
Período de prácticas efectuado del 1 de abril al 29 de...
139
Dut Chimie Departamento de ingeniería Química Universitat Politècnica de Catalunya Clément PERSON Av. Víctor Balaguer s/n. 08800 Vilanova i la Geltrú España Año 2009/2010 Período de prácticas efectuado del 1 de abril al 29 de junio de 2010 Estudio de adsorción del Boro de soluciones acuosas mediante la utilización y Síntesis de perlas de alginato de calcio y nanopartículas de quitosano modificado con azúcares. Tutor IUT Tutora de la empresa M. Arnaud Cailler Dra. Montserrat Ruiz Planas 1
Transcript of Período de prácticas efectuado del 1 de abril al 29 de...

!!
Dut Chimie Departamento de ingeniería Química Universitat Politècnica de Catalunya
Clément PERSON Av. Víctor Balaguer s/n. 08800 Vilanova i la Geltrú España Año 2009/2010
!
!!!!!!
Período de prácticas efectuado del 1 de abril al 29 de junio de 2010
!!
!!!!!!!!!!
Estudio de adsorción del Boro de soluciones acuosas mediante la utilización y
Síntesis de perlas de alginato de calcio y nanopartículas de quitosano modificado con
azúcares.
!!Tutor!IUT! Tutora!de!la!empresa!!M.!Arnaud!Cailler! Dra.!Montserrat!Ruiz!Planas!!
1!!

INDICE
AGRADECIMIENTOS Páginas
I. INTRODUCCIÓN ...................................................................................................... 6
II. PRESENTACIÓN DE LA ESCUELA ......................................................................... 9
a. Historia ......................................................................................................... 9
b. Estructura ................................................................................................... 11
III. OBJETIVO DEL PROYECTO ................................................................................. 13
a. Objetivo general ......................................................................................... 13
IV. TEORÍA EXPERIMENTAL ...................................................................................... 13
a. Boro ............................................................................................................ 13
1. Historia ............................................................................................... 13
2. Propiedades físicas y químicas .......................................................... 15
3. Toxicidad del Boro ............................................................................. 18
4. Aplicaciones industriales .................................................................... 20
b. Adsorción .................................................................................................... 22
1. Biosorción .......................................................................................... 24
2. Factores que influyen en la adsorción ................................................ 25
c. Isotermas de adsorción .............................................................................. 28
1. Método experimental de trabajo ......................................................... 31
1.1. Modelos matemáticos para las isotermas ................................ 31
d. Cinética de adsorción ................................................................................. 34
1. Modelos matemáticos para la cinética ............................................... 36
e. Adsorbentes ............................................................................................... 37
2!!

1. Tipos de adsorbentes ......................................................................... 39
2. Alginato .............................................................................................. 42
3. Interacción Boro Alginato ................................................................... 51
4. Quitosano ........................................................................................... 54
f. Desorción ................................................................................................... 61
V. DESCRIPCIÓN EXPERIMENTAL ........................................................................... 62
c. Mé 64
1. a
d. Me
1. Fab
3. C c .. 77
3.1
5. iofilización de las perlas de alginato de calcio .................................. 81
VI. DISCUCIÓN DE LOS RESULTADOS
a. Introducción ................................................................................................ 62
b. Equipos y materiales .................................................................................. 62
todo analítico del Boro ...........................................................................
Método del azomethine mediante el espectrofotómetro de dsorción molecular ........................................................................... 64 1.1 Reactivos ................................................................................. 64
1.2 Método de la azomethine H ..................................................... 64
1.3 Principio general ...................................................................... 65
1.4 Procedimiento experimental .................................................... 66
1.5 Reactivos ................................................................................. 68
todología ................................................................................................ 71
ricación de las perlas de alginato de calcio .................................. 71
1.1 Perlas húmedas ..................................................................... 75
1.2 Perlas secas .......................................................................... 75
2. Estudio del equilibrio de adsorción (isotermas de adsorción) ............ 76
inéti as de adsorción .....................................................................
Cinéticas de adsorción del boro en las perlas de alginato de calcio .................................................................................. 78
4. Cinéticas de desorción ....................................................................... 79
L
..................................................................... 89
3!!

a. Resultados experimentales ........................................................................ 89
1. Introducción ........................................................................................ 89 2. Isotermas de adsorción de las perlas de alginato de calcio ............... 89
2.1 Isoterma de adsorción de las perlas húmedas ........................ 90
2.2 Isoterma de adsorción de las perlas secas .............................. 91
2.3 Conclusión ............................................................................... 94
3. Cinéticas de adsorción de las perlas de alginato de calcio ................ 95
3.1 Cinéticas de adsorción de las perlas húmedas ....................... 96
3.2 Cinéticas de adsorción de las perlas secas ............................. 98
3.3 Conclusión ............................................................................. 101
4. Cinéticas de desorción de las perlas de alginato de calcio .............. 102
4.1 Cinéticas de desorción de las perlas húmedas ..................... 104
4.2 Cinéticas de desorción de las perlas secas ........................... 105
4.3 Conclusión ............................................................................. 106
5. Estudio del pH .................................................................................. 107
5.1 En la isoterma ........................................................................ 107
5.2 En la cinética ......................................................................... 109
VII. CONCLUSIÓN FINAL ........................................................................................... 111
VIII. GLOSARIO ........................................................................................................... 113
IX. BIBLIOGRAFÍA ..................................................................................................... 116
X. ANEXOS ............................................................................................................... 121
! !
4!!

Agradecimientos
Me gustaría brindar este proyecto a mis tutores, a Dra. Montserrat Ruiz
Planas por darme la oportunidad de ser su proyectista y brindarme la
oportunidad de conocer un mundo de la química desconocido hasta el
momento.
A mis profesores de Lille especialmente mi tutor M. Arnaud Cailler
responsable de las relaciones internacionales que sin su dedicación y esfuerzo
esto no sería posible, gracias a él todo esto es posible.
A mi compañero del laboratorio M. Hary Demey porque sin sus aportes
nada de esto sería posible, dar las gracias a la EPSEVG que me abrió estas
puertas para mi período de prácticas, en particular a la equipa de trabajo de las
relaciones internacionales dirigido por Mme. Mònica Casamián.
! A mi familia que me animó a hacer un período de prácticas en el
extranjero. Especialmente a mi hermano que tuvo la amabilidad de prestarme
su coche para el viaje, sin él no hubiera conocido todas las peripecias actuales.
! A la región del Nord-Pas-de-Calais que me permitió de financiar este
proyecto y a la comisión Europea ERASMUS que gracias a la cual pude tener
una beca de estudios.
! A Jesús Barrón que nos guió en los experimentos desde México, a
Carlos y Alberto por sus investigaciones anteriores que me permitió entender
mi proyecto.
Y para acabar a Laia, por su ayuda en el laboratorio que fue muy
importante.
Gracias a todos, porque todos vosotros habéis hecho posible esto,
porque todos habéis aportado cada grano para que este proyecto llegue a su
final. Muchas gracias.
5!!

A todos vosotros muchísimas gracias por hacerme feliz!
I. INTRODUCCIÓN
Actualmente la conservación de los recursos naturales, entre ellos el
agua, se presenta como un tema que cada día ocupa más la atención de
científicos, técnicos, políticos y en general, de muchos de los habitantes del
planeta.
En efecto la escasez de este líquido vital obliga a reiterar nuevamente
una llamada a la moderación de consumo por parte de la población a nivel
mundial. Sólo muy poca agua es utilizada para el consumo del hombre, ya que:
el 90 % es agua de mar y tiene sal, el 2 % es hielo y está en los polos, y sólo el
1 % de toda el agua del planeta es dulce, encontrándose en ríos, lagos y
mantos subterráneos.
Aproximadamente 1.100 millones de personas, es decir, el 18 por ciento
de la población mundial, no tienen acceso a fuentes seguras de agua potable, y
más de 2.400 millones de personas carecen de saneamiento adecuado.
La carencia de agua potable se debe tanto a la falta de inversiones en
sistemas de agua como a su mantenimiento inadecuado. Cerca del 50 por
ciento del agua en los sistemas de suministro de agua potable en los países en
desarrollo se pierde por fugas, conexiones ilegales y vandalismo.
En algunos países, el agua potable es altamente subsidiada para
aquellos conectados al sistema, generalmente personas en una mejor situación
económica, mientras que la gente pobre que no está conectada al sistema
depende de vendedores privados costosos o de fuentes inseguras.
En efecto, hay muchos países que no tienen la posibilidad de beber del
agua potable. Uno de los problemas que presenta la utilización del agua del
mar es la presencia de boro. El boro se encuentra en el agua marina en
concentraciones estimadas en 4,6 ppm.
6!!

El boro llega a la hidrosfera de forma natural desde los continentes
mediante el ciclo del agua y por procesos de erosión de rocas, y desde la
corteza oceánica por circulación hidrotermal, además también procede de la
precipitación atmosférica, pero también puede aparecer como consecuencia de
la contaminación provocada por actividades humanas.
El boro es un elemento que se encuentra principalmente en forma de
ácido bórico o boratos. El ácido bórico y las sales de boro son usados
ampliamente en la fabricación de vidrio, jabones y detergentes, retardantes de
flama, cosméticos, pieles y en fertilizantes para el tratamiento de suelos
deficientes en boro.
Se usan también en la industria nuclear como absorbedores de protones
y para la preservación de alimentos y maderas. Los compuestos de boro han
encontrado también uso como combustibles de alta energía, en catálisis y en la
fabricación de fluidos de corte que generan gran cantidad de residuos ricos en
boro.
Aunque el boro en pequeñas cantidades es necesario para el
crecimiento de las plantas, en cantidades superiores a 1 mg/L es dañino para la
mayoría de ellas. En los humanos, un consumo prolongado de agua y
alimentos con un gran contenido de boro puede provocar disfunción en los
sistemas: cardiovascular, nerviosa, digestiva y sexual.
Motivados por la problemática que crea el boro en el medio ambiente, los
profesores y los estudiantes de la Escuela Politécnica Superior de Ingeniería de
Vilanova i la Geltrú especialidad en química industrial han orientado son
orientados sus investigaciones hacia la eliminación del boro presente en
soluciones acuosas, mediante la utilización de perlas de alginato de calcio y de
nanopartículas de quitosano modificado con azúcares.
7!!

Actualmente la técnica más utilizada para la eliminación del boro de las
aguas son las resinas de intercambio iónico sintéticas.
En este proyecto se propone la adsorción y la desorción sobre polímeros
naturales, en particular el alginato de calcio.
La eliminación del boro de productos residuales industriales durante los
últimos diez años ha adquirido gran importancia debido a las numerosas
aplicaciones de este elemento. En efecto los vertidos industriales y urbanos es
una operación dificultosa por lo que muchas industrias que utilizan el boro no
respetan los valores permitidos por la legislación.
Sabemos que el boro es un elemento que hasta hace unos años no se
tenía apenas información. Es un elemento especial puesto que posee una
configuración electrónica que le confiere más orbitales de valencia que
electrones lo que hace posible la formación de complejos con elementos
capaces de donar electrones.
Además el crecimiento del conocimiento sobre las diferentes
aplicaciones del boro ha sido de gran importancia tanto en el aspecto comercial
como en el aspecto medioambiental.
8!!

II. PRESENTACIÓN DE LA ESCUELA
La Universidad Politécnica de Cataluña (Universitat Politècnica de
Catalunya, en catalán) es una universidad estatal española de que tiene como
objetivos prioritarios el estudio, la docencia, la investigación y la transferencia
de tecnología.
a. Historia
El primer antecedente de la Universidad Politécnica de Cataluña (UPC)
se halla en la fundación, en el año 1968, del Instituto Politécnico Superior, que
agrupó las escuelas técnicas estatales ya existentes en Barcelona.
Estas escuelas tenían una gran tradición y sus orígenes se remontan, en
algún caso, a mediados del siglo XIX como, por ejemplo, la Escuela de
Ingenieros Industriales de Barcelona, que empezó las actividades el mes de
octubre de 1851. Víctor de Buen, presidente del Instituto Politécnico Superior,
fue el primer rector de la Universidad Politécnica [29].
En marzo de 1971, se constituye la Universidad Politécnica de Barcelona
(UPB), que inicialmente está formada por las escuelas técnicas superiores de
Ingenieros Industriales de Barcelona (ETSEIB) y de Terrassa (ETSEIAT), la
Escuela Técnica Superior de Arquitectura de Barcelona (ETSAB) y algunos
institutos de investigación.
El mismo año 1971 se crea la Escuela Técnica Superior de Ingenieros de
Telecomunicación (ETSETB).
Poco antes, el 4 de agosto de 1970, se había aprobado la Ley general de
educación y de financiación de la reforma educativa.
Esta Ley comprendía todo el sistema educativo, desde la enseñanza
básica hasta la enseñanza universitaria, y establecía que las universidades
9!!

dispondrían de autonomía y determinarían los procedimientos de control y
verificación de conocimientos, el sistema de enseñanza y el régimen de
docencia e investigación.
Asimismo, las universidades asumirían la gestión y la administración de
sus centros y sus servicios. Además, la Ley preveía también la incorporación a
la estructura universitaria de las escuelas universitarias de Ingeniería Técnica y
de Arquitectura Técnica [29].
Así pues, en 1972 se incorporan a la Universidad Politécnica de
Barcelona las escuelas universitarias de Ingeniería Técnica Industrial de
Terrassa (EUETIT) y de Vilanova i la Geltrú (actual EPSEVG), la Escuela
Universitaria de Arquitectura Técnica de Barcelona (actual EPSEB) y la
Escuela Universitaria de Ingeniería Técnica de Minas de Manresa (actual
EUPM).
Este mismo año empiezan las actividades en las escuelas universitarias
de Ingeniería Técnica Agrícola de Gerona y de Lérida, que ya habían sido
creadas por disposición oficial, pero que no se habían acabado de poner en
marcha [29].
El año 1972 es nombrado rector de la Universidad Politécnica de
Barcelona el catedrático Gabriel Ferraté Pascual, que ocupó el cargo hasta
1976, cuando fue nombrado director general de Universidades en Madrid. Para
sustituirlo es elegido el profesor Julià Fernández Ferrer, que fue rector hasta
1978.
Tras unas elecciones, aquel mismo año es elegido de nuevo Gabriel
Ferraté, que ocupó el cargo de rector de la Universidad Politécnica de
Barcelona y, posteriormente, de la Universidad Politécnica de Cataluña hasta el
año 1994, momento en el que deja el cargo para construir la nueva Universitat
Oberta de Catalunya y ser su primer rector.
10!!

En 1994 es elegido rector Jaume Pagès i Fita, que ejerció el cargo hasta
2002, año en qué tuvieron lugar elecciones y en qué fue elegido un nuevo
rector, Josep Ferrer Llop. El 2006 se celebraron nuevas elecciones, accediendo
al cargo de rector el catedrático Antoni Giró Roca [29].
La Escuela Politécnica Superior de Ingeniería de Vilanova i la Geltrú,
imparte estudios del ámbito de la ingeniería desde hace más de 100 años, con
una constante renovación y modernización de sus enseñanzas. En este
sentido, fue pionera en Cataluña en la implantación de los estudios de
Ingeniería Técnica de Telecomunicación y en la implantación de la educación
en alternancia, innovadora modalidad educativa que consiste en compaginar
periodos lectivos con periodos en las empresas [4].
b. Estructura
La EPSEVG cuenta entre sus activos con el Centro Tecnológico de
Vilanova i la Geltrú (CTVG), que promueve la innovación tecnológica de su
entorno industrial y canaliza toda la actividad de investigación, desarrollo y
transferencia tecnológica que se desarrolla en la Escuela.
La Escuela tiene la mayor biblioteca de la ciudad (1.540 m2), una gran
residencia universitaria a 25 m de la Universidad y otros servicios (óptica
universitaria, tienda de informática, etc.), integrados en un gran campus.
Disponemos de una Área de Relaciones Externas muy activa. Esto
posible de estudiar en nuestra Escuela, y de poder completado su formación
mediante practicando remunerados en empresa, disfrutar de convenios de
movilidad internacional mediante acuerdos bilaterales de intercambio con
universidades de Alemania, Inglaterra, Austria, Dinamarca, Francia, Grecia,
Holanda, Irlanda, Italia, Rumanía y Suecia.
La preocupación por la sostenibilidad, el medio ambiente y la igualdad de
oportunidades han sido una constante en la EPSEVG. La Cátedra de
11!!

accesibilidad, el Centro de Estudios Tecnológicos para la Dependencia o el
Laboratorio de Aplicaciones Bioacústicas (LAB) - que trabaja para la
conservación de los mamíferos marinos del Mediterráneo-, todos con sede en
la Escuela, son algunos ejemplos [4].
En el organigrama 1 podemos ver:
El director ejerce la representación del centro y las funciones de
dirección y gestión ordinarias de la EPSEVG. Es elegido por sufragio universal,
libre, secreto y ponderado por los miembros de la comunidad universitaria de la
Escuela. Su mandato es de tres años y no puede ejercer más de dos mandatos
consecutivos.
Y su equipo Directivo está constituido por el director, los subdirectores,
las subdirectoras y el secretario académico (nombrados por el director), y el
administrador del Centro [24].
Organigrama 1. Estructura de la escuela.
Director:Enric Trullols i
Farreny
Subdirector de política académica y
calidad: EnricMartin Fuentes
Subdirector de estudios: Rafael Morillas Varón
Subdirector de relaciones
internacionales y sostenibilidad: Jordi
Segalàs y Coral
Subdirector promoción y relaciones
universidad-empresa: Ignasi PeratBenavides
Subdirector de recursos, nuevas
tecnologías y infraestructuras: Sergi Sànchez
López
Subdirector de investigación : Pere Andrada
Gascón
Administrador: Jose Miguel
Quiñones Ruiz
Subdirectora de planificación estratégica:
Ariadna Llorens García
Secretario académico: Xavier
Ruiz i Jové
12!!

III. BJETIVO DEL PROYECTO
a. Objetivo general
Estudiar la adsorción y la desorción del boro sobre el biopolímero
alginato de calcio bajo diferentes condiciones y paralelamente se desarrollado
la síntesis de nanopartículas de quitosano modificado con azúcares con el
objetivo de ser utilizado como adsorbente para el boro.
! Síntesis de perlas de alginato de calcio con la finalidad de utilizarlos
como adsorbentes.
! Realización del estudio cinético de las perlas de alginato de calcio a
diferentes concentraciones.
! Desarrollo de las isotermas de adsorción utilizando perlas de alginato de
calcio a diferentes concentraciones.
! Realización de un procedimiento para la síntesis de nanopartículas de
quitosano modificado con azúcares.
IV. TEORÍA EXPERIMENTAL
a. Boro
1. Historia
En 1747 Barón señaló que el bórax era una combinación del entonces
denominado ácido sedativo y de la sosa. Las investigaciones de J.L. Gay-
Lussac y L.T. Thémard en Francia por una parte y de H. Davy en Inglaterra por
otra, lograron en 1808 el aislamiento casi simultáneo de una forma impura del
elemento no libre en la naturaleza, mediante reducción de su óxido por el
potasio.
13!!

Este elemento recibió el nombre de boro. En 1909, Weintraub obtuvo por
primera vez boro fundido, al calentar una mezcla de BCI3 e H2 con un arco de
corriente alterna [3].
La naturaleza blanda del bórax, fácil de trabajar, unida a su presencia en
yacimientos superficiales, originaron, sin duda, que su existencia fuese pronto
conocida por los primitivos pobladores del planeta.
Se cree que los babilonios lo utilizaron en joyería como fundente y está
comprobado su uso por las civilizaciones mesopotámica y egipcia con
propósitos medicinales y como ingrediente de algunas fórmulas secretas para
embalsamar y momificar. De igual manera es conocida la utilización del bórax
para vidriar en China, 300 años a. de C. Aparentemente los persas y los
árabes, también, lo conocieron y utilizaron.
Se supone que la palabra bórax deriva del arábigo "borac" o "boraq" que
aparece en estudios anteriores o contemporáneos a Cristo.!Otros autores creen
que tiene su origen en la palabra persa "burah". En sánscrito se denomina
"tincana" y en la actualidad la palabra tincal es sinónimo de bórax [3].
La Europa medieval tuvo conocimiento del bórax a través de Marco Polo
(s. XIII) quien introdujo su utilización como fundente entre los joyeros
venecianos. Durante siglos el bórax fue objeto, en moderadas cantidades, de
un comercio regular a través de la ruta de las caravanas.
La "era moderna" del boro se puede decir que comienza a mediados del
siglo XIX con la explotación de los yacimientos de Chile, Perú y Turquía,
produciéndose un incremento en el comercio de estas sustancias.
Hoy, las múltiples aplicaciones de los versátiles boratos y de sus
derivados han generado un considerable comercio mundial.
14!!

2. Propiedades físicas y químicas
El boro se utiliza en un amplio rango de aplicaciones industriales, la
mayoría de las cuales incluyen compuestos de boro-oxígeno. Estos
compuestos poseen una estructura química muy variada, presentándose como
sólidos cristalinos y no cristalinos de importancia industrial, y mostrando una
compleja química en solución [2].
En la introducción hemos visto que el boro es un elemento que hasta
hace unos años no se tenía apenas información. Es un elemento especial
puesto que posee una configuración electrónica que le confiere más orbitales
de valencia que electrones lo que hace posible la formación de complejos con
elementos capaces de donar electrones.
El boro ocupa el primer lugar del grupo III-A de la tabla periódica de los
elementos y es un no metal ligero de número atómico 5. Se le atribuye un peso
atómico de 10,811 al presentarse en la naturaleza dos isótopos estables: el
5B11 (19,78 %) y el 5B11 (80,22 %) [3].
El boro presenta la configuración electrónica [He] 2s2 2sp1, utilizando los
electrones de valencia para combinarse con otros elementos para formar
compuestos planos trigonales del tipo BR3 mediante enlaces covalentes. Una
de las principales características es su pequeño tamaño, lo que le confiere un
elevado potencial iónico y una particular afinidad por los elementos
electronegativos.
Posee potenciales de ionización muy elevados, siendo muy difícil
arrancarle sus electrones de valencia por lo que no existe química asociada
con la especie libre B+3. El estado de oxidación estable es +III que presenta
una alta carga unida a un tamaño muy pequeño (radio iónico de 0,20 Å según
Pauling 1960); esta situación facilita la inmediata polarización de los átomos
vecinos y la transferencia de la densidad electrónica hacia el boro, es decir, la
situación favorecida energéticamente es la compartición electrónica.
15!!

Debido a que el boro posee más orbitales valencia (s, px, py, pz) que
electrones de valencia, los compuestos trivalentes del boro tienen una marcada
tendencia a formar complejos con donadores de electrones. Así, se facilita la
constitución de tres enlaces covalentes.
Al boro le faltan todavía, dos electrones (2pz0) para completar el octeto y
formar estructura de gas noble; al tener un orbital totalmente vacío, tiene
tendencia a actuar como aceptor para formar un cuarto enlace, se comporta
como un ácido de Lewis.
Por lo tanto, los compuestos con enlace trivalente pueden formar
estructuras tetracoordinadas dotadas de una carga variable según la naturaleza
de los ligandos.
Cuando el boro forma solamente tres enlaces covalentes, éstos están
dirigidos hacia los vértices de un triángulo equilátero en el mismo plano que el
boro. Cuando se forma un cuarto enlace, la disposición en el espacio es
tetraédrica (figura 1).
Figura 1 Representación de la configuración tetraédrica. [3]
16!!

Esto incluye reacciones reversibles con bases de Lewis aniónicas para
formar aniones tetraédricos en los cuales el boro tiene una carga formal
negativa.
Numerosos estudios sobres los compuestos B-H (boranos) han
demostrado la llamada “deficiencia electrónica” del boro. Estos compuestos a
menudo muestran enlaces multicéntricos.
Sin embargo, la electrofilia del boro juega un papel dominante en la
química del boro, llevando a la formación de complejas unidades estructurales
de BO3 y BO4.
Esta electrofilia junto con la tendencia a formar esteres a través de la
condensación con compuestos portadores de grupos hidroxi, incluyendo B-OH,
justifica la gran utilización industrial del boro, así como la importancia del papel
biológico del mismo [2-3].
El boro es inodoro e insípido; es insoluble en agua, alcoholes y
soluciones de álcalis; es soluble en ácido nítrico y sulfúrico y en muchos
metales fundidos como el aluminio, el calcio, el cobre, el hierro y el magnesio;
el boro no se ve afectado por el aire a temperatura ambiente pero a
temperaturas elevadas forma el nitruro BN y el óxido B2O3; a temperatura
ambiente reacciona con el flúor y si calentamos reacciona con el cloro, bromo y
azufre.
El enorme punto de fusión del boro (2300 °C) es una propiedad ligada a
los fuertes enlaces existentes entre los átomos. Su densidad es 2,34 g/cm3 y su
dureza extraordinaria (raya el rubí).
17!!

3. Toxicidad del Boro
De forma general se puede decir que la tendencia del boro a acumularse
en los tejidos animales y vegetales constituye un riesgo potencial para la salud
de aquellos que consuman alimentos y aguas con altos contenidos en boro [3].
La toxicología general del boro es poco pronunciada, ciertos derivados
son irritantes especialmente las para los ojos y las mucosas gástricas. Los
boratos no son ni mutagénicos ni carcinogénicos [3].
!
Además, en el hombre, se puede decir que los compuestos de boro no
son metabolizables y que no se acumulan en el organismo a excepción de
leves depósitos en los huesos [3].
Los síntomas de intoxicación por el boro son en particular unas náuseas,
vómitos, la diarrea, irritaciones cutáneas y una descamación de la epidermis, e
una estimulación del sistema nervioso central luego de depresión [6].
En los casos graves, la muerte habitualmente sobreviene en cinco días
por colapso cardiovascular [7].
Según las estimaciones, la dosis mortífera aguda de ácido bórico varía
de 15 a 20 g en el caso del adulto, de 5 a 6 g en el caso del joven niño y de 1 a
3 g en el caso de él bebé [8-9]
Los niños, los viejos y las personas que sufren de problemas renales son
particularmente vulnerables a los efectos tóxicos agudos del boro.
18!!

4. Aplicaciones industriales
El compuesto de boro de mayor importancia económica es el bórax que
se emplea en grandes cantidades en la fabricación de fibra de vidrio aislante y
perborato de sodio [5].
En la actualidad, la investigación se está conduciendo en la producción
de combustible en forma de hidrógeno con la interacción del agua y de un
hidruro de boro (tal como NaBH4).
El motor funcionaría mezclando el hidruro de boro con agua para
producir el hidrógeno según lo necesitado, de modo que solucionen algunas
dificultades de aplicar el hidrógeno con seguridad en el transporte y su
correspondiente almacenaje.
El ácido bórico y sus derivados se utilizan como materia prima portadora
de boro en un gran número de procesos industriales, esto es posible por las
buenas y variadas propiedades de dichos compuestos.
En la tabla 1 se indican las propiedades más sobresalientes, desde un
punto de vista industrial, y la posible aplicación asociada [3].
19!!

Tabla 1 Relación propiedades versus aplicaciones [3]
En la fabricación del vidrio de borosilicato y otros similares, es esencial el
B2O3 contenido en el bórax para facilitar la fusión, el afinado y la conformación;
para evitar la desvitrificación, para comunicarles el bajo coeficiente de
dilatación que les es característico (vidrio tipo pyrex) y para mejorar algunas
propiedades como el color, la durabilidad, la dureza, el brillo y la resistencia de
los artículos acabados.
20!!

El óxido bórico es igualmente esencial en la fabricación de vidrio óptico
para poder obtener el índice de refracción necesario. Para mantener la relación
adecuada álcali - ácido se añaden cantidades sustanciales de ácido bórico,
además del bórax [3].
Si clasificamos las aplicaciones del boro en función de sus riesgos
medioambientales, podemos distinguir tres casos:
! Sin riesgos para el medioambiente. Son las aplicaciones en las
aleaciones, los vidrios, esmaltes, cerámicas y materiales compuestos,
en general.
! Débil riesgo de liberación en el medioambiente. En este grupo están
los adhesivos y los retardantes del fuego.
! Alto potencial contaminante. En este grupo se encuadran el resto de
las aplicaciones.
• Formulaciones de detergentes y perboratos
• Baños electrolíticos
• Fertilizantes, insecticidas y herbicidas
• Compuestos anticorrosión
• Conservantes de cosméticos
• Productos farmacéuticos
21!!

b. Adsorción
Se cree que el término adsorción fue introducido por primera vez por
Kayser en 1881 para describir sus observaciones sobre la condensación de
gases sobre una superficie, fenómeno descubierto por Scheele en 1773 y
Fontana en 1777 independientemente. [10]
La adsorción también se puede diferenciar por el medio o la forma en
que se produce la atracción del sorbato a través del solvente. Según la fuerza
de atracción o la reacción que se experimenta podemos distinguir entre la
fisiosorción y la quimiosorción:
La adsorción física está causada principalmente por las fuerzas de Van
der Waals y electrostáticas, dándose éstas entre las moléculas del adsorbato y
los átomos que componen la superficie del adsorbente.
Estos adsorbentes están caracterizados principalmente por las
propiedades de la superficie, como su área superficial y polaridad. El ión es
adsorbido por el sólido dependiendo de la carga relativa entre ambos.
Este proceso puede ser lento o rápido, dependiendo mucho de la
composición del adsorbente, del adsorbato y de la temperatura.
La adsorción química o quimiosorción es debida a fuerzas de naturaleza
química, como por ejemplo compartiendo electrones entre el contaminante y el
sólido formando enlaces.
Fundamentalmente es un proceso que depende de la temperatura, de la
naturaleza química del sólido y de la concentración de la especie.
22!!

Figura 3 Diferentes procesos de sorción. [3]
Generalmente todos los procesos de adsorción se complementan con
una etapa de desorción ya que la regeneración del sólido suele ser preferible
antes que su vertido.
Los métodos típicos pueden conllevar lavado químico, purgas a elevadas
temperaturas, cambios de presión en el caso de gases, etc. [13].
Múltiples sólidos pueden ser utilizados como adsorbentes, pero cuando
el origen de estos es de carácter biológico, entonces se habla de la biosorción.
23!!

1. Biosorción
La biosorción es un fenómeno físico-químico de determinados tipos de
biomása con la capacidad de adsorber de forma eficiente diferentes especies.
Los fenómenos de biosorción se caracterizan por la retención del sorbato
mediante mecanismos físico-químicos como la absorción o el intercambio
iónico que proporcionan determinados ligandos a través de la superficie celular.
Esta interacción se produce con grupos funcionales como carboxilos,
hidroxilos y fosfatos, expuestos hacia la parte externa de la célula,
interaccionando con otras partes de moléculas que son componentes de las
paredes celulares.
El proceso de biosorción involucra la relación entre una fase sólida
(solvente) y una fase líquida (solvente), que contiene diferentes especies
disueltas que serán posteriormente adsorbidas (sorbato).
La afinidad del sorbente por las especies del sorbato determina la
capacidad de adsorción y la distribución entre la fase sólida y liquida.
La capacidad del sorbente es directamente proporcional a la cantidad de
sorbato que puede atraer y retener de forma inmovilizada.
Diferentes factores y parámetros condicionan directamente la actividad y
la capacidad del sorbente, tales como la preparación de la biomasa para
producir el sorbente o la concentración de especies metálicas en la solución.
Estas variables pueden determinar las condiciones físico-químicas del
proceso y variar la inmovilización del sorbato. Hay que destacar que la
capacidad de biosorción, del solbente puede ser manipulada mediante
tratamientos para activar los biosorbentes [3].
24!!

2. Factores que influyen en la adsorción
Factores de capacidad, factores cinéticos y factores fluido dinámicos.
Los factores de capacidad son aquellos que determinan la distribución de
equilibrio de los solutos adsorbidos entre la fase líquida y sólida. Esta influencia
vendrá representada por la isoterma de adsorción. Los factores cinéticos son
aquellos que gobiernan la velocidad de transferencia de soluto de una fase a
otra.
Finalmente, los factores fluido dinámicos pueden deberse al tipo de flujo
del fluido o a sus propiedades que afectarán al tiempo de residencia y por
tanto, al proceso de adsorción [3].
Es por tanto que podríamos hablar de cuatro factores:
! Área superficial
La adsorción es un fenómeno superficial, como tal, el grado de adsorción
es proporcional al área superficial específica. El área superficial puede definirse
como la porción de área total que está disponible para la adsorción.
Como el grado de una reacción superficial varía con el área superficial
disponible, la velocidad de adsorción tendría que evidenciar un aumento
gradual en función de la inversa del diámetro de las partículas adsorbentes.
La velocidad y grado de adsorción para partículas de un determinado
tamaño tendrían que variar de forma aproximadamente lineal con la
dosificación de adsorbente sobre un rango de dosificación que no da lugar a
grandes diferencias en la concentración de soluto que permite, en la masa
principal de la disolución [3].
! Naturaleza del adsorbente
Cuando se considera la adsorción de una solución, se tiene que tener en
cuenta el hecho de que la solubilidad del soluto influye, en gran parte, con el
25!!

control del equilibrio de adsorción. Para que la adsorción tenga lugar, es
preciso romper una especie de posible enlace entre el soluto y el disolvente.
Cuando más grande es la solubilidad, más fuerte es el enlace soluto
disolvente y menor es el grado de adsorción.
En general, la solubilidad de cualquier compuesto orgánico en el agua
disminuye cuando aumenta la longitud de la cadena, esto se debe a que el
compuesto es más parecido a un hidrocarburo [3].
Esto constituye la segunda regla principal en relación entre adsorción y
naturaleza del soluto. El tamaño molecular también tiene importancia, ya que
está relacionado con la adsorción de los solutos orgánicos.
Esta dependencia de la velocidad con el tamaño sólo se puede esperar
en reactores discontinuos de elevado grado de agitación. Cuanto mayor sea el
peso molecular del adsorbato, mayor será también la adsorción.
Las observaciones principales relativas a los efectos de la ionización
sobre la adsorción llegan a la conclusión que, mientras los compuestos tengan
una estructura simple, la adsorción es mínima para las especies neutras.
A medida que los compuestos son más complejos, el efecto de la
ionización tiene menos importancia [3].
Para los compuestos anfóteros, es decir, que tienen la capacidad de
actuar tanto de ácido como de base, los estudios realizados indican una
adsorción máxima en su punto isoeléctrico, o sea, cuando el pH en el cual los
extremos ácidos o bases de los compuestos están ionizados, y el compuesto
tiene una carga neta igual a cero.
26!!

! pH
El pH de la disolución en que tiene lugar la adsorción influye en el grado
de adsorción por varias razones.
Debido que los grupos hidroxílicos adsorben de forma bastante fuerte, la
adsorción de otros iones viene influenciada por el pH de la solución.
Además, el pH influye en la adsorción ya que también influye en el grado
de ionización de los compuestos ácidos o básicos, el cual es un factor
determinante de la adsorción.
! Temperatura
Las reacciones de adsorción son normalmente exotérmicas; por tanto el
grado de adsorción acostumbra a aumentar cuando disminuye la temperatura.
Hay que tener en cuenta que pequeñas variaciones de temperatura no alteran
mucho el proceso de adsorción.
La variación del contenido calorífico del sistema en que sucede la
adsorción, es decir, la cantidad de calor desarrollada en la adsorción de una
cantidad definida de soluto sobre un adsorbente se llama calor de adsorción.
La dependencia de la velocidad de adsorción está expresada en función
de la energía de activación. La velocidad de adsorción está relacionada con la
energía de activación por medio de la ecuación de Arrhenius [12-13].
27!!

c. Isotermas de adsorción
Históricamente se han descrito siempre en primer lugar las isotermas de
adsorción para sistemas gas-sólido.
Prácticamente todas esas expresiones pueden aplicarse por extensión a
sistemas líquidos reemplazando el término de presión por el de concentración y
teniendo en cuenta los correspondientes cambios en las unidades de los
parámetros [14].
La adsorción en fase líquida, en general, es un fenómeno más complejo
que la adsorción en fase gas. Por ejemplo, aunque se pueda suponer que
existe adsorción en monocapa, en fase líquida las moléculas adsorbidas no
están necesariamente empaquetadas con idéntica orientación.
La adsorción aparente del soluto también se denomina capacidad de
adsorción del adsorbente (menospreciando cualquier cambio de volumen en la
solución) siendo:
! " #$%&$'()* +, - (Ec.I)
Donde:
. / 01234 5, es la masa del soluto adsorbido (mg) 60 , es la masa del adsorbente (g) 7 2 , es la capacidad de adsorción del adsorbente (mg/g)
Esto es válido para soluciones diluidas, cuando es pequeña la fracción
del disolvente original que puede adsorberse. La adsorción aparente de un
soluto depende de la concentración del soluto, de la temperatura, del disolvente
y del tipo de adsorbente.
28!!

! Desde un punto de vista práctico, la concentración en la fase sólida debe
medir todo el soluto identificable o recuperable que queda dentro de los límites
exteriores de las partículas, sea cual fuere su estado químico o físico.
Para mayor generalidad, las ecuaciones de isotermas se expresan en
concentraciones a dimensionales, formuladas como las razones del valor real
de C ó q a un valor de referencia de C ó q, utilizando las mismas unidades [3].
En el caso de la adsorción de un soluto, C de referencia es por lo general
la concentración de fase fluida más alta que se encuentra y q de referencia es
la concentración de fase sólida en equilibrio que coexiste con C de referencia
[3].
La presencia de moléculas de disolvente constituye otra complicación.
Por esto, no es prudente tratar de buscar un significado físico estricto a
las expresiones de la isoterma; deben ser tomadas simplemente como
expresiones empíricas que representan unos puntos experimentales dentro de
unos rangos de concentración determinados [3].
En la Figura 4 se representa algunas de las formas típicas de isotermas.
La isoterma lineal pasa por el origen de coordenadas y la cantidad
adsorbida es proporcional a la concentración en el fluido. Las isotermas que
son convexas hacia arriba se denominan favorables, ya que se puede obtener
una carga relativamente elevada del sólido para una concentración baja de
fluido [3].
29!!

Figura 4 Representación de las diferentes isotermas. [3]
Con el fin de representar la concentración del contaminante en frente de
la capacidad de adsorción de un adsorbente, se derivaron diferentes
expresiones matemáticas expresadas en forma de gráficas (figura 4), para
poder observar la actuación y efectividad del proceso.
30!!

1. Método experimental de trabajo
Hay un método experimental de trabajo que se llama “Batch system” es
un sistema discontinuo que consiste en un montaje cerrado, es decir; se crea
un sistema cerrado de variables determinadas y de este manera se puede
observar la evolución de ellas a partir de los datos conocidos.
1.1. Modelos matemáticos para las isotermas
El equilibrio se describe con las ecuaciones de las isotermas. Anteriores
estudios muestran que los modelos de Langmuir y Freundlich (figura 5) son los
mejores que se ajustan a los datos experimentales; seguidamente se utilizan
las ecuaciones para el modelado de los resultados [1].
Figura 5 Representación de los diferentes modelos matemáticos [3].
! Isoterma de Langmuir
Este modelo se basa en las hipótesis de que todas las superficies de
adsorción en la superficie del adsorbente son equivalentes, y que por tanto
existe un centro activo en el adsorbente que podría adsorber tan solo una
molécula de adsorbato.
31!!

Esto es causado por la energía de adsorción, la fuerza de los enlaces
entre la superficie y las moléculas que se adsorben es constante para todas las
superficies, y por tanto no existen interacciones entre las moléculas adsorbidas
[3].
La denominada isoterma de Langmuir que se aplica a la adsorción en
superficies completamente homogéneas en donde la interacción de las
moléculas adsorbidas es despreciables; Q es la máxima concentración
asintótica en la fase sólida y corresponde a la cantidad de adsórbalo necesario
para formar completamente una monocapa en la superficie de adsorbente, a su
vez KL es la constante de equilibrio [3].
! " 89:$'(;<9:$'(
(Ec.II)
g adsorbato/g adsorbente. 2:
=: Máxima cantidad de adsorbato que puede adsorberse por g
adsorbente.
: Constante de equilibrio de adsorción. >?
012: Concentración en equilibrio.
La ecuación de Langmuir puede utilizarse para describir condiciones de
equilibrio de adsorción y calcular los diferentes parámetros con los cuales se
puede comparar el comportamiento de la adsorción de diferentes sistemas
adsorbato-adsorbente, o para otras condiciones dentro de un sistema dado [12,
13, 15].
Para cantidades de adsorción muy pequeñas, el modelo de Langmuir
resulta lineal, siendo q directamente proporcional a c.
32!!

Langmuir fue el primero en proponer una teoría coherente, basada en un
punto de vista cinético, para la adsorción sobre una superficie plana [3].
! Isoterma de Freundlich
Para corregir los defectos de la isoterma de Langmuir, sobre todo para
determinadas regiones de concentración, se han propuesto otras muchas
isotermas, entre ellas la isoterma de Freundlich.
La isoterma de Freundlich, corresponde a una distribución exponencial
de los calores de adsorción, KF y n son constantes empíricas que dependen de
la naturaleza del sólido adsorbente y de la temperatura; KF expresa la
capacidad del adsorbente y n es una medida de la heterogeneidad.
Esta isoterma muestra resultados más exactos que la isoterma de
Langmuir para sistemas de adsorción heterogéneos [3].
! " @AB; CD (Ec.III)
Masa de metal absorbido por unidad de adsorbente, mg/g. 2:
E y F: Constantes de Freundlich. > G: Concentración en equilibrio.
La representación gráfica da una línea recta que sirve para evaluar los
valores de Q y KL en la isoterma de Langmuir.
Análogamente, la representación de log y frente a log x en la isoterma de
Freundlich da una línea recta que permite calcular el coeficiente de Freundlich.
Además, para la isoterma de Langmuir se define un "coeficiente de
separación" r que nos indica si la curva experimental obtenida se ajusta o no, a
una isoterma favorables a la adsorción. Se ha observado que valores de r
33!!

inferiores a la unidad dan isotermas favorables para la captación, en tanto que
las curvas con r > 1 son desfavorables [3].
H " ;;<9:IJ'K
(Ec.IV)
Las isotermas se determinan experimentalmente poniendo en contacto
cantidades crecientes de adsorbente con la disolución del adsorbato, durante
un periodo de tiempo suficientemente grande como para alcanzar el equilibrio,
posteriormente se determina la cantidad de soluto que queda en la disolución
remanente [3].
d. Cinética de adsorción
La adsorción establece una relación entre el sorbente y el sorbato,
dependiendo de la afinidad y características de uno hacia el otro observamos
una evolución el proceso de adsorción. Un parámetro importante, al igual que
la capacidad de adsorción, es la cinética de adsorción [3].
Figura 6 Etapa de adsorción [3].
34!!

La cinética de adsorción determina la velocidad a la cual el sorbente es
capaz de inmovilizar las diferentes especies en relación.
La velocidad de adsorción con que las sustancias orgánicas son
eliminadas de las soluciones acuosas por los adsorbentes sólidos es un factor
muy importante para la aplicación de este proceso en el control de la calidad
del agua [16].
El mecanismo global de adsorción consta de las siguientes fases:
1) Transporte de materia externo: transporte del soluto desde la disolución
hasta la capa límite, la superficie externa del adsorbente.
2) Difusión externa: transporte a través de la capa límite hasta la superficie del
sólido.
3) Difusión interna: transporte desde la superficie hasta los centros activos en
el interior de la partícula.
4) Adsorción o intercambio iónico: fijación del soluto sobre estos centros
activos.
Si se considera que las tensiones interfaciales disminuyen por los
compuestos químicos representativos de los materiales contaminantes se
puede observar que el proceso de adsorción no es probablemente el que
controla la velocidad, y tiene que existir un proceso mucho más lento que
controle la velocidad global de adsorción.
35!!

1. Modelos matemáticos para la cinética
El estudio de la cinética determina el tiempo necesario, a parámetros
determinados, que necesita el adsorbente para realizar su atracción, y así
definir un intervalo de actuación de la sustancia. Con el fin de desarrollar las
cinéticas, existen dos métodos de tratamiento de los datos de la cinética [3].
Según la modelización escogida obtenemos la linealización de nuestros
datos cinéticos, de esta manera se obtiene la ecuación correspondiente a la
gráfica. Una vez extraída la ecuación cinética, se puede definir nuestro proceso
de cinética bajo esa ecuación, es decir, define el transcurso de nuestras
variables al paso del tiempo y de su reacción.
Pseudo-primer-orden
LM#!NO / !P) " LM# !NO) / @QRS (Ec.V)
Pseudo-segundo-orden
" ;TUO'(
V PO'(
POW
(Ec.VI)
La modelización de una cinética por medio de uno de los modelos
anteriores también determina como transcurre la adsorción en ese sistema. La
elección del pseudo-primer-orden nos informa que la adsorción de ese sistema
será a partir de la fisisorción, mientras que la elección del otro modelo, pseudo-
segundo-orden, afirma que la adsorción será controlada por el fenómeno de la
quimiosorción [3].
36!!

e. Adsorbentes
Los adsorbentes más empleados son carbón activo, tamices
moleculares, gel de sílice, polímeros orgánicos y alúminas activadas.
En los diferentes periodos de la historia, el adsorbente mayoritario ha ido
variando: antes de la Primera Guerra Mundial lo que más se empleaban eran
los carbones; en el periodo entre las Guerras Mundiales, se trabajó con
carbones activos, gel de sílice y óxidos de aluminio; después de la Segunda
Guerra Mundial, se produjo un progreso revolucionario con el descubrimiento y
aplicación de numerosos tipos de zeolitas [10].
Los materiales sólidos empleados como adsorbentes son productos
naturales o sintéticos. En cualquier caso, el proceso de fabricación ha de
asegurar un gran desarrollo superficial mediante una elevada porosidad.
Los adsorbentes naturales (arcillas, zeolitas) tienen pequeñas
superficies. Los adsorbentes industriales y los carbones activados de buena
calidad pueden llegar a tener entre 1.000 y 1.500 m2/g [17].
Otras características importantes que debe reunir un buen adsorbente
son las siguientes:
! Alta capacidad de adsorción. La relación dé equilibrio entre las fases
influye en la eficacia con que se alcanza la capacidad final y, en muchos casos,
controla la capacidad real del soluto [17].
Como quiera que los mecanismos de unión son muy complejos y no se
han determinado con precisión aún, no se dispone de una norma satisfactoria
mediante la cual puedan preverse, a priori las afinidades relativas entre un
material poroso y una sustancia.
37!!

! Propiedades físicas y tamaño de partícula adecuados para garantizar la
necesaria resistencia mecánica y facilidad de manejo, produciendo la menor
pérdida de carga posible tanto en lechos fijos como en los móviles o
fluidizados.
! Coste bajo, tanto de la materia prima como del proceso de fabricación.
! Fácil regeneración; por desorción, especialmente en el caso de los
procesos continuos [17].
La selección del adsorbente más indicado para una aplicación concreta
puede estar sujeta a diversos criterios Entre las propiedades que destacan
encontramos su capacidad, coste, regeneración y vida.
Aunque estos factores dependen del sistema a secar, podemos hacer
algunas comparaciones cualitativas para el caso de la adsorción de agua: la
alúmina y el gel de sílice, con pocas excepciones, destacan con mayores
capacidades que las zeolitas para elevadas concentraciones de agua y
requieren menores temperaturas de regeneración (100-200 ºC frente a 200-300
ºC).
! Los precios de estos dos desecantes en los tipos comerciales genéricos
son aproximadamente la cuarta parte o la mitad del precio de los tamices
moleculares respectivamente.
Por otro lado, las zeolitas proporcionan grandes capacidades para bajos
niveles de humedad en un amplio rango de temperatura.
La información sobre la vida o pérdida de actividad es algo confusa; pero
parece que cualquiera de los tres adsorbentes pierde una media de 1,5-2% de
su capacidad inicial de uso al año.
38!!

1. Tipos de adsorbentes
La mayoría de sustancias orgánicas se utiliza carbón activo o zeolitas,
pero durante el transcurso de nuestro estudio utilizaremos alginato, y siendo
este con el que centraremos nuestras explicaciones más adelante.
Carbón activo
El carbón activo fue el primer adsorbente empleado en la historia;
algunas aplicaciones se han encontrado descritas en papiros egipcios del año
3750 a.C [10] Atendiendo a la cantidad, el carbón activo es el adsorbente más
empleado.
Su proceso de fabricación con lleva distintas etapas: partiendo de alguna
sustancia carbonosa como madera, carbón, etc. se carboniza. A continuación,
este material es gasificado parcialmente con un gas ligeramente oxidante como
CO2 o vapor.
Este proceso de activación crea la porosidad y área superficial
deseadas. Está generalmente aceptado que la capacidad de adsorción para el
carbón activo depende de la naturaleza del adsorbato, las condiciones de
adsorción (tales como temperatura, pH, etc.) y de la naturaleza del adsorbente
(a los grupos funcionales formados en la superficie del carbón.
Algunas de sus múltiples aplicaciones son: purificación de gases,
máscaras, recuperación de disolventes, control de la contaminación,
decoloración de azúcar, purificación de alcohol, adsorción de vapores de
gasolina procedentes de automóviles, etc.
La superficie del carbón activo es esencialmente no polar, por ello no
adsorbe fuertemente agua, así que puede ser utilizado para purificación de
aguas o en procesos con gases húmedos. En cualquier caso, el agua se puede
adsorber, pero de un modo muy débil.
39!!

Generalmente presenta gran capacidad para adsorber compuestos
orgánicos no polares o de baja polaridad.
Por eso, una de las aplicaciones más importantes de la adsorción con
carbón activo es la purificación de efluentes acuosos. En este campo se
dispone de abundante bibliografía.
Zeolitas
Las zeolitas son aluminosilicatos cristalinos porosos que responden a la
fórmula general:
Cx/n [(Al O2)x (SiO2)y] . z H2O (Form.I)
En esta fórmula x, y, n, z son números enteros e y ! x. n es la valencia
del catión C mientras que z es el número de moléculas de agua por unidad
estructural cristalina [3].
Existen zeolitas naturales que se comercializan; pero también existen
muchas que se pueden sintetizar.
Estos compuestos son muy útiles en algunas ocasiones ya que se puede
disponer de una estructura en tres dimensiones rígida y de dimensiones
exactas en la que sólo quepan moléculas de un tamaño determinado y las
mayores no se adsorberán.
Este tipo de adsorbente se emplea, por ejemplo, para el secado de
gases o líquidos, eliminación de CO2 o H2S, separaciones de alcohol y agua o
N2 y O2 del aire, etc [3].
40!!

Gel de sílice
El gel de sílice es un sólido amorfo compuesto de partículas esféricas de
sílice, SiO2. Se produce a partir de la siguiente reacción:
Na2SiO3 + 2 HCl + n H2O " 2 NaCl + SiO2 . n H2O + H2O (Form.II)
La superficie de la sílice tiene gran afinidad por el agua por lo que el uso
más habitual del gel de sílice es para secar gases y líquidos.
Además, es más barata y fácil de regenerar que las zeolitas, aunque no
es óptima para secar gases a altas temperaturas.
Es recomendable evitar su contacto con agua líquida porque puede
dañarla.
Dentro de las aplicaciones del gel de sílice no sólo aparece la adsorción
de agua sino también la adsorción de compuestos orgánicos como el benceno
u otros.
Alúmina activada
La alúmina activada, Al2O3, se obtiene deshidratando hidratos de
aluminio. Así su superficie presenta una gran afinidad por el agua y además no
resulta dañada si es introducida en agua líquida. La aplicación más habitual es
el secado de gases y líquidos, aunque también puede adsorber otros
compuestos.
Otros adsorbentes
En la actualidad, se están realizando muchas investigaciones en el
desarrollo de nuevos adsorbentes basados en polímeros. Las resinas
poliméricas orgánicas se han venido usando durante años en intercambio
iónico.
41!!

Con el paso del tiempo, estas resinas se han usado para la adsorción de
compuestos orgánicos disueltos en agua, aplicación en la que compiten con el
carbón activo; y aunque en algunos casos sean más caros, el coste global de la
operación puede ser menor en el caso de tratar corrientes residuales
concentradas [18].
Otro nuevo tipo de materiales porosos, muy útiles para adsorción y
catálisis, son un tipo de arcillas denominadas PILC.
Son semejantes a las arcillas clásicas pero se les han intercalado
cationes metálicos u óxidos entre sus capas, con lo que se consigue un tipo de
porosidad muy práctica para ser empleada en procesos de adsorción [10].
Los cristales microporosos (especialmente “Vycors”) son otros
adsorbentes inorgánicos que destacan como adsorbentes de oxígeno.
2. Alginato
El alginato fue extraído de las algas pardas por tratamiento en medio
alcalino y por primera vez fue estudiado, a finales del siglo XIX, por el químico
E.C. Stanford, que lo llamó “algin” [3].
Las algas pardas de la familia de las "feofíceas" constituyen la materia
prima principal en la producción de alginato. El mismo, es un componente de la
pared celular de tales organismos y se encuentra formando un complejo
insoluble de ácido algínico y sus sales cálcica, magnésica y de metales
alcalinos en varias proporciones [20].
Las algas pardas crecen en todas las regiones de aguas frías del mundo,
en los hemisferios norte y sur.
42!!

Tal como ocurre con las plantas y árboles terrestres, entre ellas existe
una enorme variedad de especies que varían en tamaño, forma, así como en el
porcentaje y calidad del alginato que producen [20].
De interés para su aplicación industrial, podemos mencionar especies de
los géneros: Lessonia (Nigrescens, Flavicans, Trabeculata), Macrocystis
Pyrifera, Durvillea Antártica, Laminaria (Digitata, Saccharina y Cloustoni),
Ascophyllum, Fucus, etc.
Corresponden a organismos de grandes tamaños, conocidas también
como Macroalgas o Kelp, que alcanzan 1 a 2,5 metros de longitud (especies de
los géneros Lessonia, Laminaria, etc.) y algunas de hasta 8 metros o más del
género Macrocystis [20].
Dichas algas marinas, recursos de naturaleza subantártica (temperatura
del agua entre 13º y 20º C), viven y crecen constantemente en la zona costera
ínter y submareal (entre y bajo el nivel de las mareas respectivamente) hasta
los 20 o 30 metros de profundidad.
Son organismos fotosintéticos que sin embargo no están catalogados
como vegetales verdaderos y tienen altas tasas de crecimiento y renovación
anual, lo que las hace un recurso natural renovable de gran importancia [20].
Una de las algas pardas más importantes de la cual se extraen grandes
cantidades de alginato es la Macrocystis pyrifera debido a que cumple dos de
las principales características para ser seleccionada como especie de
importancia industrial: existe en cantidades abundantes y los alginatos
obtenidos de ella pueden aplicarse, por sus propiedades, en diversas industrias
[3].
Todas las algas contienen entre el 20% y el 30% de alginato sobre su
peso seco [19].
43!!
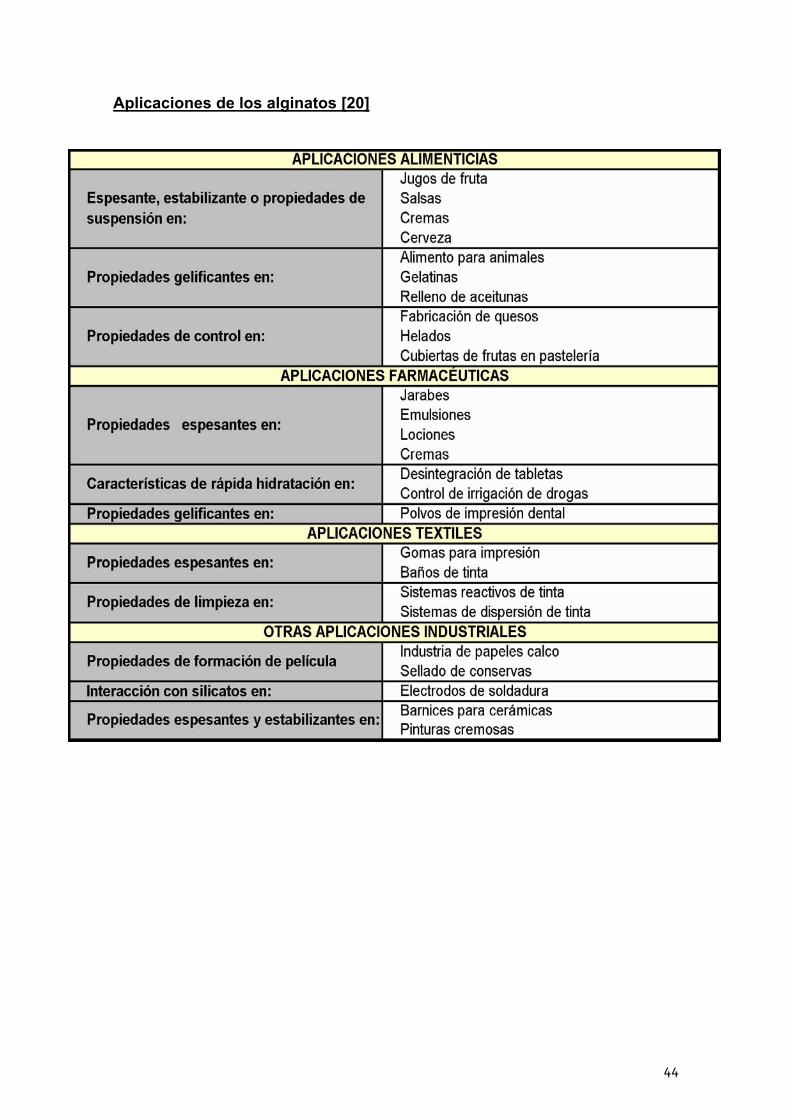
Aplicaciones de los alginatos [20]
44!!

Procedencia de los alginatos
Los alginatos son polisacáridos extraídos de algas marinas pardas
(macromoléculas naturales). Los polisacáridos más importantes extraídos de
las algas son: alginatos, agar, laminarina, fucoidina, galactanos y carragenina
que tienen diferentes usos, pero entre estos polisacáridos destacan los
alginatos y el agar por sus múltiples aplicaciones prácticas e industriales.
La algina es un término de referencia para el ácido algínico y sus
derivados de sodio, amonio, potasio, propilenglicol, etc. Se presenta en casi
todas las algas pardas como principal constituyente de su pared celular.
Las alginas se extraen por medio de un álcali y luego de blanquearlas se
precipitan, filtran, coagulan, neutralizan, deshidratan y muelen. Sus
propiedades varían según la materia prima y método del proceso de extracción
[3].
Las algas pardas crecen en todas las regiones de aguas frías del mundo
en los hemisferios norte y sur y existe una gran variedad de especies que
varían en tamaño, forma, así como en el porcentaje y calidad del alginato que
producen.
Las especies de los géneros que tienen interés para su aplicación
industrial son: Lessonia (Nigrescens, Flavicans, Trabeculata), Macrocystis
(Pyrifera,Durvillea, Antártica), Laminaria (Digitata, Saccharina, Cloustoni,
hyperborea), Ascophyllum,Fucus, etc.
El tamaño de las macroalgas del género Lessonia y Laminaria oscila
entre 1 a 2.5 metros de longitud y las algas del género Macrocystis alcanzan
los 8 metros. Estas algas crecen en la zona costera y bajo el nivel de las
mareas a una temperatura del agua entre 13 y 20 °C respectivamente y hasta
los 20 ó 30 metros de profundidad.
45!!

Estos organismos realizan la fotosíntesis, crecen rápidamente y por lo
tanto son un recurso natural que es renovable y de gran importancia económica
[3].
El mar peruano cuenta con una gran variedad de algas pardas las cuales
tienen una gran importancia económica principalmente porque de ellas se
extrae ácido algínico.
Entre estas algas tenemos a la Lessonia Trabeculata Villouta & Satelices
que es una feofita localizada en el Puerto de Ilo, cuyo valor de biomasa está en
el orden de 172 kg/m2 en zonas protegidas (5-7 m de profundidad) y en zonas
semi-expuestas hasta 136 Kg/m2 (6-12m).Muchas de estas algas son
exportadas al extranjero sin ser procesadas [3].
Estructura química de los alginatos
Los alginatos son las sales del ácido algínico, polisacárido lineal
constituido por dos unidades monoméricas, el ácido " -D-manurónico (M) y el
ácido # -L-gulurónico (G). Estos se agrupan en bloques de secuencias MM,
MG, unidos por enlaces glucosídicos " (1-4); y bloques GG, GM, unidos por
enlaces glucosídicos # (1-4).
Las fórmulas clásicas de Haworth para los dos monómeros se muestran
en la figura 7, mientras la figura 8 ilustra las llamadas fórmulas de silla que
permiten ver en forma más clara el arreglo tridimensional de las moléculas:
Figura 7 Fórmulas clásicas de las dos unidades monoméricas del ácido algínico [20].
46!!

Figura 8 Fórmulas en formas de silla [20].
Como se mencionó anteriormente, la gran variedad de aplicaciones de
estos productos se basa en la habilidad natural que poseen en el control del
comportamiento del agua, lo que científicamente se conoce como propiedad
hidrocoloide, y en su reactividad frente al calcio; ambas consecuencia de la
geometría molecular;
Ha sido demostrado que la cadena polimérica que constituye el ácido
algínico y sus sales se compone de tres tipos de regiones o bloques.
Los bloques G contienen solo unidades derivadas del ácido L-gulurónico,
los M se basan enteramente en ácido D-manurónico y las regiones MG, que
consisten en unidades alternadas de ambos ácidos.
En las figuras 9 y 10 se muestran las configuraciones espaciales que
adoptan los bloques M y G debido a los diferentes enlaces glucosídicos entre
los carbonos C-1 y C-4 de las unidades monoméricas.
Las regiones de bloques M corresponden a cadenas lineales, mientras
que los bloques G presentan una estructura en forma de bucle.
47!!

Figura 9 Bloques MM [20].
Figura 10 Bloques GG [20].
Cuando dos cadenas de bloques G se alinean lado a lado resulta un
hueco en forma de diamante, el cual tiene la dimensión ideal para acomodar en
su interior un ión calcio, formándose una estructura dimérica.
Éste modelo fue propuesto por Grant en 1973 ("egg-box model") para
explicar las propiedades gelificantes de los alginatos al reaccionar con sales
cálcicas y se ampliará posteriormente.
Lo importante por ahora es destacar que, según los porcentajes de
regiones de bloques G y M, que varían en las distintas especies de algas,
serán las características de los geles de alginatos.
Por ejemplo, el alginato obtenido de Laminaria hyperborea, con un alto
porcentaje de segmentos poligulurónicos forma geles rígidos, con baja
capacidad de unión de agua y tendencia a la sinéresis (pérdida de agua por
proceso de exudación del gel, que produce su contracción).
48!!

Por el contrario, el alginato de Macrocystis pyrifera o Ascophyllum, forma
geles elásticos, con baja tendencia a la sinéresis y capacidad de sufrir
deformación.
Obtención de los alginatos
En la producción de compuestos algínicos (ácido algínico y sus sales de
sodio, calcio, magnesio; etc.) existen dos alternativas de producción estos son:
A. Proceso del alginato de calcio
B. Proceso del ácido algínico.
Figura 11 Producción de alginato de sodio [20].
49!!

Como puede verse en la figura 11 la química de ambos procesos es
relativamente simple.
Las principales dificultades se presentan en las separaciones físicas que
se requieren, tales como la necesidad de filtrar residuos legamosos de las
soluciones viscosas o de separar precipitados gelatinosos que retienen altas
proporciones de líquido en su estructura y que resisten ambas operaciones,
filtración y centrifugación.
A simple vista, el proceso del ácido algínico parece ser el de mayor
simplicidad ya que evita una etapa, la de formación de alginato de calcio; sin
embargo, presenta ciertas desventajas en detalles de operación:
! Cuando el ácido es precipitado, lo hace en forma de una masa
gelatinosa que resulta sumamente difícil de separar, pudiendo producirse una
considerable pérdida del producto.
! El gel retiene en su estructura una elevada proporción de líquido, lo que
dificulta los procesos de escurrido y deshidratación, haciéndose necesario el
uso de alcohol como solvente para la conversión a alginato sódico.
Esto usualmente encarece el proceso, a menos que se logre un buen grado de
recuperación del solvente, lo cual suele no resultar fácil [20].
El proceso del alginato de calcio presenta las siguientes ventajas:
! Se puede precipitar el alginato de calcio en forma de fibras las cuales se
pueden separar y blanquear fácilmente.
! Posteriormente el alginato de calcio es convertido a ácido algínico el cual
conserva su estructura fibrosa, permitiendo su separación [3].
50!!

3. Interacción Boro Alginato
Al gelificar el alginato, el calcio queda atrapado en la estructura macro
reticular del biopolímero [3].
Figura 12 Estructura macroreticular del biopolímero [3].
Debido que el boro posee sólo tres electrones de valencia por lo que
cuando forma enlaces de tres pares de electrones deja un orbital p vacío en la
capa de valencia, esto crea deficiencia de electrones en muchos de los
compuestos que forma parte.
Este elemento no se encuentra libre en la naturaleza existiendo
exclusivamente enlazado a oxígeno como boratos, ácido bórico, etc.
Como se ha explicado en otro apartado, las principales fuentes de Boro
están constituidas por depósitos de diferentes minerales tales como bórax,
ulexita, colemita, kernita, etc., donde los centros de Boro en los anillos pueden
tener número de coordinación tres, cuatro o combinaciones de ambos.
La formación de estos anillos puede ser más fácilmente entendida en
términos de la disociación del ácido bórico en agua:
B(HO)3 + H2O # B(HO)4- + H+
51!!

En este caso el ácido bórico es un ácido débil (Ka=5,9·10-10), utilizando
más bien la definición de Lewis que la de Bronsted-Lowry, que acepta un par
de electrones del ión OH proveniente del agua con lo que el B pasa de
hibridación sp2 a sp3 en el producto final [3]:
Una vez establecido el equilibrio anterior y el principio de Le Chatelier, en
la reacción del boro con el alginato se encuentra el ácido bórico cuando se
trabaja a pH ácidos y los boratos a pH básicos. Siendo el ácido bórico un
complejo menos estable que el borato puesto que este último es la base
conjugada del mismo [3].
Para poder explicar la reacción del boro con el alginato es necesario
explicar la definición de éster ya que es el compuesto que se forma.
Se entiende por éster la unión de un ácido carboxílico con un alcohol o
fenol. La distribución electrónica sobre el grupo carbonilo del ácido presenta
electrones incompartidos del átomo de oxígeno adyacente. La reacción se
produce de forma lineal (cis) de manera que se pueden formar diésteres si
reaccionan con polialcoholes.
52!!

En el caso del alginato no es un alcohol propiamente dicho, pero sí que
presenta los grupos hidroxilos que reaccionarán con el ácido bórico. El ácido
bórico puede tener las siguientes estructuras:
Figura 13 Estructura del ácido bórico [3].
Como se observa en las estructuras anteriores el ácido bórico puede dar
lugar a la formación de ésteres y diésteres de boratos.
En la reacción del boro con el alginato no se forman diésteres puesto
que los grupos OH del alginato están demasiado distantes para que
reaccionen.
Además como se ha visto anteriormente, la formación de ésteres y
diésteres se da linealmente por lo que es imposible que se formen diésteres de
borato con el alginato por su propia distribución de los grupos hidroxilos [3].
Cuando se trabaja a pH básicos la forma existente del boro son los
boratos, de manera que si se tratase de un alcohol puro se tendrían que formar
diésteres de boratos puesto que la reacción siempre se da linealmente, es decir
en forma cis, pero el alginato al no ser un alcohol y debido a su estructura
molecular no puede formar diésteres:
Figura 14 Alginato Borato éster de borato [3].
53!!

Por lo tanto a pH ácidos el alginato reacciona intermolecularmente con el
ácido bórico dando lugar a ésteres de boratos tal y como se muestra en la
siguiente figura:
Figura 15 Reacciona intermolecularmente con el ácido bórico [3].
4. Quitosano
Breve historia sobre el quitosano
Por su amplia distribución en la naturaleza la quitina es el segundo
polisacárido en abundancia, después de la celulosa.
Fue descubierta por Braconnot en 1811 cuando estudiaba las sustancias
derivadas del Agaricus volvaceus y otros hongos.
Posteriormente Odier, en un artículo sobre insectos reportó que había
encontrado en algunos insectos la misma sustancia que forma la estructura de
las plantas, llamándola “quitina” (del griego tunic, envoltura).
54!!

Payen, en 1943, inició una controversia que duró más de cien años
sobre las diferencias entre la quitina y la celulosa, en parte porque se pensaba
que la presencia de nitrógeno reportada en algunas investigaciones se debía a
residuos de proteínas que no podían ser completamente eliminados de las
muestras.
El nombre sistemático de la quitina es $(1-4)-2-acetamido-2-desoxi-D-
glucosa [21].
Se encuentra principalmente en las conchas de crustáceos y formando
parte del exoesqueleto de los insectos, así como también en las paredes
celulares de muchos hongos, levaduras y algas [22].
La quitina es completamente insoluble en agua o en medio ácido. En la
figura 16 podemos ver su estructura química.
Figura 16 Unidad repetitiva de la estructura química del quitina [21].
Por su parte, el quitosano es también un polisacárido que se encuentra
en estado natural en las paredes celulares de algunos hongos; sin embargo, su
principal fuente de producción es la hidrólisis de la quitina en medio alcalino,
usualmente hidróxido de sodio o de potasio, a altas temperaturas.
55!!

El quitosano fue descubierto por Rouget en 1859, quien encontró que al
tratar quitina con una solución caliente de hidróxido de potasio se obtiene un
producto soluble en ácidos orgánicos.
Esta “quitina modificada”, como él la llamó, se tornaba de color violeta en
soluciones diluidas de ioduro y ácido, mientras la quitína era verde.
Más tarde, en 1894, fue estudiada por Hoppe-Seyler quién la denominó
“quitosano” (también se conoce como quitosana en algunos lugares, chitosan
en inglés).
En el siguiente esquema se aprecia los pasos elementales en la
obtención del quitosano:
Figura 17 Esquema elemental de la producción de los derivados de la quitina [21].
La desacetilación completa de la quitina produce un material totalmente
soluble en medio ácido conocido como quitano; sin embargo, cuando la
desacetilación del material de partida es incompleta se crea una mezcla de
cadenas que tienen distintas proporciones de unidades $(1-4)- 2-acetamido-2-
desoxi-D-glucosa y $(1-4)-2-amino-2-desoxi-D-glucosa, cuya relación depende
de las condiciones de reacción y que, obviamente, genera materiales con
distintas propiedades denominados quitosanos [21].
56!!

La diferencia en las propiedades de estos materiales puede llegar a ser
notable, como por ejemplo la distinta solubilidad en medio acuosa que puede
llegar a tener. Las estructuras químicas del quitano y el quitosano se muestran
a continuación:
Figura 18 Unidad repetitiva de la estructura del Quitano [21].
Figura 19 Unidad repetitiva de la estructura del Quitosano [21].
Nanopartículas de quitosano.
Este sistema está constituido por partículas reticuladas (de estructura
matricial) de tamaño submicrométrico, formadas por quitosano, o una
combinación de quitosano y otros compuestos (como ciclodextrina, ácido
hialurónico, poloxamero,…), además del principio activo [25].
57!!

Las tecnologías de nanopartículas de quitosano están diseñadas para la
administración de principios activos hidrofílicos, como péptidos, proteínas,
polisacáridos y plásmidos de ADN o siRNA, que son protegidos al ser
encapsulados en su interior (figura 20).
Las nanopartículas de quitosano son mucoadhesivas y tienen un tamaño
sub-micrométrico permitiendo una mayor superficie de contacto con las
mucosas lo que favorece la absorción del principio activo y su biodisponibilidad
[25].
Figura 20 Estructura esquemática (izquierda) quitosano (verde) y principio activo (rojo) e imagen de Microscopía de Transmisión Electrónica [25] (derecha) de una
nanopartícula de quitosano.
Quitosanos nanopartículas han ganado más atención como portadores
de entrega de medicina debido su mejor estabilidad, toxicidad baja, método de
preparación simple y suave, y suministro de rutas versátiles de administración
[23].
El uso para entrega mucosa también facilitada por absorción quitosano
mejora de efecto. Además, quitosano nanopartículas también mostró para ser
un adyuvante bueno para vacunas.
Por lo tanto, los objetivos de esta revisión son resumir la preparación
disponible las técnicas implicaron nanopartículas quitosano, el uso de
nanopartículas exploradas quitosano, el mecanismo de entrada de célula [23].
58!!

Las nanopartículas son partículas sólidas coloidales con diámetros en los
límites 1-1000 nm.
Ellos consisten en materiales macromoleculares y pueden ser usados
terapéuticamente como adyuvantes en vacunas o portadores de medicina en
cual el activo el ingrediente es disuelto, atrapado, encapsulado, adsorbido o
químicamente conectado.
Los polímeros solían formarse las nanopartículas pueden ser tanto
polímeros sintéticos como naturales [23].
Las nanopartículas a base de quitosano se forman de acuerdo a una
aproximación de tipo ‘bottom-up’ como resultado de procesos de auto-
asociación o entrecruzamiento en virtud de los cuales las cadenas poliméricas
se ordenan en estructuras nanoscópicas ya sea por interacciones inter o
intramoleculares de tipo covalente o no covalente.
En estas nanopartículas o nanoesferas el fármaco puede ser atrapado o
ligado a la matriz polimérica sólida (o semi-sólida) [26].
Las metodologías más comunes mediante las cuales es posible preparar
nanopartículas a base de quitosano se presentan en la Figura 21.
A menudo, se utiliza una combinación de métodos; por ejemplo,
entrecruzamiento covalente de la superficie de nanopartículas previamente
obtenidas por coacervación [26].
59!!

Figura 21 Métodos para preparar nanopartículas de quitosano [26].
Es importante señalar que a pesar de la diversidad de métodos
documentados y agentes químicos disponibles para obtener nanopartículas a
base de quitosano, sólo unos cuantos de éstos ofrecen potencial real para el
desarrollo de aplicaciones farmacéuticas debido al estricto marco regulatorio
que deben cumplir los excipientes para la liberación de fármacos [26].
60!!

f. Desorción
En producto químico procesos de la separación, el pelar también se
refiere como desorción mientras que un componente de una corriente líquida
se mueve cerca transferencia total en una fase del vapor a través del interfaz
del líquido-vapor.
La desorción es el proceso por el cual nuestro adsorbente es capaz he
regenerarse y ser reutilizado. Al igual que la adsorción, la desorción es de
carácter muy importante en la valoración de un buen adsorbente, ya que si es
capaz de regenerase, su utilidad y coste se reduce, y su aplicación se amplía
[3].
Para realizar la desorción de un adsorbente es necesario utilizar un
eluyente que sea compatible y afín a las características y propiedades del
adsorbente. El efluente normalmente se produce a partir de una sal básica,
capaz de extraer el sorbato del adsorbente y así quedar preparado para una
nueva adsorción [3].
El eluyente tiene la intención de liberar el boro inmovilizado por el
adsorbente, a causa de este efecto, el boro es extraído [3].
La elección del mejor eluyente es una de las razones más importantes
para elegir un adsorbente eficaz, debido a que si el adsorbente puede ser
regenerado, sus usos son múltiples y de coste mínimo.
Al igual que las isotermas de adsorción, para la desorción también
existen isotermas que describen la evolución de la desorción de un adsorbente.
Por medio de ellas, conocemos el rendimiento de este y de su correspondiente
eluyente [3].
61!!

V. DESCRIPCIÓN EXPERIMENTAL
a. Introducción
En este apartado explicaremos las condiciones en las que hemos
realizado este estudio, de eliminación de boro.
Y por ello relataremos los métodos analíticos que hemos utilizado para la
medición de boro.
Los análisis fueron analizados por un método, mediante un método
colorimétrico por medición en un espectrofotómetro de absorción molecular.
b. Equipos y materiales
De acuerdo con los experimentos y ensayos se deben utilizar diferentes
equipos del laboratorio para el obtener los resultados de la adsorción o para
poder realizar nuestros correspondientes experimentos, así como para obtener
una correcta ilustración de los resultados.
• Cronómetro
• Balanza BX-320H
• Balanza D-620
• Espectrofotómetro de UV SHIMADZU
• pHmetro GLP 21
• Agitador magnético simple Aginatic-N
• Agitador Rotabit
• Agitador múltiple Multimix DMM91
• Bomba peristáltica WATSON-MARLOW 323
• GILSON FC203B
62!!

El Espectrofotómetro de absorción molecular imagen 0 es de la marca:
XS UV-1603. Longitud de onda de 415 nm. Se realizan 3 lecturas de cada
muestra.
Imagen 0 Espectrofotómetro UV Shimadzu
Así mismo para el correcto funcionamiento de los experimentos
realizados se contaba con el soporte de material de laboratorio tales como:
• Botes de plástico (250 ml)
• Cuentagotas
• Pipetas y micropipetas (5 ml, 1000 %l, 100 %l)
• Tubos de ensayo
• Vasos de precipitados (100, 250, 500, 1000, 2000, 4000 ml)
• Matraces aforados (25, 500, 1000 ml)
• Colador
• Vidrios de reloj
• Espátula
• Parafilm
• Imán / varilla imantada
• Escalimetro
• Pie de Rey (digital/150 mm) MILUTOYO
63!!

c. Método analítico del boro
1. Método del azomethine mediante el espectrofotómetro de absorción molecular
1.1 Reactivos
Ácido bórico
Azomethine H
Acido acético
Ácido Ascórbico
Acetato de amonio
Amoniaco
1.2 Método de la azomethine H.
La azomethine H corresponde al ácido 8-hidroxi-(2-hidroxibencildiamino)
naftalen-3,6- disulfónico que se representa en la. Figura 22.
Figura 22 Estructura de la azomethine H
Es un sólido de color anaranjado claro que se descompone a 300 °C
dando aldehído salicílico. Es soluble en agua, poco soluble en metanol y etanol
e insoluble en éter, benceno, tolueno, cloroformo, acetona y tetracloruro de
carbono [3].
64!!
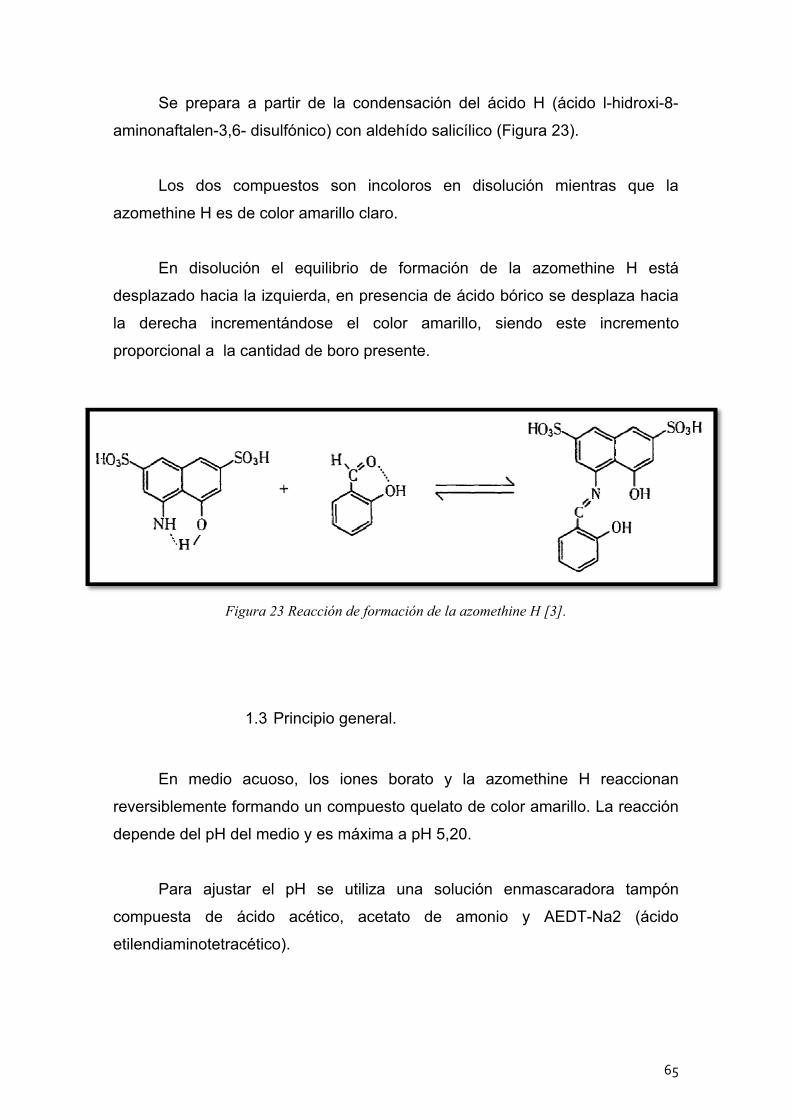
Se prepara a partir de la condensación del ácido H (ácido l-hidroxi-8-
aminonaftalen-3,6- disulfónico) con aldehído salicílico (Figura 23).
Los dos compuestos son incoloros en disolución mientras que la
azomethine H es de color amarillo claro.
En disolución el equilibrio de formación de la azomethine H está
desplazado hacia la izquierda, en presencia de ácido bórico se desplaza hacia
la derecha incrementándose el color amarillo, siendo este incremento
proporcional a la cantidad de boro presente.
Figura 23 Reacción de formación de la azomethine H [3].
1.3 Principio general.
En medio acuoso, los iones borato y la azomethine H reaccionan
reversiblemente formando un compuesto quelato de color amarillo. La reacción
depende del pH del medio y es máxima a pH 5,20.
Para ajustar el pH se utiliza una solución enmascaradora tampón
compuesta de ácido acético, acetato de amonio y AEDT-Na2 (ácido
etilendiaminotetracético).
65!!

En la figura 24 se puede observar la estructura de dos posibles quelatos
formados entre el boro y la azomethine H, en la estructura I el boro presenta
hibridación sp3 y por lo tanto es la estructura preferida.
Figura 24 Estructura de dos posibles quelato formados entre!el boro y la azomethine H [3].
1.4 Procedimiento experimental.
Hay dos procedimientos experimentales diferentes distintos:
! el análisis de la cinética
! el análisis de la isoterma
Para la cinética:
Para una solución inicial de 50 ppm de Boro, en un tubo de ensayo tomar
una muestra de 1 mL exacto de la solución a analizar, aumentar 1 mL de agua
destilada, 1,5 mL de tampón y 1,5 ml de solución de azomethine H recién
preparada.
Se agita para mezclar bien el contenido y después de 120 minutos se lee
la absorbancia a 415 nm frente a un blanco. Paralelamente se construye una
recta de calibrado siguiendo el mismo procedimiento.
66!!

Para la isoterma:
Trabajamos con una gamma de concentración diferente de 5 a 4000 ppm
por eso necesitamos de cambiar los valores de volumen por qué la dilución
cambia, en efecto podemos ver en la tabla 2 los diferentes diluciones a hacer
para preparar la análisis.
! Dilución inicial Dilución Análisis [B] mL Muestra mL Agua mL Muestra mL Agua
!!!!!5! no! ! ! no 1 ! 1
! 10! no! ! ! no 1 ! 1
! 20! no! ! ! no 1 ! 1
! 50! no! ! ! no 1 ! 1
! 75! no! ! ! no 0,5 ! 1,5
! 100! no! ! ! no 0,3 ! 1,7
! 150! no! ! ! no 0,3 ! 1,7
! 200! no! ! ! no 0,2 ! 1,8
! 300! no! ! ! no 0,15 ! 1,85
! 500! no! ! ! no 0,1 ! 1,9
! 750! 0,5! ! ! 7 0,5 ! 1,5
! 1000! 0,5! ! ! 7 0,5 ! 1,5
! 1500! 0,4! ! ! 7 0,5 ! 1,5
! 2000! 0,3! ! ! 7 0,3 ! 1,7
! 4000! 0,3! ! ! 7 0,3 ! 1,7
!
Tabla 2 Representación de diferentes diluciones
Además solo para las concentraciones de 750 ppm a 4000 ppm hay una
dilución de más. Para hacer estas diluciones tenemos que tomar una cuantidad
explicado en la tabla 2 de una muestra de un tubo de ensayo e introducir esta
cuantidad en un nuevo tubo de ensayo con 7 mL de agua destilada.
Cuando todos los diluciones han hecho en de tubos de ensayo diferentes
podemos aumentar 1,5 mL de tampón y 1,5 mL de azomethine H recién
preparada.
67!!

Se agita para mezclar bien el contenido y después de 120 minutos se lee
la absorbancia a 415 nm frente a un blanco. Paralelamente se construye una
recta de calibrado siguiendo el mismo procedimiento.
1.5 Reactivos
Solución Tampón 4.5
Disolveremos 100 g de acetato de amonio en un vaso de precipitados
que contiene 160 mL de ácido acético glacial y 20 mL de agua destilada.
Si no se disuelve, añadir agua destilada hasta su completa disolución.
Ajustar el pH de la solución a 4.5 añadiendo ácido acético o amoniaco.
La solución tampón, podemos guardarla como máximo 14 días.
Solución colorante
Disolver 5,00 g de azomethine H y 10,00 g de ácido ascórbico en 250 mL
de agua caliente (que no debe supere los 70 ºC).
Enrasar en un matraz de 500 mL con agua destilada.
Preparar esta solución para utilizar en el mismo día.
Solución madre
Preparar una solución de 50 mg/L de boro y enrasar con agua destilada.
Preparación de los patrones en tubos de ensayo de 5 mL.
Preparar los patrones adicionando en tubos ensayo 5 mL el volumen de
cada solución preparada que indica la tabla 3, así que la cantidad de agua
destilada indicado para tener un volumen final de 2 mL.
68!!

ppm solucion!madre!mL !agua!destilada!mL0,0 0,0 2,02,0 0,2 1,84,0 0,4 1,66,0 0,6 1,48,0 0,8 1,210,0 1,0 1,0
Tabla 3 Patrones para espectrofotómetro molecular
En la izquierda podemos ver las concentraciones que queremos para
nuestros patrones, y a la derecha los mL que cogeremos con la pipeta de
nuestras soluciones madres de 50 ppm de boro y de agua destilada, que
hemos preparado anteriormente.
Preparación de las muestras
En un tubo de ensayo de 5 mL adicionar la cantidad estimada de
muestra.
El rango de análisis de muestra del espectrofotómetro en la curva de
calibrado es de 0 a 10 ppm de boro (en tubo de ensayo de 5mL) por esto se
prepararan las muestras realizando los cálculos necesarios que contengan
aproximadamente 5 ppm de boro.
Añadir en cada matraz 1,5 mL de solución tampón y 1,5 mL de solución
colorante. Se agita para mezclar bien el contenido.
Los patrones y las muestras se han de preparar a la vez y luego se ha de
dejar reposar todo durante 120 minutos, antes de medir el boro, para permitir el
desarrollo del color.
La medición de las muestras en el espectrofotómetro se hace en una
longitud de onda de 415 nm. (Ver imagen 1)!!
69!!

Imagen 1 Presentación de los tubos con azomethine H
Procedimiento de análisis
Encender espectrofotómetro de absorción molecular.
Instalar los dos adaptares metálicos para las cubetas pequeñas.
Utilizar las cubetas pequeñas del volumen total de 360 $L.
Limpiar bien las cubetas antes de usarlas con agua destilada.
Introducir una cantidad de 360%$L de la solución cero o de la muestra con
la pipeta (escala 100 a 1000%$L).
Rellenar las dos cubetas de cuarzo con el patrón 0 y hacer el auto cero.
(Vaciarlas sobre un vaso de 400 mL y limpiarlas, de cada patrón o muestra con
papel las paredes de la cubeta y de la punta).
Dejar la cubeta del final con patrón 0 puesta que es la de referencia.
En la otra cubeta primero se irán introduciendo uno a uno los patrones
para hacer la recta de calibrado.
Para ello, primero se debe hacer un lavado de la cubeta interior, con el
propio patrón o muestra que se vaya a leer. Los patrones y las muestras se
leen tres veces y después se hace una media con los valores obtenidos.
70!!

d. Metodología
El objetivo principal de este trabajo experimental ha sido el estudio del
comportamiento de la adsorción de boro sobre el adsorbente de alginato en
forma de perlas, para ello se preparo un plan de trabajo divido en varias partes
fundamentales.
1. Fabricación de las perlas de alginato
Para efectuar todos los estudios se realizaron perlas de alginato para
llevar a cabo los experimentos de absorción de boro.
Antes pero explicaremos que para la realización de los experimentos se
realizaron diferentes lotes a diferentes concentraciones, 3,2 %, 2,4% y 1,6% y a
su vez variando la configuración manteniendo la misma concentración de
alginato, ahora hay dos configuraciones diferentes las primeros lotes serán
perlas húmedas y las segundos lotes serán perlas secas.
El procedimiento de fabricación de las perlas es el mismo para todos los
diferentes concentraciones, agilizar el escrito solo concretaremos un caso por
escrito, los demás solo detallaremos las diferencias.
Por ejemplo preparamos un lote de perlas de alginato de concentración
3,2 %.
Prepararemos una disolución de 3,2 g de alginato en 100 mL de agua
destilada. Esta disolución la dejaremos agitando a 800 rpm durante un tiempo
mínimo de 5 horas para que quede completamente homogenizada. (Ver
Imagen 2 y 3)
71!!

Imagen 2 Solución de alginato de sodio esta agitando antes de estar homogenizada.
Imagen 3 Solución de alginato de sodio donde falta 30 min de homogenización.
Transcurrido este tiempo se debe bajar la velocidad de agitación a unas
200 rpm para que el aire se disipe y evitar que se produzcan burbujas de aire,
hecho que dificultaría la correcta fabricación de las perlas.
La disolución debe agitarse durante 24 horas. Una vez realizada la
disolución, se preparan 1500 ml de nitrato de calcio 0,05M.
Una vez respetado los tiempos de espera se realizara el montaje para la
fabricación de las perlas. Consistiendo en una bomba peristáltica que bombea
gota a gota la solución de alginato sobre la de nitrato de calcio tal como
podemos ver en la imagen 4 y la figura 25.
72!!

Imagen 4 Imagen real de la producción de lotes de alginato de calcio.
Una vez que el alginato entra en contacto con la solución de nitrato de
calcio forma un gel transparente de alginato de calcio. Las perlas que se
obtienen se dejan madurar durante 24 h en agitación.
La agitación de la solución de nitrato de calcio se hace a 500 rpm
durante todo el tiempo de formación de las perlas. También se debe tener en
cuenta la velocidad del agitador de la solución de nitrato de calcio ha de ser
baja porque sino las perlas que se forman se romperían.
73!!

Figura 25 Esquema de la fabricación de las perlas de alginato [2].
A medida que las perlas van madurando desprenden agua y se observa
que se vuelven más opacas y esféricas.
Para el proceso de fabricación hemos tenido en cuenta en mantener una
altura (h) de 15 cm constante para la fabricación de todos los lotes de las
perlas y la misma velocidad en la bomba peristáltica solo 3 rpm. (Ver Imagen 4)
Cuando la formación de las perlas ha acabado, tenemos que poner
sobre el lote un parafilm y quedar las perlas en la nevera durante 24 horas.
Imagen 5 Fotografía real de las perlas de alginato [3].
En la tabla 3 podremos ver las diferencias a la hora de fabricación de los
diversos lotes de perlas normales. (Masa molecular de Ca(NO3)2, 4 H20=
236,15 g/mol)
74!!

3,2 3,20 100,00 17,71125 1500,00 0,0502,4 2,40 100,00 17,71125 1500,00 0,0501,6 1,60 100,00 17,71125 1500,00 0,050
[conc]!mol/L% g Alginato mL!H20 g!Ca(NO3)2,!4!H20 mL!Ca(NO3)2,!4!H20
Tabla 3 Descripción para la realización de lotes a diferentes concentraciones
1.1 Perlas húmedas
Para obtener perlas húmedas a partir de los lotes de perlas está muy
fácil.
Se debe agregar agua destilada para limpiar la superficie de la perla de los
restos de solución de nitrato de calcio.
Después se debe pesar la cuantidad final de las perlas limpias húmedas.
1.2 Perlas secas
Para obtener perlas secas a partir de las perlas húmedas, se debe
utilizar el proceso que anterior y poner las perlas encima de una estufa durante
24 horas para secar todas las perlas. Después se debe pesar la cuantidad final
de las perlas secas.
75!!

2. Estudio del equilibrio de adsorción (isotermas de adsorción)
Una isoterma representa la cantidad de boro en función de la
concentración residual en solución y permite evaluar la separación del boro
entre las dos fases (líquido y sólido) en una temperatura determinada.
Seguidamente, las isotermas serán modelizadas a partir de los modelos
de Langmuir y Freundlich para poder relacionar y ajustar la experimentación a
la teoría.
Para la realización de la isoterma es necesario crear varios puntos en la
gráfica, para relacionar la concentración de boro y la capacidad de adsorción
del adsorbente.
Con este objetivo, se prepararon diferentes ensayos a concentraciones
diferentes de boro, pero con la igual masa de adsorbente.
Estos ensayos constan de 15 recipientes de polietileno, con distintas
concentraciones de boro, entre 5 ppm y 4000 ppm (ver tabla 2) en un volumen
de 100 mL.
Pesamos la masa total de las perlas y dividirla para cada recipiente. Para
ello antes se prepararon soluciones madres de 4000 mg/L de boro (2 litros)
para llevar a cabo las isotermas.
Ajustaremos el pH a 11 con hidróxido de sodio y acidó clorhídrico,
después poner una muestra de 5 mL de cada recipiente con la ayuda de una
pipeta (escala 0,5 a 5%mL) en un tubo de ensayo, finalmente añadir una masa
de perlas alginato (húmedas o secas depende de este que queremos hacer)
calculada anteriormente sabiendo que para una isoterma la masa de alginato
de sodio es de 0,7 g para cada recipiente.
76!!

Estos se dejaron agitar durante 72 h y a 50 rpm, hasta llegar al equilibrio.
Después de las 72h, cogeremos le volumen según la tabla 2 de las
muestras para poder hacer las lecturas en tal como se describen
anteriormente.
Repetiremos el mismo procedimiento para las perlas secas y para cada
concentración de alginato de sodio diferente.
3. Cinéticas de adsorción
Los estudios de cinética de adsorción permiten determinar el tiempo
necesario para alcanzar el equilibrio y poner en evidencia mecanismos
limitantes (mecanismos de difusión). En efecto es la velocidad en función de las
condiciones externas a la partícula.
El adsorbente así como también las condiciones propias a la relación
soluto – adsorbente. Los resultados serán modelizad os con los modelos
pseudo-primer-orden y pseudo-segundo-orden, ajustando así nuestros datos
experimentales a los valores teóricos con el cálculo previo de estos modelos.
Una de las características importantes de la utilización de un adsorbente,
es la velocidad a cual es capaz de adsorber el soluto, es decir, de su cinética
de adsorción. Para conseguir un buen estudio sobre la cinética de adsorción
del boro, realizamos:
77!!

3.1 Cinéticas de adsorción del boro en las perlas de alginato
de calcio
Otra manera o forma para desarrollar y conocer una cinética de
adsorción, es por medio de la variación de masa del adsorbente. A mayor o a
menor cantidad de adsorbente, la reacción se verá afectada y mostrara
variaciones en su velocidad y adsorción.
Este estudio es importante para establecer la cantidad optima de
adsorbente requerido para la extracción de boro, debemos encontrar la dosis
de adsorbente a partir de la cual los propiedades o cualidades de adsorción
permanezcan constantes, así se realizara un uso racional del adsorbente, lo
cual afecta directamente los costes de la técnica.
Para la realización de estas cinéticas es el siguiente (ver Imagen 6):
preparamos una solución de 1 Litro de concentración de boro a 50 mg/L a partir
de la solución de 4000 mg/L preparado anteriormente para la isoterma.
Introducir 250 g exacto de solución madre de boro a 50 ppm en un vaso de 400
mL.
Imagen 6 Presentación de la realización de una cinética
78!!

Ajustar el pH a los niveles requeridos (pH=11) con hidróxido de sodio y
acidó clorhídrico.
Prepara 25 tubos de ensayo de cristal con numerosos para poner las
muestras de la cinética.
Encender el agitador a 50 rpm tomar una muestra de 1000 $L para el
tiempo inicial (0 min), y cogeremos el pH de cada muestra durante la cinética.
Directamente después el cero añadimos la masa de las perlas (húmedas
o secas depende de este que queremos hacer) calculada anteriormente
sabiendo que para una cinética la masa de alginato de sodio es de solo 1,75 g.
Encender el cronometro (“start”) y tomar una muestra de 1000 $L de la
solución de la cinética siguiente una tabla de tiempo durante dos días.
El agitador magnético utilizado será de 40 mm.
Repetiremos el mismo procedimiento con las mismas condiciones para
las perlas secas y para cada concentración de alginato de sodio diferente.
4. Cinéticas de desorción
Desorción es a fenómeno por el que una sustancia esté lanzada o a
través de una superficie. Proceso es el contrario de adsorción. Esto ocurre en
un sistema que está en el estado del equilibrio de la adsorción entre la fase a
granel y una superficie absorbente [27].
Cuando la concentración (o la presión) de la sustancia en la fase a
granel se baja, algo de la sustancia absorbida cambia al estado a granel [27].
79!!

En la química, especialmente cromatografía, la desorción es la
capacidad para que un producto químico se mueva con la fase móvil.
Cuanto más un producto químico desorbe, cuanto menos probablemente
fija por adsorción, así en vez de pegarse a la fase inmóvil, el producto químico
se levanta con el frente solvente [27].
Primero, antes de hacer una cinética de desorción necesitamos hacer
una cinética de adsorción.
El procedimiento de la desorción no está muy diferente de este de la
adsorción mientras tenemos que explicar algunas cosas para trabajar en las
mejores condiciones.
Por un lado elegir el vaso de la cinética de adsorción que queremos
desorber. Mesurar el pH de la solución anterior y tomar una muestra de 1000
$L con la pipeta (escala 100 $L a 1000 $L) para conocer la concentración de
boro de la solución a desorber.
Por otro lado preparar un vaso de 400 mL con el mismo diámetro que
antes con 250 g de agua destilada. Normalmente no hay de boro en la solución
de agua.
Después, filtrar la solución de la cinética de adsorción anterior, guardar la
cuantidad total de las perlas y lavarlas con un poquito de agua destilada (el
mínimo de cuantidad).
Preparar 25 tubos de ensayo con los numerosos del tiempo para poner
las muestras de la solución de la desorción.
Tomar 1000 $L de la solución inicial (al tiempo 0 min) sin boro con la
pipeta y notar el pH inicial.
80!!

Introducir toda la cantidad exactamente de las perlas limpias guardadas
en el vaso de 400 mL de agua.
Encender el cronometro (“start”), tomar una muestra de 1000 $L de la
solución de la cinética en función una tabla de tiempo y notar el pH de la
solución para cada tiempo durante dos días. (Ver imagen 6).
5. Liofilización de las nanopartículas de quitosano y de las perlas
de alginato de calcio
Recuerda sobre el procedimiento de las nanopartículas de quitosano
modificado con azúcares (ver Figura 26).
Chitosan 0,25%(0,5!g!en!198!ml!
H2O)
Agitar24!h
Añadir!2!ml!Ácido!acético!1!%
Filtrar!Agregar!agente!!precipitante!(Na2SO4,!10%)
Agitar1!h
Agitar24!h
Añadir!azúcar!(2,7!g)
Añadir!Borohidruro
1%
Agitar1!h
Agitar24!h!en!agitador!convencional
Centrifugar!(1!h)!y!lavar
Catalizador2!Veces!
por!lo!menos
Añadir!1!ml!por!cada!50!ml!de!solución!de!Chitosan
Figura 26 Esquema del el procedimiento de las nanopartículas de quitosano modificado con azúcares.
Ahora que conocemos el procedimiento de las nanopartículas de
quitosano modificado con azúcares y de las perlas de alginato de calcio
podemos estudiar el proceso de liofilización de estos compuestos pero:
81!!

¿Qué significa la liofilización?
Proceso utilizado para la eliminación del agua mediante desecación al
vacío y a muy bajas temperaturas. Utilizado principalmente en la industria
alimentaria y farmacéutica, aunque también se puede utilizar para fabricar
materiales como el aerogel [28].
La liofilización es un proceso en el que se congela el alimento y una vez
congelado se introduce en una cámara de vacío para que se separe el agua
por sublimación. De esta manera se elimina el agua desde el estado sólido del
alimento al gaseoso del ambiente sin pasar por el estado líquido [28].
Para acelerar el proceso se utilizan ciclos de congelación-sublimación
con los que se consigue eliminar prácticamente la totalidad del agua libre
contenida en el producto original.
Es una técnica bastante costosa y lenta si se le compara con los
métodos tradicionales de secado, pero resulta en productos de una mayor
calidad, ya que al no emplear calor, evita en gran medida las pérdidas
nutricionales y organolépticas.
En general, el café instantáneo o las sopas instantáneas no son
liofilizados, el alto precio de los liofilizadores y su relativamente baja capacidad,
hacen que esta técnica no sea muy atractiva para tratar grandes cantidades de
producto [28].
Sin embargo la liofilización si es usada en café instantáneo de una mejor
calidad, pero a un mayor precio para el consumidor.
82!!

Como proceso industrial se desarrolló en los años 50 del siglo XX, pero
sus principios eran ya conocidos y empleados por los incas.
El procedimiento ancestral consistía en dejar por la noche que los
alimentos se congelasen por la acción del frío de los Andes y gracias a los
primeros rayos de sol de la mañana y la baja presión atmosférica de las
elevadas tierras andinas se producía la sublimación del agua que se había
congelado. Este proceso es conocido como liofilización natural [28].
Presentación del liofilizador
El liofilizador consta dos equipos: una bomba de vacío y el liofilizador
que están posible de ver respectivamente en las imágenes 7 y 8. El liofilizador
tiene una pantalla táctil muy fácil de utilización (ver Imagen 8).
Imagen 7 La bomba de vacío por el liofilizador.
La bomba de vacío (ver Imagen 7): Con el objeto de eliminar los vapores
incondensables del producto y así evitar que pueda contaminarse el aceite de
la bomba, el sistema integra la función Gas-Ballast.
La bomba de vacío está provista de un filtro en la expulsión, cuya la
misión es la de retener los vapores de aceite.
83!!

La conexión eléctrica de la bomba debe conectarse a la base situada en
la parte lateral del equipo. Además, se debe conectar la admisión de la bomba
a la unidad base mediante el tubo suministrado.
Para canalizar los vapores hacia el exterior de la bomba, hay que
conectar la canalización a la expulsión del grupo de vacío y eliminar el filtro
instalado.
Imagen 8 El liofilizador.
El liofilizador (ver Imagen 8): Para encender el equipo, se tiene que
presionar el interruptor situado encima de la conexión eléctrica (ver Imagen 8),
de forma que quede en posición I. Se encenderá el equipo y aparecerá la
pantalla de presentación.
Hay dos modos de liofilización: el modo de funcionamiento
semiautomático y el modo de funcionamiento automático.
El modo automático permite establecer los procesos, los parámetros y el
tiempo mediante una receta. Pero trabajaremos solo con el modo
semiautomático.
Y para utilizar este modo: apoyar en el segundo botón de la pantalla principal.
84!!

En la pantalla del modo semiautomático hay un selector para seleccionar
el proceso a realizar:
! Paro
! Congelación
! Frío + Vacío
! Calefacción de bandejas
En ningún momento el sistema realiza rampas de temperatura ni de
presión ya que el tiempo no está establecido.
El botón de set-point de temperatura sirve para establecer la temperatura
a la que se calentarán las bandejas en el proceso “calefacción de bandejas”.
El botón de set-point de vacío sirve para establecer el valor de vacío
deseado en los procesos “calefacción de bandejas” y “frío + vacío”.
El botón inferior derecho sirve para volver a la pantalla principal.
Explicaciones de los diferentes procesos del modo semiautomático
! Paro
Parar el proceso pulsando PARO, con lo que se detiene la bomba y el
grupo frigorífico. Abrir la entrada de aire por la válvula lateral de la cámara. La
entrada de aire se tiene que abrir muy lentamente para evitar la formación de
turbulencias que provocarían el arrastre de polvo del frasco o bandeja.
Seguidamente se activa el descarche.
Al final del proceso, la bomba y el grupo frigorífico se detienen (la parada
de los compresores está retrasada para que estos recojan todo el gas que hay
en el circuito).
A continuación se tiene que abrir alguna de las válvulas de conexión de la
válvula de venteo situada en la tapa superior de la cámara para que entre aire
en el equipo.
Antes de empezar una nueva operación verificar que el condensador
está totalmente descongelado, que no quedan restos de agua en el tubo de
desagüe, y que se ha cerrado de nuevo la válvula de desagüe.
85!!

! Congelación
El producto se puede introducir previamente congelado en el equipo o se
puede congelar en el condensador de hielo.
En este segundo caso, se coloca el producto en el castillo de bandejas y
se enfría el condensador seleccionando la opción “congelación” en la pantalla
táctil.
La temperatura de congelación se determina en función del punto de
solidificación del producto. La velocidad de congelación influye en el tamaño de
los cristales y por lo tanto en la velocidad de sublimación.
Es por lo tanto esencial encontrar para cada producto parámetros de
congelación, más apropiados.
Es aconsejable congelar los productos a una temperatura por debajo los
- 20°C y durante un tiempo de 3 a 4 horas.
Además se pone en marcha el sistema de refrigeración y se enfría el
condensador.
El sistema de refrigeración requiere un tiempo de preparación (5 minutos
aprox.) en el cual la temperatura puede subir ligeramente.
Después, la temperatura bajara rápidamente. Este paso debe tener una
duración superior a 30 min para permitir al sistema alcanzar la temperatura
mínima.
86!!

! Frío + Vacío
Se pone en marcha el sistema refrigeración, se enfría el condensador y
se pone en marcha el grupo de vacío. Se inicia el vacío cuando la temperatura
de condensador está por debajo del parámetro determinado (típicamente -
40°C).
La regulación de nivel de vacío dependerá del Set Point programado. El
sistema necesitará más de 20 min para alcanzar el nivel de vacío mínimo.
! Calefacción de bandejas
Se activan las resistencias eléctricas de las bandejas para calentar hasta
la temperatura programada.
También se pone en marcha el grupo de vacío y se inicia el vacío
cuando la temperatura del condensador está por debajo del parámetro
determinado (típicamente -40 °C). La regulación del nivel de vacío dependerá
del Set Point programado.
Liofilización del producto
El producto se puede liofilizar en el interior del condensador o en la cámara:
En el interior del condensador: Se recomienda cargar son producto sólo
la bandeja inferior y la intermedia. La bandeja superior, no recibe frío de igual
manera que las otras dos bandejas puesto que la toma de vacío está situada
en la parte superior del condensador.
El equipo permite programar recetas de manera que se puede realizar un
ciclo completo de liofilización sin tener que realizar ninguna acción manual.
87!!

En el exterior del condensador: Está recomendado cuando la cantidad de
producto permite cargar las tres bandejas. Se recomienda congelar el producto
en el interior del condensador, ya que esto permite tener las bandejas
calefactables frías y realizar rampas de calefacción desde temperaturas
negativas.
Una vez congelado el producto, hay que extraer el castillo de bandejas del
condensador y montarlo en la cámara.
También se puede cargar un producto que haya sido congelado en otro
equipo. El equipo permite programar recetas de manera que se puede realizar
un ciclo de liofilización completo sin tener que realizar ninguna acción manual,
una vez el producto ya está congelado y la cámara montada.
88!!

VI. DISCUCIÓN DE LOS RESULTADOS
a. Resultados experimentales
1. Introducción
A partir de nuestra experimentación y ensayos realizados en el
laboratorio, procederemos a relatar la información que podemos extraer sobre
nuestro proceso y mecanismo de adsorción.
Nuestros resultados serán comparados y definiremos una causa o
explicación de su respuesta. En nuestro caso, y para el Boro, realizaremos la
explicación y comprensión de los resultados obtenidos.
2. Isotermas de adsorción de las perlas de alginato de calcio
En este apartado se estudian los diferentes equilibrios de adsorción entre
el boro y las perlas de alginato mediante las isotermas de adsorción realizadas
a diferentes concentraciones del alginato, así como también a diferente
configuración de las perlas del mismo (húmedas y secas) con un pH inicial de
11.
La metodología del estudio se hizo 48 horas, consiste en la preparación
de soluciones de boro de concentraciones variables entre 5 y 4000 mg/L y la
adición de una cantidad fija de alginato de sodio de (0,7 g). Para la realización
de esta gráfica se aplica la Ec.III.
Para mayor facilidad los dados se han tratado con el Solver del Excel.
Los modelos matemáticos que se aplican en este proyecto para construir
la isoterma teórica son los modelos de Langmuir y Freundlich, cuyas
ecuaciones son respectivamente:
! " 89:$'(;<9:$'(
(Ec.II) ! " @AB; CD (Ec.III)
89!!

Seguidamente, se muestran los resultados de los parámetros y
constantes que se han obtenido de las linealizaciones de cada concentración
de alginato.
2.1 Isoterma de adsorción de las perlas húmedas
0,0
20,0
40,0
60,0
80,0
100,0
0 500 1000 1500 2000 2500 3000 3500 4000
3,2% 2,4% 1,6%
q (m
g/g)
[B]e ppm Gráfico 1 Comparación a diferente concentración de alginato perlas húmedas
(pHinicial=11, 50 rpm, 48 horas, temp. y pres.=ambiente)
Podemos ver en el gráfico 1 que la concentración que tiene la mejor
capacidad de adsorción (q) es la concentración de alginato de las perlas
húmedas de 1,6%.
Mientras a partir de la concentración de boro de 2000 ppm la
concentración de alginato de 3,2 % tiene una capacidad de adsorción
ligeramente mayor que los otros.
90!!

2.2 Isoterma de adsorción de las perlas secas
0,0
20,0
40,0
60,0
80,0
100,0
0 500 1000 1500 2000 2500 3000 3500 4000
3,2% 2,4% 1,6%
q (m
g/g)
[B]e ppm Gráfico 2 Comparación a diferente concentración de alginato perlas secas.
(pHinicial=11, 50 rpm, 48 horas, temp. y pres.=ambiente)
Podemos ver en el gráfico 2 una segunda vez que por las perlas secas la
concentración de alginato de 1,6% tiene la mejor capacidad de adsorción.
Pero hay una diferencia por las perlas secas porqué la tendencia de los
puntos esta diferente y los puntos de la concentración de alginato de 3,2 %
parecen más alto que por los puntos de la concentración de alginato de 2,4 %.
En los siguientes gráficos podemos ver la comparación entre misma
concentración de alginato y la configuración de las perlas (húmedas o secas).
91!!

0,0
20,0
40,0
60,0
80,0
100,0
0 500 1000 1500 2000 2500 3000 3500 4000
3,2% Humedas
3,2% secas
q
[B]e
Gráfico 3 Comparación de las perlas a diferente configuración a 3,2% de alginato.
(pHinicial=11, 50 rpm, 48 horas, temp. y pres.=ambiente)
0,0
20,0
40,0
60,0
80,0
100,0
0 500 1000 1500 2000 2500 3000 3500 4000
2,4% Humedas
2,4% secas
q (m
g/g)
[B]e Gráfico 4 Comparación de las perlas a diferente configuración a 2,4% de alginato.
(pHinicial=11, 50 rpm, 48 horas, temp. y pres.=ambiente)
92!!
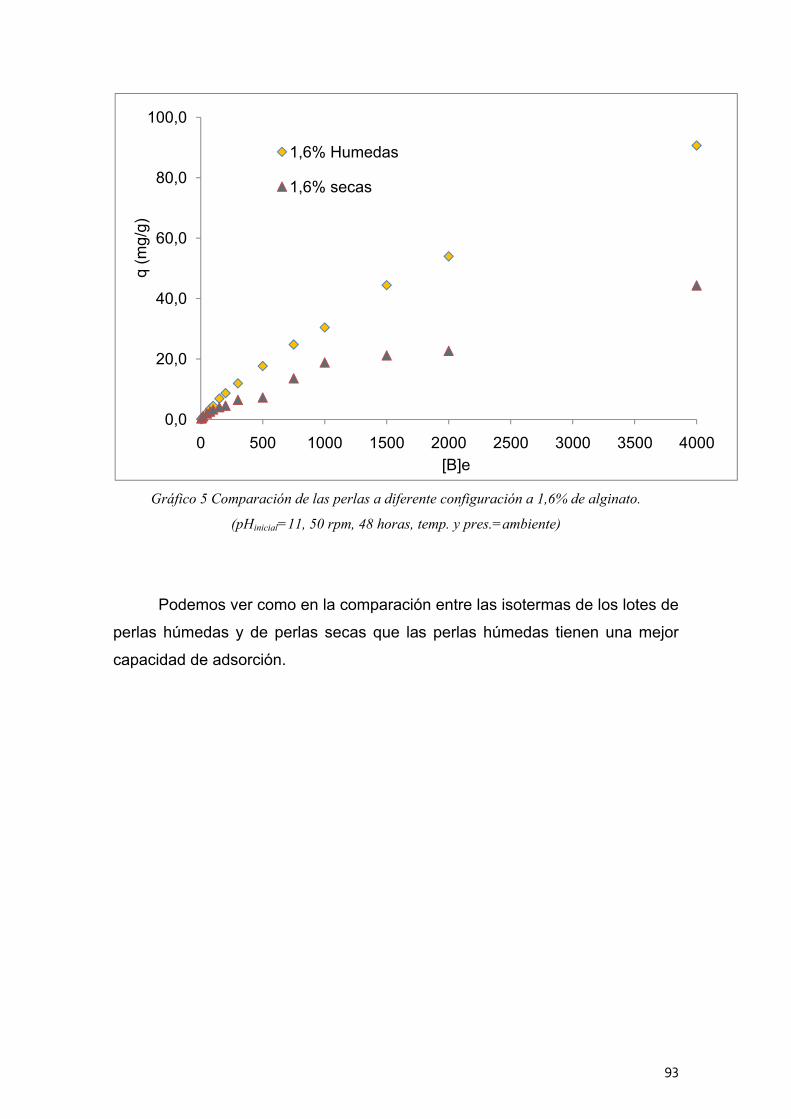
0,0
20,0
40,0
60,0
80,0
100,0
0 500 1000 1500 2000 2500 3000 3500 4000
1,6% Humedas
1,6% secas
q (m
g/g)
[B]e Gráfico 5 Comparación de las perlas a diferente configuración a 1,6% de alginato.
(pHinicial=11, 50 rpm, 48 horas, temp. y pres.=ambiente)
Podemos ver como en la comparación entre las isotermas de los lotes de
perlas húmedas y de perlas secas que las perlas húmedas tienen una mejor
capacidad de adsorción.
93!!

2.3 Conclusión
Los gráficos 1 y 2 respectivamente nos dan una explicación importante
sobre la mejor concentración de alginato a utilizar para obtener la máxima de
capacidad de adsorción. Sabiendo que la mejor configuración de las perlas
para obtener un máximum de capacidad de adsorción son las perlas húmedas.
Podemos concluir que a pH 11 el mejor procedimiento de adsorción
utilizando los modelos de Langmuir y Freundlich es las perlas de alginato de
calcio de una concentración de 1,6 %.
94!!

3. Cinéticas de adsorción de las perlas de alginato de calcio
El conocimiento y estudio de las cinéticas de adsorción son muy
importantes y describen parámetros o propiedades de gran ayuda y
comprensión. Para poder conocer y poder compararlas a partir de las
diferentes experiencias cinéticas, seguidamente describimos los resultados
obtenidos.
Para este estudio todos los condiciones serán iguales (mismo vaso,
mismo agitador…etc.) se utiliza 1,75 g de alginato de sodio y 250 mL de una
solución de boro a 50 ppm, utilizando perlas del mismo tamaño y a diferentes
configuraciones (húmedas o secas) con un pH inicial de 11 y una agitación de
50 rpm.
Además podemos ver los condiciones de trabajo por la cinética son igual
al condiciones de la isoterma, es decir, pH inicial 11, agitación de 50 rpm y
duración 48 horas.
Para la realización de esta gráfica y para calcular la capacidad de adsorción de
adsorbente se aplica la Ec.VIII que hemos visto anteriormente con la isoterma.
! " #$%&$W)* +, - , X (Ec.VIII)
Donde:
c e adsorción (mg/g) !, apacidad d
Y / ZP) +, -, es la masa del soluto adsorbido (mg) #Z ZP, la concentración de boro durante la cinética en función del tiempo
(mg/L)
, la concentración de boro inicial (0 min) (mg/L) ZY , es la masa del adsorbente (g) [ X, es un factor de dilución
Para la cinética V= 250 mL, m= 1,75 g y k= 5
95!!

Para mayor facilidad los dados se han tratado con el Solver del Excel.
Ahora podemos hacer las representaciones gráficas de la capacidad de
adsorción en función del tiempo (min).
3.1 Cinética de adsorción de las perlas húmedas
El gráfico siguiente representa las diferentes cinéticas de adsorción de
las perlas húmedas a diferentes concentraciones.
0,00
0,50
1,00
1,50
2,00
2,50
3,00
3,50
0 500 1000 1500 2000 2500 3000
1,6% HUM 2,4% HUM 3,2% HUM
q (m
g/g)
Tiempo (min)
Gráfico 6 Comparación a diferente concentración de alginato perlas húmedas.
(pHinicial=11, 50 rpm, 48 horas, [B]inicial= 50 ppm, temp. y pres.=ambiente)
Como podemos ver en la gráfica 6 la velocidad con se llega al equilibrio
es muy rápida en todo los casos y que el equilibrio se forma al mismo tiempo
(aproximadamente 90 minutos).
96!!

Los resultados obtenidos, nos indican que la concentración de las perlas
que tienen una mayor capacidad de adsorción es las de 2,4%.
Esto hecho nos muestra que las perlas de 2,4% son los que tienen una
mejor accesibilidad a los centros activos. Las perlas de 1,6% son perlas que
presentan una gran hidratación dificultado la entrada del boro por el contrario
las perlas de 3,2 % al tener más centros activos en el mismo volumen diferente
la accesibilidad del boro a todos ellos especiales.
Podemos observar también en la gráfica 6 que para la variación del
porcentaje de alginato en las perlas implica una variación de la capacidad de
adsorción en las perlas así como una ligera variación de la parte inicial de las
curvas obtenidas al realizar las cinéticas.
Sin embargo la sección inicial de las curvas controladas por la difusión
externa es menos influenciada que la segunda parte de las curvas controladas
por la difusión intraparticular.
97!!

3.2 Cinética de adsorción de las perlas secas
Ahora el gráfico siguiente representa la cinética de adsorción de las
perlas de concentraciones diferentes como en el caso anterior pero con las
perlas secas.
0,00
0,50
1,00
1,50
2,00
2,50
0 500 1000 1500 2000 2500 3000
3,2 % SEC 1,6% SEC 2,4 % SEC
q (m
g/g)
Tiempo (min)
Gráfico 7 Comparación a diferente concentración de alginato perlas secas.
(pHinicial=11, 50 rpm, 48 horas, [B]inicial= 50 ppm, temp. y pres.=ambiente)
Podemos ver una vez de más en la gráfica 7 la velocidad con se llega al
equilibrio es muy rápida en todo los casos.
Los resultados obtenidos, nos indican al igual que la cinética anterior que
las perlas en que se consiguen una mayor capacidad de absorción es las de
2,4%.
Mientras el equilibrio parece más tarde aproximadamente a partir de 600
horas.
98!!
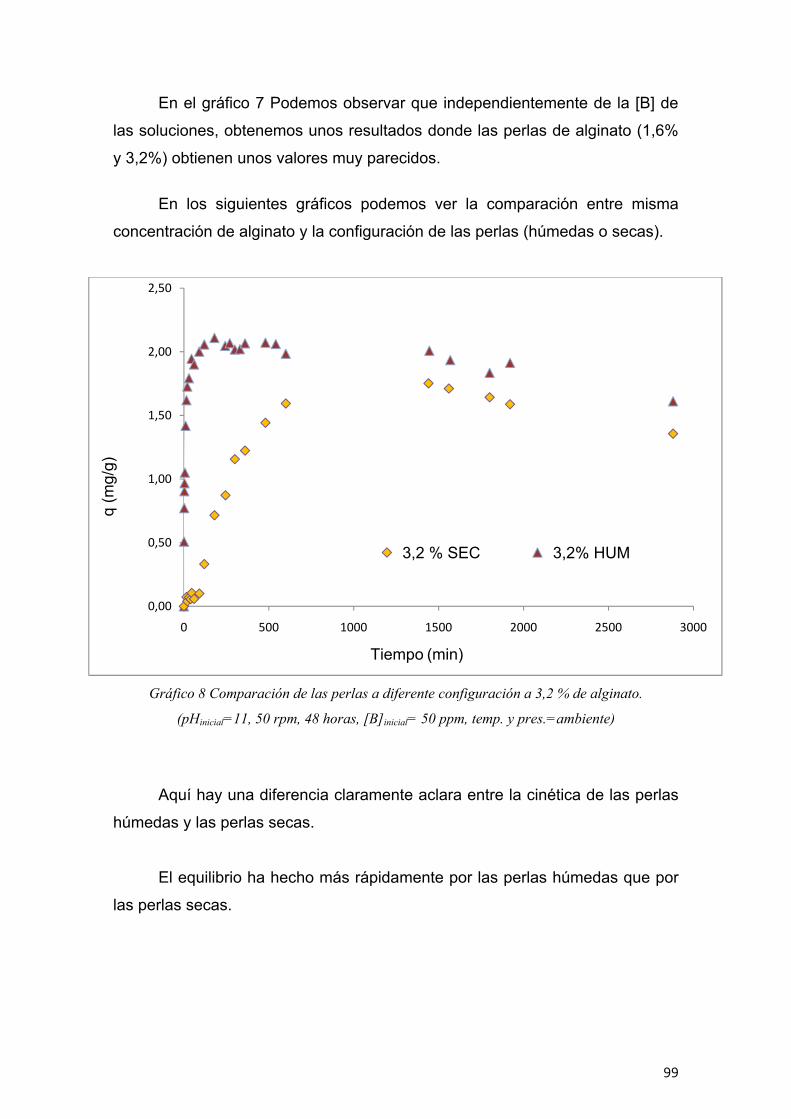
En el gráfico 7 Podemos observar que independientemente de la [B] de
las soluciones, obtenemos unos resultados donde las perlas de alginato (1,6%
y 3,2%) obtienen unos valores muy parecidos.
En los siguientes gráficos podemos ver la comparación entre misma
concentración de alginato y la configuración de las perlas (húmedas o secas).
0,00
0,50
1,00
1,50
2,00
2,50
0 500 1000 1500 2000 2500 3000
3,2 % SEC 3,2% HUM
Tiempo (min)
q (m
g/g)
Gráfico 8 Comparación de las perlas a diferente configuración a 3,2 % de alginato.
(pHinicial=11, 50 rpm, 48 horas, [B]inicial= 50 ppm, temp. y pres.=ambiente)
Aquí hay una diferencia claramente aclara entre la cinética de las perlas
húmedas y las perlas secas.
El equilibrio ha hecho más rápidamente por las perlas húmedas que por
las perlas secas.
99!!

Además la tendencia de los dos representaciones están igual en efecto a
la misma concentración de alginato de sodio añadido inicialmente el fenómeno
de adsorción se efectúa de la misma manera.
Sabiendo que los parámetros utilizados en las cinéticas de reacción son
los mismos, solamente la configuración de las diferentes perlas hace variar la
velocidad de adsorción de las perlas.
Podemos verle sobre los otros ejemplos.
0,00
0,50
1,00
1,50
2,00
2,50
3,00
3,50
0 500 1000 1500 2000 2500 3000
2,4% HUM 2,4 % SEC
q (m
g/g)
Tiempo (min) Gráfico 9 Comparación de las perlas a diferente configuración a 2,4 % de alginato.
(pHinicial=11, 50 rpm, 48 horas, [B]inicial= 50 ppm, temp. y pres.=ambiente)
100!!

0,00
0,50
1,00
1,50
2,00
2,50
3,00
0 500 1000 1500 2000 2500 3000
1,6% HUM 1,6% SEC
q (m
g/g)
Tiempo (min)
Gráfico 10 Comparación de las perlas a diferente configuración a 1,6 % de alginato.
(pHinicial=11, 50 rpm, 48 horas, [B]inicial= 50 ppm, temp. y pres.=ambiente)
Podemos llevar a la conclusión que la configuración húmeda tiene una
mejor capacidad de adsorción que la configuración seca.
3.3 Conclusión
Hemos visto para todos las cinéticas una tendencia semejante
estableciéndose el equilibrio rapidamente.
Podemos concluir que la cinética que tiene la mejor capacidad de
adsorción son las cinéticas de adsorción de las perlas de alginato húmedas, en
efecto como en el caso de la isoterma las perlas de alginato húmedas adsorben
más el boro que para las secas.
Además podemos ver que a partir de 2500 minutos los valores de las
cinéticas no están verdaderamente fiables porqué la capacidad de adsorción
tiende a disminuir, es posible que se dé un fenómeno de desorción que
aumentan las valores de la concentración a el equilibrio.
101!!

4. Cinéticas de desorción de las perlas de alginato de calcio
A partir de las cinéticas que hemos hecho podemos hacer
experimentación sobre la desorción, es decir el proceso inverso de la
adsorción, el proceso que permite de recuperar el boro de las perlas de
alginato.
Entonces, trabajamos siempre con las mismas condiciones, hay solo dos
cambios de factores, la concentración inicial de boro y el pH inicial.
Los resultados siguientes serán obtenidos a partir de la misma tabla de
tiempo que para la adsorción.
Hay otro punto a saber muy importante, el cálculo importante para la
desorción es conocer a partir de qué tiempo tenemos la máxima concentración
de boro liberado.
Podemos calcular el porcentaje de boro adsorbido de la cantidad total de
las perlas a partir de la masa de adsorbente y la masa de boro adsorbido.
Luego podremos hacer un gráfico representando el porcentaje de
desorción en función de la concentración de alginato de sodio.
La ecuación matemática es la siguiente: (Ec.IX)
\+]^_`HBabc+ " + $Kdefg+h'ijJkdje#$%&$Kdefg+fhijJkdje)
, lmm (Ec.IX)
Donde:
]^_B`HBabc, porcentaje de adsorbente \+, concentración inicial de boro de la cinética de adsorción (mg/L) ZY
bC, concentración de boro final de la cinética de adsorción (mg/L) ZnoCQp+QRqrsIoZnoCQp+RNqrsIobC, concentración de boro final de la cinética de desorción (mg/L)
102!!
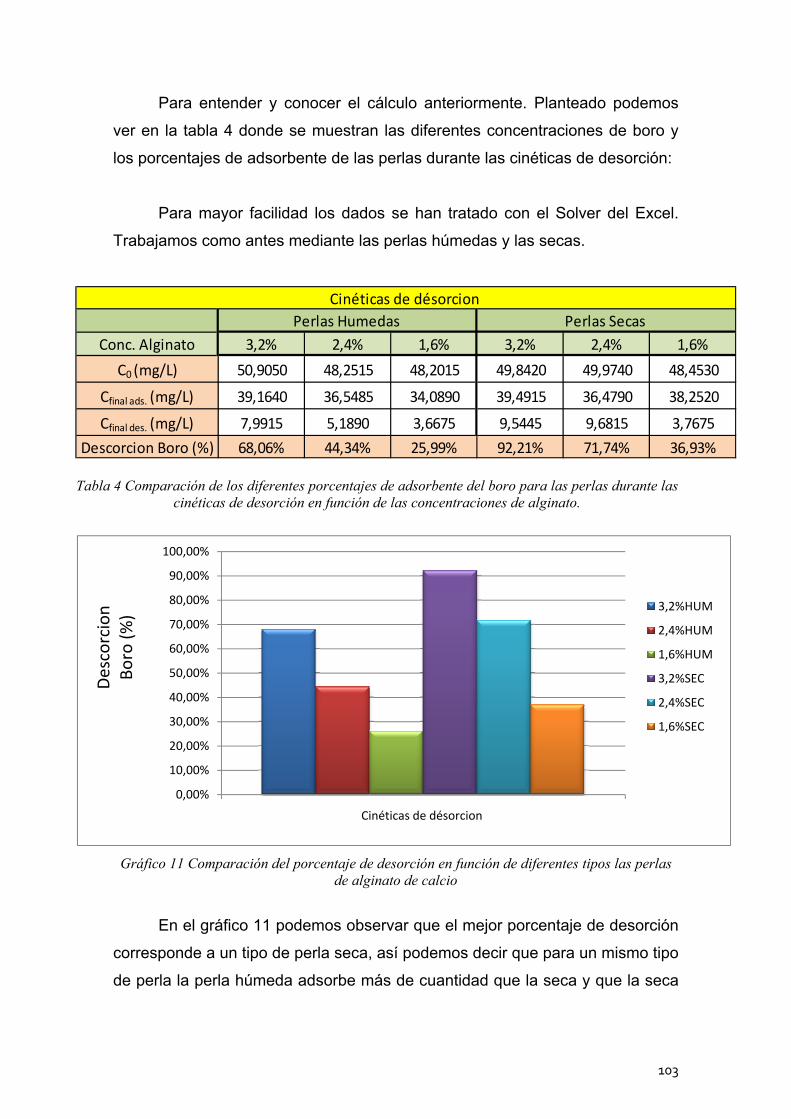
Para entender y conocer el cálculo anteriormente. Planteado podemos
ver en la tabla 4 donde se muestran las diferentes concentraciones de boro y
los porcentajes de adsorbente de las perlas durante las cinéticas de desorción:
Para mayor facilidad los dados se han tratado con el Solver del Excel.
Trabajamos como antes mediante las perlas húmedas y las secas.
Conc.!Alginato 3,2% 2,4% 1,6% 3,2% 2,4% 1,6%
C0!(mg/L) 50,9050 48,2515 48,2015 49,8420 49,9740 48,4530
Cfinal!ads.!(mg/L) 39,1640 36,5485 34,0890 39,4915 36,4790 38,2520
Cfinal!des.!(mg/L) 7,9915 5,1890 3,6675 9,5445 9,6815 3,7675
Descorcion!Boro!(%) 68,06% 44,34% 25,99% 92,21% 71,74% 36,93%
Perlas!Humedas! Perlas!Secas!Cinéticas!de!désorcion!
Tabla 4 Comparación de los diferentes porcentajes de adsorbente del boro para las perlas durante las
cinéticas de desorción en función de las concentraciones de alginato.
0,00%
10,00%
20,00%
30,00%
40,00%
50,00%
60,00%
70,00%
80,00%
90,00%
100,00%
Cinéticas!de!désorcion!
3,2%HUM
2,4%HUM
1,6%HUM
3,2%SEC
2,4%SEC
1,6%SEC
Descorcion!
Boro!(%
)
Gráfico 11 Comparación del porcentaje de desorción en función de diferentes tipos las perlas
de alginato de calcio
En el gráfico 11 podemos observar que el mejor porcentaje de desorción
corresponde a un tipo de perla seca, así podemos decir que para un mismo tipo
de perla la perla húmeda adsorbe más de cuantidad que la seca y que la seca
103!!

desorbe más rápidamente de la cuantidad que la húmeda porqué a una peor
adsorción corresponde una mejor desorción.
Podemos verificar esta hipótesis sobre los gráficos siguientes.
4.1 Cinéticas de desorción de las perlas húmedas
El gráfico 12 siguiente representa las cinéticas de desorción a diferentes
concentraciones en la configuración de la perlas húmedas.
0,0
2,0
4,0
6,0
8,0
0 500 1000 1500 2000 2500 3000
3,2% HUM 2,4% HUM 1,6% HUM
Tiempo (min)
[B]e
Gráfico 12 Comparación a diferente concentración de alginato perlas húmedas.
(50 rpm, 48 horas, temp. y pres.=ambiente)
104!!
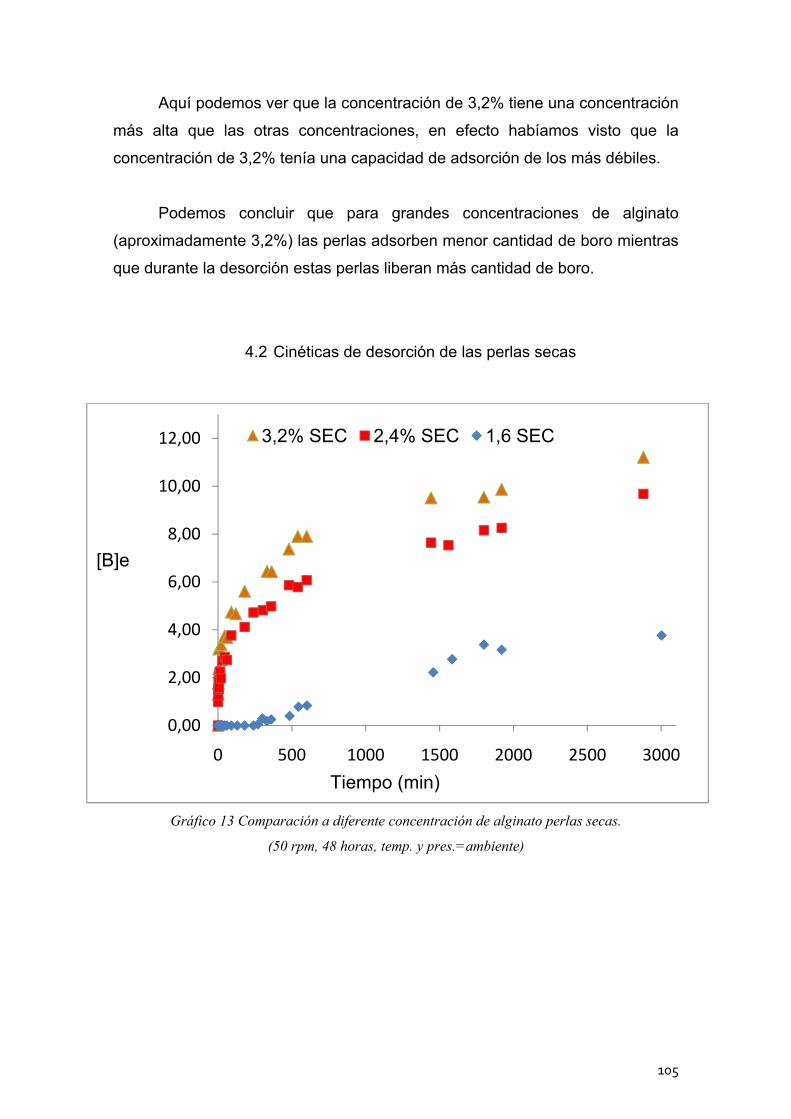
Aquí podemos ver que la concentración de 3,2% tiene una concentración
más alta que las otras concentraciones, en efecto habíamos visto que la
concentración de 3,2% tenía una capacidad de adsorción de los más débiles.
Podemos concluir que para grandes concentraciones de alginato
(aproximadamente 3,2%) las perlas adsorben menor cantidad de boro mientras
que durante la desorción estas perlas liberan más cantidad de boro.
4.2 Cinéticas de desorción de las perlas secas
0,00
2,00
4,00
6,00
8,00
10,00
12,00
0 500 1000 1500 2000 2500 3000
3,2% SEC 2,4% SEC 1,6 SEC
Tiempo (min)
[B]e
Gráfico 13 Comparación a diferente concentración de alginato perlas secas.
(50 rpm, 48 horas, temp. y pres.=ambiente)
105!!

4.3 Conclusión
Como podemos observar en las cinéticas de adsorción, las perlas secas
de 1,6 % absorben rápidamente mostrando una difusión muy alta, por la cual
es de esperar que la desorción no sea buena.
A partir de 270 minutos se produce una pequeña desorción que tiende a
equilibrarse al llegar a los 2000 minutos.
Es importante destacar que la desorción se ha llevado a cabo mediante
agua destilada sin necesidad de utilizar disolvente que contaminan.
106!!

5. Estudio del pH
Este estudio se ha realizado para observar cómo influye el pH en la
adsorción del boro; el procedimiento experimental es el mismo que se sigue en
el apartado anterior.
Para realizar la siguiente gráfica se ha de verificar el pH durante la
cinética i al final de la isoterma.
Entonces vamos a observar en los gráficos siguientes la variación del pH
(inicial y final) en función de las concentraciones del boro de la isoterma y del
tiempo de la cinética.
5.1 En la isoterma
Perlas húmedas
0
2
4
6
8
10
12
14
5 10 20 50 75 100 150 200 300 500 750 1!000 1!500 2!000 4!000
Isoterma!perlas!Humedas!2,4%
pHipH
pHf
[B]!(ppm)
107!!

Perlas secas
0
2
4
6
8
10
12
14
5 10 20 50 75 100 150 200 300 500 750 1!000 1!500 2!000 4!000
pHi
pHf
Isoterma!perlas!secas!2,4%
[B]!(ppm)
pH
Aquí podemos ver un ejemplo con la concentración de 2,4% de alginato
porqué los estudios anteriores muestran que la concentración de 2,4% tiene los
mejores capacidad de adsorción.
La variación del pH muestra en la isoterma de las perlas húmedas que
para las concentraciones altas hay prácticamente no de variación entre el pH
inicial y el pH final.
Mientras que para las perlas secas podemos ver que hay muchos
valores no fiables porqué el pH final esta superior que el pH final para por
ejemplo (300 ppm).
108!!

5.2 En la cinética
Perlas húmedas
Perlas secas
0
2
4
6
8
10
12
14
0 1 2 3 4 5 10 15 20 31 45 60 90 120
188
267
270
300
330
360
486
540
600
1!44
11!56
01!80
01!92
02!88
0
Cinética!perlas!Humedas!2,4%
pHipH
pHf
Tiempo!(min)
0
2
4
6
8
10
12
14
0 1 2 3 4 5 10 15 20 30 45 60 91 120
180
240
270
300
330
358
480
540
600
1!44
01!56
01!80
01!86
01!92
02!98
1
pHi
pHf
Cinética!perlas!secas!2,4%
Tiempo!(min)
pH
109!!

En estos gráficos se observan que el tiempo aumenta a medida que
disminuye el pH.
Si miramos la cinética de adsorción anteriormente podemos ver que el
equilibrio ha alcanzado a partir de 90 minutos para las perlas húmedas y 600
minutos para las perlas secas por eso no deberíamos ver una variación del pH
después los equilibrios, aquí no es el caso, en efecto al final de la isoterma
tenemos un pH más ácido que al principio y la adsorción disminuye con el
tiempo.
Entonces a pH ácidos la adsorción es pequeña mientras que para pH
básicos la capacidad de adsorción es mucho más elevada.
110!!

XI. CONCLUSIÓN FINAL
El estudio de la adsorción del boro en las perlas de alginato de calcio, es
influenciado por varios parámetros:
! Porcentaje de alginato de calcio en el interior de las perlas. Las perlas de
2,4% de alginato presenta la mayor adsorción, debido a mejor
disponibilidad de los centros activos. La difusión intraparticular se ve
influenciada por el diferente porcentaje de alginato en las perlas, siendo
en este caso también las perlas de 2,4% en alginato las que muestran el
mejor comportamiento.
! El tipo de las perlas. Las perlas húmedas presentan una mejor
capacidad de adsorción que las perlas secas, debido a la mejor
accesibilidad del boro a los centros activos.
! El pH no demuestra que el pH óptimo es 11. Estudios anteriores nos
indican que a pH más acido (pH 6) la capacidad de adsorción más débil.
Para concluir obtenemos ahora para la cinética una capacidad máxima
de adsorción (q) (mg/g) de boro de las perlas de concentraciones de alginato
de sodio de 2,4 % húmedas de diámetros de 2,27 mm igual a 3,04 mgB/g.
El estudio de la desorción del boro en las perlas de alginato de calcio, es
influenciado por varios parámetros:
! Porcentaje de alginato de calcio en el interior de las perlas. Las perlas de
3,2% de alginato presenta la mayor desorción. El estudio realizado de la
desorción con diferentes tipos de partícula nos demuestran, como era de
esperar, que la desorción aumenta al aumentar la concentración de
alginato, es decir para las perlas secas. Siendo en este caso las perlas
de 3,2% en alginato las que muestran el mejor comportamiento.
! El tipo de las perlas. Las perlas secas presentan una mejor capacidad de
desorción que las perlas húmedas.
111!!

La utilización del proceso en continuo, trabajando a bajas
concentraciones de boro, no comporta un marcado aumento de la capacidad
de adsorción del boro bajo las condiciones experimentales utilizadas en nuestro
estudio, siendo necesaria la realización de un estudio más completo.
Vías futuras de investigación.
Continuar este estudio utilizando de perlas de alginato liofilizadas, hemos
hecho una experiencia durante el periodo de prácticas con los mismos
condiciones para saber si las perlas liofilizadas serán un mejor tipo de
configuración que húmedas o secas para obtener un mayor capacidad de
adsorción.
Podemos observar los resultados de esta experiencia diferente sobre el
siguiente grafico de comparación de las cinéticas de adsorción de las perlas
húmedas.
0,00
0,50
1,00
1,50
2,00
2,50
3,00
0 500 1000 1500 2000 2500 3000
1,6%!HUM 2,4%!HUM3,2%!HUM 3,2%!LIOFILISEq
(mg/
g)
Tiempo!(min)
La cinética de adsorción de las perlas 3,2 % liofilizadas es mayor que la
cinética 3,2 % de las perlas húmedas podríamos en el futuro hacer las mismas
experiencias con perlas liofilizadas. Quizás la capacitad de adsorción será
mayor.
112!!

XII. GLOSARIO
Todas las palabras siguientes son difíciles a entender para una mejor
compresión he escribido en el texto anterior estas palabras en itálica y ahora
hay una definición:
Hidrotermal: Un hidrotermal es un filón cuyos minerales provienen de la
precipitación en una cavidad de la roca de sustancias contenidas en disolución
por aguas profundas muy calientes y sometidas a fuertes presiones.
Isotopos: Los isótopos, (del griego: &'(), isos = mismo; *+,(), tópos = lugar)
son todos los tipos de átomos de un mismo elemento, que se encuentran en el
mismo sitio de la tabla periódica pero tiene diferente número másico (A).
Los átomos que son isótopos entre sí son los que tienen igual número atómico
(número de protones en el núcleo) pero diferente número másico (suma del
número de neutrones y el de protones en el núcleo). Por lo tanto difieren en el
número de neutrones [30].
Electrones de valencia: Los electrones de valencia son los electrones que se
encuentran en los mayores niveles de energía del átomo, siendo éstos los
responsables de la interacción entre átomos de distintas especies o entre los
átomos de una misma.
Gas noble: Los gases nobles o gases inertes son un grupo de elementos
químicos con propiedades muy similares: bajo condiciones normales, son
gases monoatómicos inodoros, incoloros y presentan una reactividad química
muy baja. Se sitúan en el grupo 18 (8A) de la tabla periódica (anteriormente
llamado grupo 0). Los seis gases nobles que se encuentran en la naturaleza
son helio (He), neón (Ne), argón (Ar), kriptón (Kr), xenón (Xe) y el radioactivo
radón (Rn).
113!!

Ligandos: Los ligandos son iones o moléculas que rodean a un metal,
formando un complejo metálico. Un ligando enlazado a un ion central se dice
que está coordinado al ion.
Ácidos de Lewis: Las substancias que pueden aceptar pares de electrones.
Bases de Lewis: Las sustancias que pueden ceder pares de electrones.
Hidrocoloide: Estos hidrocoloide son unas macromoléculas hidrosolubles que,
en solución acuosa, molestan la movilidad del agua y determinan así el
comportamiento reológico del producto [30].
Fisisorción: Fisisorción o adsorción física es un tipo especial de adsorción. En
química se denomina adsorción al acumulo o depósito de material (adsorbato)
en un plano o superficie (a diferencia de la absorción que es un proceso
volumétrico).
El proceso físico de la fisisorción es aquel por medio del cual, un
elemento o compuesto químico, se adhiere a una superficie, que puede estar
formada por el mismo tipo de compuesto o por alguno diferente, y en el que la
especie adsorbida (fisisorbida) conserva su naturaleza química.
El otro tipo de adsorción es la quimisorción, en el cual la especie
adsorbida (quimisorbida) da lugar a una especie química distinta.
Cuando una molécula en fase vapor, se aproxima a una superficie y se
encuentra a distancias atómicas, sentirá una atracción debida a la interacción
con las moléculas de la superficie. En el caso de la fisisorción, las fuerzas que
ligan el ión o molécula a la superficie provienen de interacciones dipolares, es
decir fuerzas de Van der Waals o fuerzas de dispersión London.
114!!

Esto sucede igual para moléculas simétricas o gases inertes, cualquiera
que tenga momento dipolar. Esas moléculas o átomos actúan como dipolos
oscilantes y crean las interacciones dipolares mencionadas. [31].
Principio de “Le Chatelier”: El Principio de Le Châtelier, postulado por Henri-
Louis Le Châtelier (1850-1936), un químico industrial francés, establece que: Si
un sistema químico en equilibrio experimenta un cambio en la concentración,
temperatura, volumen, o la presión parcial, entonces el equilibrio se desplaza
para contrarrestar el cambio impuesto. Este principio es equivalente al principio
de la conservación de la energía [32].
Fuerzas de Van Der Waals: la fuerza de van der Waals (o interacción de van
der Waals), denominada así en honor al científico holandés Johannes Diderik
van der Waals, es la fuerza atractiva o repulsiva entre moléculas (o entre partes
de una misma molécula) distintas a aquellas debidas al enlace covalente o a la
interacción electrostática de iones con otros o con moléculas neutras [33].
Fotosíntesis: La fotosíntesis (del griego antiguo "luz", y, "unión") es la
conversión de energía luminosa en energía química estable, siendo el
adenosín trifosfato (ATP) la primera molécula en la que queda almacenada esa
energía química. Con posterioridad, el ATP se usa para sintetizar moléculas
orgánicas de mayor estabilidad [34].
Bronsted-Lowry:! Clasificación de ácidos y bases encaminada a facilitar la
comprensión y discusión de las reacciones básicas o ácidas, formulada por
Bronsted [35].
115!!

XIII. BIBLIOGRAFÍA
[1] Seira Ibáñez, Juana. Adsorción de boro mediante perlas de alginato. PFC,
UPC, Departamento de Ingeniería química, 2008 [Biblioteca EPSEVG].
[http://upcommons.upc.edu/pfc/bitstream/2099.1/5529/1/ART%C3%8DCULO.p
df].
[2] Juana Seira Ibáñez, Adsorción De Boro Mediante Perlas de alginato. PFC,
UPC, Departamento de Ingeniería química, 2008 [Biblioteca EPSEVG].
[http://upcommons.upc.edu/pfc/bitstream/2009.1/5529/2/MEMORIA.pdf].
[3] Carlos Tobalina Ramirez, Adsorción de boro por medio de perlas de
alginato. PFC, UPC, Departamento de Ingeniería química, 2009 [Biblioteca
EPSEVG].
[http://upcommons.upc.edu/pfc/bitstream/2099.1/8015/1/PFC_Carlos%20Tobali
na%20Ramirez.pdf].
[4] Este documento disponible en la página internet de la UPC siguiente:
[http://www.upc.edu/unitat/fitxa_unitat.php?id_unitat=124&lang=esp].
[5] Este documento disponible en la página internet de Wikipédia siguiente:
[http://es.wikipedia.org/wiki/Boro].
[6] Final report on the safety assessment of sodium borate and boric acid. J.
Am. Coll. Toxicol., 2: 87 (1983).
[7] Harvey, S.C. Antiseptics and disinfectants; fungicides, ecto-parasiticides. En:
Pharmacological basis of therapeutics. L.S. Goodman, A. Gilman, A.G. Gilman
et G.B. Koelle (dir. de publ.). Macmillan, Toronto. p. 994 (1975).
[8] Siegel, E. et Wason, S. Boric acid toxicity. Pediatr. Clin. North Am., 33: 363
(1986).
116!!

[9] Dixon, R.L., Lee, I.P. et Sherins, R.J. Methods to assess reproductive effects
of environmental chemicals: Studies of cadmium and boron administered orally.
Environ. Health Perspect., 13: 59 (1976).
[10] Dabrowski, K.. Ascorbic Acid in Aquatic Organisms: Status and
Perspectives. Boca Raton, FL: CRC Press. 2001.
[11] John J McKetta, William Aaron Cunningham. Encyclopedia of chemical
processing and design Ed: illustrated Pub: CRC Press, 1976.
[12] Pujante Rosell, Isaac. Adsorción de Paraquat. PFC, UPC, Departamento
de Ingeniería química, 2006 [Biblioteca EPSEVG].
[13] El-Rahman Abd [et al]. Modeling the sorption kinetics of cesium and
strontium ions on zeolita A. Journal of Nuclear and Radiochemical Sciences,
2006, vol. 7, núm. 2, p. 21-27.
[14] Jaroniec and Madey, 1988 M. Jaroniec and R. Madey, Physical Adsorption
on Heterogeneous Solids, Elsevier, Amsterdam (1988).
[15] Serrarlos Font, J. Adsorbió d’or i zinc amb resines impregnades XAD-2.
Superficie d’equilibri, un nou concepte per a l’adsorció. Tesis doctoral, UdG.
Departament IMA
(Informàtica i Matemàtica Aplicada), 2001.
[16] Chatjigakis, AK; Pappas, C.; Proxenia, N.; Kalantzi, O. ; Rodis, P. ;
Polissiou, M. FT-IR spectroscopic determination of the degree of esterification
of cell wall pectins from stored peaches and correlation to textural changes.
Carbohydrate Polymers 37 (1998) 395-408.
117!!

[17] Este documento disponible en la página internet siguiente:
[http://www.textoscientificos.com/quimica/carbon-activo/materiales-
adsorbentes].
[18] M. Ruiz, A.M. Sastre, M.C. Zikan, E.Guibal, Palladium sorption on
glutaraldehyde crosslinked chitosan in dynamic systems. Journal of Applied
Polymer Science, 81(2001)153-165.
[19] Seira Ibáñez, Juana. Adsorción de boro mediante perlas de alginato. PFC,
UPC, Departamento de Ingeniería química, 2008 [Biblioteca EPSEVG].
[20] Alginatos, María Silvia Dalla Lasta, Ingeniera Química.
Este documento disponible en la página internet siguiente:
[http://www.monografias.com/trabajos12/alginato/alginato.shtml#_Fuente_de_o
btenci%C3%B3n:].
[21] Algunos usos del quitosano en sistemas acuosos, Cristóbal Lárez
Velásquez, Universidad de los Andes, Facultad de Ciencias. La Hechicera,
Departamento de Química, Grupo de Polímeros. Mérida (Venezuela) Volumen
4(2), Abril 2003. Este documento es disponible en la página internet siguiente:
[http://www.ehu.es/reviberpol/pdf/ABR03/Cristobal2003.pdf].
[22] Muzzarelli R. “Chitin”. Editorial Pergamon Press. Primera edición, p. 2
(1974).
[23] Chitosan Nanoparticles : A Promising System for Drug Delivery Waree
Tiyaboonchai, Department of Pharmaceutical Technology, Faculty of
Pharmaceutical Sciences, Naresuan University, Phitsanulok 65000, Thailand
Corresponding author.
E-mail address: [email protected] (W. Tiyaboonchai) Received 2 May 2003;
accepted 9 September 2003.
118!!

[24] Este documento disponible en la página Internet de la esuela EPSEVG
siguiente: Organització de l'escola ÒRGANS UNIPERSONALS,
[http://www.epsevg.upc.edu/coneix-lepsevg/organitzacio-de-lescola].
[25] Nanopartículas de quitosano y otros compuestos, pagina internet:
[http://www.advancell.net/site/index.php?pt1=74&pt2=77&pt3=181&pt4=181].
[26] Calvo, P., Remuñán-López, C., Vila-Jato, J.L. & Alonso, M.J. (1997) Novel
hydrophilic chitosan-polyethylene oxide nanoparticles as protein carriers. J.
Appl. Polym. Sci. 63: 125-132.
[27] Desorción, Esto física- el artículo relacionado es a trozo. Usted puede
ayuda Wikipedia cerca ampliarlo. Pagina internet
[http://www.worldlingo.com/ma/enwiki/es/Desorption].
[28] Liofilización, El texto está disponible bajo la Licencia Creative Commons
Reconocimiento Compartir Igual 3.0; podrían ser aplicables cláusulas
adicionales. [http://es.wikipedia.org/wiki/Liofilizaci%C3%B3n].
[29] Universidad Politécnica de Cataluña, 2010 texto disponible en la página
internet siguiente:
[http://es.wikipedia.org/wiki/Universidad_Polit%C3%A9cnica_de_Catalu%C3%B
1a].
[30] Este documente disponible en la página Internet siguiente:
[http://fr.wikipedia.org/wiki/Hydrocollo%C3%AFde].
[31] Este documente disponible en la página Internet siguiente:
[http://es.wikipedia.org/wiki/Fisisorci%C3%B3n].
[32] Este documente disponible en la página Internet siguiente:
[http://es.wikipedia.org/wiki/Principio_de_Le_Ch%C3%A2telier]
119!!

[33] Este documente disponible en la página Internet siguiente:
[http://es.wikipedia.org/wiki/Fuerzas_de_van_der_Waals]
[34] Este documente disponible en la página Internet siguiente:
[http://es.wikipedia.org/wiki/Fotosintesis].
[35] Este documente disponible en la página Internet siguiente:
[http://es.wikipedia.org/wiki/Bronsted].
120!!

XIV. ANEXOS
Tabla Anexo.1 isoterma 3,2% Húmedas
[Alg] = 0,7 g 388 perlas[PQ]o = 767285,0 mg/LpHo = 11V = 100 mlt agitación = 2 díasv agitacón = 50 rpm
Isoterma variando la concentración de B
pH = 11[B]o Vo pHi pHf [B] FD [B]o [B] FD [B]e q (mg/g)
5 100 11 6,55 1,1173 5 5,5865 0,9468 5 4,7 0,121810 100 11 7,46 2,155 5 10,775 1,6114 5 8,1 0,388320 100 11 7,93 4,2047 5 21,0235 3,3892 5 16,9 0,582550 100 11 9,52 10,518 5 52,59 7,6236 5 38,1 2,067475 100 11 9,49 7,1479 10 71,479 5,3364 10 53,4 2,5879
100 100 11 9,57 5,6085 16,6666667 93,475 4,0292 16,6666667 67,2 3,7602150 100 11 9,76 10,123 16,6666667 168,716667 8,0663 16,6666667 134,4 4,8969200 100 11 9,88 8,7023 25 217,5575 7,1729 25 179,3 5,4621300 100 11 9,89 9,8485 33,3333333 328,283333 8,8131 33,3333333 293,8 4,9305500 100 11 10,24 11,253 50 562,65 10,2620 50 513,1 7,0786750 100 11 10,73 5,4372 150 815,58 4,7451 150 711,8 14,83071000 100 11 10,61 6,3208 150 948,12 5,2498 150 787,5 22,95001500 100 11 10,66 7,92 185 1465,2 6,6572 185 1231,6 33,37402000 100 11 10,83 5,5632 405,555556 2256,18667 4,4734 405,555556 1814,2 63,13924000 100 11 10,95 10,129 405,555556 4107,87222 8,3613 405,555556 3391,0 102,4144
Curva de calibracion
Patrón Abs0 02 0,1714 0,3436 0,5328 0,704
10 0,869
y = 0,0874xR² = 0,9997
-0,01
0,39
0,79
1,19
1,59
1,99
0 5 10 15
121!!
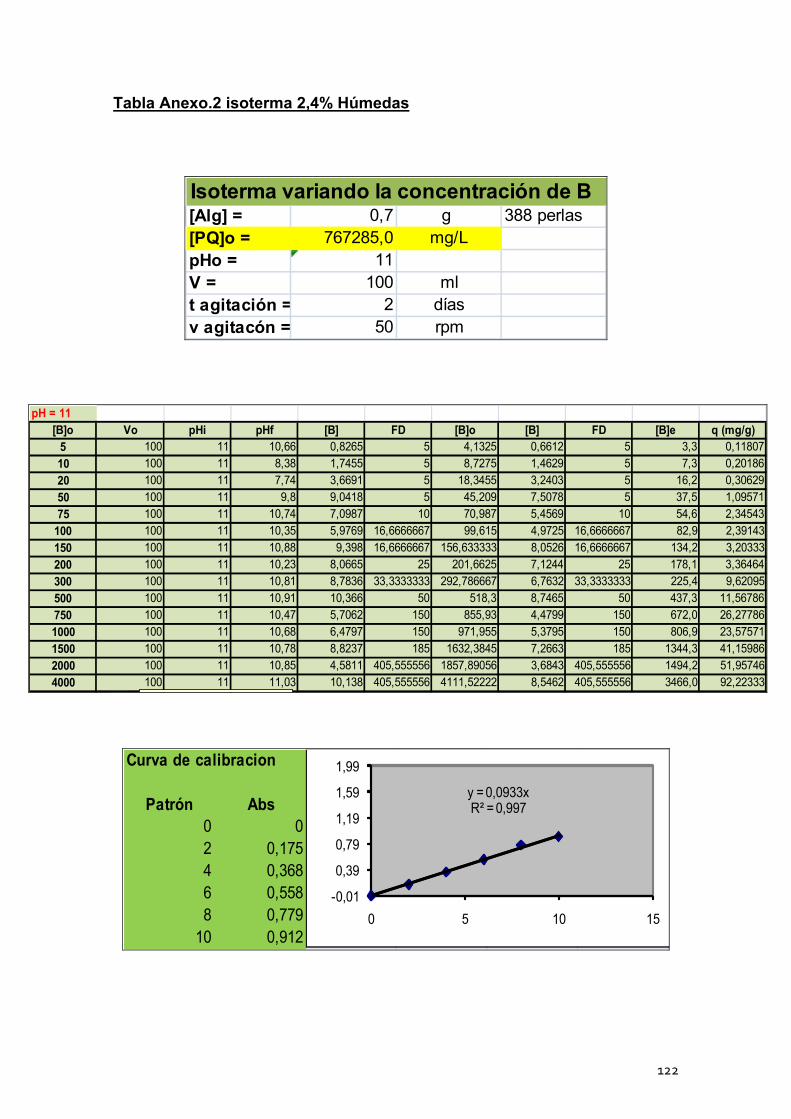
Tabla Anexo.2 isoterma 2,4% Húmedas
Isoterma variando la concentración de B[Alg] = 0,7 g 388 perlas[PQ]o = 767285,0 mg/LpHo = 11V = 100 mlt agitación = 2 díasv agitacón = 50 rpm
pH = 11
[B]o Vo pHi pHf [B] FD [B]o [B] FD [B]e q (mg/g)5 100 11 10,66 0,8265 5 4,1325 0,6612 5 3,3 0,1180710 100 11 8,38 1,7455 5 8,7275 1,4629 5 7,3 0,2018620 100 11 7,74 3,6691 5 18,3455 3,2403 5 16,2 0,3062950 100 11 9,8 9,0418 5 45,209 7,5078 5 37,5 1,0957175 100 11 10,74 7,0987 10 70,987 5,4569 10 54,6 2,34543
100 100 11 10,35 5,9769 16,6666667 99,615 4,9725 16,6666667 82,9 2,39143150 100 11 10,88 9,398 16,6666667 156,633333 8,0526 16,6666667 134,2 3,20333200 100 11 10,23 8,0665 25 201,6625 7,1244 25 178,1 3,36464300 100 11 10,81 8,7836 33,3333333 292,786667 6,7632 33,3333333 225,4 9,62095500 100 11 10,91 10,366 50 518,3 8,7465 50 437,3 11,56786750 100 11 10,47 5,7062 150 855,93 4,4799 150 672,0 26,277861000 100 11 10,68 6,4797 150 971,955 5,3795 150 806,9 23,575711500 100 11 10,78 8,8237 185 1632,3845 7,2663 185 1344,3 41,159862000 100 11 10,85 4,5811 405,555556 1857,89056 3,6843 405,555556 1494,2 51,957464000 100 11 11,03 10,138 405,555556 4111,52222 8,5462 405,555556 3466,0 92,22333
Curva de calibracion
Patrón Abs0 02 0,1754 0,3686 0,5588 0,779
10 0,912
y = 0,0933xR² = 0,997
-0,01
0,39
0,79
1,19
1,59
1,99
0 5 10 15
122!!

Tabla Anexo.3 isoterma 1,6% Húmedas
Isoterma variando la concentración de B[Alg] = 0,7 g 388 perlas[PQ]o = 767285,0 mg/LpHo = 11V = 100 mlt agitación = 2 díasv agitacón = 50 rpm
pH = 11[B]o Vo pHi pHf [B] FD [B]o [B] FD [B]e q (mg/g)
5 100 11 7,01 0,9996 5 4,998 0,8250 5 4,1 0,1247110 100 11 7,54 1,9524 5 9,762 1,6107 5 8,1 0,2440720 100 11 8,94 4,0566 5 20,283 2,7297 5 13,6 0,9477950 100 11 9,38 10,061 5 50,305 6,9542 5 34,8 2,2191475 100 11 9,62 7,5498 10 75,498 5,1119 10 51,1 3,48271
100 100 11 9,88 6,07 16,6666667 101,166667 4,1920 16,6666667 69,9 4,47143150 100 11 10,61 9,4946 16,6666667 158,243333 6,6028 16,6666667 110,0 6,88524200 100 11 10,14 8,0718 25 201,795 5,6406 25 141,0 8,68286300 100 11 10,48 8,9618 33,3333333 298,726667 6,4566 33,3333333 215,2 11,92952500 100 11 11,22 10,27 50 513,5 7,7974 50 389,9 17,66143750 100 11 11,4 5,805 150 870,75 4,6482 150 697,2 24,788571000 100 11 11,11 6,6094 150 991,41 5,1889 150 778,3 30,439291500 100 11 11,03 8,9479 185 1655,3615 7,2667 185 1344,3 44,431712000 100 11 10,22 4,7651 405,555556 1932,51278 3,8335 405,555556 1554,7 53,973654000 100 11 10,88 9,4389 405,555556 3827,99833 7,8740 405,555556 3193,3 90,66484
Curva de calibracion
Patrón Abs0 0,0012 0,1864 0,3656 0,5508 0,732
10 0,911
y = 0,0914xR² = 1
-0,010
0,390
0,790
1,190
1,590
1,990
0 5 10 15
123!!

Tabla Anexo.4 isoterma 3,2% Secas
Isoterma variando la concentración de B[Alg] = g [PQ]o = mg/LpHo =V = mlt agitación = díasv agitacón = rpm50
0,7767285,0
111002
pH = 11[B]o Vo pHi pHf [B] FD [B]o [B] FD [B]e q (mg/g)
5 100 11 8,81 0,9226 5 4,613 0,5805 5 2,9 0,244410 100 11 10,43 1,9433 5 9,7165 1,0966 5 5,5 0,604820 100 11 9,13 3,8656 5 19,328 2,9018 5 14,5 0,688450 100 11 10,15 9,1866 5 45,933 7,6514 5 38,3 1,096675 100 11 10,07 7,3743 10 73,743 5,6173 10 56,2 2,5100
100 100 11 9,87 5,9984 16,6666667 99,9733333 4,7321 16,6666667 78,9 3,0150150 100 11 10 8,9384 16,6666667 148,973333 7,2673 16,6666667 121,1 3,9788200 100 11 10,29 7,6583 25 191,4575 6,2502 25 156,3 5,0289300 100 11 10,55 9,2263 33,3333333 307,543333 7,5727 33,3333333 252,4 7,8743500 100 11 10,67 10,544 50 527,2 9,1121 50 455,6 10,2279750 100 11 10,64 4,9868 150 748,02 4,4321 150 664,8 11,88641000 100 11 10,81 6,5894 150 988,41 5,6151 150 842,3 20,87791500 100 11 10,79 7,8893 185 1459,5205 6,9751 185 1290,4 24,16102000 100 11 10,89 4,9862 405,555556 2022,18111 4,4082 405,555556 1787,8 33,48734000 100 11 10,93 10,585 405,555556 4292,80556 9,2812 405,555556 3764,0 75,5376
Curva de calibracion
Patrón Abs0 0,0012 0,184 0,3756 0,5598 0,747
10 0,922
y = 0,0928xR² = 0,9998
-0,01
0,39
0,79
1,19
1,59
1,99
0 2 4 6 8 10 12
124!!

Tabla Anexo.5 isoterma 2,4% Secas
Isoterma variando la concentración de B[Alg] = g [PQ]o = mg/LpHo =V = mlt agitación = díasv agitacón = rpm
0,7
10011
250
767285,0
pH = 11[B]o Vo pHi pHf [B] FD [B]o [B] FD [B]e q (mg/g)
5 100 11 9,53 0,5723 5 2,8615 0,0235 5 0,1 0,392010 100 11 8,82 1,6359 5 8,1795 0,8915 5 4,5 0,531720 100 11 10,25 3,4778 5 17,389 1,7328 5 8,7 1,246450 100 11 10,3 9,5324 5 47,662 5,9595 5 29,8 2,552175 100 11 10,9 7,2689 10 72,689 4,9597 10 49,6 3,2989
100 100 11 10,79 5,1347 16,6666667 85,5783333 3,6490 16,6666667 60,8 3,5374150 100 11 10,75 9,119 16,6666667 151,983333 7,1910 16,6666667 119,9 4,5905200 100 11 10,42 7,8417 25 196,0425 6,1143 25 152,9 6,1693300 100 11 11,61 8,9039 33,3333333 296,796667 7,5700 33,3333333 252,3 6,3519500 100 11 10,82 9,7207 50 486,035 8,6481 50 432,4 7,6614750 100 11 11,63 5,1069 150 766,035 4,4707 150 670,6 13,63291000 100 11 11,35 6,0691 150 910,365 5,3609 150 804,1 15,17571500 100 11 11,28 8,5535 185 1582,3975 7,7516 185 1434,0 21,19312000 100 11 11,17 4,4059 405,555556 1786,83722 3,8672 405,555556 1568,4 31,21044000 100 11 11,04 9,0822 405,555556 3683,33667 8,1039 405,555556 3286,6 56,6793
Curva de calibracion
Patrón Abs0 02 0,1824 0,3656 0,5468 0,719
10 0,819
y = 0,0899xR² = 0,9998
-0,01
0,39
0,79
1,19
1,59
1,99
0 5 10 15
125!!

Tabla Anexo.6 isoterma 1,6% Secas
[Alg] = g [PQ]o = mg/LpHo =V = mlt agitación = díasv agitacón = rpm
250
Isoterma variando la concentración de B0,7
767285,011100
pH = 11[B]o Vo pHi pHf [B] FD [B]o [B] FD [B]e q (mg/g)
5 100 11 9,43 0,8924 5 4,462 0,4842 5 2,4 0,291610 100 11 10,95 1,9233 5 9,6165 1,1475 5 5,7 0,554120 100 11 10,87 3,789 5 18,945 2,3624 5 11,8 1,019050 100 11 10,79 9,7724 5 48,862 6,9946 5 35,0 1,984175 100 11 10,7 7,4207 10 74,207 5,6037 10 56,0 2,5957
100 100 11 10,82 5,9651 16,6666667 99,4183333 4,5930 16,6666667 76,6 3,2669150 100 11 10,81 9,7764 16,6666667 162,94 8,0809 16,6666667 134,7 4,0369200 100 11 10,85 7,7892 25 194,73 6,5140 25 162,9 4,5543300 100 11 9,93 9,1523 33,3333333 305,076667 7,7930 33,3333333 259,8 6,4729500 100 11 11,08 9,9984 50 499,92 8,9831 50 449,2 7,2521750 100 11 11,01 5,4604 150 819,06 4,8266 150 724,0 13,58141000 100 11 11,12 6,2749 150 941,235 5,3958 150 809,4 18,83791500 100 11 11,4 9,2222 185 1706,107 8,4211 185 1557,9 21,17192000 100 11 11,12 5,0351 405,555556 2042,01278 4,6436 405,555556 1883,2 22,68214000 100 11 11,07 9,1857 405,555556 3725,31167 8,4201 405,555556 3414,8 44,3562
Curva de calibracion
Patrón Abs0 02 0,1834 0,3716 0,5518 0,730
10 0,905
y = 0,0911xR² = 0,9998
-0,01
0,39
0,79
1,19
1,59
1,99
0 5 10 15
126!!
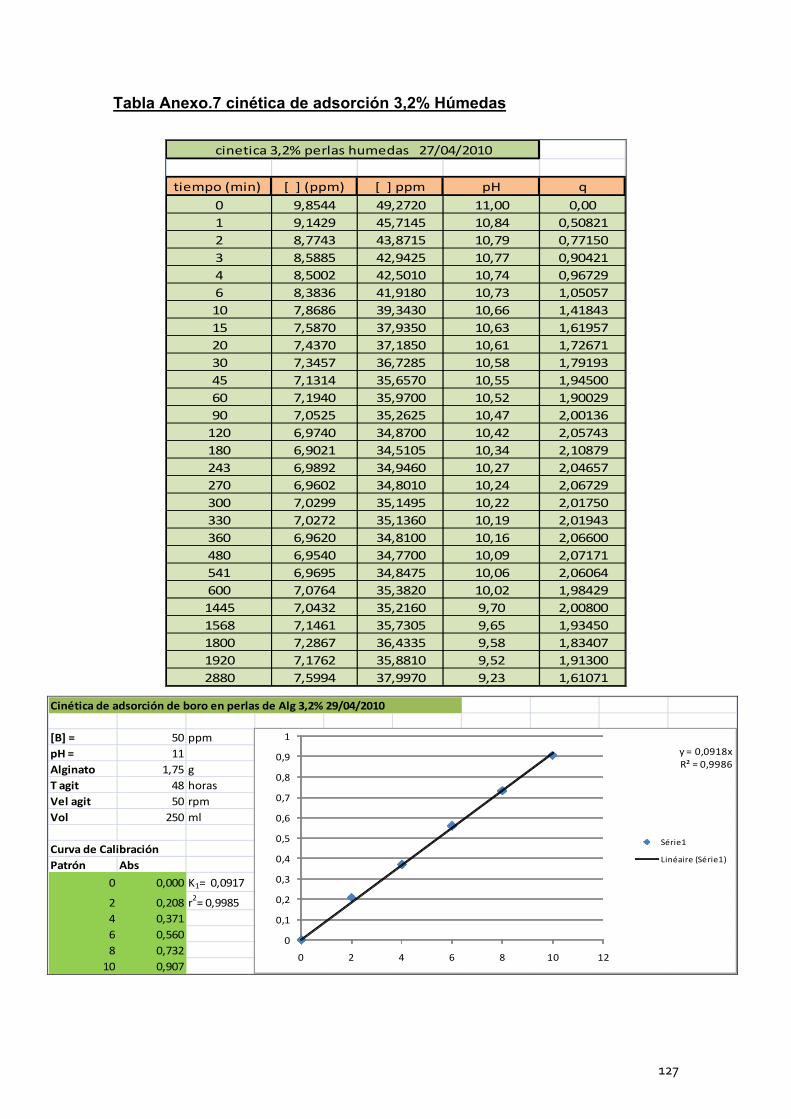
Tabla Anexo.7 cinética de adsorción 3,2% Húmedas
tiempo!(min)! [!!]!(ppm) [!!]!ppm pH q0 9,8544 49,2720 11,00 0,001 9,1429 45,7145 10,84 0,508212 8,7743 43,8715 10,79 0,771503 8,5885 42,9425 10,77 0,904214 8,5002 42,5010 10,74 0,967296 8,3836 41,9180 10,73 1,0505710 7,8686 39,3430 10,66 1,4184315 7,5870 37,9350 10,63 1,6195720 7,4370 37,1850 10,61 1,7267130 7,3457 36,7285 10,58 1,7919345 7,1314 35,6570 10,55 1,9450060 7,1940 35,9700 10,52 1,9002990 7,0525 35,2625 10,47 2,00136120 6,9740 34,8700 10,42 2,05743180 6,9021 34,5105 10,34 2,10879243 6,9892 34,9460 10,27 2,04657270 6,9602 34,8010 10,24 2,06729300 7,0299 35,1495 10,22 2,01750330 7,0272 35,1360 10,19 2,01943360 6,9620 34,8100 10,16 2,06600480 6,9540 34,7700 10,09 2,07171541 6,9695 34,8475 10,06 2,06064600 7,0764 35,3820 10,02 1,984291445 7,0432 35,2160 9,70 2,008001568 7,1461 35,7305 9,65 1,934501800 7,2867 36,4335 9,58 1,834071920 7,1762 35,8810 9,52 1,913002880 7,5994 37,9970 9,23 1,61071
cinetica!3,2%!perlas!humedas!!!27/04/2010
Cinética!de!adsorción!de!boro!en!perlas!de!Alg!3,2%!29/04/2010
[B]!= 50 ppmpH!= 11 !Alginato 1,75 gT!agit 48 horasVel!agit 50 rpmVol 250 ml
Curva!de!CalibraciónPatrón Abs
0 0,000 K1=!!0,0917
2 0,208 r2=!0,99854 0,3716 0,5608 0,73210 0,907
y!=!0,0918xR²!=!0,9986
0
0,1
0,2
0,3
0,4
0,5
0,6
0,7
0,8
0,9
1
0 2 4 6 8 10 12
Série1
Linéaire!(Série1)
127!!

Tabla Anexo.8 cinética de adsorción 2,4% Húmedas
tiempo!(min)! [!!]!(ppm) [!!]!(ppm) pH q0 9,9440 49,7200 11,00 0,001 8,8058 44,0290 10,77 0,813002 8,2460 41,2300 10,71 1,212863 7,9590 39,7950 10,67 1,417864 7,7042 38,5210 10,64 1,599865 7,4664 37,3320 10,61 1,7697110 6,8843 34,4215 10,54 2,1855015 6,6416 33,2080 10,51 2,3588620 6,4137 32,0685 10,49 2,5216431 6,1414 30,7070 10,46 2,7161445 6,0353 30,1765 10,43 2,7919360 5,8933 29,4665 10,40 2,89336
90 5,7689 28,8445 10,35 2,98221
120 5,8298 29,1490 10,31 2,93871
188 5,7590 28,7950 10,24 2,98929267 5,8101 29,0505 10,17 2,95279270 5,6936 28,4680 10,16 3,03600300 5,7165 28,5825 10,13 3,01964330 5,7648 28,8240 10,11 2,98514360 5,7572 28,7860 10,07 2,99057486 5,8235 29,1175 9,96 2,94321540 5,8159 29,0795 9,93 2,94864600 5,9359 29,6795 9,89 2,862931441 6,0756 30,3780 9,60 2,763141560 6,1584 30,7920 9,56 2,704001800 6,3116 31,5580 9,48 2,594571920 6,3828 31,9140 9,44 2,543712880 6,7294 33,6470 9,26 2,29614
cinetica!2,4%!perlas!humedas!!!04/05/2010
Cinética!de!adsorción!de!boro!en!perlas!de!Alg!2,4%!06/05/2010
[B]!= 50 ppmpH!= 11 !Alginato 1,75 gT!agit 48 horasVel!agit 50 rpmVol 250 ml
Curva!de!CalibraciónPatrón Abs
0 0,000 K1=!!0,0909
2 0,182 r2=!0,99984 0,3686 0,5508 0,73010 0,901
y!=!0,0908xR²!=!0,9998
0
0,1
0,2
0,3
0,4
0,5
0,6
0,7
0,8
0,9
1
0 5 10 15
Série1
Linéaire!(Série1)
128!!

Tabla Anexo.9 cinética de adsorción 1,6% Húmedas
tiempo!(min)! [!!]!(ppm) [!!]!(ppm) pH q0 9,8601 49,3005 11,00 0,001 8,4889 42,4445 10,79 0,979432 8,2249 41,1245 10,76 1,168003 8,0290 40,1450 10,74 1,307934 7,9193 39,5965 10,72 1,386295 7,6937 38,4685 10,71 1,5474310 7,0815 35,4075 10,67 1,9847115 6,9352 34,6760 10,65 2,0892120 6,7634 33,8170 10,63 2,2119330 6,5082 32,5410 10,60 2,3942145 6,4957 32,4785 10,57 2,40314
60 6,3360 31,6800 10,53 2,51721
90 6,3105 31,5525 10,48 2,53543
120 6,3230 31,6150 10,45 2,52650170 6,3318 31,6590 10,36 2,52021245 6,3216 31,6080 10,28 2,52750270 6,2628 31,3140 10,25 2,56950300 6,3702 31,8510 10,23 2,49279330 6,3105 31,5525 10,20 2,53543360 6,2934 31,4670 10,18 2,54764494 6,3249 31,6245 10,07 2,52514540 6,3536 31,7680 10,04 2,50464600 6,3901 31,9505 10,00 2,478571443 6,6268 33,1340 9,71 2,309501560 6,4999 32,4995 9,68 2,400141806 6,5874 32,9370 9,62 2,337641920 6,6791 33,3955 9,60 2,272143010 6,9588 34,7940 9,37 2,07236
cinetica!1,6%!perlas!humedas!6/05/2010
Cinética!de!adsorción!de!boro!en!perlas!de!Alg!1,6%!!!10/05/2010
[B]!= 50 ppmpH!= 11 !Alginato 1,75 gT!agit 48 horasVel!agit 50 rpmVol 250 ml
Curva!de!CalibraciónPatrón Abs
0 0,001 K1=!0,0895
2 0,178 r2=!0,99964 0,3586 0,5338 0,69610 0,877
y!=!0,0878xR²!=!0,9998
0
0,1
0,2
0,3
0,4
0,5
0,6
0,7
0,8
0,9
1
0 5 10 15
Série1
Linéaire!(Série1)
129!!

Tabla Anexo.10 cinética de adsorción 3,2% secas
tiempo!(min)! [!!]!(ppm) [!!]!(ppm) pH q0 9,7973 48,9865 11,00 0,001 9,9684 49,8420 10,96 "0,122212 9,8381 49,1905 10,96 "0,029143 9,8201 49,1005 10,95 "0,016294 9,9394 49,6970 10,95 "0,101505 9,8508 49,2540 10,94 "0,0382115 9,6961 48,4805 10,91 0,0722920 9,7417 48,7085 10,89 0,0397130 9,7004 48,5020 10,86 0,0692140 9,7184 48,5920 10,82 0,0563646 9,6505 48,2525 10,81 0,1048660 9,7154 48,5770 10,76 0,0585091 9,6566 48,2830 10,66 0,10050120 9,3330 46,6650 10,57 0,33164180 8,7954 43,9770 10,43 0,71564245 8,5757 42,8785 10,31 0,87257300 8,1785 40,8925 10,22 1,15629360 8,0847 40,4235 10,15 1,22329480 7,7782 38,8910 10,04 1,44221600 7,5661 37,8305 9,95 1,593711440 7,3441 36,7205 9,61 1,752291560 7,4015 37,0075 9,59 1,711291800 7,4980 37,4900 9,52 1,642361920 7,5747 37,8735 9,48 1,587572880 7,8983 39,4915 9,23 1,35643
cinetica!3,2%!perlas!secas
Cinética!de!adsorción!de!boro!en!perlas!de!Alg!Secas!3,2%!!19/04/2010
[B]!= 50 ppmpH!= 11 !Alginato 1,75 gT!agit 48 horasVel!agit 50 rpmVol 250 ml
Curva!de!CalibraciónPatrón Abs
0 0,001 K1=!!0,0928
2 0,180 r2=!0,99984 0,3756 0,5598 0,74710 0,922
y!=!0,0928xR²!=!0,9998
0
0,1
0,2
0,3
0,4
0,5
0,6
0,7
0,8
0,9
1
0 5 10 15
Série1
Linéaire!(Série1)
130!!

Tabla Anexo.11 cinética de adsorción 2,4% secas
tiempo!(min)! [!!]!(ppm) [!!]!(ppm) pH q0 9,9948 49,9740 11,00 0,001 10,0260 50,1300 10,97 "0,022292 9,9310 49,6550 10,97 0,045573 10,0260 50,1300 10,96 "0,022294 9,9087 49,5435 10,95 0,061505 9,9356 49,6780 10,95 0,0422910 9,9000 49,5000 10,92 0,0677115 9,7384 48,6920 10,88 0,1831420 9,7079 48,5395 10,85 0,2049330 9,6959 48,4795 10,78 0,2135045 9,4691 47,3455 10,70 0,3755060 9,2997 46,4985 10,61 0,4965091 9,1391 45,6955 10,46 0,61121120 8,4662 42,3310 10,36 1,09186180 7,8901 39,4505 10,21 1,50336240 7,4527 37,2635 10,11 1,81579270 7,4500 37,2500 10,08 1,81771300 7,3972 36,9860 10,04 1,85543330 7,2533 36,2665 10,00 1,95821358 7,2505 36,2525 9,97 1,96021480 7,0334 35,1670 9,87 2,11529540 6,9131 34,5655 9,83 2,20121600 6,8705 34,3525 9,78 2,231641440 6,7474 33,7370 9,50 2,319571560 6,7956 33,9780 9,47 2,285141800 6,7590 33,7950 9,37 2,311291860 6,7817 33,9085 9,36 2,295071920 6,9191 34,5955 9,33 2,196932981 7,2958 36,4790 9,07 1,92786
cinetica!2,4%!perlas!secas!!5/05/2010
[B]!= 50 ppmpH!= 11 !Alginato 1,75 gT!agit 48 horasVel!agit 50 rpmVol 250 ml
Patrón Abs
0 0 K1=!!0,0879
2 0,166 r2=!0,99984 0,3536 0,5278 0,70810 0,878
Curva!de!Calibración
Cinética!de!adsorción!de!boro!en!perlas!de!Alg!Secas!2,4%
y!=!0,0880xR²!=!0,9998
0
0,1
0,2
0,3
0,4
0,5
0,6
0,7
0,8
0,9
1
0 5 10 15
Abs
131!!

Tabla Anexo.12 cinética de adsorción 1,6% secas
tiempo!(min)! [!!]!(ppm) [!!]!(ppm) pH q0 9,6906 48,4530 11,00 0,001 9,7335 48,6675 10,99 "0,030642 9,7732 48,8660 10,98 "0,059003 9,6657 48,3285 10,97 0,017794 9,7777 48,8885 10,97 "0,062215 9,6860 48,4300 10,96 0,0032910 9,6255 48,1275 10,95 0,0465015 9,7290 48,6450 10,92 "0,0274320 9,7244 48,6220 10,90 "0,0241430 9,7583 48,7915 10,85 "0,0483645 9,6129 48,0645 10,78 0,0555060 9,4728 47,3640 10,72 0,1555790 9,3201 46,6005 10,61 0,26464120 8,9899 44,9495 10,52 0,50050180 8,5020 42,5100 10,39 0,84900240 8,3041 41,5205 10,31 0,99036300 7,9834 39,9170 10,25 1,21943368 7,7670 38,8350 10,18 1,37400480 7,5696 37,8480 10,10 1,51500600 7,4833 37,4165 10,02 1,576641440 7,2506 36,2530 9,70 1,742861560 7,2199 36,0995 9,64 1,764791800 7,3902 36,9510 9,57 1,64314
1920 7,4214 37,1070 9,55 1,62086
2891 7,6504 38,2520 9,30 1,45729
cinetica!1,6%!perlas!secas
[B]!= 50 ppmpH!= 11 !Alginato 1,75 gT!agit 48 horasVel!agit 50 rpmVol 250 ml
Curva!de!CalibraciónPatrón Abs
0 0,000 K1=!!0,0901
2 0,182 r2=0,99994 0,3606 0,5418 0,72610 0,895
Cinética!de!adsorción!de!boro!en!perlas!de!Alg!Secas!1,6%
y!=!0,0901xR²!=!0,9999
0,000
0,100
0,200
0,300
0,400
0,500
0,600
0,700
0,800
0,900
1,000
0 5 10 15
132!!
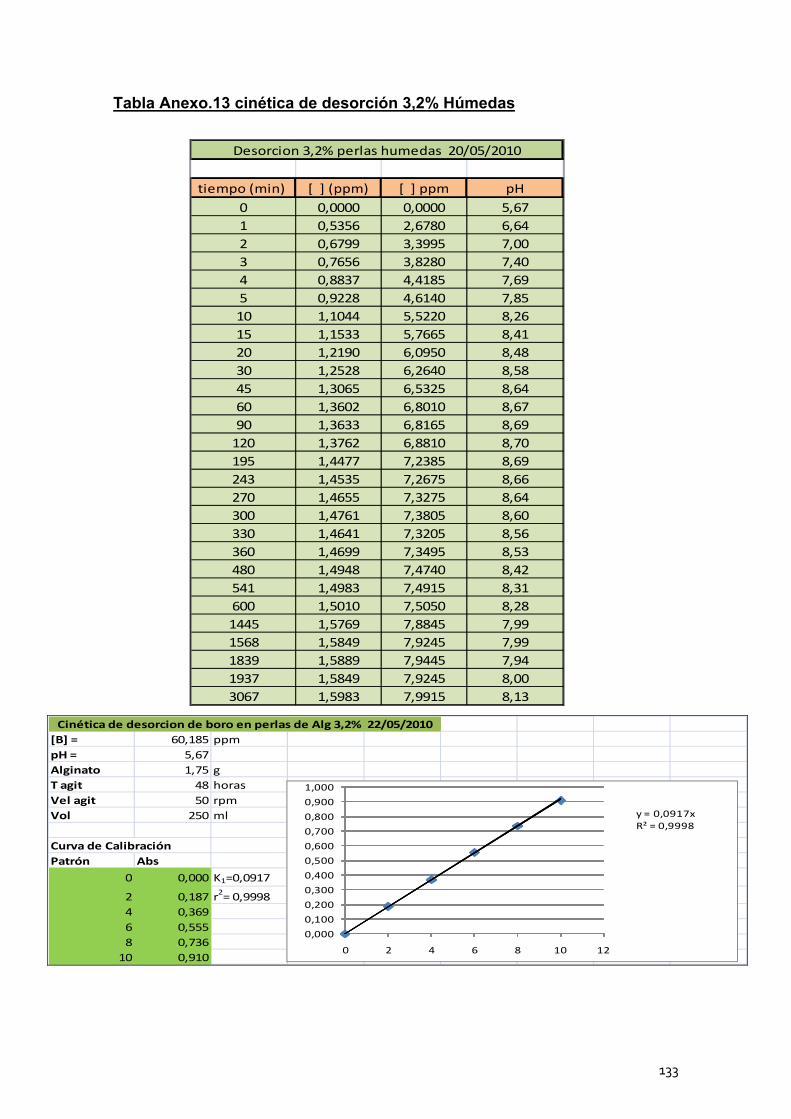
Tabla Anexo.13 cinética de desorción 3,2% Húmedas
tiempo!(min)! [!!]!(ppm) [!!]!ppm pH0 0,0000 0,0000 5,671 0,5356 2,6780 6,642 0,6799 3,3995 7,003 0,7656 3,8280 7,404 0,8837 4,4185 7,695 0,9228 4,6140 7,8510 1,1044 5,5220 8,2615 1,1533 5,7665 8,4120 1,2190 6,0950 8,4830 1,2528 6,2640 8,5845 1,3065 6,5325 8,6460 1,3602 6,8010 8,6790 1,3633 6,8165 8,69120 1,3762 6,8810 8,70195 1,4477 7,2385 8,69243 1,4535 7,2675 8,66270 1,4655 7,3275 8,64300 1,4761 7,3805 8,60330 1,4641 7,3205 8,56360 1,4699 7,3495 8,53480 1,4948 7,4740 8,42541 1,4983 7,4915 8,31600 1,5010 7,5050 8,281445 1,5769 7,8845 7,991568 1,5849 7,9245 7,991839 1,5889 7,9445 7,941937 1,5849 7,9245 8,003067 1,5983 7,9915 8,13
Desorcion!3,2%!perlas!humedas!!20/05/2010
[B]!= 60,185 ppmpH!= 5,67 !Alginato 1,75 gT!agit 48 horasVel!agit 50 rpmVol 250 ml
Curva!de!CalibraciónPatrón Abs
0 0,000 K1=0,0917
2 0,187 r2=!0,99984 0,3696 0,5558 0,73610 0,910
Cinética!de!desorcion!de!boro!en!perlas!de!Alg!3,2%!!22/05/2010
y!=!0,0917xR²!=!0,9998
0,000
0,100
0,200
0,300
0,400
0,500
0,600
0,700
0,800
0,900
1,000
0 2 4 6 8 10 12
133!!

Tabla Anexo.14 cinética de desorción 2,4% Húmedas
tiempo!(min)! [!!]!(ppm) [!!]!ppm pH0 0,0000 0,0000 6,691 0,2495 1,2475 8,462 0,3287 1,6435 8,553 0,3763 1,8815 8,614 0,4537 2,2685 8,655 0,4623 2,3115 8,6710 0,5764 2,8820 8,7615 0,6285 3,1425 8,8020 0,7068 3,5340 8,8430 0,7399 3,6995 8,8560 0,7340 3,6700 8,8890 0,7322 3,6610 8,87120 0,7843 3,9215 8,80180 0,7924 3,9620 8,71390 0,8418 4,2090 8,46420 0,8956 4,4780 8,40450 0,9029 4,5145 8,37540 0,9187 4,5935 8,25600 0,8839 4,4195 8,211562 0,8929 4,4645 8,081800 0,9355 4,6775 8,091920 0,9735 4,8675 8,122880 1,0378 5,1889 8,11
Desorcion!2,4!%!perlas!humedas!!24/05/2010
[B]!= 46,38 ppmpH!= 6,69 !Alginato 1,75 gT!agit 48 horasVel!agit 50 rpmVol 250 ml
Curva!de!CalibraciónPatrón Abs
0 0,000 K1=0,0899
2 0,179 r2=!14 0,3576 0,5388 0,72110 0,898
Cinética!de!desorcion!de!boro!en!perlas!de!Alg!2,4%!!26/05/2010
y!=!0,0898xR²!=!1
0,000
0,100
0,200
0,300
0,400
0,500
0,600
0,700
0,800
0,900
1,000
0 2 4 6 8 10 12
134!!
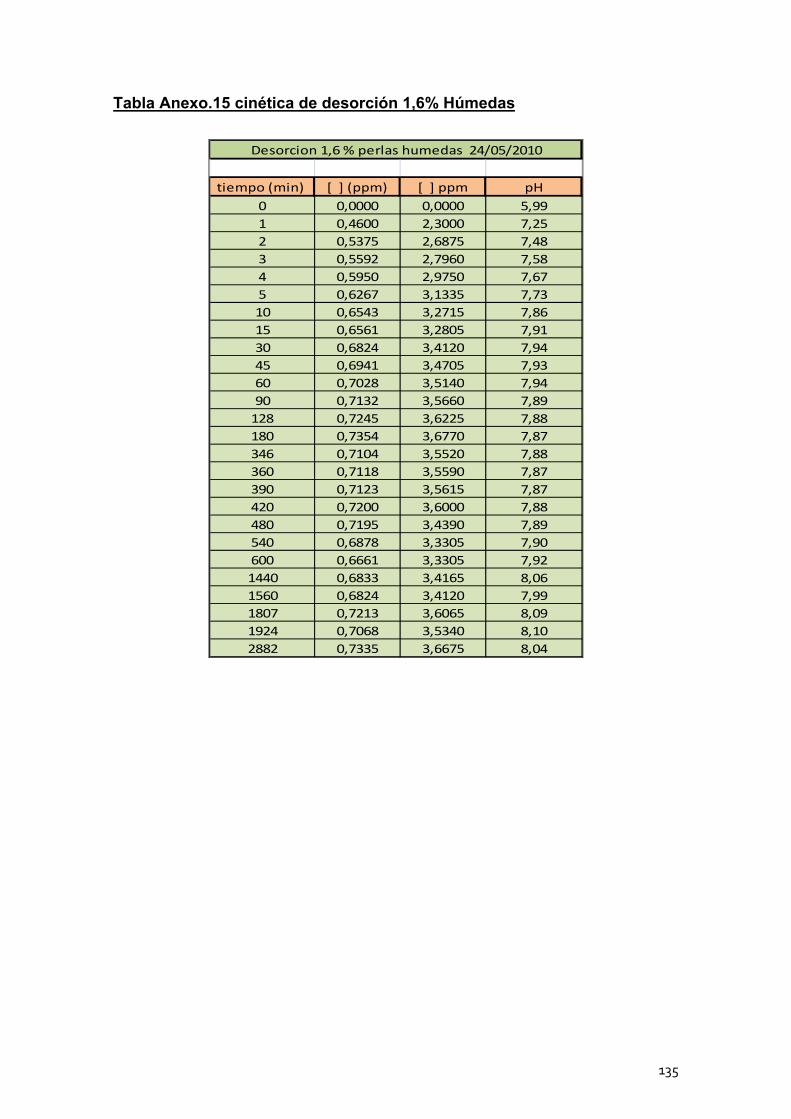
Tabla Anexo.15 cinética de desorción 1,6% Húmedas
tiempo!(min)! [!!]!(ppm) [!!]!ppm0 0,0000 0,00001 0,4600 2,30002 0,5375 2,68753 0,5592 2,79604 0,5950 2,97505 0,6267 3,133510 0,6543 3,271515 0,6561 3,280530 0,6824 3,412045 0,6941 3,470560 0,7028 3,514090 0,7132 3,5660128 0,7245 3,6225180 0,7354 3,6770346 0,7104 3,5520360 0,7118 3,5590390 0,7123 3,5615420 0,7200 3,6000480 0,7195 3,4390540 0,6878 3,3305600 0,6661 3,33051440 0,6833 3,41651560 0,6824 3,41201807 0,7213 3,60651924 0,7068 3,53402882 0,7335 3,6675
Desorcion!1,6!%!perlas!humedas!!
pH5,997,257,487,587,677,737,867,917,947,937,947,897,887,877,887,877,877,887,897,907,928,067,998,098,108,04
24/05/2010
135!!

[B]!= 47,1775 ppmpH!= 5,99 !Alginato 1,75 gT!agit 48 horasVel!agit 50 rpmVol 250 ml
Curva!de!CalibraciónPatrón Abs
0 0,000 K1=0,0899
2 0,179 r2=!14 0,3576 0,5388 0,72110 0,898
Cinética!de!desorcion!de!boro!en!perlas!de!Alg!1,6%!!26/05/2010
y!=!0,0898xR²!=!1
0,000
0,100
0,200
0,300
0,400
0,500
0,600
0,700
0,800
0,900
1,000
0 2 4 6 8 10 12
Tabla Anexo.16 cinética de desorción 3,2% secas
tiempo!(min)! [!!]!(ppm) [!!]!ppm0 0,0000 0,00001 0,2600 1,30002 0,3538 1,76903 0,3515 1,75754 0,4755 2,37755 0,6394 3,197010 0,6440 3,220015 0,8758 4,379021 0,6747 3,373530 0,5558 2,779045 0,7495 3,747560 0,7355 3,677590 0,9487 4,7435120 0,9352 4,6760180 1,1223 5,6115240 1,3220 6,6100272 1,0230 5,1150300 0,9942 4,9710332 1,2886 6,4430363 1,2872 6,4360480 1,5272 7,3740540 1,4748 7,9055600 1,5811 7,90551442 1,9024 9,51201560 1,7506 8,75301800 1,9089 9,54451920 1,9744 9,87202880 2,2419 11,2095
Desorcion!3,2!%!perlas!Secas!!
pH6,236,616,756,997,598,198,849,029,159,249,419,439,449,459,449,429,409,399,419,379,319,289,248,848,608,378,358,31
26/05/2010
136!!
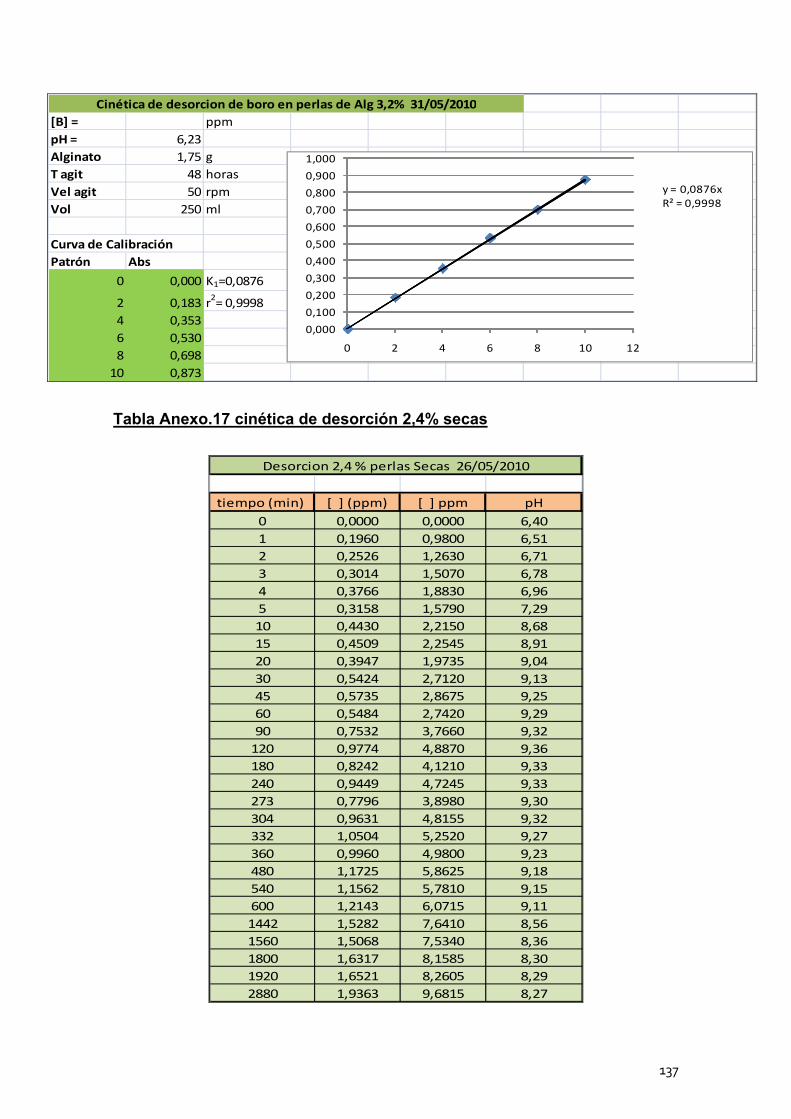
[B]!= ppmpH!= 6,23 !Alginato 1,75 gT!agit 48 horasVel!agit 50 rpmVol 250 ml
Curva!de!CalibraciónPatrón Abs
0 0,000 K1=0,0876
2 0,183 r2=!0,99984 0,3536 0,5308 0,69810 0,873
Cinética!de!desorcion!de!boro!en!perlas!de!Alg!3,2%!!31/05/2010
y!=!0,0876xR²!=!0,9998
12
0,000
0,100
0,200
0,300
0,400
0,500
0,600
0,700
0,800
0,900
1,000
0 2 4 6 8 10
Tabla Anexo.17 cinética de desorción 2,4% secas
tiempo!(min)! [!!]!(ppm) [!!]!ppm0 0,0000 0,00001 0,1960 0,98002 0,2526 1,26303 0,3014 1,50704 0,3766 1,88305 0,3158 1,579010 0,4430 2,215015 0,4509 2,254520 0,3947 1,973530 0,5424 2,712045 0,5735 2,867560 0,5484 2,742090 0,7532 3,7660120 0,9774 4,8870180 0,8242 4,1210240 0,9449 4,7245273 0,7796 3,8980304 0,9631 4,8155332 1,0504 5,2520360 0,9960 4,9800480 1,1725 5,8625540 1,1562 5,7810600 1,2143 6,07151442 1,5282 7,64101560 1,5068 7,53401800 1,6317 8,15851920 1,6521 8,26052880 1,9363 9,6815
Desorcion!2,4!%!perlas!Secas!!
pH6,406,516,716,786,967,298,688,919,049,139,259,299,329,369,339,339,309,329,279,239,189,159,118,568,368,308,298,27
26/05/2010
137!!
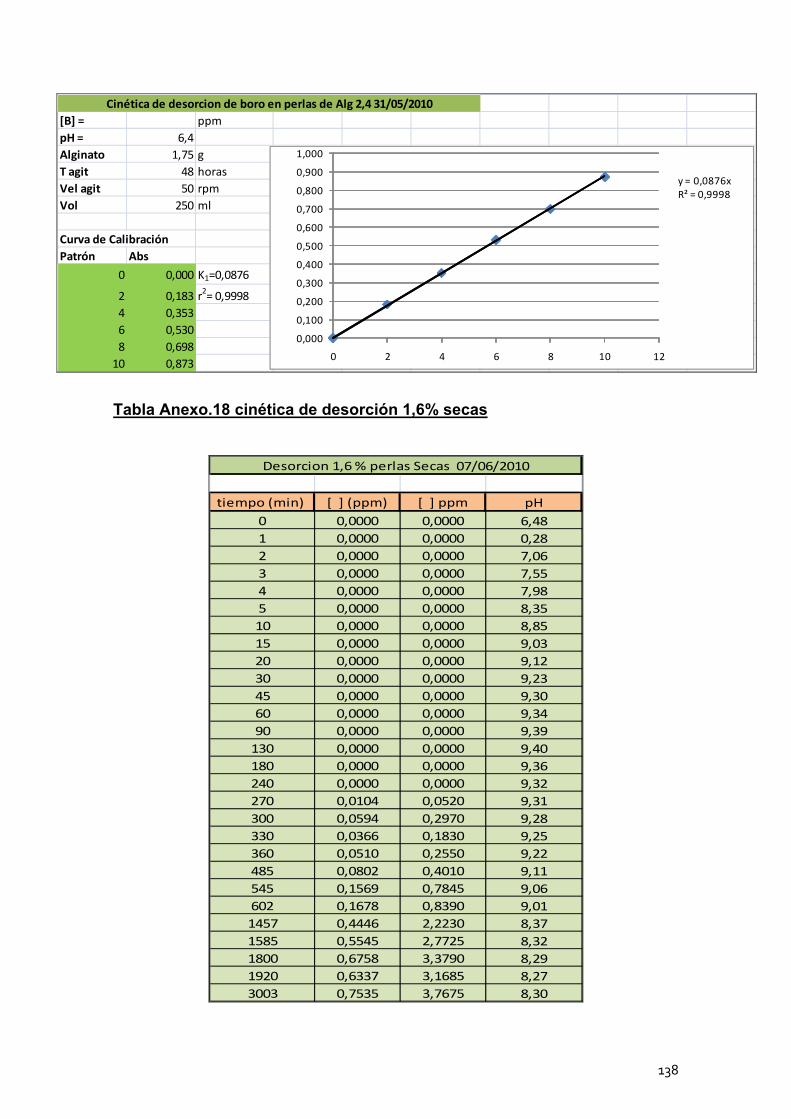
[B]!= ppmpH!= 6,4 !Alginato 1,75 gT!agit 48 horasVel!agit 50 rpmVol 250 ml
Curva!de!CalibraciónPatrón Abs
0 0,000 K1=0,0876
2 0,183 r2=!0,99984 0,3536 0,5308 0,69810 0,873
Cinética!de!desorcion!de!boro!en!perlas!de!Alg!2,4!31/05/2010
y!=!0,0876xR²!=!0,9998
12
0,000
0,100
0,200
0,300
0,400
0,500
0,600
0,700
0,800
0,900
1,000
0 2 4 6 8 10
Tabla Anexo.18 cinética de desorción 1,6% secas
tiempo!(min)! [!!]!(ppm) [!!]!ppm0 0,0000 0,00001 0,0000 0,00002 0,0000 0,00003 0,0000 0,00004 0,0000 0,00005 0,0000 0,000010 0,0000 0,000015 0,0000 0,000020 0,0000 0,000030 0,0000 0,000045 0,0000 0,000060 0,0000 0,000090 0,0000 0,0000130 0,0000 0,0000180 0,0000 0,0000240 0,0000 0,0000270 0,0104 0,0520300 0,0594 0,2970330 0,0366 0,1830360 0,0510 0,2550485 0,0802 0,4010545 0,1569 0,7845602 0,1678 0,83901457 0,4446 2,22301585 0,5545 2,77251800 0,6758 3,37901920 0,6337 3,16853003 0,7535 3,7675
Desorcion!1,6!%!perlas!Secas!!
pH6,480,287,067,557,988,358,859,039,129,239,309,349,399,409,369,329,319,289,259,229,119,069,018,378,328,298,278,30
07/06/2010
138!!

139!!
[B]!= 46,8415 ppmpH!= 6,48 !Alginato 1,75 gT!agit 48 horasVel!agit 50 rpmVol 250 ml
Curva!de!CalibraciónPatrón Abs
0 0,000 K1=0,0822
2 0,167 r2=!0,99994 0,3266 0,4958 0,65510 0,823
Cinética!de!desorcion!de!boro!en!perlas!de!Alg!1,6!!12/06/2010
y!=!0,0822xR²!=!0,9999
12
0,000
0,100
0,200
0,300
0,400
0,500
0,600
0,700
0,800
0,900
0 2 4 6 8 10


















