
REVISTA CHILENA DE EPILEPSIArevistachilenadeepilepsia.cl/wp-content/uploads/2015/04/201403_re... ·...
54
1 REVISTA CHILENA DE EPILEPSIA Publicación Oficial de la Sociedad de Epileptología de Chile Capítulo Chileno de la ILAE http: //www.epilepsiadechile.com / E-mail: [email protected] Año 14, Nº 3, Diciembre 2014 ISSN 0717-5337 versión impresa / ISSN 0719-5397 versión en línea Editorial / Editorial Epilepsia refractaria. Epilepsias de difícil control. Refractory epilepsies. Difficult epilepsies control. Roberto Caraballo 3 Trabajos Originales / Original Works • Tratamiento compasivo y de acompañamiento con cannabis en niños con epilepsia resistente. Una presentación de 2 pacientes y revisión de la literatura. Compassionate and accompaniment treatment with cannabis in children with refractory epilepsy. A presentation in two children and update. Marcelo Devilat, Carla Manterola, Juan Luis Moya 6 • Experiencia en Cirugía de Epilepsia Paliativa en Instituto de Neurocirugía Asenjo: Estimulador de Nervio Vago (VNS). Experience in Epilepsy Surgery Palliative Asenjo Institute of Neurosurgery: Vagus Nerve Stimulator. Hernán Acevedo, Osvaldo Olivares, Bárbara Garcés, Lientur Taha, Viviana Venegas 18 • Caracterización de un grupo de pacientes con convulsiones febriles. Characterization of a group of patients with febrile seizures. Patricia Alfaro, Marcelo Devilat 24 Actualizaciones / Updates • Actualización del Síndrome de West. Update West Syndrome Vanessa García, Sergio Meneses, Perla David 34 • Actualización del Síndrome de Lennox-Gastaut. Update Lennox-Gastaut Syndrome Vanessa García, Sergio Meneses, Perla David 42 Cronica / Chronicle • Cursos congresos y Actividades 2014. Congress Courses and Activities 2014 46 • Noticias poéticas 25.12.14. Poetic news 25.12.14 47 • Nuevos Socios. New members 48 • Jornadas Invernales de Epilepsia 2015. Winter days of epilepsy 49 • Declaración de intereses. Declaration of interests 51 • Fe de erratas. Erratum 53 • Sugerencias para contribuciones a los autores. Suggestion to authors of contributions 54
Transcript of REVISTA CHILENA DE EPILEPSIArevistachilenadeepilepsia.cl/wp-content/uploads/2015/04/201403_re... ·...

1
REVISTA CHILENA DE EPILEPSIAPublicación Oficial de la Sociedad de Epileptología de Chile
Capítulo Chileno de la ILAEhttp: //www.epilepsiadechile.com / E-mail: [email protected]
Año 14, Nº 3, Diciembre 2014ISSN 0717-5337 versión impresa / ISSN 0719-5397 versión en línea
Editorial / EditorialEpilepsia refractaria. Epilepsias de difícil control.Refractory epilepsies. Difficult epilepsies control.Roberto Caraballo 3
Trabajos Originales / Original Works• Tratamiento compasivo y de acompañamiento con cannabis en niños con epilepsia resistente. Una presentación de 2 pacientes y revisión de la literatura. Compassionate and accompaniment treatment with cannabis in children with refractory epilepsy. A presentation in two children and update. Marcelo Devilat, Carla Manterola, Juan Luis Moya 6• Experiencia en Cirugía de Epilepsia Paliativa en Instituto de Neurocirugía Asenjo: Estimulador de Nervio Vago (VNS). Experience in Epilepsy Surgery Palliative Asenjo Institute of Neurosurgery: Vagus Nerve Stimulator. Hernán Acevedo, Osvaldo Olivares, Bárbara Garcés, Lientur Taha, Viviana Venegas 18• Caracterización de un grupo de pacientes con convulsiones febriles. Characterization of a group of patients with febrile seizures. Patricia Alfaro, Marcelo Devilat 24
Actualizaciones / Updates • Actualización del Síndrome de West. Update West Syndrome Vanessa García, Sergio Meneses, Perla David 34• Actualización del Síndrome de Lennox-Gastaut. Update Lennox-Gastaut Syndrome Vanessa García, Sergio Meneses, Perla David 42
Cronica / Chronicle• Cursos congresos y Actividades 2014. Congress Courses and Activities 2014 46• Noticias poéticas 25.12.14. Poetic news 25.12.14 47• Nuevos Socios. New members 48• Jornadas Invernales de Epilepsia 2015. Winter days of epilepsy 49• Declaración de intereses. Declaration of interests 51• Fe de erratas. Erratum 53• Sugerencias para contribuciones a los autores. Suggestion to authors of contributions 54

2
Revista Chilena de Epilepsia Año 14, Nº 3, Diciembre 2014
Directorio ILAE 2013 - 2017
PresidentEmilio Perucca
Vice PresidentTatsuya Tanaka
Secretary- GeneralHelen Cross
TreasurerSam Wiebe
Past PresidentSalomon Moshé
IBE PresidentAthanasios Covanis
IBE Secretary-GeneralSari Tervonen
IBE TreasurerRobert Cole
Dirección:Av. Providencia 2315, Of. 215Fonos: 2231 0172, 2235 1470, Fax: 2234 0671Providencia, Santiago, Chile.E-Mail: [email protected] [email protected]
Diseño Gráfico:Juan Silva / [email protected] / 9799 5964
SOCIEDAD DE EPILEPTOLOGÍA DE CHILE
Capítulo Chileno de la Liga Internacional contra la EpilepsiaFundada el 13 de Marzo de 1999
Directorio de la Sociedad de Epileptología de Chile
PresidenteDr. Darío Ramírez
VicepresidenteDr. Cayetano Napolitano
Secretaria GeneralDra. Carolina Gallegos
TesoreroDr. Juan Moya
DirectorDr. Marcelo Devilat
Past PresidentDra. Perla David
Editores de PublicacionesDra. Perla DavidDra. Ledia TroncosoDr. Marcelo Devilat
Delegados ANLICHEDr. Tomás MesaDr. Jorge Förster
Comité EditorialDr. Fernando Ivanovic ZuvicDr. Cayetano NapolitanoDr. Juan SalinasDra. Julia SantinDra. Alejandra HernándezDra. Verónica BurónDr. Juan MoyaDra. Francisca LópezDra. Scarlet WittingDra. Loreto Ríos
Comité InternacionalProf. Dr. Roberto Caraballo, ArgentinaProf. Dr. Neurocirujano Hugo Pomata, ArgentinaProf. Dr. Pedro Serrano, EspañaProf. Dr. Eduardo Barragán, MéxicoProf. Dr. Jaderson Da Costa, BrasilProf. Dra. Magda Lahorges, BrasilProf. Dra. Elza Yacubian, BrasilProf. Dr. Juan Aicardi, FranciaProf. Dr. Alexis Arzimanouglu, Francia
Comité Revisión de ParesDra. Ledda Aguilera, Universidad de ChileDr. Jaime Godoy, Universidad Católica de ChileDr. Rodrigo Salinas, Min. de Salud / Inst. de Salud Pública

3
Epilepsia Refractaria. Epilepsias de difícil control.Roberto CaravalloMédico Principal. Hospital Garrahan, Buenos Aires, Argentina.
Editorial
En alrededor de 80% de los pacientes con epilepsia las crisis pueden ser controladas con antiepilépti-cos y más del 60% de todos los casos remiten en forma permanente. Los restantes pacientes son va-riablemente resistentes al tratamiento y las crisis se repiten a pesar de la medicación adecuada. Sin embargo, no existe una definición aceptada de epi-lepsia refractaria y los criterios varían de modo con-siderable. En general, se toma como base la falta de respuesta al uso adecuado de los antiepilépticos en las máximas dosis toleradas, durante un período mí-nimo necesario. Se ha estimado que la prevalencia de epilepsia es de 5/1.000 personas en la población general y de 1/1.000 en la población infantil.
Las epilepsias aparentemente refractarias o de di-fícil control pueden dividirse en dos grandes cate-gorías:1) Epilepsias seudoincontrolables: corresponden
a las que son incorrectamente tratadas debido a causas del paciente, deficiencias sanitarias o error u omisión del médico.
2) Epilepsias verdaderamente refractarias: las cri-sis epilépticas persisten a pesar del uso adecua-do de antiepilépticos, y de la instrumentación correcta de los procedimientos diagnósticos y de otros tratamientos.
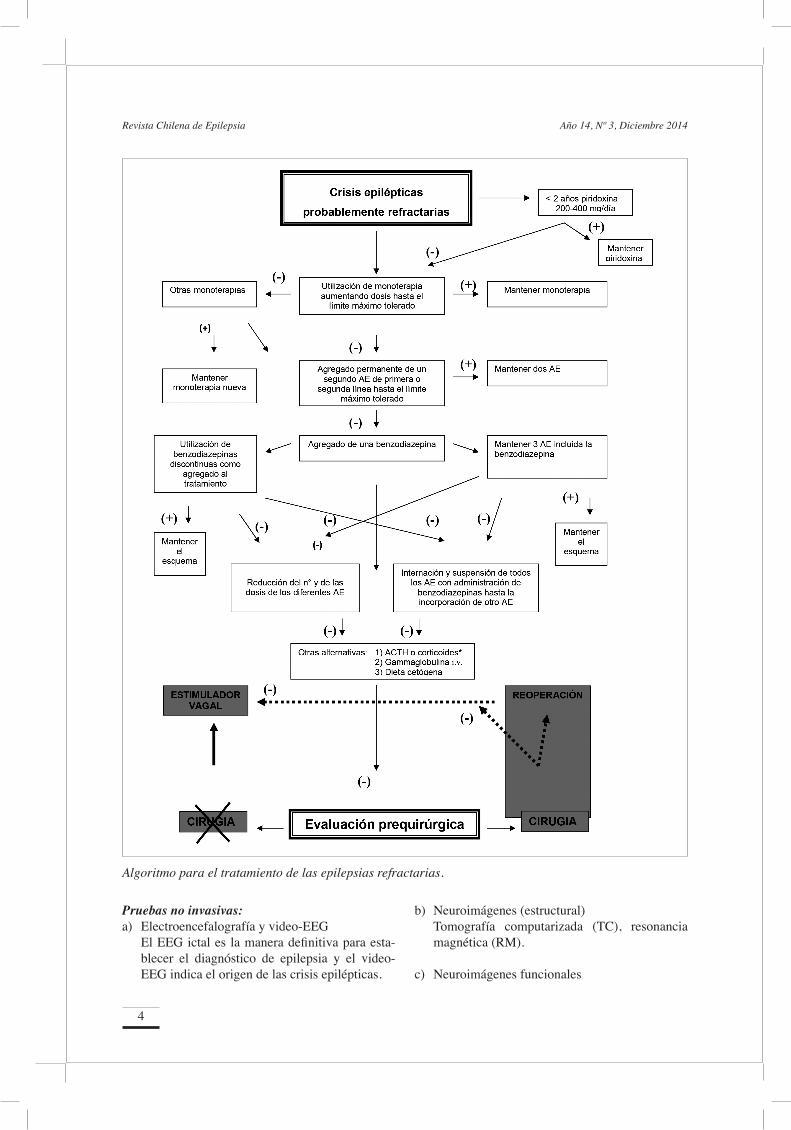
En la figura se muestra un algoritmo para el trata-miento de las epilepsias refractarias. Cabe aclarar que este esquema proporciona una orientación ge-neral y que, sin duda, es necesario considerar cada paciente en particular.
Tratamiento de las Epilepsias Refractarias
Los pacientes con epilepsias refractarias y sus fami-lias presentan una mala calidad de vida por lo cual es crucial ofrecerles otras alternativas terapéuticas no farmacológicas tales como dieta cetógena, ciru-gía, estimulador vagal y otros.
Si bien la mayoría de los niños con epilepsia están tratados con antiepilépticos (AE), la cirugía de la epilepsia se utiliza cada vez con mayor frecuencia en los pacientes resistentes al tratamiento médico. El objetivo principal es controlar las crisis epilép-ticas y mejorar la calidad de vida de los pacientes. En este sentido, es importante reducir el número de crisis, atenuar las manifestaciones clínicas (p. ej., desaparición de caídas o generalización secundaria) o disminuir los efectos adversos de los AE.
Consideramos imprescindible el estudio sistemáti-co de los pacientes con epilepsias de difícil control, por lo cual creemos que un número considerable de pacientes se beneficiarán si se realiza un diagnós-tico correcto y un tratamiento integral adecuado. Una vez que ha sido decidido que un paciente tiene una epilepsia verdaderamente refractaria a la medi-cación y que la cirugía de la epilepsia podría ser una alternativa terapéutica, el paso siguiente es realizar la evaluación prequirúrgica (EP). Esta evaluación decidirá si la cirugía es o no posible.
Tratamiento Quirúrgico
Evaluación PrequirúrgicaUna vez que se ha comprobado que un paciente es refractario a la medicación y se ha decidido llevar a cabo la cirugía de la epilepsia, el paso siguiente es realizar la evaluación prequirúrgica (EP).
Ésta debe efectuarla un equipo multidisciplinario compuesto por especialistas en neurología, neuro-fisiología, neurocirugía, psiquiatría y neuropsicolo-gía, con la intervención de enfermeros, técnicos de electroencefalografía y asistentes sociales.
La identificación de la zona epiléptógena es el ob-jetivo final en el proceso de EP. Se define como el área necesaria y suficiente para el comienzo de una crisis epiléptica y cuya resección o desconexión es imprescindible para la abolición de ésta.
4
Revista Chilena de Epilepsia Año 14, Nº 3, Diciembre 2014
Algoritmo para el tratamiento de las epilepsias refractarias.
Pruebas no invasivas:a) Electroencefalografía y video-EEG El EEG ictal es la manera definitiva para esta-
blecer el diagnóstico de epilepsia y el video-EEG indica el origen de las crisis epilépticas.
b) Neuroimágenes (estructural) Tomografía computarizada (TC), resonancia
magnética (RM).
c) Neuroimágenes funcionales

5
Epilepsia refractaria. Epilepsias de difícil control. Editorial
La tomografía con emisión de positrones (PET) muestra defectos localizados en el metabolismo cerebral. Los defectos focales a menudo corres-ponden a la zona epileptógena. Es un estudio de utilidad en los pacientes con epilepsia del lóbulo temporal, que ha mejorado la localiza-ción preoperatoria y evita tener que recurrir a estudios invasivos. También en el caso de las epilepsias generalizadas severas, como el sín-drome de West, ayuda a encontrar las áreas de hipometabolismo focal.
La tomografía computarizada por emisión de fotón único (SPECT) pone en evidencia altera-ciones en la perfusión en los estados interictal, ictal y postictal, que ayudan a identificar el sitio para la resección. En la práctica, sólo tiene valor localizador definido la SPECT ictal, sobre todo en las epilepsias neocorticales.
La resonancia magnética con espectroscopia puede evidenciar déficits localizados lateraliza-dos en las epilepsias focales.
La resonancia magnética funcional es un estudio no invasivo que permite el estudio de las funcio-nes cerebrales; también es útil para el diagnós-tico y la evaluación del manejo quirúrgico de las epilepsias. En la evaluación prequirúrgica ha sido muy beneficiosa para elaborar la estrategia correcta, pues permite reconocer áreas elocuen-tes del cerebro e identificar con certeza la latera-lidad del lenguaje. Es un estudio de sumo valor en el futuro, pues está reemplazando al test de Wada y podría ofrecer aún más beneficios en el abordaje diagnóstico y en el tratamiento de las epilepsias, sobre todo, las refractarias.
d) Pruebas neuropsicológicas La evaluación neuropsicológica contribuye al
diagnóstico en los pacientes que son candidatos a cirugía de la epilepsia. En este caso particular, las evaluaciones neuropsicológicas antes y des-pués de la cirugía constituyen una parte impor-tante en el abordaje integral del paciente.
Las funciones neuropsicológicas investigadas son:
- Habilidad intelectual general. - Habilidades perceptivas y psicomotoras. - Lateralidad y especialización hemisférica.
- Lenguaje. - Aprendizajes escolares. - Memoria y aprendizaje memorístico. - Atención y función directiva. - Conducta emocional y social.
Los objetivos principales de la evaluación neu-ropsicológica son:- Potencialidad educacional y de rehabilitación.- Confirmación de la lateralización y probable localización del área epileptógena.
- Riesgo de compromiso de la memoria y otras funciones cognitivas a largo plazo.
- Control evolutivo.
e) Evaluación psiquiátrica Puede condicionar el pronóstico y los trastornos
mayores podrían contraindicar la cirugía.
f) Test de Wada Esta prueba determina la dominancia hemisféri-
ca del lenguaje y evalúa la contribución de cada hemisferio cerebral para la memoria a largo pla-zo.
g) Otra Otras técnicas, como la magnetoencefalografía,
se utilizan en centros que disponen de la tecno-logía adecuada para una evaluación prequirúrgi-ca más completa.
Pruebas invasivasSe reconocen distintas técnicas que implican la introducción de electrodos en el cráneo: los elec-trodos profundos o intracerebrales, los electrodos subdurales y los electrodos epidurales. Una vez realizada la colocación de los electrodos intracra-neanos con la consiguiente electrografía, se puede practicar también el mapeo funcional, es decir, la estimulación cortical para definir las funciones de la corteza implicada en relación con el comienzo ictal. Este es de suma importancia en las epilepsias de localización frontal posterior, parietal anterior, occipital mesial y de resecciones temporales pos-teriores (en especial del hemisferio dominante). En este punto es necesario responder a varios interro-gantes: ¿es única la zona ictal?, ¿dónde está locali-zada?, ¿cuál es su extensión?, ¿está cercana a una lesión estructural o funcional?, ¿cuál es la función de la corteza en esa región?, ¿su resección llevará a déficits permanentes?

6
Revista Chilena de Epilepsia Año 14, Nº 3, Diciembre 2014Trabajo Original
ABSTRACT
Drug-resistant epilepsy in children is a condition leading to severe psychosocial, economic struggle among the families of these patients. The current status of social networks contributes to the develop-ment of a growing demand from the parents of these children, many of whom start to seek non-traditio-nal therapeutic alternatives. One of those options is treatment based on marijuana-derived substances, specifically cannabidiol, which has been used for therapeutic purposes since 2,700 b.C. Cannabidiol use has been assumed as a therapeutic possibility for many patients suffering from orphan, difficult-to-manage conditions. Research in children with refractory epilepsy has begun recently, derived from the approval by the FDA of investigation projects based on purified drugs containing cannabidiol. Nevertheless, there is a certain previous experience in isolated cases and small series of patients, with some methodolo-gical defects (difficulty determining the exact com-position of marijuana products, as well as the ap-propriate dosage). This experience arises as a real therapeutic chance in some children. Review of the available sources allows us to explore an extensive list of neurophysiologic, biochemical investigations that generate some certainty about the mechanism of action of cannabidiol-based pro-ducts and its eventual uses in animals and humans. Besides, it is well known that the list of countries and U.S. states that have legalized marijuana for medical use is increasing day after day. We present two children, carrying severe refractory epilepsy, who receive cannabidiol at the initiative of
their own parents. We have followed their clinical evolution, by accompanying the use of cannabidiol as a compassionate use treatment in these patients. Preliminary effects of cannabidiol have been re-markable. Although cannabidiol has not achieved a seizure.free clinical course, the frequency of epi-sodes has diminished in a proportion enough to im-prove the quality of life of their families.Key words: children, epilepsy, cannabidiol.
RESUMEN
La epilepsia resistente en los niños es una forma de epilepsia que conlleva a severos sufrimientos psico-sociales y económicos a las familias. Por lo ante-rior, y en base a la fluidez actual de las redes so-ciales, existe una creciente presión por parte de los padres de estos niños a buscar alternativas terapéu-ticas no tradicionales. Una de ellas es el tratamiento con derivados de la marihuana, específicamente el cannabidiol cuyo uso terapéutico se puede remon-tar a 2.700 años antes de Cristo. En varios cuadros huérfanos y de difícil manejo se ha aceptado que el uso del cannabidiol es una posibilidad terapéutica y de alivio para los pacientes. En epilepsia resistente la investigación en niños recién comienza con la aprobación de la FDA de proyectos que se iniciaran en Enero de 2015, con un fármaco en base a cannabidiol puro. A pesar de lo anterior existe cierta experiencia de casos aislados y pequeñas series con carencias metodológicas que se inician con la dificultad de determinar la composi-ción exacta de los productos y en parte con las dosis a utilizar, pero que permiten avizorar una posibili-dad terapéutica real en algunos pacientes. La revisión de la literatura permite conocer una ex-tensa investigación bioquímica y neurofisiológica que origina algunas certezas sobre el mecanismo de acción del producto y su manejo en animales y
Tratamiento compasivo y de acompañamiento con Cannabis en niños con Epilepsia Resistente. Una pre-sentación de 2 pacientes y revisión de la literatura.Marcelo Devilat, Carla Manterola, Juan Luis MoyaCentro de Epilepsia Infantil. Serv. de Neurología y Psiquiatría. Hosp. L.C. Mackenna. Santiago, Chile.Correo: [email protected]
Recibido 1 de Dic. Aceptado 15 de Dic. 2014Los autores declaran no tener conflictos de intereses.

7
Tratamiento compasivo y de acompañamiento con Cannabis en niños con epilepsia resistente. Marcelo Devilat et al.
humanos. Así mismo se sabe que cada día existen más países y estados de USA que permiten el uso del cannabidiol medicinal.
Se presenta aquí dos niños con epilepsias muy resistentes que por iniciativa de sus padres reci-ben cannabidiol, a los que nosotros, dentro de un “tratamiento compasivo” y de “acompañamiento”, hemos podido seguir. El efecto del cannabidiol, en ellos, ha sido excelente pues si bien no los ha dejado libres de crisis, ellas se han reducido de tal manera que han permitido mejorar la calidad de vida de sus familias.Palabras clave: niños, epilepsia, cannabidiol.
1. INTRODUCCIÓN
1.1 La epilepsia
La epilepsia es la enfermedad neurológica más fre-cuente con una prevalencia en Chile de 17 x 1000 (1), por lo que en nuestro país existirían 289.000 personas con epilepsia. Un tercio de ellas está cons-tituido por menores de 15 años (2), con lo cual ha-brían en Chile 96.333 niños con epilepsia, de los cuales un tercio sería resistente a las terapias farma-cológicas (3, 4), por lo que en nuestro país habrían 32.111 niños con epilepsia refractaria. Este tipo de epilepsia, no está cubierta por el Plan GES (5). Lo anterior origina severas consecuencias psicosocia-les: compromiso cognitivo y psiquiátrico, deterioro económico, injurias físicas, crisis familiares, aban-dono, aislamiento, rechazo social, estigma y otras (6) así como también mayor riesgo de suicidio y de mortalidad (7). Aunque hay muchas definicio-nes de epilepsia resistente, aquí se entenderá por tal a aquellas epilepsias que tratadas con al menos 2 antiepilépticos (AE), bien indicados, a dosis re-comendadas y/o niveles plasmáticos terapéuticos o tóxicos, continúan con crisis, dentro de un lapso de 6 meses. 1.2 La epilepsia resistente y su tratamiento El tratamiento de los pacientes con epilepsia resis-tente es variado e incluye AE, cirugía, dieta ceto-génica (8) y últimamente, estimulación vagal, es-timulación transcraneal (9), musicoterapia (10) y otros. Un grupo de niños persiste con crisis, a pesar del esfuerzo de los profesionales y de la industria farmacéutica, que en el último tiempo ha ofrecido numerosos nuevos AE. En consideración a lo dra-
mático que resulta tener a un niño con crisis epilép-ticas descontroladas, los padres influenciados por los medios y por lo expedito de las comunicaciones están demandando, de manera urgente, otras formas de tratamiento para sus hijos, como son los deriva-dos de la marihuana.
1.3 Tratamiento con cannabis y otras hierbas en la Historia
Las preparaciones medicinales de flores y aceites de cannabis han sido usadas en China desde hace aproximadamente 2.700 años aC para tratar, entre otras enfermedades, los episodios de ausencias. En la Edad Media, los médicos árabes usaron la can-nabis para tratar la epilepsia, así como náuseas y vómitos, inflamaciones, dolores y fiebre (11,12). Terminada la Edad Media, en 1542 Leonhardt Fu-chs fue el primero que denominó a la planta como cannabis sativa y W.B O’Shaughnessy la introdujo en la práctica médica en Inglaterra en el siglo 19. En ese siglo, más específicamente en 1854, se introdujo en Estados Unidos (13). Posteriormente la medicina occidental usó ampliamente la cannabis, y antes de la aspirina, era el más común analgésico utilizado; también se usaba para el glaucoma, dolor, náuseas y vómitos, espasmos musculares, insomnio, ansiedad y epilepsia. Otras indicaciones eficaces incluyen el dolor asociado a la neuropatía sensorial por VIH, dolor crónico, náuseas y vómitos por quimiotera-pia, y espasmos asociados a esclerosis múltiple. Es-pecíficamente, su uso moderno se inició en 1868, cuando Russell Reynolds indicó cannabis indica, otra variedad de cannabis, asociada con bromuro de potasio, en un paciente de 40 años con crisis tónico-clónicas generalizadas, el cual quedó libre crisis por 6 meses y recayó al suspender la cannabis (11). La declinación de su uso se inició en el siglo 20, debido en parte, a la aparición de medicinas sinté-ticas que la reemplazaron. Se inicia su uso “recrea-cional” especialmente en Egipto, África del Sur y USA, hasta que la Liga de las Naciones en 1924 la limitó sólo como medicina. Más tarde, en USA, a través del Acta de 1970, se declaró a la droga como tipo I, por lo que su uso se volvió ilegal. Así, la can-nabis entró hasta la actualidad a un estatus gris y de paria (13). La medicina hierbática (14) ha ofrecido una amplia gama de hierbas para la epilepsia y desde el siglo 17 se han descrito 221 plantas para uso de la epilep-

8
Revista Chilena de Epilepsia Año 14, Nº 3, Diciembre 2014
sia. Las plantas pueden ser clasificadas de acuerdo a su tipo estructural como alcaloides, flavonoides, saponinas, cumarinos y terpenos. Al último grupo pertenecen los cannabinoides: cannabidiol (CBD) y el tetrahidrocannabinol (THC) y existe la opinión de que, a pesar de la insuficiente información, hay estudios de casos o pequeños ensayos en humanos que permitirían que la ciencia básica pueda contri-buir a desarrollarlas como AE. La medicina mapu-che también tiene su aporte y ofrece el “alweshkos-kel” como preventivo de las “crisis epilépticas y las convulsiones” (15).
1.4 La cannabis para la epilepsia en el mundo actual
A pesar de las prohibiciones legales, en la mitad de los estados de USA, en Canadá, Holanda, e Israel, han autorizado el uso de cannabis para la epilepsia resistente (6). La Republica Checa autorizó el uso medicinal de cannabis el 2 de abril 2014 (16). El es-tado de Sao Paulo, en Brasil, autorizó el tratamiento con cannabis sativa a niños con epilepsia resistente, y ha permitido que el médico pueda recetar el pro-ducto que será importado por un organismo estatal (17).
La OMS se pronunció a favor del uso medicinal de cannabis para dolor, náuseas y vómitos y anorexia en inmunodeprimidos, así como en la espasticidad de la esclerosis múltiple (18).
La FDA dio su aprobación para efectuar estudios con marihuana en niños con epilepsia intratable. El medicamento será Epidiolex®, un cannabidiol puro de GW Pharmaceutical. También el fármaco ha sido autorizado para ensayo en niños con síndrome de Dravet y epilepsias resistentes (19, 20). En Chile, a pesar de las dudas de algunas socieda-des médicas sobre las posibilidades terapéuticas de la marihuana (21), el Servicio Agrícola y Ganadero autorizó la siembra de marihuana para ser utilizada con fines terapéuticos para unos 200 pacientes que presentan cáncer y epilepsia (22). 1.5 La marihuana como una fitoterapia
1.5.1 La plantaDos variantes de la planta de marihuana, la canna-bis sativa y la cannabis indica (6,12), contienen cerca de 489 compuestos, de los cuales existen
aproximadamente 70 diferentes cannabinoides. De éstos, el psicoactivo tetrahidrocannabinol (THC, aislado en 1964) y el no-psicoactivo cannabidiol (CBD, aislado en 1963), son los más importantes y se encuentran en variables concentraciones en las diferentes cepas de marihuana. La razón entre am-bos determina los efectos terapéuticos y los efectos psicoactivos (6, 23, 24). Con respecto al THC, se ha sugerido que la euforia que origina, sería debida a una depresión de la actividad de inhibición en el septum, cerebelo y tálamo (13). Ambos compuestos actúan en el sistema endocannabinoide.
1.5.2 El sistema endocannabinoide y el mecanis-mo de acciónEl sistema endocannabinoide está compuesto por algunos cannabinoides endógenos como la ananda-mina y el 2-araquidonoilglicerol, y por sus recep-tores, siendo CB1 y CB2 los más conocidos. El re-ceptor CB1 está altamente expresado en el sistema nervioso central, y puede ser encontrado en seres humanos desde la novena semana de gestación. Su distribución es amplia en periodos tempranos de la vida, pero presenta una localización más selectiva en la adolescencia (25). Se ubica en diferentes ti-pos neuronales, principalmente en los ganglios de la base, la sustancia nigra, el globo pálido, el cerebelo, el hipocampo y la corteza cerebral (6, 11, 13).
El receptor CB1 es una proteína transmembrana que se encuentra en la membrana presináptica. Cuando la neurona postsináptica es altamente estimulada, hay un aumento del calcio intracelular, que induce la biosíntesis de endocannabinoides. Éstos pasan li-bremente por la membrana celular, alcanzando a la neurona presináptica. La activación de CB1 por los endocannabinoides lleva a una inhibición del AMP cíclico, activación de proteínas kinasas, regulación de canales de calcio y apertura de canales de pota-sio, lo que a su vez lleva a inhibición en la libera-ción de neurotransmisores al espacio intersináptico. Es así como los endocannabinoides tienen un rol fundamental en el control de la estimulación y mo-dulación neuronal. Dicho mecanismo se denomina “depolarización que induce supresión de la inhibi-ción” si actúa en neuronas inhibitorias, e “hiperpo-larización que induce supresión de excitación” si actúa en neuronas excitatorias.(11, 26).
Nuestro conocimiento del receptor CB2 es mucho más limitado. Se sabe que está presente en células inmunes, tanto del sistema nervioso central como

9
Tratamiento compasivo y de acompañamiento con Cannabis en niños con epilepsia resistente. Marcelo Devilat et al.
en la periferia. Su rol aún no se ha establecido cla-ramente. Adicionalmente, se conocen otros recepto-res que tendrían relación con un rol antinociceptivo y vías metabólicas variadas (6, 11, 13).El THC se aisló en 1964 como compuesto, y 4 años más tarde se logró su síntesis. Se sabe que bioquí-micamente actúa como un agonista parcial de los receptores CB1 y CB2. Se hipotetiza que a través de esta vía lograría su efecto anti-inflamatorio, como estimulante del apetito, antiemético, de relajante muscular, analgésico y psicógeno. El efecto psicó-geno ha sido centro de gran investigación, por su importancia en salud. Se sabe que el uso de mari-huana se correlaciona estadísticamente con cuadros agudos de paranoia y psicosis; su uso crónico se ha asociado a un riesgo aumentado de trastornos psi-cóticos, entre los que se incluye la esquizofrenia, en personas con predisposición a dicha patología. El CBD es un cannabinoide que no tiene un efecto psicogénico, por lo que ha sido propuesto como un potencial agente terapéutico. Su mecanismo de ac-ción aun no es bien conocido, pero se sabe que mo-dula la concentración de calcio en la célula a través de una inhibición de los canales de calcio. Junto al CBD se ha usado en modelos animales el cannabidivarin (CBDV) que es una variante propi-lo del CBD (27). Ha probado sus habilidades anti-convulsivas y de buena tolerancia en modelos con MES y PTZ, lo que se explica más adelante, siendo también asociado a ácido valproico y etosuximida. El mecanismo por el cual CBD y CBDV ejercen su acción anticonvulsiva no está determinado, aunque varios de sus puntos de acción han sido estudiados. Entre ellos se puede citar la modulación intracelu-lar del Calcio, la proteína G-acoplada al receptor 55 (GPR55) o el canal de proteína 1 (VDAC1) selecti-vo dependiente de aniones. CBD y sus compuestos asociados pueden reducir la excitabilidad neuronal y la transmisión neuronal (11). Es además antiapop-tótico, neuroprotector y antiinflamatorio (6). Como resumen (28), el estudio de los mecanismos de acción de los cannabinoides, “ha originado un nuevo sistema de comunicación entre las células, especialmente relevante en el SNC, donde el siste-ma contiene puntos de acción activados por CBD, los cuales no sólo actúan a través de la activación, sino también facilitando su bloqueo o inhibición”. Lo anterior sugiere un “nuevo sistema susceptible de manipulación farmacológica” (28).
1.6 Modelos preclínicos
Como análisis previo es necesario destacar que, ya en 1978, ratas sometidas a electroshock máximo (MES) que recibieron una mezcla de cannabinoide, con 17% de THC y 83% de CBD, expresaron un efecto anticonvulsivo (29). Por otra parte, un perro traqueostomizado, que recibió una dosis subcon-vulsivante de penicilina y humo de cannabis (que contenía 6 mg de THC) en forma repetida, presentó crisis tónico-clónicas generalizadas y espiga-onda en la corteza occipital y frontal en el EEG. Los au-tores sugieren que el THC reduce el umbral con-vulsivo o aumenta la permeabilidad de la barrera hematoencefálica (30).
Posteriormente, se ha publicado abundante infor-mación acerca del efecto beneficioso del CBD, en modelos preferentemente de ratas sometidas a MES (que origina crisis epilépticas agudas). Sin embar-go, la información acerca del efecto en epilepsia crónica es menos evidente. De manera interesante se ha observado que el CBD no tiene efecto en crisis epilépticas focales secundariamente generalizadas, en modelos en los que se les ha implantado cobalto o que han recibido estricnina (11). Otro modelo animal utilizado ha sido en ratas con una mutación knockout para el receptor CB1. Se observó que las ratas mutadas presentan crisis de mayor duración ante un modelo epileptogénico con ácido kaínico (26). Otro estudio, que expone ratas a un modelo epileptogénico con pentilenetetrazol, mostró que las tratadas con cannabidiol presentan una disminución significativa de las crisis epilépti-cas y de la mortalidad.
En conclusión, si bien hay estudios en animales que apoyan el uso de cannabinoides en un modelo de epilepsia, y existe un mecanismo fisiológico que se relaciona con el control de estimulación neuronal, aun no hay claridad de este fenómeno en humanos (27).
1.7 Farmacología del cannabidiol en humanos Los estudios de CBD sintético o de extractos de plantas, aislado o en combinación con el THC, han entregado suficiente información acerca de la farmacología del CBD, que permite su manejo en ensayos y tratamientos clínicos (6, 11, 13, 31). La forma medicinal se administra oralmente en forma

10
Revista Chilena de Epilepsia Año 14, Nº 3, Diciembre 2014
de aceite en una cápsula, la que por su baja solubi-lidad en agua determina una absorción errática La biodisponibidad se estima cercana al 6%, debido a su paso por el hígado. En adultos voluntarios, des-pués de una dosis única de 10mg de CBD y THC, se ha demostrado un máximo de concentración de 3,0 +- 3,1 ug y un máximo tiempo de 2.8 +- 1.3 hr. La distribución está gobernada por su alta lipofilicidad (K 6-7) y alto volumen de distribución (+- 32K/kg), con rápida distribución en cerebro, grasa y otros tejidos. El CBD es altamente unido a proteí-nas del plasma y el 10% circula unido a glóbulos rojos; por lo que en la administración crónica puede acumularse en pacientes con alta adiposidad.
Metabolismo, eliminación y vida mediaComo todos los cannabinoides, es altamente meta-bolizado en hígado e hidroxilado a 7-OH-CBD por el citocromo P450 (CYP), predominantemente por CYP3A y CYP2C, y excretado por heces y esca-samente en orina. La vida media es de 18 a 32 hrs después de una dosis única en usuarios crónicos de CBD y el clearance de 960 a 1.560 cc/min. La seguridad estudiada en múltiples trabajos de CBD con placebo, controlados y ensayos abiertos, han demostrado que “es bien tolerado a través de una amplia gama de dosis (11). No hay efectos ad-versos sobre SNC, o sobre signos vitales o humor, a dosis sobre 1,500mg/día oral o 30mg EV, tanto en administración aguda o crónica”. Sin embargo, existe una limitada seguridad a largo plazo, aunque hay muchos pacientes-año de exposición a nabixi-mol ® después de su aprobación en varios países de Europa y Canadá. Se ha informado de algún riesgo teórico de inmunosupresión, ya que CBD muestra supresión de producción de interleukina 8 y 10 e inducción de apoptosis de linfocitos in vitro (11). Existe escasa información sobre las interacciones. El CBD, por ser un potente inhibidor de isoenzimas de CYP, primariamente CYP2C y CYP3A (in vitro y en modelos animales, pero no humanos), podría modificar los niveles plasmáticos de AE concomi-tantes. Específicamente, la repetida administración de CBD puede inducir isoenzimas de CYP2B en modelos animales, lo cual puede tener implica-ciones en personas con epilepsia que toman ácido valproico (AV) y clobazam (CLOB), dado que son metabolizados mediante esas enzimas. Finalmen-te, dado que el CBD es metabolizado en gran parte por CYP3A4, es probable que enzimas inductoras
comunes de AE (tales como CBZ y FNT) puedan reducir los niveles séricos de CBD. Actualmente existen a disposición varios fármacos cannabinoides, que tienen distintas concentraciones de CBD y THC. Éstos son indicados en esclerosis múltiple, cáncer, dolor crónico, náuseas y vómi-tos, trastornos del sueño y algunas enfermedades psiquiátricas. Aprobados en diferentes países (en-tre otros, Alemania, España, Dinamarca, Canadá y Nueva Zelanda), tienen un costo relativamente alto. Entre ellos se puede citar al Sativex ®, Cesamet ®, Marinol® y Cannador® (13, 23, 28). Las personas con epilepsia aun no tienen un canná-bico probado, pero el Epidiolex ® (un cannábico no psicoactivo, con CBD al 98%) iniciará pronto un ensayo, aprobado por la FDA (como ya se comen-tó más arriba). El Epidiolex® está designado como fármaco “huérfano”, especialmente indicado en el síndrome de Dravet y síndrome de Lennox-Gastaut, entre otros cuadros (6, 11, 13).
2. LO REALIZADO Y LO QUE VIENE
2.1 Lo realizado 2.1.1 Existe un creciente interés entre los familiares de los niños con epilepsia resistente y entre las per-sonas adultas que sufren esta condición en explorar la posibilidad terapéutica con cannabis. El 80% de los 32.111 niños con epilepsia resistente calculados para Chile, pertenecen mayoritariamente al grupo A y B beneficiario de Fonasa, lo que incrementa sus dificultades y sufrimientos; por cuanto deben adquirir medicamentos de segunda generación, que -como se destacó más arriba- no son propor-cionados por el Plan de Garantías Explícitas (GES) del Ministerio de Salud. De acuerdo a un reciente estudio efectuado en nuestro Servicio, las familias que deben recibir AE de segunda generación gastan mensualmente $ 48.989 en promedio, lo que repre-senta un deterioro adicional en personas especial-mente menoscabadas (32). Lo anterior es relevante, por cuanto un tratamiento intensivo, de alto costo y que requiere experiencia profesional, puede hacer posible que los niños con epilepsia resistente que-den libres de crisis en alrededor de un 20% (8, 33). Queremos destacar que desde hace muchos años hemos estado interesados en mejorar el diagnóstico y el tratamiento de estos pacientes (3, 8) y buscar ayuda internacional para optimizar su manejo (34).
11
Tratamiento compasivo y de acompañamiento con Cannabis en niños con epilepsia resistente. Marcelo Devilat et al.
Debido a la irrupción de la cannabis en la literatura médica internacional y en la prensa, la Sociedad de Epileptología de Chile, capítulo chileno de la ILAE, organizó una conferencia sobre el tema en sus XIV Jornadas Invernales de Epilepsia, a sugerencia de uno de nosotros (MD). Ésta fue dictada por la Dra. Carla Manterola y la Dra. Carolina Gallegos, el día 7 de Junio de 2014. Con lo anterior, se pretendió iniciar una discusión sobre el tema desde el punto de vista académico, a pesar de las restricciones y te-mores ambientales. Por otra parte, en Junio pasado, el Servicio de Neurología y Psiquiatría del Hospital Luis Calvo Mackenna dictó en la Reunión Clínica semanal del Servicio de Pediatría una conferencia sobre la cannabis y la experiencia clínica obtenida hasta ese momento. Desde el punto de vista internacional, la revista Epi-lepsia, órgano oficial de la ILAE (Sociedad interna-cional que acoge a la mayoría de los países del orbe, y que tiene mas de 100 años de vida), dedicó parte de su número de Junio pasado al tema “Cannabis y Marihuana Médica para la Epilepsia”, lo que es un apoyo para que los están “interesados en trabajar sobre el tema” (35)
2.1.2 Los pacientes.
Nuestra experiencia.Los autores han tenido la oportunidad de controlar 5
niños con epilepsias superresistentes. Bajo decisión y responsabilidad de sus propios padres, éstos han dado aceite de cannabis a sus hijos. Se ha decidido excluir a 3 de ellos, por cuanto el tiempo de evolu-ción es muy reducido, la información no es clara o no se respetan las indicaciones médicas. Además controlamos otros 6 pacientes que están en fase de línea basal. Las historias de los 2 pacientes se entre-gan a continuación:
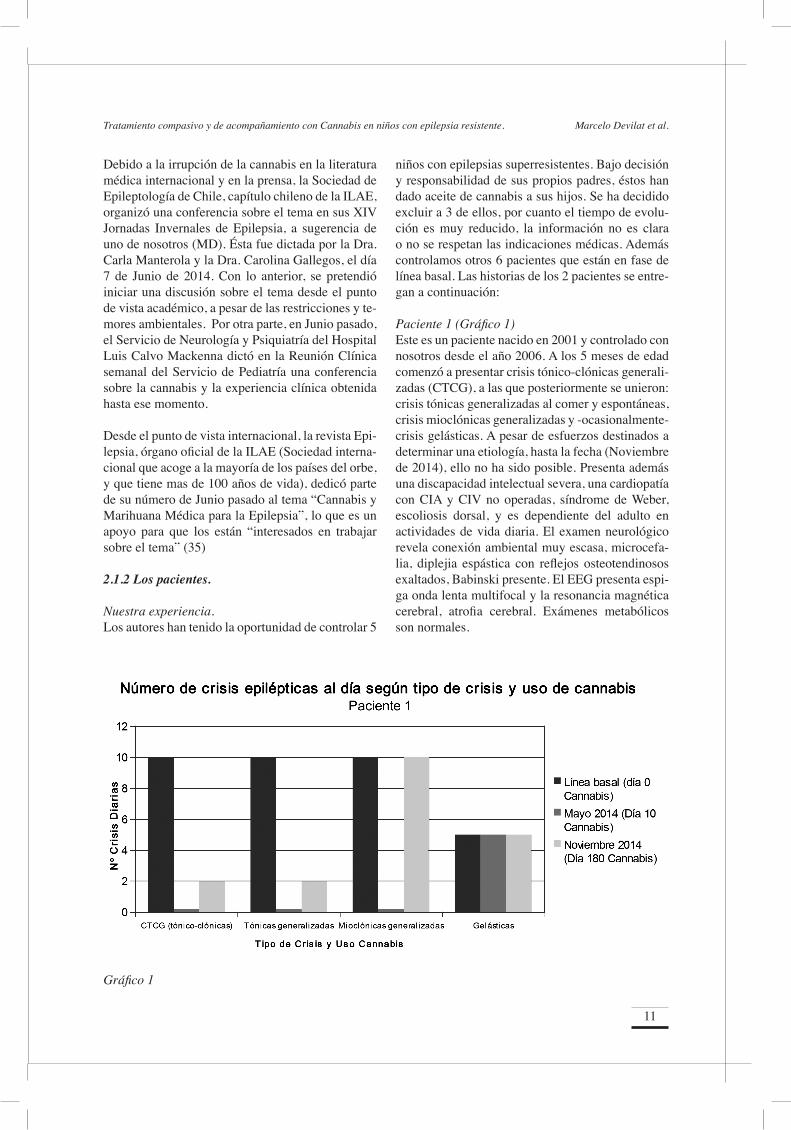
Paciente 1 (Gráfico 1)Este es un paciente nacido en 2001 y controlado con nosotros desde el año 2006. A los 5 meses de edad comenzó a presentar crisis tónico-clónicas generali-zadas (CTCG), a las que posteriormente se unieron: crisis tónicas generalizadas al comer y espontáneas, crisis mioclónicas generalizadas y -ocasionalmente- crisis gelásticas. A pesar de esfuerzos destinados a determinar una etiología, hasta la fecha (Noviembre de 2014), ello no ha sido posible. Presenta además una discapacidad intelectual severa, una cardiopatía con CIA y CIV no operadas, síndrome de Weber, escoliosis dorsal, y es dependiente del adulto en actividades de vida diaria. El examen neurológico revela conexión ambiental muy escasa, microcefa-lia, diplejia espástica con reflejos osteotendinosos exaltados, Babinski presente. El EEG presenta espi-ga onda lenta multifocal y la resonancia magnética cerebral, atrofia cerebral. Exámenes metabólicos son normales.
Gráfico 1
12
Revista Chilena de Epilepsia Año 14, Nº 3, Diciembre 2014
Las crisis generalizadas son de duración variable (de 3 hasta 10 minutos) y la frecuencia flúctua entre 2 a 10, a incontables al día; en tanto que las focales son de corta duración con frecuencia muy variable. Nunca ha estado un día completo sin crisis. Ha re-cibido, sin éxito, el siguiente tratamiento disponi-ble: fenobarbital, fenitoína, carbamazepina, ácido valproico, primidona, topiramato, levetiracetam, oxcarbazepina, lamotrigina, lacosamida, clobazam, zonisamida, pregabalina, clorazepato, dieta cetóge-na y callosotomía total. En Mayo de 2014, a propia iniciativa y responsabi-lidad de los padres, ellos inician la administración de cannabis (dosis equivalente a 2 lentejas al día) de aceite de cogollo de la cepa denominada “Juanita la Milagrosa”; sin modificar las dosis de levetiracetam (42 mg/kg/día) y primidona (14 mg/kg/día) para 35 kilos de peso. Retrospectivamente, se determina para el mes de Abril de 2014 (30 días), una línea de base para las crisis epilépticas. La línea basal de CTCG y crisis tónicas espontáneas es de 10 al día; para las crisis mioclónicas la línea de base marca incontables al día, y las gelásticas son ocasionales. Para las crisis tónicas generalizadas al comer, la lí-nea de base la constituye cada comida. La evolución de las crisis generalizadas no miocló-nicas (CTCG y tónicas), revela que en Mayo estuvo 10 días sin crisis (tiempo máximo sin crisis en su vida); pero posteriormente, en ese mes y en Junio, Julio y Agosto, se presentan en cantidad de 1 a 2 al día, o bien ninguna al día. El resto de crisis no ex-
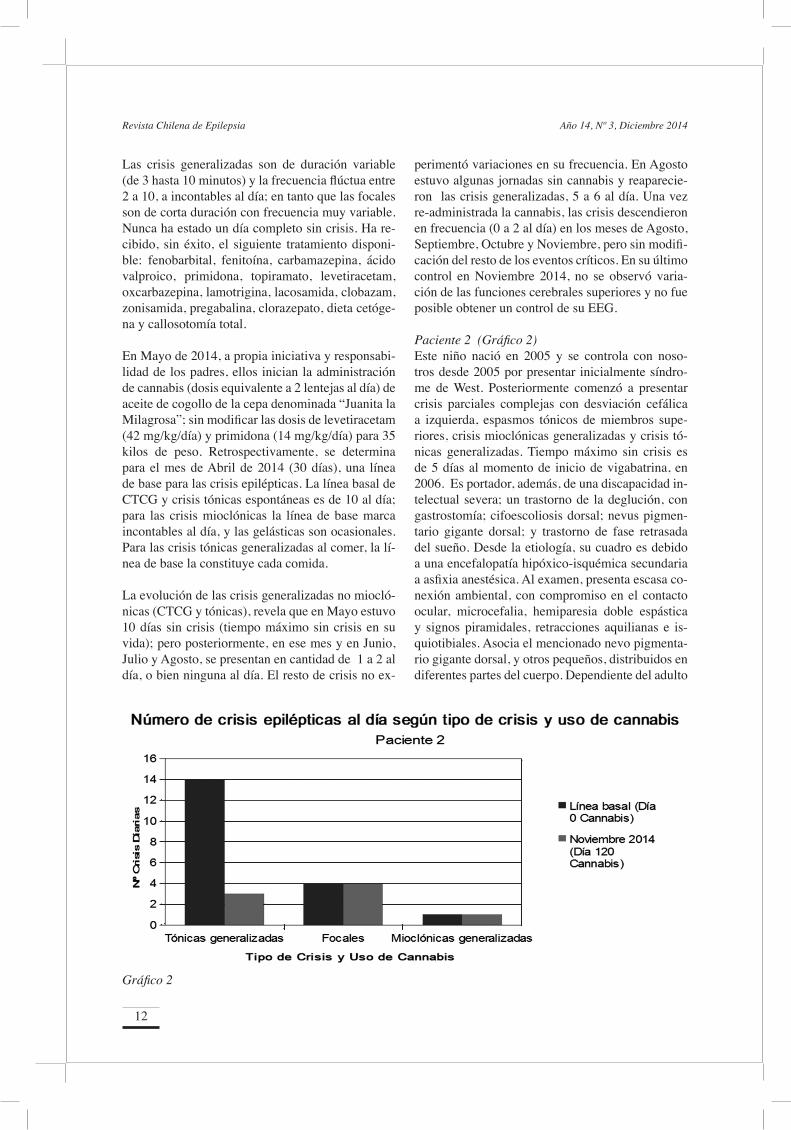
perimentó variaciones en su frecuencia. En Agosto estuvo algunas jornadas sin cannabis y reaparecie-ron las crisis generalizadas, 5 a 6 al día. Una vez re-administrada la cannabis, las crisis descendieron en frecuencia (0 a 2 al día) en los meses de Agosto, Septiembre, Octubre y Noviembre, pero sin modifi-cación del resto de los eventos críticos. En su último control en Noviembre 2014, no se observó varia-ción de las funciones cerebrales superiores y no fue posible obtener un control de su EEG. Paciente 2 (Gráfico 2)Este niño nació en 2005 y se controla con noso-tros desde 2005 por presentar inicialmente síndro-me de West. Posteriormente comenzó a presentar crisis parciales complejas con desviación cefálica a izquierda, espasmos tónicos de miembros supe-riores, crisis mioclónicas generalizadas y crisis tó-nicas generalizadas. Tiempo máximo sin crisis es de 5 días al momento de inicio de vigabatrina, en 2006. Es portador, además, de una discapacidad in-telectual severa; un trastorno de la deglución, con gastrostomía; cifoescoliosis dorsal; nevus pigmen-tario gigante dorsal; y trastorno de fase retrasada del sueño. Desde la etiología, su cuadro es debido a una encefalopatía hipóxico-isquémica secundaria a asfixia anestésica. Al examen, presenta escasa co-nexión ambiental, con compromiso en el contacto ocular, microcefalia, hemiparesia doble espástica y signos piramidales, retracciones aquilianas e is-quiotibiales. Asocia el mencionado nevo pigmenta-rio gigante dorsal, y otros pequeños, distribuidos en diferentes partes del cuerpo. Dependiente del adulto
Gráfico 2

13
Tratamiento compasivo y de acompañamiento con Cannabis en niños con epilepsia resistente. Marcelo Devilat et al.
en actividades de vida diaria. El EEG presenta es-piga onda lenta multifocal; en la actualidad, exhibe espiga-onda lenta generalizada de alto voltaje, con-tinua, en más del 50% del sueño no REM. La reso-nancia magnética revela lesiones hipóxicas difusas y atrofia cerebral. Los exámenes metabólicos han sido normales.
Las crisis tónicas generalizadas son de variable du-ración (1 a 2 minutos) y se presentan diariamente, entre 5 a 14 al día. Las crisis focales son de menos de un minuto de duración, y se presentan 3 a 4 veces al día. Las crisis mioclónicas generalizadas ocurren al menos una vez al día. Ha recibido sin éxito, el siguiente tratamiento disponible: vigabatrina, feno-barbital, ácido valproico, primidona, levetiracetam, lamotrigina, lacosamida, dieta cetógena, gabapenti-na, tioridazina, baclofeno, melatonina y toxina bo-tulínica.
Desde el 23 de Julio de 2014, por propia iniciativa y responsabilidad de los padres, ellos inician la ad-ministración de cannabis (dosis diaria equivalente a 2 lentejas) de aceite de cogollo de la cepa llamada “Juanita la Milagrosa”, sin modificar el resto de la medicación diaria: vigabatrina a (90 mg/kg/día), le-vetiracetam (90 mg/kg/día), fenobarbital (4,5 mg/kg/día), lacosamida (4,5 mg/kg/día) y tioridazina (2,3 mg/kg/día), para 22 kilos de peso. Retrospecti-vamente, se determina para los meses de Junio y Ju-lio una línea de base de crisis: para las crisis tónicas de 14 al día, para las crisis focales de 2 a 4 al día, y para las crisis mioclónicas 1 a 2 al día.
La evolución durante los meses de Agosto, Sep-tiembre, Octubre y Noviembre informa que las cri-sis tónicas generalizadas son 3 o menos al día, pero las crisis focales y las mioclónicas persisten con la frecuencia de la línea de base. En su último con-trol en Noviembre 2014, su trastorno de deglución había mejorado significativamente, observándose además una leve mejoría de la alerta y conexión con el ambiente. Un control de EEG no experimen-tó cambios. ComentarioSe trata de 2 niños controlados por nosotros por un largo tiempo, que iniciaron su epilepsia muy pre-cozmente, con múltiples y muy frecuentes tipos de crisis (generalizadas y focales), que persistieron a
pesar de los numerosos AE recibidos (incluso con dieta cetógena y callosotomía en el paciente 1). Ambos cursaron con severo daño cerebral, discapa-cidad intelectual severa, microcefalia y síndromes piramidales. Los 2 niños presentan atrofia cerebral y los EEGs, específicos para epilepsia, están seve-ramente alterados. Lo único que los diferencia neu-rológicamente es la etiología: en un niño es impes-quisable y en el otro es secundaria a encefalopatía hipóxica. Ambos reciben de sus padres la misma cepa de cannabis, logran una línea basal retrospectiva de al menos 30 días y ambos presentan una signifi-cativa disminución de las crisis generalizadas (del tipo CTCG y tónicas), después de un período de observación de 6 y 4 meses, respectivamente. Am-bos pacientes no responden a crisis parciales y mio-clónicas, sugiriendo que la cannabis es específica para las crisis generalizadas (11), apreciación que requiere el análisis de un mayor número de pacien-tes en trabajos bien controlados. Una de las diferen-cias entre ambos después de recibir cannabis, es que uno pareciera haber mejorado su contacto y el otro no. Se ha mencionado en la literatura este tipo de mejoría, que podría deberse a la eventual presencia de THC en el aceite de uno de los pacientes. Lo anterior también podría haberse debido a la dismi-nución de las crisis en ese niño. Otra diferencia está marcada por la dosis que ambos pacientes reciben, pues los dos reciben la misma “dosis” total, pero sus pesos son diferentes. Aunque tenemos la evidencia de una significativa disminución de las crisis generalizadas en ambos enfermos, el tiempo de observación no es lo sufi-cientemente prolongado como para determinar de-finitivamente el rol de la cannabis en ello. Por otra parte, la evolución de la frecuencia de crisis en la epilepsia resistente no es lineal, sino que pueden incluso desaparecer por varios meses (en una evo-lución intermitente) o bien aparecer nuevos tipos de eventos críticos. En conclusión, a pesar del resultado relativamente favorable con el uso de la cannabis en nuestros pa-cientes, es urgente y necesario realizar estudios más aceptables técnicamente, tiempos de observación más prolongados, determinar la composición exacta del producto y las dosis que deben emplearse.

14
Revista Chilena de Epilepsia Año 14, Nº 3, Diciembre 2014
2.1.3 Pacientes tratados. Una revisión de la lite-ratura.
La literatura a disposición proporciona hasta la fe-cha un “débil evidencia” acerca de la eficacia de la cannabis y sus componentes en epilepsia resis-tente. Existe una falta de ensayos bien diseñados, controlados, longitudinales, randomizados y doble ciego (36). Sin embargo, tenemos a disposición numerosas comunicaciones disponibles de casos anecdóticos, así como ensayos limitados, que sus-tentarían los beneficios de la planta en las personas con epilepsia resistente, especialmente niños. Por otra parte, las autorizaciones para su uso aumentan consistentemente y son numerosos los países que han accedido a su uso terapéutico, como fue indi-cado más arriba. Hasta donde tenemos conocimiento, la primera se-rie publicada, en 1978, corresponde a Mechoulam y Carlini (11), en la que 4 personas con epilepsia resistente recibieron por 3 meses, 200mg diarios de cannabidiol. Como contraparte, 5 sujetos recibieron placebo. El resultado indica que 2 pacientes queda-ron libres de crisis, 1 tuvo mejoría parcial y en otro no hubo modificación de la frecuencia de las crisis. No hubo toxicidad. Este trabajo tiene limitaciones, por cuanto: no se conoce el tipo de crisis y la línea basal de ellas; no se define el concepto de mejoría parcial; no se determinó el manejo de otros AE; el número de pacientes de la muestra fue escaso, con limitado poder; no hubo randomización ciega; y no se contó con información sobre los placebos. Cunha et al en 1980 (11), publicaron un grupo de 15 personas con epilepsia resistente del lóbulo tempo-ral. Siete actuaron como placebo y 8 recibieron 200 a 300 mg de cannabidiol al día, por un período de 3 a 18 semanas. Siete de estos últimos mostraron una disminución en la frecuencia de las crisis, aunque hubo somnolencia en algunos pacientes. No se deta-lla tolerancia, y un placebo se trasladó a la muestra, lo que pervierte al universo. No se informó del ma-nejo de los otros AE, si es que los hubo. Ames y Cridland en 1986 (11), dan cuenta de 12 su-jetos con retardo mental y epilepsia resistente, divi-didos en un grupo de 6 pacientes. De ellos, algunos recibieron 300mg al día de cannabidiol, durante 1 semana; otros recibieron 200mg diarios del fárma-co, por 3 semanas. Otro grupo de 6 pacientes actuó como placebo. El resultado revela que no hubo di-
ferencia ente el grupo tratado y el grupo placebo y algunos tuvieron somnolencia, sin señalar el núme-ro. Esta presentación tiene numerosas fallas meto-dológicas y faltas de información, pues es una carta al director. Trembly y Sherman en 1990 (11) informan en un póster de congreso, y después en un libro de su au-toría, sobre 10 o 12 pacientes con epilepsia resis-tente tratados con cannabidiol, con 3 meses de línea basal y 6 meses de placebo. Se randomiza cannabis y placebo durante 6 meses por muestra, y después se realiza un “cross over” por 6 meses. Como resul-tado informan que no hubo cambio en frecuencia de crisis, ni en los test cognitivos y conductuales, y tampoco toxicidad. Los 4 trabajos mencionados presentan severas fallas metodológicas por lo que es imposible obtener de ellos alguna conclusión de utilidad. “The Cochrane Collaboration” publicó una revisión de la bibliografía entre 1978 y 1990, sobre estos 4 últimos trabajos (que suman 48 pacientes). Como se ha comentado más arriba, tienen una baja calidad científica, aunque sugieren que la dosis de CBD en adultos fluctúa entre 200 a 300mg al día, la que es bien tolerada (37). En 2005 se presentó un poster (38) con 18 niños con epilepsia resistente tratados con AE tradicionales y con una solución de acei-te de CBD al 2,5%, a una dosis de 20 gotas al día como un estudio abierto. Una niña disminuyó la frecuencia e intensidad de las crisis, mejoró la aler-ta y el lenguaje, todo lo cual permitió disminuir la ingesta de fenobarbital. Otra niña (portadora de sín-drome de Lennox-Gastaut) recibió 30 gotas al día; disminuyó sus crisis y mejoró su conducta. El resto de los 16 niños, todos con epilepsias sintomáticas resistentes, recibieron CBD. Sólo 9 de ellos mantu-vieron el CBD. El resto -a pesar de la mejoría en la frecuencia de las crisis, la conducta y la alerta- no pudo seguir pagando los 300 euros al mes que cos-taba el tratamiento. No hubo efectos adversos que requirieran retiro de CBD, y “a pesar de las bajas dosis administradas”, la mayoría disminuyó sus cri-sis en más del 25%. En todos mejoró la alerta y la espasticidad. Aunque la información es anecdótica y con fallas en la metodología, pareciera haber una respuesta favorable en relación a las crisis, a lo con-ductual y cognitivo, así como la ausencia de efectos adversos de relevancia. El aspecto económico pue-de ser un factor relevante que impida el manejo de los casos que lo requieran. En este estudio, tal como

15
Tratamiento compasivo y de acompañamiento con Cannabis en niños con epilepsia resistente. Marcelo Devilat et al.
en las citas anteriores, no se informa acerca de la concentración del cannabidiol indicado.
En el año 2013 se publicó (31) un trabajo sobre ni-ños con epilepsia resistente tratados con cannabi-diol en aceite fabricado en forma artesanal. Estos niños pertenecían a un grupo de padres que com-partía información por Facebook, y 19 de ellos fue-ron captados por los investigadores. La información disponible revela que antes del uso de cannabis, los niños habían recibido 12 AE cada uno en promedio. El tipo de epilepsia incluyó las siguientes: Síndro-me de Dravet, 13 pacientes; Síndrome de Doose, 4; Síndrome de Lennox-Gastaut, 1; y epilepsia idiopática, 1 paciente. Los investigadores encon-traron disminución de la frecuencia de las crisis en 16 niños; en 3 no hubo variación. De los 16 con reducción de crisis, 2 quedaron libres de crisis; en 8 hubo reducción de ellas en más del 80%; y en 6 hubo una reducción del 25 al 60 %. De los 19 ni-ños, se observó una disminución de las conductas de auto estimulación en 6; mejoría del hábito de sueño, en 12; aumento de la alerta, en 13; y mejor humor en 14 pacientes. Como efectos adversos se reportó disminución del apetito, en 1 niño; fatiga en 3 y somnolencia en 7 niños. A pesar de las fallas metodológicas (contacto por Facebook, ausencia de línea de base, no-informa-ción sobre los AE que recibían, no-randomización, no-uso de placebo, ausencia de datos sobre dosis/concentración), este trabajo permite concluir que algunos pacientes responden a la ingesta de canna-bis, incluso quedando sin crisis, como sucedió en 2 niños. Sin embargo, no conocemos el tiempo de observación post inicio del tratamiento, y tampoco se conoce cuanto era el lapso máximo que habían estado sin crisis antes de iniciar la cannabis (a obje-to de descartar que el efecto sea una evolución na-tural de la enfermedad). Por otra parte, un grupo de pacientes, 8 de 19, tuvieron una reducción de crisis superior al 80%. No hay que olvidar que los niños eran portadores de epilepsias de difícil manejo y en-cefalopáticas, y las investigaciones en este campo aprueban los AE si reducen las crisis en más de un 50%. Hay que destacar también los efectos positi-vos sobre el sueño, el humor y la alerta. Como no se sabe la composición y la concentración de CBD y THC que ingerían los pacientes, no es posible de-terminar el responsable de los efectos beneficiosos colaterales. Entre los efectos adversos solo la som-
nolencia tiene un número relevante, pero también la pueden originar otros AE.
El caso de Charlotte Figi, de 5 años, (12) se pre-senta aquí por constituir un caso emblemático pu-blicado en la revista Epilepsia. Esta paciente, por-tadora de un síndrome de Dravet confirmado con estudio del gen SCN1A, presentaba hasta 50 crisis tónico-clónicas generalizadas al día, resistentes a los AE. Se inició tratamiento sublingual de extracto con alta concentración de cannabis, a dosis de 4mg/lb, con lo se redujeron los episodios, a 2 o 3 crisis nocturnas. Al bajar la dosis a 2mg/lb reaparecieron 5 a 10 CTCG al día, con lo cual se retornó a la do-sis anterior y resultados anteriores. Al efectuar la misma reducción de dosis en 2 ocasiones distintas separadas por tiempo, se originaba el mismo efecto. El control total para observar efecto de cannabis fue de 20 meses. El caso ilustra la sensibilidad de la cannabis a las CTCG, así como la dependencia de la dosis para obtener un buen resultado. Además, sugiere trabajar con extractos de plantas farmacoló-gicamente elaboradas. Otro trabajo fue recientemente publicado por GW Pharmaceutical, (39) siendo realizado en el Centro Médico Langone de la Universidad de Nueva York y la Universidad de California, por el Dr. Orrin De-vinsky. Informa de 62 pacientes, que se redujeron a 27, mayoritariamente niños con epilepsia resistente (9 con síndrome de Dravet), con una edad promedio de 10,2 años. Estos pacientes recibieron, con una línea de base de 4 semanas, el fármaco Epidiolex® (cannabidiol puro, aún no aprobado por la FDA). Los resultados revelaron que, tras 12 semanas, la muestra total disminuyó sus crisis en al menos 44%, y 4 enfermos estaban libres de crisis. Los 9 niños portadores de síndrome de Dravet disminuyeron sus crisis convulsivas en un 52%, y al final del estudio 3 pacientes estaban sin crisis. Los efectos adver-sos (en 62 pacientes) fueron: somnolencia en 40%, fatiga en 26%, diarrea en 16%, disminución del apetito en 11% y aumento del apetito en 10%. En la mayoría de los pacientes (80%) los efectos ad-versos fueron leves, y aunque en 7 fueron serios, no hubo retiro de medicamentos. Un paciente presentó muerte súbita inexplicable (SUDEP) no atribuible al Epidiolex®. Como defectos a mencionar, la co-municación no revela la dosis, no hay placebo ni doble ciego, no informa la composición exacta del producto y no hubo randomización.

16
Revista Chilena de Epilepsia Año 14, Nº 3, Diciembre 2014
2.2 Perspectivas a futuro del cannabidiol María Roberta Cilio, de la Universidad de Cali-fornia (6) y Orrin Devinsky de la Universidad de Nueva York (11), han recibido aprobación por la FDA para realizar una investigación con Epidiolex ® en niños de 1 a 18 años con epilepsia refractaria, síndrome de Dravet y de Lennox-Gastaut. El tra-bajo lo realizarán 6 instituciones hospitalarias con 25 pacientes cada una, con el objetivo de determi-nar la tolerabilidad, seguridad, frecuencia de crisis, dosis óptima del medicamento, e interacciones. El inicio del ensayo será en Enero de 2015. Estudio será randomizado y doble ciego, con cannabidiol (GWP42003-P) vs placebo. Los autores plantean estrictos criterios de inclusión y exclusión, así como los relacionados con el seguimiento primario y se-cundario de los niños. (19, 20, 40, 41, 42, 43). La autorización otorgada en Nueva York al trabajo precedente se acompañó del otorgamiento en Enero de 2014 de un programa que permitiría a 20 hospi-tales recetar marihuana, para uso médico en cáncer y otras enfermedades graves. (44). En conclusión, la epilepsia refractaria en niños es una patología a la que la medicina tradicional no ha logrado dar una respuesta satisfactoria, lo que im-plica un enorme sufrimiento de quienes la padecen y sus familias. Entre otras múltiples alternativas te-rapéuticas algunos de los padres de estos niños han decidido utilizar, como medida compasiva, canna-bidiol. Existe evidencia a nivel de ciencias básicas que relaciona el uso del cannabidiol con una dis-minución de crisis epilépticas y de su mortalidad. En relación a la experiencia clínica y evidencia en humanos, sólo disponemos de estudios pequeños, con importantes fallas metodológicas; y de reportes de casos, que mostrarían un efecto beneficioso del cannabidiol como tratamiento coadyudante de la epilepsia. Estudios de mayor peso metodológico es-tán siendo llevados a cabo. Paralelo a la investiga-ción científica, muchos padres de niños con epilep-sia refractaria han decidido, por su propia voluntad, administrar cannabidiol. Nos parece fundamental seguir estas líneas de investigación y, mientras no tengamos mayores respuestas médico-científicas, poder acompañar a nuestros pacientes y sus fami-lias que han optado por alternativas terapéuticas di-ferentes a la medicina tradicional.
REFERENCIAS
1 Chiofalo N, Kirshbaum A, Schoenberg B, Oli-vares, Valenzuela B et al.: Estudio epidemioló-gico de las enfermedades neurológicas en San-tiago Metropolitano. Rev Chil Neuro-Psiquiat 1992; 30: 335-341.
2 Grupo Normativo Nacional para la Epilepsia en Chile. 2002 Política y Plan Nacional para la Epilepsia en Chile. Coordinador: Marcelo De-vilat. Ministerio de Salud. Santiago de Chile. 2002.
3 Devilat M, Carrizosa J. Epilepsia refractaria en niños. Visión retrospectiva sobre su diagnós-tico y manejo. Rev Chil Neuro-Psiquiat 1996; 34: 241-246.
4 Kwan P, Arzimanoglou A, Berg AT, Brodie M, Hauser WA et al. Definition of drug resistant epilepsy: Consensus proposal by the ad hoc Task Forceo f the ILAE Commission on Thera-peutic Strategies. Epilepsia 2010; 51(6): 1069-1077.
5 Ministerio de Salud. Garantías Explícitas en Salud. Guía Clínica. Epilepsia No Refractaria en Personas desde 1 año y menores de 15 años. Santiago: Minsal 2008.
6 Cilio MR, Thiele EA, Devinsky O. The case for assessing cannabidiol in epilepsy. Epilepsia 2014; 55(6): 787-790.
7 Devilat M, Rivera G, Gómez V, Sepúlveda JP. Mortalidad en niños con epilepsia. Estudio prospectivo. Rev Neurol 2004; 38: 607-614.
8 Devilat M, Gómez V. Tratamiento de 60 niños con epilepsia resistente. Rev Chil Epilepsia 2010; 10(2): 5-13.
9 Boon P, Vonck K, De Herch, V, Van Dycke A, Goethals M et al. Deep Brain Stimulation in Pa-tients with Refractory Temporal Lobe Epilepsy. Epilepsia 2007; 48(8): 1551-1560.
10 Lin LC, Lee WT, Wang CH, Chen HL, Wu HC, et al. Mozart K. 448 acts as potencial add-on therapy in children with refractory epilepsy. Epilepsy Behav 2011; 20: 490-493.
11 Devinsky O, Cilio MR, Cross H, Fernández-Ruiz J, French Ch et al. Cannabidiol: Phar-macology and potencial therapeutic role in epilepsy and other neuropsychiatric disorders. Epilepsia 2014; 55(6): 791-802.
12 Maa E, Figi P. The case for medical marijuana in epilepsy. Epilepsia 2014; 55(6): 783-786.

17
Tratamiento compasivo y de acompañamiento con Cannabis en niños con epilepsia resistente. Marcelo Devilat et al.
13 Robson PJ. Therapeutic potencial of cannabi-noid medicines. Drug Test Analysis 2013; DOI 10.1002/dta 1529.
14 Zhu HL, Wan JB, Wang YT, Li BC, Xiang C et al. Medicinal compounds with antiepileptic/an-ticonvulsant activities. Epilepsia 2014; 55(1): 3-16.
15 www.farmaciamapuche.cl16 www.repcheca.com17 www.telesurtv.net 18 www.OMS.com 19 www.medscape.com20 Clinicaltrials.gov21 Declaración conjunta sobre Cannabis.
29.11.2014.22 www.sag.cl23 Koppel BS, Brust JC, Fife T. Systematic re-
view: Efficacy and safety of medical marijua-na in selected neurologic disorders: Report of the Guideline Development Subcommittee of American Academy of Neurology. Neurology 2014; 82: 1556-1563
24 Gordon E, Devinsky. Alcohol and marijuana: Effects on the epilepsy and use by patients with epilepsy. Epilepsia 2001; 42(10): 1266-1272.
25 Laprairie R., Kelly M., Denovan-Wright E. The dynamic nature of type 1 cannabinoid receptor (CB1) gene transcription. Brit J Pharmacol 2012; 167: 1583-1595
26 Marsicano G, Goodenough S, Monory K, Her-mann H, Eder M et al. CB1 Cannabinoid recep-tors and on-demand defense against exitotoxi-city. Science 2003; 302: 84-88
27 Hill T, Cascio M, Romano B, Duncan M, Pert-wee R. et al. Cannabidivarin-rich cannabis ex-tracts are anticonvulsant in mouse and rat via CB1 receptor-independent mechanism. Brit J Pharmacol 2013; 170: 679-692.
28 Fernández-Ruiz J. Fármacos cannabinoides para las enfermedades neurológicas : ¿que hay detrás? Rev Neurol 2012; 54(10): 613-628.
29 Devinsky O, Ghosh P, Bhattacharya SK. Anti-convulsant action of cannabis in the rat: role of monoaminas in the brain. Psychopharmacology 1978; 59: 293-297.
30 Segal M, Edelstein EL, Lerer B. Interaction between delta-6-tetrahidrocannabidiol and li-lium at the blood brain barrier in rats. Expe-rientia 1978; 34: 629.
31 Porter B, Jacobson C. Report of a parent sur-vey of cannabidiol-enriched cannabis use in pediatric treatment-resistant epilepsy. Epilepsy Behav 2013; 29: 574-577.
32 Quintanilla C, Devilat M. Costo del tratamien-to de la epilepsia tratada con antiepilépticos de segunda generación, no proporcionados por el Hospital. Poster en XXXII Congreso de la So-ciedad de Psiquiatría y Neurología de la Infan-cia y Adolescencia. Punta Arenas 2014.
33 Devilat M, Riffo C, Cuadra L. Evaluación de niños con epilepsia resistente a los antiepilépti-cos derivados a cirugía. 1996 a 2010. Rev Chil Epilepsia 2013; 13 (1): 40-49.
34 Devilat M, Troncoso M. Epilepsia con retardo mental en mujeres. Rev Chil Epilepsia 2014: 14(1): 4-13.
35 Matthern G, Nehlig A, Sperling M. Cannabi-diol and medical marijuana for the treatment of epilepsy. Epilepsia 2014; 55(6): 781.
36 Szaflarsky JP, Bebin EM. Cannabis, cannabi-diol, and epilepsy. Epilepsy Behav 2014 DOI. org. /10.1016/j.yebeh.2014.08.135.
37 Gloss D, Vickrey B. Cannabinoids for epi-lepssy. Cochrane Database Syst Rev 2012; 6: CD009270)
38 Pellicia A, Grassi G, Romano A, Crocchialo P. Treatment with CBD in oily solution og drug resistant pediatric epilepsias 2005. www.canna-bis-med.org
39 www.gwpharm.com40 www.jacksarmy.org 41 www.ucsf.edu 42 www.nj.com43 www.thestar.com44 www.elcomercio.pe45 Rahbari M, Rahbari NN. Uso compasivo de
productos medicinales en Europa: situación ac-tual y perspectivas. Boletín de la Organización Mundial de la Salud 2011; 89: 163. www.who.int

18
Revista Chilena de Epilepsia Año 14, Nº 3, Diciembre 2014Trabajo Original
Experiencia en Cirugía de Epilepsia Paliativa en Ins-tituto de Neurocirugía Asenjo: Estimulador de Nervio Vago (VNS).Hernán Acevedo1, Osvaldo Olivares2, Bárbara Garcés3, Lientur Taha4, Viviana Venegas5.Correo Hernán Acevedo: [email protected]
ABSTRACT
At the Institute of Neurosurgery Asenjo, the epilepsy surgery program currently underway in both adult and pediatric team, the latter being a vast experien-ce. Within palliative neurosurgical options, we have mainly callosotomy and vagus nerve stimulator. This paper presents part of our initial results, in the interval between the years 2012 to 2014 patients evaluated under protocol period, analyzing results of 17 patients, average age of 19.8 years (range 6-41 years), mean follow up of 16.2 months (range 5-34 months). The results of crisis control mana-gement >70% was obtained in 7 patients, >50% <70% in 6 cases, inadequate control or greater number of crises in 4 patients.
This is the first report of results in epilepsy surgery using vagal stimulator of our team. Current expe-rience obtained with this technique, we hope to be contribution to global growth theme refractory epi-lepsy, especially in our public system.
RESUMEN
En el Instituto de Neurocirugía Asenjo, se desarro-lla actualmente el programa de cirugía de epilepsia, tanto en el equipo adulto como de pediatría, siendo este último de una vasta trayectoria. Dentro de las opciones neuroquirúrgicos de carácter paliativo, tenemos principalmente la callosotomía y el esti-mulador de nervio vago. Este trabajo presenta parte de nuestros resultados iniciales, dentro del periodo comprendido entre los años 2012 al 2014 de pacien-tes evaluados bajo protocolo, analizando resultados
de 17 pacientes, promedio de edad de 19.8 años (rango de 6 a 41 años), seguimiento promedio de 16.2 meses (rango de 5 a 34 meses). Los resultados de control de crisis >70% se obtuvo en 7 pacientes, >50% <70% en 6 casos, control inadecuado o ma-yor número de crisis en 4 pacientes. Este es el primer informe de resultados en cirugía de epilepsia con el uso de estimulador vagal de nues-tro equipo. La experiencia actual, obtenida con esta técnica, esperamos sea aporte para el crecimiento global del tema de epilepsia refractaria, principal-mente en nuestro sistema público.
INTRODUCCIÓN
Aproximadamente un 30% de los pacientes epilép-ticos sufren de refractariedad a fármacos antiepilép-ticos (FAE) (1, 2). Algunos pacientes son candida-tos a cirugías resectivas, tales como lesionectomías o lobectomías temporales, cuando hay clínica, estu-dio electroencefalográfico e imagenológico compa-tible (2). El estimulador del nervio vago (VNS) es una alternativa de tratamiento quirúrgico paliativo para los pacientes con epilepsia refractaria que no son candidatos a cirugía resectiva, describiéndose su eficacia en epilepsias parciales y generalizadas, tanto en adultos como en población pediátrica. EL VNS consiste en la estimulación eléctrica intermi-tente del nervio vago izquierdo a nivel cervical, que se lleva a cabo a través de una cirugía que no reviste mayor complejidad para un Neurocirujano entrenado, y que tiene un bajo porcentaje de com-plicaciones, la mayoría de ellas locales y transito-rias (infecciones, disfonía, ronquera, bradicardia). El mecanismo de acción exacto del VNS no está completamente dilucidado, sin embargo se postula la desincronización del circuito tálamo cortical, a través de aferencias desde el núcleo del tracto soli-tario, con modulación de neurotransmisores, princi-palmente norepinefrina y serotonina, como una de las principales vías implicadas (1,2,3,4). En cuanto
1. Neurocirujano, Instituto de Neurocirugía Asenjo.2. Neurólogo, Instituto de Neurocirugía Asenjo3. Becada de Neurocirugía, Instituto de Neurocirugía Asenjo.4. Neurocirujano, Instituto de Neurocirugía Asenjo5. Neurología, Hospital San Juan de DiosRecibido 3 de Dic. Aceptado 27 de Dic. 2014Los autores declaran no tener conflictos de intereses.

19
Experiencia en cirugía de epilepsia paliativa en Instituto de Neurocirugía Asenjo. Hernán Acevedo et al.
a la tasa de reducción de crisis convulsivas, existen 2 estudios clase I de evidencia (5, 6) que demuestran un 30.9% de promedio de reducción de crisis (5), y un 28% (6) en el grupo de pacientes que recibieron una alta estimulación del VNS. Posteriormente se han realizado múltiples estudios clase II y III, cuyos resultados apuntan a un porcentaje de reducción de frecuencia de crisis de aproximadamente un 50% a 5 años, y de un 6% a un 13% de ausencia de crisis a un año de seguimiento (7). Se ha reportado que los resultados de reducción de crisis aumentan pro-gresivamente con los años post-implantación. En un estudio con 440 adultos con epilepsia parcial, la tasa de >50% de reducción de crisis aumentaba 7% por año desde el año 1 al 3 post operatorio (8). Se han descrito algunos factores predictores de buena respuesta, como es la presencia de malformaciones del desarrollo cortical, y la ausencia de descargas epileptiformes interictales bilaterales (9). Además se ha implicado al VNS con la mejoría del ánimo de los pacientes operados, corroborado por scores va-lidados, hecho descrito como un beneficio adicional de esta técnica (7).
En el Instituto de Neurocirugía se lleva a cabo este procedimiento en adultos y pacientes pediátricos evaluados por el equipo de cirugía de Epilepsia. Es así como nos planteamos describir los resultados de los pacientes operados de VNS en nuestra institu-ción.
MATERIALES Y MÉTODOS
Se describen 17 pacientes, adultos y pediátricos, operados con instalación de estimulador de nervio vago en el Instituto de Neurocirugía Asenjo con diagnóstico previo de epilepsia refractaria, eva-luados por el equipo de Cirugía de Epilepsia, que cumplan con criterios de refractariedad y estudios pre-quirúrgicos acorde a protocolo, en el periodo de diciembre del 2012 hasta mayo del 2014, con ante-cedentes y seguimiento completo en ficha clínica.
Criterios de selección y características clínicas
Dentro de las variables presentes en nuestros pa-cientes es que sean refractarios a tratamiento médi-co, según definición de ILAE, estudio electrofisio-lógico standard y video monitoreo electrofisiológico con grabación de crisis de al menos 2, resonancia cerebral con protocolo de epilepsia, evaluación pre-via por neurólogo y neurocirujano con formación
en epilepsia, evaluaciones fonoaudiológicas y kine-siológica habituales.
Se objetivaron variables como tipo y frecuencia de crisis, trauma craneal o status asociado a cri-sis, número de fármacos, comorbilidad asociada, antecedente de cirugía de epilepsia, descripción de resonancia cerebral y del estudio electrofisiológico.
La activación del VNS, los controles clínicos y ajustes electrofisiológicos fueron realizados por 2 neurólogos epileptólogos (adulto y pediatras) en nuestro hospital una vez por mes hasta la fecha de evaluación de este trabajo, en coordinación con em-presa que vende VNS, quienes poseen dispositivos técnicos para control.
La respuesta a tratamiento quirúrgico fue objetiva-da con revisión de ficha clínica, además de entre-vista vía telefónica a tutores o padres del paciente, haciendo referencia a número de crisis previo y pos-terior a instalación del estimulador hasta el tiempo de la entrevista (número absoluto y porcentaje de reducción con respecto a basal), complicaciones asociados a procedimiento o posterior a uso de VNS, tipo de crisis predominante post instalación de VNS, apreciación subjetiva de tutores relaciona-do con capacidades de relación interpersonal o de-terioro o mejoría cognitiva del paciente y si existe variación en el peso pre y post quirúrgico.
Finalmente se preguntó a tutores, dado su experien-cia personal actual con uso del VNS, y si aprobarían o no instalación de VNS en su familiar.
RESULTADOS
El grupo de pacientes evaluado (n: 17) presenta edad entre 6 a 41 años, promedio 19,8 años,12 pa-cientes mayores de 16 años, 5 pacientes menores de 12 años. Seguimiento promedio de 16 meses (5 a 34 meses). Dentro de los antecedentes a destacar se ob-tuvo Sd. West, neuroinfección, hemorragia neona-tal y hemofilia, sólo 1 paciente no presentó retardo cognitivo en su ficha (16 presentaban retardo mo-derado o severo). En estudio imagenológico, sólo 3 pacientes con resonancia magnética (RM) normal. Se evidencia una variada presentación y superposi-ción de tipos de crisis, siendo principalmente crisis tónico clónico generalizado o CTCG (31%), atóni-cas (12%) y crisis parcial complejas o CPC (24%) las de mayor frecuencia, existiendo además presen-

20
Revista Chilena de Epilepsia Año 14, Nº 3, Diciembre 2014
cia de crisis tipo espasmos, crisis parcial secunda-riamente generalizada, tónicas y miclonías. Sólo en 4 pacientes tutores describieron un solo tipo de cri-sis (CTCG). En el 100% de los pacientes existió uso de 2 o más fármacos antiepilépticos, y en múltiples combinaciones.
El 82% (N:14) presentaba crisis diarias (incluso un caso >300 crisis diarias) y el 53%, tenía como ante-cedente status en algún momento de su evolución.El 59% de nuestros pacientes había sido operado de cirugía de epilepsia, y de este grupo, el 55% fue ca-llosotomía y el 45% alguna variante de cirugía tipo resectiva. La principal actividad electrofisiológica evidenciada fue actividad irritativa bilateral o gene-ralizada. Sólo una paciente inicia epilepsia a los 17 años, todos los demás inicio de enfermedad antes de los 12 años.
Grafico nº1
Gráfico nº 2
Técnica quirúrgica y complicaciones inmediatas
En relación a la cirugía, se realizó técnica quirúrgi-ca descrita en la literatura, con sugerencias descritas por empresa generadora de VNS (Cyberonics). El procedimiento fue realizado por neurocirujanos de equipo adulto y pediátrico, según grupo etario de pacientes, con experiencia en cirugía de epilepsia. Habitualmente la hospitalización consideró 2 días, sin necesidad de UCI o cama intermedio, sólo post operado y sala básica.
No existieron complicaciones infecciosas que pro-vocaran retiro de VNS, se registró un caso de hema-toma pectoral en paciente con hemofilia, sin ser de resorte quirúrgico, y un caso de disfonía transitoria post-quirúrgica.

21
Experiencia en cirugía de epilepsia paliativa en Instituto de Neurocirugía Asenjo. Hernán Acevedo et al.
Resultados post-quirúrgicos
En relación a resultados post quirúrgicos, se obtuvo frecuencia y tipo de crisis a través de revisión de fi-chas y entrevista telefónica con encuesta uniforme a tutores o padres (2 entrevistadores), el porcentaje de reducción de crisis fue calculado a partir de número de crisis pre-quirúrgico.
El tiempo de seguimiento fue de 16,2 meses, en rango de 5 a 34 meses. Se observó una reducción de crisis mayor a 70% en 7 pacientes, con un segui-miento de 17.1 meses (rango 5 a 34 meses), reduc-ción entre 50% y 69% en 6 casos, con seguimiento promedio 16.8 meses, rango 10 a 24 meses, dos pa-cientes sin cambios, con seguimiento promedio de 12 meses (7 y 17 meses), y finalmente, evolución con mayor frecuencia de crisis, en 2 casos promedio de seguimiento 15 meses (12 y 18 meses).
Se obtuvo información sobre mejoría cognitiva apreciada subjetivamente por tutores, como tam-bién si existió evidencia objetiva de reducción de peso. El 65% de los padres o tutores describió me-joría cognitiva evidente y en el 29% existió reduc-ción de peso al menos de 5 kg.
Tabla nº 1
% Reducción de crisis Nº de pacientes
>=70% 7<70% >=50% 6<50% 0IGUAL 2PEOR 2
Grafico nº3
El 100% de los pacientes mantiene 2 o más fárma-cos durante su evolución post-quirúrgica.
En relación a la pregunta si aceptarían nuevamente la realización del procedimiento quirúrgico de ins-talación de VNS, teniendo la información y expe-riencia actual, la respuesta fue positiva en el 82% de los encuestados (tutores de pacientes).
Gráfico nº4
DISCUSIÓN
Este es el primer trabajo con datos del sistema pú-blico que describe la experiencia en el uso de es-timulador de nervio vagal en población adulta y pediátrica, generado de la unidad de cirugía de epi-lepsia del Instituto de Neurocirugía Asenjo basados en protocolo disponible en nuestro hospital. En el grupo evaluado podemos destacar que el inicio de presentación de crisis es fundamentalmente en edad pediátrica, y asociado a esto, el 94% presentó retar-do cognitivo como antecedente, con uso de 2 o más fármacos en el 100% de ellos. Estos datos coinciden con observaciones de autores que han relacionado la persistencia de crisis como factor crónico de de-terioro neurológico (10) (Chen et al, 2012), afecta-do por la crisis en sí mismas, la actividad interictal

22
Revista Chilena de Epilepsia Año 14, Nº 3, Diciembre 2014
(11) (Holmes et al, 2013) y, el uso de fármacos en forma prolongada.
La indicación de VNS inicialmente fue aprobado por la FDA el año 1997 para pacientes mayores de 12 años y epilepsia focal, pero su uso en población pediátrica hoy por hoy no tiene grandes distractores, siendo una recomendación de la academia america-na de neurología (agosto 2013, Epilepsy Currents) basado en 14 estudios clase III, por lo tanto no exis-te contraindicación en su uso.
En el análisis de nuestra población, tanto adulta como pediátrica, en relación al tipo de crisis, se ob-servaron diversos tipos de presentación clínica, no afectando los resultados finales de la respuesta al dispositivo VNS, siendo estos resultados compati-bles con datos descritos por Englot en meta-análisis publicado en el 2011 (12) (dic, Journal of Neurosur-gery), donde el análisis de 787 pacientes demostró disminución de crisis en el 53,7% de ellos.
En el 59% de nuestros pacientes presentó antece-dente de cirugía de epilepsia, siendo el 55% callo-sotomía. Amar y Apuzzo (13) (Neurosurgery, 2008) compararon grupos de cohorte no consecutivos, la efectividad de tratamiento VNS para pacientes sin (n:3822) y con (n:921) cirugía de epilepsia previa, no existiendo gran diferencia entre ellos (control de crisis >50% en el 55.1% con cirugía previa, y sin antecedente, en el 62.2%).
El grupo de datos registrados en este grupo de pa-cientes, tales como el inicio de crisis, diversidad ti-pos de crisis, uso de múltiple fármacos y anteceden-tes de cirugía de epilepsia, nos permiten concluir que el grupo analizado corresponde a un segmen-to de la población no solamente refractarios, sino que además catastróficos ya que no han podido ser controlados pese a múltiples estrategias médicas o quirúrgicas realizadas. Pese a esto, los resultados de respuesta de control de crisis superior al 50% fue-ron observados en el 76% de la población evaluada.
El tiempo en seguimiento de estos pacientes es un factor clave que debemos considerar obligatoria-mente, considerando que la mayoría de los traba-jos y observaciones en el mundo describen que el tiempo óptimo para el control de crisis es posterior al año de seguimiento, con mejoría incluso a los 2 y tres años. La serie de Seul en Korea (14) (n:20, pu-blicados en Journal of Epilepsy Research), alcanzan
control de crisis superior a 50% al 1ª, 2ª y 4ª año, de 40%, 50% y 70% respectivamente; en la serie de Uthman de 48 pacientes (15) (una de las series con seguimiento más prolongada), la respuesta fue me-jor con el tiempo, presentaron disminución de crisis en un 26%, 30% y 52% al primer, quinto y doceavo año de tratamiento con VNS; por lo tanto, espera-mos que el control del crisis sea, probablemente, aún mejor mientras más tiempo transcurra. Esta observación presenta además, un punto importante para nuestra práctica clínica, que radica en pacien-cia y cautela de médicos y pacientes para no reali-zar cambios abruptos en perfil electrofisiológico de programa del VNS o bien racionalizar muy bien la decisión de apagar el estimulador si no existen com-plicaciones de importancia vital y falta de respuesta esperada. Con respecto a las diferentes modalidades de ajuste de VNS, será objetivo de publicación en actual desarrollo.
Nuestras complicaciones están acorde a experiencia internacional, siendo ronquido y disfonía las prin-cipales, no debiendo retirar ningún VNS por infec-ción.
Los fracasos de control de crisis, están dentro de las posibilidades estadísticas, dado que es un me-dida paliativa, pero el tiempo de observación aún es precoz motivo por el cual se mantendrá su se-guimiento (tiempo promedio de evolución de los no respondedores fue de 12 meses).
En conclusión, consideramos que el estimulador de nervio vago es una medida paliativa para pacien-tes epilépticos refractarios, y en general, catastró-ficos, es una herramienta útil, de fácil intervención y manejo peri-operatorio, pero que en la actualidad dado su costo y difícil acceso para el sistema pú-blico, y el privado, por restricciones económicas, debe estar disponible en centros con equipos mul-tidisciplinarios formados para la toma decisión so-bre que paciente será el más beneficiado, tomado en consideración riesgos, beneficios y planificación de inversión de recursos, siempre escasos, y que consideren que la instalación es solo el principio de un largo camino de seguimiento y decisiones caso a caso.
Esperamos seguir analizando datos para otorgar a autoridades que determinan políticas de distribu-ción de recursos, información con datos locales y de esa forma, adopten las mejores decisiones.

23
Experiencia en cirugía de epilepsia paliativa en Instituto de Neurocirugía Asenjo. Hernán Acevedo et al.
BIBLIOGRAFÍA
1. Mapstone T. Vagus nerve stimulation: current Concepts. Neurosurg Focus 25 (3):E9, 2008.
2. Connor D. Vagal nerve stimulation for the treatment of medically refractory epilepsy: a review of the current literature. Neurosurg Fo-cus 32 (3):E12, 2012.
3. Lulic D. Ahmadian A. Vagus nerve stimulation. Neurosurg Focus 27 (3):E5, 2009.
4. Ben-Menachem E. Vagus-nerve stimulation for the treatment of epilepsy. Lancet Neurology 2002; 1: 477–82.
5. Ben-Menachem E, Manon-Espaillat R, Ris-tanovic R, et al. Vagus nerve stimulation for treatment of partial seizures: 1, a controlled study of effect on seizures. Epilepsia 1994; 35: 616–26.
6. Handforth A, De Giorgio CM, Schachter SC, Uthman BM, Naritoku DK, Tecoma ES, et al: Vagus nerve stimulation therapy for partial-on-set seizures: a randomized active-control trial. Neurology 51:48–55, 1998.
7. Morris G, et al. Evidence-Based Guideline Up-date: Vagus Nerve stimulation for the Treatment of Epilepsy. Epilepsy Currents, Vol. 13, No. 6 (November/December) 2013 pp. 297–303.
8. Morris GL III, Mueller WM; for Vagus Ner-ve Stimulation Study Group E01-E05. Long-
term treatment with vagus nerve stimulation in patients with refractory epilepsy. Neurology 1999;53:1731–1735.
9. Ghaemi K, Elsharkawy AE, Schulz R, Hop-pe M, Polster T, Pannek H, et al: Vagus nerve stimulation: outcome and predictors of seizu-re freedom in long-term follow-up. Seizure 19:264–268, 2010.
10. Chen CY et al, Child Neurology Society VNS study group. Short-term results of vagus nerve stimulation in pediatric patients with refractory epilepsy. Pediatrics. Neonatol. 2012;53:184—7.
11. Holmes et al, Epilepsia 54, Supl. 52, 60-62, 2013
12. Englot et al, Meta-analysis Long-Term Outco-me of Vagus Nerve Stimulation for Refractory Epilepsy: A Longitudinal 4 year Follow-up, J Neurosurg 115:1248–1255, 2011
13. Amar et al, Vagal nerve stimulation theraphy after failed cranial surgery for intractable epi-lepsy, Feb,62, Suppl 2: 506-13, Neurosurgery 2008
14. Su Jung Choi, Study in Korea, Journal of Epi-lepsy Research Vol. 3, No. 1, 2013
15. Uthman et al, Neurology 2004, Effectiveness of vagus nerve stimulation in epilepsy patients: a 12 year obervation. September 28, 2004, vol.63 nº 6 1124-1126.

24
Revista Chilena de Epilepsia Año 14, Nº 3, Diciembre 2014Trabajo Original
Caracterización de un grupo de pacientes con convul-siones febrilesPatricia Alfaro1, Marcelo Devilat2
Centro de Epilepsia infantil. Servicio de Neurología y Psiquiatría, Hospital Luis Calvo Mackenna. Santia-go, Chile.Correo: [email protected]
ABSTRACT Febrile seizures are common in childhood. The ob-jective of this publication was to characterize pa-tients diagnosed with febrile seizure controlled at our Center.
Retrospective study in which the available medical records of one hundred twenty five children with fe-brile seizures controlled between July 2005 and July 2012 were reviewed. The time control was on ave-rage 3.4 years (range 6 months to 12 years). Two ty-pes of treatment were considered: acute treatment of seizures and treatment of recurrences (prophylaxis of recurrences). The latter was divided into two ty-pes: intermittent (at the beginning of each fever) and continuous (permanent use of valproic acid).
Sixty-five patients (52%) were men and sixty (48%) were women. Thirty-six (29%) had a history of re-latives with epilepsy or febrile seizure. Patients had five crisis on average (range 1-8 crisis), presenting the first episode on average seventeen months. Ten patients (8%) had more than five attacks in total du-ring the observation time. Seventy-nine (63%) had only simple febrile seizures, thirty-two (26%) only complex and fourteen (11%), both types of crisis. Thirteen patients (10%) had crisis more than ten minutes at some point in their evolution, which were managed as febrile status epilepticus. Fourteen (11%) had delayed psychomotor development. One hundred twenty-two children (97%) had a normal EEG. Eighty-four patients (67%) were treated with rectal diazepam in case of crisis, thirteen (10%) with intermittent rectal diazepam prophylaxis and twenty eight (22%) with valproic acid. Of all pa-tients, seventy-four (59%) completed the treatment properly. Forty-six patients (37%) had recurrences
in a time of 3.4 years median follow. Of these, 48% were treated only with rectal diazepam in case of crisis. Of all children, three (2%) progressed with epilepsy and six (5%) with febrile seizures plus. Thirty (24%) were discharged, fifty-one (41%) dis-continued controls and forty-four (35%) are still on track.
Although most patients had simple febrile seizures, a third of the group had recurrences. One in ten pre-sented febrile status epilepticus and a low number evolved with epilepsy or febrile seizures plus.
Keywords: febrile seizures, treatment, epilepsy.
RESUMEN
Las convulsiones febriles son frecuentes en la in-fancia. El objetivo general de esta publicación fue caracterizar a los pacientes con diagnóstico de con-vulsión febril controlados en el Centro de Epilepsia Infantil del Hospital Luis Calvo Mackenna.
Estudio retrospectivo en el cual se revisaron las fi-chas clínicas disponibles de ciento veinticinco niños con convulsiones febriles controlados entre Julio de 2005 y Julio de 2012. El tiempo de control fue en promedio de 3,4 años (rango 6 meses a 12 años). Se consideraron dos tipos de tratamiento: tratamiento agudo de las crisis y tratamiento de las recurrencias (profilaxis de las recurrencias). El último se dividió en dos tipos: intermitente (al inicio de cada cuadro febril) y continua (uso permanente de ácido valproi-co como fármaco antiepiléptico).
Sesenta y cinco pacientes (52%) fueron hombres y sesenta (48%) fueron mujeres. Treinta y seis (29%) tenían antecedentes de familiares directos con epi-lepsia o convulsión febril. Los pacientes tuvieron cinco crisis en promedio (rango 1-8 crisis), presen-tándose el primer episodio en promedio a los dieci-siete meses de vida. Diez pacientes (8%) tuvieron más de cinco crisis en total durante el tiempo de ob-
1. Residente Neurología pediátrica. Universidad de Chile2. Neurólogo pediátrico.Recibido15 Dic. Aceptado 28 Diciembre 2014.Los autores declaran no tener conflictos de intereses.

25
Caracterización de un grupo de pacientes con convulsiones febriles. Patricia Alfaro et al.
servación. Setenta y nueve (63%) presentaron sólo convulsiones febriles simples, treinta y dos (26%) sólo complejas y catorce (11%), ambos tipos de cri-sis. Trece enfermos (10%) tuvieron crisis de más de diez minutos en algún momento de su evolución, los que fueron manejados como estatus epiléptico febril. Catorce (11%) tenían retardo del desarrollo psicomotor. Ciento veintidós niños (97%) presen-taron un EEG normal. Ochenta y cuatro pacientes (67%) fueron tratados con diazepam rectal en caso de crisis, trece (10%) con diazepam rectal como profilaxis intermitente y veintiocho (22%) con áci-do valproico. Del total de enfermos, setenta y cua-tro (59%) cumplió adecuadamente el tratamiento. Cuarenta y seis enfermos (37%) tuvieron recurren-cias en un tiempo de control promedio de 3,4 años. De éstos, el 48% estaba en tratamiento sólo con dia-zepam rectal en caso de crisis. Del total de niños, tres (2%) cursaron con epilepsia y seis (5%) con convulsión febril plus. Treinta (24%) fueron dados de alta, cincuenta y uno (41%) abandonaron contro-les y cuarenta y cuatro (35%) se encuentran aún en seguimiento.
Aunque la mayoría de los pacientes tuvo convul-siones febriles simples, un tercio del grupo presen-tó recurrencias. Uno de cada diez presentó estatus epiléptico febril y un escaso número cursó poste-riormente con epilepsia o con convulsiones febriles plus.
Palabras clave: convulsiones febriles, tratamiento, epilepsia.
INTRODUCCIÓN
Una convulsión febril (CoFe) se define como una crisis epiléptica en un niño mayor de un mes du-rante un episodio febril (1). Las crisis epilépticas asociadas a fiebre corresponden al trastorno neuro-lógico más común en la población pediátrica, afec-tando al 2-4% de todos los niños menores de cinco años. Además, alrededor del 2% de las consultas en los servicios de urgencia pediátrico son debidos a CoFe (2).
Clásicamente se les ha dividido en dos tipos: CoFe simples y CoFe complejas. Las CoFe simples se caracterizan porque tienen una duración menor a quince minutos, son primariamente generalizadas y porque la crisis no se repite durante las siguien-tes 24 horas. Por otra parte, las CoFe complejas
se caracterizan porque tienen una duración mayor a quince minutos, son de inicio focal, la crisis se repite en las siguientes 24 horas y por la presencia de una crisis prolongada, de cinco a quince minutos de duración, que se resuelve con el uso de fármacos anti epilépticos (3).
En términos generales, el pronóstico de las CoFe es bueno, por lo que la gran parte de los pacientes no presentan secuelas desde el punto de vista neuroló-gico (1-3). Sin embargo, también es conocido que una parte de los pacientes afectados puede persistir con crisis epilépticas después de los cinco años de edad, por ejemplo, aquellos pacientes que evolu-cionan hacia el grupo de las epilepsias del espectro GEFS+ (Generalized Epilepsy and Febrile Seizures plus) (4), entre otros.
OBJETIVOS
Objetivo general
Caracterizar a los pacientes con diagnóstico de con-vulsión febril controlados en el centro de Epilepsia Infantil del Hospital Luis Calvo Mackenna.
Objetivos específicos
1. Identificar las características generales de las convulsiones febriles.
2. Identificar el tipo de tratamiento utilizado en el manejo a largo plazo de los pacientes con con-vulsiones febriles.
3. Determinar la relación entre el tratamiento utili-zado y las recurrencias.
4. Determinar la evolución a largo plazo de los pa-cientes con convulsiones febriles.
MATERIAL Y MÉTODOS
Estudio retrospectivo. Se revisaron las fichas clíni-cas de ciento veinticinco pacientes con diagnóstico de CoFe controlados en el Centro de Epilepsia In-fantil del Hospital Luis Calvo Mackenna entre Julio de 2005 y Julio de 2012. El tiempo de control fue en promedio de 3,4 años (rango 6 meses a 12 años).
El total de pacientes con diagnóstico de CoFe se obtuvo de los registros de electroencefalograma (EEG) y del registro de estadística diario de nuestro Centro.

26
Revista Chilena de Epilepsia Año 14, Nº 3, Diciembre 2014
Se analizaron las siguientes variables: sexo, ante-cedente de familiares de primer grado con epilepsia o CoFe, edad promedio en el momento de la pri-mera crisis, número promedio de crisis, pacientes con más de cinco crisis, tipo de CoFe, presencia de crisis de más de diez minutos de duración, logro de hitos del desarrollo psicomotor (DSM), caracterís-ticas del electroencefalograma (EEG), tipo de trata-miento, cumplimiento del tratamiento, recurrencias, evolución a largo plazo y estado actual de segui-miento.
Se consideraron dos tipos de tratamiento: trata-miento agudo de las crisis y tratamiento de las recurrencias (profilaxis de las recurrencias o trata-miento a largo plazo). El tratamiento agudo de la crisis se hizo con diazepam por vía rectal en dosis de 0,5 mg/Kg dosis. Por otra parte, el tratamiento profiláctico de las recurrencias incluyó a dos grupos de pacientes: los tratados con profilaxis intermiten-te con diazepam rectal (en igual dosis que para el tratamiento agudo de las crisis) y los tratados con profilaxis continua con ácido valproico en dosis de 15-50 mg/Kg día. Se define profilaxis intermiten-te como el uso de diazepam por vía rectal al inicio de cada cuadro febril. Se define profilaxis continua como el uso de ácido valproico por vía oral en for-ma continua o permanente.
Para facilitar el análisis de los resultados con res-pecto al tratamiento, los pacientes fueron clasifi-cados en seis grupos: los tratados sólo con ácido valproico (AV), los tratados con ácido valproico y diazepam en caso de crisis (AV + DR-C), los trata-dos con ácido valproico y con diazepam como pro-filaxis intermitente (AV + DR-P), los tratados sólo con diazepam en caso de crisis (DR-C), los trata-dos sólo con diazepam como profilaxis intermitente (DR-P) y los tratados con diazepam en caso de cri-sis y diazepam como profilaxis intermitente (DR-C + DR-P).
Se considera que ha cumplido adecuadamente el tratamiento cuando el paciente sigue las prescrip-ciones indicadas por el médico tratante, en cuanto a dosis, horario de administración y forma de pre-sentación del fármaco. Por otra parte, se considera que el paciente presenta recurrencia de crisis cuan-do tiene recaídas aún cuando está usando fármacos antiepilépticos.
Para los pacientes que presentaron recurrencias se
analizó además cuántos de ellos tenían buena adhe-rencia a tratamiento.
RESULTADOS
Las características generales de los pacientes con convulsión febril (CoFe) están resumidas en la Ta-bla 1.
De los ciento veinticinco pacientes estudiados se-senta y cinco fueron hombres (52%) y sesenta fue-ron mujeres (48%). Treinta y seis pacientes del total de la muestra (29%) tuvieron antecedentes de fa-miliares directos con epilepsia y/o CoFe. El primer episodio de CoFe se presentó en promedio a los die-cisiete meses de vida (rango 3-60 meses) y los pa-cientes tuvieron cinco crisis en promedio (rango 1-8 crisis). Diez pacientes (8%) tuvieron más de cinco crisis en el total de tiempo de observación.
Setenta y nueve pacientes (63%) presentaron sólo CoFe simples, Treinta y dos pacientes (26%) sólo CoFe complejas, y catorce pacientes (11%) presen-taron ambos tipos de crisis durante su evolución. Trece pacientes (10%) presentaron crisis de más de diez minutos de duración, los que fueron cataloga-dos y manejados como estatus febril.
Por otra parte, ciento once pacientes (89%) pre-sentaron un DSM normal según su edad y catorce (11%) cursaron con retraso en la adquisición de los hitos del DSM, con compromiso indistinto de los componentes de lenguaje y motor.
Ciento veintidós pacientes (97%) tuvieron un EEG normal y tres pacientes (3%) tuvieron un registro de EEG alterado: en dos pacientes se describe acti-vidad epiléptica tipo espiga y espiga onda y en un paciente se describe un registro con lentitud basal difusa y sharp waves en regiones posteriores.
En cuanto al tratamiento, en ochenta y cuatro pa-cientes (67%) se indicó sólo DR-C y en trece pa-cientes (10%) se indicó sólo DR-P. Veintiocho pa-cientes (22%) fueron tratados con AV: veintiséis de ellos fueron tratados además con DR-C y los otros dos pacientes fueron tratados además con DR-P. No hubo pacientes tratados sólo con AV y tampoco hubo pacientes tratados con DR-C + DR-P (Tabla 2).
Del total de pacientes, setenta y cuatro (59%) cum-

27
Caracterización de un grupo de pacientes con convulsiones febriles. Patricia Alfaro et al.
plieron adecuadamente el tratamiento durante el tiempo de observación. El 61% de ellos estaba en tratamiento sólo con DR-C, el 32% con AV + DR-C, el 4% sólo con DR-P y el 3% estaba en trata-miento con AV + DR-P (Tabla 3).
Cuarenta y seis pacientes (37%) tuvieron recurren-cias en un tiempo de control promedio de 3,4 años (rango 6 meses a 12 años). De estos, el 48% estaba en tratamiento sólo con DR-C, el 26% con AV + DR-C, el 24% sólo con DR-P y el 2% con AV + DR-P.
Del total de pacientes con recurrencias veintidós tuvo un adecuado cumplimiento del tratamiento, nueve de los cuales estaban en tratamiento con AV + DR-C (Tabla 4 y Tabla 5).
Seis pacientes (5%) evolucionaron con una clínica concordante con convulsión febril plus y tres pa-cientes (2%) cursaron con epilepsia (dos pacien-tes con epilepsia parcial, con crisis parcial simple y parcial simple con generalización secundaria a crisis tónico clónico generalizada; un paciente con epilepsia generalizada, con crisis tónico clónico ge-neralizadas).
Hasta Mayo de 2014, del total de ciento veinticin-co pacientes hay cuarenta y cuatro (35%) pacientes en control. Cincuenta y uno (41%) abandonaron el tratamiento y los controles y treinta (24%) fueron dados de alta.
En el momento del corte (Julio de 2012) habían pre-sentado recurrencias: veinticinco de los cuarenta y cuatro pacientes en control, quince de los cincuen-ta y uno que abandonaron tratamiento y seis de los treinta pacientes que fueron dados de alta.
DISCUSIÓN
Se define una CoFe como una crisis epiléptica aso-ciada a fiebre (considerándose fiebre una tempe-ratura axilar de 37,5º C o más) que ocurre en un paciente de rango etáreo entre seis meses y cinco años, en el cual no hay síntomas ni signos que su-gieran una infección del sistema nervioso central (1). Además, la definición considera que no debe haber antecedentes de crisis afebriles previas ni de anomalías neurológicas pre existentes (1, 2).
Resulta interesante destacar que en nuestra muestra
un 63% de los pacientes presentó sólo CoFe sim-ples, 26% sólo CoFe complejas y un 11% presentó ambos tipos de crisis. Además, hubo un 10% del total de pacientes que presentó un estatus febril en algún momento de su evolución, cifra que es su-perior a lo que se describe en la literatura (4-5%). Esto puede explicarse porque nuestro Hospital con-trola sólo población infantil y además porque en el Centro de Epilepsia Infantil se realiza un policlínico que atiende a pacientes con epilepsia refractaria de-rivados desde todo el país, por lo que este hallazgo muy probablemente podría corresponder a un sesgo de la muestra.
Una de las aristas importantes al hablar de CoFe son sus factores de riesgo. Clásicamente se ha reportado que el principal factor de riesgo es el antecedente de familiares directos con historia de Cofe, repor-tándose que hasta un 25-40% de los pacientes tiene este antecedente en la familia (1, 5-8). En nuestra revisión, treinta y seis de los ciento veinticinco pa-cientes (29%) tuvieron antecedente de familiares directos con CoFe o con epilepsia, lo que concuerda con lo reportado en la literatura.
Por otra parte, también es interesante destacar que un 8% de nuestros pacientes presentó más de cinco crisis durante el tiempo total de observación, aspec-to que pone énfasis en la necesidad del seguimiento y control periódico para estos pacientes.
Basándonos en las guías clínicas disponibles (9, 13, 14), los estudios complementarios se restringen sólo para las CoFe complejas y para el estatus febril estando indicado en ellos realizar EEG y neuroimá-genes, sobre todo en los pacientes con crisis de du-ración prolongada y en las de inicio focal (1, 7, 8). Sin embargo, no existe claridad ni consenso acerca de cuál es el verdadero rol que cumple el EEG en el estudio de las CoFe complejas, así como tampoco acerca de cuál es el mejor momento para realizarlo (1, 2). Además, el reporte de anomalías encontradas en los EEG de pacientes con CoFe es extremada-mente variable (2-86%), lo que se explica básica-mente por lo heterogéneo de los estudios (3). En nuestra revisión tres pacientes (3%) tuvieron un EEG informado con alteraciones, habiendo presen-tando los tres pacientes episodios de CoFe simple. En el resto de los pacientes el EEG fue normal.
Con respecto al tratamiento, se debe destacar que en la etapa aguda la mayoría de las CoFe tienen

28
Revista Chilena de Epilepsia Año 14, Nº 3, Diciembre 2014
una duración auto limitada la que generalmente es menor de diez minutos, no siendo necesario en mu-chos casos el uso de fármacos antiepilépticos que yugulen la crisis (13,14). Al respecto, las normas actuales recomiendan el uso de benzodiacepinas por vía rectal, endovenosa (diazepam, lorazepam) o intranasal (midazolam) en aquellos casos en los cuales la crisis febril tiene una duración mayor o igual a cinco minutos. No hay evidencia que avale el uso de otros fármacos antiepilépticos en la fase aguda (1, 2, 9, 13, 14).
Por otra parte, también debe considerarse el tra-tamiento farmacológico para el manejo de las re-currencias (profilaxis de las recurrencias). Actual-mente existe consenso en que las CoFe simples no requieren tratamiento a largo plazo: primero por el carácter auto limitado (y en general de buen pro-nóstico) del cuadro y segundo porque, pese a que el beneficio en la disminución de las recurrencias ha sido bien demostrado para los pacientes que usan fenobarbital y ácido valproico, los potenciales efec-tos adversos de estos fármacos limitarían su uso (13,14). Sin embargo, existe evidencia a favor de tratarlas en algunas circunstancias, ya que su uso disminuiría las recurrencias (1, 5, 6). En este con-texto, se han descrito dos modalidades terapéuticas a largo plazo:- Tratamiento continuo o permanente: estaría in-
dicado en aquellos pacientes que cursan con una alta frecuencia de crisis (tres o más en seis meses o cuatro o más en un año) (5), ansiedad y/o inade-cuado manejo ambulatorio por parte de la fami-lia, presencia de tres o más factores de riesgo de recurrencia y duración prolongada de las crisis. Los fármacos cuya eficacia ha sido demostrada en la disminución de las recurrencias son feno-barbital y ácido valproico, prefiriéndose el uso de este último dado su menor frecuencia de efectos adversos, sobre todo de la esfera cognitiva. No hay estudios que avalen el uso de otros fármacos en CoFe simples (13, 14).
- Tratamiento intermitente: referido al uso de fár-macos antiepilépticos en las primeras 24 horas de cada cuadro febril. Existen diversos reportes en la literatura que justifican su administración para los mismos casos en los que se usa tratamiento continuo (5-7), debiendo considerarse además en este contexto la mayor frecuencia de efectos deletéreos de los fármacos antiepilépticos al ser usados en forma permanente (8). Su administra-ción, por lo tanto, no sólo disminuiría las recu-
rrencias a largo plazo sino que también disminui-ría el requerimiento de tratamiento permanente y por lo tanto, también disminuiría la probabilidad de efectos adversos secundarios a estos fármacos (8). En esta modalidad de tratamiento los fárma-cos que se sugiere usar son los del grupo de las benzodiacepinas (por vía rectal, endovenosa o in-tranasal). No existe evidencia que avale el uso de otro tipo de fármacos antiepilépticos para el trata-miento intermitente de las Cofe simples (1, 2, 13, 14). En este punto es importante recalcar que en los casos en los que se decida usar tratamiento in-termitente la vigilancia del paciente debe ser aún más estricta, para evitar enmascarar los síntomas de una infección del sistema nervioso central (1). Reportes recientes (9) plantean el probable uso de clobazam como tratamiento profiláctico. Sin em-bargo, su eficacia aún está en estudio.
Con respecto a las CoFe complejas, existen pocos reportes que avalen el uso de tratamiento a largo plazo, por lo que en general también se sugiere no tratarlas. Sin embargo, existen estudios disponibles que proponen tratarlas siguiendo los mismos pará-metros y usando los mismos fármacos ya discutidos para las CoFe simples, agregándose las crisis de ini-cio focal. Esto se justifica además por el hecho de que las CoFe complejas tienen más riesgo de cursar en el largo plazo con crisis afebriles y con epilepsia. En este contexto, existe un estudio recientemente publicado (1) que sugiere el uso de levetiracetam como tratamiento continuo a largo plazo para dis-minuir las recurrencias en las Cofe complejas.
Por otra parte, toda la evidencia disponible plantea que el uso de fármacos antipiréticos (antiinflama-torios no esteroidales, tales como ibuprofeno o pa-racetamol) no disminuye las recurrencias ni de las CoFe simples ni de las CoFe complejas, por lo que su uso está justificado sólo como manejo sintomá-tico (9, 13, 14).
En nuestra casuística, al 88% de nuestros pacientes se le indicó diazepam por vía rectal para el manejo de las crisis de cinco o más minutos de duración. De estos, en el 67% esta benzodiacepina fue la única modalidad de tratamiento durante todo el tiempo de observación.
Con respecto al tratamiento a largo plazo, en el 12% de los casos (quince pacientes) se indicó diazepam como profilaxis intermitente y en el 23% (veintio-

29
Caracterización de un grupo de pacientes con convulsiones febriles. Patricia Alfaro et al.
cho pacientes) se indicó tratamiento permanente con ácido valproico. Dos pacientes del total de la muestra estaban en tratamiento al mismo tiempo con ácido valproico en forma continua y con diaze-pam rectal como profilaxis intermitente.
Con respecto a las recurrencias, está bien estipula-do en la literatura que el riesgo de recurrencia para las CoFe en general es de alrededor de un 30-40% (1, 2) y que alrededor del 50% de los pacientes que recurren lo hacen en los primeros seis meses (8). En nuestra muestra el riesgo de recurrencia fue de 30%, lo que concuerda con lo ya descrito. Además, en nuestra casuística el porcentaje de recurrencias fue prácticamente el mismo para los pacientes que estaban en tratamiento sólo con diazepam como profilaxis intermitente que para los pacientes que estaban en tratamiento con ácido valproico en for-ma continua. Tampoco encontramos diferencias en-tre los pacientes que usaron tratamiento profiláctico para las recurrencias (diazepam profiláctico y ácido valproico) y los que no usaron (diazepam rectal sólo en caso de crisis). Observamos un porcentaje de recurrencias mucho menor para los pacientes que usaron ácido valproico continuo y diazepam inter-mitente como manejo a largo plazo, sin embargo, el número de pacientes que usó esa combinación de tratamiento representó sólo el 2% del total de la muestra, por lo que no es factible deducir conclu-siones extrapolables.
Como ya se ha descrito anteriormente, las Cofe tienen un curso clínico en general favorable, evo-lucionando con recuperación completa y sin com-plicaciones tanto desde el punto de vista pediátrico general como desde el punto de vista neurológico (1, 2). En este contexto, es conocido que el riesgo de desarrollar epilepsia para los pacientes con an-tecedente de CoFe simple es del 1-1,5%, mientras que en las CoFe complejas es de alrededor del 4% (1). En nuestra muestra seis pacientes evoluciona-ron con convulsión febril plus, tres pacientes con epilepsia y catorce pacientes con RDSM, mientras que el resto de los pacientes evolucionaron asinto-máticos, lo que concuerda con lo expresado en la literatura.
Como conclusión podemos establecer que las CoFe son entidades frecuentes durante la infancia, que si bien en términos generales tienen un curso favo-rable y de buen pronóstico existe un porcentaje de pacientes que pueden evolucionar hacia otras enti-
dades tales como las epilepsias del espectro GEFS+ entre otras (4), por lo que es necesario un segui-miento minucioso a largo plazo.
Por otra parte, también es importante destacar que si bien existen publicaciones que sugieren no usar tratamiento a largo plazo, también existe eviden-cia de que en ciertas circunstancias se justifica usar fármacos antiepilépticos para el manejo de estos pacientes, ya sea en forma continua como en forma intermitente, por lo que la decisión de tratarlas o no tratarlas debe ser tomada según las característi-cas clínicas de cada paciente y de cada familia en particular y por supuesto según la experiencia del médico.
BIBLIOGRAFÍA
1. Patel A, Vidaurre J. Complex febrile seizures: a practical guide to evaluation and treatment. Journal of Child Neurology 2013; 28: 762.
2. Shah PB James, Elayaraja S. EEG for children with complex febrile seizures. Review. Cochrane Database of systematic Reviews 2014; Issue 1. Art. No.: CD009196. DOI: 10.1002/14651858.CD009196.pub2.
3. Jeong K, Hee Han M, Hye Lee E, Chung S. Early postictal electroencephalography and correlation with clinical findings in children with febrile seizures. Korean J Pediatr 2013; 56(12):534-539.
4. Juan L Moya-Vilches, Marcelo Devilat. Genera-lized epilepsy and febrile seizures plus (GEFS+) in a series of 22 patients. Journal of Pediatric Neurology 2014; 3: 93-106
5. Capovilla G, Mastrangelo M, Romeo A, Vige-vano F. Recommendations for the management of febrile seizures: Ad hoc Task Force of LICE Guidelines Commission. Epilepsia 2009; 50: 2–6.
6. Pavlidou E, Hagel C, Panteliadis C. Febrile seizures: recent developments and unanswered questions. Childs Nerv Syst 2013; 29: 2011–2017.
7. Lux A. Treatment of febrile seizures: historical perspective, current opinions, and potential fu-ture directions. Brain and Development 2010; 32: 42–50.
8. Devilat M, Masafierro MP. Profilaxis selecti-va discontinua en domicilio de recurrencias de convulsiones febriles con Diazepam rectal. Rev Chil Ped 1989; 60(4): 195-597

30
Revista Chilena de Epilepsia Año 14, Nº 3, Diciembre 2014
9. Offringa M, Newton R. Prophylactic drug ma-nagement for febrile seizures in children. Re-view. Cochrane Database of Systematic Re-views 2012, Issue 4. Art. No.: CD003031. DOI: 10.1002/14651858.CD003031.pub2.
10. Mastrangelo M, Midulla F, Moretti C. Actual insights into the clinical management of febri-le seizures. Eur J Pediatr 2014. DOI 10.1007/s00431-014-2269-7.
11. Ahmad S, Marsh E. Febrile status epilepticus: current state of clinical and basic research. Se-min Pediatr Neurol 2010; 17: 150-154.
12. Karimzadeh P, Rezayi A, Togha M, Ahmadaba-di F, Derakhshanfar H, Azargashb E, Khodaei F. The best time for EEG recording in febrile
seizure. Iran J Child Neurol 2014; 1: 20-25. 13. Subcommittee on febrile seizures. Febrile seizu-
res: a guideline for the neurodiagnostic evalua-tion of the child with a simple febrile seizure. Pediatrics 2011; 127: 389.
14. Steering committee on quality improvement and management, Subcommittee on febrile seizures. Febrile seizures: clinical practice guideline for the long-term management of the child with simple febrile seizures. Pediatrics 2008; 121: 1281.
15. Oluwabusia T, Sood S. Update on the manage-ment of simple febrile seizures: emphasis on minimal intervention. Curr Opin Pediatr 2012; 24:259-265.
31
Caracterización de un grupo de pacientes con convulsiones febriles. Patricia Alfaro et al.
ANEXOS
Tabla 1: Resumen características generales de los pacientes con convulsiones febriles
Características generales de los pacientes con convulsiones febriles
Características Número total % (En relación al total de pacientes)
Sexo 65 hombres 52% 60 mujeres 48%
Antecedentes familiares de primer grado 36 pacientes 29%con epilepsia o convulsión febril Crisis:Edad promedio primera crisis 17 meses (3-60 meses) -No total de crisis en promedio 5 (1-8 crisis) -Pacientes con más de 5 crisis 10 8% Adquisición hitos DSM:-DSM normal 111 89%-Retraso DSM 14 11%
Tipo de convulsión febril:-Simple 79 pacientes 63%-Compleja 32 pacientes 26%-Ambas 14 pacientes 11%
Crisis de más de 10 minutos de duración 13 pacientes 10%(status febril) Características EEG:-Registro normal 122 97%-Registro alterado 3 3%
Evolución a largo plazo:-Convulsión febril plus (Cofe+) 6 5%-Epilepsia 3 2%-Sin recurrencias, epilepsia ni Cofe+ 116 (93%)-Muerte 0 0
Recurrencias 46 37%
Condición actual:-En control 44 35%-Abandono de tratamiento 51 41%-Alta 30 24%

32
Revista Chilena de Epilepsia Año 14, Nº 3, Diciembre 2014
Tabla 2: Resumen del tratamiento de los pacientes con convulsiones febriles
Características del tratamiento de los pacientes con convulsiones febriles Tipo de tratamiento Total de pacientes NO total y % (% en relación al total de pacientes)
Diazepam (DZP) rectal
DZP-C 84 (67%)Sólo DZP-P 13 (10%)DZP-C + DZP-P 0
TOTAL en tratamiento con DZP 97 pacientes (77%)
Ácido valproico (AV)
Sólo AV 0AV + DZP-C 26 (21%)AV + DZP-P 2 (2%)
TOTAL en tratamiento con AV 28 pacientes (23%)
TOTAL de pacientes (DZP + AV) 125 pacientes (100%)
Tabla 3: cumplimiento del tratamiento
Características del cumplimiento del tratamiento de los pacientes con convulsiones febriles Tipo de tratamiento Total de pacientes (NO total y %) (% en relación a los pacientes que cumplen adecuadamente)
Diazepam (DZP) rectal
DZP-C 45 (61%)Sólo DZP-P 3 (4%)DZP-C + DZP-P 0
TOTAL con DZP que cumplen adecuadamente 48 pacientes (65%)
Ácido valproico (AV)
Sólo AV 0AV + DZP-C 24 (32%)AV + DZP-P 2 (3%)
TOTAL con AV que cumplen adecuadamente 26 pacientes (35%)
TOTAL de pacientes que cumplen 74 pacientes (100%)adecuadamente el tratamiento

33
Caracterización de un grupo de pacientes con convulsiones febriles. Patricia Alfaro et al.
Tabla 4: Resumen de las recurrencias de los pacientes con convulsiones febriles
Características de las recurrencias de los pacientes con convulsiones febriles Tipo de tratamiento Total de pacientes (NO total y %) (% en relación a los pacientes que tuvieron recurrencias)
Diazepam (DZP) rectal
DZP-C 22 (48%)Sólo DZP-P 11 (24%)DZP-C + DZP-P 0
TOTAL con DZP que tuvieron recurrencias 33 pacientes (72%)
Ácido valproico (AV)
Sólo AV 0AV + DZP-C 12 (26%)AV + DZP-P 1 (2%)
TOTAL con AV que tuvieron recurrencias 13 pacientes (28%)
TOTAL de pacientes que tuvieron recurrencias 46 pacientes (100%)
Tabla 5: pacientes con recurrencias que tuvieron un adecuado cumplimiento del tratamiento
Pacientes con recurrencias que tuvieron un adecuado cumplimiento del tratamiento Tipo de tratamiento NO total de pacientes
Diazepam (DZP) rectal
DZP-C 7Sólo DZP-P 5DZP-I + DZP-P 0
TOTAL DZP con recurrencias que cumplen tratamiento 12
Ácido valproico (AV)
Sólo AV 0AV + DZP-C 9AV + DZP-P 1
TOTAL AV con recurrencias que cumplen tratamiento 10
TOTAL de pacientes con recurrencias que cumplen 22 pacientesadecuadamente con el tratamiento

34
Revista Chilena de Epilepsia Año 14, Nº 3, Diciembre 2014Actualizaciones
Actualización del Síndrome de WestVanessa García, Sergio Meneses, Perla David Clinica Dávila, Universidad de los Andes, Universidad de Valparaíso, Universidad de los Andes, Santiago, Chile.Correo: [email protected]
ABSTRACT
West syndrome (WS) is a kind of catastrophic epi-lepsy in the first year of life, characterized by the electroclinical triad of clinical spasms, psychomo-tor retardation and EEG pattern of hypsarrhyth-mia. Its pathophysiology remains unknown and presents a wide range of etiologies, under which can be classified into: genetic, structural/metabolic and unknown cause (cryptogenic). Unlike another epilepsies, SW usually is refractory to conventional antiepileptic drugs, and the first line treatments are adrenocorticotropic hormone (ACTH) and vigaba-trin, although there is no consensus regarding the optimal dose and duration of treatment. The prog-nosis is poor; 75-90% of patients have mental retar-dation, 50-60% have recurrent seizures at five years of age, while the mortality rate ranges from 3-33%. All of this things implies the SW like a therapeutic challenge that remains to the present, where new diagnostic modalities and treatment options are being studied to improve the morbidity and morta-lity of this disease.
RESUMEN
El Síndrome de West (SW) es un tipo de epilepsia catastrófica del primer año de vida, caracterizada por la tríada electroclínica: espasmos clínicos, re-tardo del desarrollo psicomotor y patrón electroen-cefalográfico de hipsarritmia. Su fisiopatología per-manece desconocida y presenta una amplia gama de etiologías, según las cuales se puede clasificar en: genético, estructural/metabólica y de causa desco-nocida (criptogénico). El SW, a diferencia de otras epilepsias, suele ser refractario a los fármacos anti-epilépticos convencionales, siendo los tratamientos de primera línea la hormona corticotropina (ACTH) y la vigabatrina, aunque no hay consenso en cuanto a las dosis óptimas y la duración del tratamiento.
El pronóstico es pobre; un 75-90% de los pacien-tes presentan retraso mental, un 50-60% presentan convulsiones recurrentes a los cinco años de edad, mientras que la tasa de mortalidad varía entre un 3-33%. Todo esto implica que el SW sea un desafío terapéutico que permanece hasta la actualidad, don-de nuevas modalidades diagnósticas y opciones de tratamiento están en estudio para mejorar la morbi-mortalidad de esta patología.
INTRODUCCIÓN
En el año 1841 un médico británico, el Dr. William West, describió por primera vez los espasmos infan-tiles, una forma severa de epilepsia de la primera in-fancia, que se caracteriza por la presencia de grupos o “racimos” de breves contracciones musculares axiales, asociadas a un patrón electroencefalográ-fico típico conocido como “hipsarritmia”, descrito en 1952 por Gibbs y Gibbs. En años posteriores se acuñó el término Síndrome de West (SW) para la tríada clásica de espasmos infantiles, hipsarritmia y detención o regresión del desarrollo psicomotor (1,2,3). Debido a esto último, sumado a su alta tasa de mortalidad y refractariedad a los fármacos anti-epilépticos convencionales, se le considera un tipo de “epilepsia catastrófica” (2,4). Debido a que los espasmos infantiles son los signos más representativos del SW, muchos médicos no distinguen entre ambas entidades, considerándolos como sinónimos y utilizando sus nombres indistin-tamente. Sin embargo en la terminología actual se considera al SW un subtipo dentro del Síndrome de Espasmos Infantiles, un espectro de trastornos epi-lépticos que incluyen además otras variantes como espasmos únicos, espasmos sin hipsarritmia e hip-sarritmia sin espasmos (1). Los espasmos infantiles y el SW representan el tipo más frecuente de epilepsia en el primer año de vida. La incidencia varía de 2 a 5/10.000 nacidos vivos, Recibido 2 de Dic. Aceptado 24 de Dic. 2014
Los autores declaran no tener conflictos de intereses.

35
Actualización del Síndrome de West Vanessa García et al.
con inicio en el primer año de vida en el 90% de los afectados. Esto se traduce en aproximadamente 2.000 a 2.500 nuevos casos por año en los Estados Unidos (3). En Europa, la prevalencia al nacer es-timada hasta la fecha es de 3,7/100.000 niños (5).
La edad peak de aparición es entre los 3 y 7 meses; es poco frecuente el inicio después de los 18 meses, aunque se ha reportado aparición en niños de hasta 4 años. En un 85% los espasmos cesan a los 5 años de edad, pero se pueden presentar otros tipos de cri-sis en hasta un 60% de los casos. El SW se presenta en niños de todos los grupos étnicos, siendo leve-mente predominante en hombres (3:2) (1,3).
CLASIFICACIÓN
Tradicionalmente el SW se clasifica en sintomático y criptogénico. En el primer caso existe una etiolo-gía identificada y/o un significativo retraso en el de-sarrollo en el momento del inicio de los espasmos; mientras que en el segundo no se logra identificar una etiología precipitante (1,4). Posteriormente se agregó un grupo idiopático para incluir aquellos pacientes sin causa conocida y sin síntomas ni al-teraciones neurológicas (4). Esta clasificación fue revisada el 2010 por la comisión ILAE sobre Clasi-ficación y Terminología, sustituyendo los términos “idiopático”, “sintomático” y “criptogénico”, por “genético”, “estructural/metabólico” y “de causa desconocida”, respectivamente (6).
ETIOLOGÍA
La etiología del SW es amplia y variada, reportán-dose en la literatura más de 200 posibles causas (7), lográndose identificar en un 60-90% de los casos (8). De acuerdo al estudio UKISS (United Kingdom Infantile Spams Study) realizado el año 2010, las causas más comunes de espasmos infantiles en or-den de frecuencia fueron la encefalopatía hipóxico-isquémica (10%), anomalías cromosómicas (8%), malformaciones (8%), accidente cerebrovascular perinatal (8%), complejo de esclerosis tuberosa (7%) y leucomalacia periventricular o hemorragia cerebral (5%) (9). Otras etiologías incluyen la dis-plasia cortical, meningoencefalitis, trauma, otros trastornos neurocutáneos como nevo sebáceo li-neal, incontinencia pigmentaria, síndrome de Ito y neurofibromatosis tipo I, infecciones congénitas (TORCH) y errores innatos del metabolismo (4). En los niños, el síndrome de West también puede
ser causado por mutaciones del gen ARX, a menudo asociado con genitales ambiguos (10). Como resul-tado del aumento en el uso de resonancia magnética y PET (Positron Emission Tomography), un menor número de espasmos (aproximadamente 10-15%) son etiquetados como criptogénicos y más son atri-buidos a alguna anomalía cerebral subyacente no detectada previamente (2). PATOGÉNESIS
Pese a que han transcurrido más de 150 años desde el reconocimiento clínico de los espasmos infanti-les, su base biológica aún no está completamente determinada. Se postula que son resultado de una injuria no específica en un punto crítico del desarro-llo ontogenético del cerebro, donde varios factores etiopatogénicos, ya sean estructurales, metabólicos, genéticos, inflamatorios e incluso neuroinmunes, parecen estar implicados (4,8). El hecho de que este fenotipo específico de epilepsia se asocie a diversas etiologías apunta hacia una “vía final común”, aun-que históricamente no existe ningún modelo animal adecuado que lo explique (11). Actualmente existe evidencia creciente de que el mecanismo subyacente sea debido a alteraciones en las vías claves de regulación genética del desa-rrollo del cerebro, específicamente la red genética reguladora del desarrollo dorsal-ventral del cerebro anterior GABAérgico y anormalidades en el gen ex-presado en la sinapsis. Los niños con estas asocia-ciones genéticas también tienen fenotipos más allá de la epilepsia, incluyendo rasgos dismórficos, au-tismo, trastornos del movimiento y malformaciones sistémicas (1,10). Sin embargo, la respuesta terapéutica a la ACTH y altas dosis de esteroides, así como informes clínicos ocasionales de anomalías de citoquinas o de etio-logías infecciosas y autoinmunes, han planteado la posibilidad de que al menos en cierta cantidad de pacientes pudieran estar involucradas vías neuroin-flamatorias. La vía de mTOR parece tener un papel central en la convergencia de varios mecanismos de señalización y puede representar una vía promete-dora para futuras intervenciones terapéuticas (8). CLÍNICA
El SW se caracteriza por la tríada electroclínica de espasmos epilépticos, retardo del desarrollo psico-

36
Revista Chilena de Epilepsia Año 14, Nº 3, Diciembre 2014
motor y patrón electroencefalográfico de hipsarrit-mia.
El espasmo epiléptico o espasmo clínico, consiste en una breve contracción brusca de menos de 2 se-gundos de duración, seguida de una contracción tó-nica sostenida de menor intensidad, habitualmente de 2 a 10 segundos de duración, que involucra los grupos musculares del cuello, tronco y extremi-dades, habitualmente de forma simétrica y sincró-nica, aunque pueden haber varios patrones clínicos (2,11,12). Con menor frecuencia la fase de contrac-ción dura menos de 0,5 segundos y ocurre sin la fase tónica (12). Los espasmos pueden ser de tipo flexor, extensor o mixto, sin que esto sugiera etiología ni influya en el pronóstico, pudiendo presentarse más de uno en el mismo paciente. Sin embargo, cuando son asimétri-cos (5-25%) orientan a una anomalía estructural del cerebro (11). Alrededor del 80% de los espasmos se presentan en grupos o “racimos” de varios minutos de duración, donde pueden presentarse de 2 a 125 en un episo-dio, con un promedio de 13 por minuto. Habitual-mente se presentan previo al despertar o antes de dormir, con una frecuencia similar entre el día y la noche, aunque raramente ocurren durante el sueño (1,11,12). En general no se encuentra una circuns-tancia o estímulo precipitante evidente, aunque al-gunos factores que se postulan como inductores de crisis son la dificultad para conciliar el sueño y los despertares nocturnos, durante la manipulación del niño, los ruidos fuertes, la alimentación, infeccio-nes, fiebre, emoción, hambre y temperaturas am-bientales extremas (2,11). Durante la convulsión se pueden observar varios fe-nómenos clínicos, tales como cambios conductua-les, desviación de la mirada, nistagmus o cambios en el patrón respiratorio. Frecuentemente el niño puede presentar un grito o llanto posterior a la fase ictal, aunque no se consideran parte de la convul-sión. Luego de la crisis puede haber irritabilidad o hiporreactividad transitoria. Además, en un 30-50% de los pacientes pueden presentarse otro tipo de convulsiones, como crisis parciales, mioclónicas, tónicas y tónico-clónicas (1,2,11,12).
El retardo o regresión en el neurodesarrollo habi-tualmente es severo, con manifestaciones motoras y
cognitivas evidentes en la mayoría de los pacientes, pudiendo incluir retraso mental y del habla, rasgos autistas y dispraxia visomotora. El retardo psico-motor puede preceder el inicio de los espasmos, pero también puede ocurrir simultáneamente o ser tardío (1,2,12). Características electroencefalográficas
El sello distintivo del EEG en SEI/SW es la hip-sarritmia, un patrón interictal desorganizado que consiste en ondas lentas y puntas de alto voltaje, aleatorias, en todas las áreas corticales. Este patrón se observa mientras el niño está despierto y en sue-ño no-REM, mientras que durante el sueño REM hay una marcada disminución e incluso puede des-aparecer. Además del patrón clásico ya descrito, se han descrito variantes de hipsarritmia que se han agrupado con el término de “hipsarritmia modifica-da” o “atípica”, la que se caracteriza por una mayor sincronización hemisférica y simetría y se observa en alrededor de dos tercios de los pacientes. Estas variantes incluyen hipsarritmia con aumento de la sincronización interhemisférica, patrón asimétrico o unilateral, descargas anormales por anomalías focales, episodios de atenuación de la actividad y patrón de ondas lentas de alto voltaje sin puntas (2,11,12). El patrón ictal más frecuente consiste en una onda lenta trifásica, positiva y de gran amplitud, que se correlaciona con el espasmo actual, seguida de un aplanamiento por una abrupta atenuación generali-zada de la actividad. No existe correlación entre el patrón ictal y el tipo de espasmo (2,11). Se debe te-ner en cuenta que hallazgos focales o lateralizados al EEG pueden indicar la presencia de una lesión cerebral estructural (12).
Diagnóstico diferencial
Los diagnósticos diferenciales del SW son aquellas patologías que se pueden confundir con los espas-mos clínicos, tales como el reflujo gastroesofágico, cólicos, sobresaltos excesivos, reflejos de Moro exagerados, mioclono neonatal benigno del sueño, convulsiones tónicas reflejas de la primera infancia y epilepsias mioclónicas benignas y graves. Se debe tener en cuenta que estas condiciones también pue-den ocurrir en pacientes que previamente han teni-do o tienen actualmente SW (2,12).

37
Actualización del Síndrome de West Vanessa García et al.
Enfoque diagnóstico
La evaluación inicial comienza con una historia clí-nica y examen físico detallados. El diagnóstico se realiza con un cuadro clínico compatible confirma-do por EEG (2,12). El método recomendado es realizar un video mo-nitoreo EEG de 24 horas, para lograr capturar un evento ictal e incluir un ciclo de sueño-vigilia com-pleto, aunque en algunos casos puede ser suficiente un estudio de dos o cuatro horas. En caso de que la evaluación inicial no sea diagnóstica se puede pro-longar el monitoreo o repetirlo dentro de 1-2 sema-nas (11,12) Una vez establecido el diagnóstico, la evaluación siguiente debe enfocarse en el estudio de la etio-logía subyacente, fundamental para determinar el tratamiento y el pronóstico. En este sentido, el es-tudio con neuroimagen es esencial, ya que permite confirmar la etiología en aproximadamente un 70% de los casos, siendo la técnica inicial de elección la resonancia magnética (RM), ya que permite de-tectar malformaciones cerebrales, atrofia cerebral, retraso en la mielinización y otras lesiones focales no visibles en la tomografía computada (TC). Tam-bién puede proporcionar información sobre el pro-nóstico, especialmente en relación con el desarrollo motor; los pacientes con RM sin alteraciones tienen mejores resultados en comparación con aquellos con lesiones (2,11). En casos donde el examen clínico o la neuroimagen permiten sospechar un trastorno genético, se reco-mienda realizar pruebas genéticas, por ejemplo en casos de síndrome de Down o lisencefalia. Si no se logra identificar una etiología luego de la evalua-ción clínica, EEG y RM, o en casos de espasmos de inicio temprano, asociación con otros tipos de con-vulsiones y retardo mental severo en el inicio, o un mal resultado después del control de los espasmos, se recomienda realizar un cribado metabólico; de-terminar niveles de transaminasas, lactato, piruvato, amoníaco, ácidos orgánicos en orina y aminoácidos séricos, así como análisis del líquido cefalorraquí-deo para medir niveles de glucosa, proteínas, ami-noácidos, lactato y piruvato. La combinación de resultados de estudio metabólico (suero y líquido cefalorraquídeo) con pruebas genéticas puede de-terminar la etiología en un 10 % adicional de los casos. Se debe considerar la detección de nuevas
anomalías genéticas (CDKL5 y ARX mutación, del 1p36 e inv dup) si los estudios de primera línea son negativos. Además, la RM puede repetirse después de los 2 años con el fin de detectar anomalías no diagnosticadas en la imagen inicial, como por ejem-plo la displasia focal. (2,6,11,12). Otro estudio recomendado es el PET, útil en la iden-tificación de anomalías no detectadas en RM o TC, y considerado en casos médicamente intratables cuando la presencia de anomalías focales en el EEG o hallazgos al examen clínico sugieren posibles zo-nas epileptogénicas que pudieran ser susceptibles de resección quirúrgica (2,11). Actualmente siguen en desarrollo nuevas pruebas diagnósticas, principalmente enfocadas en identifi-car los genes involucrados en la producción de SW, esto debido a su importancia tanto en el pronósti-co de la enfermedad como por su implicancia en la consejería genética (6). Tratamiento
Hasta la fecha, el tratamiento del SW sigue siendo en gran medida empírico, lo que se atribuye princi-palmente a la mala comprensión de su fisiopatología (13) y a la resistencia a los fármacos antiepilépticos (FAE) convencionales. La estrategia terapéutica ac-tual busca lograr un tratamiento efectivo del trastor-no, definido como el cese completo de los espasmos más la abolición de la hipsarritmia (respuesta �todo o nada”) (7). I) Terapias de primera línea.
En general el tratamiento se divide en dos grandes grupos; los niños portadores de esclerosis tuberosa (ET), donde el fármaco de preferencia es la vigaba-trina, y los portadores de todas las otras etiologías, donde la terapia de elección es la hormona cortico-tropina (ACTH) (2,7,14).
Estudios recientes sugieren que la pronta iniciación del tratamiento, ya sea con terapia hormonal o vi-gabatrina, mejora los resultados cognitivos a largo plazo. Además, se ha observado que la ACTH es superior a la vigabatrina en cuanto al resultado cog-nitivo en grupos criptogénicos (7).
ACTHEs el tratamiento más eficaz a corto plazo y de elec-

38
Revista Chilena de Epilepsia Año 14, Nº 3, Diciembre 2014
ción desde 1958 (7), pese a lo cual aún se desconoce su mecanismo de acción. Se postula un efecto anti-convulsivante directo por supresión de la hormona liberadora de ACTH (CRH), un neuropéptido endó-geno que puede provocar convulsiones en el cere-bro inmaduro, el cual sería liberado junto con otros mediadores como respuesta al estrés provocado por las diversas etiologías atribuidas al SW (15).
Aún no existe consenso entre los expertos con respecto a la dosis óptima y la duración del trata-miento. El espectro de dosificación varía entre 20 y 120 UI/L i.m o s.c., aunque estudios recientes han mostrado que las dosis bajas pueden ser tan eficaces como las dosis altas, con menores efectos adversos. Se desconoce la duración exacta del tratamiento hormonal, pero si el resultado es satisfactorio pue-de detenerse después de apenas 1 mes, ya que se logra un efecto permanente luego de 2-4 semanas de tratamiento (1,7). Existe acuerdo en cuanto a evi-tar tratamientos prolongados (como orientación, no más de 6 semanas), en razón de los severos efectos adversos y mortalidad observados con tratamientos de larga duración (14). Los efectos secundarios más frecuentemente observados son irritabilidad, au-mento del apetito y características cushingoides, sin embargo pueden presentarse también hipertensión, hipokalemia, opacidades en la retina, trastornos del crecimiento y, en casos raros, infecciones fulmi-nantes secundarias a la inmunosupresión (1,15). La recomendación de la Academia Americana de Neu-rología (AAN) en 2012 es que la terapia con dosis bajas de ACTH debe ser considerada como una al-ternativa a la dosis alta (Nivel B) (7).
En Chile, la ACTH también es la primera opción de tratamiento para pacientes con SW no portado-res de ET. La dosificación recomendada es de 0,05 mg/kg/dosis i.m., día por medio por 2 semanas. En pacientes con SW de causa precisada y con estudio metabólico normal, se inicia simultáneamente ácido valproico como FAE de mantención, en dosis de 20 mg/kg/d, 3 dosis al día, cada 8 horas, hasta alcan-zar los 50 mg/kg/d. Si en el control clínico y EEG a los 14 días se logra un tratamiento efectivo, se finaliza la cura con ACTH y se deja el ác. valproi-co de mantención; si la respuesta no es completa se recomienda realizar una segunda cura con ACTH de iguales características que la primera y controles semanales, manteniendo el ác. valproico. En caso de fracaso de tratamiento se continúa con la segun-da opción; la vigabatrina. Como terceras opciones
se pueden considerar el uso de topiramato o leve-tiracetam y, por último, la dieta cetogénica como opción no farmacológica (14).
VigabatrinaEs un FAE que actúa como inhibidor de la GABA-transaminasa (1). En general se considera como un tratamiento efectivo de primera línea, sin embargo en estudios comparativos con ACTH y predniso-lona, éstos demuestran mayor efectividad que la vigabatrina, con excepción de los pacientes porta-dores de ET. La dosis óptima y la duración del tra-tamiento tampoco están completamente definidos. En la actualidad, la dosificación típica consiste en una dosis inicial de 50 mg/kg/d, escalando hasta al-canzar los 100 a 150 mg/kg/d, donde habitualmente se alcanza la respuesta clínica, con evaluación de la eficacia a las dos semanas después del cambio de dosis. Con respecto a la duración, se sugiere que los pacientes que responden al tratamiento continúen el medicamento durante seis meses, con vigilancia continua por posibles efectos adversos secundarios a su toxicidad, que incluyen defectos del campo vi-sual periférico y disfunción irreversible de la retina. Además, recientemente se han reportado anomalías estructurales en la RM cerebral de niños tratados con vigabatrina (7,15).
En Chile, la terapia con vigabatrina es de elección en los pacientes con SW portadores de ET, y su do-sis es de 100 mg/kg/d cada 12 horas, con controles a los 7, 14 y 30 días. Si se logra un tratamiento efec-tivo en el primer control se mantiene el tratamien-to hasta 6 meses, considerando los posibles daños retinianos; si persisten crisis y/o EEG alterado se aumenta la dosis a 150 mg/kg/d cada 12 hrs. Si en el segundo control aún no se logra una respuesta efectiva, se aumenta la dosis a 200 mg/kg/d cada 12 hrs. En caso de resistencia al tratamiento en el control de los 30 días se sugiere continuar con las otras opciones (topiramato, levetiracetam, dieta ce-togénica) (14)
II) Terapias de segunda línea
Diferentes corticosteroides y FAEs se pueden utili-zar como alternativas de segunda línea cuando los medicamentos de primera línea son ineficaces o es-tán contraindicados (7).
Corticosteroides Incluyen hidrocortisona, prednisona, prednisolona

39
Actualización del Síndrome de West Vanessa García et al.
y dexametasona. Son un tipo de tratamiento hormo-nal menos costoso y más fácil de administrar que la ACTH, pero hasta la fecha no hay evidencia su-ficiente como para recomendar su uso como alter-nativa de primera línea. Los efectos adversos son generalmente similares a los de la ACTH, pero ésta tiene un menor efecto supresor sobre el eje hipota-lámico-hipofisiario-adrenal (HHA) (7,15)
En Marzo de 2014 el grupo de Wanigasinghe et al. publicó el primer ensayo clínico aleatorizado reali-zado para mostrar la superioridad de la prednisolo-na oral sobre la ACTH en el SW. El estudio conclu-ye que las dosis moderadas a altas de prednisolona oral parecen ser más efectivas que las dosis bajas a moderadas de ACTH para el tratamiento inicial del SW (13), sin embargo se debe tener en cuenta que los resultados fueron evaluados sólo desde la pers-pectiva de mejoría de la hipsarritmia.
FAEsLa evidencia de otros FAEs es muy limitada en comparación con la vigabatrina. Hay algunos es-tudios de fase IV de topiramato y ácido valproico, fase I de sultiamo y fase IV de zonisamida, piridoxi-na, nitrazepam, levetiracetam, lamotrigina y dieta cetogénica (7).
El topiramato es un tratamiento popular de segunda línea, que a menudo se utiliza como primera línea, especialmente donde no hay acceso a vigabatrina. Se ha reportado respuesta de alrededor de un 40%, y sus efectos adversos son somnolencia, oligohidro-sis, síntomas gastrointestinales, cálculos renales, además de potenciales alteraciones cognitivas y del aprendizaje (7,15).
El ácido valproico se utiliza a menudo como trata-miento de segunda línea y ha sido usado como pri-mera línea con 39% de respuesta completa o “bue-na” (7). Sin embargo, los reportes de tratamiento con ácido valproico tienen resultados inconsisten-tes, hasta un 40-70% de pacientes refractarios a ACTH pueden beneficiarse, pero la respuesta apa-rente puede reflejar la historia natural del SW más que el efecto del tratamiento (15)
El sultiamo es un FAE que se ha probado en es-tudios pequeños y asociado a piridoxina, donde ha mostrado mejor resultado que el placebo, pero tiene varias desventajas; es una opción poco disponible fuera de E.E.U.U, sus efectos adversos pueden ser
severos y la eficacia a largo plazo es desconocida (7,15)
La zonisamida, un derivado de las sulfonamidas, ha mostrado cierta efectividad tanto como monotera-pia como complemento a la piridoxina en series de casos de pacientes recientemente diagnosticados, aunque se observó recurrencia en algunos casos, y aquellos que no tuvieron respuesta permanecieron refractarios otros tratamientos, incluyendo ACTH y vigabatrina. Sus efectos adversos son similares a los del topiramato (7,15).
Otro fármaco, la piridoxina, no tiene ensayos con-trolados aleatorios donde se estudie su utilidad en el tratamiento del SW. El resultado a largo plazo en es-tudios abiertos parece ser favorable en los pacientes que responden a la piridoxina, sin embargo faltan estudios con mediciones objetivas (15).
Las benzodiazepinas, tales como nitrazepam, clo-nazepam y diazepam, se han utilizado anecdótica-mente en el SW, sin embargo, no hay evidencia de que sean tan eficaces como ACTH o corticosteroi-des (15).
III) Otras alternativas terapéuticas
Ganaxolona Es un análogo sintético de neuroesteroides, modu-lador de los receptores GABA-A, que se ha postu-lado como alternativa en casos refractarios, por su similitud de acción con la vigabatrina. En modelos experimentales en animales se han observado re-sultados promisorios en los modelos criptogénicos (16). En un estudio realizado en niños refractarios al tratamiento convencional, la frecuencia de los es-pasmos se redujo al menos un 50% en el 33% de los pacientes (fase IV). El fármaco fue bien tolerado, pero sólo mostró una eficacia modesta y no signi-ficativa, y no se informó su efecto sobre la hipsa-rritmia (17)
Dieta cetogénica Una dieta alta en grasa, adecuada en proteínas y baja en carbohidratos, que se utiliza como alter-nativa a los otros tratamientos, especialmente en casos resistentes a los tratamientos con FAEs. Es-tudios observacionales han mostrado reducción en la frecuencia de espasmos en casos refractarios, sin embargo no existe evidencia de que sea mejor que los compuestos mencionados anteriormente y ade-

40
Revista Chilena de Epilepsia Año 14, Nº 3, Diciembre 2014
más para los pacientes no es fácil de seguir, ya que a menudo presentan complicaciones como diarrea, hipovitaminosis, pérdida de peso y cálculos renales (1,7).
Nuevas opcionesUna opción prometedora para SW refractario a ACTH son los pulsos de rapamicina, un inhibidor de la vía mTOR; en un modelo de ratón refractario a ACTH se obtuvieron buenos resultados en el con-trol de convulsiones, mientras que en otro modelo animal se observó mejoría en el resultado cognitivo (1,7).
Otras terapias nuevas incluyen; a) el uso de fluna-rizina, que podría tener efectos neuroprotectores en niños con SW sin etiología conocida, pero faltan es-tudios de largo plazo (18); b) el rol potencial del fac-tor de crecimiento tipo insulina (IGF) -1, un factor esencial para el desarrollo sináptico, donde se han observado peores resultados cognitivos en pacien-tes con niveles bajos en el líquido cefalorraquídeo (19); c) los agonistas del receptor de melanocortina, que reducen la producción de agentes proinflamato-rios en las células cerebrales luego de una injuria, y tendrían un rol en la neuroprotección (20); y d) la terapia de combinación de tratamiento hormonal con vigabatrina, que actualmente se está probando en el estudio ICISS (International Infantile Spasms Study), donde se postula que puede ser más efectiva y con mejores resultados en el neurodesarrollo que la terapia hormonal por sí sola (21).
En un futuro próximo el estándar de oro podría ser el desarrollo de nuevas terapias que se dirijan a las vías específicas de la patogénesis (1).
IV) Tratamiento quirúrgico.
Alrededor de un 25-40% de los pacientes siguen teniendo espasmos pese a la farmacoterapia inten-siva, y expresan retraso psicomotor, de modo que podrían ser candidatos para cirugía (7). Las resec-ciones quirúrgicas corticales focales han dado como resultado el cese de los espasmos y la normaliza-ción del desarrollo. Se han realizado hemisferec-tomías funcionales exitosas en casos de anomalías hemisféricas graves, como el síndrome de Sturge Weber, hemimegalencefalia, o derrame cerebral he-misférico con hemiparesia asociada. Sin embargo no se considera la opción quirúrgica si la resección cortical puede crear un nuevo déficit neurológico
inaceptable, en casos de daño cerebral difuso o la presencia de alguna enfermedad degenerativa o me-tabólica (2,7,15). PronósticoA pesar de los avances para lograr un diagnóstico precoz y tratamiento adecuado, el pronóstico del SW sigue siendo pobre; 75-90% de los pacientes presentan retraso mental y el 50-60% de los niños tienen convulsiones recurrentes a los cinco años de edad (6). Aproximadamente un 27-50% desarrollan el síndrome de Lennox-Gastaut, una forma seve-ra de epilepsia. La tasa de mortalidad varía entre 3-33% (15).
Los factores pronósticos reportados como favora-bles incluyen etiología criptogénica, edad de inicio mayor de 4 meses, ausencia de espasmos atípicos y crisis parciales, ausencia de anormalidades asi-métricas en el EEG, inicio precoz del tratamiento, y una respuesta temprana y sostenida a éste. Pese a no haber evidencia suficiente como para concluir que el tratamiento exitoso efectivamente mejora el pronóstico a largo plazo, sí existen datos observa-cionales actuales que lo sugieren (15,22).
REFERENCIAS
1. Pavone P., Striano P., Falsaperla R., Pavone L., Ruggieri M. Infantile spasms syndrome, West syndrome and related phenotypes: What we know in 2013. Brain & Development 2014; 36: 739–751.
2. Werz M., Pita I. Infantile Spasms/West Syndro-me. En: Epilepsy Syndromes,Chapter 6. Ed. El-sevier, 2011: 27-32
3. Wheless J., Gibson P., Rosbeck K., Hardin M., O’Dell C., et al. Infantile spasms (West syndro-me): update and resources for pediatricians and providers to share with parents. BMC Pediatrics 2012; 12:108
4. Glaze D. Etiology and pathogenesis of infantile spasms. UpToDate 2014.
5. Prevalence of rare diseases: Bibliographic data, Informes Periódicos de Orphanet, Serie Enfer-medades Raras, Mayo 2014, Número 2: Lista por orden de prevalencia decreciente o por nú-mero de casos publicados. Disponible en: http://www.orpha.net/orphacom/cahiers/docs/ES/Pre-valencia_de_las_enfermedades_raras_por_pre-valencia_decreciente_o_casos.pdf
6. Poulat A., Lesca G., Sanlaville D., Blanchard

41
Actualización del Síndrome de West Vanessa García et al.
G., et al. A proposed diagnostic approach for in-fantile spasms based on a spectrum of variable aetiology. European Journal of Paediatric Neu-rology 2014; 18: 176-182
7. Riikonen R. Recent Advances in the Pharma-cotherapy of Infantile Spasms. CNS Drugs 2014; 28:279–290.
8. Pardo C., Nabbout R., Galanopoulou A. Mecha-nisms of Epileptogenesis in Pediatric Epileptic Syndromes: Rasmussen Encephalitis, Infantile Spasms, and Febrile Infection-related Epilepsy Syndrome (FIRES). Neurotherapeutics 2014; 11: 297–310
9. Osborne J., Lux A., Edwards S., Hancock E., Johnson A., et al. The underlying etiology of infantile spasms (West syndrome): Information from the United Kingdom Infantile Spasms Stu-dy (UKISS) on contemporary causes and their classification. Epilepsia 2010; 51(10): 2168–2174
10. Kliegman R., Stanton B., Geme J., Schor NBe-hrman R. Seizures in Childhood. En: Nelson Textbook of Pediatrics, Chapter 586. Ed. Else-vier, 2011: 2013-2039.
11. Swaiman K., Ashwal S., Ferriero D., Schor N. Myoclonic Seizures and Infantile Spasms. En: Swaiman’s Pediatric Neurology: Principles and Practice, Chapter 56. Ed. Elsevier, 2011: 774-789.
12. Glaze D. Clinical features and diagnosis of in-fantile spasms. UpToDate 2014.
13. Wanigasinghe J., Arambepola C., Ranganathan S., Sumanasena S. The Efficacy of Moderate-to-High Dose Oral Prednisolone versus Low-to-Moderate Dose Intramuscular Corticotropin for Improvement of Hypsarrhythmia in West Syndrome: A Randomized, Single-Blind, Pa-rallel Clinical Trial. Pediatric Neurology 2014; 51: 24-30.
14. Mesa T, López I, Förster J, Carvajal M, David P. Consenso Chileno de Manejo de Fármacos Antiepilépticos en algunos Síndromes Electro-Clínicos y otras Epilepsias en Niños y Adoles-
centes. Rev Soc Psiquiatr Neurol Infanc Ado-lesc 2011; 22(3):232-274.
15. Glaze D. Management and prognosis of infanti-le spasms. UpToDate 2014.
16. Yum M., Lee M., Ko T., Velisek L. A potential effect of ganaxolone in an animal model of in-fantile spasms. Epilepsy Research 2014; 108: 1492—1500.
17. Kerrigan J., Shields W., Nelson T., Bluesto-ne D., Dodson W., et al. Ganaxolone for trea-ting intractable infantile spasms: a multicenter, open-label, add-on trial. Epilepsy Res. 2000; 42: 133–139.
18. Bitton J., Sauerwein H., Weiss S., Donner E., Whiting S. A randomized controlled trial of flu-narizine as add-on therapy and effect on cogni-tive outcome in children with infantile spasms. Epilepsia 2012; 53(9):1570–1576.
19. Riikonen R., Jaaskelainen J., Turpeinen U. Insulin-like growth factor-1 is associated with cognitive outcome in infantile spasms. Epilep-sia 2010; 51(7):1283–1289.
20. Catania A. Neuroprotective actions of melano-cortins: a therapeutic opportunity. Trends Neu-rosci. 2008; 31(7):353–360.
21. ICISS (unpublished data only) International Collaboration Infantile Spasms Study. Ongoing study. May 2007. Disponible en: http://www.controlled-trials.com/ISRCTN54363174.Lee J., Lee J., Yu H., Lee M. Prognostic factors of infantile spasms: Role of treatment options in-cluding a ketogenic diet. Brain & Development 2013; 35: 821–826.
22. Johannes R. Lemke, MD,1,2 Rik Hendrickx, BSc,3,4 Kirsten Geider, MSc,5, Bodo Laube, PhD,5 Michael Schwake, PhD,6 Robert J. Har-vey, PhD GRIN2B Mutations in West Syndrome and Intellectual Disability with Focal Epilepsy. West Syndrome Epilepsy Annal neurol 2014, 75(1) 147.
23. Ghasemi M, Schachter SC. The NMDA receptor complex as a therapeutic target in epilepsy: a re-view. Epilepsy Behav 2011;22:617–640.

42
Revista Chilena de Epilepsia Año 14, Nº 3, Diciembre 2014Actualizaciones
Síndrome de Lennox-Gastaut, una revisión actualizada.David P.1, García V.2, Meneses S3.Correo: [email protected]
ABSTRACT
The Lennox-Gastaut Syndrome is a severe form of chronic epileptic encephalopathy, characterized cli-nically by the triad: slow spike-wave pattern of the EEG, multiple types of seizures and cognitive decli-ne. Corresponds to 3-6% of children with epilepsy, with male predominance and has a peak of onset between 3 and 5 years. The LGS is associated with a wide variety of etiologic factors, although up to 40% of the causes are unknown, and its pathophy-siological substrate remains uncertain. Treatment is lifelong and aims to improve the quality of life of patients by decreasing the frequency of seizures, since there is no optimal therapy for complete re-mission. Drugs that have proven useful are Valproic Acid (first line AED at Chile), Lamotrigine, Topira-mate, Felbamate and Rufinamide. Other treatment options for refractory cases are the ketogenic diet, vagus nerve stimulation and surgery. The LGS has a poor long-term prognosis, with a mortality of 10% before 11 years of age.
RESUMEN
El Síndrome de Lennox-Gastaut es una forma seve-ra de encefalopatía epiléptica crónica, clínicamente caracterizada por la triada: patrón de espiga-onda lenta al Electroencefalograma, múltiples tipos de crisis epilépticas y deterioro cognitivo. Correspon-de al 3-6% de los niños con epilepsia, con predomi-nio masculino y presenta un peak de inicio entre los 3 y 5 años. El SLG se asocia con una amplia varie-dad de factores etiológicos, aunque hasta en un 40% la causa es desconocida, y su substrato fisiopatoló-gico permanece incierto. El tratamiento es de por vida y su objetivo es mejorar la calidad de vida del
paciente a través de la disminución de la frecuencia de las crisis, ya que no existe terapia óptima para la remisión total. Los fármacos que han demostrado utilidad son Ácido Valproico (FAE de primera línea en Chile), Lamotrigina, Topiramato, Rufinamida y Felbamato. Otras alternativas de tratamiento para los casos refractarios son la dieta cetogénica, la es-timulación del nervio vago y el tratamiento quirúr-gico. El SLG presenta un pobre pronóstico a largo plazo, con una mortalidad de un 10% antes de los 11 años de edad.
INTRODUCCIÓN
El Síndrome de Lennox-Gastaut (SLG), descrito originalmente por Lennox en 1949 y en la mayor serie de 100 pacientes por Gastaut en 1966 (1), co-rresponde a una forma severa de encefalopatía epi-léptica crónica que se manifiesta habitualmente en la primera infancia, con un peak de inicio entre los 3 y 5 años (2), y corresponde aproximadamente al 3 a 6% de los niños con epilepsia, con claro predo-minio masculino (5:1) (3). El término definitivo fue introducido en 1969 por Niedermeyer.
Este síndrome se caracteriza por presentarse con un curso refractario, tipos de crisis características, de-terioro cognitivo marcado y prominentes anorma-lidades electroencefalográficas interictales. A pesar de esta homogeneidad en su presentación, el SLG se asocia con una amplia variedad de factores etio-lógicos y su substrato fisiopatológico permanece incierto (4).
ETIOLOGÍA
El SLG se ha asociado a múltiples causas, entre las que se incluyen trastornos genéticos, síndromes neurocutáneos (ej, esclerosis tuberosa), encefalopa-tías post lesiones hipóxico-isquémicas, meningitis, traumas cefálicos, lesiones cerebrales focales o di-fusas, e incluso se ha observado en pacientes sin le-
1. Clínica Dávila, Universidad de los Andes2. Interna Medicina Universidad de los Andes3. Interno Medicina Universidad de ValparaísoRecibido 21 de Nov. Aceptado 20 de Dic. 2014Los autores declaran no tener conflictos de intereses.

43
Síndrome de Lennox-Gastaut, una revisión actualizada. Perla David et al.
sión cerebral evidente. Además puede desarrollarse a partir de un síndrome de West en hasta un 20-30% de los casos.
Pese a todo, aproximadamente el 25-40% de los pa-cientes tienen una etiología desconocida, aunque en estos niños cada vez se encuentran más trastornos genéticos, particularmente síndromes cromosómi-cos o mutaciones de novo (4,5).
PATOGÉNESIS
La patogénesis es poco conocida, pero puede impli-car una influencia genética en el desarrollo del sis-tema nervioso inmaduro, particularmente durante el desarrollo tálamo-cortical, provocando patología cerebral estructural o funcional. La teoría más acep-tada es que el SLG es el resultado de una injuria cortical difusa o multifocal que ocurre en un deter-minado momento en el cerebro en desarrollo, lo que conllevaría a una mayor excitabilidad del sistema nervioso inmaduro, que a su vez pone en marcha complejos cambios neuropatológicos secundarios y establece un difuso sistema epileptogénico hiperex-citable, responsable de las descargas epileptiformes bilaterales y de una epilepsia médicamente refrac-taria. El grave deterioro de las funciones cognitiva, sensorial y/o motora se cree que ocurre secundario a las frecuentes convulsiones severas y actividad epi-léptica interictal persistente que son mantenidas por este sistema de circuitos cerebrales disfuncionales (4).
CLÍNICA
El SGL se caracteriza fundamentalmente por una triada clásica: múltiples tipos de crisis epilépticas, patrón de espiga-onda lenta al Electroencefalogra-ma (EEG) y deterioro mental.
I) Crisis epilépticas. Se presentan con alta fre-cuencia y son de diversos tipos; las más ca-racterísticas son las crisis tónicas y atónicas, comúnmente llamadas “crisis de caídas” (drop attacks), las que son casi patognomónicas (92% de los pacientes). También se pueden mani-festar con crisis atónicas (26-56%), ausencias atípicas (20-65%), estatus epiléptico no con-vulsivo (50-66%), crisis tónico-clónicas, crisis parciales, espasmos y crisis mioclónicas.
II) Anormalidades características en el EEG. La
actividad de base del trazado es desorganiza-da y lenta, con un patrón ictal o interictal ca-racterizado por descargas de espiga-onda lenta de frecuencia menor a 2,5 Hz con predominio frontal EEG de vigilia, así como la presencia de paroxismos de actividad espicular rítmica rápi-da de 10 a 20 Hz durante el sueño; conocido como un patrón de “ondas y espigas atípicas”.
III) Deterioro cognitivo con retardo mental. Se presenta en el 95% de los pacientes y oca-sionalmente es progresivo. Habitualmente se presentan asociados graves trastornos de com-portamiento, tales como hiperactividad, agresi-vidad y tendencias autistas, y trastornos de la personalidad. Además, es frecuente que aparez-can síntomas psicóticos. A menudo, el desarro-llo neurológico es normal antes de la primera convulsión (3,5,6,7).
TRATAMIENTO
Dado su carácter de síndrome crónico el SLG re-quiere tratamiento de por vida, y los pacientes casi siempre desarrollan déficits conductuales y psicoló-gicos progresivos. A diferencia de la mayoría de los trastornos convulsivos, donde el objetivo del trata-miento es lograr la remisión completa de las crisis, en el SLG esto raramente se consigue por su natu-raleza resistente y los diversos tipos de crisis con-vulsivas que experimentan los pacientes, de modo que el tratamiento se enfoca en mejorar la calidad de vida del paciente a través de la disminución de la frecuencia de las crisis, con el menor número posi-ble de eventos adversos (2).
La terapia óptima para el SLG es incierta y puede depender en parte de la etiología subyacente (5). En la actualidad no existen evidencias suficientes que permitan recomendar un tratamiento estándar, por lo que las recomendaciones para su tratamiento se basan en opiniones de expertos.
I) Tratamiento farmacológico. Dada la variedad de tipos de convulsiones asociadas con la LGS, se necesitan fármacos con un amplio espectro de eficacia para el tratamiento de este síndro-me. En revisiones sistemáticas de ensayos alea-torios controlados se ha concluido que ningún fármaco ha demostrado ser altamente efectivo, aunque el Ácido valproico, la Lamotrigina, el Topiramato, la Rufinamida, el Felbamato y el

44
Revista Chilena de Epilepsia Año 14, Nº 3, Diciembre 2014
Clobazam pueden ser útiles. En contraste, el uso de Carbamazepina puede precipitar drop attacks en niños (5).
En Chile, en el año 2010, se decidió establecer un consenso de especialistas chilenos sobre el uso de fármacos antiepilépticos (FAE) en algu-nos Síndromes Epilépticos y Epilepsias de ni-ños y adolescentes, de mayor frecuencia o más difícil manejo, en el cual se fijaron propuestas de tratamiento para el SLG:
i) Como FAE de primera línea se prefiere el Ácido Valproico, dada la gran experiencia acumulada de su uso en diferentes tipos de crisis.
ii) Como segunda opción se sugiere asociar Clobazam (CLB), Lamotrigina (LTG) o Topiramato (TPM). Entre las benzodiace-pinas se prefiere CLB por tener menor efecto sedativo y por su eficacia en drop attacks, pero a dosis altas (1 mg/kg/d) (8). La recomendación de LTG o TPM se basa en la experiencia de su uso en otras epilep-sias refractarias.
iii) Como tercera opción se sugieren algunos fármacos que no se encuentran disponibles en Chile, tales como Felbamato, Rufinami-da y Zonisamida (3).
II) Otras alternativas terapéuticas. Debido a que las convulsiones son a menudo médicamente refractarias, ocasionalmente se consideran otras opciones de tratamiento, tales como la dieta ce-togénica, que ha sido útil en algunos niños con SLG, y la estimulación del nervio vago, que también parece ser eficaz en algunos pacien-tes ya que conduce a una reducción de más del 50% en la frecuencia de las crisis (en particular para convulsiones tónicas y atónicas), así como reducción de la duración de las crisis y dismi-nuye el número de FAE prescritos. Además, la psicoterapia y las intervenciones psiquiátricas pueden ser útiles en el tratamiento de los tras-tornos psiquiátricos (7).
III) Tratamiento quirúrgico. En los casos en que la etiología del SLG se debe a lesiones corticales localizadas, tales como la displasia cortical, es-clerosis tuberosa cortical, tumores, heterotopía en banda y malformaciones vasculares, los pa-cientes son susceptibles de recibir tratamiento
quirúrgico temprano con un impacto significa-tivo en el control de las crisis y el desarrollo cognitivo (9).
Otras opciones quirúrgicas, incluyendo callo-sotomía o cirugía del foco epileptogénico en pacientes con hamartoma hipotalámico, pueden ser consideradas en algunos casos refractarios (5).
PRONÓSTICO
Los niños con este síndrome tienen un pobre pro-nóstico a largo plazo; aunque con frecuencia la epi-lepsia mejora, es raro lograr una remisión completa de las crisis y por el contrario los trastornos menta-les y psiquiátricos tienden a empeorar con el tiempo (7).
Los pacientes sin una causa identificable pueden tener un fenotipo algo más suave y un deterio-ro funcional y neurológico menos profundo en la edad adulta. La presencia de espiga-onda lenta en el EEG en niños con múltiples tipos de crisis predi-ce la coexistencia de retraso mental profundo y en general son más propensos a tener parálisis cerebral que aquellos con múltiples tipos de crisis pero sin espiga-onda lenta en el EEG. Además, el riesgo de lesiones graves asociadas a drop attacks es alta.
La mortalidad también es alta (tasa de mortalidad estandarizada = 14); hasta el 10% de los niños con SLG mueren antes de los 11 años de edad (5,10).
REFERENCIAS
1. Widdess-Walsh P, Dlugos D, Fahlstrom R, Joshi S, Shellhaas R, et al. Lennox-Gastaut syndrome of unknown cause: Phenotypic characteristics of patients in the Epilepsy Phenome/Genome Pro-ject. Epilepsia 2013; 54(11):1898–1904.
2. Ng Y, Conry J, Paolicchi J, Kernitsky L, Mit-chell W, et al. Long-term safety and efficacy of clobazam for Lennox–Gastaut syndrome: In-terim results of an open-label extension study. Epilepsy & Behavior 2012; 25:687–694
3. Mesa T, López I, Förster J, Carvajal M, David P. Consenso Chileno de Manejo de Fármacos Antiepilépticos en algunos Síndromes Electro-Clínicos y otras Epilepsias en Niños y Adoles-centes. Rev Soc Psiquiatr Neurol Infanc Ado-lesc 2011; 22(3):232-274.

45
Síndrome de Lennox-Gastaut, una revisión actualizada. Perla David et al.
4. Badawy R, Macdonell R, Vogrin S, Lai A, Cook M. Cortical excitability decreases in Lennox-Gastaut syndrome. Epilepsia 2012; 53(9); 1546-1553.
5. Wilfong A. Epilepsy syndromes in children. Up-ToDate 2014.
6. Sociedad Española de Neurología. Capítulo 28: Actitud Terapéutica en el Síndrome de Lennox-Gastaut. Guía para el Diagnóstico y Tratamiento de la Epilepsia 2008:219-222.
7. Hancock E, Cross J. Treatment of Lennox-Gas-taut syndrome. Cochrane Database of Systema-tic Reviews 2013; 2: 1-12.
8. Cramer J, Sapin C, Francois C. Indirect com-
parison of clobazam and other therapies for Lennox–Gastaut syndrome. Acta Neurol Scand 2013; 128: 91–99
9. Liu S, An N, Fang X, Singh P, Oommen J, et al. Surgical Treatment of Patients with Lennox-Gastaut Syndrome Phenotype. The Scientific World Journal 2012: 1-10.
10. Trevathan E. Lennox-Gastaut Syndrome. Wash-ington University School of Medicine and St. Louis Children’s Hospital, Department of Neu-rology 2006-2012. Artículo disponible en www.neuro.wustl.edu/patientcare/clinicalservices/pediatricepilepsycenter/patientfamilyphysician/lennoxgastautsyndrome.

46
Revista Chilena de Epilepsia Año 14, Nº 3, Diciembre 2014
Calendario de Reuniones Año 2014Crónica
REUNION DE TRABAJO Nº 143Sábado 12 de Abril de 2014 a las 9.30 hrs.“Síndrome Dress. A propósito de un caso clínico”Dra. Carolina GallegosNeuróloga Hospital de Carabineros
REUNION DE TRABAJO Nº 144Sábado 10 de Mayo de 2014 a las 9.30 hrs.“Síndrome de Dravet”Dra. Joanna BoraxNeuróloga InfantilHospital de Carabineros
REUNION DE TRABAJO Nº 145Sábado 12 de Julio de 2014 a las 9.30 hrs.“Presentación Caso Clínico: Status Convulsivo y Porfiria” Dr. Luis Espinoza MartínezNeurólogoClínica Bicentenario
REUNION DE TRABAJO Nº 146Sábado 09 de Agosto de 2014 a las 9.30 hrs.“Cirugía de Epilepsia Lóbulo Temporal sin Lesión, presentación de un caso con monitoreo invasivo” Dra. Karina Rosso A.Neurología InfantilPontificia Universidad Católica
REUNION DE TRABAJO Nº 147Sábado 13 de Septiembre de 2014 a las 9.30 hrs.“Guias Prácticas de Epilepsia” Dra. Lilian Cuadra O.Ministerio de Salud
REUNIÓN DE TRABAJO N°148Sábado 11 de Octubre de 2014 a las 9.30 hrs.
“Cirugía de la Epilepsia en edad pediátrica”.Dr. Christian Cantillano M.NeurocirujanoHospital Sótero del Río.
REUNIÓN DE TRABAJO N°149Sábado 08 de Noviembre de 2014 a las 9.30 hrs.“Epilepsia y Música”Dra. Claudia Riffo A.Neuropediatra / Neuróloga InfantilUniversidad Católica de Chile
REUNION DE TRABAJO Nº 150Sábado 13 de Diciembre de 2014 a las 9.30 hrs.“Epilepsia y Enfermedades Mitocondriales”Dr. Juan MoyaNeurólogo InfantilHospital Luis Calvo Mackenna
REUNION DE TRABAJO Nº 151Sábado 10 de Enero de 2015 a las 9.30 hrs.“Actualización en el tratamiento de Apnea de Sueño y Epilepsia”Dr. Mauricio BravoHospital Militar
“Epilepsia súper resistente. Tratamiento compa-sivo y de acompañamiento con cannabis. Presen-tación de un caso”.Dr. Marcelo Devilat, Dra. Carla Manterola, Dr. Juan Moya.Servicio de Neurología y Psiquiatría Hospital Luis Calvo Mackenna.
REUNIÓN DE TRABAJO N°152Asamblea General Ordinaria, Sábado 14 de Marzo 2015.Elección de Directorio.Conmemoración XVI años Sociedad de Epilep-tología de Chile.

47
Noticias Poéticas 25.12.14Crónica
“En las noches de mis días maullando mendigo un trocito de luna. ¿Y qué he conseguido?
(Clemencia Tariffa)*
Clemencia Tariffa es una poeta, (como ella deseaba autonominarse, y no poetisa) nacida en 1959 en Codazzi, Colombia y que falleció en una clínica mental en Santa Marta en 2009. Clemencia estuvo internada los diez últimos años de su vida pues sufría una epilepsia, probablemente con crisis tónico-clónicas generalizadas, que fueron “llenando su rostro de cicatrices” y que posteriormente derivó en un serio compromiso neuro-psiquiátrico. Por su carácter rebelde y desapego familiar, no tuvo un tratamiento adecuado después que falleció su madre en 1999, lo que la llevó prematuramente a la muerte. A través del libro que comentamos, sabemos que su trabajo poético contiene dos volúmenes: “El ojo de la noche” (1987) y “Cuartel” (2007). En ellos podemos destacar entre muchas otras, la pieza titulada “Claridad” (pág. 93) en la que con maestría y delicadeza nos comunica un sentimiento de aislamiento y soledad, posiblemente derivado de las experiencias con su enfermedad, pero que no se niega a que su situación se puede revertir a través del amor:
“Yo poseo la autoridadde veinte mariposashaciendo el amor,yo sueño con libertadaleteando desde mi cama,
y también sé destilar veneno;claro que en usted es una lágrima en arena
Yo, con hoja y lápizle escribiré una cartacon ilusiones y color,si aún puede ser.
Yo quiero amarlo muchoen una mañana clara como ayer,Ven.Que estás acostumbrado a huiryo no quiero más soledady temor a la vez”.
Clemencia, doblemente premiada en su país, se fue haciendo dependiente de su madre, y ella misma nos relata (pág. 129) que:” mi madre me hace los vestidos….me cocina, me suministra las medicinas, me cuida. Desde hace muchos años nadie me da un trabajo porque yo no sé hacer trabajo alguno”. ¿Qué papel habrá desempeñado la epilepsia en esta dependencia y en este rechazo? Nos parece que debió haber sido relevante el estigma social que cayó sobre ella, lo que fue asociado a su propia inseguridad cuando dice:…”ni siquiera me he atrevido a tener un hijo, por que no sabría cómo criarlo”. A pesar de anterior, la poeta no se deja abatir y se refugia en su poesía cuando dice. “Como puedes ver, en mi caso, escribir poesía es salvarme, es vivir”. Y da prueba de ello en su bellísima y cantarina poesía Amistad (pág. 91):
“Si nuestra amistadno fuese tan puracomo el amorque hacemos clandestinamente,o tan claracomo tus ojillos color castañoo tan firmecomo esta luna de marfil
* Del libro “Difícil hablar con las sombras. Poesía reunida”.Ediciones Exilio. Bogotá. Zapatoca. Santa Marta. 2014. Ejem-plar enviado por nuestro amigo y colega Dr. Jaime Carrizosa, colombiano.
Con autorización Sr. Hernán Vargascarreño, editor.

48
Revista Chilena de Epilepsia Año 14, Nº 3, Diciembre 2014
que revolotea en mi país,de veras, no escribiría esta noche.Yo solo quiero tu amistad
La lectura de las poesías de Clemencia nos emocionan y nos sobrecogen por que expone sus sentimientos y deseos sin barreras, con autoridad y transparencia, pero a excepción de dos o afirmaciones indirectas sobre su enfermedad, en ninguna de sus poesías o comentarios aparece una mención directa sobre su epilepsia, designándola como tal. Así mismo, ninguno de sus comentaristas aluden a ello ni describen sus males con la palabra epilepsia. Lo anterior es llamativo y sugiere como una concertación, a pesar de las evidencias de vida de la poeta, de ignorar el vocablo, con lo cual solo se obtiene profundizar el estigma y el rechazo a las personas con epilepsia como misma confiesa que lo sufre. Grandes escritores y poetas tuvieron epilepsia y acompañan a Clemencia, entre ellos: Dostoievsky, Lord Byron, Edgar Allan Poe, Dickens y Flaubert. Este último como nuestra poeta se encerró en sí mismo y se replegó con todas sus energías en la escritura. En todos los citados como en Clemencia, la epilepsia marcó sus vidas y su repertorio creativo con la antigua estigmatización que lamentablemente
Nuevos Socios Año 2014
El Año 2014 se incorporaron como nuevos socios:
• Dra. Carolina Gallegos (Abril 2014)
• Dra. Joanna Borax (Mayo 2014)
• Dra. Carla Manterola (Junio 2014)
• Dr. Luis Espinoza (Julio 2014) • Dra. Karina Rosso (Agosto 2014)
• Dr. Christian Cantillano (Octubre 2014)
• Dra. Claudia Riffo (Noviembre 2014)
Dra. Carla ManterolaSecretaria General
Dr. Cayetano NapolitanoPresidente
persiste hasta nuestros. ¿Qué podemos hacer para demostrar que la epilepsia es una enfermedad como cualquier otra?
Dr. Marcelo Devilat Barros

49
Jornadas Invernales de Epilepsia. Santiago, Chile, 05 y 06 de Junio de 2015.“Epilepsia desde una mirada moderna”
Crónica
VIERNES 05 JUNIO 2015
08:15-08:30 Introducción. Dra. Ledia Troncoso.
Módulo I: Epileptogénesis: desde la neurocien-cia a la clínica.
08:30-09:00 Epigenética-optogenética. Dr. Andrés Barrios.
09:00-09:30 Nuevos síndromes epilépticos, nue-vos genes, canalopatías.
Dr. Alvaro Retamales.
09:30-10:00 En la búsqueda etiológica: afecciones heredometabólicas y genéticas con epilepsia como síntoma marcador.
Dr. Felipe Castro.
10:00-10:30 Café
Módulo II: Visión actualizada en el estudio de las epilepsias.
10:30-11:00 Electrofisiología. Dr. Roberto Caraballo.
11:00-11:30 Neuroimagenología. Dr. Salvador Camelio.
11:30-12:00 Mesa redonda - módulos 1-11. Preside: Dr. Roberto Caraballo. Participan: Expositores de ambos
módulos.
13:00-14:00 Simposio almuerzo. Neuroestimula-ción: estimulador del Nervio Vago, estimulador de núcleo anterior talá-mico, hipotermia cerebral, perfusión de drogas localizadas, estimulación óptica Cortex cerebral.
Dra. Loreto Ríos, Dr. Hernán Aceve-do.
14:00-15:30 Presentación de pósters. Dirigen: Dra. Maritza Carvajal, Dr.
Enzo Rivera, Dra. Lilian Cuadra, Dra. Viviana Venegas.
Módulo III: Manejo moderno de las epilepsias.
15:30-16:00 Monoterapia en fármacos antiepilép-ticos de segunda generación.
Dra. Daniela Triviño.
16:00-16:30 Politerapia: fármacos antiepilépticos y uso racional.
Dra. Daniela Aguilera.
16.30-17:00 Fármacos antiepilépticos de tercera generación: Lacosamida, Rufinami-da, Ezogabina, Retigabina, Perampa-nel.
Dr. Roberto Caraballo.
17:00-17:30 Cafe
17:30-18:00 Terapia con inmunomoduladores. Dr. Reinaldo Uribe.
18:00-18:30 Real utilidad de antiepilépticos vía parenteral (endov.) vs vía oral / Dieta cetogénica.
Dr. Juan Moya.
18.30-19.45 Una mirada moderna del manejo te-rapéutico del paciente con epilepsia y trastornos psiquiátricos.
Dr. Jaime Godoy / Dr. Fernando Iva-novic-Zuvic.

50
Revista Chilena de Epilepsia Año 14, Nº 3, Diciembre 2014
SÁBADO 06 DE JUNIO 2015
Módulo III (continuación): Cirugía de la Epi-lepsia.
09:00-09:30 Estimulación cerebral profunda. Dr. David Aguirre.
09:30-10:00 Gammaknife en epilepsia. Dr. Claudio Lühr.
10:00-10:30 Cirugía no lesional. Dr. Christian Cantillano. 10:30-11:00 Mesa redonda. Preside: Dr. Arturo Zuleta. Participan: Dr. Manuel Campos, Dr.
Claudio Lühr, Dr. Christian Cantilla-no, Dr. David Aguirre.
Módulo IV: Una mirada moderna desde la clí-nica.
11:00-11:30 Convulsiones neonatales: diagnósti-coy manejo actualizdo.
Dra. Karla Henríquez.
11:30-12:00 Esclerosis tuberosa: un modelo de epilepsia refractaria.
Dra. María Francisca López.
12:30-13:00 Estado epiléptico súper-refractario. Dr. Cayetano Napolitano.
13:00-13:15 Cierre. Comité organizador XV Jornadas In-
vernales de Epilepsia.

51
Declaración de InteresesCrónica
SUBSECRETARÍA DE SALUD PÚBLICADIVISIÓN DE PREVENCIÓN Y CONTROL DE
ENFERMEDADES
Las consideraciones de salud pública tienen una im-portancia primordial en todo el trabajo técnico del Ministerio de Salud. Es preciso que se adopten me-didas para garantizar que se efectúe la mejor eva-luación posible de los datos científicos, en una at-mósfera independiente exenta de presiones directas o indirectas. Por lo tanto, para preservar la integri-dad técnica y la imparcialidad del trabajo del Minis-terio de Salud, es necesario prevenir situaciones en las cuales el resultado de ese trabajo pudiera verse afectado por intereses financieros o de otra índole.
Por consiguiente, se pide a cada experto(a) que de-clare si es parte interesada en algo que, en lo refe-rente a su participación en el trabajo que realiza en el Ministerio de Salud, podría dar lugar a un con-flicto real, potencial o aparente de intereses entre (1) entidades comerciales y el participante perso-nalmente o (2) entidades comerciales y la unidad administrativa para la cual trabaja el participante. Por “entidad comercial” se entiende cualquier em-presa, asociación, organización u otra entidad, sea cual fuere su naturaleza, que tenga intereses comer-ciales.
¿Qué es un conflicto de intereses?
Hay conflicto de intereses si:
1. El experto(a) o su pareja (“por pareja” se entien-de un cónyuge u otra persona con la cual el experto mantiene una estrecha relación personal de natura-leza semejante), o la unidad administrativa para la cual trabaja el experto, tienen un interés financiero o de otra índole que podría afectar indebidamente a la posición del experto, en lo concerniente al asunto que se está considerando.2. Hay conflicto aparente de intereses cuando un in-
terés, que no necesariamente influiría en el experto, podría dar lugar a que otros cuestionasen la objeti-vidad de éste.
3. Existe un conflicto potencial de intereses toda vez que una persona razonable se pregunta si debe o no informar acerca de un interés.
Se puede prever diferentes tipos de intereses finan-cieros o de otra índole, bien sea personal o rela-cionado con la unidad administrativa para la cual trabaja el experto, y la siguiente lista, que no es ex-haustiva, puede servir de orientación. Por ejemplo, deben declararse los siguientes tipos de situaciones:
a. toda participación patrimonial vigente en una sustancia, una tecnología o un proceso (por ejemplo la propiedad de una patente), que se examinarán en la reunión o en el trabajo o que están relacionados de otra manera con el tema correspondiente;
b. todo interés financiero vigente, por ejemplo la posesión de valores bursátiles tales como acciones u otros títulos de una entidad comercial que sea par-te interesada en el asunto por examinar en la reu-nión o el trabajo (Ej.: Industria Farmacéutica);
c. todo empleo, consultoría, cargo de dirección u otra posición, remunerados o no, en el curso de los 4 años precedentes en cualquier entidad comercial que sea parte interesada en el tema de la reunión/trabajo, o una negociación en curso sobre un posi-ble empleo u otra asociación con una entidad co-mercial semejante;
d. todo trabajo o investigación remunerados realiza-dos en el curso de los 4 últimos años por encargo de una entidad comercial que sea parte interesada en el tema de tas reuniones o del trabajo;
e. todo pago u otra forma de apoyo recibidos en el curso de tos 4 últimos años, o cualquier expectativa

52
Revista Chilena de Epilepsia Año 14, Nº 3, Diciembre 2014
de apoyo futuro de una entidad comercial que sea parte interesada en el tema de tas reuniones o del trabajo, aunque no beneficie al experto personal-mente sino a su puesto o a la Unidad Administrativa para la cual trabaja el experto, por ejemplo una sub-vención, una beca u otro tipo de pago, por ejemplo para financiar un puesto o una consultoría,
En relación con lo anterior, se debe declarar igual-mente si uno es parte interesada en una sustancia, una tecnología o un proceso competidores, o en al-gún trabajo realizado para, en asociación con o con apoyo de una entidad comercial que tenga un inte-rés competidor directo.
Cómo se rellena esta declaración:
Debe declarar cualquier interés financiero o de otra índole que pudiera dar lugar a situaciones de con-flicto real, potencial o aparente de intereses:
1) En relación con usted mismo o su pareja, así como2) en relación con la unidad administrativa para la cual trabaja usted.
Debe revelar solamente el nombre de la entidad co-mercial y la naturaleza del interés; no es necesario especificar ninguna cantidad (aunque usted lo pue-de hacer si considera que esa información es perti-nente para evaluar el interés). En lo concerniente a los puntos 1 y 2 de la lista precedente, el interés sólo se debe declarar si es vigente. Con respecto a los puntos 3, 4 y 5, se debe declarar cualquier interés existente en el curso de los 4 últimos años. Si el interés ya no es vigente, sírvase declarar el año en que dejó de serlo.
Declaración:
¿Tiene usted o tiene su pareja un interés financiero o de otra índole en el tema de la reunión o en el traba-jo en el cual usted participará, y puede considerarse que ello dará lugar a un conflicto real, potencial o aparente de intereses?
Sí: No: En caso afirmativo, sírvase especificar.____________________________________________________________________________________________________________________________________________________________________
___________________________________________________________________________________________________________________________
Por favor responda las siguientes preguntas, en rela-ción a los últimos 24 meses:
¿Ha recibido usted honorarios por dictar conferen-cias?
Sí: No:En caso afirmativo, sírvase especificar._______________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
¿Ha recibido apoyos económicos e invitaciones para asistir a congresos y otras actividades cien-tíficas?
Sí: No:En caso afirmativo, sírvase especificar._______________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
¿Ha recibido fondos para realizar investigaciones?
Sí: No:En caso afirmativo, sírvase especificar._______________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
¿Ha recibido honorarios por consultorías?
Sí: No: En caso afirmativo, sírvase especificar.__________________________________________________________________________________

53
Declaración de Intereses Crónica
_____________________________________________________________________________________________________________________________________________________________________________________________________________
¿Hay algo más que podría afectar a su objetividad o independencia o en el trabajo que Ud. Realiza en el Ministerio de Salud, o la impresión que otros podrían tener de la objetividad e independencia de usted?_______________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
Por la presente, declaro que la información revelada es correcta y que no tengo conocimiento de ninguna otra situación de conflicto real, potencial o aparente de intereses. Me comprometo a notificar cualquier cambio al respecto, o incluso si se plantea una cues-
tión pertinente durante el curso mismo del trabajo que realizó en el Ministerio de Salud.
Nombre:__________________________________________________________________________________
Especialidad:__________________________________________________________________________________
Institución:__________________________________________________________________________________
Firma:
_________________________________________
Fecha:_________________________________________
FE DE ERRATAS:La Comorbilidad Psiquiátrica en Epilepsia fue presentada en las Jornadas Invernales del año 2004 por
la Dra. Inés Lackington y publicada en Revista Chilena de Epilepsia.

54
Revista Chilena de Epilepsia Año 14, Nº 3, Diciembre 2014
Sugerencias para las contribuciones a los autores
Las contribuciones podrán tener la forma de trabajos originales de investigación clínica o experimental, de medicina social y salud pública relacionadas con las epilepsias, revisiones de temas, casos clínicos, crónica y cartas al editor.
Las colaboraciones deberán ser enviadas a la secreta-ría de la Sociedad de Epileptología de Chile y revisa-das por el Comité Editorial.
Los artículos se entregarán mecanografiados en papel tamaño carta con doble espacio, con un máximo de 26 líneas por página, con un margen de 2.5 cm en todos sus bordes, escritos con letra Arial nivel 12. La exten-sión máxima para los artículos originales y de revisión será de 16 páginas, de 8 para los casos clínicos y de 3 para los artículos de crónica y cartas al editor. Se incluirá un original con dos fotocopias y un archivo en CD utilizando programa Word para PC.
Se aceptarán figuras (dibujos y gráficos) enviados en forma de copia fotográfica en papel satinado blanco y negro de 10 x 15 cm. La lectura de las figuras se hará en hoja separada. En el dorso de cada figura se marcará el número que la identifica y una flecha con su orienta-ción con lápiz de carbón. En el texto se indicará dónde debe ser intercalada.
Las tablas (cuadros o tablas) se enviarán mecanogra-fiados y numerados según orden de aparición en el tex-to, en el cual se señalará su ubicación.
Se aceptará un máximo de 5 elementos (figuras o ta-blas) por artículo.
El título deberá ser claro y conciso. Se incluirá el nombre de los autores con el primer apellido, el título profesional de cada uno de ellos y el lugar donde se realizó el trabajo. Las referencias bibliográficas deben limitarse a un máximo de 15. Se sugiere referir y citar bibliografía latinoamericana y chilena y al terminar mencionar el e-mail del autor principal.
Clasificación de las contribuciones:1. Trabajo original. Realizado según el siguiente es-
quema: a) Introducción, donde se plantea la situación ge-
neral del problema; b) Objetivos, donde se plan-
tean los antecedentes y los problemas que se quie-re resolver; c) Material o Pacientes y Métodos, en el que se hacen explícitas las características del universo y cómo se instrumentalizó; d) Resulta-dos, donde se expone la situación obtenida; e)Dis-cusión, en la que se comentan los resultados con relación a los problemas planteados o a la informa-ción proporcionada por otros autores; f) Resumen de 200 palabras en español e inglés.
2. Trabajos de revisión. Se trata de una revisión bi-bliográfica acerca de un tema específico, presen-tado según las instrucciones de longitud y referen-cias bibliográficas ya señaladas.
3. Casos clínicos. Presentación de casos de interés práctico, según el esquema de trabajo original.
4. Actualidades. Revisión de capítulos de interés especial, realizadas por profesionales que tengan experiencia en el tema y contribuyan a clarificar conceptos.
5. Crónica. Espacio destinado a noticias de interés en el campo de la clínica, neurofisiología, imágenes, Salud Pública o administración. Presentación se-gún instrucciones detalladas más arriba.
6. Cartas al editor, cuyo objetivo es ser una tribuna abierta de la Revista a sus lectores.
7. Enviar resumen en español e inglés.8. Debe consignar fecha de envío del trabajo ya que
será recibida y enviada a dos revisores expertos anónimos, para revisión aprobación y/o rechazo o modificación.
9. Se debe declarar conflictos de intereses de los au-tores.
Presentación de las referencias bibliográficasDeben enumerarse en el texto en forma consecutiva, en el mismo orden en que aparecen citadas por primera vez, y acompañarse la lista total de ellas. En caso de haber más de 5 autores, se colocará la palabra “et al” para incluir los restantes. Cada referencia de revista debe anotarse en el orden siguiente: Apellido paterno del autor con la primera inicial del nombre, título del trabajo, revista en que aparece el artículo según “In-dex Medicus”, año, volumen, página inicial y final del texto. Las referencias de libros se anotarán así: título del libro, ciudad en que fue publicado, editorial, año. Se usarán comas para separar a los autores entre si. Ejemplos: Pérez J, Santos G. Serotonina humana. Rev Med Chile 1967;45:12-14.
Crónica


















