que la combinación de tiramina con IMAO puede provocar hemorragias cerebrales debido a aumentos bruscos de la presión arterial.
Figura 3.17. Estructura de la tiramina
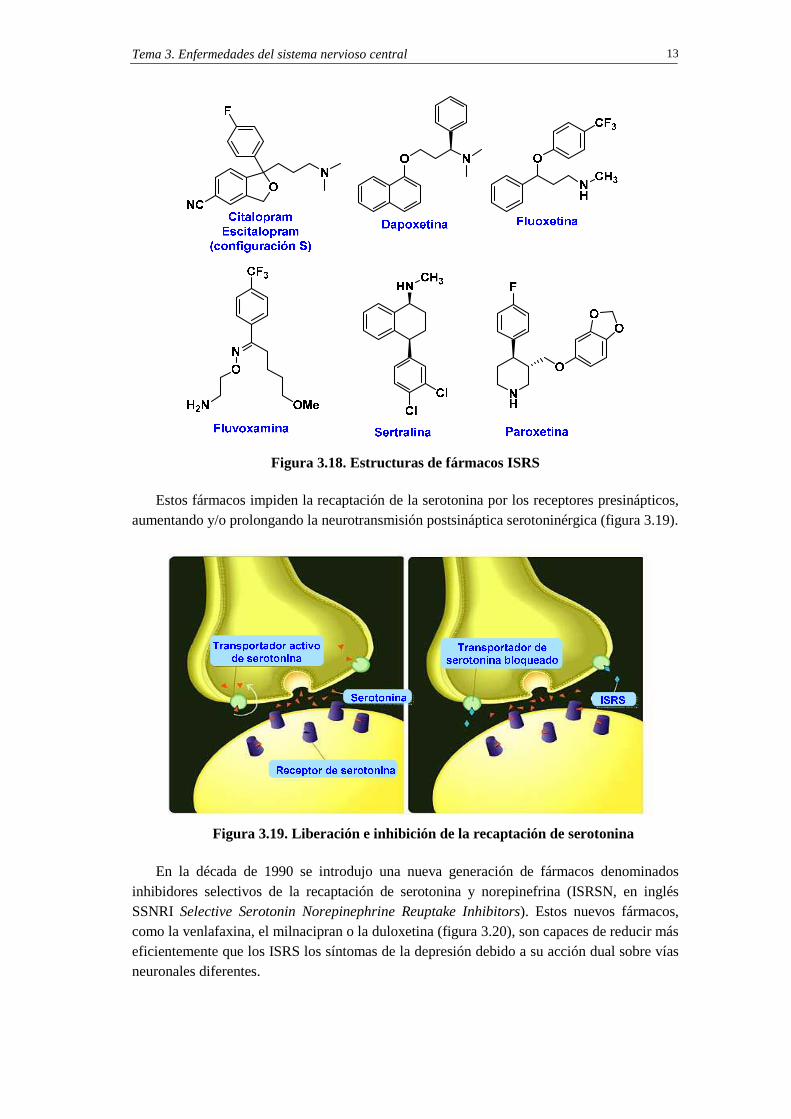
Las estructuras de fármacos inhibidores selectivos de la recaptación de la serotonina (ISRS) se indican en la figura 3.18.
Tema 3. Enfermedades del sistema nervioso central 13
Figura 3.18. Estructuras de fármacos ISRS
Estos fármacos impiden la recaptación de la serotonina por los receptores presinápticos, aumentando y/o prolongando la neurotransmisión postsináptica serotoninérgica (figura 3.19).
Figura 3.19. Liberación e inhibición de la recaptación de serotonina
En la década de 1990 se introdujo una nueva generación de fármacos denominados inhibidores selectivos de la recaptación de serotonina y norepinefrina (ISRSN, en inglés SSNRI Selective Serotonin Norepinephrine Reuptake Inhibitors). Estos nuevos fármacos, como la venlafaxina, el milnacipran o la duloxetina (figura 3.20), son capaces de reducir más eficientemente que los ISRS los síntomas de la depresión debido a su acción dual sobre vías neuronales diferentes.
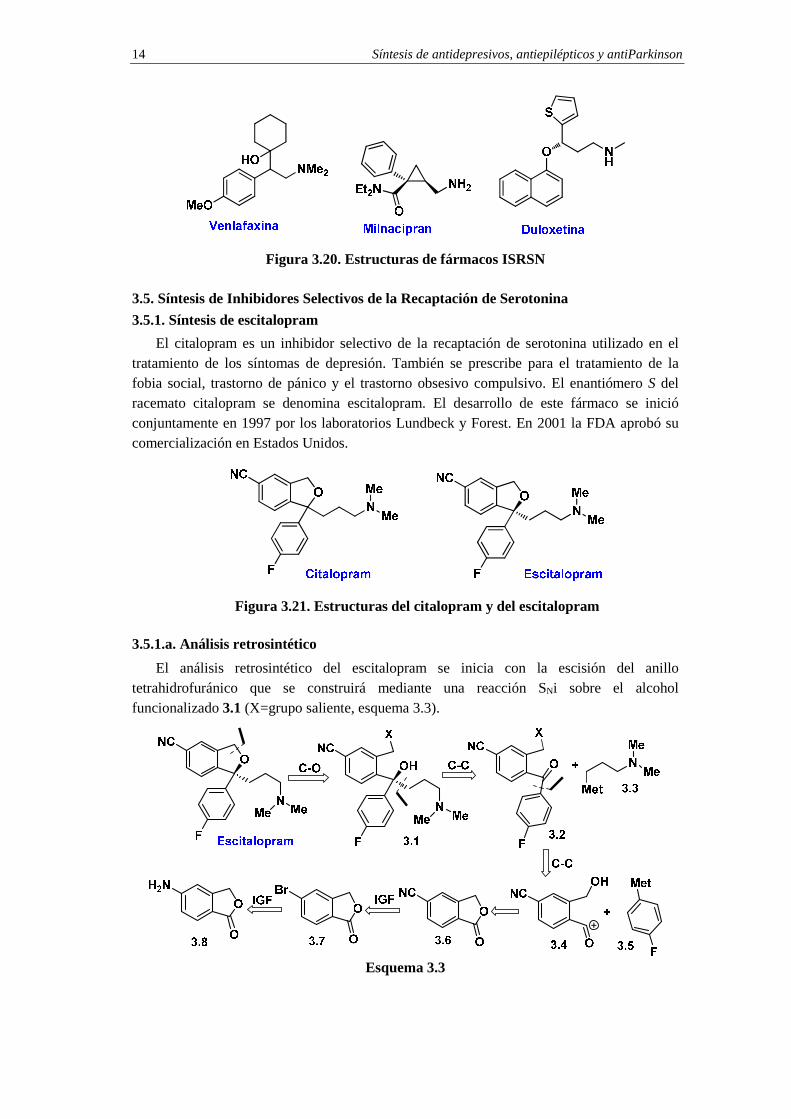
Síntesis de antidepresivos, antiepilépticos y antiParkinson 14
Figura 3.20. Estructuras de fármacos ISRSN
3.5. Síntesis de Inhibidores Selectivos de la Recaptación de Serotonina
3.5.1. Síntesis de escitalopram
El citalopram es un inhibidor selectivo de la recaptación de serotonina utilizado en el tratamiento de los síntomas de depresión. También se prescribe para el tratamiento de la fobia social, trastorno de pánico y el trastorno obsesivo compulsivo. El enantiómero S del racemato citalopram se denomina escitalopram. El desarrollo de este fármaco se inició conjuntamente en 1997 por los laboratorios Lundbeck y Forest. En 2001 la FDA aprobó su comercialización en Estados Unidos.
Figura 3.21. Estructuras del citalopram y del escitalopram
3.5.1.a. Análisis retrosintético
El análisis retrosintético del escitalopram se inicia con la escisión del anillo tetrahidrofuránico que se construirá mediante una reacción SNi sobre el alcohol funcionalizado 3.1 (X=grupo saliente, esquema 3.3).
Esquema 3.3
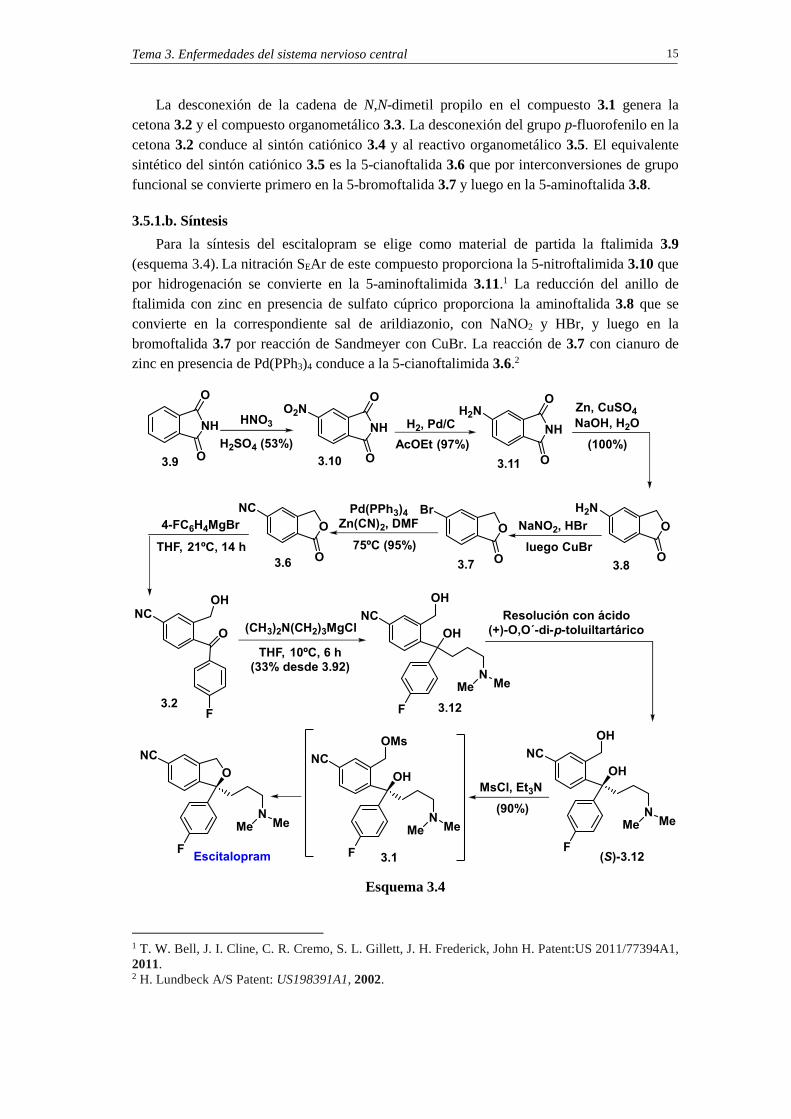
Tema 3. Enfermedades del sistema nervioso central 15
La desconexión de la cadena de N,N-dimetil propilo en el compuesto 3.1 genera la cetona 3.2 y el compuesto organometálico 3.3. La desconexión del grupo p-fluorofenilo en la cetona 3.2 conduce al sintón catiónico 3.4 y al reactivo organometálico 3.5. El equivalente sintético del sintón catiónico 3.5 es la 5-cianoftalida 3.6 que por interconversiones de grupo funcional se convierte primero en la 5-bromoftalida 3.7 y luego en la 5-aminoftalida 3.8.
3.5.1.b. Síntesis
Para la síntesis del escitalopram se elige como material de partida la ftalimida 3.9 (esquema 3.4). La nitración SEAr de este compuesto proporciona la 5-nitroftalimida 3.10 que por hidrogenación se convierte en la 5-aminoftalimida 3.11.1 La reducción del anillo de ftalimida con zinc en presencia de sulfato cúprico proporciona la aminoftalida 3.8 que se convierte en la correspondiente sal de arildiazonio, con NaNO2 y HBr, y luego en la bromoftalida 3.7 por reacción de Sandmeyer con CuBr. La reacción de 3.7 con cianuro de zinc en presencia de Pd(PPh3)4 conduce a la 5-cianoftalimida 3.6.2
NC
O
OBr
O
O
O
NH
3.6 3.7 3.8
O
H2N
O
O
3.9
O2N
O
NH
O
3.10
H2N
O
NH
O
3.11
H2SO4 (53%)
H2, Pd/C
AcOEt (97%)
Zn, CuSO4
NaOH, H2O
(100%)
HNO3
NaNO2, HBr
luego CuBr75ºC (95%)
(CH3)2N(CH2)3MgCl
Pd(PPh3)4Zn(CN)2, DMF
THF, 21ºC, 14 h
4-FC6H4MgBr
NC
OH
F
NC
O
F
OH
NMe Me
3.2
THF, 10ºC, 6 h(33% desde 3.92)
OH
3.12
Resolución con ácido(+)-O,O´-di-p-toluiltartárico
NC
OH
F
NMe Me
OH
(S)-3.12
NC
O
FEscitalopram
NMe Me
MsCl, Et3N
NC
OH
F
NMe Me
OMs
(90%)
3.1
Esquema 3.4
1 T. W. Bell, J. I. Cline, C. R. Cremo, S. L. Gillett, J. H. Frederick, John H. Patent:US 2011/77394A1, 2011. 2 H. Lundbeck A/S Patent: US198391A1, 2002.

Síntesis de antidepresivos, antiepilépticos y antiParkinson 16
Cuando el compuesto 3.6 se trata con bromuro de 4-fluorofenilmagnesio se obtiene la hidroxicetona 3.2, que por reacción con el bromuro de (3-dimetilaminopropil)magnesio se transforma en el aminodiol 3.12. La resolución de este compuesto se consigue con el ácido (+)-O,O -di-p-toluiltartárico, lo que permite la obtención del (S)-3.12. Cuando este compuesto se trata con cloruro de mesilo se genera el mesilato 3.1 que se convierte en escitalopram por reacción SNi.3
3.5.1.c. Cuestiones 1) ¿Por qué la reducción de 3.11 para dar 3.8 es regioselectiva?
2) Explique mecanísticamente la conversión de 3.7 en 3.6.
3.5.2. Síntesis de dapoxetina
La dapoxetina es un inhibidor selectivo de la recaptación de serotonina de corta duración de acción. Su poca eficacia como antidepresivo llevó a investigar otras aplicaciones terapéuticas como el tratamiento de la eyaculación precoz.
3.5.2.1a. Análisis retrosintético
En el esquema 3.5 de indica un análisis retrosintético de la dapoxetina que se inicia con la desconexión del sistema naftalénico basado en una reacción SNAr. La operacicón retrosintética conduce al aminoalcohol 3.13 (el nucleófilo de la reacción SNAr) y el α-halonaftaleno 3.14. La operación de intercambio de grupo funcional en el aminoalcohol 3.13 genera el β-aminoéster 3.15 que se sintetizará mediante adición conjugada Michael de la dimetilanina al éster conjugado 3.16.
Esquema 3.5
3.5.2.1b. Síntesis
La síntesis de la dapoxetina se inicia con la adición conjugada de la dimetilamina al cinamato de etilo 3.16 lo que proporciona el β-aminoéster 3.15 (esquema 3.6).4 Este compuesto, por reducción con LiAlH4 se convierte en el aminoalcohol 3.13. Cuando este
3 Para patentes relacionados con el citalopram y el escitalopram véase: (a) K. P. Boegesoe, J. Perregaard, Patente: US4943590 1990. (b) K. P. Boegesoe, A. S. Toft, Patente: US4136193 1979. (c) H. Ahmadian, H. Petersen, Patente: WO03051861 2003. (d) K. P. Boegesoe, Patente: US4650884 1987. 4 W. J. Wheeler, D. D. O’Bannon. J. Labelled. Compd. Radiopharm. 1992, 31, 305-315.
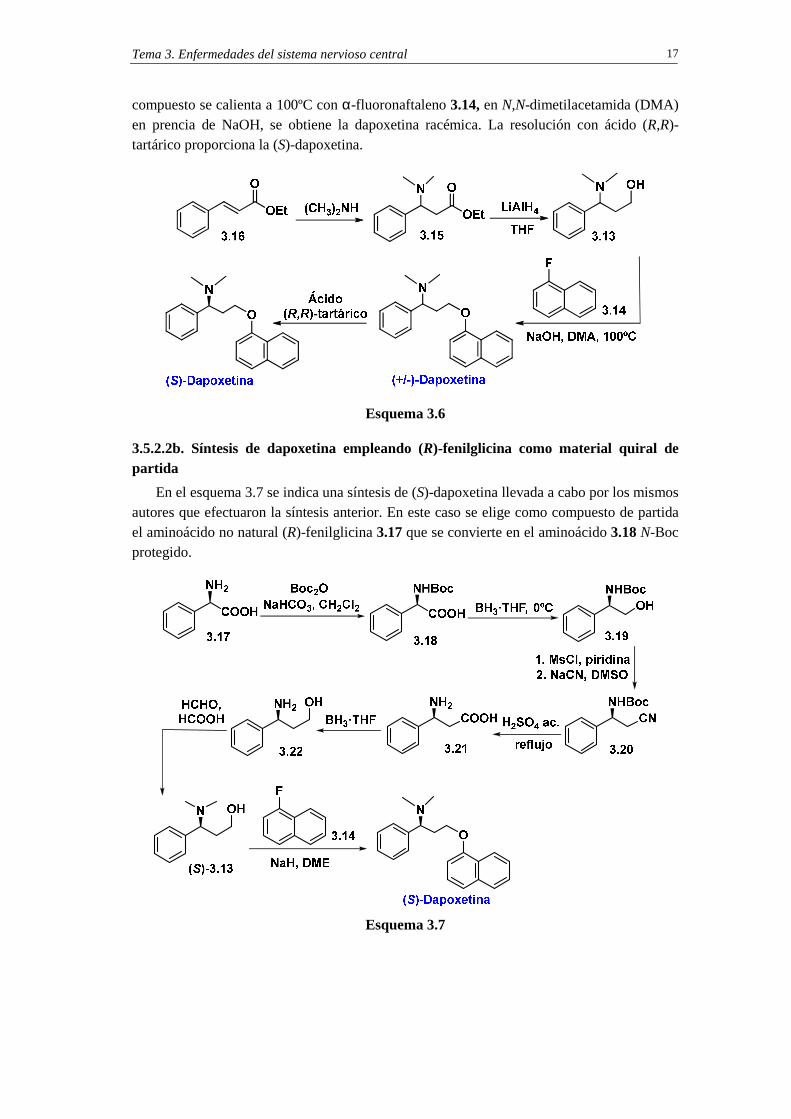
Tema 3. Enfermedades del sistema nervioso central 17
compuesto se calienta a 100ºC con α-fluoronaftaleno 3.14, en N,N-dimetilacetamida (DMA) en prencia de NaOH, se obtiene la dapoxetina racémica. La resolución con ácido (R,R)-tartárico proporciona la (S)-dapoxetina.
Esquema 3.6
3.5.2.2b. Síntesis de dapoxetina empleando (R)-fenilglicina como material quiral de partida
En el esquema 3.7 se indica una síntesis de (S)-dapoxetina llevada a cabo por los mismos autores que efectuaron la síntesis anterior. En este caso se elige como compuesto de partida el aminoácido no natural (R)-fenilglicina 3.17 que se convierte en el aminoácido 3.18 N-Boc protegido.
Esquema 3.7

Síntesis de antidepresivos, antiepilépticos y antiParkinson 18
La reducción de 3.18 con borano proporciona el alcohol 3.19 el cual, mediante mesilación y reacción con cianuro sódico, se convierte en el nitrilo 3.20. La hidrólisis de este compuesto conduce al aminoácido 3.21 que por reducción con borano forma el aminoalcohol 3.21. La metilación de Eschweiler-Clarke de 3.21 lo convierte en el N,N-dimetilaminoalcohol (S)-3.13. La (S)-dapoxetina se obtiene mediante reacción SNAr de (S)-3.13 con α-fluronaftaleno en 1,2-dimetoxietano (DME) en presencia de hidruro sódico.
3.5.2.2c. Cuestiones
1) Explique mecanísticamente la reacción de metilación de Eschweiler-Clarke que permite la obtención de (S)-3.13 a partir de 3.22.
3.5.3. Síntesis de fluoxetina (Prozac)
La fluoxetina se comercializa en forma de racemato, puesto que ambos enantiómeros presentan similar actividad in vitro. Sin embargo, el enantiómero con mayor poder terapéutico es la (S)-fluoxetina debido a que es eliminada más lentamente que el enantiómero (R). Investigaciones recientes han apuntado la posibilidad de que la duración prolongada del enantiómero (S) sea la causante de las contraindicaciones del fármaco. 3.5.3.a. Análisis retrosintético
La fluoxetina (Prozac) se comercializó por primera vez en 1986. A pesar de que ya lleva en el mercado 25 años, sigue siendo uno de los fármacos antidepresivos más recetados.
En el esquema 3.8 se indica un análisis retrosintético para la fluoxetina. El proceso de desconexión comienza con la escisión del enlace C-O. La escisión del enlace C-O conduce al p-trifluorometilfenol 3.23 y la amina funcionalizada 3.24 (X=grupo saliente), cuyo precursor será el aminoalcohol 3.25. El aumento del estado de oxidación de la función hidroxilo genera la β-aminocetona 3.26, cuya desconexión, basada en una reacción de tipo Mannich, conduce a la acetofenona 3.27, el formaldehído 3.28 y la amina 3.29.
Esquema 3.8

Tema 3. Enfermedades del sistema nervioso central 19
3.5.3.b. Síntesis
La síntesis de la fluoxetina se describe en el esquema 3.9 y comienza con la reacción de Mannich entre la acetofenona 3.27, el formaldehído 3.28 y la dimetilamina 3.30.5 La reacción de Mannich proporciona la β-aminocetona 3.31, que se transforma en el aminoalcohol 3.32 mediante reducción de la función cetónica. La transformación del alcohol 3.32 en el cloruro 3.33, seguida de reacción SN2 con p-triflurometilfenóxido, generado in situ a partir del p-trifluorometilfenol, conduce al compuesto 3.34. Para conseguir la conversión de 3.34 en fluoxetina se debe llevar a cabo la N-desmetilación reductiva del grupo N,N-dimetilamino. Esta operación se consigue en una secuencia de dos pasos. En primer lugar el N,N-dimetilamino, compuesto 3.34, se convierte en la N-metil-N-cianoamina 3.35 por reacción con bromuro de cianógeno (BrCN). A continuación, el compuesto 3.35 se transforma en fluoxetina por descianación reductiva con KOH acuoso en etilenglicol.
3.283.27
HO
CF3
3.23
O
NCH3
O
CH3 HNCH3
O
H H +
3.30CH3CH3
3.31
B2H6, THF
23ºC, 16 h
OH
NCH3
CH33.32
SOCl2, HCl
CHCl3, reflujo
Cl
NCH3
CH3NaOH, MeOH, reflujo
O
NCH3
CF3
FluoxetinaCH3
3.33
3.34
BrCN, PhMe
rt, 16 h
O
NCN
CF3
CH3
KOH, H2O
130ºC, 20 h
etilenglicol
3.35
+
Esquema 3.9
La fluoxetina preparada según la síntesis que se describe en el esquema 3.9 se obtiene en forma racémica. Conviene indicar que ambos enantiómeros presentan una actividad similar in vitro. Así, la constante de inhibición Ki (concentración de fármaco necesaria para inhibir la mitad de la actividad enzimática in vitro) para la serotonina, en cortex de rata, es de 21 nM para el enantiómero (S)-(+) y de 33 nM para el enantiómero (R)-(-). El enantiómero S es el que lleva a cabo la mayor parte del efecto terapéutico, ya que este enantiómero es eliminado más lentamente que el enantiómero R.
3.5.3.c. Cuestiones
1) Explique mecanísticamente la formación del compuesto 3.31 mediante la reacción de Mannich. ¿Por qué se utiliza dimetilamina en lugar de metilamina en esta reacción?
5 (a) B. B. Molloy, K. K. Schmiegel, US Patent 1982, 4,314,081. (b) J. Saunders Top Drugs. Top Synthetic Routes. Ed. Oxford University Press 2000.

Síntesis de antidepresivos, antiepilépticos y antiParkinson 20
2) ¿Cuál es el mecanismo que explica la conversión del compuesto 3.34 en la N-cianoamina 3.35?
3) Explique mecanísticamente la conversión de la N-cianoamina 3.35 en la fluoxetina.
3.5.4. Síntesis de sertralina
La sertralina, también conocida por las marcas comerciales Zoloft®, Altruline®, Sertex® o Besitrán®, es un antidepresivo perteneciente al grupo de los ISRS (Inhibidores Selectivos de la Recaptación de Serotonina). Actúa inhibiendo la recaptación de la serotonina en el espacio intersináptico por parte de la neurona emisora, lo cual aumenta la disponibilidad de la misma. La sertralina no tiene afinidad sobre el bloqueo de la recaptación de la noradrenalina (norepinefrina) y dopamina.6
La sertralina se utiliza principalmente en el tratamiento de la depresión, esté o no asociada con estados de ansiedad, en el tratamiento del trastorno por estrés postraumático, en el trastorno obsesivo compulsivo, en los ataques de pánico, en el trastorno esquizoide de la personalidad y en la fobia social.
3.5.4.a. Análisis retrosintético
La sertralina se desarrolló en los laboratorios Pfizer en los cuales se había descubierto que una serie de trans-1-amino-4-ariltetralinas presentaban potente actividad biológica en la reabsorción de norepinefrina. La actividad era altamente específica para el trans-(1R,4S) (figura 3.22). El enantiómero trans-(1S,4R) era mucho menos activo y el racemato cis era inactivo en el bloqueo de la reabsorción de norepinefrina. Posteriormente a estos descubrimientos se encontró que los isómeros cis eran potentes inhibidores de la reabsorción de serotonina.
Figura 3.22
El compuesto (1S,4S)-cis-1-amino-4-ariltetralina se denominó sertralina. Este enantiómero dextrogiro es mucho más potente en la inhibición de la reabsorción de serotonina que el enantiómero levogiro (1R,4R). Por tanto, y en contraposición a la fluoxetina, la sertralina se comercializa únicamente en su forma de enantiómero (1S,4S).
En el esquema 3.10 se indica un análisis retrosintético para la sertralina que se inicia con una operación de intercambio del grupo funcional metilamina por cetona. La dihidronaftalenona 3.36 genera en la operación IGF se convierte en el ácido 4-arilfenilbutanoico 3.37 mediante una operación retrosintética basada en una reacción SEAr intramolecular. El análisis se continúa con la adición de un doble enlace en el punto de
6 W. M. Welch, A. R. Kraska, R. Sarges, B. K. Koe. J. Med. Chem. 1984, 27, 1508-1515.

Tema 3. Enfermedades del sistema nervioso central 21
ramificación de la estructura 3.37. Esta operación genera la olefina 3.38 que se desconecta en el doble enlace a la cetona 3.39 y al sintón nucleofílico 3.40. Este sintón no tiene existencia real y su equivalente sintético es el anión 3.41, derivado del succinato de dialquilo.
Esquema 3.10
3.5.4.b. Síntesis
La síntesis de sertralina se inicia con la condensación de Stobbe entre la diarilcetona 3.39 y el succinato de dietilo 3.42 (esquema 3.11).7 La condensación de Stobbe se lleva a cabo en presencia de t-butóxido de potasio como base y proporciona el acidoéster insaturado 3.43. El tratamiento del compuesto 3.243 con HBr en ácido acético provoca la hidrólisis de la función éster y la subsiguiente descarboxilación. El resultado final es la formación del ácido insaturado 3.38, que por hidrogenación se convierte en el ácido 4-arilfenilbutanoico 3.37. La construcción del anillo de dihidronaftalenona se consigue mediante reacción SEAr intramolecular del cloruro de ácido derivado de 3.37. El producto de la ciclación, compuesto 3.36, se convierte en la N-metilimina 3.44 por condensación con metilamina en presencia de TiCl4. La hidrogenación de 3.44, en presencia de Pd/C al 10%, proporciona una mezcla racémica de cis- y trans-aminas diastereoisoméricas (+/-)-3.45 y (+/-)-3.46, en relación 70:30 respectivamente. La cis-amina racémica (+/-)-3.45 se separa de la mezcla mediante cristalización fraccionada en forma de clorhidrato. Finalmente, la sertralina se obtiene en forma ópticamente activa por resolución óptica de la mezcla racémica (+/-)-3.45 con ácido D-(-)-mandélico (ácido (R)-α-hidroxifenilacético).
7 (a) M. Williams, G. Quallich. Chem. & Ind. (London) 1990, 10, 315-319. (b) G. J. Quallich, T. M. Woodall, Tetrahedron 1992, 48, 10239. (c) E. J. Corey, T. G. Gant. Tetrahedron Lett. 1994, 35, 5373-5376.
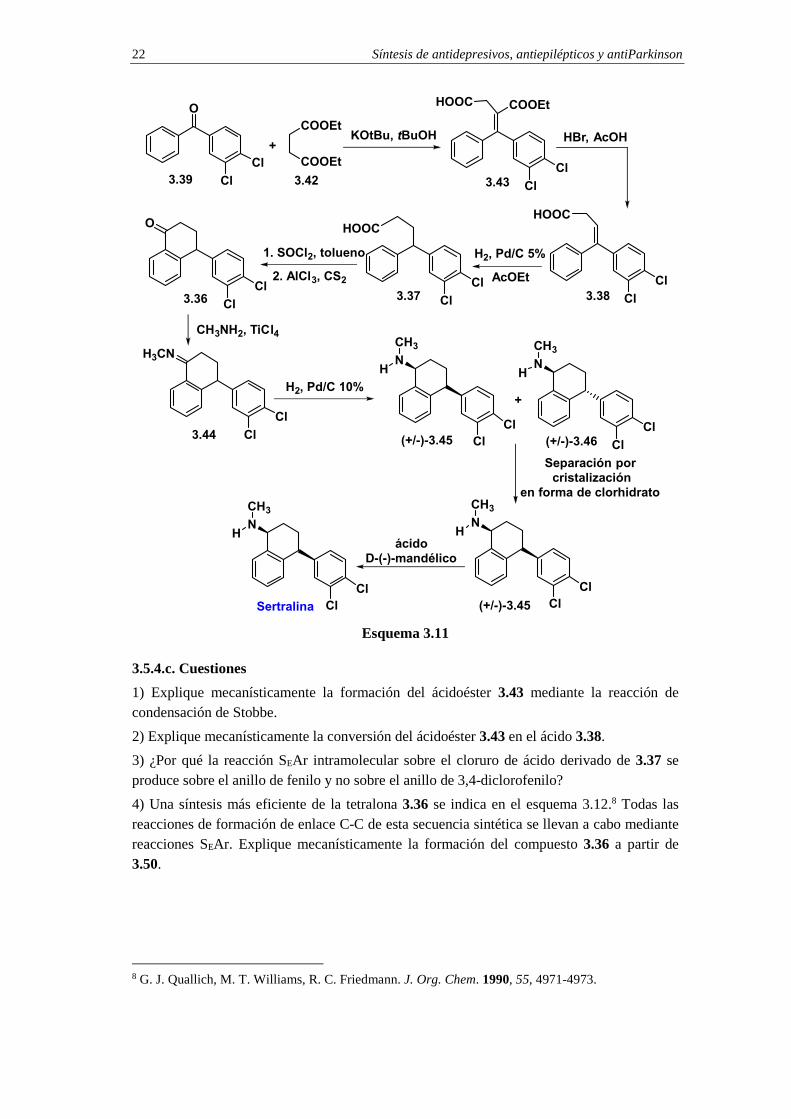
Síntesis de antidepresivos, antiepilépticos y antiParkinson 22
O
Cl
Cl3.39 3.42
COOEt
COOEt
+KOtBu, tBuOH
HOOC
Cl
Cl
COOEt
3.43
HBr, AcOH
HOOC
Cl
Cl3.38
H2, Pd/C 5%
AcOEt
HOOC
Cl
Cl3.37
1. SOCl2, tolueno
2. AlCl3, CS2
Cl
Cl
O
CH3NH2, TiCl4
Cl
Cl
H3CN
3.36
3.44
H2, Pd/C 10%
ClCl
N
(+/-)-3.45
Separación porcristalización
en forma de clorhidrato
Cl
Cl
N
(+/-)-3.46
ácido D-(-)-mandélico
Sertralina
+
ClCl
N
(+/-)-3.45
H
CH3
H
CH3
H
CH3
ClCl
NH
CH3
Esquema 3.11
3.5.4.c. Cuestiones
1) Explique mecanísticamente la formación del ácidoéster 3.43 mediante la reacción de condensación de Stobbe.
2) Explique mecanísticamente la conversión del ácidoéster 3.43 en el ácido 3.38.
3) ¿Por qué la reacción SEAr intramolecular sobre el cloruro de ácido derivado de 3.37 se produce sobre el anillo de fenilo y no sobre el anillo de 3,4-diclorofenilo?
4) Una síntesis más eficiente de la tetralona 3.36 se indica en el esquema 3.12.8 Todas las reacciones de formación de enlace C-C de esta secuencia sintética se llevan a cabo mediante reacciones SEAr. Explique mecanísticamente la formación del compuesto 3.36 a partir de 3.50.
8 G. J. Quallich, M. T. Williams, R. C. Friedmann. J. Org. Chem. 1990, 55, 4971-4973.

Tema 3. Enfermedades del sistema nervioso central 23
Esquema 3.12
5) En el esquema 3.13 se describe una síntesis enantioselectiva de la tetralona (S)-3.36. En esta síntesis se parte del cetoácido 3.49 que se convierte en el t-butiléster 3.51.9 El paso clave en la secuencia sintética es la reducción enantioselectiva del carbonilo cetónico de 3.51 que se lleva a cabo con BH3 y una oxazaborolidina quiral derivada de prolina. La reacción proporciona el hidroxiéster 3.52 con un rendimiento químico del 100% y con un exceso enantioselectivo del 90%. La mesilación del hidroxilo forma el mesilato 3.53 que experimenta una reacción de tipo SN2, con inversión de la configuración, por reacción con el difenilcianocuprato de dilitio (Ph2Cu(CN)Li2). Esta reacción conduce al diariléster 3.54 que se convierte en la tetralona quiral (S)-3.36 mediante reacción SEAr intramolecular promovida por ácido trifluoroacético. La tetralona (S)-3.36 se transforma en sertralina mediante la secuencia de reacciones indicada en el esquema 3.11.
Cl
Cl
O
OOH
3.49 Cl
Cl
O
OOtBu
isobutileno, H2SO4
diclorobenceno(92%)
3.51
CBS/BH3
(100%, 90%ee)
Cl
Cl
O
HOOtBu
MsCl, Et3N
Cl
Cl
O
MsOOtBu
Ph2Cu(CN)Li2
Cl
Cl
O
OtBu
3.52
3.533.54
TFA(94%)
Cl
Cl
O
sertralina
(S)-3.36
(70% 2 pasos)
Esquema 3.13
El método de reducción enantioselectiva de cetonas, aplicado en la síntesis del cetoéster 3.51 se debe a E. J. Corey, R. K. Bakshi y S. Shibata y se conoce como método CBS, por
9 G. J. Quallich, T. M. Woodall. Tetrahedron, 1992, 48, 10239-10248.

Síntesis de antidepresivos, antiepilépticos y antiParkinson 24
las iniciales de estos tres autores.10 Este método permite la reducción enantioselectiva de determinado tipo de cetonas por reacción con BH3, en presencia de oxazaborolidinas quirales, que se preparan a partir de L-prolina. En el esquema 3.14 se describe la preparación de la oxazaborolidina quiral (S)-3.55 a partir de L-prolina.
Esquema 3.14
La oxazaborolidina enantiomérica (R)-3.55 se obtiene mediante un proceso similar que implica una etapa de resolución (esquema 3.15).
Esquema 3.15
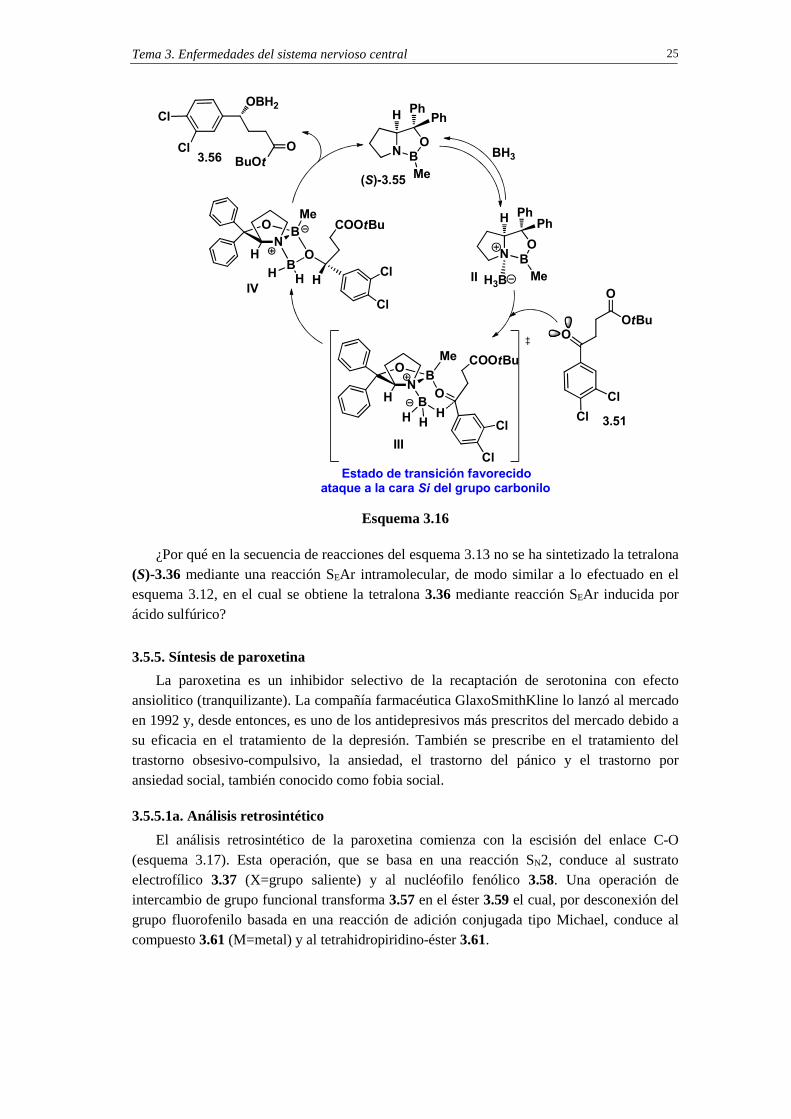
En el esquema 3.16 se indica el ciclo catalítico de la reducción enantioselectiva con el método CBS. En el primer paso se produce la coordinación del borano al átomo de nitrógeno de la oxazaborolidina (S)-3.55. Esta coordinación activa el BH3 como dador de hidruro y aumenta la acidez de Lewis del boro endocíclico del catalizador. A continuación, el átomo de boro del catalizador se coordina a la cetona 3.51 por el par de electrones no enlazantes estéricamente más accesibles, que es el par electrónico en sin con respecto del sustituyente estéricamente menos impedido. Esta coordinación disminuye las interacciones estéricas entre la cetona y el catalizador, puesto que el sustituyente más voluminoso de la cetona está orientado en trans, y por tanto alejado del grupo metilo del catalizador. En el esquema 3.16 se dibuja la conformación del estado de transición (estructura III ), en la que se observa cómo el carbonilo cetónico y el borano adquieren un orientación que permite la transferencia favorable de hidruro desde la cara Si de la cetona mediante la intervención de un estado de transición de seis eslabones. La transferencia de hidruro produce el alcoxiborano 3.34, que por hidrólisis ácida proporciona el alcohol 3.30.
10 (a) E. J. Corey, S. Shibata, R. K. Bakshi. J. Org. Chem. 1988, 53, 2861-2863. (b) E. J. Corey, K. Bakshi, S. Shibata, C. Chen, V. K. Singh. J. Am. Chem. Soc. 1987, 109, 7925-7926.
Tema 3. Enfermedades del sistema nervioso central 25
BH3
N BO
H PhPh
MeH3B
Estado de transición favorecidoataque a la cara Si del grupo carbonilo
(S)-3.55
II
III
IV
NO
B
Me
OBH
HH H
NO
B
Me
OB
H
HH H
N BO
H PhPh
Me
Cl
Cl
O
OOtBu
3.51
Cl
Cl
COOtBu
COOtBu
Cl
Cl
Cl
Cl O
OBH2
BuOt3.56
Esquema 3.16
¿Por qué en la secuencia de reacciones del esquema 3.13 no se ha sintetizado la tetralona (S)-3.36 mediante una reacción SEAr intramolecular, de modo similar a lo efectuado en el esquema 3.12, en el cual se obtiene la tetralona 3.36 mediante reacción SEAr inducida por ácido sulfúrico?
3.5.5. Síntesis de paroxetina
La paroxetina es un inhibidor selectivo de la recaptación de serotonina con efecto ansiolitico (tranquilizante). La compañía farmacéutica GlaxoSmithKline lo lanzó al mercado en 1992 y, desde entonces, es uno de los antidepresivos más prescritos del mercado debido a su eficacia en el tratamiento de la depresión. También se prescribe en el tratamiento del trastorno obsesivo-compulsivo, la ansiedad, el trastorno del pánico y el trastorno por ansiedad social, también conocido como fobia social.
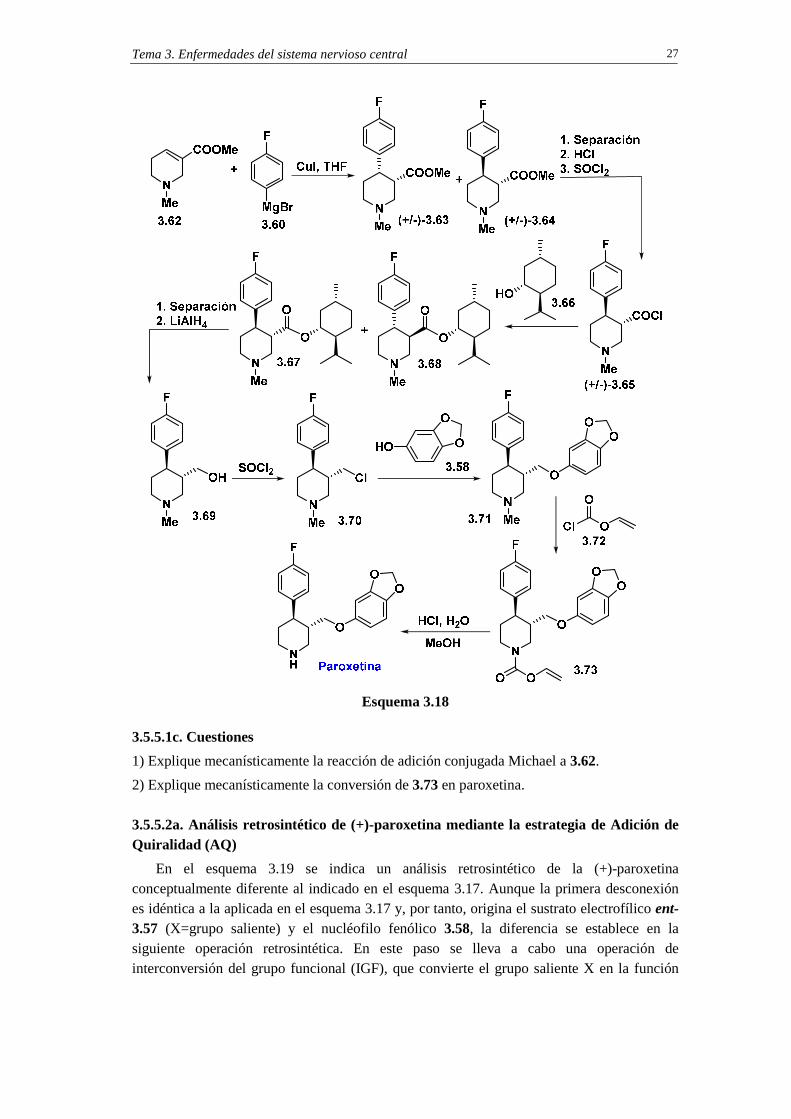
3.5.5.1a. Análisis retrosintético
El análisis retrosintético de la paroxetina comienza con la escisión del enlace C-O (esquema 3.17). Esta operación, que se basa en una reacción SN2, conduce al sustrato electrofílico 3.37 (X=grupo saliente) y al nucléofilo fenólico 3.58. Una operación de intercambio de grupo funcional transforma 3.57 en el éster 3.59 el cual, por desconexión del grupo fluorofenilo basada en una reacción de adición conjugada tipo Michael, conduce al compuesto 3.61 (M=metal) y al tetrahidropiridino-éster 3.61.

Síntesis de antidepresivos, antiepilépticos y antiParkinson 26
Esquema 3.17
3.5.5.1b. Síntesis
Para la síntesis de la paroxetina se elige como material de partida la arecolina 3.62 (esquema 3.18), un alcaloide de origen natural que se obtiene de la nuez de areca, una palmera originaria de Indonesia. La adición conjugada a la arecolina del p-flurorofenilcuprato de litio, generado in situ a partir del bromuro de p-flurorofenilmagnesio 3.50, proporciona una mezcla de los diastereoisómeros cis y trans, ambos obtenidos como racematos (compuestos (+/-)-3.63 y (+/-)-3.64).11
La separación de la mezcla diastereoisomérica permite obtener el diastereoisómero trans puro (+/-)-3.64 el cual, por hidrólisis de la función éster y reacción con cloruro de tionilo, se transforma en el cloruro de ácido (+/-)-3.65. En este punto se lleva a cabo el proceso de separación de enantiómeros. Para ello, la mezcla racémica (+/-)-3.65 se esterifica con (-)-mentol (3.66) y la mezcla de diastereoisómeros formada por los ésteres 3.67 y 3.68 se separa por destilación fraccionada, lo que permite la obtención del compuesto 3.67 puro. La reducción de 3.67 con LiAlH4 conduce al alcohol 3.69, que se activa frente al proceso SN2 mediante conversión en el cloruro 3.70. La reacción SN2 de 3.70 con sesamol 3.58 proporciona la N-metilparoxetina 3.71. La paroxetina se obtiene en dos pasos a partir de 3.71: en el primero de ellos se produce la transformación de 3.71 en el vinilcarbamato 3.73 y en el segundo la metanolisis ácida del carbamato.
11 J. A. Christensen, R. F. Squires. US Patent 1977, 4,007,196.
Tema 3. Enfermedades del sistema nervioso central 27
Esquema 3.18
3.5.5.1c. Cuestiones
1) Explique mecanísticamente la reacción de adición conjugada Michael a 3.62.
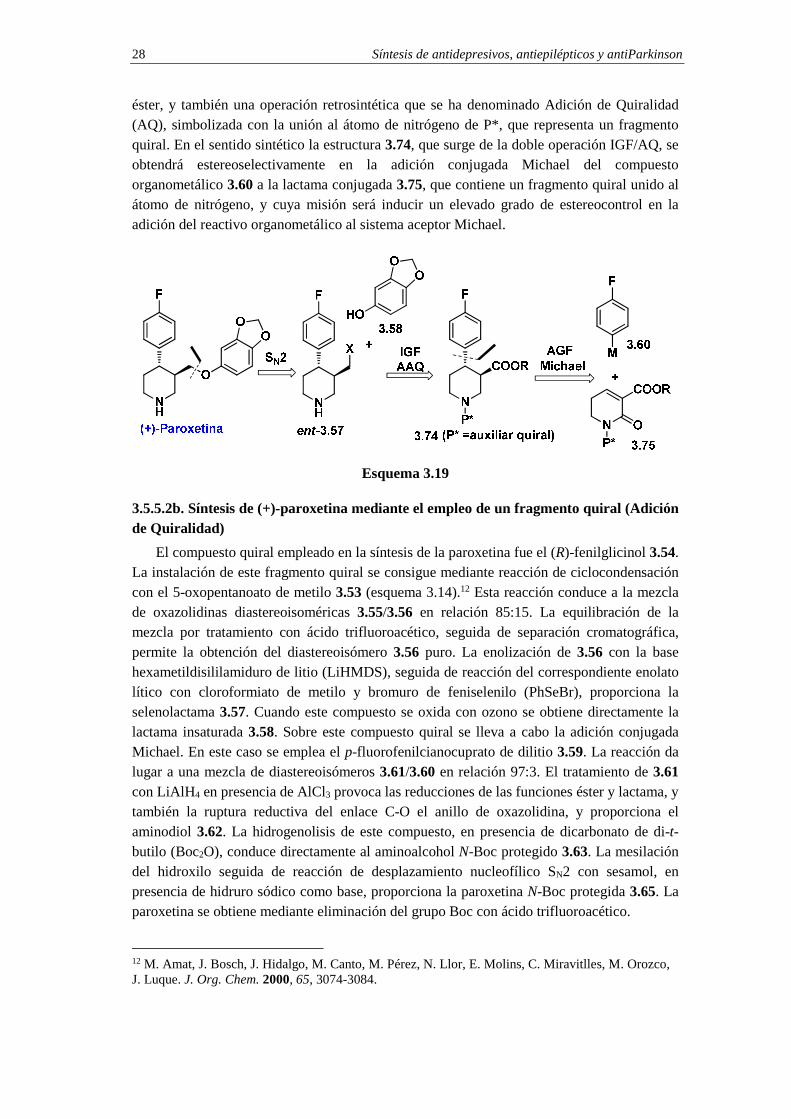
2) Explique mecanísticamente la conversión de 3.73 en paroxetina. 3.5.5.2a. Análisis retrosintético de (+)-paroxetina mediante la estrategia de Adición de Quiralidad (AQ)
En el esquema 3.19 se indica un análisis retrosintético de la (+)-paroxetina conceptualmente diferente al indicado en el esquema 3.17. Aunque la primera desconexión es idéntica a la aplicada en el esquema 3.17 y, por tanto, origina el sustrato electrofílico ent-3.57 (X=grupo saliente) y el nucléofilo fenólico 3.58, la diferencia se establece en la siguiente operación retrosintética. En este paso se lleva a cabo una operación de interconversión del grupo funcional (IGF), que convierte el grupo saliente X en la función
Síntesis de antidepresivos, antiepilépticos y antiParkinson 28
éster, y también una operación retrosintética que se ha denominado Adición de Quiralidad (AQ), simbolizada con la unión al átomo de nitrógeno de P*, que representa un fragmento quiral. En el sentido sintético la estructura 3.74, que surge de la doble operación IGF/AQ, se obtendrá estereoselectivamente en la adición conjugada Michael del compuesto organometálico 3.60 a la lactama conjugada 3.75, que contiene un fragmento quiral unido al átomo de nitrógeno, y cuya misión será inducir un elevado grado de estereocontrol en la adición del reactivo organometálico al sistema aceptor Michael.
Esquema 3.19
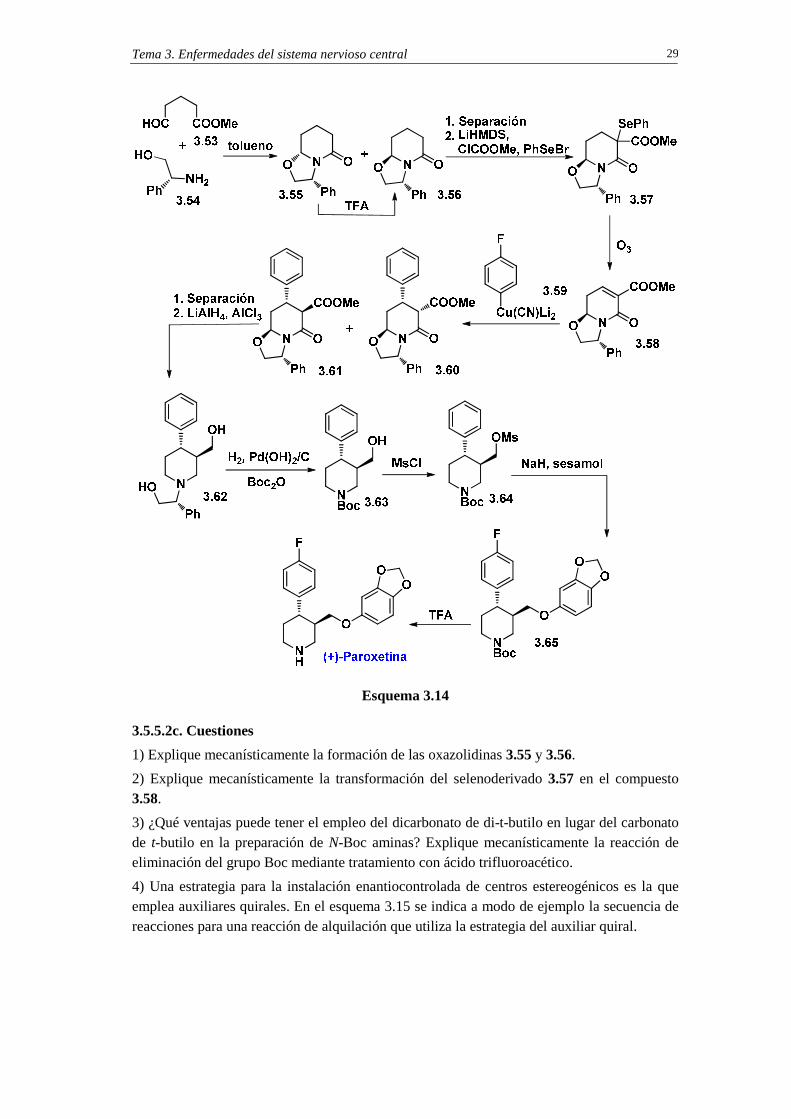
3.5.5.2b. Síntesis de (+)-paroxetina mediante el empleo de un fragmento quiral (Adición de Quiralidad)
El compuesto quiral empleado en la síntesis de la paroxetina fue el (R)-fenilglicinol 3.54. La instalación de este fragmento quiral se consigue mediante reacción de ciclocondensación con el 5-oxopentanoato de metilo 3.53 (esquema 3.14).12 Esta reacción conduce a la mezcla de oxazolidinas diastereoisoméricas 3.55/3.56 en relación 85:15. La equilibración de la mezcla por tratamiento con ácido trifluoroacético, seguida de separación cromatográfica, permite la obtención del diastereoisómero 3.56 puro. La enolización de 3.56 con la base hexametildisililamiduro de litio (LiHMDS), seguida de reacción del correspondiente enolato lítico con cloroformiato de metilo y bromuro de feniselenilo (PhSeBr), proporciona la selenolactama 3.57. Cuando este compuesto se oxida con ozono se obtiene directamente la lactama insaturada 3.58. Sobre este compuesto quiral se lleva a cabo la adición conjugada Michael. En este caso se emplea el p-fluorofenilcianocuprato de dilitio 3.59. La reacción da lugar a una mezcla de diastereoisómeros 3.61/3.60 en relación 97:3. El tratamiento de 3.61 con LiAlH4 en presencia de AlCl3 provoca las reducciones de las funciones éster y lactama, y también la ruptura reductiva del enlace C-O el anillo de oxazolidina, y proporciona el aminodiol 3.62. La hidrogenolisis de este compuesto, en presencia de dicarbonato de di-t-butilo (Boc2O), conduce directamente al aminoalcohol N-Boc protegido 3.63. La mesilación del hidroxilo seguida de reacción de desplazamiento nucleofílico SN2 con sesamol, en presencia de hidruro sódico como base, proporciona la paroxetina N-Boc protegida 3.65. La paroxetina se obtiene mediante eliminación del grupo Boc con ácido trifluoroacético.
12 M. Amat, J. Bosch, J. Hidalgo, M. Canto, M. Pérez, N. Llor, E. Molins, C. Miravitlles, M. Orozco, J. Luque. J. Org. Chem. 2000, 65, 3074-3084.
Tema 3. Enfermedades del sistema nervioso central 29
Esquema 3.14
3.5.5.2c. Cuestiones
1) Explique mecanísticamente la formación de las oxazolidinas 3.55 y 3.56.
2) Explique mecanísticamente la transformación del selenoderivado 3.57 en el compuesto 3.58.
3) ¿Qué ventajas puede tener el empleo del dicarbonato de di-t-butilo en lugar del carbonato de t-butilo en la preparación de N-Boc aminas? Explique mecanísticamente la reacción de eliminación del grupo Boc mediante tratamiento con ácido trifluoroacético.
4) Una estrategia para la instalación enantiocontrolada de centros estereogénicos es la que emplea auxiliares quirales. En el esquema 3.15 se indica a modo de ejemplo la secuencia de reacciones para una reacción de alquilación que utiliza la estrategia del auxiliar quiral.

Síntesis de antidepresivos, antiepilépticos y antiParkinson 30
Esquema 3.15
En una primera etapa el auxiliar quiral (Xq* en el esquema anterior) se une de forma covalente al sustrato aquiral, compuesto 3.66 del esquema anterior. Esta reacción proporciona el compuesto 3.67, que ya es quiral debido a que ha incorporado en su estructura el auxiliar quiral. Si, por ejemplo, lo que se pretende es llevar a cabo una reacción de alquilación asimétrica, el sustrato quiral 3.67 se enoliza y el correspondiente enolato se trata con el agente alquilante (R´X en el esquema 3.15). Como el enolato es quiral provocará inducción asimétrica en la reacción de alquilación y el resultado será la formación diastereoselectiva del compuesto alquilado 3.68. En una etapa posterior el auxiliar quiral se elimina del sustrato, obteniéndose el producto de alquilación 3.69 de forma enantioselectiva.
Las condiciones que debe cumplir un auxiliar quiral son las siguientes: a) Se debe poder instalar en el sustrato a enolizar con alto rendimiento y pureza óptica. b) Debe ser estable a las condiciones de enolización y alquilación. c) Debe inducir una elevada selectividad diastereofacial. d) Debe ser fácilmente recuperado mediante desinstalación del sustrato en condiciones que no provoquen pérdida de pureza óptica.
A tenor de todo lo explicado anteriormente ¿se puede calificar el empleo del (R)-fenilglicinol en la síntesis de la paroxetina como un ejemplo de aplicación de la estrategia de auxiliar quiral?
3.5.5.3a. Análisis retrosintético de paroxetina mediante una estrategia de desimetrización
En el esquema 3.16 se indica un análisis retrosintético de paroxetina que emplea el concepto de desimetrización. La primera desconexión es similar a la efectuada en los dos análisis retrosintéticos precedentes y, por tanto, origina el sustrato electrofílico 3.35 (X=grupo saliente) y el nucléofilo fenólico 3.36. En el segundo paso de la retrosíntesis se llevan a cabo simultáneamente dos operaciones retrosintéticas. Una de ellas es una adición del grupo funcional carbonilo (AGF) y convierte la amina en lactama. La otra es una operación de interconversión de grupo funcional en la cual se transforma la parte del grupo saliente en un grupo funcional éster. El resultado es la generación de la estructura 3.70 la cual, por escisión del grupo alcoxicarbonilo, conduce a la lactama 3.71. La desconexión del enlace lactámico en 3.71 forma el aminoéster 3.72 que derivará del éster funcionalizado 3.73 (X=grupo saliente) que es un compuesto quiral. Una operación IGF en este último compuesto conduce al diéster aquiral 3.74. Para lograr la síntesis enantioselectiva de la paroxetina el diéster aquiral 3.74 se deberá someter a un proceso de desimetrización mediante, por ejemplo, la hidrólisis enantioselectiva de uno de los dos grupos éster.
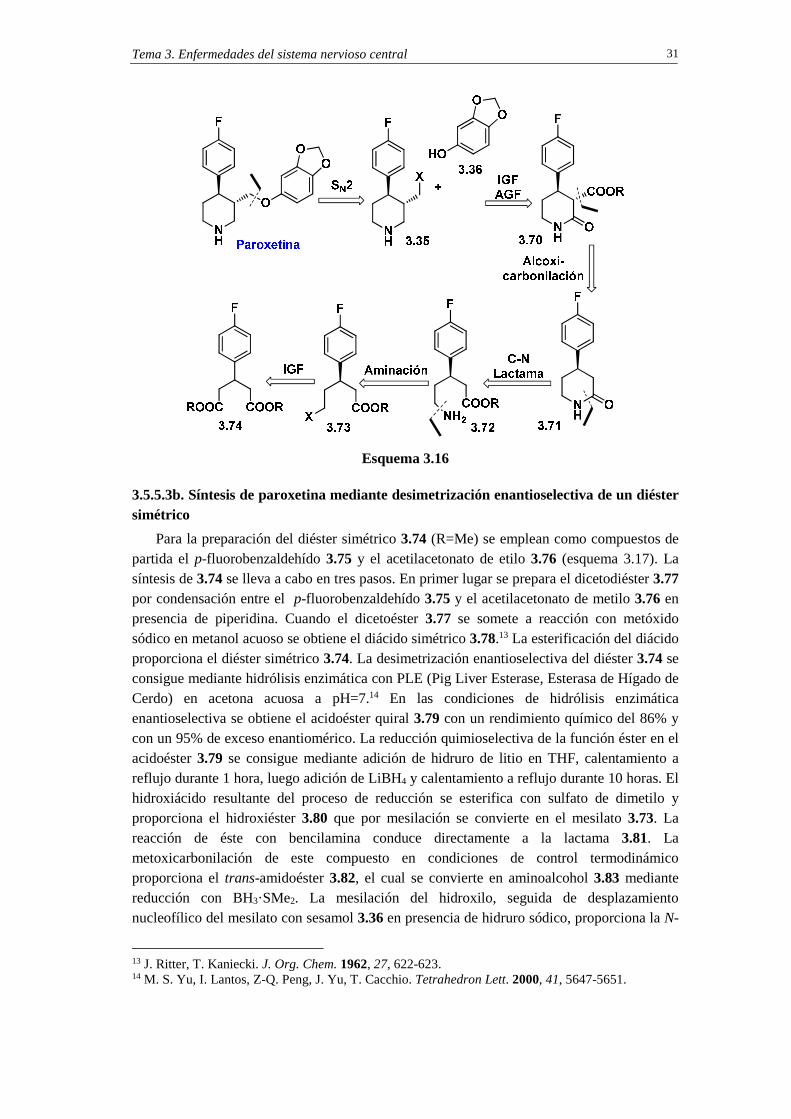
Tema 3. Enfermedades del sistema nervioso central 31
Esquema 3.16
3.5.5.3b. Síntesis de paroxetina mediante desimetrización enantioselectiva de un diéster simétrico
Para la preparación del diéster simétrico 3.74 (R=Me) se emplean como compuestos de partida el p-fluorobenzaldehído 3.75 y el acetilacetonato de etilo 3.76 (esquema 3.17). La síntesis de 3.74 se lleva a cabo en tres pasos. En primer lugar se prepara el dicetodiéster 3.77 por condensación entre el p-fluorobenzaldehído 3.75 y el acetilacetonato de metilo 3.76 en presencia de piperidina. Cuando el dicetoéster 3.77 se somete a reacción con metóxido sódico en metanol acuoso se obtiene el diácido simétrico 3.78.13 La esterificación del diácido proporciona el diéster simétrico 3.74. La desimetrización enantioselectiva del diéster 3.74 se consigue mediante hidrólisis enzimática con PLE (Pig Liver Esterase, Esterasa de Hígado de Cerdo) en acetona acuosa a pH=7.14 En las condiciones de hidrólisis enzimática enantioselectiva se obtiene el acidoéster quiral 3.79 con un rendimiento químico del 86% y con un 95% de exceso enantiomérico. La reducción quimioselectiva de la función éster en el acidoéster 3.79 se consigue mediante adición de hidruro de litio en THF, calentamiento a reflujo durante 1 hora, luego adición de LiBH4 y calentamiento a reflujo durante 10 horas. El hidroxiácido resultante del proceso de reducción se esterifica con sulfato de dimetilo y proporciona el hidroxiéster 3.80 que por mesilación se convierte en el mesilato 3.73. La reacción de éste con bencilamina conduce directamente a la lactama 3.81. La metoxicarbonilación de este compuesto en condiciones de control termodinámico proporciona el trans-amidoéster 3.82, el cual se convierte en aminoalcohol 3.83 mediante reducción con BH3·SMe2. La mesilación del hidroxilo, seguida de desplazamiento nucleofílico del mesilato con sesamol 3.36 en presencia de hidruro sódico, proporciona la N-
13 J. Ritter, T. Kaniecki. J. Org. Chem. 1962, 27, 622-623. 14 M. S. Yu, I. Lantos, Z-Q. Peng, J. Yu, T. Cacchio. Tetrahedron Lett. 2000, 41, 5647-5651.
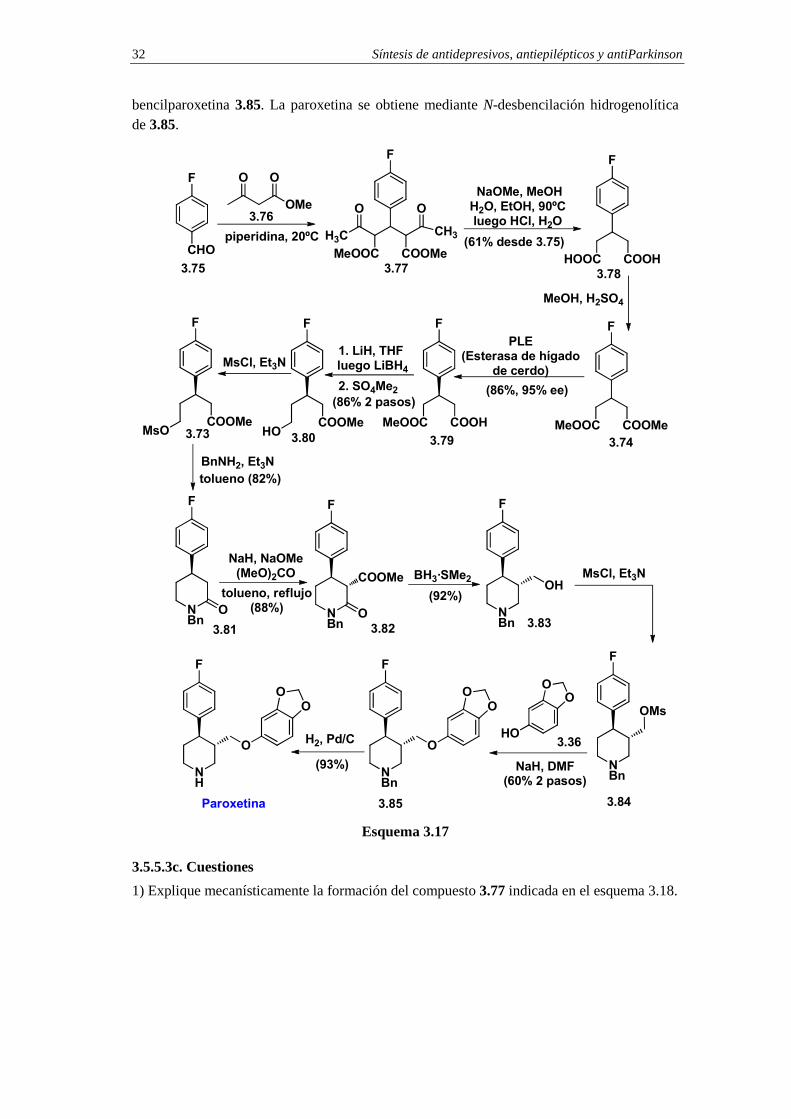
Síntesis de antidepresivos, antiepilépticos y antiParkinson 32
bencilparoxetina 3.85. La paroxetina se obtiene mediante N-desbencilación hidrogenolítica de 3.85.
CHO
F
OMe
O O
piperidina, 20ºCMeOOC COOMe
F
O
CH3
O
H3C
NaOMe, MeOHH2O, EtOH, 90ºCluego HCl, H2O
(61% desde 3.75)HOOC COOH
F
3.75 3.77 3.78
MeOH, H2SO4
MeOOC COOMe
F
3.74
PLE(Esterasa de hígado
de cerdo)
(86%, 95% ee)
MeOOC COOH
F
3.79
1. LiH, THFluego LiBH4
COOMe
F
HO 3.80COOMe
F
MsO 3.73
BnNH2, Et3Ntolueno (82%)
NH
F
O
OO
Paroxetina
HO
OO
3.36
NBn
F
O
3.81
NaH, NaOMe(MeO)2CO
tolueno, reflujo(88%) N
Bn
F
O
COOMe
3.82
BH3·SMe2
(92%)NBn
F
OHMsCl, Et3N
NBn
F
OMs
3.83
3.84
NaH, DMF(60% 2 pasos)
NBn
F
O
OO
H2, Pd/C
(93%)
3.85
3.76
MsCl, Et3N
2. SO4Me2(86% 2 pasos)
Esquema 3.17
3.5.5.3c. Cuestiones
1) Explique mecanísticamente la formación del compuesto 3.77 indicada en el esquema 3.18.
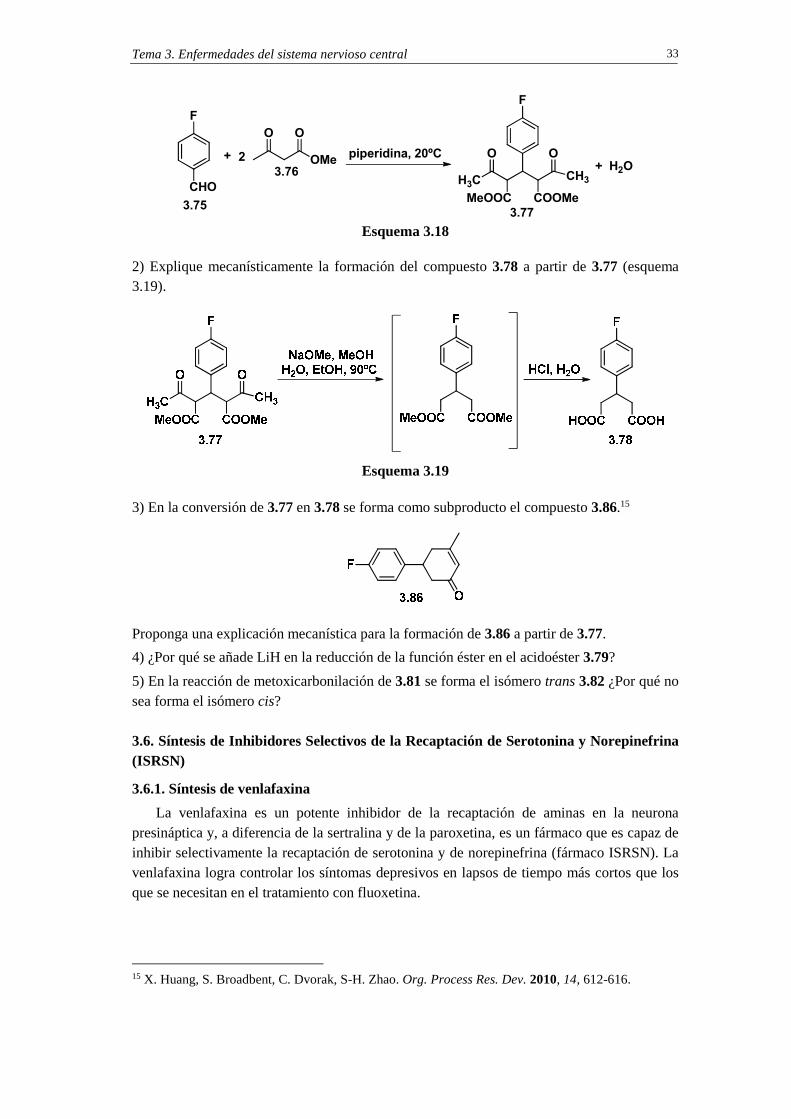
Tema 3. Enfermedades del sistema nervioso central 33
CHO
F
OMe
O O
piperidina, 20ºC
MeOOC COOMe
F
O
CH3
O
H3C
3.753.77
3.76+ 2
+ H2O
Esquema 3.18
2) Explique mecanísticamente la formación del compuesto 3.78 a partir de 3.77 (esquema 3.19).
Esquema 3.19
3) En la conversión de 3.77 en 3.78 se forma como subproducto el compuesto 3.86.15
Proponga una explicación mecanística para la formación de 3.86 a partir de 3.77.
4) ¿Por qué se añade LiH en la reducción de la función éster en el acidoéster 3.79?
5) En la reacción de metoxicarbonilación de 3.81 se forma el isómero trans 3.82 ¿Por qué no sea forma el isómero cis? 3.6. Síntesis de Inhibidores Selectivos de la Recaptación de Serotonina y Norepinefrina (ISRSN)
3.6.1. Síntesis de venlafaxina
La venlafaxina es un potente inhibidor de la recaptación de aminas en la neurona presináptica y, a diferencia de la sertralina y de la paroxetina, es un fármaco que es capaz de inhibir selectivamente la recaptación de serotonina y de norepinefrina (fármaco ISRSN). La venlafaxina logra controlar los síntomas depresivos en lapsos de tiempo más cortos que los que se necesitan en el tratamiento con fluoxetina.
15 X. Huang, S. Broadbent, C. Dvorak, S-H. Zhao. Org. Process Res. Dev. 2010, 14, 612-616.

Síntesis de antidepresivos, antiepilépticos y antiParkinson 34
3.6.1.a. Análisis retrosintético
En el esquema 3.20 se indica un análisis retrosintético para la venlafaxina que se inicia con la desconexión de los grupos metilo de la parte de dimetilamina. Este proceso conduce a la hidroxiamina 3.101 que por interconversión del grupo amino en grupo ciano se convierte en el hidroxinitrilo 3.102. Este compuesto se obtendrá mediante adición a la ciclohexanona 3.103 de la base conjugada derivada del 4-metoxifenilacetonitrilo 3.102.
OH
MeO
N
Venlafaxina
OH
MeO
H2N
N-metilación
IGF
OH
MeO
C
N
MeO
C
N
O
+
3.100
3.1013.1033.102
Esquema 3.20
3.6.1.b. Síntesis
La venlafaxina se comercializa en forma de racemato, por tanto no es necesario llevar a cabo una síntesis enantioselectiva de este fármaco. La síntesis de la venlafaxina comienza con la adición de la base conjugada derivada del 4-metoxifenilacetonitrilo 3.102 a la ciclohexanona 3.103 (esquema 3.21).16 La adición se lleva a cabo en presencia de NaOH y Bu4NBr en metanol-agua y proporciona el hidroxinitrilo 3.101 con un 96% de rendimiento. La hidrogenación del nitrilo con hidrógeno molecular a 10 atmósferas de presión, en una mezcla de MeOH/NH3 y en presencia de Ni-Raney como catalizador, genera el aminoalcohol 3.100. Después de acabada la hidrogenación se añade formaldehído acuoso a la mezcla de reacción, se agita durante 3 horas, se filtra el catalizador, se evapora el metanol y se añade hexano. Este procedimiento experimental proporciona la oxacina 3.104, que es un compuesto sólido estable que se obtiene sin necesidad de ninguna separación cromatográfica. La transformación de la oxazina 3.104 en la venlafaxina se consigue mediante reacción de N-metilación con formaldehído acuoso en presencia de ácido fórmico como reductor. La venlafaxina obtenida en la reacción anterior se convierte en el correspondiente clorhidrato por reacción con HCl en isopropanol.
16 (a) B. C. V. Kavitha, K. S. Rangappa. Bioorg. Med. Chem. Lett. 2004, 14, 3279-3281. (b) J. P. Yardley, J. G. E. M. Husbands, G. Stack, J. Butch, J. Bicksler, J. A. Moyer, E. A. Muth, T. Andree, H. Fletcher, M. N. G. James, A. R. Sieleckit. J. Med. Chem. 1990, 33, 2899-2905

Tema 3. Enfermedades del sistema nervioso central 35
Esquema 3.21
3.6.1.c. Cuestiones
1) Explique mecanísticamente la formación de la venlafaxina a partir de la oxazina 3.104.
3.6.2. Síntesis de desvenlafaxina
Uno de los principales metabolitos de la venlafaxina es el derivado O-desmetilado (desvenlafaxina) que presenta mayor eficacia y perfil de seguridad que aquélla. La desvenlafaxina fue aprobada por la FDA en 2008 para el tratamiento del Trastorno Depresivo Mayor.
3.6.2.a. Análisis retrosintético
La conversión de la función amina de la desvenlafaxina en amida conduce al compuesto 3.105 (esquema 3.22). Este compuesto se puede preparar por adición del reactivo nucleofílico 3.106 a la ciclohexanona.

Síntesis de antidepresivos, antiepilépticos y antiParkinson 36
Esquema 3.22
3.6.2.b. Síntesis
El material de partida para la síntesis de la desvenlafaxina es el ácido 4-benciloxifenilacético 3.107.17 Este compuesto se convierte en la N,N-dimetilamida 3.108 la cual, por ionización con LiHMDS y reacción con ciclohexanona, se transforma en la hidroxiamida 3.105. Finalmente, la desvenlafaxina se obtiene por reducción de la amida 3.105 a la amina 3.109 seguida de hidrogenolisis de la parte de benciléter.
OH
BnO
N O
3.105
BnO
COOH
3.107
1) SOCl2, DMF
(90% 2 pasos)
2) Me2NH·HCl, Et3N
BnO
N O
3.108
LiHMDS, THF. -70ºCluego ciclohexanona
(82%)
BH3·THF, THF
(66%)
OH
BnO
N
H2, Pd/C
EtOH (87%)
3.109
OH
HO
N
Desvenlafaxina
Esquema 3.23
3.6.3. Síntesis de milnacipran
El antidepresivo milnacipran inhibe la recaptación de serotonina y de norepinefrina (fármaco ISRSN) en una relación aproximada de 1:3. Su uso se aprobó por primera vez en Francia (nombre comercial Ixel®) en diciembre de 1996. En enero de 2009 la FDA aprobó el empleo de este fármaco para el tratamiento de la fibromialgia.
3.6.3.a. Análisis retrosintético
La primera desconexión en el análisis retrosintético del milnacipran es la escisión del enlace C-N (esquema 3.24). Esta operación genera amoniaco y el fragmento electrofílico 3.110 (X=grupo saliente). La siguiente operación retrosintética no es evidente y para seguir el proceso de desconexión de enlaces se ha indicado el mismo mediante flechas. Así, la
17 V. G. Gore, V. S. Kulkarni, V. S. Wakchaure, M. G. Hublikar, S. R. Wavhal, Patente: WO 08093142 A1, 2008.

Tema 3. Enfermedades del sistema nervioso central 37
ruptura intramolecular del anillo ciclopropánico, por ataque nucleofílico del grupo X origina el anión heterociclopropánico 3.111. En el sentido sintético este anión, generado a partir de la amida 3.110, formará el compuesto ciclopropánico 3.111 mediante apertura nucleofílica intramolecular del anillo heterociclopropánico. El análisis retrosintético se continua con la desconexión del fragmento metilenheterociclopropánico en la estructura 3.112. Esta operación genera el compuesto 3.113 y el anión 3.114. Un equivalente sintético de este anión puede ser el anión derivado de fenilacetonitrilo 3.115, fácilmente generable a partir del propio fenilacetonitrilo 3.116.
Esquema 3.24
3.6.3.b. Síntesis
El milnacipran se comercializa en forma de racemato, por tanto no es necesario llevar a cabo una síntesis enantioselectiva de este fármaco. La síntesis se inicia con la reacción entre la base conjugada del fenilacetonitrilo 3.116 y el clorometiloxirano 3.117 (esquema 3.25).18 Este proceso proporciona una mezcla cis/trans de los compuestos hidroxinitrilo-ciclopropánicos 3.118. La hidrólisis del grupo nitrilo conduce a la mezcla cis/trans de los hidroxiácidos ciclopropánicos 3.119. La convergencia de la mezcla de diastereoisómeros 3.119 en la lactona ciclopropánica 3.120 se consigue mediante calentamiento de aquélla a 150ºC. En el siguiente paso sintético se emplea la ftalimida potásica 3.121 como equivalente sintético de amoniaco (síntesis de Gabriel). Así, la reacción de la ftalimida potásica 3.121 con la lactona ciclopropánica 3.120 proporciona el derivado ftalimidociclopropánico 3.122. La función dietilamida se instala por conversión del ácido carboxílico en cloruro de ácido y reacción subsiguiente con dietilamina. Esta secuencia de dos pasos conduce a la amida 3.123 que por aminólisis de la parte de ftalimida con metilamina se convierte en el milnacipran neutro. La adición de HCl proporciona el clorhidrato de milnacipran.
18 B. Bonnaud, H. Cousse, G. Mouzin, M. Briley, A. Stenger, F. Fauran, J-P. Couzinier. J. Med. Chem. 1987, 30, 318-325.
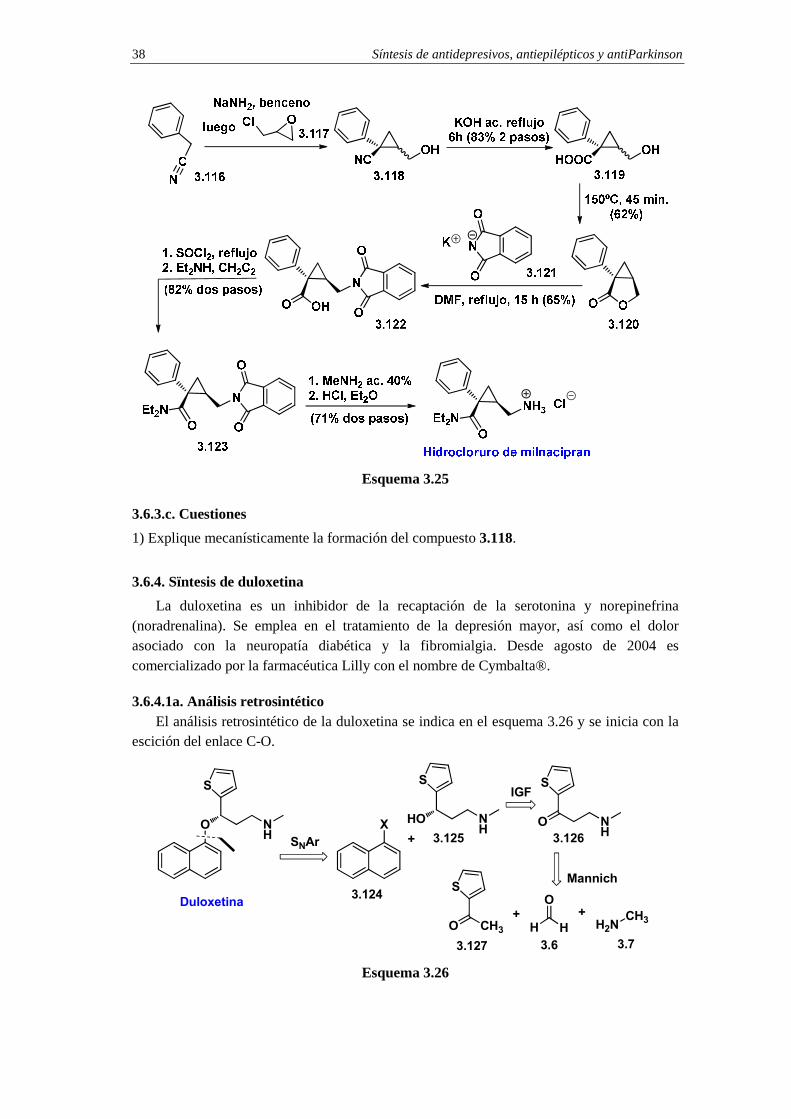
Síntesis de antidepresivos, antiepilépticos y antiParkinson 38
Esquema 3.25
3.6.3.c. Cuestiones
1) Explique mecanísticamente la formación del compuesto 3.118.
3.6.4. Sïntesis de duloxetina
La duloxetina es un inhibidor de la recaptación de la serotonina y norepinefrina (noradrenalina). Se emplea en el tratamiento de la depresión mayor, así como el dolor asociado con la neuropatía diabética y la fibromialgia. Desde agosto de 2004 es comercializado por la farmacéutica Lilly con el nombre de Cymbalta®.
3.6.4.1a. Análisis retrosintético El análisis retrosintético de la duloxetina se indica en el esquema 3.26 y se inicia con la
escición del enlace C-O.
Duloxetina
O NH
S
X HO NH
S
3.124
3.125+
IGF
O NH
S
3.126
Mannich
O CH3
S
H2NCH3
O
H H+ +
3.127 3.73.6
SNAr
Esquema 3.26

Tema 3. Enfermedades del sistema nervioso central 39
La desconexión del enlace C-O operación se basa en una reacción SNAr y conduce al 1-halonaftaleno 3.124 (X=halógeno), el componente electrofílico de la reacción SNAr, y al aminohidroxitiofeno 3.125, que por ionización del hidroxilo generará el componente nucleofílico de la reacción SNAr. Una operación de intercambio de grupo funcional transforma el compuesto 3.125 en el cetoaminotiofeno 3.126. El sistema de β-aminocetona de 3.126 proporciona, mediante una desconexión basada en la reacción de Mannich, la metil tiofenil cetona 3.127, formaldehído y metilamina (para una desconexión similar véase el análisis retrosintético de la fluoxetina en el esquema 3.2)
3.6.4.1b. Síntesis
La síntesis de la duloxetina se inicia con la reacción de Mannich entre la metil tiofenil cetona 3.108, el clorhidrato de metilamina y paraformaldehído (esquema 3.27).
O N
S
Me
F
3.124
O N
S
Me
3.129
O CH3
S
+ +
3.127 3.83.128
(CH3)2NHHCl 2N, EtOH
Me
NaBH4, K2CO3H2O, MeOH
HO N
S
Me
Me
·HCl(CHO)n
(+/-)-3.130
Resolución conácido (S)-(+)-mandélico
HO N
S
Me
Me3.131
NaH, DMSO luego
50ºC, 72 h(91%)
Me
3.132
O
Cl O
Cl
Cl
Cl3.133
benceno
O N
S
Me
O
OCl
Cl
Cl
Zn, HCOOH 2.5%
DMF (82% 2 pasos)
O N
S
Me
H
Duloxetina
·HCl
3.134
Esquema 3.27

Síntesis de antidepresivos, antiepilépticos y antiParkinson 40
La reacción de Mannich se lleva a cabo en etanol en presencia de HCl) y proporciona el clorhidrato 3.129.19 La reducción del carbonilo cetónico con NaBH4 conduce al aminoalcohol racémico (+/-)-3.130, que por resolución con ácido (S)-(+)-mandélico (ácido (S)-α-hidroxifenilacético) proporciona el aminoalcohol 3.131 ópticamente puro. El tratamiento de 3.131 con NaH en dimetilsulfóxido genera el correspondiente alcóxido sódico que reacciona con el 1-fluoronaftaleno 3.124 para dar el compuesto 3.132. La obtención de la duloxetina mediante N-desmetilación de 3.132 se consigue en dos pasos. En primer lugar 3.132 se transforma en el tricloetilcarbamato 3.134 por reacción con el cloroformiato de 2,2,2-tricloroetilo 3.133. Luego el carbamato 3.134 se convierte en duloxetina por reacción con zinc en presencia de ácido fórmico.
3.6.4.1c. Cuestiones
1) Explique mecanísticamente la conversión de 3.132 en 3.134.
2) Explique mecanísticamente la conversión de 3.134 en duloxetina.
3.6.4.2a. Análisis retrosintético de duloxetina mediante reducción enantioselectiva
En el esquema 3.28 se indica un análisis retrosintético de la duloxetina que se inicia también con la escisión del enlace C-O y la generación de los fragmentos 3.124 y 3.125. El análisis se continúa mediante una operación de intercambio de grupo funcional que transforma el grupo metilamino en grupo X, siendo X un grupo saliente, por ejemplo halógeno. En el sentido sintético la función metilamina se instalará mediante reacción de desplazamiento SN2 de X en el sustrato 3.135.
Duloxetina
O NH
S
SNuAr
X
HO NH
S
3.1243.125
+
O OH
S
3.135 (X=halógeno)
HO X
S
O X
S
O
S
3.138 3.137 3.136
C-C AED IGF
IGF
Esquema 3.28
19 F. P. Bymaster,a E. E. Beedle, J. Findlay, P. T. Gallagher, J. H. Krushinski, S. Mitchell,b D. W. Robertson, D. C. Thompson, L. Wallace, D. T. Wong. Bioorg. Med. Chem. Lett. 2003, 13, 4477-4480.

Tema 3. Enfermedades del sistema nervioso central 41
La siguiente operación retrosintética aumenta el estado de oxidación del alcohol 3.115 y lo convierte en la halocetona 3.136. En el sentido de la síntesis este será el paso clave puesto que la reducción de 3.136 se deberá efectuar de manera enantioselectiva. La halocetona 3.136 se obtendrá de la vinilcetona 3.137 mediante Adición Electrofílica a Doble enlace (AED) de XCl. La vinilcetona 3.137 se sintetizará a partir del ácido 2-tiofenocarboxílico 3.138.
3.6.4.2b. Síntesis de duloxetina mediante reducción enantioselectiva
La síntesis de la duloxetina de acuerdo con el esquema retrosintético anterior comienza con la obtención de la vinilcetona 3.137 a partir del ácido tiofenocarboxílico 3.138, por conversión en el cloruro de ácido 3.139 seguida de reacción de acoplamiento de Stille con Bu3SnCH=CH2 en presencia del catalizador Bn(Ph3P)2ClPd(II) en 1,3-dimetil-3,4,5,6-tetrahidro-2(1H)-pirimidona (DMPU), un disolvente polar aprótico (esquema 3.29). La reacción de Stille proporciona la vinilcetona 3.137 que se convierte en la clorocetona 3.136 mediante adición regioselectiva de HCl al doble enlace. El paso de reducción enantioselectiva se lleva a cabo mediante la aplicación del método CBS con BH3 en presencia de la (R)-oxazaborolidina quiral. Esta reacción proporciona el cloroalcohol 3.135 ópticamente activo. El desplazamiento nucleofílico del cloruro con yoduro, seguido del desplazamiento del yoduro con metilamina conduce al aminoalcohol 3.125. La reacción del aminoalcohol con NaH genera el correspondiente alcoxilato que proporciona la duloxetina por reacción SNAr con el 1-fluorofenol 3.124. La adición de HCl permite obtener la fluoxetina en forma de clorhiddrato.
Esquema 3.29
Síntesis de antidepresivos, antiepilépticos y antiParkinson 42
3.6.4.2c. Cuestiones
1) Proponga un mecanismo para el acoplamiento de Stille que transforma el cloruro de ácido 3.139 en la vinilcetona 3.137.
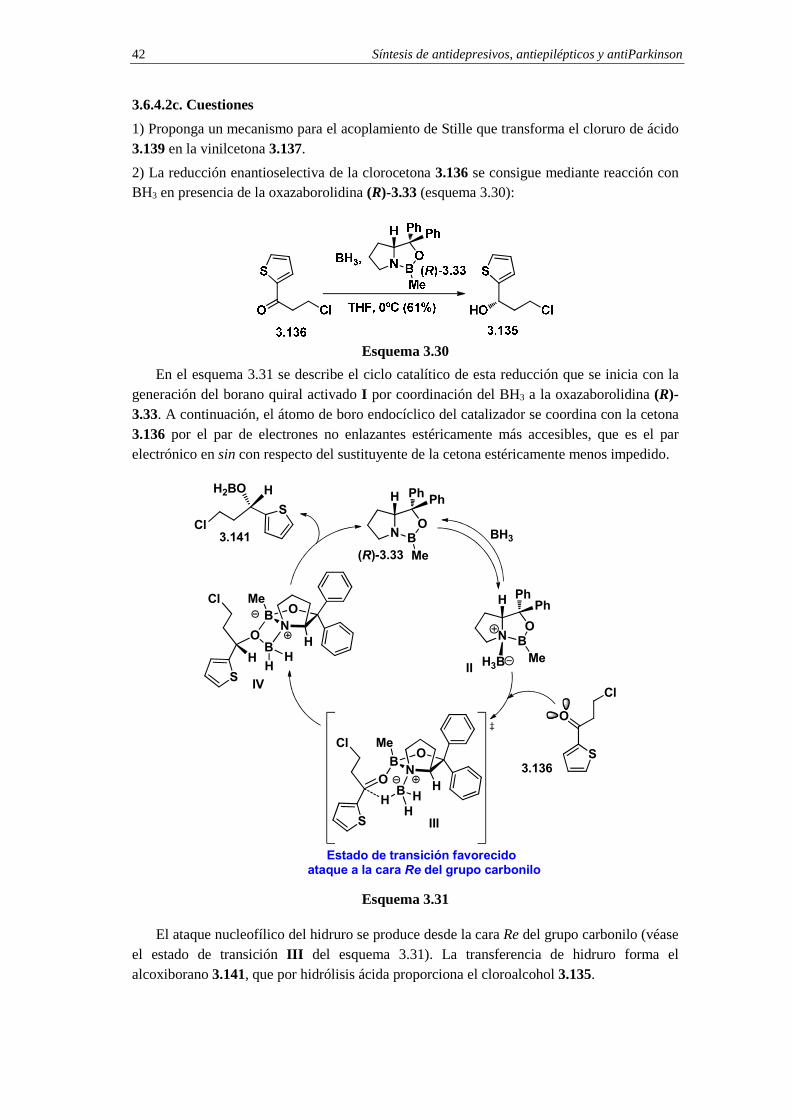
2) La reducción enantioselectiva de la clorocetona 3.136 se consigue mediante reacción con BH3 en presencia de la oxazaborolidina (R)-3.33 (esquema 3.30):
Esquema 3.30
En el esquema 3.31 se describe el ciclo catalítico de esta reducción que se inicia con la generación del borano quiral activado I por coordinación del BH3 a la oxazaborolidina (R)-3.33. A continuación, el átomo de boro endocíclico del catalizador se coordina con la cetona 3.136 por el par de electrones no enlazantes estéricamente más accesibles, que es el par electrónico en sin con respecto del sustituyente de la cetona estéricamente menos impedido.
BH3
N BO
H PhPh
MeH3B
Estado de transición favorecidoataque a la cara Re del grupo carbonilo
(R)-3.33
II
III
IV
N BO
H PhPh
Me
Cl
O
SB
Me
ON
O
HBH
HH
Cl
S
BMe
ON
O
HBH
HH
Cl
S
H2BO H
ClS
3.136
3.141
Esquema 3.31
El ataque nucleofílico del hidruro se produce desde la cara Re del grupo carbonilo (véase el estado de transición III del esquema 3.31). La transferencia de hidruro forma el alcoxiborano 3.141, que por hidrólisis ácida proporciona el cloroalcohol 3.135.

Tema 3. Enfermedades del sistema nervioso central 43
3.6.4.3a. Análisis retrosintético de duloxetina mediante esterificación enzimática enantioselectiva
El tercer análisis retrosintético de la duloxetina se indica en el esquema 3.32 y se basa en la preparación enantioselectiva de un hidroxitiofeno quiral obtenido mediante la esterificación enzimática enantioselectiva de la correspondiente mezcla racémica. El primer paso del análisis retrosintético es similar a los dos precedentes y genera los fragmentos 3.124 y 3.125. La interconversión del grupo metilamino en nitrilo conduce al hidroxinitrilo 3.142, que se obtendrá en forma ópticamente activa mediante esterificación enzimática enantioselectiva (operación EEE) del racemato (+/-)-3143. La desconexión del grupo nitrilo forma el alcohol 3.144 (X=halógeno) que por aumento del estado de oxidación de la función hidroxilo proporciona la halocetona 3.145. Este compuesto se obtendrá mediante reacción de acilación de tipo Friedel-Crafts del tiofeno 3.147 con el haluro de haloacetilo 3.146.
Esquema 3.32
3.6.4.3b. Síntesis de duloxetina mediante esterificación enzimática enantioselectiva
La preparación de la duloxetina según el análisis retrosintético indicado en el esquema 3.33, comienza con la reacción Friedel-Crafts del tiofeno 3.147 con el cloruro de cloroaceetilo 3.148 en presencia de AlCl3 (esquema 3.33).20 La reducción del carbonilo cetónico, seguida de reacción SN2 con cianuro sódico, proporciona el cianoalcohol racémico (+/-)-3143. Para la esterificación enzimática enantioselectiva de (+/-)-3.143 se ensayaron los enzimas lipasa de Pseudomonas cepacia inmovilizada sobre partículas cerámicas modificadas, lipasa de Pseudomonas cepacia inmovilizada sobre diatomita (Lipasa PD), lipasa de Pseudomonas cepacia (PS), Lipasa de páncreas de cerdo (Pig Pancreactic Lipase, PPL), lipasa de Candida cylindracea (CCL), lipasa de Candida rugosa (CRL) y lipasa inmovilizada de Mucor meihei. El mejor resultado se obtiene cuando se lleva a cabo la esterificación del racemato (+/-)-3.143 con acetato de vinilo en presencia de la lipasa Pseudomonas cepacia inmovilizada sobre diatomita. Después de 14 horas de agitación se
20 A. Kamal, G. B. R. Khanna, R. Ramu, T. Krishnaji. Tetrahedron Lett. 2003, 44, 4783-4787.
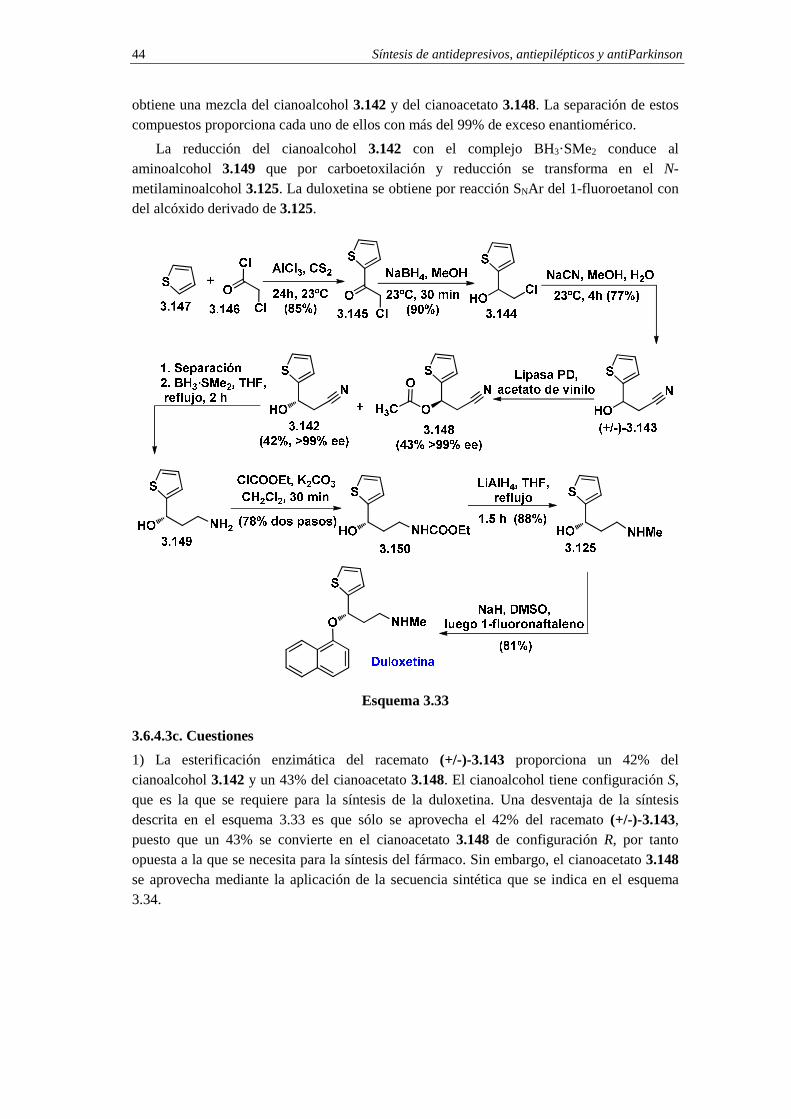
Síntesis de antidepresivos, antiepilépticos y antiParkinson 44
obtiene una mezcla del cianoalcohol 3.142 y del cianoacetato 3.148. La separación de estos compuestos proporciona cada uno de ellos con más del 99% de exceso enantiomérico.
La reducción del cianoalcohol 3.142 con el complejo BH3·SMe2 conduce al aminoalcohol 3.149 que por carboetoxilación y reducción se transforma en el N-metilaminoalcohol 3.125. La duloxetina se obtiene por reacción SNAr del 1-fluoroetanol con del alcóxido derivado de 3.125.
Esquema 3.33
3.6.4.3c. Cuestiones
1) La esterificación enzimática del racemato (+/-)-3.143 proporciona un 42% del cianoalcohol 3.142 y un 43% del cianoacetato 3.148. El cianoalcohol tiene configuración S, que es la que se requiere para la síntesis de la duloxetina. Una desventaja de la síntesis descrita en el esquema 3.33 es que sólo se aprovecha el 42% del racemato (+/-)-3.143, puesto que un 43% se convierte en el cianoacetato 3.148 de configuración R, por tanto opuesta a la que se necesita para la síntesis del fármaco. Sin embargo, el cianoacetato 3.148 se aprovecha mediante la aplicación de la secuencia sintética que se indica en el esquema 3.34.
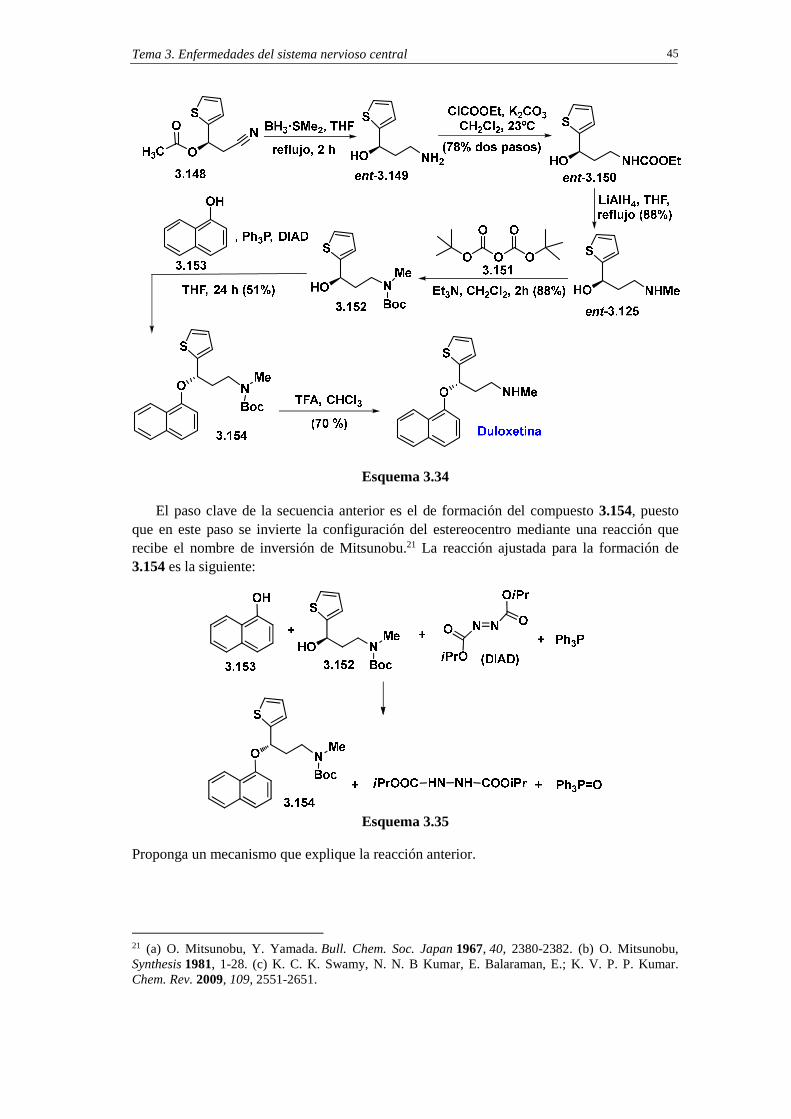
Tema 3. Enfermedades del sistema nervioso central 45
Esquema 3.34
El paso clave de la secuencia anterior es el de formación del compuesto 3.154, puesto que en este paso se invierte la configuración del estereocentro mediante una reacción que recibe el nombre de inversión de Mitsunobu.21 La reacción ajustada para la formación de 3.154 es la siguiente:
Esquema 3.35
Proponga un mecanismo que explique la reacción anterior.
21 (a) O. Mitsunobu, Y. Yamada. Bull. Chem. Soc. Japan 1967, 40, 2380-2382. (b) O. Mitsunobu, Synthesis 1981, 1-28. (c) K. C. K. Swamy, N. N. B Kumar, E. Balaraman, E.; K. V. P. P. Kumar. Chem. Rev. 2009, 109, 2551-2651.
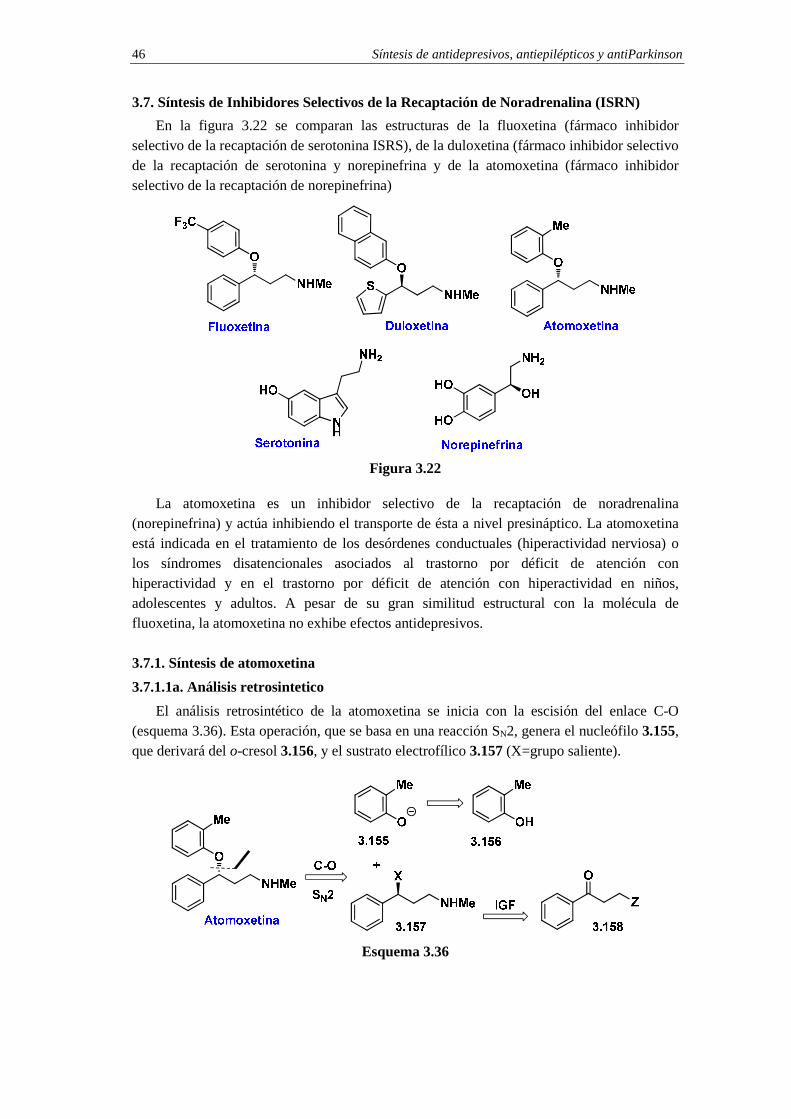
Síntesis de antidepresivos, antiepilépticos y antiParkinson 46
3.7. Síntesis de Inhibidores Selectivos de la Recaptación de Noradrenalina (ISRN)
En la figura 3.22 se comparan las estructuras de la fluoxetina (fármaco inhibidor selectivo de la recaptación de serotonina ISRS), de la duloxetina (fármaco inhibidor selectivo de la recaptación de serotonina y norepinefrina y de la atomoxetina (fármaco inhibidor selectivo de la recaptación de norepinefrina)
Figura 3.22
La atomoxetina es un inhibidor selectivo de la recaptación de noradrenalina (norepinefrina) y actúa inhibiendo el transporte de ésta a nivel presináptico. La atomoxetina está indicada en el tratamiento de los desórdenes conductuales (hiperactividad nerviosa) o los síndromes disatencionales asociados al trastorno por déficit de atención con hiperactividad y en el trastorno por déficit de atención con hiperactividad en niños, adolescentes y adultos. A pesar de su gran similitud estructural con la molécula de fluoxetina, la atomoxetina no exhibe efectos antidepresivos.
3.7.1. Síntesis de atomoxetina
3.7.1.1a. Análisis retrosintetico
El análisis retrosintético de la atomoxetina se inicia con la escisión del enlace C-O (esquema 3.36). Esta operación, que se basa en una reacción SN2, genera el nucleófilo 3.155, que derivará del o-cresol 3.156, y el sustrato electrofílico 3.157 (X=grupo saliente).
Esquema 3.36

Tema 3. Enfermedades del sistema nervioso central 47
La configuración del estereocentro en 3.157 debe ser opuesta a la de la atomoxetina, puesto que la reacción SN2 sobre aquél provocará la inversión de la configuración. El sustrato quiral 3.157 se obtendrá mediante reducción enantioselectiva de la cetona funcionalizada 3.158.
3.7.1.1b. Síntesis
Para la síntesis de la atomoxetina se elige como compuesto de partida la clorocetona proquiral 3.158 (esquema 3.37).
Esquema 3.37
La reducción de la clorocetona 3.158 con (+)-Ipc2BCl proporciona, después de la recristalización, el cloroalcohol quiral 3.159 con mas del 99.5% de exceso enantiomérico.22 La instalación de la parte de o-cresol se consigue mediante reacción de Mitsunobu del cloroalcohol 3.159 con el o-cresol 3.156 en presencia de azodicarboxilato de dietilo (DEAD) y de trifenilfosfina. El producto de la reacción, el cloroéter 3.137, se convierte en la atomoxetina mediante desplazamiento del cloruro con metilamina.
En el esquema 3.38 se indica la estructura del (+)-Ipc2BCl y el estado de transición para la reducción enantioselectiva de cetonas proquirales (Rl= grupo voluminoso, Rs= grupo pequeño).
22 M. Srebnick, P. Ramachandran, H. C. Brown. J. Org. Chem. 1988, 53, 2916-2920.
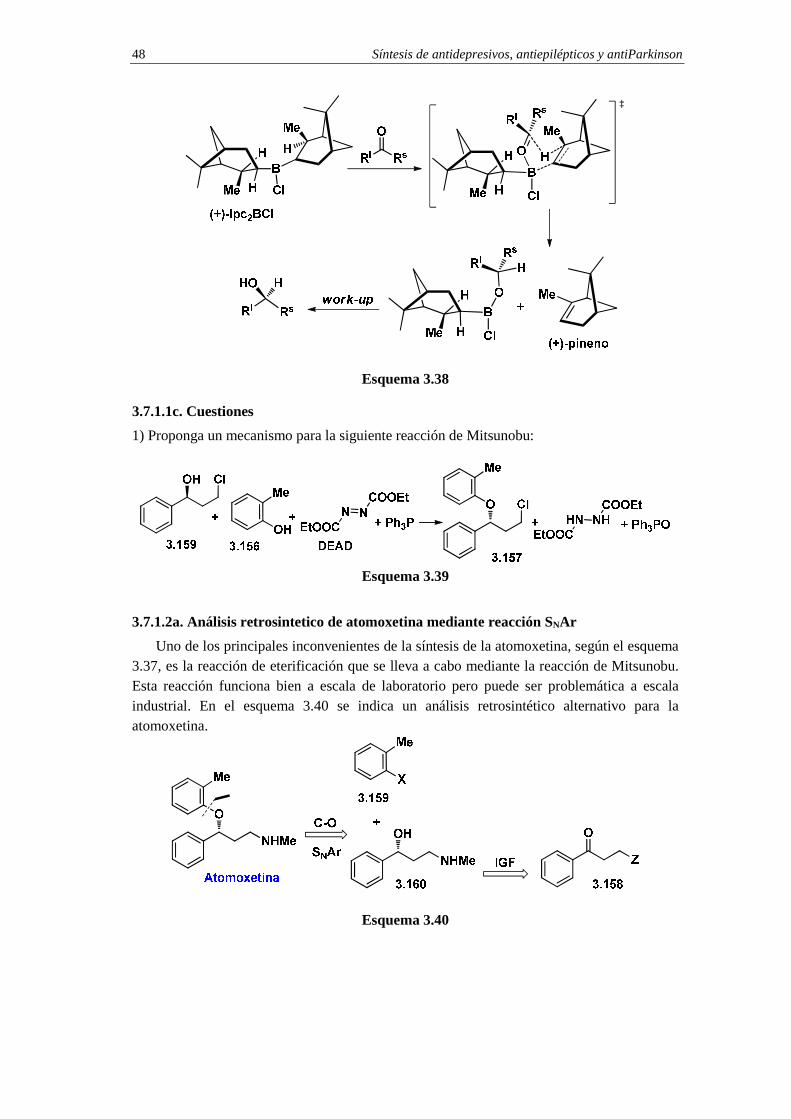
Síntesis de antidepresivos, antiepilépticos y antiParkinson 48
Esquema 3.38
3.7.1.1c. Cuestiones
1) Proponga un mecanismo para la siguiente reacción de Mitsunobu:
Esquema 3.39
3.7.1.2a. Análisis retrosintetico de atomoxetina mediante reacción SNAr
Uno de los principales inconvenientes de la síntesis de la atomoxetina, según el esquema 3.37, es la reacción de eterificación que se lleva a cabo mediante la reacción de Mitsunobu. Esta reacción funciona bien a escala de laboratorio pero puede ser problemática a escala industrial. En el esquema 3.40 se indica un análisis retrosintético alternativo para la atomoxetina.
Esquema 3.40

Tema 3. Enfermedades del sistema nervioso central 49
La retrosíntesis se inicia también con la escisión del enlace C-O. Sin embargo en este caso la eterificación se llevará a cabo mediante la aplicación de una reacción SNAr entre el compuesto nucleofílico 3.160, o su equivalente sintético, y el sustrato electrofílico 3.159 (X=grupo saliente). Finalmente, una doble operación de intercambio de grupo funcional convierte el compuesto 3.160 en la cetona proquiral 3.158.
3.7.1.2b. Síntesis de atomoxetina mediante reacción SNAr La síntesis alternativa se inicia con la reducción enantioselectiva de la clorocetona 3.158
(esquema 3.41).23 La reducción enantioselectiva de la clorocetona se consigue mediante la aplicación del método CBS. Así, la reducción de 3.158 con borano en presencia de cantidades catalíticas de la oxazaborolidina quiral (S)-CBS proporciona el cloroalcohol 3.161 con el 99% de rendimiento químico y con el 94% de exceso enantiomérico (esquema 3.41). El desplazamiento SN2 del cloruro, por reacción de 3.161 con dimetilamina, conduce al aminoalcohol 3.162. La reacción de eterificación se lleva a cabo mediante ionización del aminoalcohol 3.162 con hidruro sódico en dimetilsulfóxido seguida de reacción SNAr con la fluoroimina 3.163. Este proceso conduce al iminoéter 3.164 el cual, por hidrólisis de la función imina a aldehído, seguida de reducción a alcohol y reacción con cloruro de tionilo, se convierte en el clorocompuesto 3.165.
Cl
O
3.158 (99%, 94% ee)
(S)-CBS, BH3 Cl
OH
3.161
Me2NH, EtOH
reflujo (90%)
NMe2OH
3.162
N
F
NaH, DMSO luego
3.163NMe2O
N
3.164
NMe2O
3.165
1. AcOH, H2O2. NaBH43. SOCl2(96% 3 pasos) (98%)
Cl
Zn, AcOH
(95%)
NMe2O
Me
3.166
Cl O
O
Ph 3.167
Et3N, tolueno
luego HCl ac. AcOEtreflujo (98%)
NHMe
O
Me
.HCl
Clorhidrato de atomoxetina
Esquema 3.41
Cuando el compuesto 3.165 se somete a descloración reductiva, mediante reacción con zinc en ácido acético acuoso, se obtiene el aminoéter 3.166. Finalmente, la reacción de 3.166
23 P. Heath, A. Ratz, L. Weigel. Patente: WO 00/58262, 2000 (para Eli Lilly).
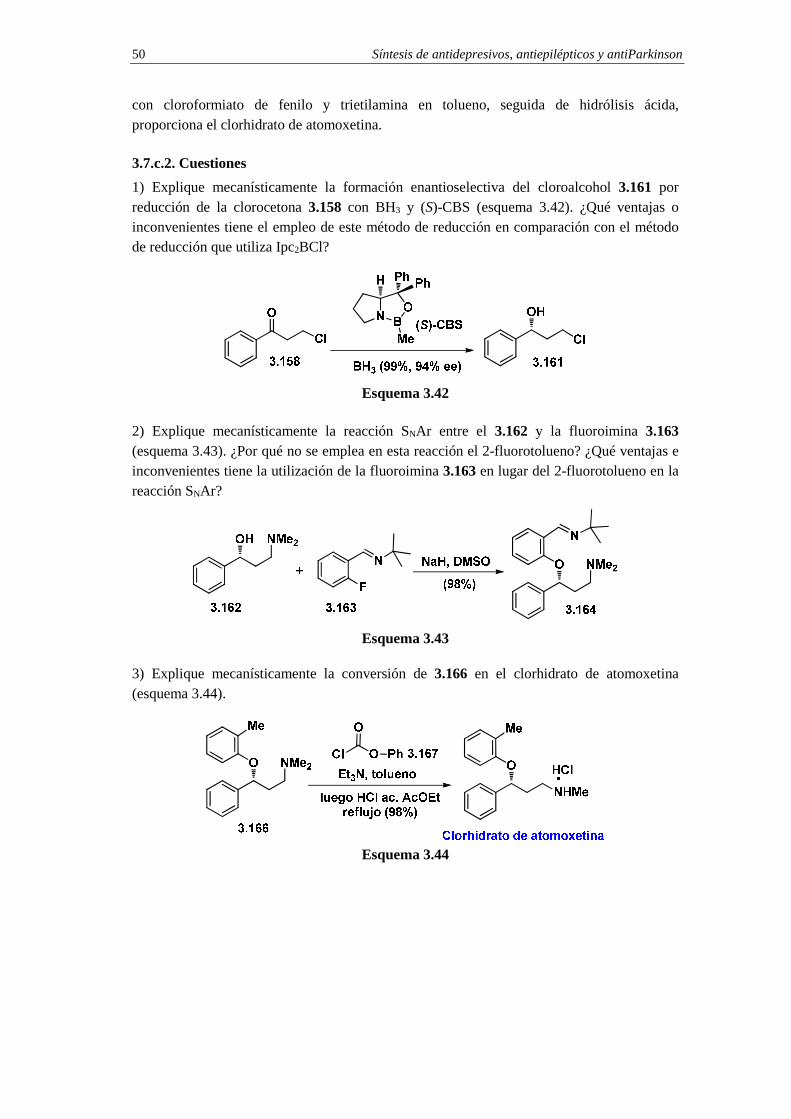
Síntesis de antidepresivos, antiepilépticos y antiParkinson 50
con cloroformiato de fenilo y trietilamina en tolueno, seguida de hidrólisis ácida, proporciona el clorhidrato de atomoxetina.
3.7.c.2. Cuestiones
1) Explique mecanísticamente la formación enantioselectiva del cloroalcohol 3.161 por reducción de la clorocetona 3.158 con BH3 y (S)-CBS (esquema 3.42). ¿Qué ventajas o inconvenientes tiene el empleo de este método de reducción en comparación con el método de reducción que utiliza Ipc2BCl?
Esquema 3.42
2) Explique mecanísticamente la reacción SNAr entre el 3.162 y la fluoroimina 3.163 (esquema 3.43). ¿Por qué no se emplea en esta reacción el 2-fluorotolueno? ¿Qué ventajas e inconvenientes tiene la utilización de la fluoroimina 3.163 en lugar del 2-fluorotolueno en la reacción SNAr?
Esquema 3.43
3) Explique mecanísticamente la conversión de 3.166 en el clorhidrato de atomoxetina (esquema 3.44).
Esquema 3.44

Tema 3. Enfermedades del sistema nervioso central 51
3.8. Síntesis de antidepresivos tricíclicos
Los antidepresivos tricíclicos reciben su nombre a la presencia de un sistema tricíclico en su estructura química. De entre esta clase de fármacos merece la pena destacar a a la amitriptilina, la imipramina, la clomipramina y la nortriptilina (véase la figura 3.23).
N
Amitriptilina
N
N
Imipramina
N
N
Clomipramina
Cl
NH
Nortriptilina Figura 3.23
Los antidepresivos tricíclicos son muy en el tratamiento médico de los trastornos del estado de ánimo (como los trastornos bipolares). El primer antidepresivo tricíclico fue la imipramina, descubierta accidentalmente en los años 1950 cuando se investigaba el desarrollo de nuevos compuestos antipsicóticos. Los antidepresivos tricíclicos impiden la recaptación de la serotonina y la noradrenalina, lo que da lugar, por tanto, a un aumento de sus niveles en el encéfalo. Se utilizan para impedir la depresión posterior provocada por la ingestas de drogas como el MDMA (3,4-metilendioximetanfetamina).
3.8.1. Síntesis de amitriptilina
La amitriptilina es un antidepresivo que inhibe la recaptación de serotonina y de norepinefrina en casi la misma proporción. Es el antidepresivo tricíclico más ampliamente usado y tiene, al menos, igual eficacia contra la depresión que los nuevos ISRS. Es también útil en el tratamiento de migrañas, cefaleas por tensión, ataques de ansiedad yen algunos síntomas esquizofrénicos. También se emplea en el tratamiento de determinados tipos de fibromialgia.
3.8.1.a. Análisis retrosintético
El análisis retrosintético de la amitriptilina se inicia con la desconexión de la parte de dimetilamina basada en una reacción SN2 sobre el compuesto 3.168 (esquema 3.45).
Esquema 3.45

Síntesis de antidepresivos, antiepilépticos y antiParkinson 52
El doble enlace que contiene el compuesto 3.168 se formará mediante deshidratación del haloalcohol 3.169 (X=halógeno). La cadena de halopropilo que contiene 3.169 procederá de un sistema ciclopropánico como el que contiene el compuesto 3.170. La desconexión del anillo ciclopropánico conduce a la cetona 3.171 que se sintetizará a partir del ácido 3.173 mediante reacción SEAr intramolecular. 3.8.1.b. Síntesis
La síntesis de la amitriptilina se inicia con la conversión del ácido 2-fenetilbenzoico 3.173 en el correspondiente cloruro de ácido (esquema 3.46). La subsiguiente reacción SEAr intramolecular, en presencia de la resina ácida Nafión-H (R-CF2-SO3H), proporciona la cetona tricíclica 3.171.24 Cuando este compuesto se trata con bromuro de ciclopropilmagnesio se obtiene el alcohol 3.170, que se convierte en el cloroderivado 3.168 mediante tratamiento con HCl en ácido acético. Po último, La reacción de 3.168 con dimetilamina permite la obtención de la amitriptilina base.25
N
AmitriptilinaCl
3.168
HOO
BrMg
OHO
3.1703.171
3.172
3.173
1. SOCl2
2. Nafión-Hxileno, reflujo
(90%)
HCl, AcOH
THF, reflujo, 1 h(100%)
30 min, 10ºC(98%)
(CH3)2NH
98ºC, 18 h
Esquema 3.46
3.8.1.c. Cuestiones
1) Explique mecanísticamente la conversión del alcohol 3.170 en el cloroderivado 3.168 mediante reacción con HCl en AcOH.
2) Proponga una síntesis para el ácido 2-fenetilbenzoico 3.173 a partir del 2-metilbenzoato de metilo.
3.8.2. Síntesis de imipramina
La imipramina es un fármaco antidepresivo que se utiliza en psiquiatría desde mediados de los años 50 del siglo XX. Su nombre comercial más conocido es Tofranil® y su acción terapéutica se basa en la inhibición de la recaptación de serotonina y noradrenalina. A diferencia de los antidepresivos más modernos, también tiene numerosos efectos sobre receptores de otros muchos neurotransmisores, lo que explica sus efectos adversos.
Está indicado en el tratamiento de todas las formas de depresión siendo sus resultados más evidentes en las formas endógenas (melancolía). También ha demostrado utilidad en los
24 T. Yamato, J. C. Hideshima, G. K. Surya Prakash, G. A. Olah. J. Org. Chem. 1991, 56, 3955-3957. 25 R. D. Hoffsommer, D. Taub, N. L. Wendler. J. Org. Chem. 1962, 27, 4134-4137.
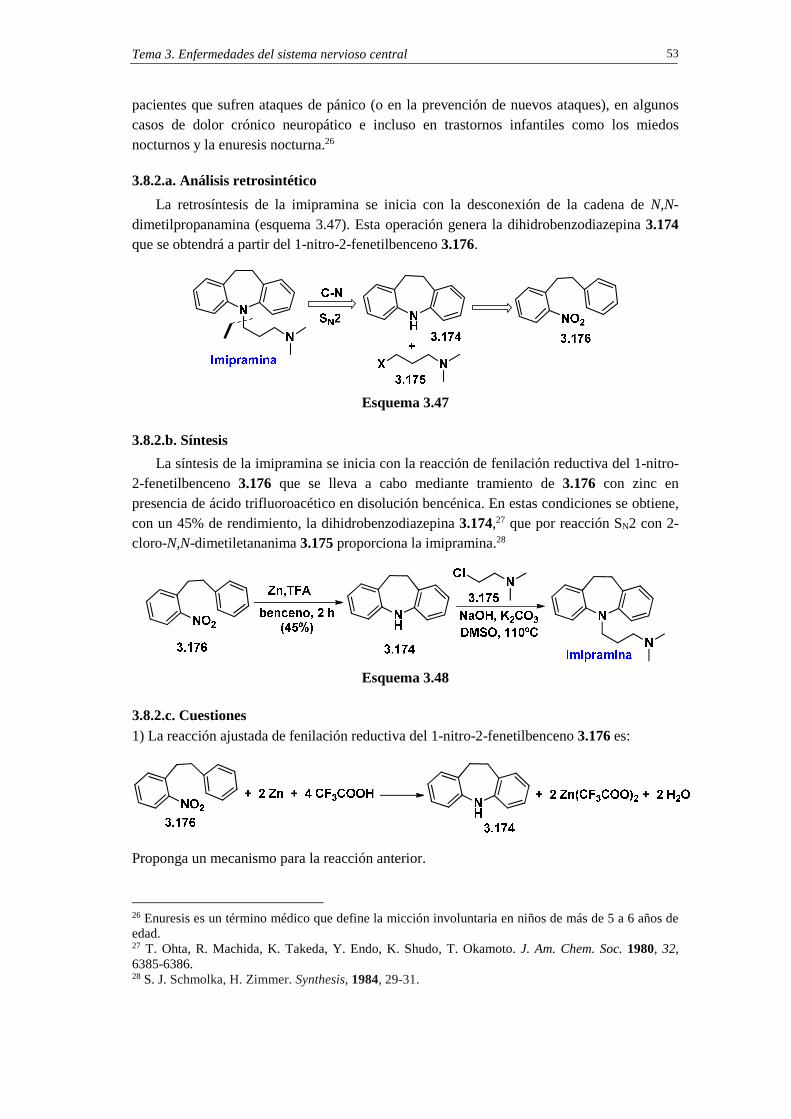
Tema 3. Enfermedades del sistema nervioso central 53
pacientes que sufren ataques de pánico (o en la prevención de nuevos ataques), en algunos casos de dolor crónico neuropático e incluso en trastornos infantiles como los miedos nocturnos y la enuresis nocturna.26
3.8.2.a. Análisis retrosintético
La retrosíntesis de la imipramina se inicia con la desconexión de la cadena de N,N-dimetilpropanamina (esquema 3.47). Esta operación genera la dihidrobenzodiazepina 3.174 que se obtendrá a partir del 1-nitro-2-fenetilbenceno 3.176.
Esquema 3.47
3.8.2.b. Síntesis
La síntesis de la imipramina se inicia con la reacción de fenilación reductiva del 1-nitro-2-fenetilbenceno 3.176 que se lleva a cabo mediante tramiento de 3.176 con zinc en presencia de ácido trifluoroacético en disolución bencénica. En estas condiciones se obtiene, con un 45% de rendimiento, la dihidrobenzodiazepina 3.174,27 que por reacción SN2 con 2-cloro-N,N-dimetiletananima 3.175 proporciona la imipramina.28
Esquema 3.48
3.8.2.c. Cuestiones 1) La reacción ajustada de fenilación reductiva del 1-nitro-2-fenetilbenceno 3.176 es:
Proponga un mecanismo para la reacción anterior.
26 Enuresis es un término médico que define la micción involuntaria en niños de más de 5 a 6 años de edad. 27 T. Ohta, R. Machida, K. Takeda, Y. Endo, K. Shudo, T. Okamoto. J. Am. Chem. Soc. 1980, 32, 6385-6386. 28 S. J. Schmolka, H. Zimmer. Synthesis, 1984, 29-31.

Síntesis de antidepresivos, antiepilépticos y antiParkinson 54
3.9. Epilepsia
La epilepsia es una enfermedad crónica que se caracteriza por la presencia de episodios críticos recurrentes denominados crisis epilépticas. La crisis epiléptica se origina en un núcleo neuronal o foco epiléptico, cuya actividad bioeléctrica se incrementa de manera violenta y fuera de control. La crisis epiléptica es muy variable en duración e intensidad, pudiendo quedar circunscrita al foco epiléptico o propagarse a áreas vecinas. La sintomatología de una crisis epiléptica puede afectar al estado de consciencia y/o a la actividad motora o sensorial, según sea el área del cerebro donde se genera el foco epiléptico.
Una persona que tiene una crisis tónico-clónica, también llamada de gran mal, puede gritar, perder el sentido y desplomarse, ponerse rígido y sufrir espasmos musculares.
Otro tipo de crisis epiléptica es la denominada crisis parcial compleja, en la que el paciente puede parecer confundido o aturdido sin que pueda responder a preguntas o seguir instrucciones.
Otras personas tienen ataques muy leves que ni siquiera son notados por otros. Algunas veces, la única manifestación de la crisis epiléptica es un parpadeo rápido o algunos segundos de mirada perdida con desconexión del medio. A este tipo de crisis epiléptica se lo denomina ausencia y es relativamente frecuente en la infancia.
La epilepsia puede estar causada por lesiones cerebrales tales como traumatismos craneales, secuelas de meningitis, tumores, etc, aunque también puede tener su origen en una predisposición de tipo genético. La epilepsia condicionada por una predisposición genética se denomina epilepsia idiopática. Se sospecha que algunos de los factores desencadenantes de una crisis epiléptica se deben a cambios iónicos extracelulares e intracelulares, que modifican la excitabilidad neuronal, ya sea por cambios bruscos en el potencial de reposo, o por desequilibrio en el balance de los mecanismos neuronales de excitación e inhibición provocados por un incremento de la actividad excitatoria o por depresión de la actividad inhibitoria.
3.10. Fármacos antiepilépticos
Bajo el nombre de fármacos antiepilépticos (o anticonvulsivos) se engloban aquéllos fármacos destinados a combatir, prevenir o interrumpir las convulsiones o los ataques epilépticos. La administración de fármacos antiepilépticos reduce de manera importante la frecuencia de las crisis en un 60% de los pacientes, y en un 20% de los casos se consigue alguna mejora.
La administración de los fármacos antiepilépticos, también denominados fármacos AED (en inglés Anti Epileptic Drugs) puede provocar efectos secundarios adversos debido a la alta dosis requerida para el control de las crisis. Por ello es absolutamente necesaria una vigilancia facultativa regular de la terapia.
Los compuestos antiepilépticos pueden ser divididos en ocho grupos principales: 1) Bloqueadores de los canales de sodio de activación repetitiva: fenitoína, carbamazepina, oxcarbazepina y rufinamida.
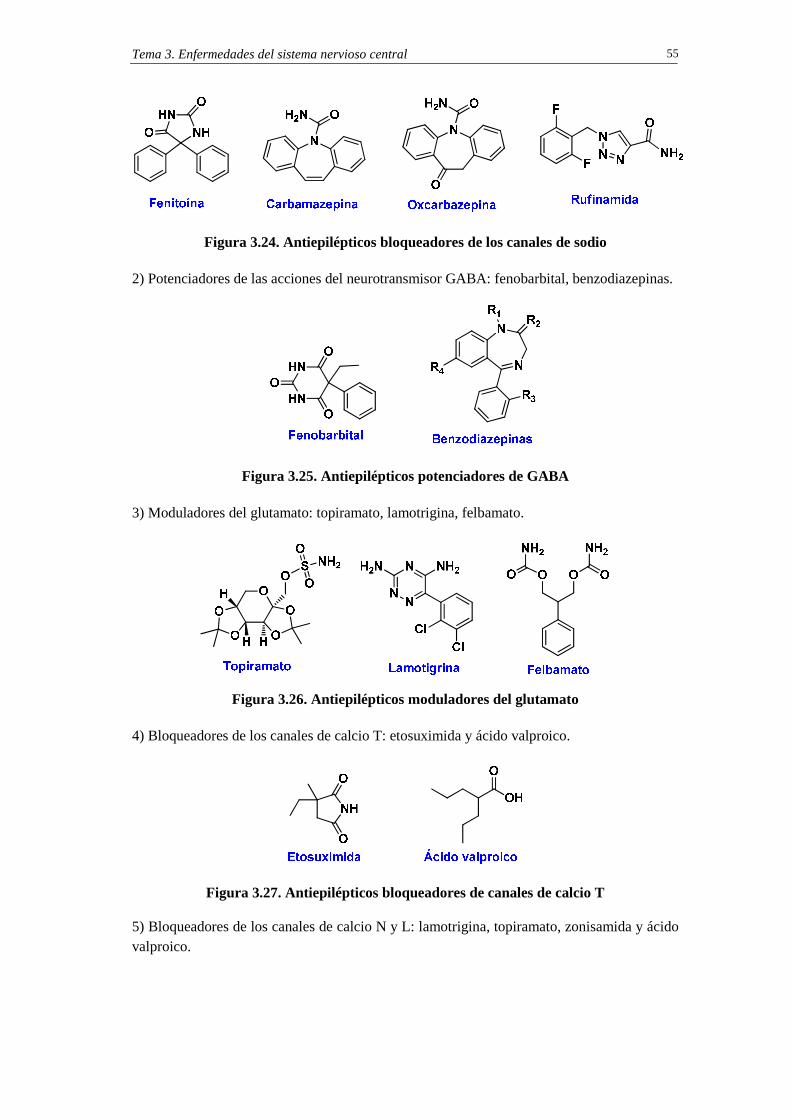
Tema 3. Enfermedades del sistema nervioso central 55
Figura 3.24. Antiepilépticos bloqueadores de los canales de sodio
2) Potenciadores de las acciones del neurotransmisor GABA: fenobarbital, benzodiazepinas.
Figura 3.25. Antiepilépticos potenciadores de GABA
3) Moduladores del glutamato: topiramato, lamotrigina, felbamato.
Figura 3.26. Antiepilépticos moduladores del glutamato
4) Bloqueadores de los canales de calcio T: etosuximida y ácido valproico.
Figura 3.27. Antiepilépticos bloqueadores de canales de calcio T 5) Bloqueadores de los canales de calcio N y L: lamotrigina, topiramato, zonisamida y ácido valproico.
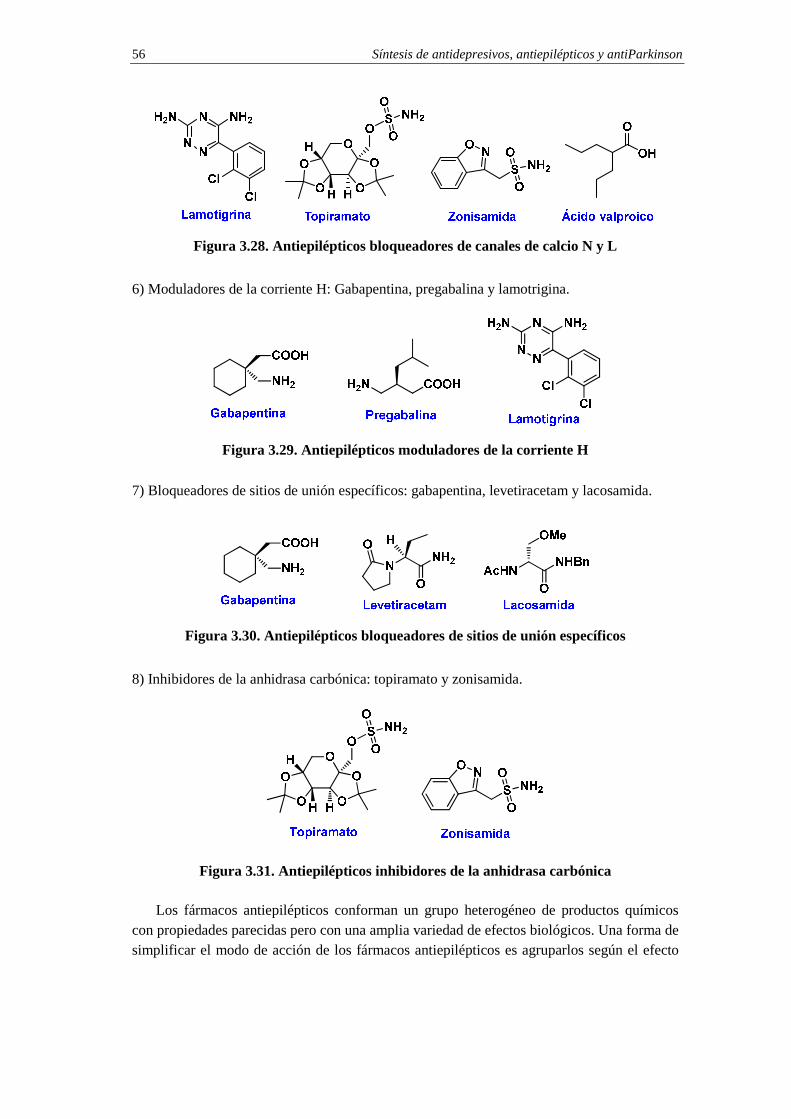
Síntesis de antidepresivos, antiepilépticos y antiParkinson 56
Figura 3.28. Antiepilépticos bloqueadores de canales de calcio N y L
6) Moduladores de la corriente H: Gabapentina, pregabalina y lamotrigina.
Figura 3.29. Antiepilépticos moduladores de la corriente H
7) Bloqueadores de sitios de unión específicos: gabapentina, levetiracetam y lacosamida.
Figura 3.30. Antiepilépticos bloqueadores de sitios de unión específicos
8) Inhibidores de la anhidrasa carbónica: topiramato y zonisamida.
Figura 3.31. Antiepilépticos inhibidores de la anhidrasa carbónica
Los fármacos antiepilépticos conforman un grupo heterogéneo de productos químicos
con propiedades parecidas pero con una amplia variedad de efectos biológicos. Una forma de simplificar el modo de acción de los fármacos antiepilépticos es agruparlos según el efecto

Tema 3. Enfermedades del sistema nervioso central 57
bloqueador de los canales iónicos y de los mecanismos implicados en el daño neuronal, del siguiente modo:
1) Inhibición de la activación repetitiva y sostenida de los canales de Na+.
2) Inhibición mediada a por GABA.
3) Atenuación de actividad de canales de Ca2+ voltaje-dependientes.
4) Disminución en la excitación mediada por el glutamato. 3.10.1. GABA: un neurotransmisor inhibitorio cerebral
Usualmente se percibe al sistema nervioso central como un conjunto de células excitadas, sin embargo, las células nerviosas no solamente excitan a sus vecinas sino que también las inhiben. La inhibición está mediada por el ácido γ-aminobutírico (GABA, figura 3.32), que fue identificado como constituyente químico único del encéfalo y considerado como transmisor inhibitorio desde 1950.
Figura 3.32. Estructura del GABA
El ácido γ-aminobutírico modula la actividad de otros neurotransmisores como la dopamina, la serotonina y la norepinefrina, también provoca la inhibición de GnRH (Hormona Liberadora de Gonadotropinas), ayuda a la recuperación muscular en deportistas y, junto con la ornitina, mejora el sueño.
El glutamato es un pariente excitatorio del GABA. Es el neurotransmisor más común en el sistema nervioso central y es especialmente importante en relación con la memoria. Curiosamente, el glutamato es tóxico para las neuronas, y un exceso de este neurotransmisor provoca la destrucción de las mismas. Así, el daño cerebral provocado por un traumatismo craneoencefálico puede llevar a un exceso de glutamato y a la destrucción por éste de las células neuronales. La Esclerosis Lateral Amiotrófica, más comúnmente conocida como enfermedad de Lou Gehrig, está provocada por una producción excesiva de glutamato.
Los fármacos que aumentan la producción del GABA disminuyen los eventos excitatorios regulados por glutamato, actuando como un freno de los neurotransmisores excitatorios que llevan a la ansiedad. Si el GABA está ausente en algunas partes del cerebro, se produce la epilepsia.
El GABA es sintetizado mediante descarboxilación del glutamato mediada por la enzima Glutamato Descarboxilasa (GAD) (véase el esquema 3.49). Una vez sintetizado, el GABA es introducido en vesículas y, cuando se produce el estímulo nervioso, el GABA es liberado de

Síntesis de antidepresivos, antiepilépticos y antiParkinson 58
la neurona presináptica y llega hasta la neurona postsináptica, donde es reconocido por los receptores GABA-A y GABA-B.
El GABA que no interacciona con los receptores es recaptado por la célula presináptica o por las células gliales. Una vez allí, es degradado por el enzima GABA Aminotransferasa (GABA-AT) al semialdehído succínico. Este metabolito se puede convertir en ácido succínico mediante acción de la enzima SSADH (Succínico Semialdehído DesHidrogenasa), o se puede transformar en el ácido γ-hidroxibutírico (GHB) mediante intervención de la enzima SSR (Succínico Semialdehído Reductasa).
H2N COOH
COOH
GlutamatoDescarboxilasa (GAD)
Ácido Glutámico
GABA
GABA aminotransferasa(GABA-AT)
H2N COOH
OHCCOOH
Semialdehídode ácido succínico
SSADHHOOC
COOH
Ácido succínico
SSR HO COOH
GHB
Esquema 3.49
En la figura 3.33 se representa la estructura de la enzima GABA-AT. Las líneas en rojo indican las entradas a los centros activos.
Figura 3.33. Representación de la enzima GABA-AT
La enzima glutamato descarboxilasa (GAD) se halla en interneuronas, riñón, hígado, páncreas, ganglios autónomos, epífisis e hipófisis posterior; mientras que la distribución de
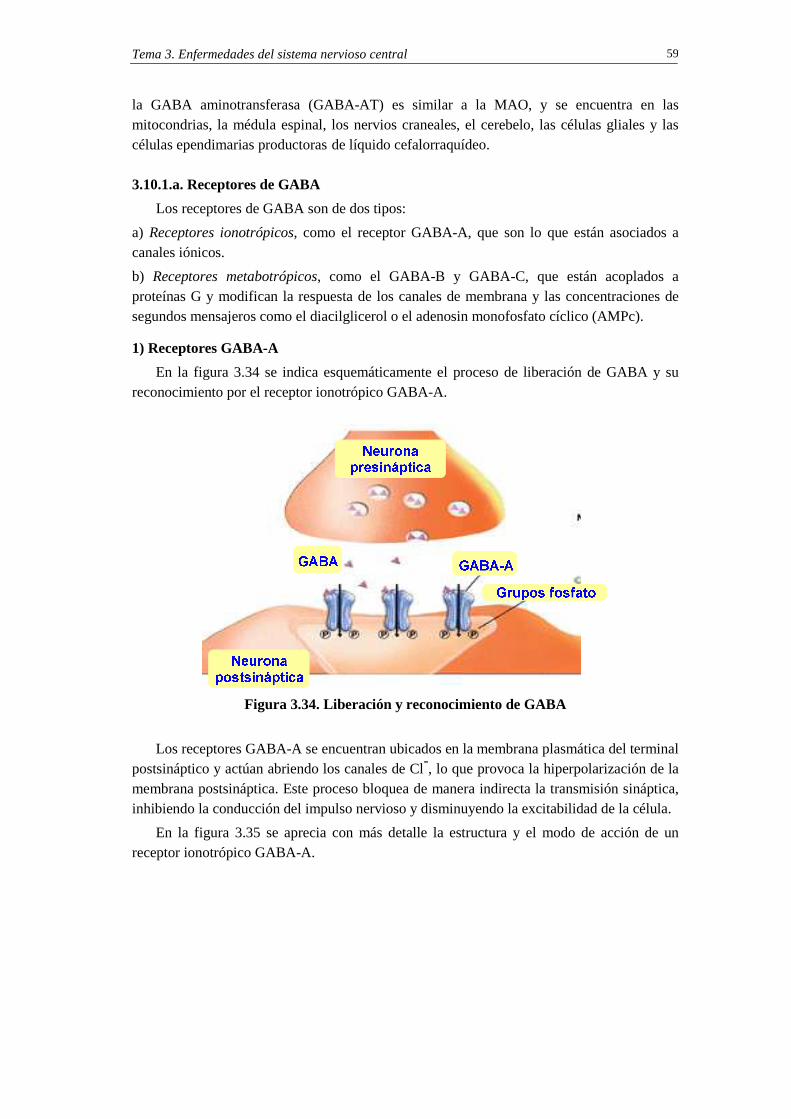
Tema 3. Enfermedades del sistema nervioso central 59
la GABA aminotransferasa (GABA-AT) es similar a la MAO, y se encuentra en las mitocondrias, la médula espinal, los nervios craneales, el cerebelo, las células gliales y las células ependimarias productoras de líquido cefalorraquídeo.
3.10.1.a. Receptores de GABA
Los receptores de GABA son de dos tipos:
a) Receptores ionotrópicos, como el receptor GABA-A, que son lo que están asociados a canales iónicos.
b) Receptores metabotrópicos, como el GABA-B y GABA-C, que están acoplados a proteínas G y modifican la respuesta de los canales de membrana y las concentraciones de segundos mensajeros como el diacilglicerol o el adenosin monofosfato cíclico (AMPc).
1) Receptores GABA-A
En la figura 3.34 se indica esquemáticamente el proceso de liberación de GABA y su reconocimiento por el receptor ionotrópico GABA-A.
Figura 3.34. Liberación y reconocimiento de GABA
Los receptores GABA-A se encuentran ubicados en la membrana plasmática del terminal postsináptico y actúan abriendo los canales de Cl-, lo que provoca la hiperpolarización de la membrana postsináptica. Este proceso bloquea de manera indirecta la transmisión sináptica, inhibiendo la conducción del impulso nervioso y disminuyendo la excitabilidad de la célula.
En la figura 3.35 se aprecia con más detalle la estructura y el modo de acción de un receptor ionotrópico GABA-A.

Síntesis de antidepresivos, antiepilépticos y antiParkinson 60
Espacio intersináptico
Figura 3.35. Representación del modo de acción de un receptor GABA-A
El receptor GABA-A es el blanco principal de la acción de muchos fármacos antiepilépticos (benzodiacepinas, fenobarbital, topiramato, etc.). Cada uno de estos fármacos aumenta la frecuencia de apertura de los canales de cloro o la duración de dicha apertura.
En la parte de la izquierda de la figura 3.36 se representa la estructura de la subunidad de un receptor GABA-A con indicación de las cuatro α-hélices transmembrana. El puente disulfuro, característico de la familia de receptores cis-loop, está colocado en el dominio C-teminal extracelular y dibujado con una línea amarilla. En la parte de la derecha se dibujan las cinco subunidades que constituyen el receptor, que están colocadas simétricamente alrededor del canal de cloruro (en esta parte de la figura no se han dibujado los bucles extracelulares). Los segmentos transmembranales M2 de las cinco subunidades (en color amarillo en la figura 3.35) se encuentran en estrecha cercanía y son los que constituyen propiamente el canal. La especificidad para el paso de un ión y no de otros, depende de los aminoácidos que constituyen el canal. Se ha demostrado que el cambio de aminoácidos, mediante mutagénesis dirigida, perturba la selectividad iónica.
Figura 3.36. Representación de la estructura de un receptor GABA-A

Tema 3. Enfermedades del sistema nervioso central 61
2) Receptores GABA-B
Los receptores metabotrópicos GABA-B se encuentran en la membrana plasmática tanto del terminal presináptico como del terminal postsináptico.
La unión del neurotransmisor al receptor GABA-B presináptico disminuye la entrada de Ca2+ en la célula presináptica, provocando de esta forma una menor liberación de glutamato y de monoaminas.
Por otro lado, la activación de los receptores GABA-B postsinápticos inicia un proceso mediado por un segundo mensajero que es considerablemente mas lento que el proceso de activación mediado por el canal iónico GABA-A, ya que los receptores GABA-B no están emparentados con canales de cloruro, como el receptor GABA-A, sino que modulan indirectamente canales de Ca2+ y de K+ por interacción con la proteína G y la adenilciclasa. La unión de un agonista al receptor GABA-B postsináptico aumenta la salida de potasio al medio extracelular produciendo un potencial inhibitorio lento.
En la figura 3.37 se representa esquemáticamente el modo de acción de los receptores metabotrópicos tipo GABA-B. Así, la unión del neurotransmisor al receptor activa al trímero de proteína G que se disocia activando segundos mensajeros cuya misión es la apertura de los canales iónicos de Ca2+ y de K+ .
Mensajerosintracelulares
Neuro-transmisor
1. Unión delneurotransmisor
Receptor
Proteína G
2. Activación dela proteína G
Proteína efectora
4. Aperturadel
canal iónico
Iones
5. Flujo de ionesa través de la
membrana
3. La subunidad de la proteína Go mensajeros intracelulares
modulan canales iónicos
Figura 3.37. Transducción de señales en un receptor metabotrópico tipo GABA-B
En la figura 3.38 se indica esquemáticamente el mecanismo de liberación y reconocimiento de GABA por receptores de tipo GABA-B.
Paso 1: La glutamina ubicada en la célula presináptica se convierte en glutamato por acción de la enzima glutaminasa.
Paso2: El glutamato se convierte en GABA, que es almacenado en las vesículas.
Paso 3: El proceso de exocitosis libera el GABA, que sale al espacio intersináptico.
Paso 4: El GABA se une a los receptores GABA-B modulando los canales de Ca2+ y de K+ por interacción con la proteína G y la adenilciclasa.

Síntesis de antidepresivos, antiepilépticos y antiParkinson 62
Paso 5: El GABA es reabsorbido por los receptores presinápticos.
Paso 6: Alternativamente, el GABA es reabsorbido por la glía. Las células gliales, conocidas también genéricamente como glía o neuroglía, son células nodriza del sistema nervioso y desempeñan, principalmente, la función de soporte de las neuronas. Las células gliales, intervienen activamente en el procesamiento cerebral de la información y controlan el microambiente celular en lo que respecta a la composición iónica, los niveles de neurotransmisores y el suministro de citoquinas y otros factores de crecimiento.
Paso 7: El GABA que penetra en la glía experimenta el proceso de transaminación a α-cetoglutarato catalizado por la enzima GABA-transaminasa, regenerándose el glutamato que, a su vez, es convertido en glutamina. Finalmente, este metabolito es reintroducido en la neurona presináptica.
Glutamina
Glutamato
GABA
Célulapostsináptica
Célula delglial
GABA
Vesícula con GABA
Receptor GABA-B
Transportador
Canal iónico
Terminalpresináptico
Glutamina
Glutamato
Figura 3.38. Liberación, reconocimiento y recaptación de GABA
El ácido glutámico se convierte en glutamina mediante la enzima glutaminasa de los terminales nerviosos. De hecho, la regulación de este enzima está estrechamente ligada a la actividad de la terminación nerviosa: si los niveles intraterminales de glutamato son altos la glutaminasa se inactiva. Tras la llegada de un potencial de acción a la célula presináptica la glutaminasa se activa y se inicia la formación de glutamato, lo que restaura los niveles del neurotransmisor en el terminal sináptico.
La conversión del glutamato en glutamina se lleva a cabo en dos pasos. En el primero de ellos se produce la fosforilación por reacción del glutamato con ATP mediada por la enzima
Tema 3. Enfermedades del sistema nervioso central 63
glutamina sintetasa (esquema 3.50). A continuación, el glutamato fosforilado reacciona con el amoníaco y forma la glutamina:
Esquema 3.50
La glutamina es transportada a la célula neuronal y convertida en glutamato por acción de la enzima glutaminasa (esquema 3.51). El glutamato es convertido en GABA por acción del enzima glutamato descarboxilasa.
Esquema 3.51
3.10.2. Fármacos antiepilépticos: gabapentina y pregabalina
Ya se ha indicado anteriormente que una de las causas de epilepsia es la disminución de los niveles de GABA en el cerebro. Sin embargo, la administración de GABA, ya sea por vía oral o intravenosa, no sirve para tratar la epilepsia, ya que el GABA no puede atravesar la barrera hematoencefálica, una membrana compuesta por células endoteliales fuertemente empaquetadas que protege al cerebro de agentes químicos extraños que puedan ser transportados por la sangre que llega a aquél.
Una forma de aumentar los niveles de GABA en el cerebro es mediante la administración de fármacos que puedan atravesar la barrera hematoencefálica y sean capaces de inhibir la acción de la enzima GABA-AT, que es la encargada de la conversión del GABA en el semialdehído succínico (véase el esquema 3.49). Al mismo tiempo los fármacos inhibidores de GABA-AT no tienen que inhibir la acción de la enzima ácido glutámico descarboxilasa (GAD), puesto que la inhibición del GAD disminuiría los niveles de GABA, provocándose el efecto contrario del que se desea obtener con el fármaco.
En un proyecto de investigación llevado a cabo en la universidad Northwestern (Illinois, USA) liderado por el profesor Richard Bruce Silverman se prepararon diversos 3-alquilderivados del ácido γ-aminobutítico (figura 3.39). Todos estos compuestos eran incapaces de inhibir la enzima GABA-aminotransferasa (GABA-AT). Sin embargo, y contrariamente a lo esperado, todos los 3-alquilderivados se mostraron eficaces en la
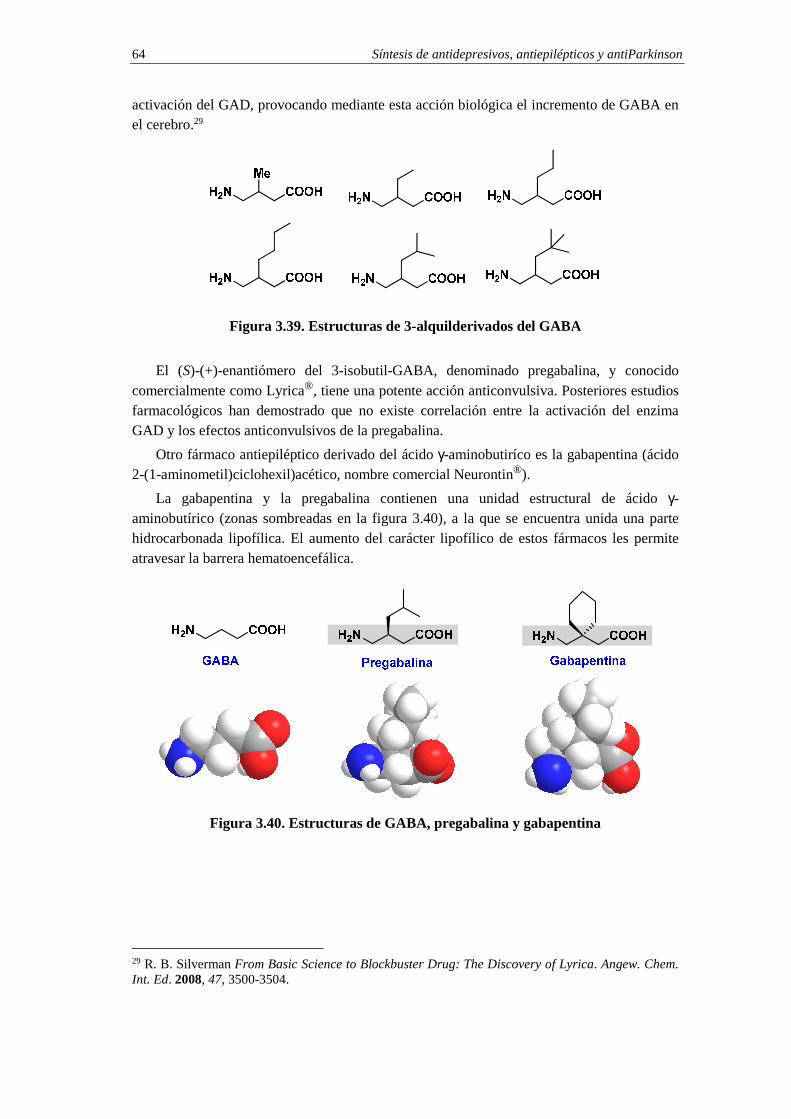
Síntesis de antidepresivos, antiepilépticos y antiParkinson 64
activación del GAD, provocando mediante esta acción biológica el incremento de GABA en el cerebro.29
Figura 3.39. Estructuras de 3-alquilderivados del GABA
El (S)-(+)-enantiómero del 3-isobutil-GABA, denominado pregabalina, y conocido comercialmente como Lyrica®, tiene una potente acción anticonvulsiva. Posteriores estudios farmacológicos han demostrado que no existe correlación entre la activación del enzima GAD y los efectos anticonvulsivos de la pregabalina.
Otro fármaco antiepiléptico derivado del ácido γ-aminobutiríco es la gabapentina (ácido 2-(1-aminometil)ciclohexil)acético, nombre comercial Neurontin®).
La gabapentina y la pregabalina contienen una unidad estructural de ácido γ-aminobutírico (zonas sombreadas en la figura 3.40), a la que se encuentra unida una parte hidrocarbonada lipofílica. El aumento del carácter lipofílico de estos fármacos les permite atravesar la barrera hematoencefálica.
Figura 3.40. Estructuras de GABA, pregabalina y gabapentina
29 R. B. Silverman From Basic Science to Blockbuster Drug: The Discovery of Lyrica. Angew. Chem. Int. Ed. 2008, 47, 3500-3504.

Tema 3. Enfermedades del sistema nervioso central 65
3.10.3. Modo de acción de la pregabalina
La actividad anticonvulsiva de la pregablina, así como la de la gabapentina, se debe principalmente a su unión selectiva a la subunidad α2δ de los canales de calcio voltaje-dependientes. En la figura 3.41 se muestra esquemáticamente un canal de calcio. El poro que forma el canal (en blanco en la figura 3.41) tiene una longitud de entre 1.800-2.300 aminoácidos, y está compuesto por cuatro dominios (I-IV). En la figura 3.40 sólo se dibujan los bucles transmembrana del dominio I.
Figura 3.41. Estructura de un canal de calcio voltaje-dependiente
La denominada subunidad extracelular α2 (en naranja en la figura 3.41) está compuesta por 9.300 aminoácidos, y la subunidad transmembrana δ (en verde en la figura 3.41) está compuesta por unos 150 aminoácidos.
La subunidad β (en azul claro en la figura 3.41) está compuesta por 480-600 aminoácidos y se une al bucle intracelular de un dominio denominado AID (en rojo en la figura 3.41).
Unido al canal de calcio se encuentra también la proteína de bajo peso molecular denominada calmodulina (CaM) compuesta por 148 aminoácidos, uno de cuyos sitios de acción es el denominado dominio IQ, que se localiza en el segmento C-terminal citoplasmático de la subunidad α1.
La unión de la pregabalina a la subunidad α2δ disminuye el flujo de Ca2+ en la neurona provocando indirectamente una disminución de la liberación de glutamato, de noradrenalina y de la sustancia P (undecapéptido con capacidad neurotransmisora).
En la figura 3.42 se muestra la zona de unión de la pregabalina al dominio α2 de un canal de calcio.

Síntesis de antidepresivos, antiepilépticos y antiParkinson 66
Figura 3.42. Estructura de un canal de calcio y zona de unión de la pregabalina
La pregabalina también aumenta los niveles neuronales de GABA mediante el incremento de la actividad de la enzima GAD (ácido glutámico descarboxilasa).
De entre los 3-alquilderivados de GABA indicados en la figura 3.39, el (S)-(+)-enantiómero del 3-isobutil-GABA es el que exhibe mayor capacidad anticonvulsiva, debido a que es un sustrato para el sistema de transporte L y por tanto es eficientemente transportado hacia el cerebro. El sistema de transporte L se encarga de introducir, entre otros, el aminoácido L-leucina en el cerebro. La gran similitud estructural entre este aminoácido y el (S)-(+)-enantiómero del 3-isobutil-GABA (véase la figura 3.43) explica por qué este último es también un sustrato para el sistema de transporte L.
Figura 3.43
Los otros 3-alquilderivados de GABA que se indican en la figura 3.39 no son sustratos para el sistema de transporte L y, por tanto, no pueden atravesar eficazmente la barrera hematoencefálica, por lo que su acción anticonvulsiva es mucho menor.

Tema 3. Enfermedades del sistema nervioso central 67
3.11. Sintesis de fármacos antiepilépticos
3.11.1. Síntesis de gabapentina
3.11.1.1a Análisis retrosintético
La retrosíntesis de la gabapentina se inicia con la conversión de la gabapentina en el cianoácido 3.177 que se consigue mediante conversión del grupo funcional amino en grupo nitrilo (esquema 3.52). El grupo nitrilo ocupa una posición β con respecto al grupo de ácido carboxílico y por tanto su desconexión, basada en una reacción de adición conjugada Michael, genera el ácido conjugado 3.178 y el anión cianuro. Por último, la desconexión del doble enlace conduce a la ciclohexanona 3.179 y al sintón aniónico 3.180.
Esquema 3.52
3.11.1.b.1. Síntesis
Como equivalente sintético del sintón aniónico 3.180 se elige el malonato de dietilo 3.181 (esquema 3.53). Así, la reacción de condensación de Knoevenagel de la ciclohexanona 3.179 con el malonato de dietilo 3.181, en presencia de TiCl4 en piridina, proporciona el ciclohexilidenmalonato de dietilo 3.182.30 La reacción de este compuesto con KCN en etanol y HCl conduce al cianodiéster 3.183 mediante adición conjugada del anión nitrilo. Cuando el compuesto 3.183 se hidrogena a 145 psi de presión, en presencia de Ni como catalizador, se provoca la reducción del grupo nitrilo a amino y la lactamización concomitante y se obtiene la lactama 3.184, que sometida a hidrólisis ácida se convierte en el clorhidrato de gabapentina.
O
3.179
COOEt
COOEt
3.171
TiCl4, piridina(56%)
COOEtEtOOC
3.182
NC
COOEt
COOEtKCN, HCl
EtOH (88%)
3.183
H2 (145 psi), NiEtOH, 90ºC (88%)
COOEt
HNO
3.184
HCl, H2O
reflujo (75%)
COOHNH2HCl.
Clorhidrato de Gabapentina
Esquema 3.53
30 G. Griffths, H. Mettier, L. S. Mills, F. Previdoli. Helv. Chim Acta 1991, 74, 309-314.
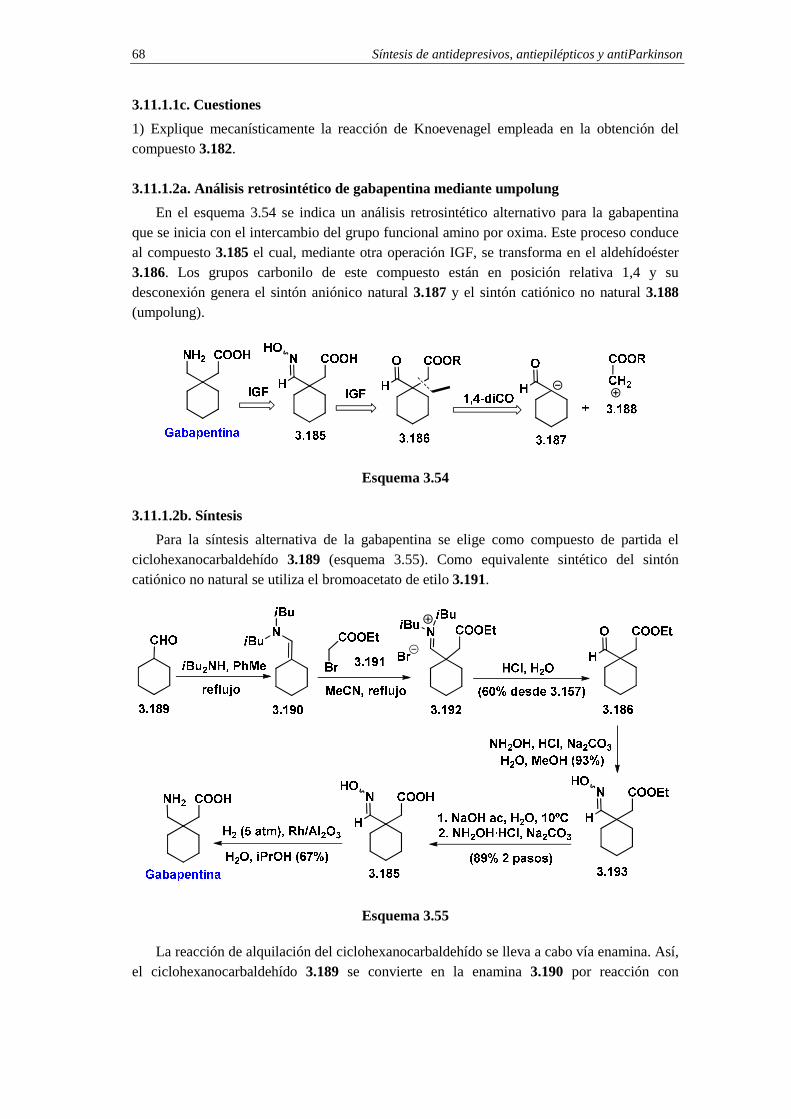
Síntesis de antidepresivos, antiepilépticos y antiParkinson 68
3.11.1.1c. Cuestiones
1) Explique mecanísticamente la reacción de Knoevenagel empleada en la obtención del compuesto 3.182.
3.11.1.2a. Análisis retrosintético de gabapentina mediante umpolung
En el esquema 3.54 se indica un análisis retrosintético alternativo para la gabapentina que se inicia con el intercambio del grupo funcional amino por oxima. Este proceso conduce al compuesto 3.185 el cual, mediante otra operación IGF, se transforma en el aldehídoéster 3.186. Los grupos carbonilo de este compuesto están en posición relativa 1,4 y su desconexión genera el sintón aniónico natural 3.187 y el sintón catiónico no natural 3.188 (umpolung).
Esquema 3.54
3.11.1.2b. Síntesis
Para la síntesis alternativa de la gabapentina se elige como compuesto de partida el ciclohexanocarbaldehído 3.189 (esquema 3.55). Como equivalente sintético del sintón catiónico no natural se utiliza el bromoacetato de etilo 3.191.
Esquema 3.55
La reacción de alquilación del ciclohexanocarbaldehído se lleva a cabo vía enamina. Así, el ciclohexanocarbaldehído 3.189 se convierte en la enamina 3.190 por reacción con

Tema 3. Enfermedades del sistema nervioso central 69
diisobutilamina en tolueno a reflujo. La reacción de la enamina con bromoacetato de etilo 3.191, en acetonitrilo a reflujo, genera la sal de iminio 3.192, que por hidrólisis ácida proporciona el aldehído éster 3.186. Este compuesto se convierte en la oxima 3.193 por reacción con hidroxilamina. Durante la hidrólisis del grupo éster con NaOH acuoso un 25% de la función oxima es hidrolizada a aldehído, por lo que la mezcla de reacción se trata con hidroxilamina a pH 5 y se convierte en la oxima-ácido 3.185. Finalmente, la hidrogenación de 3.185, a 9 atmósferas de presión en presencia de rodio depositado sobre alúmina, proporciona la gabapentina.
3.11.2. Sintesis de pregabalina
La pregabalina es un fármaco anticonvulsivo empleado en el tratamiento del dolor neuropático. Estudios recientes han demostrado que la pregabalina también es efectiva en el tratamiento de la ansiedad y la fibromialgia.
3.11.2.1a. Análisis retrosintético
El análisis retrosintético para la pregabalina racémica se indica en el esquema 3.56. El proceso comienza con la conversión del grupo amino de la pregabalina en grupo nitro. Esta operación IGF genera el nitroácido 3.194 que por escisión de enlace C-C conduce al anión de nitrometano 3.195 y al ácido conjugado 3.196.
Esquema 3.56
3.11.2.1b. Síntesis
La síntesis de la pregabalina racémica se inicia con la adición conjugada del anión de nitrometano al éster 3.195. Esta reacción se lleva a cabo en presencia de la base 1,1,3,3-tetrametilguanidina (TMG) y forma el aducto Michael 3.198 (esquema 3.57).31
Esquema 3.57
31 (a) R. Andruszkiewicz, R. B. Silverman. Synthesis 1989, 12, 953-955. (b) P.-W. Yuen, G. D. Kanter, C. P. Taylor, Bioorg. Med. Chem. Lett. 1994, 4, 823. (c) M. S. Hoekstra, D. M. Sobieray, M. A. Schwindt, T. A. Mulhern, T. M. Grote, B. K. Huckabee, V. S. Hendrickson, L. C. Franklin, G. L. Karrick, G. L. Org. Proc. Res. Dev. 1997, 1, 26.

Síntesis de antidepresivos, antiepilépticos y antiParkinson 70
La hidrogenación del nitroéster genera el correspondiente aminoéster que lactamiza en el seno de la reacción y conduce a la lactama 3.199. La hidrólisis ácida de la lactama proporciona la pregabalina racémica.
La (S)-pregabalina se obtiene mediante resolución de la mezcla racémica con ácido (S)-mandélico, lo que permite la separación de la sales diastereoisoméricas (esquema 3.58). A continuación, el tratamiento de la sal 3.200 con THF acuoso, seguido de cristalización en etanol, proporciona la (S)-pregabalina ópticamente pura.
Esquema 3.58
3.11.2.1c. Cuestiones
1) Proponga una síntesis para el éster 3.197.
3.11.2.2a. Análisis retrosintético de (S)-pregabalina mediante el empleo de un auxiliar quiral
En el esquema 3.59 se indica un análisis retrosintético para la (S)-pregabalina. El proceso se inicia con una doble operación IGF que genera el azidoéster 3.201, el cual, por escisión del enlace C-N basada en una reacción SN2, forma el anión azida y el compuesto 3.202 (X=grupo saliente). La operación IGF en el sustrato 3.202 conduce al compuesto 3.203 en el cual Xq representa una parte estructural quiral.
H2N COOH
Pregabalina
N3 COOR
3.201 X
COORN3 +
O
COORXq
O
H2CCOORXq 1,4-diCO
+
O
Xq
3.202
3.2033.188
3.2043.205
C-NIGF
IGF
Esquema 3.59
El compuesto 3.203 contiene una relación 1,4-dicarbonílica que se desconecta al sintón catiónico no natural 3.188 y al sintón aniónico natural 3.204. Este compuesto se generará de 3.205 mediante reacción de ionización con base. En el sentido sintético el fragmento quiral
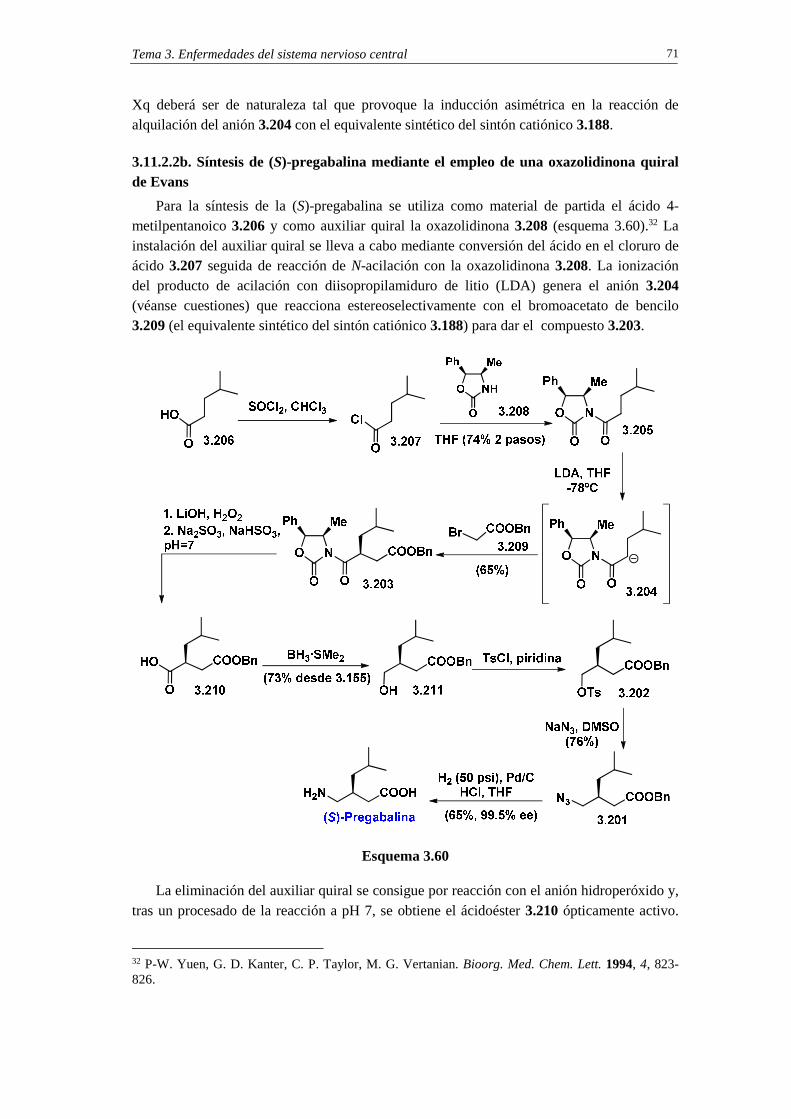
Tema 3. Enfermedades del sistema nervioso central 71
Xq deberá ser de naturaleza tal que provoque la inducción asimétrica en la reacción de alquilación del anión 3.204 con el equivalente sintético del sintón catiónico 3.188.
3.11.2.2b. Síntesis de (S)-pregabalina mediante el empleo de una oxazolidinona quiral de Evans
Para la síntesis de la (S)-pregabalina se utiliza como material de partida el ácido 4-metilpentanoico 3.206 y como auxiliar quiral la oxazolidinona 3.208 (esquema 3.60).32 La instalación del auxiliar quiral se lleva a cabo mediante conversión del ácido en el cloruro de ácido 3.207 seguida de reacción de N-acilación con la oxazolidinona 3.208. La ionización del producto de acilación con diisopropilamiduro de litio (LDA) genera el anión 3.204 (véanse cuestiones) que reacciona estereoselectivamente con el bromoacetato de bencilo 3.209 (el equivalente sintético del sintón catiónico 3.188) para dar el compuesto 3.203.
Esquema 3.60
La eliminación del auxiliar quiral se consigue por reacción con el anión hidroperóxido y, tras un procesado de la reacción a pH 7, se obtiene el ácidoéster 3.210 ópticamente activo.
32 P-W. Yuen, G. D. Kanter, C. P. Taylor, M. G. Vertanian. Bioorg. Med. Chem. Lett. 1994, 4, 823-826.

Síntesis de antidepresivos, antiepilépticos y antiParkinson 72
La síntesis de la (S)-pregabalina se completa del siguiente modo. El ácidoéster 3.210 se reduce quimioselectivamente al hidroxiéster 3.211 con el complejo BH3·SMe2 y, a continuación, el hidroxiéster se convierte en el tosilato 3.202. El desplazamiento SN2 del tosilato con el anión azida proporciona el azido éster 3.201. La hidrogenación de este compuesto provoca la hidrogenolisis del benciléster y la reducción de la función azida a amina y proporciona la (S)-pregabalina. Este compuesto se obtiene al final de la secuencia sintética con un exceso enantiomérico del más del 99.5%.
3.11.2.2c. Cuestiones
1) Las oxazolidinonas desarrolladas por Evans a partir de aminoácidos han demostrado ser auxiliares quirales muy útiles en reacciones de alquilación asimétrica. A partir del (S)-valinol se obtiene, por reacción con fosgeno o carbonato de dietilo, la (S)-4-isopropil-oxazolidin-2-ona 3.212. Del mismo modo a partir del (S)-fenilalaninol y de la norefedrina se obtiene la (S)-4-bencil-oxazolidin-2-ona 3.214 y la (4R,5S)-4-metil-5-fenil-oxazolidin-2-ona 3.216, respectivamente. La N-acilación de estos compuestos conduce a las correspondientes N-aciloxazolidinonas (esquema 3.61).
NH2HONO
O O
R
(S)
NHO
O
(S)-valinol
COCl2
óO
EtO OEt
O
ClR
base
NH2HONO
O O
R
Bn(S)
NHO
O
BnBn
(S)-fenilalaninol
COCl2
óO
EtO OEt
O
ClR
base
NH2HO
Me
NO
O O
R
Ph MePh
COCl2
ó
O
EtO OEt
(S) (R)
NHO
O
Ph Me
O
ClR
base
norefedrina
3.212 3.213
3.214 3.215
3.216 3.217
Esquema 3.61
Los enolatos derivados de las N-aciloxazolidinonas de Evans reaccionan con electrófilos activados, como yoduros de alquilo, bromuros de alilo o bencilo, o bromuros de acetato para dar lugar a los correspondientes productos de C-alquilación con elevados excesos diastereoselectivos.33
En el esquema 3.62 se indica el proceso de alquilación para la N-aciloxazolidinona 3.213, que se inicia con la enolización mediante reacción con bases como LDA o NaHDMS.
33 D. A. Evans, M. D. Ennis, J. D. Mathre. J. Am. Chem. Soc. 1982, 104, 1737.
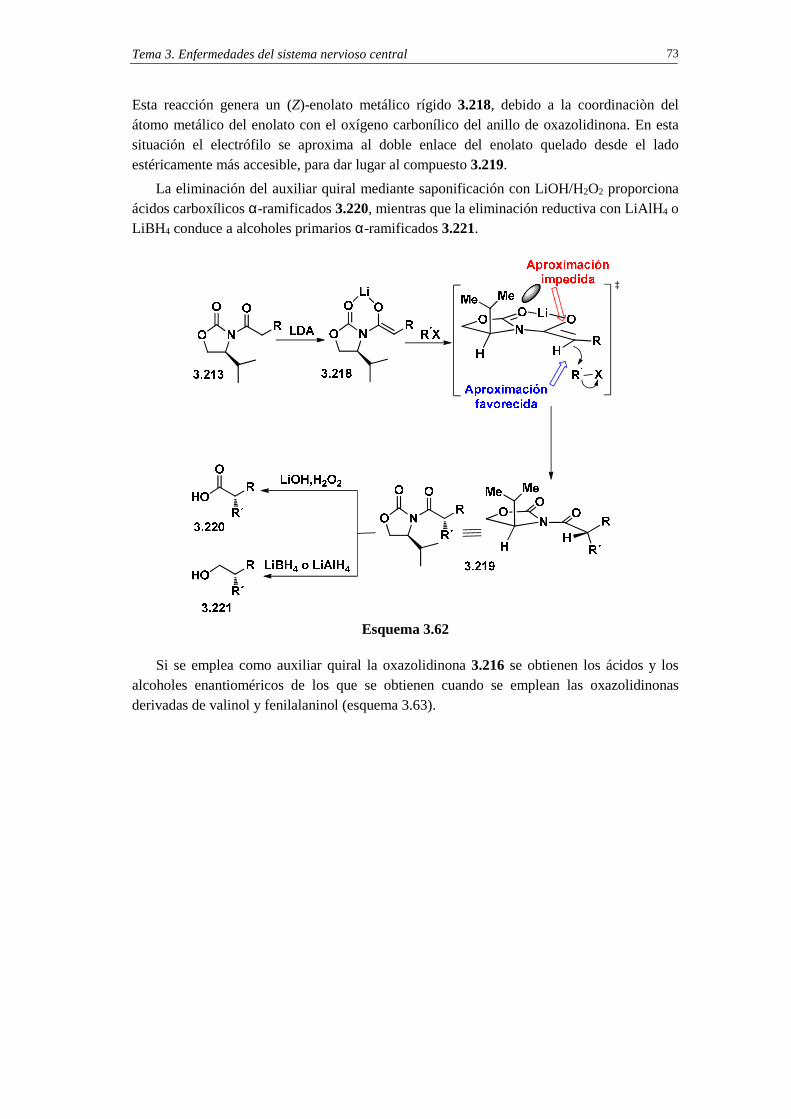
Tema 3. Enfermedades del sistema nervioso central 73
Esta reacción genera un (Z)-enolato metálico rígido 3.218, debido a la coordinaciòn del átomo metálico del enolato con el oxígeno carbonílico del anillo de oxazolidinona. En esta situación el electrófilo se aproxima al doble enlace del enolato quelado desde el lado estéricamente más accesible, para dar lugar al compuesto 3.219.
La eliminación del auxiliar quiral mediante saponificación con LiOH/H2O2 proporciona ácidos carboxílicos α-ramificados 3.220, mientras que la eliminación reductiva con LiAlH4 o LiBH 4 conduce a alcoholes primarios α-ramificados 3.221.
Esquema 3.62
Si se emplea como auxiliar quiral la oxazolidinona 3.216 se obtienen los ácidos y los alcoholes enantioméricos de los que se obtienen cuando se emplean las oxazolidinonas derivadas de valinol y fenilalaninol (esquema 3.63).
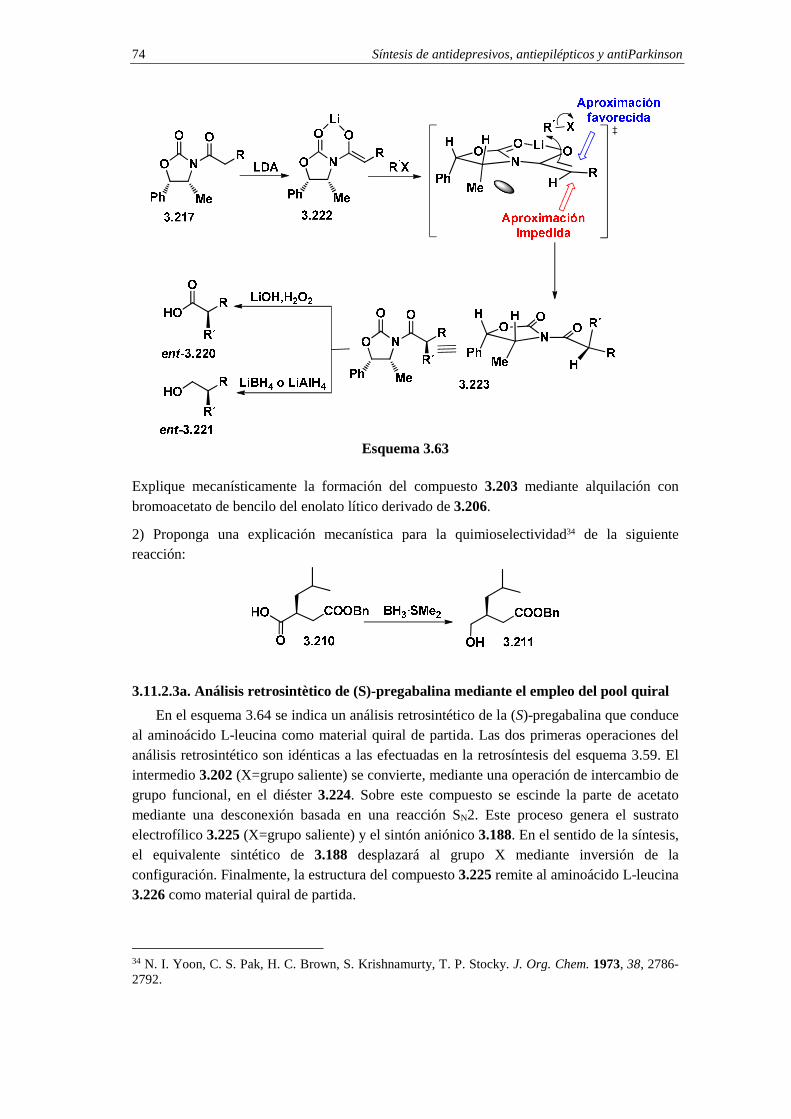
Síntesis de antidepresivos, antiepilépticos y antiParkinson 74
Esquema 3.63
Explique mecanísticamente la formación del compuesto 3.203 mediante alquilación con bromoacetato de bencilo del enolato lítico derivado de 3.206.
2) Proponga una explicación mecanística para la quimioselectividad34 de la siguiente reacción:
3.11.2.3a. Análisis retrosintètico de (S)-pregabalina mediante el empleo del pool quiral
En el esquema 3.64 se indica un análisis retrosintético de la (S)-pregabalina que conduce al aminoácido L-leucina como material quiral de partida. Las dos primeras operaciones del análisis retrosintético son idénticas a las efectuadas en la retrosíntesis del esquema 3.59. El intermedio 3.202 (X=grupo saliente) se convierte, mediante una operación de intercambio de grupo funcional, en el diéster 3.224. Sobre este compuesto se escinde la parte de acetato mediante una desconexión basada en una reacción SN2. Este proceso genera el sustrato electrofílico 3.225 (X=grupo saliente) y el sintón aniónico 3.188. En el sentido de la síntesis, el equivalente sintético de 3.188 desplazará al grupo X mediante inversión de la configuración. Finalmente, la estructura del compuesto 3.225 remite al aminoácido L-leucina 3.226 como material quiral de partida.
34 N. I. Yoon, C. S. Pak, H. C. Brown, S. Krishnamurty, T. P. Stocky. J. Org. Chem. 1973, 38, 2786-2792.
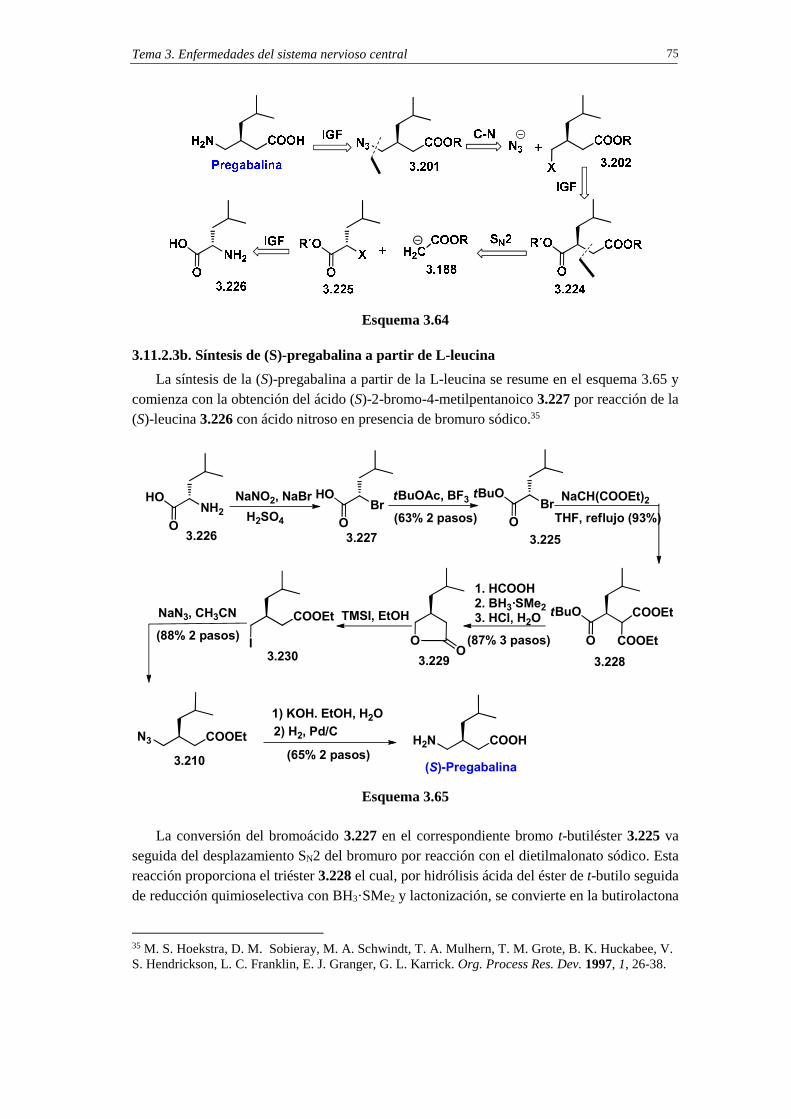
Tema 3. Enfermedades del sistema nervioso central 75
Esquema 3.64
3.11.2.3b. Síntesis de (S)-pregabalina a partir de L-leucina
La síntesis de la (S)-pregabalina a partir de la L-leucina se resume en el esquema 3.65 y comienza con la obtención del ácido (S)-2-bromo-4-metilpentanoico 3.227 por reacción de la (S)-leucina 3.226 con ácido nitroso en presencia de bromuro sódico.35
N3 COOEt
3.210
I
COOEt
3.2253.226O
HONH2
NaNO2, NaBr
H2SO4 O
HOBr
tBuOAc, BF3
(63% 2 pasos) O
tBuOBr
NaCH(COOEt)2
THF, reflujo (93%)
O
tBuO
COOEt
COOEt
3.227
3.228
1. HCOOH2. BH3·SMe23. HCl, H2O
(87% 3 pasos)OO
3.229
TMSI, EtOHNaN3, CH3CN
(88% 2 pasos)
1) KOH. EtOH, H2O
2) H2, Pd/C
(65% 2 pasos)H2N COOH
(S)-Pregabalina
3.230
Esquema 3.65
La conversión del bromoácido 3.227 en el correspondiente bromo t-butiléster 3.225 va seguida del desplazamiento SN2 del bromuro por reacción con el dietilmalonato sódico. Esta reacción proporciona el triéster 3.228 el cual, por hidrólisis ácida del éster de t-butilo seguida de reducción quimioselectiva con BH3·SMe2 y lactonización, se convierte en la butirolactona
35 M. S. Hoekstra, D. M. Sobieray, M. A. Schwindt, T. A. Mulhern, T. M. Grote, B. K. Huckabee, V. S. Hendrickson, L. C. Franklin, E. J. Granger, G. L. Karrick. Org. Process Res. Dev. 1997, 1, 26-38.

Síntesis de antidepresivos, antiepilépticos y antiParkinson 76
3.229. Cuando este compuesto se hace reaccionar con yoduro de trimetilsililo en etanol se obtiene el yodoéster 3.230 que se transforma en el azidoéster 3.210 por reacción con azida sódica. El compuesto 3.210 se convierte en (S)-pregabalina mediante saponificación seguida de hidrogenación.
3.11.2.3c. Cuestiones
1) Explique mecanísticamente la formación del bromoácido 3.226 a partir de la L-leucina 3.225.
2) Explique mecanísticamente la formación del t-butiléster 3.227 a partir del bromoácido 3.226.
3) Para la siguiente secuencia de reacciones:
a) Identifique la estructura del compuesto A y explique mecanísticamente su formación.
b) Identifique la estructura del compuesto B y explique mecanísticamente su formación a partir de A.
c) Explique mecanísticamente la conversión de B en 3.229.
4) Explique mecanísticamente la conversión de la lactona 3.229 en el yodoéster 3.230.

Tema 3. Enfermedades del sistema nervioso central 77
3.11.3. Síntesis de rufinamida
La rufinamida es un fármaco antiepiléptico que bloquea los canales de sodio y reduce la recuperación del potencial de acción neuronal dependiente de este catión. El fármaco fue desarrollado por Novartis para el tratamiento de la epilepsia asociada al síndrome de Lennox-Gastaut.
De alguna manera las membranas biológicas contribuyen a que se mantenga un exceso relativo de cargas negativas en el interior celular con respecto al medio extracelular. Los potenciales de membrana, o potenciales de acción, son cambios rápidos de polaridad a ambos lados de la membrana celular que separa el interior del exterior de una célula. Duran menos de 1 milisegundo y se generan por la diferencia de concentración iónica a ambos lados de la membrana celular.
En el medio extracelular (o líquido intersticial), el anión más abundante es el anión cloruro y el catión más abundante es el sodio y el calcio.
En el medio intracelular (o citoplasma), los aniones más abundantes son las proteínas, que en las condiciones del pH celular interno están ionizadas negativamente, y el catión más abundante es el potasio.
El desequilibrio iónico que produce la polarización de la membrana es debido a la distinta permeabilidad que presenta frente a cada uno de los diferentes iones. El ión potasio atraviesa la membrana libremente; la permeabilidad para el sodio es menor, y además es expulsado por medio de un transporte activo llamado bomba de sodio-potasio. Las proteínas, debido a su tamaño, no pueden atravesar libremente la membrana. Toda esta dinámica establece una diferencia de potencial en condiciones de reposo. Así, en reposo el potencial de membrana es, normalmente, negativo en la zona intracelular con un valor de unos -70 mV. El potencial de membrana no es el mismo en todas las células encontrándose células que tienen -90 mV y otras, como por ejemplo las musculares, que oscilan entre -50 y 60 mV.
Cuando una neurona recibe un estímulo se abren los canales de sodio de la membrana y el Na+ entra en la célula a favor del gradiente de concentración, provocándose la despolarización de la membrana al cambiar el potencial a positivo.
Si la despolarización alcanza un determinado valor umbral se genera un potencial de acción provocándose la apertura de los canales de potasio y el cierre de los canales de sodio, lo que conduce a la repolarización de la membrana.
Los canales de sodio están constituidos por proteínas de membrana dependientes del voltaje. El poro del canal de sodio contiene un filtro de selectividad en su zona intermedia. En esta zona se encuentran una serie de aminoácidos cargados negativamente que cumplen la función de atraer los iones positivos y repeler los iones negativos, a su vez el poro se vuelve más estrecho (0.2-0.3 nm) hacia el interior. En esta zona se encuentra un ácido glutámico que filtra el tamaño del ión sodio y permite el paso de éste y no de otros cationes. En esta zona también ocurre la deshidratación del catión. A la salida del catión del canal de sodio se produce la rehidratación del mismo.
Los canales de sodio poseen al menos tres estados: desactivado (cerrado), activado (abierto) e inactivado (cerrado). En el estado normal los canales se encuentran en estado desactivado encontrándose el canal cerrado a la conducción de iones. Cuando ocurre un

Síntesis de antidepresivos, antiepilépticos y antiParkinson 78
cambio en el potencial de membrana el canal se abre (paso 1 de la figura 3.44). La apertura del canal es de corta duración (aproximadamente 2-5 milisegundos) y una vez abierto el canal comienza el proceso de inactivación que consiste en la oclusión del poro en la cara intracelular (paso 2 de la figura 3.44). En este estado, el canal permanece abierto pero se encuentra en un estado de no conducción iónica por lo tanto en términos prácticos está cerrado. La remoción de la inactivación ocurre una vez que la membrana se repolariza. En este proceso el canal pasa al estado desactivado, estando cerrado el poro en su parte intermedia pero abierto en la parte intracelular (paso 3 de la figura 3.44). El estado desactivado no permite el paso de iones pero permite que el canal vuelva a estar disponible para la conducción iónica frente a un cambio del potencial de membrana.
Figura 3.44. Apertura, inactivación y cierre de los canales de sodio
Cuando una parte de la membrana celular se despolariza lo suficiente como para que se
abran los canales de sodio dependientes de voltaje, los iones de sodio entran en la célula por difusión facilitada. Una vez dentro, los iones positivos de sodio impulsan los iones próximos a lo largo del axón por repulsión electrostática, y atraen los iones negativos desde la membrana adyacente. Como resultado, una corriente positiva se desplaza a lo largo del axón, sin que ningún ion se esté desplazando muy rápido. Una vez que la membrana adyacente está suficientemente despolarizada, sus canales de sodio dependientes de voltaje se abren, realimentando el ciclo. El proceso se repite a lo largo del axón, generándose un nuevo potencial de acción en cada segmento de la membrana.
3.11.3.a. Análisis retrosintético
La retrosíntesis de la rufinamida comienza con una operación de interconversión del grupo funcional amida en nitrilo (esquema 3.66). Esta operación conduce al compuesto 3.231 cuyo anillo de triazol se desconecta mediante la aplicación de una operación

Tema 3. Enfermedades del sistema nervioso central 79
denominada RetroCicloAdición (RCA). Este proceso conduce al azidometil-diflurorobenceno 3.232 y al propiolonitrilo 3.233.
Esquema 3.66
3.11.3.b. Síntesis
La proyectada construcción del anillo de triazol se lleva a cabo mediante la cicloadición entre el azidometil-diflurorobenceno 3.232 y el cloroacrilonitrilo 3.234, que actúa como equivalente sintético del propiolonitrilo (esquema 3.67).36 La reacción se lleva a cabo mediante calentamiento en agua a 80ºC durante 24 horas. El exceso de acrilonitrilo se elimina mediante destilación y el residuo resultante (compuesto 3.231) se disuelve en tolueno y se calienta durante 40 minutos a 80ºC en presencia de NaOH acuoso al 30%. Estas condiciones provocan la hidrólisis de la función nitrinilo y permiten la obtención de la rufinamida.
Esquema 3.67
3.11.3.c. Cuestiones
1) Explique mecanísticamente la formación del compuesto 3.231.
3.11.4. Síntesis de lacosamida
Muchos fármacos antiepilépticos ralentizan el proceso de inactivación de los canales iónicos reduciendo así la capacidad de las neuronas para producir potenciales de acción. Como la inactivación sólo ocurre en las neuronas que están produciendo potenciales de acción, los fármacos que modulan la inactivación rápida reducen selectivamente las descargas en las neuronas que se encuentran activas en ese momento. Los anticonvulsionantes clásicos, como la carbamacepina, la fenitoína y la lamotrigina, actúan potenciando la inactivación rápida de los canales de sodio dependientes del voltaje.37
La inactivación lenta es un proceso similar, pero su efecto dura cientos de milisegundos y no produce el bloqueo completo de los canales de sodio dependientes del voltaje.
36 R. Portmann, Patente: WO02423, 1998. 37 B. K. Beyreuther, J. Freitag, C. Heers, N. Krebsfänger, U. Scharfenecker, T. CNS Drug. Rev. 2007, 13, 21-42.

Síntesis de antidepresivos, antiepilépticos y antiParkinson 80
La lacosamida (Vimpat®) es una amida funcionalizada derivada de D-serina que actúa interaccionando sobre los canales de sodio dependientes del voltaje. Sin embargo, no actúa mediante el modo convencional estabilizando la inactivación rápida del canal, sino que más bien potencia la inactivación lenta haciendo que la inactivación tenga lugar en los potenciales de membrana menos despolarizados. De esta forma solo se ven afectadas las neuronas que están despolarizadas (activas) durante largos periodos de tiempo, como las neuronas de los focos epilépticos.
3.11.4.a. Análisis retrosintético
La estructura de la lacosamida remite a la D-serina 3.235 como material de partida (esquema 3.68).
Esquema 3.68
3.11.4.b. Síntesis
Se han descrito varias síntesis de licosamida,38 pero la descrita en el esquema 3.69 es adecuada para la preparación del fármaco a gran escala.39 Así, para la síntesis de la lacosamida se elige como compuesto de partida la D-serina N-Boc protegida 3.236. La metilación de este compuesto, con sulfato de dimetilo en condiciones de transferencia de fase, proporciona la O-metil-D-serina N-Boc protegida 3.237. La activación del ácido carboxílico con cloroformiato de isobutilo, en presencia de N-metilmorfolina (NMO), seguida de reacción con bencilamina conduce a la amida 3.238. Finalmente, la lacosamida se obtiene mediante hidrólisis ácida de la función N-Boc seguida de N-acetilación.
Esquema 3.69
38 (a) D. Choi, J. P. Stables, H. Kohn, J. Med. Chem. 1996, 39, 1907-1916. (b) K. Kohn. Patente: US 5773475, 1998. (d) H. Kohn, S. V. Andurkar, S. V. Patente: US 6048899, 2000. 39 J. Riedner, G. Dunne, Patente: US 0027137(A1), 2008.

Tema 3. Enfermedades del sistema nervioso central 81
3.11.5. Síntesis de perampanel
El perampanel (nombre comercial Fycompa®, véase su estructura en la figura 3.45) es un fármaco antiepiléptico que actúa como un antagonista selectivo no competitivo de glutamato en los receptores AMPA (acrónimo del inglés Alpha-Amino-3-hydroxy-5-methyl-4-isoxazolePropionic Acid), el mayor subtipo de receptores ionotrópicos de glutamato.
Figura 3.45
Como se acaba de indicar, el perampanel se une selectivamente a los receptores AMPA y no se une a otros receptores ionotrópicos de glutamato como el receptor NMDA (del inglés N-Methyl-D-Aspartate) o los receptores de kainato. En la figura 3.46 se representa la unión selectiva del perampanel (Fycompa®) a los receptores AMPA en presencia de otros receptores ionotrópicos de glutamato.40
Figura 3.46. Unión selectiva de Fycompa® al receptor AMPA
El perampanel es también un antagonista selectivo no-competitivo de glutamato. En la parte izquierda de la figura 3.47 se representa la unión de un antagonista competitivo de glutamato al receptor AMPA. Se puede apreciar que el antagonista va a parar a los sitios de unión que ocupa el glutamato en el receptor, impidiendo la unión del neurotransmisor. En la parte de la derecha de la figura 3.47 se puede observar cómo el Fycompa® se une al receptor AMPA pero no impide la unión del glutamato a este receptor.
Se emplea en el tratamiento de las crisis epilépticas en pacientes de 12 años y mayores.
40 http://www.fycompa.eu/mode-of-action.php
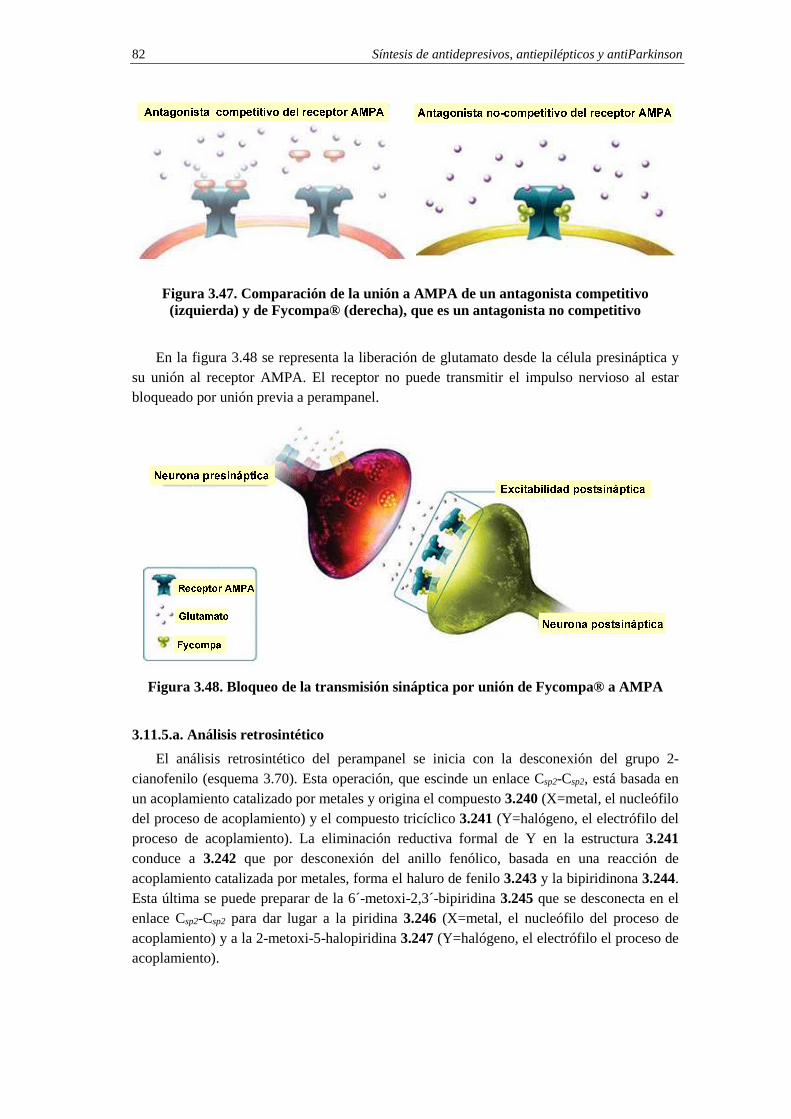
Síntesis de antidepresivos, antiepilépticos y antiParkinson 82
Figura 3.47. Comparación de la unión a AMPA de un antagonista competitivo (izquierda) y de Fycompa® (derecha), que es un antagonista no competitivo
En la figura 3.48 se representa la liberación de glutamato desde la célula presináptica y su unión al receptor AMPA. El receptor no puede transmitir el impulso nervioso al estar bloqueado por unión previa a perampanel.
Figura 3.48. Bloqueo de la transmisión sináptica por unión de Fycompa® a AMPA
3.11.5.a. Análisis retrosintético
El análisis retrosintético del perampanel se inicia con la desconexión del grupo 2-cianofenilo (esquema 3.70). Esta operación, que escinde un enlace Csp2-Csp2, está basada en un acoplamiento catalizado por metales y origina el compuesto 3.240 (X=metal, el nucleófilo del proceso de acoplamiento) y el compuesto tricíclico 3.241 (Y=halógeno, el electrófilo del proceso de acoplamiento). La eliminación reductiva formal de Y en la estructura 3.241 conduce a 3.242 que por desconexión del anillo fenólico, basada en una reacción de acoplamiento catalizada por metales, forma el haluro de fenilo 3.243 y la bipiridinona 3.244. Esta última se puede preparar de la 6´-metoxi-2,3´-bipiridina 3.245 que se desconecta en el enlace Csp2-Csp2 para dar lugar a la piridina 3.246 (X=metal, el nucleófilo del proceso de acoplamiento) y a la 2-metoxi-5-halopiridina 3.247 (Y=halógeno, el electrófilo el proceso de acoplamiento).
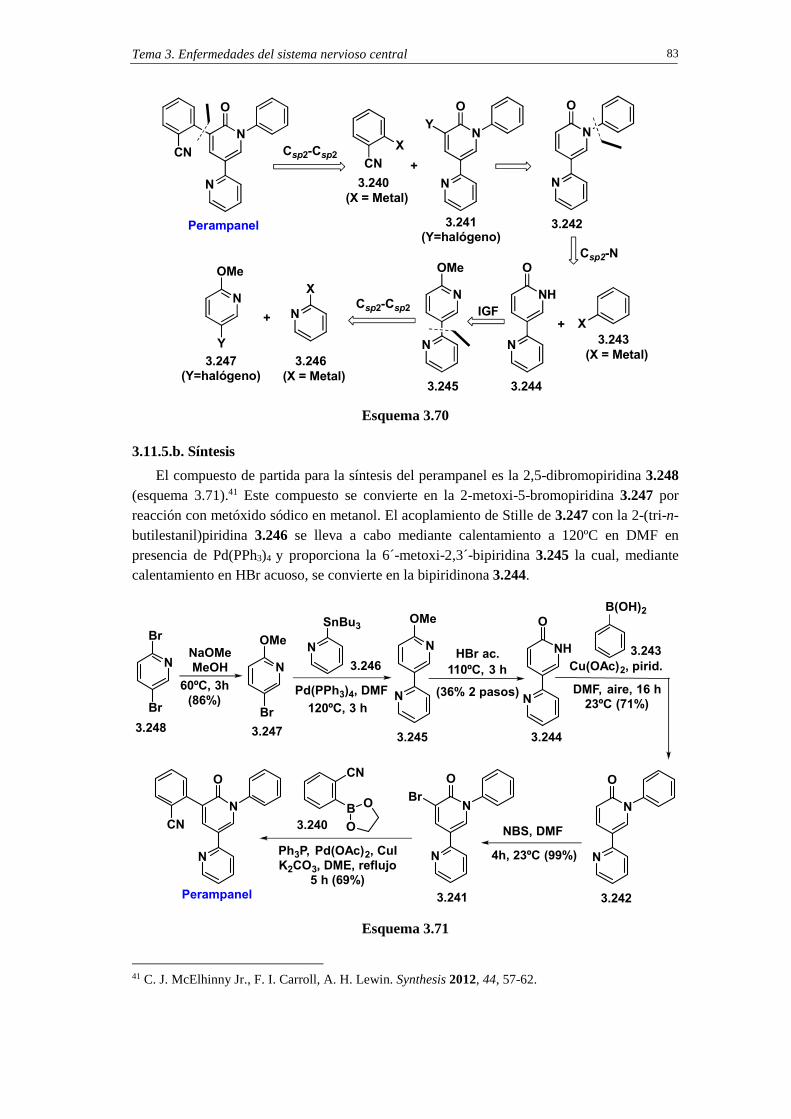
Tema 3. Enfermedades del sistema nervioso central 83
N
N
O
CN
Perampanel
Csp2-Csp2X
CN
N
N
O
Y
3.240
3.241(Y=halógeno)
+
(X = Metal)
N
N
O
Csp2-N
NH
N
O
X
3.242
+3.243
(X = Metal)
3.244
Csp2-Csp2N
N
OMe
N IGF
X
+
3.245
N
OMe
Y
3.246(X = Metal)
3.247(Y=halógeno)
Esquema 3.70
3.11.5.b. Síntesis
El compuesto de partida para la síntesis del perampanel es la 2,5-dibromopiridina 3.248 (esquema 3.71).41 Este compuesto se convierte en la 2-metoxi-5-bromopiridina 3.247 por reacción con metóxido sódico en metanol. El acoplamiento de Stille de 3.247 con la 2-(tri-n-butilestanil)piridina 3.246 se lleva a cabo mediante calentamiento a 120ºC en DMF en presencia de Pd(PPh3)4 y proporciona la 6´-metoxi-2,3´-bipiridina 3.245 la cual, mediante calentamiento en HBr acuoso, se convierte en la bipiridinona 3.244.
N
N
O
CN
Perampanel
B N
N
O
Br
3.240
3.241
N
N
O
NH
N
O
3.242
3.243
3.244
N
N
OMe
N
SnBu3
3.246N
Br
Br
3.248
N
OMe
Br
3.247
NaOMeMeOH
Pd(PPh3)4, DMF
HBr ac.Cu(OAc)2, pirid.
NBS, DMF
B(OH)2
CN
O
O
60ºC, 3h (86%)
120ºC, 3 h(36% 2 pasos)
110ºC, 3 h
3.245
DMF, aire, 16 h23ºC (71%)
4h, 23ºC (99%)Ph3P, Pd(OAc)2, CuIK2CO3, DME, reflujo
5 h (69%)
Esquema 3.71
41 C. J. McElhinny Jr., F. I. Carroll, A. H. Lewin. Synthesis 2012, 44, 57-62.
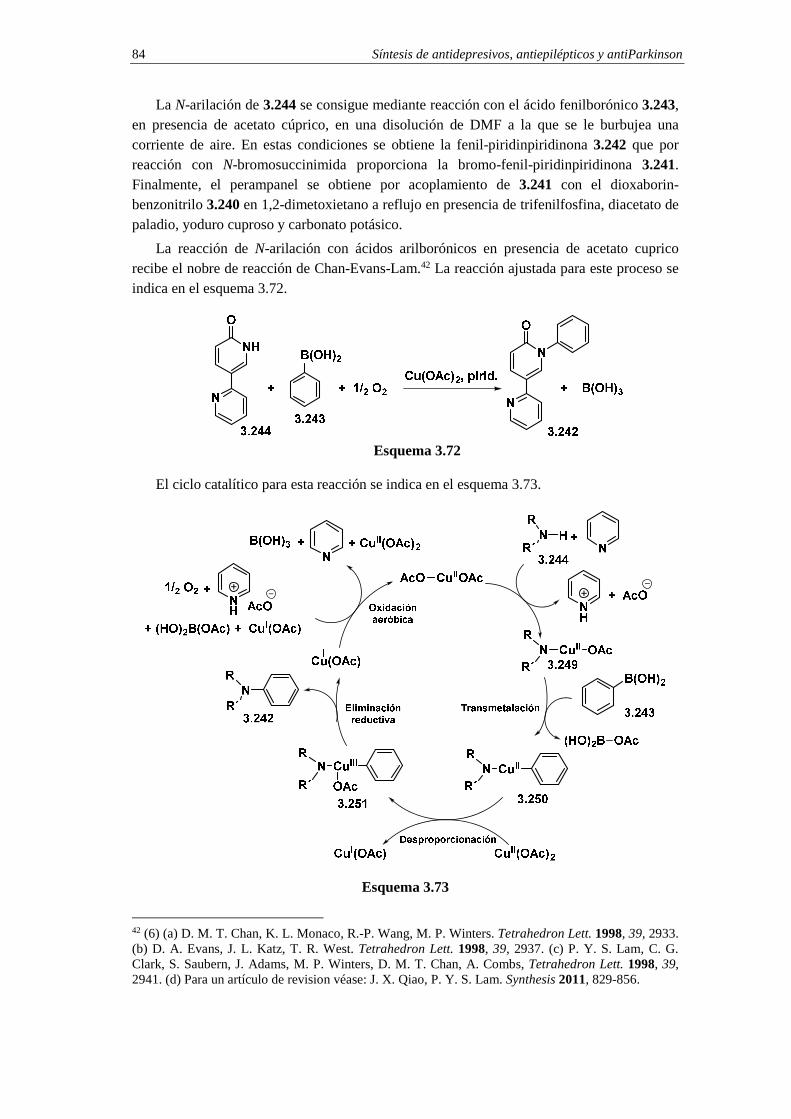
Síntesis de antidepresivos, antiepilépticos y antiParkinson 84
La N-arilación de 3.244 se consigue mediante reacción con el ácido fenilborónico 3.243, en presencia de acetato cúprico, en una disolución de DMF a la que se le burbujea una corriente de aire. En estas condiciones se obtiene la fenil-piridinpiridinona 3.242 que por reacción con N-bromosuccinimida proporciona la bromo-fenil-piridinpiridinona 3.241. Finalmente, el perampanel se obtiene por acoplamiento de 3.241 con el dioxaborin-benzonitrilo 3.240 en 1,2-dimetoxietano a reflujo en presencia de trifenilfosfina, diacetato de paladio, yoduro cuproso y carbonato potásico.
La reacción de N-arilación con ácidos arilborónicos en presencia de acetato cuprico recibe el nobre de reacción de Chan-Evans-Lam.42 La reacción ajustada para este proceso se indica en el esquema 3.72.
Esquema 3.72
El ciclo catalítico para esta reacción se indica en el esquema 3.73.
Esquema 3.73
42 (6) (a) D. M. T. Chan, K. L. Monaco, R.-P. Wang, M. P. Winters. Tetrahedron Lett. 1998, 39, 2933. (b) D. A. Evans, J. L. Katz, T. R. West. Tetrahedron Lett. 1998, 39, 2937. (c) P. Y. S. Lam, C. G. Clark, S. Saubern, J. Adams, M. P. Winters, D. M. T. Chan, A. Combs, Tetrahedron Lett. 1998, 39, 2941. (d) Para un artículo de revision véase: J. X. Qiao, P. Y. S. Lam. Synthesis 2011, 829-856.

Tema 3. Enfermedades del sistema nervioso central 85
La amina 3.244, representada como RNHR´en el esquema 3.73, reacciona con el acetato cúprico en presencia de piridina para, mediante un proceso de intercambio de ligandos, formar el complejo 3.249. Este intermedio experimenta el proceso de transmetalación con el ácido fenilborónico y origina el complejo 3.250 junto con ácido bórico monoacetato. El complejo 3.250 sufre un proceso de desproporcionación para dar lugar al complejo 3.251 (Cu(III)) y acetato cuproso (Cu(I)). El complejo de Cu(III) es el que experimenta el proceso de eliminación reductiva dando lugar al producto de N-arilación 3.242 y a acetato cuproso. La oxidación aeróbica del acetato cuproso, en presencia de acetato de piridinio y del ácido bórico monoacetato, forma ácido bórico, piridina y acetato cúprico, que inicia un nuevo ciclo catalítico.
3.11.5.c. Cuestiones
1) Explique mecanísticamente la formación de 2-metoxi-5-bromopiridina 3.247 por reacción de la 2,5-dibromopiridina 3.248 con metóxido sódico en metanol (esquema 3.74) ¿Por qué no se sustituye el bromo en la posición 5?
Esquema 3.74
2) Explique mecanísticamente la reacción de Stille que convierte la 2-metoxi-5-bromopiridina 3.247 en 6´-metoxi-2,3´-bipiridina 3.245 por reacción con la 2-(tri-n-butilestanil)piridina 3.246 (esquema 3.75).
Esquema 3.75
3) Proponga un mecanismo que explique la formación de 3.241 por reacción de 3.242 con N-bromosuccinimida (esquema 3.76).
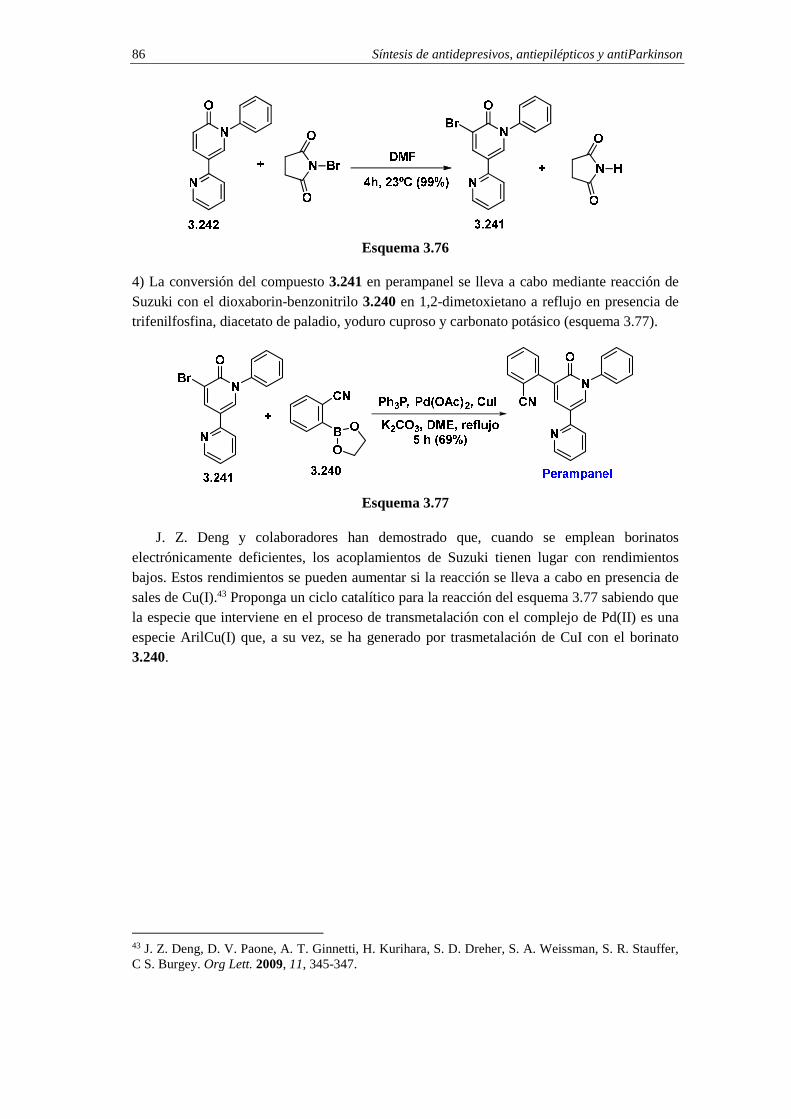
Síntesis de antidepresivos, antiepilépticos y antiParkinson 86
Esquema 3.76
4) La conversión del compuesto 3.241 en perampanel se lleva a cabo mediante reacción de Suzuki con el dioxaborin-benzonitrilo 3.240 en 1,2-dimetoxietano a reflujo en presencia de trifenilfosfina, diacetato de paladio, yoduro cuproso y carbonato potásico (esquema 3.77).
Esquema 3.77
J. Z. Deng y colaboradores han demostrado que, cuando se emplean borinatos electrónicamente deficientes, los acoplamientos de Suzuki tienen lugar con rendimientos bajos. Estos rendimientos se pueden aumentar si la reacción se lleva a cabo en presencia de sales de Cu(I).43 Proponga un ciclo catalítico para la reacción del esquema 3.77 sabiendo que la especie que interviene en el proceso de transmetalación con el complejo de Pd(II) es una especie ArilCu(I) que, a su vez, se ha generado por trasmetalación de CuI con el borinato 3.240.
43 J. Z. Deng, D. V. Paone, A. T. Ginnetti, H. Kurihara, S. D. Dreher, S. A. Weissman, S. R. Stauffer, C S. Burgey. Org Lett. 2009, 11, 345-347.

Tema 3. Enfermedades del sistema nervioso central 87
3.12. Enfermedad de Parkinson
La enfermedad de Parkinson (EP), también denominada parkinsonismo idiopático o parálisis agitante, es un trastorno neurodegenerativo crónico provocado por la destrucción de las neuronas dopaminérgicas de la sustancia negra, lo que conduce con el tiempo a lesiones en los tejidos que desembocan en la pérdida del control de los movimientos a cargo del Sistema Nervioso.
La enfermedad de Parkinson se ha clasificado como un trastorno del movimiento, que también desencadena alteraciones en la función cognitiva, en la expresión de las emociones y en la función autónoma.
Después de la enfermedad de Alzheimer, la enfermedad de Parkinson es el trastorno neurodegenerativo que afecta a un mayor número de pacientes. Esta enfermedad está extendida por todo el mundo y afecta tanto al sexo masculino como al femenino, siendo frecuente que aparezca a partir del sexto decenio de vida. Además de la variedad tardía existe otra versión precoz que se manifiesta en edades inferiores a los cuarenta años.
No se ha identificado ningún marcador biológico de esta enfermedad por lo que el diagnóstico de la misma se apoya en la detección de la característica tríada rigidez-temblor-acinesia (hipoactividad psíquica y motora o parálisis muscular).
Hasta el momento no se dispone de un método definitivo que cure la enfermedad, aunque lo síntomas de la EP se pueden paliar mediante el tratamiento farmacológico, rehabilitador e incluso quirúrgico.
3.12.1. Causas de la enfermedad de Parkinson
La enfermedad de Parkinson fue descrita y documentada en 1817 (Essay on the Shaking Palsy) por el médico británico Dr. James Parkinson. Cada 11 de abril se celebra el Día mundial del Parkinson con el objetivo de concienciar a la sociedad acerca de las necesidades de las personas aquejadas por esta dolencia. La fecha del 11 de abril fue escogida por coincidir con el nacimiento del médico James Parkinson.
Se conocen diversos procesos probablemente implicados en la producción del daño neuronal asociado a la EP. Uno de ellos es la formación de radicales libres, que reaccionan oxidando a las compuestos o elementos circundantes, especialmente metales como el hierro. Se ha demostrado que los pacientes con enfermedad de Parkinson tienen niveles elevados de hierro en el cerebro, en especial en la materia gris, y niveles decrecientes de ferritina, que sirve como mecanismo protector del hierro.
También se ha sugerido que la EP puede ser ocasionada por una toxina externa o interna que destruye selectivamente las neuronas dopaminérgicas. La teoría se apoya en el hecho de que algunas toxinas, como la 1-metil-4-fenil-1,2,3,6-tetrahidropiridina (MPTP) inducen síntomas similares a los de la enfermedad de Parkinson, así como lesiones en las neuronas de la materia gris en los seres humanos y en animales. Sin embargo, hasta la fecha, ninguna investigación ha proporcionado pruebas definitivas de que una toxina sea la causa de la enfermedad.
Una tercera hipótesis se basa en el factor genético como desencadenante de la EP. De un 15 a un 25 por ciento de los pacientes de Parkinson tienen un familiar cercano que ha

Síntesis de antidepresivos, antiepilépticos y antiParkinson 88
experimentado síntomas de esta patología, pudiendo ser el deterioro del ADN de las mitocondrias el responsable de la enfermedad.
Una cuarta teoría propone que la EP se produce por el desgaste de las neuronas productoras de dopamina.
Probablemente, una combinación de los cuatro mecanismos: daño oxidativo, toxinas ambientales, predisposición genética y envejecimiento acelerado podría ser el causante de la enfermedad.
3.13. Fármacos anti-Parkinson
El tratamiento de la EP puede ser de tres tipos: farmacológico, quirúrgico y/o rehabilitador. En los tres casos se pretende mejorar, o al menos mantener o prolongar la funcionalidad del enfermo durante el mayor tiempo posible.
Muchos de los síntomas característicos de la enfermedad de Parkinson son debidos a una deficiencia en el cerebro del neurotransmisor dopamina (figura 3.49).
Receptor dedopamina
Dopamina
Bomba dereabsorción
Figura 3.49. Representación del proceso de reconocimiento y reabsorción de dopamina
La dopamina se biosintetiza a partir del aminoácido L-tirosina mediante una secuencia de reacciones biológicas que se inicia con la conversión de la L-tirosina en levodopa por acción de la enzima Tirosina-hidroxilasa. A continuación, la descarboxilación de la levodopa, por la enzima Dopa-descarboxilasa forma la dopamina (esquema 3.78).
Esquema 3.78

Tema 3. Enfermedades del sistema nervioso central 89
El suministro de dopamina al paciente, con el objetivo de reponer las reservas agotadas, no resulta eficaz, porque la dopamina no puede atravesar la barrera hematoencefálica. Por ello, los fármacos anti-Parkinson tienen como objetivo la restitución, aunque sea de forma temporal, de la dopamina del cerebro.
Conviene señalar que ninguno de los fármacos usados en el tratamiento de la EP actúa sobre la progresión de la enfermedad. En la actualidad, los fármacos más usados son la levodopa y varios agonistas de dopamina, aunque también tienen cierta relevancia otros como la selegilina y la rasagilina (inhibidores de la MAO-B), la amantadina (liberador de dopamina) o la benztropina (antagonista del receptor muscarínico de la acetilcolina).
3.13.1. Levodopa
La levodopa es el fármaco antiparkinsoniano más efectivo en tratamiento de la enfermedad. Se introdujo en 1967 para tratar afecciones tales como la rigidez, el temblor y la hipocinesia-bradicinesia (disminución en la velocidad de los movimientos normales).
La estructura de levodopa le permite atravesar la barrera hematoencefálica convirtiéndose en dopamina en un solo paso por la enzima DOPA-descarboxilasa (véase el esquema 3.78). La levodopa se suele administrar combinada con inhibores de la enzima DOPA-descarboxilasa periférica, como la carbidopa o la benseracida. De esta forma se impide la transformación prematura de la levodopa en dopamina, lo cual permite suministrar dosis menores y minimizar los efectos secundarios gastrointestinales y cardiovasculares provocados por la dopamina liberada antes de llegar al cerebro.
En torno a un 80% de los pacientes tratados con levodopa manifiesta una mejoría inicial, sobre todo en lo referente a rigidez e hipocinesia-bradicinesia, mientras que un 20% de las personas llega a recuperar por completo la función motora.
El principal inconveniente de la levodopa es que pierde el efecto a los 3-5 años de tratamiento, apareciendo efectos secundarios como discinesias (espasmos asociados a movimientos anormales e involuntarios) o el llamado fenómeno on/off, o fluctuaciones del estado del enfermo, de duración variable e impredecible, que oscila entre ratos sin síntomas (fases "on" o fases de conexión a la levodopa) y otros en que reaparecen el temblor, la dificultad para caminar y la lentitud (fases "off" o fases de desconexión a la levodopa). En los períodos "on" pueden presentarse discinesias. El fenómeno on/off parece estar asociado a variaciones en sangre de los niveles de levodopa como consecuencia de su interacción con las proteínas de la dieta.
3.13.2. Agonistas de dopamina
La utilización de los agonistas dopaminérgicos está muy extendida en el tratamiento de los estadios tempranos de la enfermedad de Parkinson, con la finalidad de retrasar al máximo posible la administración de levodopa. En la figura 3.49 se indica esquemáticamente el modo de acción de los fármacos agonistas de dopamina.
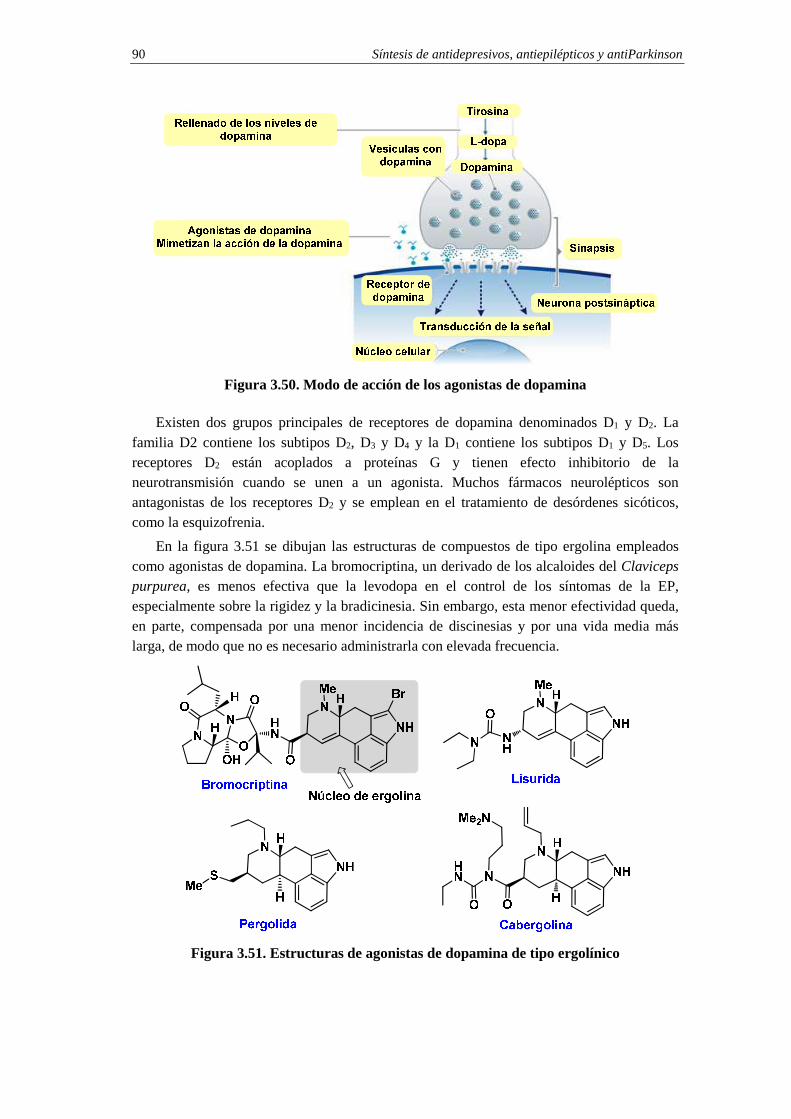
Síntesis de antidepresivos, antiepilépticos y antiParkinson 90
Figura 3.50. Modo de acción de los agonistas de dopamina
Existen dos grupos principales de receptores de dopamina denominados D1 y D2. La familia D2 contiene los subtipos D2, D3 y D4 y la D1 contiene los subtipos D1 y D5. Los receptores D2 están acoplados a proteínas G y tienen efecto inhibitorio de la neurotransmisión cuando se unen a un agonista. Muchos fármacos neurolépticos son antagonistas de los receptores D2 y se emplean en el tratamiento de desórdenes sicóticos, como la esquizofrenia.
En la figura 3.51 se dibujan las estructuras de compuestos de tipo ergolina empleados como agonistas de dopamina. La bromocriptina, un derivado de los alcaloides del Claviceps purpurea, es menos efectiva que la levodopa en el control de los síntomas de la EP, especialmente sobre la rigidez y la bradicinesia. Sin embargo, esta menor efectividad queda, en parte, compensada por una menor incidencia de discinesias y por una vida media más larga, de modo que no es necesario administrarla con elevada frecuencia.
Figura 3.51. Estructuras de agonistas de dopamina de tipo ergolínico

Tema 3. Enfermedades del sistema nervioso central 91
La lisurida es otro alcaloide de tipo ergolida que exhibe capacidad agonista parcial de los receptores de la dopamina y la serotonina. Tiene una alta afinidad por los receptores D2, D3 y D4 de la dopamina, así como por los receptores 5-HT1A y 5-HT2A/C de la serotonina. La administración de la lisurida es parenteral y actualmente está aprobada para el tratamiento de la enfermedad de Parkinson en Europa pero no en EE.UU.
La pergolida es el más potente de los fármacos de tipo ergolida. Sin embargo este medicamento fue retirado en marzo del 2007 del mercado estadounidense por su asociación con valvulopatías cardiacas.
La cabergolina es un potente agonista de los receptores de dopamina tipo D2. Tiene una larga semivida de eliminación que permite administración única diaria.
En la figura 3.51 se indican las estructuras del pramiprexol y del ropinirol, fármacos anti-Parkinson no ergolínicos con actividad agonista de dopamina.
N
SN
H
NH2NH
O
N
Pramipexol Ropinirol
Figura 3.52. Estructuras de agonistas de dopamina no ergonilicos
El pramipexol se emplea en el control del temblor y la depresión, siendo principalmente activo frente a los receptores D3.
El ropinirol se utiliza en el tratamiento de la enfermedad de Parkinson y también en el tratamiento del síndrome de piernas inquietas. Este síndrome denominado RLS, por sus siglas en inglés, Restless-Legs-Syndrome, es un trastorno neurológico caracterizado por sensaciones desagradables en las piernas y un impulso incontrolable de moverse y andar cuando se está descansando, en un esfuerzo del paciente de aliviar estas sensaciones. A los que sufren esta enfermedad se les denomina andadores nocturnos.
3.13.3. Inhibidores de la monoaminooxidasa B: selegilina y rasagilina
La selegilina se emplea en el tratamiento del Parkinson, la depresión y la demencia senil (enfermedad de Alzheimer). El mesilato de rasagilina es un potente inhibidor de la monoaminooxidasa B (MAO-B) y se emplea en el tratamiento de estadios iniciales de la enfermedad de Parkinson. Actualmente está en fase II para el tratamiento del Alzeheimer.
Figura 3.53. Estructuras de inhibidores de MAO-B

Síntesis de antidepresivos, antiepilépticos y antiParkinson 92
La selegilina y la rasagilina actúan inhibiendo el enzima MAO-B, que es la monoaminoxidasa predominante en las zonas del sistema nervioso central que tienen dopamina. Con la inhibición de la MAO-B se consigue proteger a la dopamina de la degradación intraneuronal. En la figura 3.54 se representa esquemáticamente el modo de acción de la selegilina mediante inhibición de la MAO-B.
Dopamina
Selegilina
Espaciointersináptico
Receptor dedopamina
Reabsorciónde dopamina
MAO-B
Selegilina
Neurona postsináptica
Figura 3.54. Representación del modo de acción de la selegilina 3.13.4. Liberadores presinápticos de dopamina: amantadina
La amantadina, o 1-aminoadamantano, fue aprobada como antiviral por la FDA en 1976 para el tratamiento de la gripe común y el tratamiento de la gripe tipo A en adultos. El fármaco también reduce los síntomas del Parkinson y se prescribe junto a la levodopa cuando ésta pierde efectividad por desarrollo de tolerancia.
Figura 3.55. Estructura de la amandatina
El modo de acción de la amantadina está relacionado con su capacidad para incrementar la liberación de dopamina, inhibir la recaptación de aminas y actuar directamente sobre los

Tema 3. Enfermedades del sistema nervioso central 93
receptores de dopamina. También inhibe la acción del glutamato, la sustancia química cerebral que provoca la generación de radicales libres.
La amantadina no es tan eficaz como la levodopa o la bromocriptina y su acción se ve disminuida con el transcurso del tiempo. En contraposición a esto, sus efectos secundarios son cualitativamente similares a los de la levodopa, pero ostensiblemente menos importantes. En la actualidad se utiliza asociada a levodopa, a fin de prolongar la vida útil de ésta y controlar los trastornos motores, especialmente la discinesia.
3.13.5. Antagonistas del receptor muscarínico de la acetilcolina: benztropina
La benztropina, o benzatropina, es un anticolinérgico que se emplea como fármaco de segunda línea en el tratamiento de la enfermedad de Parkinson. La administración de benztropina disminuye los temblores y la rigidez, aunque no la bradicinesia. La benztropina también se emplea en el tratamiento de la distonia, una enfermedad que causa la contracción anormal de los músculos.
Figura 3.56. Estructura de la benztropina
3.14. Síntesis de fármacos antiParkinson
3.14.1. Síntesis de pramiprexol
El pramiprexol se receta en el el tratamiento de los signos y síntomas de la enfermedad de Parkinson idiopática, sólo (sin levodopa) o en asociación con levodopa, es decir, durante el curso de la enfermedad, hasta las últimas etapas cuando el efecto de la levodopa desaparece o se convierte en irregular, produciéndose fluctuaciones del efecto terapéutico (fluctuaciones on-off). 3.14.1.a. Análisis retrosintético
El análisis retrosintético del pramipexol se inicia con el cambio del grupo funcional amina por amida (esquema 3.79).

Síntesis de antidepresivos, antiepilépticos y antiParkinson 94
N
SN
H
NH2
Pramipexol
IGF
N
SN
H
NH2O
amid.
N
SH2NNH2
CDA
HN
HSH2NNH2
O
+
3.252
3.2533.2543.255
X
Esquema 3.79
La operación IGF genera el compuesto 3.252 que por desconexión del enlace amida conduce a la amina 3.253. La siguiente operación retrosintética se encarga de la desconexión del anillo de tiazol y se ha indicado como operación de ciclodeshidratación (CDA). La operación de desconexión genera el ácido carbamidotióico 3.254 y la aminohalociclohexanona 3.255 (X=halógeno).
3.14.1.b. Síntesis
Para la síntesis del pramiprexol se elige como compuesto de partida la N-acetil-4-aminociclohexanona 3.256 (esquema 3.80).44
Esquema 3.80
El compuesto 3.256 se convierte en el tetrahidrobenzotiazol 3.258 mediante α-
halogenación con bromo en ácido acético, seguida de reacción de la correspondiente α-bromocetona con tiourea 3.257 (equivalente sintético del ácido carbamidotióico 3.254). La
44 (a) C. S. Schneider, J. Mierau. J. Med. Chem. 1987, 30, 494-498. (b) M. Zivec, B. Anzic, S. Gobec. Org. Process Res. Dev. 2010, 14, 1125.1129.

Tema 3. Enfermedades del sistema nervioso central 95
reacción de N-desacetilación mediante hidrólisis ácida conduce al compuesto racémico (+/-)-3.253 el cual, mediante resolución con ácido L-(+)-tartárico, proporciona el diamino tiazol 3.253, ópticamente activo. El tratamiento de este compuesto con anhidrido propiónico, en presencia de trietilamina, conduce a la propanamida 3.252 que se convierte en pramipexol mediante reducción con borano.
En el esquema 3.81 se indica una síntesis para la N-acetil-4-aminociclohexanona 3.256.
Esquema 3.81
3.14.1.c. Cuestiones
1) Explique mecanísticamente la formación del tetrahidrobenzotiazol 3.258 a partir de la N-acetil-4-aminociclohexanona 3.256.
2) ¿Por qué la reacción de propanoilación de 3.253 es quimioselectiva? ¿Por qué no reacciona el grupo amino unido al anillo de tiazol?
3.14.2. Síntesis de ropinirol
El ropinirol se emplea en monoterapia, para retrasar la administración de L-dopa en los estadios iniciales de la enfermedad de Parkinson y en combinación con este fármaco durante fases más avanzadas de la enfermedad, cuando el efecto de la L-dopa disminuye. También se receta en el tratamiento del síndrome de piernas inquietas idiopático de moderado a grave.
3.14.2.1a. Análisis retrosintético
La retrosíntesis del ropinirol se inicia con la apertura del anillo lactámico, lo que conduce al aminoácido 3.261, que por interconversión del grupo amino en grupo nitro se convierte en el nitroácido 3.262 (esquema 3.82).
NH
O
N
Ropinirol
C-N
Amida NH2
N
COOH IGF
NO2
N
COOH
NO2
CH3
N
AGF
NO2
CH3
N
O
Homol.
C-N
AmidaNO2
CH3
O
OH
IGF
NO2
CH3
CN
IGF
NO2
CH3
X
IGF
NO2
CH3
COOH
3.261 3.262 3.263
3.2643.2653.2663.2673.268
Esquema 3.82
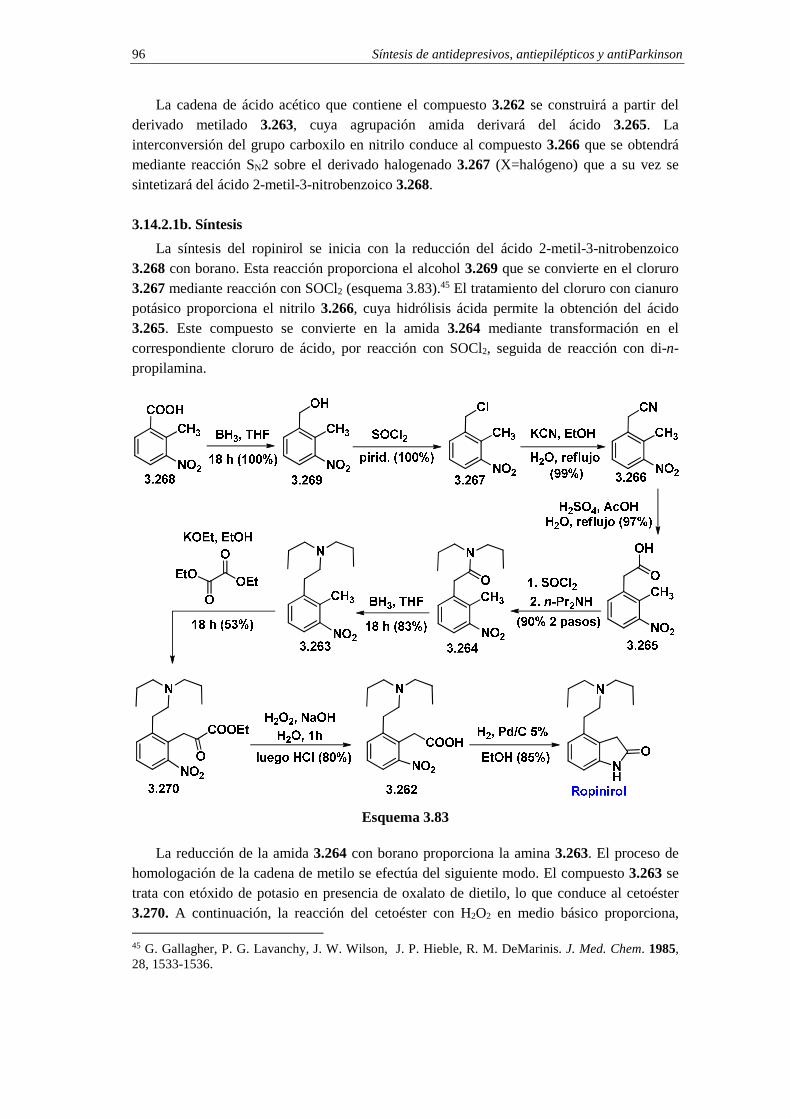
Síntesis de antidepresivos, antiepilépticos y antiParkinson 96
La cadena de ácido acético que contiene el compuesto 3.262 se construirá a partir del derivado metilado 3.263, cuya agrupación amida derivará del ácido 3.265. La interconversión del grupo carboxilo en nitrilo conduce al compuesto 3.266 que se obtendrá mediante reacción SN2 sobre el derivado halogenado 3.267 (X=halógeno) que a su vez se sintetizará del ácido 2-metil-3-nitrobenzoico 3.268.
3.14.2.1b. Síntesis
La síntesis del ropinirol se inicia con la reducción del ácido 2-metil-3-nitrobenzoico 3.268 con borano. Esta reacción proporciona el alcohol 3.269 que se convierte en el cloruro 3.267 mediante reacción con SOCl2 (esquema 3.83).45 El tratamiento del cloruro con cianuro potásico proporciona el nitrilo 3.266, cuya hidrólisis ácida permite la obtención del ácido 3.265. Este compuesto se convierte en la amida 3.264 mediante transformación en el correspondiente cloruro de ácido, por reacción con SOCl2, seguida de reacción con di-n-propilamina.
Esquema 3.83
La reducción de la amida 3.264 con borano proporciona la amina 3.263. El proceso de homologación de la cadena de metilo se efectúa del siguiente modo. El compuesto 3.263 se trata con etóxido de potasio en presencia de oxalato de dietilo, lo que conduce al cetoéster 3.270. A continuación, la reacción del cetoéster con H2O2 en medio básico proporciona, 45 G. Gallagher, P. G. Lavanchy, J. W. Wilson, J. P. Hieble, R. M. DeMarinis. J. Med. Chem. 1985, 28, 1533-1536.

Tema 3. Enfermedades del sistema nervioso central 97
después de acidificar, el ácido 3.262. La hidrogenación de este compuesto, con hidrógeno molecular en presencia de Pd/C, genera el aminoácido 3.261, que se convierte in situ en ropirinol.
3.14.2.1c. Cuestiones
1) La reacción ajustada para la conversión del compuesto 3.263 en el cetoéster 3.270 se indica a continuación (esquema 3.84). La reacción no consume etóxido de potasio, pero esta base es necesaria para que tenga lugar la reacción. Con estos datos explique mecanísticamente la formación del cetoéster 3.270.
Esquema 3.84
2) La reacción del cetoéster 3.270 con H2O2 y NaOH forma el carboxilato sódico 3.271 mediante la reacción ajustada que se indica en el esquema 3.85 (la acidificación de la mezcla de reacción protona el carboxilato 3.271 y proporciona el ácido 3.262).
Esquema 3.85
Con los datos anteriores proponga un mecanismo que explique la conversión del cetoéster 3.270 en el carboxilato 3.271.
3.14.2.2b. Síntesis a partir de isocromano
En el esquema 3.86 se indica una síntesis industrial del ropinirol a partir del isocromano 3.272.46 El proceso se inicia con la reacción de este compuesto con cloruro de benzoilo en diclorometano en presencia de cloruro de zinc. Esta reacción forma el clorobenzoato 3.273. A la mezcla que contiene este compuesto se le añade hexametilenetetramina (HMTA) y metanol y se calienta a reflujo, con eliminación de disolvente, durante 1 hora, lo que genera el cloruro de hexametilenetetraamonio 3.274. A la disolución caliente que contiene esta sal se le añade ácido acético y agua y la mezcla resultante se calienta a reflujo, con eliminación
46 (a) J. D. Hayler, S. L. B. Howie, R. G. Giles, A. Negus, P. W. Oxley, T. C. Walsgrove, M. Whiter. Org. Process Res. Dev. 1998, 2, 3-9. (b) J. D. Hayler, S. L. B. Howie, R. G. Giles, A. Negus, P. W. Oxley, T. C. Walsgrove, S. E. Walsh, R. E. Dagger, J. M. Fortunak, A. Mastrocola. J. Hetreocyclic Chem. 1975, 32, 875-882.

Síntesis de antidepresivos, antiepilépticos y antiParkinson 98
de disolvente. Luego se enfría y se extrae con metil t-butil éter (MTBE). La fase orgánica se lava secuencialmente con H2SO4 2 M y carbonato sódico acuso. La concentración de la fase de MTBE proporciona el 2-formilfenetilbenzoato 3.275. Este compuesto se disuelve en metanol y se le añade nitrometano, ácido acético y n-butilamina. La mezcla se agita a 22ºC durante 18 horas. Luego, la centrifugación seguida de lavado con isopropanol y secado proporciona el nitroestireno 3.276 con un 55% de rendimiento global a partor de isocromano.
NO
3.272
PhCOCl, ZnCl2
CH2Cl2, reflujo Cl
OCOPh
3.273
HMTA, MeOH
reflujo
OCOPh
N
N
N
Cl
AcOH, H2Oreflujo
CHO
OCOPh
3.274
3.275
CH3NO2, MeOH
30ºC, BuNH2
OCOPh
NO2
FeCl3, CH3COCl
CH2Cl2, 5ºC, 1 h (64%)
NH
O
OCOPh
NH
O
OCOPh
Ropinirol
3.276
3.277
Cl
NH
O
OH
1. TsCl, piridina
2. n-Pr2NH (85%)
3.278
NH2NH2, Pd/C al 10%reflujo, 1 h
luego NaOH ac.reflujo 30 min (85%)
(87%)
Esquema 3.86
La reacción del nitroestireno 3.276 con cloruro de acetilo y cloruro férrico, en diclorometano durante 1 h a 5ºC, proporciona el clorooxindol 3.277.47 El calentamiento de este compuesto con cloridrato de hidrazina en presencia de Pd/C al 10%, seguida de saponificación, permite la obtención del compuesto hidroxioxindol 3.278. El ropinirol se obtiene a partir de este compuesto mediante tosilación y reacción con di-n-propilamina.
3.14.2.2c. Cuestiones
1) Explique mecanísticamente la conversión del isocromano en el clorobenzoto 3.273 mediante reacción con cloruro de benzoilo en presencia de cloruro de zinc.
2) La oxidación de haluros de alquilo primarios mediante reacción con hexametilendiamina seguida de hidrólisis ácida recibe el nombre de oxidación de Sommelet. La reacción ajustada para la oxidación de cloruro de hexametilentrtramonio 3.274 se indica en el esquema 3.87. Con estos datos proponga un mecanismo para esta reacción.
47 J. Guillaumel, P. Demerseman, J-M. Clavel, R. Royer, N. Platzer, C. Brevard. Tetrahedron 1980, 36, 2459-2465.

Tema 3. Enfermedades del sistema nervioso central 99
Esquema 3.87
3) Proponga uen mecanismo para la formación del clorooxindol 3.277 por reacción del nitroestireno 3.276 con cloruro de acetilo y cloruro férrico.
3.14.3. Síntesis de selegilina
La selegilina se emplean en el tratamiento del Parkinson idiopático, como monoterapia en estadios iniciales de dicha enfermedad, o como coadyuvante de la L-Dopa (con o sin inhibidores de la descarboxilasa).
3.14.3.a. Análisis retrosintético
El análisis retrosintético de la selegilina se inicia con la desconexión del fragmento propargílico (esquema 3.88). Esta escisión genera la N-metilamina 3.279 y el haluro de propargilo 3.280 (X=halógeno). La siguiente operación retrosintética se encarga de adicionar el grupo funcional hidroxilo sobre el grupo metilo. Este proceso genera el aminoalcohol 3.281, el cual, por aumento del estado de oxidación de la función hidroxilo, se convierte en el aminoaldehído 3.282. La operación clave de la retrosíntesis es la que escinde el grupo metilamino. Esta desconexión genera el sintón aniónico natural 3.283 y el sintón catiónico no natural 3.284. En la parte de síntesis se explicará cuál es el equivalente sintético para el sintón catiónico 3.284.
Esquema 3.88
3.14.3.b. Síntesis
Para la síntesis de la selegilina se elige como compuesto de partida el 3-fenilpropanal 3.285 que se hace reaccionar con el azodicarboxilato de dibencilo 3.286, que actúa como
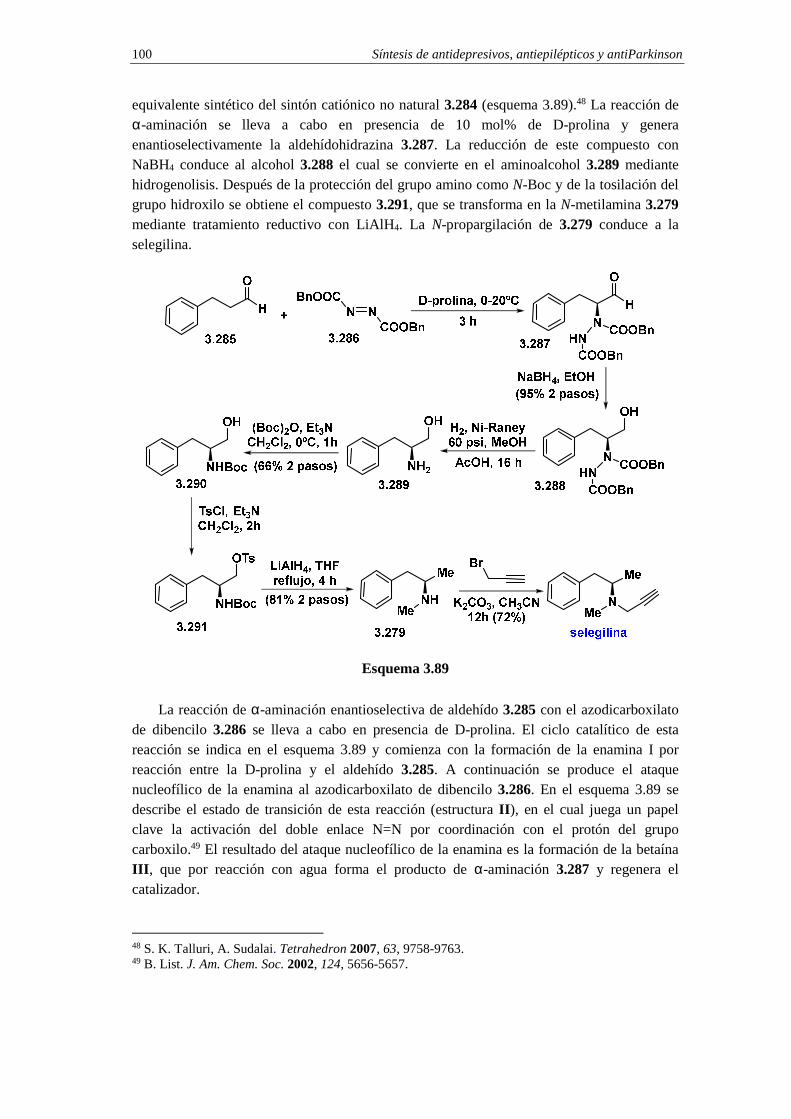
Síntesis de antidepresivos, antiepilépticos y antiParkinson 100
equivalente sintético del sintón catiónico no natural 3.284 (esquema 3.89).48 La reacción de α-aminación se lleva a cabo en presencia de 10 mol% de D-prolina y genera enantioselectivamente la aldehídohidrazina 3.287. La reducción de este compuesto con NaBH4 conduce al alcohol 3.288 el cual se convierte en el aminoalcohol 3.289 mediante hidrogenolisis. Después de la protección del grupo amino como N-Boc y de la tosilación del grupo hidroxilo se obtiene el compuesto 3.291, que se transforma en la N-metilamina 3.279 mediante tratamiento reductivo con LiAlH4. La N-propargilación de 3.279 conduce a la selegilina.
Esquema 3.89
La reacción de α-aminación enantioselectiva de aldehído 3.285 con el azodicarboxilato de dibencilo 3.286 se lleva a cabo en presencia de D-prolina. El ciclo catalítico de esta reacción se indica en el esquema 3.89 y comienza con la formación de la enamina I por reacción entre la D-prolina y el aldehído 3.285. A continuación se produce el ataque nucleofílico de la enamina al azodicarboxilato de dibencilo 3.286. En el esquema 3.89 se describe el estado de transición de esta reacción (estructura II ), en el cual juega un papel clave la activación del doble enlace N=N por coordinación con el protón del grupo carboxilo.49 El resultado del ataque nucleofílico de la enamina es la formación de la betaína III , que por reacción con agua forma el producto de α-aminación 3.287 y regenera el catalizador.
48 S. K. Talluri, A. Sudalai. Tetrahedron 2007, 63, 9758-9763. 49 B. List. J. Am. Chem. Soc. 2002, 124, 5656-5657.
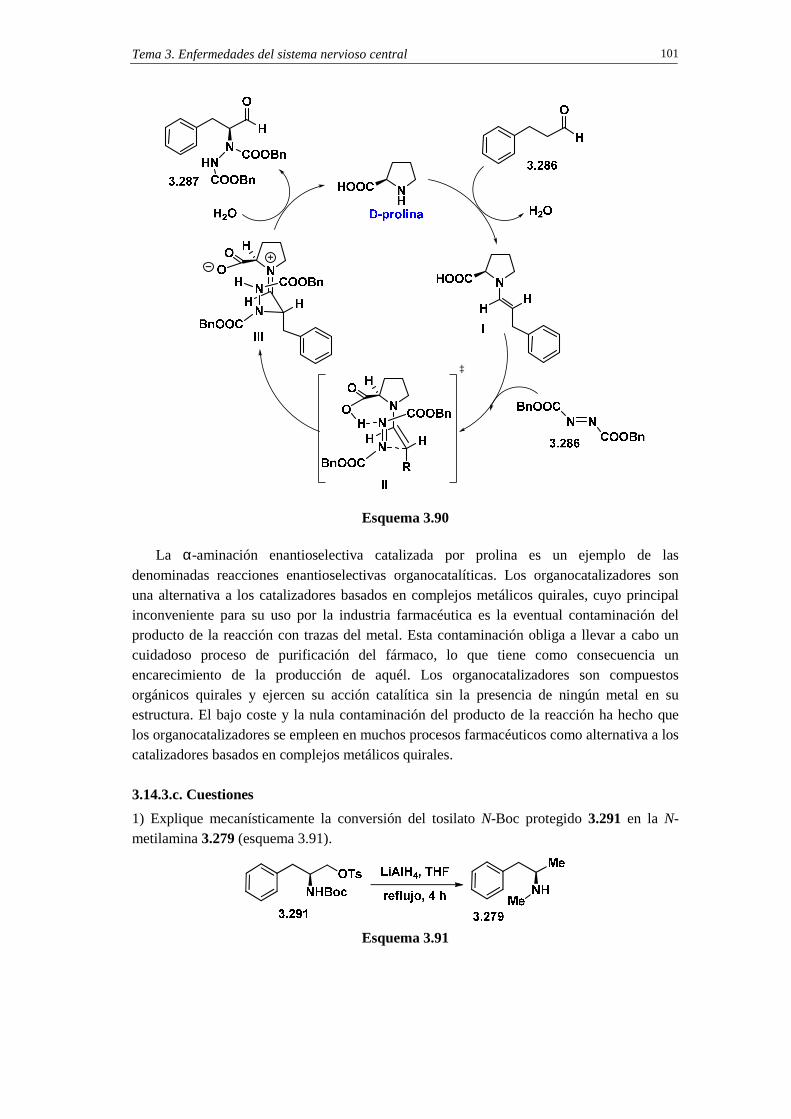
Tema 3. Enfermedades del sistema nervioso central 101
Esquema 3.90
La α-aminación enantioselectiva catalizada por prolina es un ejemplo de las denominadas reacciones enantioselectivas organocatalíticas. Los organocatalizadores son una alternativa a los catalizadores basados en complejos metálicos quirales, cuyo principal inconveniente para su uso por la industria farmacéutica es la eventual contaminación del producto de la reacción con trazas del metal. Esta contaminación obliga a llevar a cabo un cuidadoso proceso de purificación del fármaco, lo que tiene como consecuencia un encarecimiento de la producción de aquél. Los organocatalizadores son compuestos orgánicos quirales y ejercen su acción catalítica sin la presencia de ningún metal en su estructura. El bajo coste y la nula contaminación del producto de la reacción ha hecho que los organocatalizadores se empleen en muchos procesos farmacéuticos como alternativa a los catalizadores basados en complejos metálicos quirales.
3.14.3.c. Cuestiones
1) Explique mecanísticamente la conversión del tosilato N-Boc protegido 3.291 en la N-metilamina 3.279 (esquema 3.91).
Esquema 3.91

Síntesis de antidepresivos, antiepilépticos y antiParkinson 102
3.14.4. Síntesis de mesilato de rasagilina
La rasagilina se emplea en el tratamiento del Parkinson idiopático en monoterapia o en terapia coadyuvante con levodopa al final de las fluctuaciones de la dosis.
3.14.4.a. Análisis retrosintético
El análisis retrosintético del mesilato de rasagilina se inicia con la desconexión del fragmento propargílico (esquema 3.92). Esta escisión genera el haluro de propargilo 3.280 (X=halógeno) y la indenamina 3.292 que se preparará de la indanona 3.293.
HNCH3SO3H
Mesilato de rasagilina
C-N
NH2
+
3.280
X
IGF
O3.292 3.293
Esquema 3.92
3.14.4.b. Síntesis
La síntesis del mesilato de rasagilina se inicia con la condensación entre la indanona 3.293 y la bencilamina (esquema 3.93). 50 La imina resultante del proceso de condensación, compuesto 3.294, es reducida a la amina racémica 3.295 con NaBH4. La resolución del racemato se consigue mediante cristalización con ácido L-tartárico. La sal diastereoisomérica se recicla mediante racemización en condiciones básicas. La hidrogenolisis en medio básico de la sal de tartrato proporciona la indenamina 3.292. Este compuesto, mediante N-propargilación y reacción subsiguiente con ácido metanosulfónico, se convierte en el mesilato de rasagilina.
Esquema 3.93
50 S. Uruyama, E. Mutou, A. Inagaki, K Okada, S. Sugisaki, Patente: WO2006030739(A1), 2006.


















