
REVISTA CLÍNICA M · [email protected] Imagen de portada realizada por ... la...
56
REVISTA CLÍNICA de MELILLA Volumen 3, Número 1. Diciembre 2012 ISSN: 2171-9489 AMIC (Asociación Melillense de Investigación Clínica)
Transcript of REVISTA CLÍNICA M · [email protected] Imagen de portada realizada por ... la...

REVISTA CLÍNICA de MELILLA
Volumen 3, Número 1. Diciembre 2012
ISSN: 2171-9489 AMIC (Asociación Melillense de Investigación Clínica)

Ficha técnica pagina 51 y siguientes

REVISTA CLÍNICA de MELILLA
Editores
Enrique Crespillo Montes
Rafael Soler González
Santiago José Villanueva Serrano
Editor Adjunto
Rafael Javier Martín-Vivaldi Jiménez
Secretaría Administrativa, Coordina-ción de Redacción
Inmaculada Reus González
Comité Científico
Armando Fernández Ruiz
Karim Ghazi El-Hamouti
Manuel Jerónimo Requena Pou
Carlos Rodríguez Escalera
Ángel Felipe Morilla Bernal
Comité Editorial
Daniel Carballo Fernández
Daniel Castrillejo Pérez
Alicia García Olert
Pilar Marí Segura
Ana Martín Morales
Mónica Morales Rodríguez
Sergio Rodríguez Montoya
Rosa María García-Vandelwalle García
Consejo Asesor
Francisco Javier Alonso López
Fernando Díaz Otero
Jacobo Díaz Portillo
Andrea Díaz Sánchez
David Ezpeleta Echávarri
Ana Isabel Eusebio del Castillo
Antonio María García Castillo
Javier González Vereda
María Rosario Linares Valera
Ana María Matas Cobos
Carlos Martínez Agudiez
Juan José Torrero Rodríguez
Enma Navarro Guerrero
Sahily Pérez Parra
Leopoldo Rodríguez del Prado
Eva Luque Sánchez
Francisco Sánchez López
Pablo Simon Lorda
Marta Tejada Pérez
Francisco Javier de la Vega Olias
Asesoría Informática
Carlos Moreno Villar
Entidades colaboradoras
Asociación Melillense de Investigación
Neurológica y Cardiológica (AMINCAR)
Instituto Nacional de Gestión Sanitaria
(INGESA)
Publicación anual
© Copyright 2010 AMIC (Asociación
Melillense de Investigación Clínica).
Reservados todos los derechos.
Protección de datos: AMIC (Asociación
Melillense de Investigación Clínica)
declara cumplir los dispuesto por la Ley
Orgánica 15/1999 de 13 de diciembre
de Protección de Datos de Carácter
Personal.
Depósito legal: ML-31-2010
ISSN: 2171-9489
Secretaría de Redacción:
Imagen de portada realizada por
Osvaldo Estévanez Botello
Órgano Oficial de Expresión de la AMIC
(Asociación Melillense de Investigación
Clínica).
Incluida en LATINMEX folio 21928


Volumen 3, Número 1. Diciembre 2012
Editorial Cuidados paliativos: “nuestro reto” . Antonia Vázquez de la Villa.
5
Artículos originales El Equipo de la Asociación Española Contra el Cáncer en 2011: un año de cuidados paliativos en Melilla. A. Castillo Polo, M. Pérez Medina. M. González Cendán.
6-14 Estudio descriptivo y comparativo del suicidio en Melilla y Ceuta. J. M. Fernández-Millán, M. Fernández-Navas.
15-18
Brote de toxiinfección alimentaria por Salmonella entérica en dos centros de menores institucionalizados en Ceuta.
A.I. Rivas Pérez, M. Medina Vinuesa, C. Romero Esteban,
M.D. Barrientos Reyes, J.M. Cantón Gálvez, D. Castrillejo Pérez. 19-25
Revisiones Enfermedad de Alzheimer: neuroanatomía y fisiopatología. F. Molina Rueda. M. J. Molina Rueda. A. Martín-Vivaldi Jimé-nez.
26-31
Efecto de los antirresortivos sobre la calidad ósea y su in-fluencia en la decisión terapéutica. C. Rodríguez-Escalera, M. J. Requena Pou, A. Fernández Ruiz, R. J. Martín-Vivaldi Jiménez, R. Soler González, J. R. Do-mínguez Vicent.
32-38 Casos clínicos Fístula traqueo-esofágica gigante.
A. M. Matas Cobos, R. J. Martín-Vivaldi Jiménez, M. J. Re-quena Pou, E. Navarro Guerrero, C. Martínez Agudiez.
39-42 Medicina en imágenes Infección aguda sobre prótesis de cadera en paciente con enfermedad de Crohn. A. Martín-Vivaldi Jiménez, M. J. Molina Rueda, R. J. Martín-Vivaldi Jiménez.
43-45 Leucoencefalopatía con necrosis palidal bilateral debido a in-toxicación severa por monóxido de carbono. R. Soler González, E. Crespillo Montes, K. Ghazi El Hamouti, C. Martínez Agúdiez.
46-47 Fractura patológica abierta en miembro inferior de un pa-ciente con artritis reumatoide. Solución quirúrgica.
A. Martín-Vivaldi Jiménez, M. J. Molina Rueda, F. Molina Rueda..
48-49 Normas para los autores
50
Sumario
REVISTA CLÍNICA de MELILLA

ASPANIES FEAPS MELILLA C/ Músico Granados, Casa de la Juventud
Tlfno: 952 67 81 51 E-mail: [email protected]
Para la mejora de la atención en consulta de las
PERSONAS CON DISCAPACIDAD INTELECTUAL
Esta demostrado que las personas con discapacidad intelectual
o del desarrollo tienen más problemas que otras personas para ac-
ceder a servicios sanitarios, tanto de atención primaria como es-
pecialistas
Por otro lado, muchos estudios epidemiológicos afirman que las
personas con discapacidad intelectual o del desarrollo tienen más
probabilidades de tener problemas de salud como la obesidad,
problemas cardíacos, respiratorios… que otras personas
Para mejorar la calidad del servicio de los profesionales que
atienden a Personas con Discapacidad Intelectual en el ámbito sa-
nitario, Feaps ha creado la Guía: En Consulta con una Persona
con Discapacidad Intelectual”
Más información: http://www.feaps.org/archivo/centro-
documental/doc_download/17-en-consulta-con-una-persona-con-
discapacidad-intelectual-.html

Cuidados Paliativos: “Nuestro reto”
“Los que sufren no son los cuerpos, son las personas” . (Eric Cassell)
R E V I S T A C L Í N I C A d e M E L I L L A
Editorial
Rev Clin Mel, 2012;3(1): 5 5
Siguiendo las recomendaciones de la OMS, garantizar el acceso universal a cuidados paliativos de calidad es una cues-tión central de salud pública y por tanto es responsabilidad de las distintas administraciones competentes regular y organizar bien estos cuidados y prestarlos con los niveles de accesibilidad y excelencia que son exigibles en nuestras sociedades desarro-lladas. Cuidados, que deben ir orientados no sólo a los pacientes oncológicos, sino a todos los pacientes con patologías crónicas con discapacidad progresiva (sea cual sea su edad) que les abo-que a la muerte y puedan beneficiarse de una atención holística: “control de síntomas, cuestiones relacionadas con la calidad de vida, de buena comunicación, del tratamiento del impacto emo-cional” … No todas las Comunidades Autónomas ofrecen los mismos recursos en la atención a estos pacientes en el final de la vida. Por ello, si bien es cierto que todos moriremos, no lo es que pueda garantizarse una atención homogénea en nuestros últi-mos días, pues ésta puede diferir según el lugar en el que deci-das, ó coincida, que te encuentres. La atención en cuidados paliativos desde la perspectiva de atención integral de la persona, paciente ó familiar en sus as-pectos físicos, psicológicos, sociales y espirituales debe ser ga-rantizada con la correcta actuación interdisciplinar de los profe-sionales de Atención Primaria, Atención Hospitalaria y Unidad de Cuidados paliativos. La adecuada comunicación entre todos es necesaria para asegurar al paciente y a su familia un modelo de organización comunitario que les aporte el máximo beneficio en la asistencia que se les ofrece, incorporando en el mismo el abordaje psicológico en el acompañamiento de la experiencia de sufrimiento.
En nuestro medio, desde 2002, mediante un convenio entre la Asociación Española Contra el Cáncer, INGESA y la Ciudad Autónoma se constituyó la Unidad de Cuidados paliativos que goza de un reconocido prestigio social por la importante labor que a diario realizan, trabajando en ocasiones en situaciones de difícil coordinación institucional. Actualmente, en el marco del Plan Integral de Cuidados Paliativos de INGESA (2011-2014) se están estableciendo los medios para asegurar el acompaña-miento en el final de la vida de todos los pacientes, oncológicos ó no, aunando el esfuerzo y el trabajo de todos los profesiona-les y sumando los recursos disponibles. Nuestro reto es conse-guir una única mirada, la del cuidado paliativo, cuando llega el momento en el que la medicina no puede curar pero sí cuidar y acompañar. Y hacerlo desde el máximo respeto a los deseos, preferencias y expectativas de los pacientes, al ser quienes deben decidir en última instancia cómo esperan ser atendidos. Todos: responsables sanitarios, pacientes, familiares, profe-sionales y voluntarios, que con su callada labor acompañan a diario a los pacientes que necesitan cuidados paliativos y cono-cen la vulnerabilidad de estos enfermos, sabemos que el reto que tenemos por delante es más humano que técnico. Por ello, invitamos a todos los agentes implicados a la formalización de un compromiso institucional en nuestra Ciudad Autónoma que nos permita hacer realidad el ideal que nos trasmitió Cicely Saunders: “Usted importa por lo que usted es. Usted importa hasta el último momento de su vida, y haremos todo lo que esté a nues-tro alcance no sólo para ayudarle a morir en paz, sino también para que mientras viva, lo haga con dignidad”.
Antonia Vázquez de la Villa

El Equipo de la Asociación Española Contra el Cáncer en 2011: Un año de cuidados paliativos en Melilla. The Spanish Association Against Cancer Team in 2011: a year of palliative care in Melilla
A. Castillo Polo1, M. Pérez Medina.2 M. González Cendán3.
1 Médico de la Asociación Española Contra el Cáncer 2 Psicóloga de la Asociación Española Contra el Cáncer 3 Enfermera de la Asociación Española Contra el Cáncer
Autor para correspondencia: Antonio Castillo Polo ([email protected])
RESUMEN La Unidad de Cuidados Paliativos nace gracias a un conve-nio de colaboración firmado entre la Consejería de Sanidad de la Ciudad Autónoma de Melilla, Asociación Española contra el Cáncer (AECC) y el INSALUD, allá por el año 2002, y posterior-mente ratificado en 2005, con el fin de prestar atención a los pacientes oncológicos terminales. Este equipo está formado por una enfermera, una psicóloga, un médico y un equipo de voluntarios. Desde entonces se han atendido a más de 700 en-fermos y sus familias. En el momento actual se está produ-ciendo un gran cambio en la concepción de los cuidados palia-tivos y se está dando un gran impulso con la celebración de reuniones, jornadas y actividades relacionadas con la concien-ciación, tanto del colectivo sanitario como de la población. En este artículo se muestran los datos referentes a los pa-cientes atendidos por nuestro Equipo durante el año 2011. Palabras Clave: cuidados paliativos, AECC, cáncer, volunta-riado. ABSTRACT Palliative care unit was born thanks to a collaboration agreement signed between the Health´s Departament City of Melilla, the AECC (Spanish Association Against Cancer) and IN-SALUD, back in 2002, and ratified in 2005, to pay attention to
terminal cancer patients. This team consists of a nurse, a psychologist, a doctor and a team of volunteers. Since then they have treated more than 700 patients and their families. At present it is producing a major change in the concept of pa-lliative care and is being given a boost with the holding of meetings, conferences and awareness activities, both collecti-ve and population healt. In this paper, we present data on patients seen by our team in 2011. Key words: paliative care, AECC, cáncer, volunteering
PRESENTACIÓN El año 2011 se puede considerar como muy provechoso para el desarrollo de los cuidados paliativos en nuestra ciudad. Por un lado se han estado celebrando periódicamente reunio-nes con la Dirección del Hospital y el Servicio de Medicina In-terna, así como con Atención Primaria para coordinar las ac-tuaciones en materia de Paliativos y culminar con la celebra-ción de las Primeras Jornadas en Cuidados Paliativos de la ciu-dad de Melilla, con la participación de excelentes profesiona-les de dentro y fuera de nuestras fronteras. Además en dichas Jornadas participaron las autoridades
6
Artículo Original
R E V I S T A C L Í N I C A d e M E L I L L A
Artículo admitido para su publicación en noviembre de 2012
Rev Clin Mel, 2012;3(1): 6-14

Rev Clin Mel, 2012;3(1):6-14
sanitarias de INGESA en nuestra materia, concluyendo con la presentación de la estrategia en materia de cuidados paliati-vos y el deseo de su pronta puesta en marcha en las Ciudades Autónomas de Ceuta y Melilla. Aunque este proceso se está cronificando en el tiempo, son muchas las esperanzas que se han depositado en esta es-trategia, ya presentada en años anteriores, continuamos con ilusión todos los días a la espera que se produzca una mejora en la atención y en las prestaciones que tienen que recibir los pacientes en los últimos momentos de su vida. La realización del Primer Curso de Formación en materia de Cuidados Paliativos y atención al paciente terminal ha sido muy gratificante por la participación de los alumnos, el interés y el poder divulgar y sensibilizar a todos nuestros compañeros de la inmensa importancia que tiene la atención a los pacien-tes terminales y su familia. El artículo que a continuación se desarrolla, intenta refle-jar solamente en datos, el trabajo asistencial realizado por un grupo de profesionales dedicados al cuidado de pacientes en fase terminal de su enfermedad y a sus familias. Desgraciada-mente, no podemos plasmar en cifras ni en un papel la satis-facción y el enriquecimiento humano y espiritual que se gene-ra en las relaciones interpersonales que se mantienen durante la atención a los pacientes y familias, ese ejemplo de lucha an-te la adversidad, la capacidad de sufrimiento y resignación, la ausencia de egoísmo, la entrega y el amor de las familias, el instinto de superación, así como esos momentos de debilidad y cansancio, de rabia e ira contenidos, de pena y sufrimiento, de angustia y, en definitiva, todos los sentimientos humanos tan profundos, que nos hacen seguir luchando por nuestro ob-jetivo común y primero conseguir el bienestar de todos aque-llos por los que nuestro trabajo tiene sentido. INTRODUCCIÓN La Unidad de Cuidados Paliativos se formó, gracias al con-venio tripartito entre INSALUD, Consejería de Sanidad y la AECC, que fue firmado el día 2 de octubre de 2002 y que regu-la la estructura, organización y financiación. Con posterioridad se ratifica en el año 2005 ya con la participación del actual IN-GESA. Desde esa fecha la Unidad ha atendido a más de 700 pa-cientes, prestando atención fundamentalmente a nivel domi-ciliario, además de participar de forma activa en el seguimien-to a nivel hospitalario de dichos pacientes. Con una estrecha relación, como no puede ser de otra ma-nera, con el servicio de Oncología y otros servicios de nuestro hospital, queda pendiente el poder estrechar esos lazos con todos los servicios hospitalarios, así como Atención Primaria y Servicios Especiales de Urgencias, para facilitar los mecanis-mos de comunicación y poder dar mayor la calidad a la aten-ción de los pacientes terminales. Con este objetivo en mente, este ha sido un año lleno de iniciativas para llevarlo a efecto. Por un lado la realización de las Primeras Jornadas en Cuidados Paliativos de la Ciudad de Melilla, organizadas por el INGESA, que han contado con la participación de profesionales sanitarios de nuestra ciudad, el equipo de compañeros de la UCP de Ceuta y con algunas pro-fesionales de prestigio a nivel del territorio español, como es el caso entre otros, del presidente de la SECPAL (Sociedad Es-pañola de Cuidados Paliativos). Asimismo se presentó por par-
te de las autoridades de INGESA el Plan Estratégico de Aten-ción en Cuidados Paliativos para Ceuta y Melilla elaborado por INGESA, con el fin de su puesta en marcha definitiva y que ya se comentó en anteriores ocasiones. Dicho Plan convierte al equipo sanitario de atención primaria en el mayor soporte del cuidado del paciente terminal, como no puede ser de otra for-ma, eso sí, con el apoyo de las unidades específicas de Cuida-dos Paliativos dependiendo de los distintos grados de comple-jidad de los pacientes. De la misma forma surge la necesidad de la atención a nivel hospitalario y para ello la creación de unidades específicas para dichas actuaciones. Con la futura creación de un comité decisorio se pretende garantizar y hacer participativa la atención a los pacientes con enfermedades terminales en los últimos momentos de su vi-da. La puesta en marcha de los planes de formación dirigidos por INGESA al personal sanitario han tenido en este año que termina su culminación con la realización del Primer Curso de Formación en Cuidados Paliativos y Atención al paciente ter-minal, impartido por todos los miembros de la unidad. Esta experiencia ha sido muy gratificante para todo el equipo, no solo por la participación, sino por el interés mostrado desde todos los estamentos participantes, sobre todo por la Subdi-rección del Hospital en la persona de la Dra. Antonia Vázquez de la Villa, a quien aprovecho desde aquí para mostrar nues-tro agradecimiento por el interés y la dedicación que a nues-tra labor presta a diario. RECURSOS Y EQUIPO HUMANO UCP DE MELILLA Se encuentra ubicada en la primera planta del Hospital Co-marcal, frente al despacho del servicio de Oncología y junto a la Unidad del Dolor, próxima al hospital de día. Recursos humanos: equipo multidisciplinario El equipo básico de la Unidad está compuesto por: Una enfermera (DUE): María González Cendán. Una psicóloga : Marta Pérez Medina Un médico: Antonio Castillo Polo Voluntariado de la AECC La unidad dispone de un pequeño despacho para reunio-nes del equipo y una consulta para recibir y atender a pacien-tes y/o familiares, tanto por parte médica y de enfermería, co-mo por la psicóloga. OBJETIVOS DE LA UNIDAD DE CUIDADOS PALIATIVOS DE ME-LILLA Los cuidados paliativos intentan dar una respuesta profe-sional, científica y humana a las necesidades de los enfermos en fase avanzada y terminal y las de sus familiares. Los objeti-vos fundamentales son: - Atención al dolor, otros síntomas físicos y a las necesida-des emocionales, sociales y espirituales y aspectos prácticos del cuidado de enfermos y familiares. - Información, comunicación y apoyo emocional, asegu-rando al paciente ser escuchado, participar en las decisiones, obtener respuestas claras y honestas y expresar sus emocio-nes. - Asegurar la continuidad asistencial a lo largo de su evolu-ción, estableciendo mecanismos de coordinación entre todos los niveles y recursos implicados. El objetivo es el bienestar y
R E V I S T A C L Í N I C A d e M E L I L L A
7

la calidad de vida y no la duración de la misma. - Posibilitar la atención en domicilio como lugar idóneo pa-ra desarrollar los Cuidados Paliativos hasta el final si el pacien-te así lo desea. - Apoyar a la familia, si lo precisa, para una adecuada reso-lución del duelo, tratando de evitar el duelo patológico, que se comienza a prevenir con su participación activa en los cui-dados antes de fallecer el paciente. - Comprobar el grado de satisfacción de los pacientes y fa-miliares atendidos. - Aumentar la cobertura del programa, tanto en número de personas atendidas como en el tiempo de la atención (número de visitas, tiempo de las visitas, etc.) de los pacientes en programa. - Promover la identificación precoz de los pacientes tribu-tarios de tratamiento paliativo y su pronta inclusión en progra-ma, colaborando así con los servicios de tratamiento activo, ya sea atención especializada o atención primaria y ofreciendo la atención adaptada a las necesidades del paciente y familia-res. - Reducir el tiempo de hospitalización y evitar la aplicación de medidas (tratamientos y exploraciones) molestas e innece-sarias, así como el encarnizamiento terapéutico. - Ser la Unidad de referencia en nuestra Ciudad, profundi-zando en el estudio y la comprensión de las necesidades de los pacientes y familiares. Promover la investigación con intención de generar evidencia científica de nuestra tarea y favorecer la difusión de los cuidados paliativos en la Comunidad. - Divulgar la necesidad de la atención en cuidados paliati-vos a todos los profesionales relacionados con la sanidad. - Concienciar a las autoridades sanitarias de la necesidad de ampliar la cobertura de cuidados paliativos con la creación de Unidades de cuidados paliativos hospitalarios. - Conseguir la ampliación de servicios a pacientes con en-fermedades terminales no oncológicas. LOS DATOS DEL AÑO 2011 Desde su formación en el año 2002, la Unidad de Cuidados Paliativos de la AECC ha atendido a un total de 702 pacientes, lo que supone una media de aproximadamente 70,2 pacientes al año. Durante el ejercicio de 2011 han sido incluidos en el pro-grama de cuidados paliativos un total de 80 pacientes, de los cuales 39 fueron hombres, por un total de 41 mujeres, la me-dia de edad fue de 68,96 años. La media de inclusión de pa-cientes fue de 5,08 pacientes nuevos/mes, con una distribu-ción en seguimiento por la unidad, 26,25 pacientes/mes.
Los cuidadores cumplen un papel fundamental en la aten-ción a los pacientes, siendo la mayoría los hijos los que asu-men este papel. En este caso la mayor proporción de cuidado-res corresponde a los hijos de los pacientes. La distribución se muestra en la figura 1. La estancia media de pacientes en nuestra unidad fue de 149 días, con diferencias a favor del sexo femenino. Las muje-res permanecen más tiempo que los hombres. La distribución general de pacientes seguidos en nuestra unidad se muestra en la siguiente tabla (tabla 1), en la que se aprecian las diferencias en pacientes incluidos, las bajas y los fallecidos con respecto a ambos sexos. Durante el año 2011 se produjeron un total de 63 bajas, fueron 59 fallecidos y 4 bajas de programa, por pérdida de pista, traslado a otra ciudad, etc. La media de defunciones/bajas ascendió a 5,25 pacientes/mes. Como antes se comentó las defunciones durante 2011 han sido similares a los años anteriores, rondando el 75% de los pacientes incluidos, así como la media mensual, entre 4,5 y 5,5 por cada mes.
Figura 1. Distribución de cuidadores de los pacientes durante 2011 Un parámetro a tener en cuenta es el del porcentaje de fa-llecimientos en domicilio respecto a los producidos en el Hos-pital, siendo para nosotros uno de los objetivos primordiales conseguir el mayor número posible a nivel domiciliario, ya que como el propio nombre de la unidad indica es de atención do-miciliaria. Sin embargo las últimas tendencias se centran más en la calidad y prestación de los servicios que en el lugar de fa-
CONYUGES
34,78%
OTROS
CUIDADORES
10,00%
OTROS
FAMILIARES
1,25%
HERMANOS
11,25%
HIJOS
40,58%
EN PROGRAMA FALLECIDOS
EN DOMICILIO EN HOSPITAL BAJAS TOTAL
HOMBRES 7 13 17 2 39
MUJERES 10 16 13 2 41
TOTAL 17 29 30 4 80
Tabla 1. Distribución de pacientes en UCP 2011
R E V I S T A C L Í N I C A d e M E L I L L A
8 Rev Clin Mel, 2012;3(1):6-14

llecimiento de los pacientes, ya que esto tiene una dependen-cia multifactorial; como el tipo de cuidador, el sexo, la edad, el tipo de tumor, el acceso a los servicios, preferencias de pa-cientes y familias, etc. En la figura 2 se aprecia la comparativa con el resto de los años desde 2006. En el año actual ha sido del 46,96% . Si cal-culamos la media global de la Unidad desde esa fecha se apro-xima al 55%. La procedencia de los pacientes que han sido atendidos durante el año pasado fue muy variada, (figura 3) aunque, ló-gicamente el estrecho contacto con la Unidad de Oncología y el carácter de esta unidad, dependiente de la AECC, hace que sea ésta la que más enfermos nos derive a lo largo del año, con un total de 57 pacientes. Llama la atención, con respecto a años anteriores, el au-mento de las derivaciones realizadas por Medicina Interna en su conjunto (Digestivo, Neumología, Nefrología, Medicina In-
terna, Hematología...), aunque siguen faltando algunas deriva-ciones de pacientes por parte de Atención Primaria y otros servicios, con los que deben mejorarse las comunicaciones, como por ejemplo el servicio de Urología y Ginecología para evitar la falta de atención de algunos pacientes. Por lo que respecta al número de ingreso hospitalario de pacientes incluidos en programa, que suponen una media de 2,71 días de ingreso por paciente/año. De la misma forma, fueron atendidos o, precisaron acudir por descompensación a los distintos servicios de urgencias un total de 44 pacientes lo que significa un 55 % del total . La localización de los tumores se muestra en la tabla 2 y los porcentajes respecto al total en la figura 4. Se aprecia que el cáncer más frecuente fue el de pulmón con 25 casos. Por-centualmente el número de casos de cáncer de pulmón en las mujeres es similar al de mama, que ocupa el primer lugar con un 27,3 %. Si comparamos, respecto a las localizaciones de los tumo-res en los pacientes valorados en años anteriores, observamos como la proporción en cáncer de pulmón que había experi-mentado un descenso en los últimos años, ha vuelto a aumen-tar y suponer más de un 30% de la totalidad. Se ha producido un ligero descenso en tumores ginecológicos desde hace va-rios años, suponiendo al final del año 2011 un número de tu-mores del 5%. En cuanto a colon-recto, cabeza-cuello y mama, no se ha producido diferencias significativas con años anterio-res. ATENCIÓN PSICOLÓGICA Durante los meses de febrero a julio del presente año la psicóloga de la Unidad estuvo de baja laboral, siendo sustitui-da en la atención y seguimiento de pacientes de forma pun-tual por la Psicóloga de la Junta Provincial de la AECC.
51,08
44,98
55,71
69,58
58,17
46,92
0
10
20
30
40
50
60
70
80
2006
2007
2008
2009
2010
2011
NEFROLOGÍA
1,25%NEUROCIRUGÍA
1,25%
HEMATOLOGIA
1,25%
NEUMOLOGÍA
1,25%
RESIDENCIA
2,50%
U. DOLOR
2,50%
PROPIA
2,50%
CIRUGÍA
2,50%M. INTERNA
3,75%
ATENCIÓN PRIMARIA
6,25%
ONCOLOGÍA
70%
Figura 2. Porcentaje de fallecimientos en domicilio de la UCP (2006-2011)
Figura 3. Derivación de pacientes hacia la UCP 2011
R E V I S T A C L Í N I C A d e M E L I L L A
Rev Clin Mel, 2012;3(1):6-14 9

De los 80 pacientes en seguimiento durante el año 2011 por la Unidad de Paliativos, 42 recibieron atención psicológica,
21 fueron hombres y 21 mujeres con una media de edad de 64,64 años, con mínimas diferencias en cuanto a los sexos.
HOMBRES
MUJERES
VIVOS FALLECIDOS VIVAS FALLECIDAS BAJAS TOTAL
MTS. ÓSEAS 1 1
MTS. HEPÁTICAS 1 1
NO FILIADO 1 1
COLANGIOCARCINOMA 1 1
CARCM. PERITONEAL 1 1
VESÍCULA 1 1
TIROIDES 1 1
PULMÓN+AMÍGDALA 1 1
PRÓSTATA 1 1
PÁNCREAS 1 1
PIEL 1 1
RENAL 1 1 2
HÍGADO 1 1 2
GÁSTRICO 1 1 2
SNC 3 3
HEMATOLÓGICOS 3 3
VEJIGA 3 3
GINECOLÓGICOS 3 1 4
CABEZA-CUELLO 2 1 1 1 5
MAMA 4 5 1 10
COLON 7 1 2 10
PULMON 5 11 2 6 1 25
TOTAL 7 29 10 30 4 80
Tabla 2. Localización de los tumores en pacientes en programa UCP 2011
Figura 4. Distribución comparativa de tumores 2006-2011
R E V I S T A C L Í N I C A d e M E L I L L A
10 Rev Clin Mel, 2012;3(1):6-14

Además se prestó atención a 13 pacientes más que no esta-ban incluidos en la Unidad. Por tanto el servicio de psicología atendió a un total de 55 pacientes. La atención psicológica incluye también a las familias de los pacientes. Se atendió a 34 familias, un 80,95% de los 42 pa-cientes atendidos por el equipo. A un 19,04% de pacientes no se les prestó este servicio porque carecen de familia, o éstas viven en Marruecos y no acuden a Melilla, o declinan esta atención. Finalmente, como se comentó al inicio del apartado, se ha prestado atención psicológica, a 13 pacientes no incluidos en seguimiento por la Unidad, de los cuales 2 son valorados y de-rivados al servicio de atención psicológica de la AECC. Estos datos podrían explicarse por varios factores: La cer-canía de las consultas del Hospital de Día y Oncología a la de la Unidad, ser demandas de atención social o de apoyo emocio-nal sin cumplir criterios de admisión. El resumen de los datos de la atención psicológica se muestra en la tabla 3. SEGUIMIENTO DE DUELO Facilitar el duelo de las familias que han sufrido la pérdida de uno de sus miembros y la prevención del duelo patológico es una de las funciones y objetivos de la Unidad. Esta tarea se realiza de forma conjunta por todo el equipo, con un trabajo previo al fallecimiento y durante la agonía del paciente, aun-que tras el éxitus, es la psicóloga la que lleva el peso de la in-tervención. Generalmente se inicia el protocolo de duelo con una lla-mada a los familiares a partir de los diez días de la defunción,
en la que se valora la evolución del proceso y la conveniencia de hacer un seguimiento posterior con el consentimiento de la familia según el protocolo establecido. Con familias en las que se detecta que existe riesgo de duelo patológico, la intervención es más intensiva. Se atendieron un total de 37 familias durante el año 2011, 29 familias nuevas y se continuó el protocolo de duelo con otras 8 familias ya incluidas el año anterior. Durante el año 2011 se han enviado 45 cartas de atención al duelo. Dentro de cada una de ellas se incluía una encuesta de satisfacción a rellenar por los familiares implicados en el cuidado de los pacientes. Se muestran al final de la memoria en él.
VOLUNTARIADO Una de las tareas más valiosas y valoradas en la Unidad, es la presencia del voluntariado formado y coordinado por la Asociación Española Contra el Cáncer. En los últimos años, es-tas personas, con larga experiencia en el acompañamiento de pacientes y familiares, han visto aumentada su presencia gra-cias al esfuerzo de la AECC. El ingreso o estancia en el hospital por el diagnóstico de al-guna enfermedad oncológica, puede ser vivido por el paciente y su familia como un acontecimiento especialmente traumáti-co, debido a la serie de experiencias que de él se derivan. En pacientes en fase terminal, estas vivencias se intensifican. Con el programa de voluntariado hospitalario, la AECC pre-tende, en el nivel de contacto personal con el paciente y la fa-milia, aportar un suplemento de atención, de calor humano, de sostén afectivo que complemente la atención ofrecida por el personal sanitario, así como detectar situaciones o necesi-dades que requieran la intervención de personal especializa-do. Para desarrollar esta labor se ha contado en el año 2011 con 22 voluntarios de hospital que también están formados para dar apoyo a pacientes de cuidados paliativos y sus fami-lias. Durante este año los voluntarios han seguido realizando cursos on–line de formación continua autorizados por la Sede central de la AECC para reciclarse. Estos cursos son de forma-ción de Voluntariado en Domicilio, de Voluntariado de Cuida-dos Paliativos y de Voluntariado con Niños Oncológicos Hospi-talizados. En el año han sido atendidos por el voluntariado un total de 140 pacientes en hospital de día y 87 pacientes onco-lógicos ingresados en planta y sus familias, de los cuales 51
eran enfermos con criterios de terminalidad. Durante todo el año se ha efectuado el seguimiento de la labor de los voluntarios para detectar problemas surgidos y la satisfacción con la tarea realizada. Con entrevistas personales por parte de la coordinadora de voluntariado en el Hospital y con las reuniones semestrales en la sede de la asociación junto a la Coordinadora General de voluntariado. La valoración del personal sanitario del hospital es satisfac-toria. Consideran que la labor de los voluntarios con los enfer-mos y familiares es útil y que se benefician con su presencia para gestiones de acompañamiento dentro y fuera del hospital y con ahorro de tiempo.
ATENCIÓN PSICOLÓGICA UCP 2011 MEDIA MENSUAL TOTAL ANUAL
Nº PACIENTES NUEVOS ATENDIDOS 3,5 42
Nº FAMILIAS NUEVAS ATENDIDAS 3.75 25
FAMILAS NUEVAS ATENDIDAS EN DUELO 2.18 28
Nº VISITAS/DÍA 2.58 ----
DISTANCIA KM. 83,08 997
Nº LLAMADAS EMITIDAS/RECIBIDAS A PACIEN-
TES ----- 136/29
Tabla 3. Datos de atención psicológica en UCP 2011.
R E V I S T A C L Í N I C A d e M E L I L L A
Rev Clin Mel, 2012;3(1):6-14 11

FORMACIÓN Y DOCENCIA Como es propio de una disciplina en auge, la formación profesional es un aspecto crucial de su desarrollo. Conscien-tes de ello, en nuestra Unidad tenemos como prioridad la formación continuada de los profesionales que la integran, así como, la formación en Cuidados Paliativos de nuevos pro-fesionales. Durante al año pasado se realizaron por el personal de esta unidad los siguientes: - Sesiones clínicas junto al Servicio de Oncología y perso-nal de hospital de Día. - Realización del Taller: “Aspectos éticos y jurídicos de la atención sanitaria al final de la vida” Mes de Noviembre 2011. Organizado por la Escuela Andaluza de Salud Pública. - Curso de Formación en Materia de Cuidados Paliativos y atención al final de la vida, organizado por INGESA, destinado a la formación del personal sanitario de Melilla (30 horas lecti-vas). Destinado a personal de INGESA relacionado con la aten-ción al paciente terminal. - I Jornadas de Cuidados Paliativos de Melilla. - A través de las Asociaciones de Cuidados Paliativos (Sociedad Española de Cuidados Paliativos -SECPAL- y Socie-dad Andaluza de Cuidados Paliativos –SACPA-) que pretenden agrupar al número de investigadores clínicos y personal gene-ral que estudian los problemas de los Cuidados Paliativos para sumar sus actividades y ponerlas en relación con la So-ciedades Internacionales. - Reuniones con la Dirección del Hospital y Atención Pri-maria para el estudio del Borrador del Plan Integral de Cuida-
dos Paliativos de INGESA en Ceuta y Melilla. EVALUACIÓN La evaluación y seguimiento es la etapa final que nos per-mite conocer la implantación del Plan de asistencia, a la vez que detectar las posibles deficiencias del mismo. El resumen final de datos se muestra en la tabla 4.
INDICADORES De proceso. 1. Tiempo de ejecución de los procesos: Tiempo de espera de ingreso en Programa desde la solicitud de admisión hasta la primera evaluación-intervención. (Indicador inverso). Prácticamente se le valora en el mismo día de la deriva-ción, por lo que actualmente sería menor de 1 día espera. No se ha modificado con respecto a las actuaciones de años ante-riores. 2. Producción de servicios: (tabla 4) Atenciones a pacien-tes, duelo, etc. (visitas, llamadas telefónicas: realizadas, cartas enviadas). De resultados. De efectividad: * Relación número de éxitus en domicilio/hospital: 46,96% * Consumo de opioides : casi la mitad de pacientes atendi-
R E V I S T A C L Í N I C A d e M E L I L L A
12 Rev Clin Mel, 2012;3(1):6-14

dos (48,75% del total) consumieron a lo largo de alguna etapa de su proceso opioides en cualquiera de sus presentaciones y variedades. * Asistencia a urgencias (Indicador inverso): EL 55 % re-quirió a lo largo de algún momento de su enfermedad aten-ción por alguno de los servicios de urgencias. * Media de días de ingreso Hospitalario (indicador inver-so): 2,78 días por paciente y año. De satisfacción: * Encuesta de satisfacción de usuarios, a pacientes y fami-liares: Se enviaron un total de 45 cuestionarios de satisfacción a las familias de los pacientes incluidos en programa, una vez éstos habían fallecido. Fueron devueltas únicamente 10 en-cuestas, todas ellas con calificación muy positiva. FUTURO DE LA UNIDAD DE CUIDADOS PALIATIVOS Desde esta unidad se pretenden, además de los objetivos comentados al inicio de la memoria, los siguientes para el futuro. - Implantación definitiva, sensibilización y desarrollo en materia de Cuidados Paliativos. Todos los pacientes deben tener un acceso rápido y lo más directo posible, tanto desde el Hospital como desde Atención Primaria. - Creación de un comité decisorio en materia de cuidados paliativos compuesto por todos aquellos profesionales impli-cados en la atención de dichos pacientes. Presentación en Atención Especializada el funcionamiento de nuestra Unidad, así como el protocolo de sedación en la ago-nía, incluido en la Intranet hospitalaria, pero falto de uso en la actualidad. - Establecer lazos de comunicación adecuado con Atención Primaria y servicios de Urgencias (SNU, 061 SUHC), con el fin de dar mejor atención y cobertura a los pacientes y familiares. - Elaboración de folleto informativo con instrucciones sobre el funcionamiento de la Unidad, significado, intención, teléfonos, etc., con el fin de poner en conocimiento de la po-blación la existencia y funciones de nuestra unidad.
- Servir de apoyo en la implantación del Proceso Asisten-cial Integrado Cuidados Paliativos del Plan de Calidad del Siste-ma Sanitario Público de Melilla en aplicación del convenio de colaboración INGESA/AECC/Ciudad Autónoma y al Plan Estra-tégico de atención en materia de Cuidados Paliativos de IN-GESA. - Ampliar la colaboración con otras Unidades en nuestra misma situación, como sería el caso de Ceuta. Posible creación de un Instituto o Sociedad Ceuta/Melilla en materia de Onco-logía y Cuidados Paliativos. - Ampliar los cuidados paliativos a pacientes terminales con patologías no oncológicas. - Charlas de formación en materia de Cuidados Paliativos en los distintos Centros de Salud. Los temas a tratar podrían incluir: “Utilización de la vía subcutánea y vías alternativas en la administración de medicamentos en Cuidados Paliativos”. “Cuidados paliativos al final de la vida: una necesidad para todos” “Usos y mitos de la morfina y sus derivados”. - Creación del Registro de Voluntades anticipadas en el Hospital Comarcal de Melilla.
Queremos agradecer a Eduardo Triguboff (oncólogo del Hospital) y Encarni Delgado Muñoz (voluntaria de la AECC en el Hospital ), así como a todos, los que de una u otra mane-ra colaboran en la atención día a día a los pacientes con cán-cer y especialmente a los que se encuentran en la fase final de su enfermedad.
Tabla 4. Resumen de datos de la Unidad. Año 2011. General y Psicólogo
R E V I S T A C L Í N I C A d e M E L I L L A
Rev Clin Mel, 2012;3(1):6-14 13

BIBLIOGRAFIA 1.Astudillo W, Mendinueta C. La paliación en un sistema integral de salud. En: Cuidados del Enfermo en fase terminal y atención a su fami-lia. Editado por W. Astudillo y col. EUNSA, Barañain, 2002, 4ª Edición, 569-583.
2.Astudillo W, Mendinueta C. Importancia del apoyo psicosocial en la terminalidad. En: Necesidades psicosociales en la terminalidad. Edita-do por W. Astudillo, Eduardo Clavé y E. Urdaneta. Sociedad Vasca de Cuidados Paliativos. San Sebastián, 2001, 19-41.
3.Constantini M, Camoirano E, Madeddu L, Bruzzi P, Verganelli E, Henriquet F. Palliative come care and place of death among cancer patients: a population- based study. Pall. ed,1993, 7:323-31.
4.Fernández R, Pérez Suárez, MC, et. al. Lugar de preferencia para morir ante una hipotética enfermedad incurable. razones influyentes. Med Pal 1998, 5,1;35-43.
5.Cabrera C, Arenas O, Bonilla M, Cortés L, Márquez I, porta R et al. Perfil del cuidador principal del enfermo atendido por equipos de cuidados paliativos: estudio multicéntrico, descriptivo, transversal. Med Paliat 2000; (4): 140-4
6.Bruera E, Sweeney C, Russell N, Willey JS, Palmer JL. Place of death of Houston area residents with cancer over a two-year period. J Pain Symptom Manage 2003;26: 637-43.
7.Gallo WT, Baker MJ, Bradley EH. Factors associated with home ver-sus institutional death among cancer patients in Connecticut. J Am Geriatr Soc 2001;49: 771-7.
8.Lavera M. Crawley. Racial, Cultural, and Ethnic Factors Influencing End-of-Life Care. J Palliat Med. 8:supplement 1, s-58-69.
9.Catalán-Fernandez JG, Pons-Sureda O, Recober-Martinez A, Avella-
Mestre A, Carbonero-Malberti JM, Benito-Oliver E, et al. Dying of cancer. The place of death and family circumstances. Med Care 1991;29: 841-52.
10.Protocolos de la Sociedad Catalano-Balear de Cuidados Paliativos. Pendiente de Publicación 1993.
11.Gómez-Batiste Alentorn, X., Roca Casas, J., Piadevali Casellas, C., Gorchs Font, N., Guinovart Garriga, C. Atención Domiciliaria, Monogra-fias Clínicas en Atención Primaria. Ed. DOYMA, Barcelona, R M López y N Maymo (Editores), 1991 :131-149.
12.Gómez-Sancho, M., el al. Control de Síntomas en el Enfermo de Cáncer Terminal. Ed. ASTA Médica. 1992.
13.Gómez-Sancho, M., Ojeda Martin, M., Dario García-Rodriguez, E., Navarro Marrero, M A. Organización de los Cuidados del enfermo de cáncer terminal en Las Palmas - Norte. Farmacoterapia, Vol IX, Nº 4 : 203-210, 1992.
14.Sanz Ortiz, J. Principios y Práctica de los Cuidados Paliativos (Editorial) Medicina Clínica (Barcelona), 1989-92: 143 - 145.
15.Gómez-Batiste, X., Borras, J, M., Fontanais, M, D., Stjernsword, J., Trias, X. Palliative Care in Catalonia 1990-95. Palliative Medicine 1992; 6: 321 - 327.
16.Cancer Pain Relief and Palliative Care. Technical Report Series 804. Organización Mundial de la Salud. Ginebra, 1990.
17.Twycross, R, G., Lack, S, A. Therapeutics in Terminal Cancer (2 Ed.) Ed. Churchill-Livingstone, Edinburgo, 1990.
18.Saunders, C. Cuidados de la Enfermedad Maligna Terminal. Ed. Salvat, Barcelona, 1988.
R E V I S T A C L Í N I C A d e M E L I L L A
14 Rev Clin Mel, 2012;3(1):6-14

Estudio descriptivo y comparativo del suicidio en Melilla y Ceuta. Descriptive and comparative study of suicide in Melilla and Ceuta.
Juan M. Fernández-Millán 1, Marina Fernández-Navas1
1Miembro del Grupo de Intervención Psicológica en Emergencias, Crisis y Catástrofes del Colegio Oficial de Psicólogos de Melilla. Autor para correspondencia: Juan M. Fernández-Millán ([email protected])
Rev Clin Mel, 2012;3(1):15-18 15
Artículo Original
R E V I S T A C L Í N I C A d e M E L I L L A
Artículo admitido para su publicación en octubre de 2012
RESUMEN El presente trabajo muestra un estudio descriptivo sobre la evolución y la comparación del suicidio en las ciudades de Ceuta y Melilla, utilizando como variables el sexo y el método utilizado, así como las diferencias entre años desde 2007 has-ta 2010. La importancia del estudio viene del hecho de que desde el 2008 el suicidio es en España la primera causa exter-na de muerte, siendo Melilla, en ese mismo año, la 4ª autono-mía en tasa de suicidio. Todo ello apunta la necesidad de estu-diar cuales son los factores de riesgo de suicidio, así como los protectores. Palabras claves: suicidio, método, muerte. ABSTRACT This paper is a descriptive study about the suicide’s evolu-tion and comparison between two cities: Ceuta and Melilla. Variables we work with are: genre, kind of death and differ-ences between years (since 2007 to 2010). In Spain, since 2008 suicide is the first external cause of death and Melilla was, on the same year, fourth region in Spain suicide's rate. These two circumstances convert this study to a relevant work to improve knowledge about risk factors and preventative measures against suicides cases. Key words: suicide, method, death.
INTRODUCCIÓN El suicidio ha sido definido por la Organización Mundial de la Salud (OMS) como “un acto con resultado letal, deliberada-mente iniciado y realizado por el sujeto, sabiendo o esperando su resultado letal y a través del cual pretende obtener los cam-bios deseados” 1
Durkheim es uno de los más famosos estudiosos del suici-dio y, desde una perspectiva sociológica, lo definió en su obra de 1897 como “todo caso de muerte que resulte, directa o indirectamente, de un acto, positivo o negativo, realizado por la víctima misma, sabiendo ella que debía producir este resul-tado” 2. Algunos datos, ofrecidos por la OMS pueden aclararnos la importancia que el suicidio tiene a nivel mundial. Así, según esta Organización3 cada año se suicidarían casi un millón de personas, es decir, que hay una tasa de mortalidad "global" debida al suicidio de 16 por 100 000, o lo que es lo mismo y más llamativo, un suicidio cada 40 segundos. En los últimos 45 años las tasas de suicidio han aumentado en un 60% a nivel mundial. Estos datos sitúan al suicidio entre una de las tres primeras causas de defunción entre las personas de 15 a 44 años en muchos países. Por poner un ejemplo México ha mos-trado un aumento del 275% en los últimos 30 años4. Debido a este incremento de la tasa de suicidio, son nu-merosos los estudios que se han centrado en indagar sobre qué factores podrían estar determinando el comportamiento suicida5,6,7. Pero el estudio de este fenómeno o comportamiento

presenta una serie de problemas añadido a los que general-mente van unido a cualquier estudio científico. El primero y más importante es el hecho de que su trato por algunas reli-giones hace del acontecimiento un tema tabú, que dificulta su acceso cuando no es ocultado o, incluso, disimulado bajo otra nomenclatura. A ello se suma la necesaria protección de datos y el exceso de celo de algunas entidades o profesionales que obstaculizan el acceso a los mismos o a los expedientes cuya consulta es necesaria para la realización de estudios científi-cos. Otro problema es la falta de bases de datos de las que se pueda obtener información sobre variables predictivas (factores de riesgo y protectores) lo que dificulta la realización de estudios correlacionales e inferenciales. Por ello, abarcar el estudio del suicidio de forma global es una tarea complicada, pudiendo quedarse éste en cifras su-perficiales. Debido a esto, son muchos los autores que focali-zan su área de estudio, pudiendo además, concretar los facto-res que están propiciando un aumento o descenso de la tasa de suicidio en determinadas zonas. Ejemplo de ello son Ruiz-Pérez8 que restringe su área de estudio a España, Morant, Criado, García-Piña, García-Guerrero y Domper9, que trabajan con la población de Castilla – La Mancha o Iglesias y Álvarez 10 quienes centran su trabajo a Asturias. En España, y basándonos en datos del INE, el suicidio se presenta como un problema serio de salud pública al situarse desde el 2008 como la primera causa externa de defunción ante el descenso de los fallecidos en accidentes de tráfico. En 2010 se produjeron por esta causa 3.145 muertes (2.456 hom-bres y 689 mujeres). Es importante que estas cifras se traduz-can en lo que realmente son: Dolor, drama personal, proble-mas de superación del duelo y estigmatización. Centrándonos en las ciudades de estudio, en 2008 Melilla se situó como la 4ª autonomía por tasa de suicidio (10,46) mientras que Ceuta lo hacía como la última –puesto 19- (2,58)11. Creemos que los datos aportados en esta introducción son motivo suficiente para plantear la necesidad de realizar estu-dios científicos (descriptivos e inferenciales) que nos permitan conocer más profundamente el fenómeno en nuestra ciudad y, basándonos en este conocimiento, proponer medidas pre-ventivas. El presente trabajo es un estudio descriptivo mediante un código arbitrario de observación12.
MÉTODO Muestra Los datos de esta investigación se han obtenido de las estadísticas del Instituto Nacional de Estadística (INE) en las distintas páginas que ofrece a través de Internet, por tanto, la muestra utilizada es la población de Ceuta y Melilla, así como la totalidad de la población Española en los distintos años estudiados. Procedimiento Para obtener las distintas estadísticas y comparativas se han rescatado datos de distintas bases de datos del INE.
RESULTADOS 1. Número de suicidios totales y por CC.AA y año En el gráfico 1 puede compararse el número de suicidios que se produjeron en cada año (del 2007 al 2010) en Ceuta y en Melilla. Los datos de años anteriores no se han anotado dado que la metodología y clasificación utilizada por el INE era distinta. Sin embargo puede anotarse que en 2006 hubo un suicida varón en Ceuta por saltar desde un lugar alto. En 2005 el INE presenta los datos conjuntos de Ceuta y Melilla (4 varo-nes, 3 ahorcamiento, 1 objeto cortante) y en 2004 no se ofre-cen datos sobre método utilizado.
Tabla 1: Numero de suicidios totales y por CC.AA y año
Los datos traducidos a suicidios por cada 100.000 habitantes y año se presentan en la tabla 2.
Tabla 2: Suicidios por cada 100.000 habitantes y año
Puede observarse que en los dos primeros años (2007 y 2008) la tasa fue muy superior en Melilla, invirtiéndose esta situación en los dos años posteriores (2009 y 2010) sin llegar a presentar las diferencias anteriores. 2. Método utilizado para llevar a cabo el suicidio. En cuanto al método utilizado por el suicida para consu-mar la muerte destaca con mucho el ahorcamiento en todos los años (12 en el total de los 4 años estudiados) seguido de
R E V I S T A C L Í N I C A d e M E L I L L A
16 Rev Clin Mel, 2012;3(1):15-18
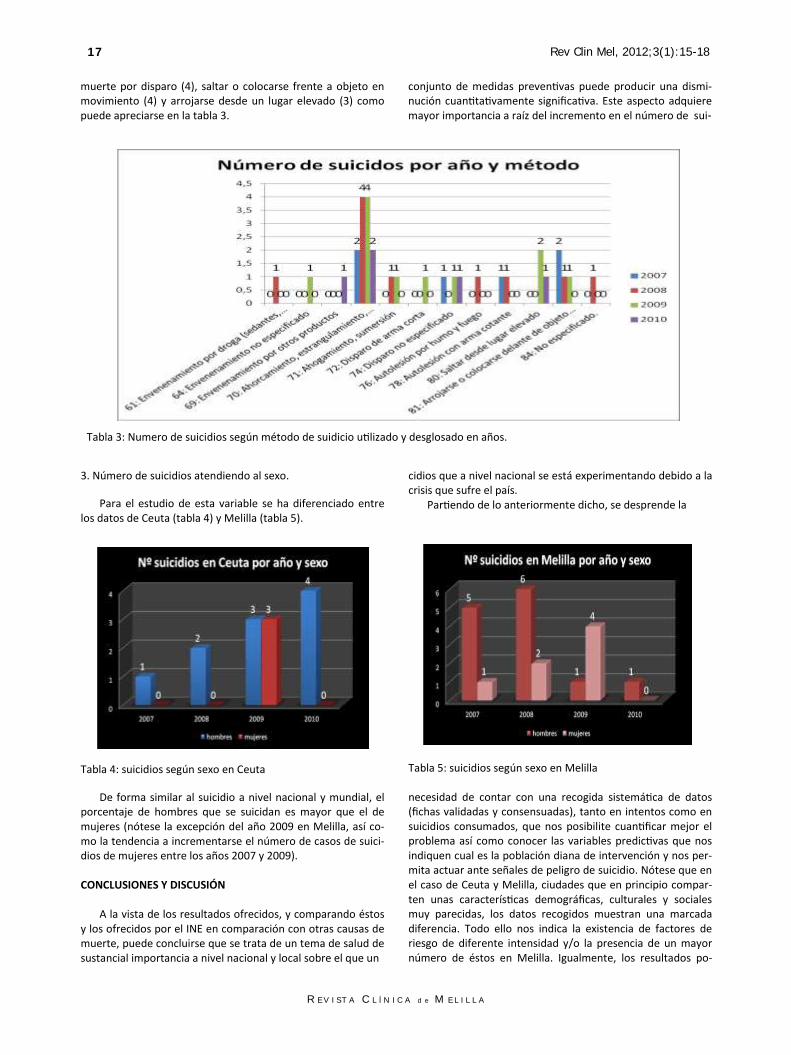
muerte por disparo (4), saltar o colocarse frente a objeto en movimiento (4) y arrojarse desde un lugar elevado (3) como puede apreciarse en la tabla 3.
conjunto de medidas preventivas puede producir una dismi-nución cuantitativamente significativa. Este aspecto adquiere mayor importancia a raíz del incremento en el número de sui-
R E V I S T A C L Í N I C A d e M E L I L L A
3. Número de suicidios atendiendo al sexo.
Para el estudio de esta variable se ha diferenciado entre los datos de Ceuta (tabla 4) y Melilla (tabla 5).
Tabla 4: suicidios según sexo en Ceuta
De forma similar al suicidio a nivel nacional y mundial, el porcentaje de hombres que se suicidan es mayor que el de mujeres (nótese la excepción del año 2009 en Melilla, así co-mo la tendencia a incrementarse el número de casos de suici-dios de mujeres entre los años 2007 y 2009). CONCLUSIONES Y DISCUSIÓN A la vista de los resultados ofrecidos, y comparando éstos y los ofrecidos por el INE en comparación con otras causas de muerte, puede concluirse que se trata de un tema de salud de sustancial importancia a nivel nacional y local sobre el que un
cidios que a nivel nacional se está experimentando debido a la crisis que sufre el país. Partiendo de lo anteriormente dicho, se desprende la
Tabla 5: suicidios según sexo en Melilla necesidad de contar con una recogida sistemática de datos (fichas validadas y consensuadas), tanto en intentos como en suicidios consumados, que nos posibilite cuantificar mejor el problema así como conocer las variables predictivas que nos indiquen cual es la población diana de intervención y nos per-mita actuar ante señales de peligro de suicidio. Nótese que en el caso de Ceuta y Melilla, ciudades que en principio compar-ten unas características demográficas, culturales y sociales muy parecidas, los datos recogidos muestran una marcada diferencia. Todo ello nos indica la existencia de factores de riesgo de diferente intensidad y/o la presencia de un mayor número de éstos en Melilla. Igualmente, los resultados po-
Tabla 3: Numero de suicidios según método de suidicio utilizado y desglosado en años.
Rev Clin Mel, 2012;3(1):15-18 17

R E V I S T A C L Í N I C A d e M E L I L L A
drían ser producidos por una riqueza de factores protectores en Ceuta. En cualquier caso, la identificación de dichos facto-res en las dos Ciudades Autónomas, facilitarían las labores de prevención. No debemos terminar sin indicar que el presente estudio presenta limitaciones que hacen necesario la realización de estudios más profundos y con muestras más amplias (en nú-mero y tiempo).
BIBLIOGRAFÍA
1. World Health Organization (WHO). Working Group on Preventive Practices in Suicide and Attempted Suicide 1986 York, UK. Copenha-gen:WHO Regional Office for Europe;1986.
2. Durkheim E. El suicidio. 6ª ed. Madrid: Akal; 2008.
3. OMS. Prevención del suicidio. SUPRE. (sitio en Internet). Disponible en: http://www.who.int/mental_health/prevention/suicide/suicideprevent/es/index.html. Acceso el 17 de diciembre de 2012.
4. Borges G, Orozco R, Benjet C, Medina-Mora ME. Suicidio y conduc-tas suicidas en México: retrospectiva y situación actual. Salud Pública de México 2010; 52: 292-304.
5. Gabilondo A, Alonso J, Pinto A. Prevalencia y factores de riesgo de las ideas, planes e intentos de suicidio en la población general españo-la: resultados del estudio ESEMeD. Medicina Clínica 2007, 129 (13): 494-500.
6. Tuesca R, Navarro E. Factores de riesgo asociados al suicidio e in-tento de suicidio. Salud Uninorte 2003, 17: 19–28.
7. Pérez Barrero SA. El suicidio, comportamiento y prevención. Santia-go de Cuba: Editorial Oriente; 1996.
8. Ruiz-Pérez I. El suicidio en la España de hoy. Gaceta Sanitaria 2006,20 (Supl 1):25.
9. Morant C, Criado JJ, García-Piña R, García-Guerrero J, Domper J. Mortalidad por suicidio en Castilla-La Mancha (1991-1998). Psiq biol, 2001; 8(4):135-140.
10. Iglesias C, Álvarez, JA. Un estudio del suicidio en Asturias: incre-mento de la frecuencia en las dos últimas décadas. Actas Esp Psiquia-tri. 1999, 27(4): 217-222.
11. Grupo de trabajo de la Guía de Práctica Clínica de Prevención y Tratamiento de la Conducta Suicida. I. Evaluación y Tratamiento. Guía de Práctica Clínica de Prevención y Tratamiento de la Conducta Suici-da. Plan de Calidad para el Sistema Nacional de Salud del Ministerio de Sanidad, Política Social e Igualdad. Axencia de Avaliación de Tecno-loxías Sanitarias de Galicia (avalia-t); 2010. Guías de Práctica Clínica en el SNS: Avalia-t 2010/02
12. Montero I, León GL. A guide for naming research studies in Psy-
chology. Int. J. Clin. Health Psychol. 2007; 7, (3): 847-62
18 Rev Clin Mel, 2012;3(1):15-18

Brote de toxiinfección alimentaria por Salmonella entérica en dos centros de menores institucionaliza-dos en Ceuta
Outbreak foodborne Salmonella enterica in two centers in institutionalized children in Ceuta. Rivas Pérez A.I.1; Medina Vinuesa M.1; Romero Esteban C.1; Barrientos Reyes MªD.1; Cantón Gálvez J.M.1; Castrillejo Pérez D.2
1Consejería de Sanidad y Consumo. Ciudad Autónoma de Ceuta 2Consejería de Sanidad y Consumo. Ciudad Autónoma de Melilla Autor para correspondencia: A. Rivas Pérez ([email protected])
Artículo Original
R E V I S T A C L Í N I C A d e M E L I L L A
Artículo admitido para su publicación en octubre de 2012
RESUMEN Objetivos: Describir un brote de toxiinfección alimentaria en dos centros de menores de Ceuta con catering común, entre el 30/06 y 02/07 de 2011. Determinar el vehículo de transmisión y factores asociados. Proponer medidas correcto-ras. Métodos: Estudio de cohortes retrospectivo en base a encuesta diseñada ad-hoc y toma de muestras clínicas a en-fermos y manipuladores de alimentos. Inspección de condicio-nes técnicas higiénico-sanitarias de las instalaciones de la empresa suministradora del catering, de los alimentos, agua y platos testigos. Resultados: La tasa de ataque global fue del 89%, 88,6 en el centro I y 75% en el centro II. La media de edad fue de 17,59 años con un rango entre 13 y 59 años. El 95% de los enfermos fueron varones y el 5% mujeres. El síntoma más frecuente fue dolor abdominal (85,8%) seguido de diarrea (84,3%). Los menores del centro I presen-taron todos los síntomas en mayor proporción que los del II. La mediana del periodo de incubación fue de 10 horas. En 4 enfermos y dos manipuladores se aisló Salmonella entérica, subespecie entérica I, serotipo Enteritidis 9,12:g,m, fagotipo 1. También se aisló Salmonella en la ensaladilla rusa de la cena del día 30 y diversos gérmenes en el agua, que presentaba una turbidez no ajustada a la legislación. En el análisis univariado de alimentos se encontró una asociación estadísticamente significativa (p 0,0001) entre la
enfermedad y el consumo de ensaladilla rusa de la cena, con un riesgo relativo (RR) de 1,28 y un intervalo de confianza (IC) al 95% entre 1,1-1,48. Discusión: La aparición de casos antes de la cena indica que además de la ensaladilla rusa existen otro u otros alimen-tos contaminados de una ingesta anterior. La tasa de ataque global tan elevada (89%) en compara-ción con otros brotes hace pensar en un inóculo importante, que puede deberse a consumir alimentos contaminados en la comida y en la cena e incluso de una ingesta anterior, además del papel amplificador de los manipuladores enfermos. La mayor afectación en el centro I puede deberse a la mayor carga microbiológica, debida al mayor tiempo de transporte de los alimentos por distancia mayor de la empresa de cate-ring al centro. La tasa de asistidos mayor en los menores del centro I que en los del II, puede deberse a las condiciones termo-higrométricas de los módulos y al número de menores por módulo. Conclusiones: Se confirmó un brote de infección alimenta-ria por Salmonella entérica, subespecie entérica I, serotipo Enteritidis 9,12:g,m, fagotipo 1. Se identificó como vehículo la ensaladilla aunque no se des-cartaron otros. La implicación de varios alimentos sugiere una contaminación cruzada a la que pudieron contribuir las defi-ciencias higiénico-sanitarias en la zona de elaboración y alma-cenamiento de alimentos y las prácticas incorrectas de mani-pulación. La existencia de manipuladores enfermos puede ser factor desencadenante o contribuyente al brote.
Rev Clin Mel, 2012;3(1):19-25 19

Palabras clave: Brote epidémico. Toxiinfección Alimentaria. Salmonella enteritidis. ABSTRACT Objectives: To describe an outbreak of food poisoning in two juvenile facilities with catering Ceuta common, between 30/06 and 02/07, 2011. Determine the vehicle of transmission and associated factors. Propose corrective measures Methods: A retrospective cohort study based on ad-hoc designed questionnaire and clinical sampling and ill food han-dlers. Inspection techniques sanitary conditions of the premis-es of the company supplying the catering, food, water and dishes witnesses. Results: The overall attack rate was 89%, 88.6 at the cen-ter I and 75% in the center II. The mean age was 17.59 years with a range between 13 and 59 years. 95% of the patients were male and 5% female. The most common symptom was abdominal pain (85.8%) followed by diarrhea (84.3%). The lower the center I had all the symptoms in a greater proportion than the II. The median incubation period was 10 hours. In 4 patients and two manipulators was isolated Salmonella enterica enterica subspecies I serotype Enteritidis 9.12: g, m, phage type 1. Salmonella was also isolated in the potato salad for dinner on 30 and various germs in the water, which had not adjusted to a turbidity legislation. In univariate analysis of food found a statistically significant association (p 0.0001) between illness and consumption of
potato salad for dinner, with a relative risk (RR) of 1.28 and a confidence interval (CI) 95% between 1.1 to 1.48. Discussion: The occurrence of cases before dinner indi-cates that in addition there are other potato salad or other foods contaminated by previous intake. The overall attack rate as high (89%) compared with other outbreaks suggests an important inoculum, which may be caused by eating contaminated food at lunch and dinner and even a previous intake, in addition to the role of the amplifier sick manipulators. The greater involvement in the center I may be due to increased microbiological burden, due to longer transport of food by the distance of the center caterer. Assist-ed rate higher in smaller than in the center I of II, may be due to thermo-hygrometric conditions of the modules and the number of children per module. Conclusions: We confirmed an outbreak of foodborne illness caused by Salmonella enterica subspecies I serotype Enteritidis 9.12: g, m, phage type 1. Vehicle was identified as the salad but not others were dis-carded. The involvement of various foods suggests cross-contamination that may have contributed to the sanitary defi-ciencies in the preparation and storage of food and bad han-dling practices. The existence of handlers can trigger ill or contributor to outbreak. Keywords: Epidemic outbreak. Food poisoning. Salmonella enteritidis.
INTRODUCCIÓN La salmonelosis es una enfermedad bacteriana de distri-bución mundial, aunque se identifica con mayor frecuencia en países desarrollados con buenos sistemas de notificación. Se clasifica como una enfermedad de origen alimentario porque los alimentos contaminados, principalmente los de origen animal, constituyen el modo predominante de transmisión. La mayoría de casos que ocurren en personas adultas son asinto-máticos, entre el 60-80% esporádicos, pero también puede aparecer en forma de pequeños brotes en la población y a veces en grandes brotes en hospitales, guarderías, geriátricos y restaurantes2. En Ceuta, el Sistema de Información Microbiológica notifi-có durante el periodo 2005-2010 una media de 46,83 aisla-mientos esporádicos de salmonella spp anuales21. De los bro-tes en la ciudad con aislamiento de salmonella en los últimos 10 años, el más importante ocurrió en 2004 con un total de 184 afectados de 7 banquetes de un mismo establecimiento de restauración colectiva. En Estados Unidos se calcula que el número real de casos de esta enfermedad es de 1-5 millones anualmente y repre-senta más del 50% de todos los brotes de gastroenteritis de causa bacteriana16. En España constituye el 50 % de los brotes de origen alimentario15.
Esta causada por un bacilo Gram negativo del género Salmonella, perteneciente a la familia de las enterobacterias. El género Salmonella está constituido por 2 especies: S. enté-rica y S. bongori. La Salmonella entérica está compuesta por 6 subespecies entre las que se encuentra la entérica. Las subes-pecies se dividen en más de 2.400 serovariedades, definidas en función de diferentes asociaciones de factores antigénicos somáticos O y flagelares H. La serotipificación permite deter-minar la prevalencia de la serovariedad en las distintas zonas geográficas y también resulta útil para el estudio de brotes y para conocer la fuente de infección y el mecanismo de trans-misión. La mayoría de serovariedades aisladas del hombre pertenecen a la subespecie entérica (I) 3. El reservorio pueden ser animales domésticos y salvajes de diverso tipo; los animales productores de carne y las aves de corral son los más implicados y de ellos el pollo y derivados son los más importantes ya que prácticamente todas las sal-monellas conocidas pueden ser patógenas para él1, 2. También puede encontrarse en la leche, verduras, frutas y agua. El ser humano puede ser reservorio, tanto enfermos como portado-res convalecientes, sobre todo casos leves o asintomáticos. El estado de portador crónico es raro en seres humanos, pero común en animales. El período de incubación oscila de 6 a 72 horas, general-
R E V I S T A C L Í N I C A d e M E L I L L A
20 Rev Clin Mel, 2012;3(1):19-25

mente entre 12 y 362. El mecanismo de transmisión es por ingestión de alimen-tos contaminados bien en origen (más frecuente) o durante su manipulación. La transmisión persona-persona es importante en hospitales y colectividades cerradas1, 2. El tamaño del inóculo necesario para producir gastroenteritis es muy varia-ble, según autores va de 10 2-3 a 10 5-8. Influyen favoreciendo la enfermedad, la aclorhidria medicamentosa o quirúrgica, el consumo de marihuana, la edad, la inmunodepresión, la admi-nistración de antibióticos de amplio espectro, etc1, 2. La enfermedad se manifiesta de manera repentina, con cefalea, dolor abdominal cólico, diarrea, náuseas y a veces vómitos. Las heces son acuosas y profusas, pueden presentar moco y trazas de sangre. Casi siempre hay fiebre. La deshidra-tación puede ser grave en lactantes y ancianos. Con frecuencia la anorexia y la diarrea persisten durante días1, 2. La gravedad de la enfermedad guarda relación con el serotipo, el número de microorganismos ingeridos y factores relacionados con el huésped. La excreción del microorganismo en heces suele persistir por días o semanas después de la fase aguda y se prolonga si se administran antibióticos2. En el presente trabajo describimos un brote ocurrido en dos centros de menores dependientes de la Consejería de Juventud, Deporte y Menores que comparten empresa de catering entre el 30 de junio y el 2 de julio de 2011:
*El centro I acoge a 105 Menores Extranjeros No Acompañados (MENA) todos varones, procedentes en su ma-yoría del norte de Marruecos. Consta de un edificio central y varios módulos entre los que se encuentran los dormitorios comunitarios.
*El centro II, en él se encuentran en la fecha del brote 24 Menores, todos varones salvo 2 mujeres. Se trata de un único edificio de varias alturas con salas comunes de dife-rentes usos y habitaciones individuales y compartidas. La alerta la realiza la jefa de Área de Menores telefónica-mente el día 1 de julio al Servicio de Inspección Veterinaria, que lo comunica a los Servicios de Epidemiología y Salud Esco-lar de la Consejería de Sanidad y Consumo. OBJETIVOS Describir el brote. Determinar origen, vehículo de transmi-sión y factores asociados. Proponer medidas correctoras. METODOLOGÍA Estudio epidemiológico que incluyó: estudio descriptivo, estudio de las condiciones técnicas e higiénico-sanitarias de las instalaciones de la empresa suministradora del catering, de los alimentos, agua y platos testigos, estudio microbiológico de las muestras clínicas de enfermos y manipuladores y estu-dio de cohortes retrospectivo. Para la recogida de información se usó la encuesta según protocolo, que incluye datos de filiación del entrevistado, consumo de alimentos, hora de inicio de síntomas, tipo y du-ración y asistencia por servicios de urgencia. Las encuestas a los menores se realizaron por personal del Servicio de Salud Escolar, de Prevención y Promoción de la Salud y de Epidemio-logía, a lo largo de los días 1, 2 y 3 de julio, en total 4 entrevis-tadoras. Los afectados en su gran mayoría no hablaban caste-
llano, por lo que fue necesario contar con la ayuda de intér-pretes entre los menores que sí conocían el idioma. Las encuestas a los adultos que habían consumido el me-nú se realizaron telefónicamente, fueron 9 enfermos. El personal de la empresa de catering que trabajó los días 29 y 30 fue entrevistado en la Consejería de Sanidad y Consu-mo; en total 10 se consideró que habían realizado algún tipo de manipulación y con algunos de ellos también hubo dificul-tades de idioma. Se creó una base de datos con el software SPSS (Stadistical Package for Social Science), Versión 18, donde se recogen todas las variables del estudio. Antes de proceder al análisis estadístico, se hizo una depuración de los datos, con el fin de corregir errores de trascripción. Se procedió a la obtención de los estadísticos descriptivos, se aplicaron pruebas paramétricas para el contraste de hipó-tesis. Para la comparación de proporciones se utilizó el Test de Ji-cuadrado (χ²) de Pearson, como prueba estadística de con-traste de hipótesis. Utilizando el Test exacto de Fisher cuando uno de los valores esperados fuese menor de 5 en la χ². Se enviaron muestras clínicas al Laboratorio de Microbio-logía del Hospital Universitario del INGESA de Ceuta, (coprocultivos de 5 enfermos y coprocultivos seriados y frotis nasal de 10 manipuladores). Se enviaron muestras de los platos testigos de los menús del día 30 a un laboratorio bromatológico acreditado y del agua al Laboratorio de Microbiología del Hospital Universitario del Ingesa de Ceuta y al laboratorio bromatológico. La hipótesis de partida fue que el consumo de uno o más alimentos del día 30 de junio causó el cuadro de gastroenteri-tis. RESULTADOS Se entrevistaron 136 personas, de las que 121 resultaron afectadas. Del total de enfermos, el 76,9% procedían del cen-tro I y el 14,9% del centro II. El resto eran trabajadores de uno u otro centro o familiares. Ver Tabla 1. La tasa de ataque global fue del 89%, 88,6 en el centro I y 75% en el centro II. La media de edad fue de 17,59 años con un rango entre 13 y 59 años. Ver Tabla 2. El 95% de los enfermos fueron varones y el 5% mujeres. Los síntomas clínicos que presentaron se distribuyeron de la siguiente manera. Ver Tabla 3. Los menores del centro I presentaron todos los síntomas en mayor proporción que los del II. La tasa de asistidos en urgencias es del 52,8% mayor en el centro I (54,8) que en el II (38,8). El periodo epidémico fue del 30 de junio al 2 de julio. El primer caso se produjo el 30 de junio a las 18:00 y el último el 2 de julio a las 16:00. El pico de la curva epidémica (Ver Figura1) se produjo entre las 22 h. del 30 de junio y la 1 de la madrugada del 1 de julio. La mediana del periodo de incubación fue de 10 horas.
La duración de la enfermedad fue de 2 hasta 12 días. Los menores del centro I presentaron todos los síntomas en mayor proporción que los del II. La tasa de asistidos en urgencias es del 52,8% mayor en el centro I (54,8) que en el II (38,8).
R E V I S T A C L Í N I C A d e M E L I L L A
Rev Clin Mel, 2012;3(1):19-25 21

Tabla 1: Procedencia de los enfermos
Tabla 2: Distribución por edad de los enfermos
Frecuencia Porcentaje válido Porcentaje acumulado
Menor Centro I 93 76,9 76,9
Menor Centro II 18 14,9 91,7
Trabajador Centro I 2 1,7 93,4
Trabajador Centro II 1 0,8 94,2
Familiar de trabajador del Cen-
tro 4 3,3 97,5
Personal ajeno al centro 1 0,8 98,3
Manipulador de alimentos 2 1,7 100,0
Total 121 100,0
Frecuencia
Porcentaje
Porcentaje
válido
Porcentaje acu-
mulado
Válidos 13 3 2,5 2,6 2,6
14 10 8,3 8,5 11,1
15 20 16,5 17,1 28,2
16 34 28,1 29,1 57,3
17 38 31,4 32,5 89,7
18 2 1,7 1,7 91,5
19 1 0,8 0,9 92,3
20 1 0,8 0,9 93,2
21 1 0,8 0,9 94,0
23 1 0,8 0,9 94,9
35 1 0,8 0,9 95,7
40 2 1,7 1,7 97,4
45 1 0,8 0,9 98,3
51 1 0,8 0,9 99,1
59 1 0,8 0,9 100,0
Total 117 96,7 100,0
Perdidos Sistema 4 3,3
Total 121 100,0
R E V I S T A C L Í N I C A d e M E L I L L A
22 Rev Clin Mel, 2012;3(1):19-25

Síntomas Nº de afectados Porcentaje (%)
Dolor abdominal 103 85,8
Diarrea 102 84,3
Fiebre 100 84,0
Cefalea 89 74,8
Vómitos 87 72,5
Tabla 3: Sintomatología
El periodo epidémico fue del 30 de junio al 2 de julio. El primer caso se produjo el 30 de junio a las 18:00 y el último el 2 de julio a las 16:00. El pico de la curva epidémica ( Ver Figura1) se produjo entre las 22 h. del 30 de junio y la 1 de la madrugada del 1 de julio. La mediana del periodo de incubación fue de 10 horas.
La duración de la enfermedad fue de 2 hasta 12 días. En la inspección del local de catering se observaron defi-ciencias en limpieza y mantenimiento en equipos e instalacio-nes en zona de elaboración de alimentos y en zona de almace-namiento. Se constató que durante los días 30 de junio y 1 de julio hubo una avería en la red de agua potable, usándose agua de un estanque artificial para lavar mesas de trabajo y termos para el transporte de comida. En las 5 muestras de enfermos se encontró flora entero-patógena, aislándose Salmonella spp. que se envió para su tipificación al Centro Nacional de Microbiología. La serovarie-dad en 4 de ellas fue Salmonella entérica, subespecie enterica
I, serotipo Enteritidis 9,12:g,m, fagotipo 1; una muestra llegó contaminada. En 2 de los manipuladores (jefe de cocina y oficial de man-tenimiento) se encontró flora enteropatógena, aislándose Salmonella spp. que se envió para su tipificación al Centro Nacional de Microbiología. La serovariedad fue Salmonella entérica, subespecie enterica I, serotipo Enteritidis 9,12:g,m,
fagotipo 1. El jefe de cocina dio positivo también a Vibrio parahemolí-tico. Se aisló salmonella en la ensaladilla rusa de la cena del día 30 y diversos gérmenes en el agua, que presentaba una turbi-dez no ajustada a la legislación. En el análisis univariado de alimentos se encontró una asociación estadísticamente significativa (p 0,0001)* entre la enfermedad y el consumo de ensaladilla rusa de la cena, con un riesgo relativo (RR) de 1,28 y un intervalo de confianza (IC) al 95% entre 1,1-1,48. Ver Tabla 4.
Curva epidémica
0
2
4
6
8
10
12
14
16
18
20
30/
06 16
-17
h
30/
06 18
-19
h
30/
06 20
-21
h
30/
06 22
-23
h
01/
07 00
-01
h
01/
07 02
-03
h
01/
07 04
-05
h
01/
07 06
-07
h
01/
07 08
-09
h
01/
07 10
-11
h
01/
07 12
-13
h
01/
07 14
-15
h
01/
07 16
-17
h
01/
07 18
-19
h
01/
07 20
-21
h
01/
07 22
-23
h
02/
07 00
-01
h
02/
07 02
-03
h
02/
07 04
-05
h
02/
07 06
-07
h
02/
07 08
-09
h
02/
07 10
-11
h
02/
07 12
-13
h
02/
07 14
-15
h
02/
07 16
-17
h
caso
s
R E V I S T A C L Í N I C A d e M E L I L L A
Figura 1: Curva epidémica (se excluyen los que no pudieron precisar hora de inicio de síntomas)
Rev Clin Mel, 2012;3(1):19-25 23
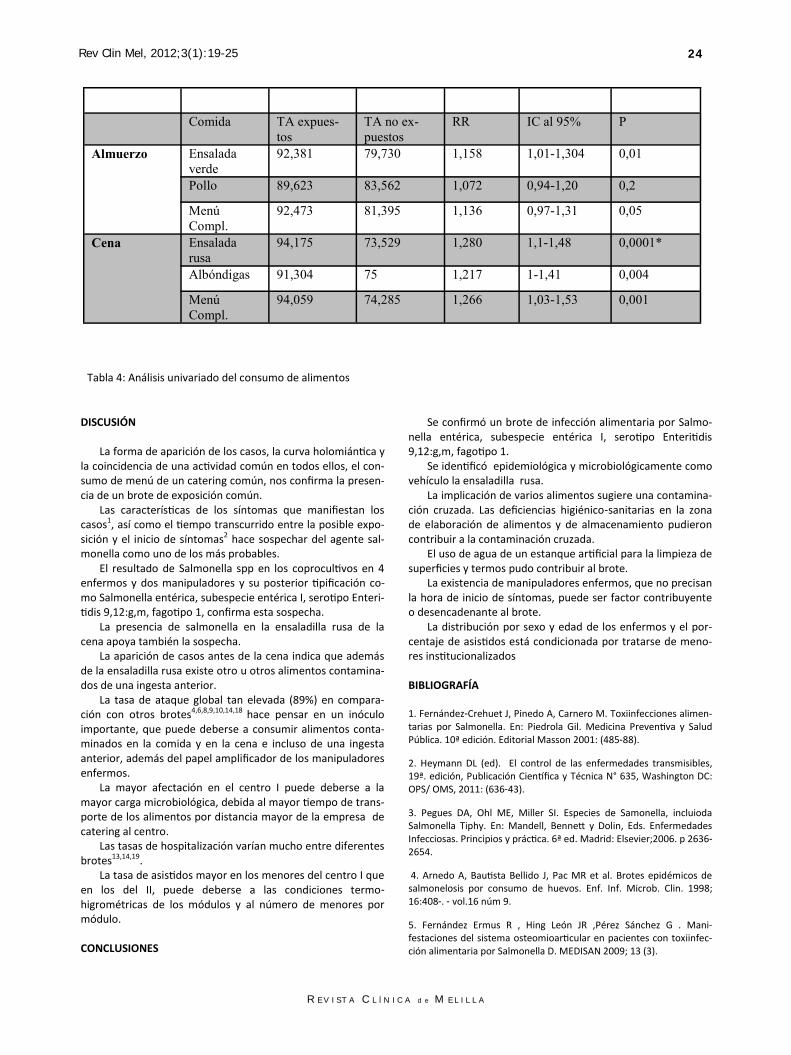
Comida TA expues-
tos
TA no ex-
puestos
RR IC al 95% P
Almuerzo Ensalada
verde
92,381 79,730 1,158 1,01-1,304 0,01
Pollo 89,623 83,562 1,072 0,94-1,20 0,2
Menú
Compl.
92,473 81,395 1,136 0,97-1,31 0,05
Cena Ensalada
rusa
94,175 73,529 1,280 1,1-1,48 0,0001*
Albóndigas 91,304 75 1,217 1-1,41 0,004
Menú
Compl.
94,059 74,285 1,266 1,03-1,53 0,001
Tabla 4: Análisis univariado del consumo de alimentos
R E V I S T A C L Í N I C A d e M E L I L L A
DISCUSIÓN La forma de aparición de los casos, la curva holomiántica y la coincidencia de una actividad común en todos ellos, el con-sumo de menú de un catering común, nos confirma la presen-cia de un brote de exposición común. Las características de los síntomas que manifiestan los casos1, así como el tiempo transcurrido entre la posible expo-sición y el inicio de síntomas2 hace sospechar del agente sal-monella como uno de los más probables. El resultado de Salmonella spp en los coprocultivos en 4 enfermos y dos manipuladores y su posterior tipificación co-mo Salmonella entérica, subespecie entérica I, serotipo Enteri-tidis 9,12:g,m, fagotipo 1, confirma esta sospecha. La presencia de salmonella en la ensaladilla rusa de la cena apoya también la sospecha. La aparición de casos antes de la cena indica que además de la ensaladilla rusa existe otro u otros alimentos contamina-dos de una ingesta anterior. La tasa de ataque global tan elevada (89%) en compara-ción con otros brotes4,6,8,9,10,14,18 hace pensar en un inóculo importante, que puede deberse a consumir alimentos conta-minados en la comida y en la cena e incluso de una ingesta anterior, además del papel amplificador de los manipuladores enfermos. La mayor afectación en el centro I puede deberse a la mayor carga microbiológica, debida al mayor tiempo de trans-porte de los alimentos por distancia mayor de la empresa de catering al centro. Las tasas de hospitalización varían mucho entre diferentes brotes13,14,19. La tasa de asistidos mayor en los menores del centro I que en los del II, puede deberse a las condiciones termo-higrométricas de los módulos y al número de menores por módulo. CONCLUSIONES
Se confirmó un brote de infección alimentaria por Salmo-nella entérica, subespecie entérica I, serotipo Enteritidis 9,12:g,m, fagotipo 1. Se identificó epidemiológica y microbiológicamente como vehículo la ensaladilla rusa. La implicación de varios alimentos sugiere una contamina-ción cruzada. Las deficiencias higiénico-sanitarias en la zona de elaboración de alimentos y de almacenamiento pudieron contribuir a la contaminación cruzada. El uso de agua de un estanque artificial para la limpieza de superficies y termos pudo contribuir al brote. La existencia de manipuladores enfermos, que no precisan la hora de inicio de síntomas, puede ser factor contribuyente o desencadenante al brote. La distribución por sexo y edad de los enfermos y el por-centaje de asistidos está condicionada por tratarse de meno-res institucionalizados BIBLIOGRAFÍA 1. Fernández-Crehuet J, Pinedo A, Carnero M. Toxiinfecciones alimen-tarias por Salmonella. En: Piedrola Gil. Medicina Preventiva y Salud Pública. 10ª edición. Editorial Masson 2001: (485-88).
2. Heymann DL (ed). El control de las enfermedades transmisibles, 19ª. edición, Publicación Científica y Técnica N° 635, Washington DC: OPS/ OMS, 2011: (636-43).
3. Pegues DA, Ohl ME, Miller SI. Especies de Samonella, incluioda Salmonella Tiphy. En: Mandell, Bennett y Dolin, Eds. Enfermedades Infecciosas. Principios y práctica. 6ª ed. Madrid: Elsevier;2006. p 2636-2654.
4. Arnedo A, Bautista Bellido J, Pac MR et al. Brotes epidémicos de salmonelosis por consumo de huevos. Enf. Inf. Microb. Clin. 1998; 16:408-. - vol.16 núm 9.
5. Fernández Ermus R , Hing León JR ,Pérez Sánchez G . Mani-festaciones del sistema osteomioarticular en pacientes con toxiinfec-ción alimentaria por Salmonella D. MEDISAN 2009; 13 (3).
24 Rev Clin Mel, 2012;3(1):19-25

R E V I S T A C L Í N I C A d e M E L I L L A
6. Hernando V, Narot Arranz L, Catalán S, Gómez P, Hidalgo C, Barrasa A, et al. Investigación de una intoxicación alimentaria en un centro penitenciario de alta ocupación. Gaceta Sanitaria vol. 21, nº 6. Nov/Dec 2007.
7. Sección de Vigilancia Epidemiológica. Informe de brotes. Aragón 2008. Dirección General de Salud Pública. Enero 2009.
8. Carbó Malonda RM, Miralles Espí MT, Sanz Bou R, Mañas Gimeno F, Guiral Rodrigo S, Pérez Pérez E. Brote de toxiinfección alimentaria por Salmonella Entérica en un establecimiento de restauración colectiva. Revista Española de Salud Pública Vol 79 nº 1.Ene/Feb 2005.
9. Yáñez Ortega J.L, Carramiñana Martínez I, Bayona Ponte M. Brote por Salmonella Enteritidis en una residencia de ancianos. Revista Española de Salud Pública Vol 75 nº 1. Ene/Feb 2001.
10. Godoy P, Artigues A, Usera MA, González JL, Pablo N, Agustí M. Brote de Toxiinfección alimentaria por consumo de espaghetis a la carbonara causado por Salmonella enteritidis. Enf. Inf. Microb. Clin. 2000; Vol.18 nº16: (257-61).
11. Hernández G, Soler P, Usera M, Tello O, Torres A. Vigilancia epide-miológica de brotes alimentarios relacionados con el consumo de huevos o derivados. España 1998-2011. Boletín Epidemiológico Sema-nal 2003; Vol.11 nº 4: (37-48).
12. Centro Nacional de Epidemiología. Protocolos de las enfermeda-des de declaración obligatoria. 2º edición. Ministerio de Sanidad y Consumo. 2001: (269-76).
13. Servicio Madrileño de Salud. Brotes epidémicos. Comunidad de Madrid. Año 2009.
14. Alcayaga Urbina S, Brantes Martínez J, Cortes Veas J, Olivari Piña F, Loyola Muñoz L, Muñoz Cariaga P. Investigación de un brote por Sal-monella entérica serotipo enteritidis, en una clínica privada, Región Metropolitana. Rev. Chil Salud Pública 2010; Vol 14 (2-3): (139-140).
15. Herrera Monteache A, Conchillo Moreno P. La cadena alimentaria como riesgo para la salud pública. Contaminación y alteración alimen-taria. En: Hernandez Rodríguez M, Sastre Gallego A. Tratado de Nutri-ción. Madrid. Díaz de Santos. 1999: (504-41).
16. Cleary T G, Ashkenazi S. Infecciones por Salmonella. En Berhfman R E, Kliegman R M, Harbin A M editores. Tratado de pediatría, 15 ed. La Habana. Ciencia Médicas, 1997,VII : (984-89).
17. European Centre for Disease Prevention and Control. Annual epidemiological report on communicable diseases in Europe. 2010:( 87-90).
18. Avila Montes G.A, Amador N, España R, Rostrán V, Orellana J, Pinel Vallecillo M, et al. Brote de gastroenteritis por Salmonella enteritidis entre trabajadores de maquila en Naco, Honduras. Rev Med Hond 2004; 72: (85-91).
19. Barahona de Figueroa J, Zúñiga J, Cruz C. Brote de Intoxicación Alimentaria por Salmonella Enteritidis en Hospital Nacional Zacamil. Marzo 2007. Crea cienc. 2007 Vol. 4(6): (18-24).
20. Uribe C, Suárez M.C. Salmonelosis no tifoidea y su transmisión a través de alimentos de origen aviar. Colombia Médica. Vol. 37 (2), 2006: (151-58).
21. Servicio de Vigilancia Epidemiológica de Ceuta. Boletín epidemio-lógico nº 10. Abril 2010. Consejería de Sanidad y Consumo de Ceuta.
Rev Clin Mel, 2012;3(1):19-25 25

Enfermedad De Alzheimer: Neuroanatomía y Fisiopatología. Alzheimer´s Disease: Neuroanatomy and Pathophysiology. F. Molina Rueda1 , M. J. Molina Rueda2, Andrés Martín-Vivaldi Jiménez3. 1Departamento de Fisioterapia, Terapia Ocupacional, Rehabilitación y Medicina Física. Facultad de Ciencias de la Salud. Universi-dad Rey Juan Carlos (Madrid). 2Servicio de Medicina Preventiva y Salud Pública. Hospital Universitario Virgen de las Nieves (Granada). 3 Servicio de Cirugía Ortopédica y Traumatología. Hospital Universitario Virgen de las Nieves (Granada). Autor para correspondencia: Francisco Molina Rueda ([email protected])
Revisión
R E V I S T A C L Í N I C A d e M E L I L L A
Artículo admitido para su publicación en noviembre de 2012 .
RESUMEN La base de una de las patologías más relevantes en el mundo occidental actual, fue publicada por Alzheimer en 1907 en un trabajo de un par de páginas. El objetivo de este trabajo reside en comprender qué estructuras neuroanatómi-cas se afectan en la enfermedad de Alzheimer, y cuáles son sus peculiaridades patológicas. A partir de este punto, se pre-tende mejorar la comprensión de las manifestaciones sinto-máticas y abogar por una intervención interdisciplinar. El lóbulo límbico, las áreas corticales de asociación y el sistema colinérgico basal anterior, representan las principales entidades anatómicas que involucionan en la enfermedad de Alzheimer. Las lesiones neuropatológicas que tienen lugar en la enfermedad de Alzheimer son: atrofia cerebral, formación de placas seniles y ovillos neurofibrilares, muerte neuronal y pérdida de sinapsis. Además, se producen trastornos bioquí-micos importantes, como el descenso de la función colinérgi-ca. Teniendo en cuenta las estructuras afectadas, resulta más fácil entender la clínica de la enfermedad, caracterizada prin-cipalmente por una pérdida de memoria reciente y dificultad para aprender cosas nuevas. Progresivamente se afectan otras funciones cognitivas, y ya en fases avanzadas, pérdida de las capacidades motoras y del juicio crítico. La discapacidad que origina la enfermedad de Alzheimer justifica un abordaje coordinado entre profesionales. Su ac-tuación debe comenzar desde los inicios de la enfermedad, incluyendo terapias respaldadas por la evidencia científica y los cambios fisiopatológicos que acontecen en la enfermedad. Palabras clave: Neurología, Neuroanatomía, Enfermedad de
Alzheimer. ABSTRACT The base of one of the most important diseases, was pub-lished by Alzheimer in 1907 in a work of a couple of pages. The aim of this work lies in understanding what neuroanatom-ical structures are affected in Alzheimer's disease and what are its pathological characteristics. At this point, it is intended to improve understanding of the manifestations symptomatic and advocate an interdisciplinary approach. The limbic lobe, the cortical areas of association and the basal cholinergic system above, represent the major anatomi-cal involution that in Alzheimer's disease. Neuropathological lesions that occur in Alzheimer's disease include cerebral atro-phy, the formation of senile plaques and neurofibrillary tan-gles, death and loss of neuronal synapses. In addition, there are biochemical disorders, such as the decrease in cholinergic function. Taking into account the structures concerned, it is easier to understand the clinical disease, characterized by loss of memory loss and difficulty learning new things. Gradually affect other cognitive functions, and in advanced stages, loss of motor skills and critical thinking. The disability caused by Alzheimer's disease warrants a coordinated approach between professionals. Its action should start from the beginning of the disease, including ther-apies backed by scientific evidence and pathophysiologic changes that occur in the disease. Keywords: Neurology, Neuroanatomy, Alzheimer's Disease.
R E V I S T A C L Í N I C A d e M E L I L L A
26 Rev Clin Mel, 2012;3(1):26-31

INTRODUCCIÓN Repaso histórico El 3 de noviembre de 1906, Alois Alzheimer, un eminente patólogo alemán, presentó en el 37mo. encuentro de los psi-quiatras de Alemania, su comunicación "Acerca de una enfer-medad peculiar del córtex cerebral"1,2; sin lugar a dudas, no imaginó el impacto que en un futuro tendría su descubrimien-to, pues representa la base de una de las patologías más rele-vantes en el mundo occidental actual. En un segundo trabajo publicado en 1911, volvió a incluir la observación ampliada del caso anterior (Augusta D) y un segundo caso (Johann F)3-5. El término, EA, fue acuñado por Emil Kraepelin, una de las mayores personalidades de la psiquiatría biológica, en 19102. Con todo, Kraepelin empleó este término para referirse a la “demencia presenil”, diferenciándola de la “demencia senil provocada”, según él, por enfermedad arterioesclerótica6. Sin embargo, más tarde se demostró la existencia de un mismo patrón clínico e idéntico sustrato anatomopatológico para la mayoría de las demencias; así, a mediados de los setenta, se pudo concluir que la EA era la causa más frecuente de demen-cia, independientemente de la edad del individuo en el mo-mento de su instauración6. Epidemiología descriptiva La enfermedad de Alzheimer constituye uno de los mayo-res problemas sanitarios y económico-sociales en los países desarrollados7. Representa la principal causa de demencia6, de hecho en EE.UU y Europa Occidental, explica el 70-75% de los casos8. En nuestro país, afecta aproximadamente a medio millón de españoles y es previsible el aumento de su prevalen-cia debido al constante envejecimiento de la población8. De hecho se puede afirmar que su prevalencia se incrementa con la edad, así, los trabajos consultados indican tasas cercanas al 1.5% a los 65 años, observándose como estas cifras se dupli-can cada cuatro años hasta alcanzar porcentajes superiores al 30% en los mayores de ochenta años6,9. La Alzheimer’s Disea-se International calcula que entre los años 2000 y 2025 la cifra de enfermos pasará de 18 a 34 millones en todo el mundo8. En cuanto a la incidencia, ésta también aumenta con la edad hasta los 85 años9, alrededor de un 1% por año6. OBJETIVOS Delimitar las estructuras anatómicas cerebrales que se afectan en la EA, así como, describir las lesiones anatomopa-tológicas que caracterizan la enfermedad. Favorecer la comprensión de cómo se manifiesta sintomá-ticamente la enfermedad, de acuerdo con su base neuropato-lógica. En definitiva, aumentar el conocimiento de las bases fisio-patológicas que ocasionan la discapacidad en la EA. NEUROANATOMÍA Y NEUROFISIOLOGÍA EN EL ALZHEIMER. Lóbulo límbico. Se refiere a la sustancia gris en las partes medial y basal del hemisferio que forma un limbo (borde) alrededor del tallo
cerebral. Es un lóbulo de síntesis donde sus partes componen-tes derivan de diferentes lóbulos del cerebro (frontal, parietal, temporal)10. Las estructuras que lo forman se sitúan en la zona medial del telencéfalo y se organizan en anillos céntricos más o me-nos completos alrededor del agujero interventricular, del diencéfalo y del cuerpo calloso [Tabla 1]. La definición de ló-bulo límbico es, en sí misma, un concepto funcional más que
anatómico11. Tabla 1. Componentes del Lóbulo Límbico (División en Anillos) El área entorrinal, ubicada en la circunvolución hipocam-pal, sirve como una compuerta importante entre la corteza cerebral y el hipocampo, representando el eslabón intermedio en la conexión del hipocampo con el resto de la corteza cere-bral. La información de muchas áreas corticales en los lóbulos frontal, temporal, parietal y occipital que trasladan informa-ción visual, auditiva y somatosensorial converge en la corteza entorrinal y el giro parahipocámpico posterior. A su vez, la corteza entorrinal propaga esta información cortical al hipo-campo. De manera recíproca, la eferencia hipocámpica se releva a la corteza entorrinal10. Los estudios que examinan los primeros cambios en el cerebro de los pacientes que presentan una forma incipiente (presintomática) de la EA, indican que las primeras lesiones tienen lugar en las neuronas de la capa II de la corteza entorri-nal del lóbulo temporal y, posteriormente, en el propio hipo-campo12. En las distintas fases de su evolución, la patología de la EA se concentra en las zonas límbicas y paralímbicas, y luego en las áreas de asociación unimodales y multimodales, con una relativa conservación de las cortezas sensorial y motora pri-marias, el cerebelo y muchas partes del tronco encefálico11,12. La distribución de la patología de la EA se asocia con una disminución de la extensión dendrítica y con una pérdida neu-
R E V I S T A C L Í N I C A d e M E L I L L A
Rev Clin Mel, 2012;3(1):26-31 27

ronal y sináptica; esto lleva a una desconexión de múltiples áreas de asociación, lo cual refleja el deterioro cognitivo glo-bal que los pacientes de la EA desarrollan12. Funciones del sistema límbico. El sistema límbico está relacionado con la conducta emo-cional y sexual, y con la motivación. Por otro lado, y de forma más concreta, una de las funciones mejor conocidas del hipo-campo es la retención de información de la memoria a corto plazo y su transferencia hacia la memoria declarativa de largo plazo. Además, desempeña la integración de respuestas ho-meoestáticas, como las relacionadas con la preservación de la especie, la lucha por el alimento y la reacción de alerta o huida10. Sistema colinérgico basal anterior. La pérdida de memoria es la alteración más precoz en la EA y por lo general, el déficit más importante. Su sustrato biológico reside en el hecho, de que los circuitos implicados en el aprendizaje y la memoria, son los que se lesionan antes y de manera más extensa. Uno de ellos, el que conecta el hipocampo con la corteza cerebral, a través del área entorri-nal, se ha explicado anteriormente; el segundo de ellos, con-forma el sistema colinérgico basal anterior. La región cerebral basal anterior está constituida por los núcleos del prosencéfalo basal (colinérgicos): núcleos del rafe, locus coeruleus o nucleo rojo y sustancia negra. Los núcleos colinérgicos representan agrupaciones neuronales que pro-yectan sus axones para dar inervación al hipocampo, a la amígdala y al córtex frontal, parietal, temporal y occipital. Atendiendo a su situación anatómica y a la región que inervan se consideran cuatro núcleos12: Un primer grupo de neuronas colinérgicas, las del septum medial y un segundo grupo, localizado en el brazo vertical de la banda diagonal de broca, envían aferencias al hipocampo y otras estructuras límbicas. Existe un tercer grupo de neuronas que inervan al bulbo olfatorio; mientras que el cuarto y último grupo, está constituido por las neuronas que componen el núcleo basal de Meynert, que se sitúa debajo del cuerpo es-triado y que da inervación al neocórtex y a la amigdala12. La mayor densidad de inervación colinérgica se encuentra normalmente en las regiones límbicas (el hipocampo y la amígdala) y paralímbicas, seguidas por las áreas corticales de asociación. Las áreas primarias corticales son las que reciben la menor densidad de fibras colinérgicas12,13. El sistema cerebral basal anterior mantiene operativa a la corteza cerebral, y desempeña un papel decisivo en los proce-sos de memoria y de atención selectiva12. ETIOPATOGENIA Sigue plenamente vigente la hipótesis de la cascada de amiloide; de hecho futuras estrategias terapéuticas siguen ensayándose en esta diana. Básicamente, las placas seniles o amiloides son productos de un catabolismo patológico de dicha cascada amiloide. Esta ruta metabólica, en condiciones fisiológicas, degrada la proteína precursora del beta-amiloide, a través de beta y gamma-secretasas, hasta obtenerse beta-amiloide.
La hipótesis de la cascada del amiloide postula que un de-sequilibrio en esta ruta metabólica por aumento de su pro-ducción o reducción de su eliminación, provoca aumento de β-amiloide, que se agrega formando placas seniles, que des-truyen tejido nervioso sano; a su vez se producirían otros fenómenos, como: disfunción sináptica por toxicidad directa, alteración de la homeostasis del calcio neuronal, inflamación y estrés oxidativo. Otro fenómeno coadyuvante y adyacente al anteriormen-te expuesto es la fosforilación patológica de la proteína Tau, que provoca los ovillos neurofibrilares. Cómo se vinculan ambos fenómenos es sujeto de contro-versia; existen muchas teorías: la más aceptada en la actuali-dad, es la hipótesis de la enzima Glucógeno Sintetasa Cinasa 3 (GSK-3), de modo que la hiperactividad de la GSK3β (la isofor-ma más abundante) provocaría no sólo aumento de β-amiloide y fosforilación de la proteína Tau, sino muerte neuro-nal y toxicidad glial. ANATOMÍA PATOLÓGICA. Aspectos Macroscópicos. La atrofia cerebral aparece en fases clínicas de la enferme-dad y afecta preferentemente al lóbulo temporal, incluyendo las regiones entorrinal y subicular, así como el hipocampo y la amígdala. Los lóbulos parietales y frontales están menos alte-rados. La atrofia afecta a la corteza cerebral, que aparece más delgada, con circunvoluciones atróficas y surcos más grandes, y a la sustancia blanca. Se caracteriza, además, por una dilata-ción de los ventrículos. El lóbulo occipital y la corteza motora no suelen afectarse18. Aspectos Microscópicos. Placas seniles. También denominadas placas amiloideas. Están constitui-das por agregados insolubles extracelulares del péptido beta amiloide (en adelante RA). Estos depósitos representan una característica invariable de la EA, sin embargo, no es exclusi-va7. La deposición gradual de AR da lugar, al principio, a una placa difusa o preamiloidea que carece de microglía y astro-glía alteradas circundantes, de neuritas distróficas y daño neuronal; luego, el RA se convierte en fibrillas constituyendo las placas neuríticas clásicas (en adelante PN)12. Éstas se carac-terizan por presentar axones y dendritas degenerados, ade-más, contienen cantidades variables de microglía activada, así como astrocitos reactivos en las áreas circundantes, desenca-denándose la subsiguiente reacción inflamatoria y daño neu-ronal (cascada amiloide). Las PN, son consideradas placas maduras asociadas a un estado avanzado de la EA, y que ge-neralmente se localizan, en grandes cantidades, en estructu-ras límbicas y en la neocorteza asociativa14,19. También se observan depósitos amiloideos en las paredes de los vasos sanguíneos cerebrales de todos los pacientes con EA. La gravedad de esta deposición vascular oscila entre las cantidades mínimas de amiloide y los depósitos masivos, que pueden alterar completamente la arquitectura de los vasos
R E V I S T A C L Í N I C A d e M E L I L L A
28 Rev Clin Mel, 2012;3(1):26-31

cerebrales (angiopatía amiloide cerebral)12. Ovillos Neurofibrilares Los ovillos neurofibrilares (en adelante ONF) son algunos de los marcadores más importantes de la EA9. Son depósitos intracelulares de elementos citoesqueléticos en el citoplasma formados por filamentos helicoidales apareados12, cuyo com-ponente básico es una forma hiperfosforilada e insoluble de la forma normal citosólica y muy soluble de la proteína asociada a microtúbulos tau7. El ONF altera el transporte intraneuronal de los componentes celulares al desplazar los elementos intra-citoplásmicos, lo que interrumpe el transporte exodendrítico y conduce, finalmente, a la muerte neuronal 9,12. Parece que los ONF se forman en las primeras etapas del curso de la enfermedad, en las regiones perirrinal y entorri-nal7; desde allí, se extienden por todo el lóbulo temporal y, luego, ascienden hacia el neocórtex12. Los ONF también se pueden ver en el envejecimiento nor-mal, y hasta un 80% de las personas mayores sin patología desarrollarán ONF en las áreas corticales entorrinales antes de los 90 años de edad. Sin embargo, en los pacientes con EA, hay una formación generalizada de PN asociadas con los ONF, incluso en las primeras etapas. El evento que inicia la forma-ción de ONF no está completamente aclarado, no obstante, algunos estudios apuntan a una relación con los depósitos de RA12. Muerte neuronal y pérdida de sinapsis. La muerte programada de las neuronas o apoptosis, es uno de los cambios estructurales finales que acontecen en la cascada neuropatológica de la EA. Un proceso en el que los depósitos amiloideos parece que juegan un papel clave. No obstante, el camino a la apoptosis es complejo y multifacto-rial12. Se ha puesto de manifiesto que el efecto tóxico de los agregados proteínicos característicos de la EA se produce a través de la unión de éstos con receptores celulares específi-cos, denominados ‘receptores basurero’ (scavenger)20. Estos receptores se encargan de interiorizar, para después digerir, las proteínas extracelulares alteradas. La unión de éstas a dichos receptores induce la formación de radicales libres21, que pueden llegar a producir la muerte celular por apoptosis. La unión del AR a las células microgliales provoca también la formación y liberación de radicales libres, de sustancias ci-totróficas (interleucinas y factores de crecimiento), que atraen y activan nuevas células gliales; y de sustancias citotóxicas (óxido nítrico y excititoxinas). De esta manera, al igual que las células de la serie blanca implicadas en los procesos inflama-torios sistémicos, las células gliales activadas amplifican el daño celular local20. Las sustancias neurotóxicas pueden provocar una activa-ción exagerada de las vías permeables a los iones de calcio, lo que produce un influjo excesivo de calcio, que conduce a la muerte neuronal. Generando un aumento de la liberación de Glutamato; su exceso, unido a la falta de regulación de sus receptores perpetúa el proceso ya iniciado, por lo que el resul-tado es un daño neuronal irreversible12. La pérdida de las neuronas, afecta particularmente a las neuronas piramidales glutaminérgicas, predominantes en el
neocortex y el hipocampo. La pérdida de las neuronas tam-bién afecta a estructuras subcorticales, como es el caso de las neuronas colinérgicas de los ganglios basales, que proveen de inervación colinérgica al córtex y al hipocampo22. Por lo tanto, conlleva la pérdida del fenotipo neuroquímico, es decir, un déficit en la inervación colinérgica de la corteza y el hipocam-po23. Con todo, la integridad sináptica, esencial para mantener las funciones intelectuales, queda alterada. La pérdida de sinapsis es significativa en las regiones del cerebro afectadas por la patología de la EA, y es el correlato estructural más significativo de la gravedad de la demencia, como se demues-tra en las biopsias corticales. Los mecanismos patológicos que conducen al daño y a la desaparición de las sinapsis no se comprenden bien12. Trastornos bioquímicos. Déficit colinérgico. La acetilcolina (en adelante Ach) es el neurotransmisor modulador más importante del cerebro y el que más se afecta en la EA. Las enzimas, CAT (Colinoacetiltransferasa) y ACE (Acetilcolinesterasa), se encargan de su síntesis e hidrólisis, respectivamente. La enzima butirilcolinesterasa (BC), que se sintetiza en la glía, participa en la degradación de la Ach en colina y acetato. Dicha enzima abunda en el córtex temporal medial, en la amígdala y en el hipocampo. La CAT se encuen-tra sólo en las neuronas colinérgicas presinápticas, mientras que la ACE se encuentra en las vías colinérgicas presinápticas y postsinápticas20. En la EA se produce una gran pérdida de neuronas colinér-gicas, especialmente las que están situadas en el núcleo basal de Meynert12. Los niveles de la enzima de síntesis CAT en la corteza y el hipocampo se mantienen en las etapas leve a moderada de la EA24,25; no descienden a niveles muy bajos hasta que la enfermedad alcanza una fase de deterioro clínico más grave26,27, donde los niveles de ACE y CAT caen un 85-90% con respecto al valor normal. Mientras, la enzima butirilcoli-nesterasa está aumentada, parece que de forma compensato-ria20. Además, existe una reducción significativa de la densidad de receptores colinérgicos N (nicotínicos)20, a diferencia de la densidad global de los receptores M (muscarínicos) que per-manece relativamente estable, aunque en algunos casos se ha encontrado una reducción del 20 al 30%12. Esta pérdida pare-ce producirse a expensas de los subtipos M1 y M2, mientras que los subtipos M3 y M4 permanecen estables12. Tal depleción, afecta a las áreas con mayor inervación colinérgica (áreas límbicas, paralímbicas y luego neocórtex de asociación). Las zonas primarias motoras y somatosensoriales y visuales y la circunvolución del cíngulo son las que presentan una menor inervación colinérgica12. El déficit colinérgico de los enfermos de Alzheimer llevó a formular la hipótesis colinérgica, que ha servido de modelo para el desarrollo de los fármacos sintomáticos actuales para la EA. El uso de los inhibidores de la acetilcolinesterasa (ACE) puede prolongar la vida media de la ACh en la sinapsis colinér-gica20,22,23. Otros trastornos bioquímicos resumidos en la tabla siguiente:
R E V I S T A C L Í N I C A d e M E L I L L A
Rev Clin Mel, 2012;3(1):26-31 29

Tabla 2. Otros trastornos bioquímicos CONCLUSIONES La discapacidad ocasionada por la EA dispone de un sus-trato anatómico. Se trata de los circuitos implicados en el aprendizaje y la memoria. Por un lado, la relación establecida entre el Hipocampo y la corteza cerebral a través del área entorrinal; y por otro, las estructuras que conforman el siste-ma colinérgico basal anterior, quiénes mantienen operativo al córtex cerebral y juegan un papel decisivo en los procesos de memoria y atención selectiva. Recientemente, grupos de investigación del Reino Unido y de Francia han descubierto tres genes relacionados con la EA (CLU, CR1 y PICALM). Este descubrimiento podría reducir en un 20% los nuevos casos de la enfermedad. Este descubri-miento muestra la revolución médica que existe en torno a esta enfermedad. Los fisioterapeutas no deben ser ajenos a la investigación y al avance biomédico, de hecho, los nuevos planes de estudio nos ofrecen la posibilidad de avanzar en nuestra formación hasta el máximo nivel, lo que permitirá la inclusión y creación de grupos de investigación de primer orden. Por tanto, una exposición detallada y al mismo tiempo, simplificada, de la patología, es fundamental para crear iniciativas e interrogan-tes, además de fortificar el conocimiento de los profesionales. La misión de este trabajo consiste en que la enfermedad de Alzheimer no pase desapercibida a los fisioterapeutas en los diversos centros sanitarios, principalmente, centros de día y residencias de mayores. El individuo con Alzheimer necesita la fisioterapia, no sólo en su tratamiento, también en la pugna por mejorar su calidad de vida a través de la investigación. El autor declara no tener conflictos de interés. BIBLIOGRAFÍA 1. LLibre J. La enfermedad de Alzheimer en los umbrales del siglo XXI. Revista Cubana de Investigaciones Biomédicas 1998; 17(3): 189.
2. Fuentes P. Enfermedad de Alzheimer: una nota histórica. Revista Chilena de Neuro-psiquiatría 2003; 41(2): 9-12.
3. Zarranz J. Del empirismo a la neurociencia en la enfermedad de Alzheimer. Revista de Neurología 2004; 39 (6): 576-582.
4. Graeber MB, Köser S, Egensperger R, Banati RB, Müller U, Bise K, et al. Rediscovery of the case described by Alois Alzheimer in 1911: his-torical, histological and molecular genetic analysis. Neurogenetics 2004; 1(1): 73-80.
5. Möller HJ, Graeber MB. The case described by Alois Alzheimer in 1911. European archives of Psychiatry and clinical neuroscience 2004; 248(3): 111-122.
6. González VM, Martín C, Martín M, González MJ, García F, Riu S. La enfermedad de Alzheimer. SEMERGEN 2004; 30(1): 18-33.
7. Rodrigo J, Martínez A, Fernández AP, Serrano J, Bentura ML, Mo-reno E, et al. Características neuropatológicas y moleculares de la enfermedad de Alzheimer. Revista española de Geriatría y Gerontolo-gía 2007; 42(2): 103-10.
8. Codina A, Boada M. Genética y enfermedad de Alzheimer. Medicina Clínica 2003; 120(20): 786-92.
9. Villar A, Molinuevo JL, Gómez-Isla T. Enfermedad de Alzheimer. JANO 2004; 67(1537): 1318-1320.
10. Kiernan J. Sistema límbico: hipocampo y amígdala. En: Kiernan J. El sistema nervioso humano: un punto de vista anatómico. México D.F: McGraw Hill Interamericana, 2006. p. 254-275.
11. Ojeda JL, Icardo JM. Anatomía macroscópica del telencéfalo. Es-tructura general. Configuración externa de la corteza cerebral: surcos, lóbulos y circunvoluciones. En: Ojeda JL, Icardo JM. Neuroanatomía humana: aspectos funcionales y clínicos. Madrid: Elsevier España, 2004. p. 45-60.
12. López OL, DeKosky ST. Neuropatología de la enfermedad de Alz-heimer y del deterioro cognitivo leve. Revista Neurología 2003; 37: 155-63.
13. De Toledo M. Inflamación y enfermedad de Alzheimer. Revista Neurología 2006; 42(7): 433-438.
14. Coria F. Avances en la patología molecular de la enfermedad de Alzheimer. Revista Neurología 2006; 42(5): 306-309.
15. Xudong MP, Cuajungco CS, Atwood MA, Hartshorn T, Graeme H, Karen S, et al. Cu (II) potentiation of Alzheimer AR neurotoxicity. The Journal of Biological Chemistry 1999; 274(82): 37111-37116.
16. Jiménez FJ, Alonso H, Ayuso T, Jabbour T. Estrés oxidativo y enfer-medad de Alzheimer. Revista Neurología 2006; 42(7): 419-427.
17. George JA. La función del estrés oxidativo en la patogénesis de laenfermedad de Alzheimer. Revista chilena de Neuro-Psiquiatría 2003; 41(2): 47-53.
18. Domenico KU, Susan JQ, Virginia MYL. Increased lipid peroxidation precedes amyloid plaque formation in an animal model of Alzheimer amyloidosis. The Journal of Neuroscience 2001; 21(12): 4183-4187.
19. Perry EK, Tomlinson BE, Blessed G, Bergmann K, Gibson PH, Perry RH. Correlation of cholinergic abnormalities with senile plaques and mental test scores in senile dementia. British Medical Journal 1978; 2: 1457-9.
20. Hoenicka J. Genes de la enfermedad de Alzheimer. Revista Neuro-logía 2006; 42(5): 302-305.
R E V I S T A C L Í N I C A d e M E L I L L A
Anatomía Núcleos dorsales del Rafe
Núcleo Rojo
Anatomía patológica
Depósitos de ONF y pérdida neuronal
Depósitos de ONF. Importante pérdida neuronal (40-80%)
Trastorno Bioquímico
Descenso de los niveles de serotoni-na
Déficit de noradre-nalina
Sintomatología asociada
Se asocia con la sin-tomatología psiquiá-trica de la EA: ansie-dad, depresión, psi-cosis, trastornos del sueño…
Suele asociarse con la depresión
30 Rev Clin Mel, 2012;3(1):26-31

21. Alberca R, López-Pousa S. Enfermedad de Alzheimer y otras de-mencias. Madrid: Panamericana. p. 147,189-211.
22. Jiménez E. Tratamiento de las demencias con fármacos anticoli-nesterásicos. En: Lozano M, Ramos JA. Utilización de los psicofárma-cos en psiquiatría. Barcelona: Masson. p. 289-293.
23. Francis PT. Neuroanatomy / pathology and the interplay of neuro-transmitters in moderate to severe Alzheimer disease. Neurology 2005; 65(6): 65-69.
24. Manzano S, De la Morena MA, Barquero MS. Neurotransmisores en la enfermedad de Alzheimer. Revista neurología 2006; 42(6): 350-
353.
25. Davis KL, Mohs RC, Marin D, et al. Cholinergic markers in elderly patients with early signs of Alzheimer’s disease. JAMA 1999; 281: 14334.
26. Tiraboschi P, Hansen LA, Alford M, Masliah E, Thal LJ, Corey-Bloom J. The decline in synapses and cholinergic activity is asynchro-nous in Alzheimer’s disease. Neurology 2000; 55: 127883.
27. Davis K, Mohs R, Marin D, Purohit D, Perl D, Lantz M, et al. Cholin-ergic markers in elderly patients with early sings of Alzheimer disease. JAMA 1999; 281(15): 1401-1407.
R E V I S T A C L Í N I C A d e M E L I L L A
Rev Clin Mel, 2012;3(1):26-31 31

Efectos de los antirresortivos sobre la calidad ósea y su influencia en la decisión terapéutica. Effects Of The Antirresortive Medicaments On The Bony Quality And Their Influence In The Therapeutic Opinion Rodríguez-Escalera C1, Requena Pou MJ2, Fernández Ruiz A2, Martín-Vivaldi Jiménez RJ3, Soler González R4, Domínguez Vicent JR2. 1Unidad de Reumatología del Hospital Comarcal de Melilla. 2Unidad de Medicina Interna, del Hospital Comarcal de Melilla. 3Unidad de Digestivo, del Hospital Comarcal de Melilla. 4 Unidad de Neurología, Hospital Comarcal de Melilla. Autor para correspondencia: Carlos Rodríguez-Escalera ([email protected])
Revisión
R E V I S T A C L Í N I C A d e M E L I L L A
Artículo admitido para su publicación en julio de 2012
RESUMEN La osteoporosis es una enfermedad esquelética caracteri-zada por una resistencia ósea disminuida que predispone al aumento del riesgo de fractura. Todas las fracturas osteopo-róticas se asocian a una morbilidad significativa, pero las fracturas vertebrales y de cadera se asocian a una mayor mortalidad. El objetivo primario del tratamiento farmacológi-co de la osteoporosis es reducir el riesgo de fractura. Los bifosfonatos son actualmente la clase de fármaco utilizados más extensamente en el tratamiento de osteoporosis. Los bifosfonatos son los únicos fármacos aprobados en el trata-miento de la osteoporosis posmenopáusica que disminuyen el riesgo de fractura de cadera. Existen claramente diferen-cias importantes entre los bifosfonatos en su potencia anti-rresortiva. La selección del fármaco debe individualizarse tomando en consideración el riesgo de fractura en cada pa-ciente, la eficacia antifractura demostrada en estudios con-trolados, la densidad mineral ósea en columna vertebral y en tercio proximal del fémur, la edad, los efectos secundarios de cada fármaco, sus contraindicaciones y el cumplimiento pre-visto. PALABRAS CLAVE: Bifosfonatos, Antiresortivos, Osteopo-rosis, Fractura, Densidad Mineral Ósea. ABSTRACT Osteoporosis is a skeletal disease which is characterized
by a bone diminished resistance that predisposes to the in-crease of the risk of fracture. All the osteoporotic fractures are associated to a significant morbidity, but the vertebral and hip fractures are associated to a major mortality. The primary aim of the pharmacological treatment of the osteo-porosis is to reduce the risk of fracture. The bisphosphonates are nowadays the class of medicine used more extensively in the treatment of osteoporosis. The bisphosphonates are the only medicine approved in the treatment of the postmeno-pausal osteoporosis that diminishes the risk of hip fracture. Important differences between the bisphosphonates exist clearly in its antiabsorption power. The medicine selection must be individualized taking in consideration the risk of frac-ture in each patient, the anti-fracture efficiency demonstrat-ed in controlled researches, the bone mineral density in the vertebral column and in third proximal of the femur, the age, the after-effects of each medicine, its contraindications and the foreseen fulfillment. KEYWORDS: Bisphosphonates, antiabsorption, osteopo-rosis, fracture, bone mineral density. DEFINICIÓN
La osteoporosis, según se ha definido en la conferencia de consenso del National Institute of Health (NIH), es una enfermedad esquelética caracterizada por una resistencia ósea disminuida que predispone al aumento del riesgo de fractura. La resistencia ósea refleja la integración de la densi-
32 Rev Clin Mel, 2012;3(1):32-38

dad y la calidad óseas. A su vez, la densidad ósea está deter-minada por el valor máximo de masa ósea y la magnitud (cantidad y velocidad) de su pérdida, mientras que la calidad ósea depende de la arquitectura, el recambio óseo, la acumu-lación de microlesiones y la mineralización1. La calidad ósea abarca un número de propiedades tisulares óseas que domina la resistencia mecánica, tales como la geometría ósea, las propiedades corticales, la microarquitectura trabecular, la mineralización de la matriz ósea, la calidad del colágeno y de los cristales de apatita, y la presencia de microfracturas. En la práctica clínica, sólo podemos medir la densidad mineral ósea (DMO). Los valores de la DMO están estrechamente asociados con el riesgo de fractura osteoporótica. Los estudios prospec-tivos indican que el riesgo de fractura osteoporótica aumenta continuamente conforme se reduce la DMO, con un aumento del riesgo de fractura de 1.5 a 3 veces por cada desviación típica de disminución de la DMO2. Sin embargo, sólo un pe-queño porcentaje del efecto antifractura de los inhibidores de la resorción es atribuible a la ganancia de la DMO (16-28% con alendronato y risedronato). FRACTURA ÓSEA La osteoporosis, por sí misma, es asintomática si no ha habido fractura. Las fracturas óseas son la consecuencia clíni-ca de la osteoporosis y pueden producirse en cualquier locali-zación, aunque las más relevantes son las de fémur proximal, el antebrazo distal y la columna vertebral. Todas las fracturas osteoporóticas se asocian a una morbilidad significativa, pero las fracturas vertebrales y de cadera se asocian a una mayor mortalidad. Aproximadamente, el 40% de las mujeres caucási-cas tendrán al menos una fractura osteoporótica después de los 50 años3-8. INCIDENCIA-COSTE Los datos ofrecidos por el Instituto de Información Sanita-ria perteneciente al Ministerio de Sanidad y Política Social, en su informe sobre la atención a la fractura de cadera en los hospitales del SNS sobre el año 2008, nos muestran: La fractura de cadera, en los pacientes ancianos, es la causa más frecuente de ingreso en el hospital en los servicios de traumatología y ortopedia. El número de hospitalizaciones por fractura de cadera que se produjeron en nuestro país fue de 47.308. La estancia media fue de 13.34 días. La tasa de mortalidad hospitalaria asociada a una fractura de cadera fue de 5.5%. El coste global de los casos de hospitalización en el Siste-ma Nacional de Salud como consecuencia de una fractura de cadera fue de 395.7 millones de euros. Los costes medios por paciente fueron de 8365.25 euros. La Organización Mundial de la Salud, en un informe reali-zado conjuntamente con la Fundación Internacional de Osteo-porosis, afirma que “se espera que el número de fracturas de cadera debido a la osteoporosis se triplique en los próximos 50 años, pasando de 1.7 millones en 1990 hasta 6.3 millones en 2050”9. TRATAMIENTO FARMACOLÓGICO
El objetivo primario del tratamiento farmacológico de la osteoporosis es reducir el riesgo de fractura10, no aumentar la DMO. En el momento actual, existen tres categorías de fárma-cos antiosteoporóticos: anticatabólicos, que actúan frenando la resorción ósea; anabólicos, cuya acción principal se produce estimulando la osteoformación, y de acción mixta, con propie-dades anticatabólicas y anabólicas.
Tabla 1. Fármacos antirresortivos Nosotros centraremos nuestra exposición en los bifosfo-natos.
BIFOSFONATOS
DEFINICIÓN Los bifosfonatos son actualmente la clase de fármaco utilizados más extensamente en el tratamiento de osteoporo-sis. Los bifosfonatos pertenecen a una clase de fármacos con una estructura central relativamente simple formada por dos moléculas de fosfato unidas por un átomo de carbono central. La terminal fosfato se une ávidamente a la hidroxiapatita cál-cica ósea, por lo que ancla los bifosfonatos a la superficie ósea donde pueden incorporarse durante el remodelado. La cade-na lateral R1 determina la afinidad de unión de la sustancia. La cadena lateral R2 determina la potencia antirresortiva del pre-parado. Desde un punto de vista funcional, los bifosfonatos
Anticatabólicos o antiresortivos
Estrógenos
Bifosfonatos Etidronatoa
Alendronato
Risedronato
Ibandronato
Zoledronato
Modulado-res Selecti-vos de los receptores estrogénicos (SERM)
Raloxifeno Bazedoxifeno
Calcitoninas
Inhibidores del RANKL
Denosumab
Anabólicos u osteoformadores
Análogos de la paratohormona (PTH)
PTH 1-34 (teriparatida)
PTH 1-84a
Acción mixta
Ranelato de estroncioa aFármacos no aprobados por la FDA
R E V I S T A C L Í N I C A d e M E L I L L A
Rev Clin Mel, 2012;3(1):32-38 33

pueden dividirse en los que contienen un átomo de nitrógeno en su cadena lateral y los que no. Los primeros son más poten-tes y dan cuenta virtualmente de todos los bifosfonatos utili-zados clínicamente en la actualidad. MECANISMO DE ACCIÓN Los bifosfonatos reducen la resorción ósea mediada por los osteoclastos aumentando la muerte celular programada (disminuye su número) e inhibiendo las enzimas en la vía bio-sintética del colesterol (reduciendo su función). En conjunto, esta clase de fármacos actúa disminuyendo la velocidad del recambio óseo. Puesto que las unidades de remodelado per-sisten durante un periodo de tiempo más prolongado, estos fármacos aumentan el grado de mineralización. Este parece ser el mecanismo más probable de los aumentos progresivos de la DMO observados en todos los ensayos clínicos efectua-dos con esta clase de fármacos. Los bifosfonatos que no contienen nitrógeno inhiben la resorción ósea produciendo análogos tóxicos del ATP que dan lugar a la muerte prematura de los osteoclastos. Los que con-tienen nitrógeno inhiben la farnesilpirofosfatasa, lo que origi-na una disminución de la actividad resortiva de los osteoclas-tos y una apoptosis acelerada (muerte celular programada). La potencia antirresortiva está relacionada directamente con la potencia en la inhibición de esta enzima. FARMACOLOGIA Los bifosfonatos administrados por vía oral, debido a su tendencia a unirse a todo residuo cargado positivamente, apenas se absorben en condiciones ideales (entre un 1-5% de la dosis oral). Existe evidencia cada vez más numerosa de que su depósito se localiza en las superficies óseas tanto activas como inactivas11. La presencia de bifosfonato sobre otras su-perficies óseas proporciona un reservorio para el fármaco que, durante los siguientes meses y años, puede envenenar gene-raciones futuras de osteoclastos que intentan resorber estas superficies. La única vía de eliminación de los bifosfonatos es la excreción renal. Aproximadamente el 70% del bifosfonato absorbido es eliminado por el riñón, y el 30% restante es in-corporado al hueso. Este porcentaje se incrementa en condi-ciones de recambio óseo alto, con una menor disminución de la excreción renal12. EFICACIA ANTIFRACTURA La capacidad de los bifosfonatos para prevenir fracturas en mujeres osteoporóticas está actualmente bien demostra-da, particularmente para el alendronato y el risedronato.
Etidronato A dosis de 400 mg/día por vía oral en ciclos de 2 semanas cada trimestre, reduce el riesgo de fracturas vertebrales en mujeres osteoporóticas, pero no hay evidencia sobre su ac-ción en las fracturas no vertebrales. Inhibe la mineralización ósea, pudiendo causar osteomalacia. Alendronato
A dosis de 10 mg/día vía oral, reduce de forma significativa el riesgo de fracturas vertebrales y no vertebrales, incluidas las de cadera, en mujeres con osteoporosis posmenopáusica con y sin fracturas. El alendronato ha demostrado que aumen-ta la DMO a nivel de la columna lumbar y de la cadera tanto en estudios de tratamiento como de prevención de mujeres osteoporóticas. Este efecto se ha observado tanto con la ad-ministración diaria como con la semanal, mostrando ambas una eficacia similar. Existen presentaciones de 70 mg/semanal y una presentación que agrega colecalciferol. Diversos estu-dios han sugerido disminuciones de las fracturas al cabo de 1 año de tratamiento con alendronato. Los efectos beneficiosos del alendronato sobre las fracturas se han demostrado en ensayos clínicos de 3 años de duración y con una pauta de administración diaria. Con la administración semanal no se ha estudiado directamente el efecto sobre las fracturas.
Risedronato A dosis de 5 mg/día por vía oral, ha demostrado su efecto beneficioso en la reducción del riesgo de fracturas vertebrales y no vertebrales, incluidas las de cadera, en mujeres posme-nopáusicas osteoporóticas con y sin fracturas. Existe una pre-sentación de 35 mg/semanal. Los efectos beneficiosos de risedronato se han demostrado con una pauta de administra-ción diaria. Con la administración semanal de 35 mg no se ha estudiado directamente el efecto sobre las fracturas. El efecto sobre la DMO se ha observado tanto con la administración diaria como con la semanal, mostrando ambas una eficacia similar. Ibandronato A dosis de 2,5 mg/día por vía oral ha demostrado eficacia en la prevención de fracturas vertebrales en mujeres posme-nopáusicas con osteoporosis establecida. Es el único bifosfo-nato de administración en dosis única mensual de 150 mg para el tratamiento de la osteoporosis posmenopáusicas. El ibandronato puede administrarse también en inyección intra-venosa mediante infusión o inyección en bolos de hasta 3 mg en 15 a 30 segundos, cada 2 o 3 meses. No disminuyen las fracturas no vertebrales. En subgrupos de alto riesgo el ibandronato podría ser eficaz en la disminución de fracturas no vertebrales. En las pacientes tratadas con 2.5 mg diarios de ibandronato la DMO aumenta en la columna lumbar y en la cadera total. Un estudio de no inferioridad respecto a esta dosis demostró la eficacia de la administración de 150 mg mensuales. La dosis de 150 aumentó la DMO más que 2.5 mg diarios. Un estudio de no inferioridad con 3 mg por vía intra-venosa cada 3 meses respecto a 2.5 mg orales diarios ha de-mostrado no sólo no inferioridad, sino que son superiores al régimen oral en su efecto sobre la DMO. Zoledronato Es el bifosfonato más potente disponible en la actualidad. Se administra de forma anual en dosis de 5 mg y su infusión se realiza en un periodo de 15 minutos. Incrementa la DMO y reduce el riesgo de fractura. Los bifosfonatos son los únicos fármacos aprobados en el
R E V I S T A C L Í N I C A d e M E L I L L A
34 Rev Clin Mel, 2012;3(1):32-38

tratamiento de la osteoporosis posmenopáusica que disminu-yen el riesgo de fractura de cadera. La preservación de la mi-croarquitectura trabecular fue demostrado primero con ri-sedronato y posteriormente con alendronato. PREVENCIÓN DE FRACTURAS EN MUJERES OSTEOPÉNICAS Los bifosfonatos tienen efectos positivos sobre la DMO en las mujeres que no tienen osteoporosis. Por ejemplo, en un estudio de mujeres que acababan de tener la menopausia, el tratamiento con alendronato a dosis de 5 mg/día durante 7 años aumentó la DMO en columna y trocánter en un 3-4%, mientras que la DMO en el cuello femoral se mantuvo13. Un estudio a 2 años con risedronato tuvo resultados parecidos14. Se ha descrito que el tratamiento con etidronato cíclico inter-mitente previene la pérdida de masa ósea en mujeres con menopausia reciente. Un tema importante no resuelto es si los bifosfonatos previenen las fracturas en individuos que aún no tienen osteoporosis. Los estudios con alendronato y ri-sedronato sugieren que no es así, aunque ninguno de estos estudios tenía un poder estadístico suficiente para abordar este tema. Algunos estudios post hoc sugieren que la reduc-ción de fracturas vertebrales con la toma de alendronato pue-de extenderse a las mujeres con osteopenia sin fracturas pre-vias. La eficacia antifractura de estos fármacos es indepen-diente de la edad, índice de masa corporal, DMO basal o ante-cedentes de fracturas. Este hallazgo representa una expansión potencialmente significativa de las indicaciones de los bifosfo-natos. La FDA ha aprobado el alendronato, ibandronato y ri-sedronato para la prevención de la pérdida de masa ósea en mujeres con menopausia reciente. DIFERENCIAS ENTRE LOS BIFOSFONATOS Existen claramente diferencias importantes entre los bi-fosfonatos en su potencia antirresortiva, siendo el zoledrona-to más de 100.000 veces más potente en algunos ensayos que el etidronato. Algunos datos objetivos apoyan las diferencias entre los diferentes compuestos. Por ejemplo, el risedronato es aproximadamente 5 veces más potente que el alendronato como inhibidor de la farnesil difosfato sintetasa, pero en clíni-ca se usa sólo a mitad de dosis del alendronato. Incluso a esta dosis produce incrementos menores de la densidad ósea y una supresión menos marcada de la resorción ósea que el alendronato15. Asimismo, las dosis intravenosas de ibandrona-to pierden su efecto en menos de 3 meses16,17, mientras que una dosis única de zoledronato puede durar más de 1 año18. Este contraste aparente puede tener 2 explicaciones. En pri-mer lugar, recientemente se ha prestado atención a la capaci-dad y afinidad de la unión de los bifosfonatos al hueso. Los estudios in vitro sugieren que el zoledronato y el alendronato tienen una unión más estrecha a la hidroxiapatita que otros bifosfonatos19. Este efecto conduciría teóricamente a una mayor captación del fármaco por el hueso y una duración de acción más prolongada. Un segundo mecanismo puede deber-se a las diferencias aparentes de la duración de acción en relación a la inhibición de la resorción ósea en los estudios clínicos. En la práctica, esto significa que, en comparación a otros bifosfonatos, el ibandronato inhibe la resorción ósea a concentraciones más reducidas y ello hace posible su adminis-
tración en dosis espaciadas. Diferentes estudios comparativos señalan lo siguiente:
El risedronato aumenta la masa ósea y suprime los mar-cadores más que el etidronato y parece disminuir más el ries-go de fractura.
El tratamiento con alendronato diario induce mayores aumentos de la DMO que el etidronato.
El alendronato semanal aumenta la DMO en columna y cadera más que el risedronato semanal. Sin embargo, no hay diferencias en la incidencia de fracturas.
En ausencia de datos consistentes en ensayos clínicos de que ibandronato reduce el riesgo de fractura de cadera, se recomienda alendronato o risedronato como primera opción de tratamiento con bifosfonato en pacientes con alto riesgo de fractura.
No hay datos de eficacia directa sobre fractura para iban-dronato endovenoso. En ausencia de estos, zoledronato es la primera opción de terapia con bifosfonatos endovenoso en pacientes que no toleran tratamiento oral.
¿CUANTO TIEMPO SE DEBEN MANTENER LOS BIFOSFONATOS EN LA OSTEOPOROSIS? Desde la introducción de los bifosfonatos ha habido preo-cupación respecto a que estos potentes agentes antirresorti-vos pudieran reducir el recambio óseo a un nivel en el que las microlesiones no pudieran repararse, lo que se traduciría en una reducción de la resistencia biomecánica. En ausencia de datos controlados convincentes, el consenso es que no hay mayores problemas dentro de los periodos cubiertos por los ensayos controlados y los estudios de extensión. Sin embargo, esto no significa que la acumulación de microlesiones no pue-da llegar a ser un problema con el tratamiento muy prolonga-do, de forma que muchos expertos creen que es razonable considerar limitar de alguna manera la duración del trata-miento con estos fármacos. La idea de realizar descansos de tratamiento o reducciones de la dosis con el uso prolongado se hace más aceptable por la evidencia de que los efectos de algunos agentes persisten tras su suspensión. En dos estudios clínicos recientes de extensión se ha docu-mentado que la administración a largo plazo de alendronato continúa manteniendo la DMO y disminuyendo el remodelado óseo durante hasta 10 años. Las tasas de fractura al término de los estudios con alendronato fueron similares a las obser-vadas en los periodos iniciales, lo que sugiere que el uso a largo plazo no produce una mayor fragilidad ósea. Se dispone de datos similares para un tratamiento de hasta 7 años con risedronato. Con risedronato a los 7 años la DMO en la colum-na continúa aumentando, y en la cadera permanece estabili-zada. En un análisis provisional a los 2 años de un estudio a 3 años en pacientes en tratamiento con zoledronato frente a placebo en mujeres posmenopáusica con osteopenia los mar-cadores de recambio óseo permanecieron bajos y la DMO permaneció alta en el grupo de zoledronato comparado con placebo20. Estos datos sugieren que zoledronato a dosis de 5 mg puede tener un beneficio de hasta 2 años. Sin embargo, la eficacia antifractura de este régimen es desconocida. El estudio de extensión del FIT, Fracture Intervention Trial Long-Term Extension Study (FLEX)21, analizó 1099 mujeres posmenopáusicas y comparó un periodo de 5 años de trata-
R E V I S T A C L Í N I C A d e M E L I L L A
Rev Clin Mel, 2012;3(1):32-38 35

miento continuado con alendronato, seguido de 5 años de placebo, con 10 años de utilización continuada. En este estu-dio sus autores documentaron que la DMO disminuye lenta-mente al interrumpir el tratamiento con alendronato y el re-modelado óseo aumenta gradualmente. Aunque las fracturas vertebrales clínicas disminuyeron entre los que interrumpie-ron el tratamiento con alendronato comparado con aquellos que lo continuaron, otras tasas de fracturas fueron similares. Estos datos sugieren que, en algunos pacientes (probablemente los de menor riesgo) después de 5 años de tratamiento con alendronato podemos considerar su suspen-sión. La suspensión del alendronato se sigue de un efecto residual en la DMO, que es mayor cuanto, a su vez, mayor (en tiempo y dosis) haya sido la administración del fármaco, y que se deja notar más claramente en la columna que en la cadera; aunque la DMO disminuya hacia valores basales, en los estu-dios en los que se ha utilizado un grupo placebo se ha compro-bado que permanece por encima de la correspondiente a éste. Las mujeres con alto riesgo de fracturas fueron exclui-das. En resumen, suspender el tratamiento con alendronato después de 5 años (vigilando la DMO y los factores de riesgo para osteoporosis) puede ser razonable en mujeres con bajo riesgo. En mujeres con alto riesgo de fractura se sugiere conti-nuar alendronato más allá de 10 años, ya que los beneficios sobre la DMO y las fracturas se mantienen sin incrementar el riesgo de efectos adversos. Cuando risedronato se suspende, sus efectos beneficiosos sobre la DMO y los marcadores de recambio óseo parecen revertirse parcial o completamente en el plazo de 1 año como se muestra en un estudio de extensión del ensayo VERT22. En este estudio tanto las mujeres que recibieron risedronato como placebo durante 3 años fueron reevaluadas un año des-pués de suspender el tratamiento. La DMO decreció en el grupo que tomaba risedronato, aunque los valores permane-cieron más alto que en los basales y que los que tomaban placebo. Sin embargo, los marcadores de recambio óseo vol-vían al estado basal y eran los mismos que en el grupo place-bo. Sin embargo, la incidencia de nuevas fracturas vertebrales en pacientes tratados previamente con risedronato permane-ció baja. En base a estos resultados, suspender risedronato después de 3 años de tratamiento puede ser una opción en algunas mujeres, pero debido a que los marcadores de recam-bio óseo vuelven en el plazo de 1 año al estado en el que se encontraban antes de iniciar el tratamiento, no recomenda-mos suspender risedronato después de 3 años de terapia, especialmente en aquellos individuos con alto riesgo de frac-tura. Atendiendo a estos hallazgos, se abren varias líneas de acción: - Una es concluir que el tratamiento durante 10 años es seguro y eficaz y continuar el tratamiento en la mayoría de los pacientes durante este periodo. - En las mujeres que están recibiendo alendronato, una segunda línea de acción es reducir la dosis de 10 mg/día a 5 mg/día (o su equivalente) tras 5 años de uso. No se puede determinar si este tipo de reducción de la dosis es adecuado para el risedronato. No es posible determinar cuál de los abordajes descritos es óptimo en términos del único criterio de valoración que realmente importa, las fracturas. La línea de actuación menos
especulativa es continuar los bifosfonatos hasta el décimo año y entonces considerar una reducción de la dosis junto con la monitorización de la DMO y de los marcadores óseos. EFECTOS ADVERSOS Todos los bifosfonatos deben administrarse en ayunas, tragados sin masticar con un vaso de agua y el paciente debe permanecer erguido. Una proporción significativa (10-30%) de pacientes trata-dos con la primera dosis intravenosa de un aminobifosfonato experimenta reacciones de fase aguda (fiebre, mialgias, linfo-penia, etc.), pero ésta rara vez se produce con la administra-ción repetida. Con la administración intravenosa rápida puede aparecer toxicidad renal de modo que el tiempo sugerido de administración para un preparado como el ácido zoledrónico es de 15 minutos. Con la administración parenteral rápida de bifosfonatos, puede desarrollarse hipocalcemia. Sin embargo, es poco fre-cuente y de carácter leve. Con alendronato se ha descrito pancreatitis. Se han publicado casos de osteonecrosis de mandíbula asociados con su utilización, pero se han limitado en particular a pacientes oncológicos, que reciben dosis frecuentes de bi-fosfonatos intravenosos. No obstante, se ha descrito también algún caso en pacientes con osteoporosis tratados con bifos-fonatos orales. Se da sobre todo en la mandíbula, y el 60% de las veces sigue a una manipulación dentaria23-26. Se han descrito casos aislados de pacientes tratadas con alendronato durante largo tiempo que han sufrido fracturas por insuficiencia27-29. Sin embargo, las mujeres tratadas 10 años con el fármaco no han presentado una mayor incidencia de fracturas que las mujeres que recibieron el fármaco los 5 primeros años y placebo otros 5. Se ha suscitado preocupa-ción de que los bifosfonatos pueden inactivar por completo el remodelado, dando lugar a un “hueso congelado” y un au-mento final de la fragilidad ósea. No se dispone de pruebas de que este fenómeno se produzca con las dosis usadas clínica-mente. En pacientes tratados crónicamente con bifosfonatos la curación de las fracturas no parece ser un problema, aun-que apenas se han publicado estudios que hayan abordado este importante problema. Están contraindicados en estenosis y acalasia esofágicas, en pacientes con insuficiencia renal grave (aclaramiento de creatinina < 35 ml/min) y en la hipocalcemia. Se usarán con precaución en individuos con una concentración sérica baja de 25-hidroxivitamina D ya que esto puede dar lugar a una hipo-calcemia aguda. Análisis retrospectivos de alendronato y ri-sedronato en pacientes con insuficiencia renal moderada y severa mostraron incrementos en la DMO y prevención de fractura vertebral frente a placebo independientemente del grado de afectación renal. La FDA recomienda que los bifosfonatos no sean usados en pacientes con esófago de Barrett. ¿CUAL ES EL FARMACO ANTIOSTEOPORICO MAS INDICADO? La selección del fármaco debe individualizarse tomando en consideración el riesgo de fractura en cada paciente, la eficacia antifractura demostrada en estudios controlados, la DMO en columna vertebral y en tercio proximal del fémur, la
R E V I S T A C L Í N I C A d e M E L I L L A
36 Rev Clin Mel, 2012;3(1):32-38

edad, los efectos secundarios de cada fármaco, sus contrain-dicaciones y el cumplimiento previsto. Es conveniente evaluar la respuesta al tratamiento me-diante densitometría. La práctica de DXA de columna y de cadera, cada 2 años, es un buen método de valoración, con-juntamente con la evaluación clínica, sobre todo de la apari-ción de nuevas fracturas. Los marcadores de remodelación ósea pueden ser útiles para controlar la evolución precoz de la eficacia. Cuando la DMO desciende más del 5% se recomienda cambiar de un bifosfonato oral a uno endovenoso. CONCLUSIONES Como clase, los bifosfonatos son los fármacos estudiados más exhaustivamente para la prevención y el tratamiento de la osteoporosis. Estos fármacos aumentan la DMO vertebral lumbar y la de cadera. En pacientes con osteoporosis establecida, la disminución del riesgo de fracturas vertebrales y no vertebrales es sustan-cial. En la actualidad, estos fármacos son el único tratamiento aprobado en la investigación sobre la reducción del riesgo de fractura de cadera en ensayos clínicos prospectivos, aleatori-zados. La reducción del riesgo de fractura se produce con relativa rapidez. Los aumentos de la DMO se mantienen durante 10 años, y no se dispone de pruebas de que el tratamiento a largo plazo aumente la fragilidad ósea. En general, estos fármacos siguen considerándose de primera línea para el tratamiento de la mayor parte de pa-cientes con osteoporosis. Aunque los bifosfonatos combinados con estrógenos o raloxifeno producen mayores beneficios en la masa ósea fren-te a la monoterapia, no puede recomendarse el uso de dos fármacos antirresortivos combinados porque no se ha demos-trado un mayor beneficio sobre el riesgo de fractura y por el aumento del coste y de los efectos adversos. La paratohormo-na añadida a un tratamiento continuado con bifosfonato re-sulta beneficiosa.
El autor declara no tener conflicto de intereses.
BIBLIOGRAFÍA
1.NIH Consensus Development Panel on Osteoporosis Prevention D, and Theraphy. Osteoporosis prevention, diagnosis, and theraphy. JAMA. 2001;285:785-95.
2.World Health Organization. Assessment of fracture risk and its ap-plication to screening for postmenopausal osteoporosis. WHOTech-nical Report Series. Geneva (Switzerland): WHO;1994.
3.Robinson CM, Royds M, Abraham A, McQueen MM, Court-Brown CM, Christie J. Refractures in patients at least forty-five years old. a
prospective analysis of twenty-two thousand and sixty patients. J Bone Joint Surg Am. 2002;84-A:1528–33.
4.World Health Organization. Guidelines for Preclinical Evaluation and Clinical Trials in Osteoporosis. 1998.
5.Melton 3rd LJ, Thamer M, Ray NF, Chan JK, Chesnut 3rd CH, Einhorn TA, et al. Fractures attributable to osteoporosis: report from the Na-tional Osteoporosis Foundation. J Bone Miner Res. 1997;12:16–23.
6.Brown JP, Josse RG. 2002 clinical practice guidelines for the diagno-sis and Management of osteoporosis in Canada. CMAJ. 2002;167 Suppl 10:S1–34.
7.Scottish Intercollegiate Guidelines Network [actualizado Mayo 2011; citado Mayo 2011]. Disponible en: www.sign.ac.uk.
8.Melton 3rd LJ. How many women have osteoporosis now? J Bone Miner Res. 1995;10:175–7.
9.Simón Méndez L, Thuissard Vasallo IJ, Gogorcena Aoiz, MA. Instituto de Información Sanitaria. Estadísticas Comentadas: La Atención a la Fractura de Cadera en los Hospitales del SNS [Publicación en Inter-net]. Madrid: Ministerio de Sanidad y Política Social; 2010. Disponible en: http://www.msps.es/estadEstudios/estadisticas/cmbdhome.htm.
10.Pérez Edo L, Alonso Ruiz A, Roig Vilaseca D, García Vadillo A, Gua-ñabens Gay N, Peris P, et al. Actualización 2011 del consenso Socie-dad Española de Reumatología de osteoporosis. Reumatol Clin. 2011;7(6):357-9.
11.Masarachia P, Weinreb M, Balena R, et al. Comparison of the dis-tribution of 3H-alendronate and 3H-etidronate in rat and mouse bones. Bone. 1996;19(3):281-90.
12.Fleisch H. Pharmacokinetics. In: Fleisch H, editor. Bisphosphonates in Bone Disease: From the Laboratory to the Patient. Berne;1993. p.50.
13.Sambrook PN, Rodriguez JP, Wasnich RD, et al. Alendronate in the prevention of osteoporosis: 7-year follow-up. Osteoporos Int 2004;15(6):483-8.
14.Mortensen L. Charles P, Bekker PJ, et al. Risedronate increases bone mass in an early postmenopausal population-two years of treat-ment plus one year of follow-up. J Clin Endrocinol Metab 1998;83(2):396-402.
15.Rosen CJ, Hochberg MC, Bonnick SL, et al. Treatment with once-weekly alendronate 70 mg compared with once-weekly risedronate 35 mg in women with postmenopausal osteoporosis: a randomized double-blind study. J Bone Miner Res 2005;20(1):141-51.
16.Recker R, Stakkestad JA, Chesnut CH, et al. Insufficiently doses intravenous ibandronate injections are associated with suboptimal antifractrure efficacy in postmenopausal osteoporosis. Bone 2004;34(5):890-9.
17.Christiansen C, Tanko LB, Warming L, et al. Dose dependent effects on bone resorption and formation of intermittently administered intravenous ibandronate. Osteoporos Int 2003;14(7):609-13.
18.Reid IR, Brown JP, Burckhardt P, et al. Intravenous zoledronic acid in postmenopausal women with low bone mineral density. N Engl J Med 2002;346(9):653-61.
19.Nancollas GH, Tang R, Phipps RJ, et al. Novel insights into actions of bisphosphonates on bone: differences in interactions with hydroxy-apatite. Bone 2006;38(5):617-27.
20.Grey A, Bolland MJ, Wattie D, et al. The antiresorptive effects of a single dose of zoledronate pesist for two years: a randomized, place-
R E V I S T A C L Í N I C A d e M E L I L L A
Rev Clin Mel, 2012;3(1):32-38 37

bo-controlled trial in osteopenic postmenopausal women. J Clin En-drocrinol Metab 2009;94:538.
21.Black DM, Schwartz AV, Ensrud KE, et al. Effects of continuing or stopping alendronate after 5 years of treatment: the Fracture Inter-vention Trial Long-term Extension (FLEX): a randomized trial. JAMA 2006;296:2927.
22.Harris ST, Watts NB, Genant HK, et al. Effects of risedronate treat-ment on vertebral and nonvertebral fractures in women with post-menopausal osteoporosis: a randomized controlled trial. Vertebral Efficacy With Risedronate Therapy (VERT) Study Group. JAMA 1999;282:1344.
23.Khosla S, Burr D, Cauley J, et al. Bisphosphonate-associated oste-onecrosis of the jaw: report of a task force of the American Society for Bone and Mineral Research. J Bone Miner Res 2007;22:1479.
24.Yarom N, Yahalom R, Shoshani Y, et al. Osteonecrosis of the jaw induced by orally administered bisphosphonates: incidence, clinical features, predisposing factors and treatment outcome. Osteoporos Int
2007;18:1363.
25.Khan AA, Sándor GK, Dore E, et al. Bisphosphonate associated osteonecrosis of the jaw. J Rheumatol 2009;36:478.
26.Favia G, Pilolli GP, Maiorano E. Osteonecrosis of the jaw correlated to bisphosphonate therapy in non-oncologic patients: clinicopatholog-ical features or 24 patients. J Rheumatol 2009;36:2780.
27.Lenart BA, Lorich DG, Lane JM. Atypical fractures of the femoral diaphysis in postmenopausal women taking alendronate. N Engl J Med 2008;358:1304.
28.Visekruna M, Wilson D, McKiernan FE. Severely suppressed bone turnover and atypical skeletal fragility. J Clin Endocrinol Metab 2008;93:2948.
29.Odvina CV, Levy S, Rao S, et al. Unusual mid-shaft fractures during
long-term bisphosphonate therapy. Clin Endocrinol (Oxf) 2010;72:161.
R E V I S T A C L Í N I C A d e M E L I L L A
38 Rev Clin Mel, 2012;3(1):32-38

Fístula traqueo-esófágica gigante. Giant tracheo-esophageal fistula. A. Matas Cobos1, R.J. Martín-Vivaldi Jiménez1, M.J. Requena Pou2, E. Navarro Guerrero3, C. Martínez Agúdiez3, A. Fernández Ruiz2
1 Aparato Digestivo. Medicina Interna. Hospital Comarcal de Melilla 2 Medicina Interna. Medicina Interna. Hospital Comarcal de Melilla 3 Medicina General. Atención Primaria. Melilla Autor para correspondencia: Ana Matas Cobos ([email protected])
Rev Clin Mel, 2012;3(1):39-42 39
Caso Clínico
R E V I S T A C L Í N I C A d e M E L I L L A
Artículo admitido para su publicación en septiembre de 2012
RESUMEN La fistula traqueo-esofágica maligna (FTEM) es una com-plicación del cáncer de esófago que provoca síntomas graves que conllevan en muchos casos la muerte. Presentamos una fístula traqueoesofágica gigante que fue diagnosticada me-diante gastroscopia, técnica inusual para su diagnóstico, que se suele realizar mediante radiología (principalmente TAC) Palabras clave: fístula traqueo-esofágica, tumor, endosco-pia, diagnóstico. ABSTRACT The malignant tracheoesophageal fistula (MTEF) is a com-plication of esophageal cancer that causes severe symptoms in many cases involving death. We present a giant trache-oesophageal fistula was diagnosed by gastroscopy, unusual technique for diagnosis, which is usually done by radiology (mainly ACT). Keywords: trachea-esophageal fistula, tumor, endoscopy, diagnostic. INTRODUCCIÓN
La fistula traqueo-esofágica maligna (FTEM) es una com-plicación del cáncer de esófago que se suele presenta en
etapas avanzadas de éste. Las principales causas son: disemi-nación tumoral e insuficiencia vascular (compresión extrínse-ca, prótesis previamente colocadas, etc.), complicaciones infecciosas agregadas o complicaciones del tratamiento (radiación, quimioterapia y cirugía)1. Los pacientes con FTEM presentan síntomas tales como tos, aspiración, disnea y dolor torácico; generalmente mueren por neumonía, sepsis y desnutrición en un plazo de días o semanas si no es tratada. CASO CLÍNICO Paciente de 54 años con antecedentes personales de Carcinoma epidermoide de esófago irresecable en estadío T4bNxMx diagnosticado hace 4 meses, en tratamiento con quimioterapia: CDDP (cis-diaminodicloroplatino)/5-FU (5-fluorouracilo)/DCT (docetaxel). El paciente acude a Urgencias por disfagia progresiva en los últimos 15 días, así como deterioro del estado general. Se procede a ingreso hospitalario y se solicito TAC tóraco-abdominal de control en el que se evidencia una marcada disminución de tamaño tumoral respecto a estudio de imagen previo (imagen 1), así como varias lesiones cavitadas pulmo-nares (no presentes en TAC previo) (imagen 2, 3), que en el contexto clínico del paciente plantean diagnóstico diferencial entre metástasis cavitadas vs origen infeccioso.
Dada la persistencia de la sintomatología del pacien-te, a pesar de la mejoría en las pruebas de imagen, se decide realizar endoscopia digestiva alta, con los siguientes hallazgos: a 28 cm de arcada dentaria se identifica gran orificio fistuloso de unos 5 cm (imagen 4, 5), que comunica esófago con trá-quea; distal a éste, a unos 2 cm aproximadamente, aprecia-mos estenosis puntiforme que corresponde a la luz esofágica (imagen 6).
Debido al gran tamaño de la fístula y a la imposibili-dad, por tanto, de terapéutica endoscópica se decide deriva-ción a Cirugía general para tratamiento quirúrgico de la mis-ma. Finalmente el paciente fallece como consecuencia de una infección respiratoria.
Imagen 1. TAC torácico en el que se identifica lesión tumo-ral esofágica
Orificio fistuloso
Luz esofágica
Imagen 4. Imagen endoscópica de fístula traqueo-esofágica.
Imagen 2. TAC torácico en el que se evidencia lesión cavitada
Imagen 3. TAC torácico en el que se evidencia lesión cavitada.
R E V I S T A C L Í N I C A d e M E L I L L A
40 Rev Clin Mel, 2012;3(1):39-42

Imagen 5. Imagen endoscópica de luz traqueal.
Orificio fistuloso
Luz esofágica
Imagen 6. Imagen endoscópica donde se aprecia estenosis puntiforne de luz esofágica.
Discusión
En pacientes con cáncer de esófago, la FTEM apare-ce en el contexto de un tumor localmente avanzado o como una complicación de la radioterapia o quimiorradioterapia.
La incidencia de una fístula traqueoesofágica duran-
te la quimiorradioterapia alcanza el 6% en algunas series pu-blicadas2 representando la mitad de todas las fístulas que se desarrollaron en los pacientes con cáncer de esófago2.
El diagnóstico generalmente es mediante técnicas de imagen (TAC, tránsito baritado), siendo en muy pocos ca-sos necesaria la endoscopia digestiva para confirma el diag-nóstico.
Existen múltiples modalidades para el cierre de la FTEM; sin embargo, actualmente se considera como trata-miento de elección la colocación endoscópica de prótesis metálica auto-expandible cubierta3. Esto es por su relativo bajo costo, facilidad de colocación, tolerabilidad, baja morbi-mortalidad del procedimiento y una tasa de oclusión de 70% a 100% con una baja tasa de complicaciones de 10% a 30%4,5. Sin embargo, el crecimiento posterior del tumor a medida que progresa la enfermedad, hace que muchos pacientes (hasta 50 por ciento) requieran intervenciones adicionales para la disfagia recurrente o eventos adversos relacionados con el stent, tal como la migración6.
Las alternativas a la colocación de stents incluyen la
exclusión esofágica con esofagostomía cervical, gastrostomía, y la colocación de un tubo de alimentación de yeyunostomía. La derivación esofágica rara vez es necesaria.
La resección paliativa del esófago con la reparación de la fístula, la reconstrucción traqueoesofágica, o la coloca-ción de un tubo de gastrostomía no está justificada, ya que es extremadamente complicado y suele estar asociada con una alta morbilidad y mortalidad.7
El tratamiento quirúrgico se reserva para casos en
los que no es posible una solución endoscópica, debido al tamaño de la fístula o a su localización, principalmente cuan-do ésta se sitúa cerca de la unión gastro-esofágica.
En resumen: la formación de una FTEM es una com-plicación grave que conlleva un pronóstico sombrío. La mayo-ría de estos pacientes tienen enfermedad recurrente o desa-rrollar esta complicación durante la quimioterapia. El trata-miento de elección consiste en la colocación endoscópica de prótesis esofágica. Sin embargo, una nueva reintervención es necesaria hasta en el 40 por ciento de los casos, presentando por tanto, una alta tasa de mortalidad a corto-medio plazo.
Bibliografía 1.Suárez-Moran, Edgardo; Mendoza-Varela, Fidel. Manejo endoscópi-co de fístula traqueo-esofágica maligna con prótesis metálica auto-expandible cubierta; informe de un caso. Endoscopia 2010; 22 :196-199.
2.Muto M, Ohtsu A, Miyamoto S, et al. Concurrent chemoradiothera-py for esophageal carcinoma patients with malignant fistulae. Cancer 1999; 86:1406.
3.Park JY, Shin JH, Song HY, et al. Airway complications after covered stent placement for malignant esophageal stricture: special reference to radiation therapy. AJR Am J Roentgenol 2012; 198:453.
R E V I S T A C L Í N I C A d e M E L I L L A
Rev Clin Mel, 2012;3(1):39-42 41

4.Van den Bongard HJ, Boot H, Baas P, Taal BG. The role of parallel stent insertion in patients with esophagorespiratory fistulas. Gastroin-test Endosc 2002; 55:110.
5.Sharma P, Kozarek R. The Practice Parameters Committee of the American College of Gastroenterology. Role of esophageal stents in benign and malignant diseases. Am J Gastroenterol 2010;105:258-273
6.Rozanes I, Poyanli A, Acunaº B. Palliative treatment of inoperable malignant esophageal strictures with metal stents: one center's experi-ence with four different stents. Eur J Radiol 2002; 43:196.
7. Verschuur EM, Kuipers EJ, Siersema PD. Esophageal stents for ma-lignant strictures close to the upper esophageal sphincter. Gastrointest Endosc 2007; 66:1082.
R E V I S T A C L Í N I C A d e M E L I L L A
42 Rev Clin Mel, 2012;3(1):39-42

Medicina En Imágenes
R E V I S T A C L Í N I C A d e M E L I L L A
Infección aguda sobre prótesis de cadera en pa-ciente con enfermedad de Crohn. Acute infection of hip prosthesis in patients with Crohn's disease.
A. Martín-Vivaldi Jiménez1*, M. J. Molina Rueda2, R. J. Martín-Vivaldi Jiménez3
1 Servicio de Cirugía Ortopédica y Traumatología. Hospital Universitario Virgen Nieves. Granada. 2 Servicio de Medicina Preventiva y Salud Pública. Hospital Universitario Virgen de las Nieves. Granada. 3 Servicio Medicina Interna. Aparato Digestivo. Hospital Comarcal de Melilla. Melilla *Correspondencia: A. Martín-Vivaldi Jiménez ([email protected])
Artículo admitido para su publicación en abril de 2012
RESUMEN Se trata de una paciente con enfermedad de Crohn cono-cida y displasia de cadera que fue intervenida para corregir las secuelas de dicha displasia (mediante artroplasita total de cadera con vástago poroso metafisario) que presenta un mes después una fístula desde fosa iliaca a cadera y que precisó amputación abdomino-perineal. Cirugías ortopédicas comple-jas pueden verse complicadas por situaciones atípicas como la que presentamos Palabras clave: enfermedad de Crohn, displasia de cadera, fistula, Enterococcus faecalis, artroplastia total cadera. ABSTRACT We present a patient with known Crohn's disease and hip dysplasia who underwent surgery to correct the consequences of such dysplasia (through a total hip arthroplasty with a po-rous metaphyseal stem). One month later she had a fistula from hip to lower abdominal quadrant, that required abdom-ino-perineal amputation. Complex orthopedic surgery can be complicated by atypical situations such as the one presented. Keywords: Crohn's disease, hip dysplasia, fistula, Entero-coccus faecalis, total hip arthroplasty INTRODUCCIÓN: El cribado habitual de la displasia de cadera en los centros de neonatología permite en la actualidad un diagnóstico pre-coz y un tratamiento eficaz1.
MATERIAL Y MÉTODO: Presentamos el caso de una paciente mujer de 26 años que es enviada a nuestro servicio con el diagnóstico de secue-la de displasia de cadera para valoración de tratamiento. Entre las patologías previas encontramos enfermedad de Crohn desde los 14 años diagnosticada tras apendicectomía. En el momento de la consulta había sufrido dos resecciones intesti-nales por actividad de su patología inflamatoria. A la exploración destaca una marcha anadeante, trende-lemburg por insuficiencia glútea, con rotaciones de cadera abolidas y dolorosas. Radiológicamente se observa una secue-la de displasia congénita en ambas caderas. Se plantea artro-plastia total de cadera con vástago poroso metafisario. RESULTADO: La paciente presenta una evolución favorable con radio-grafías satisfactorias, siendo dada de alta deambulando. Acude a urgencias cuatro semanas después por supura-ción de herida quirúrgica, necesitando lavado de urgencia y toma de cultivo. Se aisla Enterococcus faecalis, la paciente es vista por el servicio de Digestivo que diagnostica fístula desde fosa iliaca hasta la cadera (nivel hidroaéreo evidente en radio-grafía simple y en el paso de contraste durante el TAC2). Valo-rada por el servicio de Cirugía se decide amputación abdo-mino-perineal. La paciente precisa de varios lavados quirúrgi-cos. En la actualidad tras un tratamiento antibiótico prolonga-do, deambula sin problemas y mantiene parámetros analíticos dentro de la normalidad3.
Rev Clin Mel, 2012;3(1):43-45 43

CONCLUSIONES: La cirugía de corrección de secuelas de cadera displásica supone un reto para el cirujano ortopédico en busca de un buen balance pélvico. Situaciones atípicas como la presenta-da ponen en peligro el éxito de la cirugía4. BIBLIOGRAFÍA: 1.Dziewulski M, Dziewulski W, Barcińska-Wierzejska I. The importance of early diagnosis and treatment of developmental dysplasia of the hips. Ortop Traumatol Rehabil. 2001;3(3):412-7.
2.Jacquier A, Champsaur P, Vidal V, Stein A, Monnet O, Drancourt M, Argenson JN, Raoult D, Moulin G, Bartoli JM. CT evaluation of total hip prosthesis infection. J Radiol 2004;85:2005-12.
3.Gasiunas V, Plenier I, Herent S, May O, Senneville E, Migaud H. Transabdominal removal of femoral and acetabular components of a severely protruded and infected hip arthroplasty with urinary tract complications. Rev Chir Orthop Reparatrice Appar Mot 2005;91(4):346-50.
4.Treatment of infected retained implants. J Bone Joint Surg Br 2005;87(2):249-56.
Figura 1. Coxartrosis bilateral de predominio derecho como secuela de displasia congénita de caderas.
Figura 2. Prótesis total de cadera derecha que requiere osteotomía pélvica.
R E V I S T A C L Í N I C A d e M E L I L L A
44 Rev Clin Mel, 2012;3(1):43-45

R E V I S T A C L Í N I C A d e M E L I L L A
Figura 4. Fístula artroentérica desde la cadera derecha.
Figura 3. TAC abdominal que evidencia la colección enteropélvica.
Rev Clin Mel, 2012;3(1):43-45 45

Medicina En Imágenes
R E V I S T A C L Í N I C A d e M E L I L L A
Leucoencefalopatía con necrosis palidal bilateral de-bido a intoxicación severa por monóxido de carbono. Leukoencephalopathy with bilateral pallidal necrosis due to severe intoxication carbon monoxide R. Soler González1, E. Crespillo Montes2, K. Ghazi El Hamouti 3,4, C. Martínez Agudiez4. Sección de Neurología1. Medicina Interna. Hospital Comarcal de Melilla Sección de Cardiología2. Medicina Interna. Hospital Comarcal de Melilla Servicio de Urgencias3. Hospital Comarcal de Melilla Unidad Docente de MFyC4. Hospital Comarcal de Melilla. Autor para Correspondencia: Rafael Soler González ([email protected])
Artículo admitido para su publicación en abril de 2012
RESUMEN La intoxicación por monóxido de carbono es causa conoci-da de daño cerebral adquirido. Presentamos una paciente con una situación de coma e insuficiencia respiratoria severa por inhalación prolongada de monóxido de carbono, que tras 6 meses y múltiples complicaciones, permanece en estado vege-tativo. Las imágenes muestran necrosis bilateral de ambos núcleos pálidos, discretamente asimétrica, así como una leu-coencefalopatía severa, de predominio posterior.
Palabras Clave: monóxido de carbono, intoxicación, nú-cleos pálidos, leucoencefalopatia. ABSTRACT The carbon monoxide intoxication is a known cause of acquired brain injury. We present a patient with a situation of severe coma and respiratory failure by prolonged inhalation of carbon monoxide, which after six months and multiple compli-cations, remains in a vegetative state. The images show both bilateral necrosis pale nuclei, asymmetric discretely and a severe leukoencephalopathy, predominantly posterior. Keywords: carbon monoxide intoxication, pale nuclei, leukoencephalopathy
CASO CLÍNICO La intoxicación por monóxido de carbono es causa conoci-da de daño cerebral adquirido. Las imágenes que presenta-mos corresponden a una paciente de 65 años que sufrió la exposición continuada, al menos durante 24 horas, al monóxi-do de carbono, fruto de la combustión prolongada de carbo-nes vegetales en una habitación cerrada, sin ventilación, y escaso oxígeno, por tanto. La paciente ingresó en el hospital en situación de coma e insuficiencia respiratoria severa; el esposo de la paciente falleció en el lugar de la intoxicación. Después de 6 meses y múltiples complicaciones, la paciente superó el coma inicial, pero permanece en estado vegetativo, concretamente en estado de mínima conciencia, ya que la única actividad volitiva es el seguimiento ocular ocasional y breve a personas del entorno. La paciente presenta una tetra-paresia espástica, está encamada y precisa de una traqueosto-mía para ventilación y nutrición por sonda enteral. Desde el punto de vista de las imágenes, podemos obser-var la necrosis bilateral de ambos núcleos pálidos, discreta-mente asimétrica, así como una leucoencefalopatía severa, de predominio posterior. En el caso de nuestra paciente, tanto la exposición prolon-gada, como la edad, jugaron en contra de su pronóstico fun-cional, ya que los pacientes en edades intermedias de la vida suelen poseer mejor expectativa de recuperación.
46 Rev Clin Mel, 2012;3(1):46-47

R E V I S T A C L Í N I C A d e M E L I L L A
Figura 3: Corte axial secuencia Flair a nivel gangliobasal
Figura 1: Corte axial T2 a nivel gangliobasal. Se observa una imagen “en ojos” como consecuencia de la necrosis de am-bos globos pálidos
Figura 2: Corte axial en secuencias Flair, donde podemos observar las lesiones en sustancia blanca que definen la leucoencefalopatía por CO.
Rev Clin Mel, 2012;3(1):46-47 47

Medicina En Imágenes
R E V I S T A C L Í N I C A d e M E L I L L A
Fractura patológica abierta en miembro inferior de un paciente con artritis reumatoide. Solución quirúr-gica. Pathological open fracture in a lower limb of a rheumatoid arthritis patient. Surgical solution. A. Martín-Vivaldi Jiménez1*, M. J. Molina Rueda2, F. Molina Rueda3
1Servicio de Cirugía Ortopédica y Traumatología. Hospital Universitario Virgen Nieves. Granada. 2Servicio de Medicina Preventiva y Salud Pública. Hospital Universitario Virgen de las Nieves. Granada. 3Facultad de Ciencias de la Salud. Universidad Rey Juan Carlos. Madrid. *Correspondencia: A. Martín-Vivaldi Jiménez ([email protected])
Artículo admitido para su publicación en abril de 2012
RESUMEN Presentamos el caso de una paciente con artritis reuma-toide, que sufre tras una torcedura de tobillo, una fractura de estrés del tercio distal de tibia y peroné con herida circunfe-rencial de diez centímetros (grado III de Gustilo). Se trató con antibioterapia intensiva, sutura e inmovilización y posterior-mente artrodesis calcáneo-astrágalo-tibial, que evolucionó satisfactoriamente. Las fracturas de estrés son frecuentes pero su asociación con heridas amplias es muy infrecuente y empeora el pronóstico. Palabras Clave: artritis reumatoide, fractura de estrés, fractura abierta, artrodesis tibiotalo-calcánea. ABSTRACT We report a patient with rheumatoid arthritis, who suffers a stress fracture of the distal third of the tibia and fibula with a circumferential wound of ten centimeters (Gustilo grade III) after an ankle torsion. She was treated with an intensive anti-biotic therapy, suture and immobilization and subsequent tibiotalar-calcaneus fusion, which evolved satisfactorily. Stress fractures are common but their association with large wounds is very infrequently which worsens the prognosis. Keywords: rheumatoid arthritis, stress fracture, open fracture, tibiotalar-calcaneus arthrodesis.
Las fracturas de estrés son frecuentes en deportistas y se localizan en la tibia en alrededor del 20% de los casos1. Su asociación con lesiones de partes blandas es extraordinaria-mente infrecuente2. MATERIAL Y MÉTODO Presentamos el caso de una mujer de 60 años, con artri-tis reumatoide como antecedente de interés, que es remitida a urgencias tras sufrir torcedura de tobillo mientras camina-ba. Presenta una herida circunferencial en cara interna de tobillo de aproximadamente 10 cm, grado IIIA de Gustilo (figura 1). En el estudio radiológico se aprecia fractura de tercio distal de tibia y peroné sobre lesión patológica que impresiona de fractura de estrés, así como una evolucionada artropatía degenerativa a nivel de tobillo y pie (figura 2). RESULTADO La paciente ingresa con antibioterapia intravenosa reali-zándose un lavado abundante de la herida, puntos de aproxi-mación e inmovilización en el servicio de urgencias. Tras una evolución favorable de las partes blandas, se planifica inter-vención quirúrgica realizándose en un mismo tiempo una artrodesis calcáneo-astrágalo-tibial mediante una osteotomía de peroné distal y enclavado retrógrado de la fractura tibial (figura 3). Tras tres meses de evolución, la paciente presenta signos radiológicos de consolidación (figura 4) . CONCLUSIONES
48 Rev Clin Mel, 2012;3(1):48-49

La fractura de estrés en este caso, no se relaciona con una actividad deportiva de alta competición, sino con fuerzas de tensión repetidas por mal apoyo sobre una tibia con una ar-tropatía importante a nivel del tobillo y pie. La asociación con fracturas abiertas es infrecuente y aumenta la tasa de infec-ción y pseudoartrosis en estos pacientes3.
BIBLIOGRAFÍA 1.Age-related change in the damage morphology of human cortical bone and its role in bone fragility.Bone 2006;38(3):427-31.
2.Predictors of stress fracture susceptibility in young female recruits. Am J Sports Med 2006;34(1):108-15.
3. Stress fracture of the distal tibia and fibula . Am J Ind Med 2005;47
Figura 1. Fractura abierta grado IIIA de Gustilo
Figura 2. Radiografías de urgencias AP y Lateral
R E V I S T A C L Í N I C A d e M E L I L L A
Rev Clin Mel, 2012;3(1):48-49 49

REVISTA CLÍNICA DE MELILLA es una publicación científica periódica (2 números cada año) de difusión na-cional que ofrece a todos los profesionales de la salud de la Ciudad Autónoma de Melilla la posibilidad de dar a co-nocer sus trabajos de investigación clínica relacionados con la medicina, enfermería, farmacia, psicología y otros ámbitos de las ciencias de la salud.
Admite para publicación manuscritos de trabajos ori-ginales de contenido científico, clara y correctamente ex-presado, realizados mediante una metodología apropiada que proporcionen una información relevante.
REVISTA CLÍNICA DE MELILLA asume la normas de publicación enunciadas en Uniform requirements for ma-nuscripts submitted to biomedical journals (International Comitee of Medical Journal Editors), que pueden consul-tarse en http://www.icmje.org.
Los manuscritos deben entregarse en formato digital a la Secretaria de Redacción en mano o por vía electróni-ca ([email protected]). Se empleará un tipo de fuente Arial de 12 pto, con interlineado sencillo (un espacio entre líneas), sin negritas ni tabulaciones. Cada manuscrito debe incluir los siguientes apartados:
- Título
- Autores (como norma general se admitirá un máximo de 6 autores en un artículo)
- Dirección electrónica de autor para correspondencia
- Resumen en español
- Palabras clave en español (3-10)
- Resumen en inglés
- Palabras clave en inglés (3-10)
- Cuerpo del artículo
- Bibliografía
De acuerdo con las normas de Vancouver, las pala-bras clave se obtendrán del tesauro Medical Subject Hea-dings (MeSH) del Index Medicus En el caso de que se de-signen términos de reciente aparición que aún no figuren en el MeSH pueden usarse los nuevos términos. La base de datos de Descriptores en Ciencias de la Salud (DeCS), puede consultarse desde el sitio: http://Desc.bvc.br/E/homepagee.htm.
Las citas bibliográficas se enumerarán al final de cada trabajo por orden de aparición en el texto siguiendo las normas de Vancouver (disponible en: http://www.icmje.org). Los nombres de las revistas se enuncia-rán mediante las abreviaturas utilizadas en el Index Me-dicus (disponible en http://www.ncbi.nlm.nih.gov/entrez/query. fcgi?db=journals).
Los autores garantizan que el contenido de los traba-jos remitidos a la Revista para su publicación es original e inédito, que poseen sus derechos de explotación, que su publicación no vulnera los derechos de terceras perso-nas y que cumple la legislación vigente sobre protección de datos de carácter personal. Asimismo, los autores ce-den en exclusiva a la Asociación Melillense de Investiga-ción Clínica todos los derechos de explotación que pue-dan derivarse de los trabajos seleccionados para su pu-blicación en la Revista. Todos los manuscritos deben ir acompañados de una carta de presentación en la que los autores expresen su aceptación de estas normas.
Los trabajos experimentales en los que participan se-res humanos deben cumplir las normas éticas expresadas en la Declaración de Helsinki de 1975 (disponible en: http://www.wma.net/s/policy/b3.htm). No deben publi-carse imágenes, nombres, ni ningún otro dato que per-mitan la identificación de las personas incluidas en el es-tudio.
INSTRUCCIONES PARA LOS AUTORES
R E V I S T A C L Í N I C A d e M E L I L L A
50

CRESTOR: FICHA TÉCNICA PROMOCIONAL COMPLETA COMBINADA
NOMBRE DEL MEDICAMENTO. Crestor 5 mg comprimidos recubiertos con película. Crestor 10 mg comprimidos recubiertos con película. Crestor 20 mg comprimidos recubiertos con película. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA.Crestor 5 mg comprimidos recubiertos con película:Cada comprimido contiene 5 mg de rosuvastatina (como rosuvastatina de calcio). Cada comprimido contiene 94,88 mg de lactosa monohidrato.Crestor 10 mg comprimidos recubiertos con película:Cada comprimido contiene 10 mg de rosuvastatina (como rosuvastatina de calcio). Cada comprimido contiene 91,3 mg de lactosa monohidrato.Crestor 20 mg comprimidos recubiertos con película:
Cada comprimido contiene 20 mg de rosuvastatina (como rosuvastatina de calcio). Cada comprimido contiene 182,6 mg de lactosa monohidrato.Para la lista completa de excipientes, ver sección “Lista de excipientes”.FORMA FARMACÉUTICAComprimidos recubiertos con película.Crestor 5 mg comprimidos recubiertos con película: Comprimidos redondos, amarillos, con el grabado “ZD4522” y “5” en una cara y lisos por la cara inversa.Crestor 10 mg comprimidos recubiertos con película:Comprimidos redondos, rosas, con el grabado “ZD4522” y “10” en una cara y lisos por la cara inversa.Crestor 20 mg comprimidos recubiertos con película:Comprimidos redondos, rosas, con el grabado
“ZD4522” y “20” en una cara y lisos por la cara inversa.DATOS CLÍNICOS.Indicaciones terapéuticasTratamiento de la hipercolesterolemiaAdultos, adolescentes y niños de edad igual o mayor de 10 años con hipercolesterolemia primaria (tipo IIa incluyendo hipercolesterolemia familiar heterocigótica) o dislipidemia mixta (tipo IIb) como tratamiento complemen-tario a la dieta cuando la respuesta obtenida con la dieta y otros tratamientos no farmacológicos (p. ej., ejercicio, pérdida de peso) no ha sido adecuada.Hipercolesterolemia familiar homocigótica en tratamiento combinado con dieta y otros tratamientos hipolipemiantes (p. ej., aféresis de las LDL) o si dichos tratamientos no son apropiados.Prevención de
Eventos Cardiovasculares.Prevención de eventos cardiovasculares mayores en pacientes considerados de alto riesgo de sufrir un primer evento cardiovascular.(Ver sección “Propiedades farmacodinámicas”), como tratamiento adyuvante a la corrección de otros factores de riesgo.Posología y forma de administración.Antes de iniciar el tratamiento, el paciente debe someterse a una dieta estándar para reducir los niveles de colesterol que continuará durante el tratamiento. La dosis debe ser individualizada de acuerdo con el objetivo del tratamiento y la respuesta del paciente empleando las guías de tratamiento actuales.Crestor puede administrarse a cualquier hora del día, con o sin alimen-
tos.Tratamiento de la hipercolesterolemiaLa dosis inicial recomendada es 5 ó 10 mg vía oral, una vez al día tanto en pacientes que no hayan recibido estatinas como en pacientes que hayan sido tratados previamente con otro inhibidor de la HMG-CoA reductasa. En la elección de la dosis de inicio deberá tenerse en cuenta el nivel de colesterol del paciente y el posible riesgo cardiovascular, así como el riesgo potencial de reacciones adversas (ver a continuación). Si fuera necesario, tras 4 semanas puede aumentarse la dosis hasta el siguiente nivel de dosis, (ver sección “Propiedades farmacodinámicas”). Debido al aumento de notificaciones de reacciones adversas con la dosis de 40 mg en comparación con las
dosis menores (ver sección “Reacciones adversas”), solamente se considerará un ajuste final a la dosis máxima de 40 mg en pac ientes con hipercolesterolemia severa con alto riesgo cardiovascular (especialmente pacientes con hipercolesterolemia familiar) que no alcancen sus objetivos de tratamiento con 20 mg, y en los que se llevará a cabo un seguimiento rutinario (ver sección “Advertencias y precauciones especiales de empleo”). Se recomienda iniciar la dosis de 40 mg bajo la supervisión de un especialista.Prevención de Eventos CardiovascularesEn el estudio sobre reducción del riesgo de eventos cardiovasculares, la dosis utilizada fue de 20 mg al día (ver sección “Propiedades farmacodinámi-
cas”).Población pediátricaSu uso en población pediátrica debe llevarse a cabo por especialistas.Uso en niños y adolescentes de 10 a 17 años de edad, (varones en el estadio II de Tanner y superiores) y adolescentes del sexo femenino al menos un año después de la menarquia:La dosis habitual de inicio recomendada para niños y adolescentes con hipercolesterolemia familiar heterocigótica, es de 5 mg diarios. El rango de dosis recomendado es de 5-20 mg/ día por vía oral. Las dosis deben individualizarse y ajustarse de acuerdo con la respuesta y la tolerabilidad de los pacientes pediátricos, como figura en población pediátrica (ver sección “Advertencias y precauciones especiales de empleo”). Los
niños y adolescentes deberán someterse a una dieta estándar específica para reducir el colesterol antes de iniciar el tratamiento con rosuvastatina; esta dieta deberá continuarse a lo largo del tratamiento. La seguridad y eficacia de dosis superiores a 20 mg en esta población no ha sido estudiada. Los comprimidos de 40 mg no son recomendados para el uso en población pediátrica.Niños menores de 10 años de edad:La experiencia en niños menores de 10 años de edad es limitada a un número pequeño (entre los 8 y 10 años) con hipercolesterolemia familiar homocigótica. Por lo tanto, no se recomienda administrar Crestor en niños menores de 10 años de edad.Uso en ancianosEn pacientes mayores de 70
años, se recomienda una dosis de inicio de 5 mg (ver sección “Advertencias y precauciones especiales de empleo”). No es necesario ningún otro ajuste de la dosis en relación a la edad.Uso en pacientes con insuficiencia renal.No es necesario ajustar la dosis en pacientes con insuficiencia renal leve a moderada. En pacientes con insuficiencia renal moderada (aclaramiento de creatinina <60 ml/min) la dosis de inicio recomendada es de 5 mg. La dosis de 40 mg está contraindicada en pacientes con insuficiencia renal moderada. En pacientes con insuficiencia renal grave el uso de Crestor está contraindicado a cualquier dosis (ver sección “Contraindicaciones” y sección “Propiedades farmacocinéticas”). Uso
en pacientes con insuficiencia hepática No hubo aumento de la exposición sistémica a la rosuvastatina en pacientes con puntuaciones de 7 o menos en la escala de Child-Pugh. Sin embargo, sí se ha observado un aumento de la exposición sistémica en pacientes con puntuación de 8 y 9 en la escala de Child-Pugh (ver sección “Propiedades farmacocinéti-cas”). En estos pacientes debe considerarse la realización de una evaluación de la función renal (ver sección “Advertencias y precauciones especiales de empleo”). No existe experiencia en sujetos con valores de Child-Pugh superiores a 9. Crestor está contraindicado en pacientes con enfermedad hepática activa (ver sección “Contraindicaciones”).RazaSe
ha observado una exposición sistémica aumentada en pacientes de origen asiático (ver sección “Contraindicaciones”, sección “Advertencias y precauciones especiales de empleo” y sección “Propiedades farmacocinéticas”). En pacientes de origen asiático, la dosis de inicio recomendada es de 5 mg. La dosis de 40 mg está contraindicada en estos pacientes. Uso en pacientes con factores de predisposición a la miopatíaEn pacientes con factores de predisposición a la miopatía, la dosis de inicio recomendada es de 5 mg (ver sección “Advertencias y precauciones especiales de empleo”). La dosis de 40 mg está contraindicada en algunos de estos pacientes (ver sección
“Contraindicaciones”).ContraindicacionesCrestor está contraindicado:-en pacientes con hipersensibilidad a la rosuvastatina o a alguno de los excipientes.-en pacientes con enfermedad hepática activa incluyendo elevaciones persistentes, injustificadas de las transaminasas séricas y cualquier aumento de las transaminasas séricas que supere tres veces el límite superior normal (LSN).-en pacientes con insuficiencia renal grave (aclaramiento de creatinina <30 ml/min).-en pacientes con miopatía.-en pacientes con tratamiento concomitante con ciclosporina.-durante el embarazo y lactancia y en mujeres en edad fértil que no estén empleando métodos anticonceptivos apropiados.La dosis de 40 mg está
contraindicada en pacientes con factores de predisposición a la miopatía/rabdomiolisis. Dichos factores incluyen:-insuficiencia renal moderada (aclaramiento de creatinina <60 ml/min)-hipotiroidismo-historial personal o familiar de alteraciones musculares hereditarias-historial previo de toxicidad muscular con otro inhibidor de la HMG-CoA reductasa o fibrato-alcoholismo -situaciones en las que puedan darse aumentos de los niveles plasmáticos -pacientes de origen asiático -uso concomitante de fibratos.(Ver secciones “Advertencias y precauciones especiales de empleo”, “Interacción con otros medicamentos y otras formas de interacción” y “Propiedades farmacocinéticas”)Advertencias y precauciones
especiales de empleoEfectos renalesSe ha observado proteinuria, detectada mediante tira reactiva y principalmente de origen tubular, en pacientes tratados con dosis altas de Crestor, en particular 40 mg, en los que fue transitoria o intermitente en la mayoría de los casos. No se ha demostrado que la proteinuria sea indicativa de enfermedad renal aguda o progresiva (ver sección “Reacciones adversas”). La frecuencia de notificación de acontecimientos renales graves en el uso pos t-comercialización es mayor con la dosis de 40 mg. Debe considerarse realizar una evaluación de la función renal durante el seguimiento rutinario de pacientes que estén siendo tratados con dosis de 40 mg.Efectos musculoesqueléti-
cos.En pacientes tratados con Crestor se han registrado efectos sobre el músculo esquelético, por ej. mialgia, miopatía y, raramente, rabdomiolisis con todas las dosis, especialmente con dosis superiores a 20 mg. Se han registrado casos muy raros de rabdomiolisis con el uso de ezetimiba en combinación con inhibidores de la HMG-CoA reductasa. No se puede descartar una interacción farmacodinámica (ver sección “Interacción con otros medicamentos y otras formas de interacción”) y se debe tener cuidado con el uso concomitante. Al igual que con otros inhibidores de la HMG-CoA reductasa, la frecuencia de notificaciones de rabdomiolisis asociada a Crestor durante el uso post-comercialización es mayor con la
dosis de 40 mg.Medida de la Creatina cinasa. No deben medirse los niveles de creatina cinasa (CK) después de la realización de ejercicio intenso o en presencia de una posible causa alternativa del aumento de CK que pueda influir en la interpretación de los resultados. Si los valores iniciales de CK son significativamente elevados (>5xLSN) se deberá realizar de nuevo el ensayo al cabo de 5-7 días para confirmar los resultados. Si el nuevo ensayo confirma los valores iniciales de CK >5xLSN, no se deberá iniciar el tratamien-to.Antes de iniciar el tratamient. Al igual que otros inhibidores de la HMG-CoA reductasa, Crestor debe prescribirse con precaución a pacientes con factores de predisposición a
rabdomiolisis, tales como: insuficiencia renal, hipotiroidismo, historial personal o familiar de alteraciones musculares hereditarias, historial de toxicidad muscular previa con otro inhibidor de la HMG-CoA reductasa o fibrato, alcoholismo, edad > 70 años, situaciones en las que pueda producirse un aumento de los niveles plasmáticos (ver sección “Propiedades farmacocinéticas”), uso concomitante de fibratos. En dichos pacientes el riesgo del tratamiento debe considerarse en relación al posible beneficio del tratamiento y se recomienda la realización de una monitorización clínica. Si los valores iniciales de CK son significativamente elevados (>5xLSN) no se deberá iniciar el tratamiento. Durante el tratamientoDebe
pedirse a los pacientes que comuniquen inmediatamente cualquier dolor muscular, debilidad o calambres injustificados, en particular si están asociados a malestar o fiebre. Deben medirse los niveles de CK en estos pacientes. En el caso de que los niveles de CK sean notablemente elevados (>5xLSN) o si los síntomas musculares son graves y provocan malestar diario (incluso si los niveles de CK son ≤ 5xLSN), debe interrumpirse el tratamiento. Si los síntomas remiten y los niveles de CK vuelven a la normalidad, entonces puede considerarse el re-establecimiento del tratamiento con Crestor o un inhibidor de la HMG-CoA reductasa alternativo a la dosis mínima y bajo una estrecha monitorización. La
monitorización rutinaria de los niveles de CK en pacientes asintomáticos no está justificada. Se han notificado casos muy raros de una miopatía necrotizante mediada por el sistema inmune clínicamente caracterizada por una debilidad muscular proximal persistente y unos niveles elevados de creatina cinasa sérica durante el tratamiento o tras la interrupción de rosuvastatina. Pueden ser necesarios análisis serológicos y neuromusculares adicionales. Puede requerirse un tratamiento con agentes inmunosupresores. En los ensayos clínicos no hubo evidencia de un aumento de los efectos musculoesqueléticos en el reducido número de pacientes tratados con Crestor y tratamiento concomitante. Sin embargo, se ha
observado un aumento de la incidencia de miositis y miopatía en pacientes que reciben otros inhibidores de la HMG-CoA reductasa junto con derivados del ácido fíbrico incluido gemfibrozilo, ciclosporina, ácido nicotínico, antifúngicos tipo azol, inhibidores de la proteasa y antibióticos macrólidos. El gemfibrozilo aumenta el riesgo de miopatía cuando se administra de forma concomitante con algunos inhibidores de la HMG-CoA reductasa. Por lo tanto, no se recomienda la combinación de Crestor y gemfibrozilo. El beneficio de alteraciones adicionales en los niveles lipídicos por el uso concomitante de Crestor con fibratos o niacina se debe sopesar cuidadosamente frente a los riesgos potenciales de tales
combinaciones. La dosis de 40 mg está contraindicada con el uso concomitante de un fibrato (ver sección “Interacción con otros medicamentos y otras formas de interacción” y sección “Reacciones adversas”). No debe emplearse Crestor en pacientes con trastornos agudos graves sugerentes de miopatía o que predispongan al desarrollo de insuficiencia renal secundaria a rabdomiolisis (p.ej. sepsis, hipotensión, intervención quirúrgica mayor, trauma, trastornos metabólicos, endocrinos o electrolíticos graves o convulsiones no controladas). Efectos hepáticos Al igual que otros inhibidores de la HMG-CoA reductasa, Crestor debe usarse con precaución en pacientes que ingieran cantidades excesivas de
alcohol y/o presenten un historial de enfermedad hepática. Se recomienda la realización de pruebas hepáticas antes del inicio del tratamiento y 3 meses después de iniciado el tratamiento con Crestor. Si el nivel de transaminasas séricas sobrepasa 3 veces el límite superior normal se deberá interrumpir el tratamiento con Crestor o reducirse la dosis. La frecuencia de notificaciones de acontecimientos hepáticos graves (que consisten principalmente en un aumento de las transaminasas séricas) durante el uso post-comercialización es mayor con la dosis de 40 mg. En pacientes con hipercolesterolemia secundaria provocada por hipotiroidismo o síndrome nefrótico, la enfermedad subyacente debe ser tratada antes
de iniciar el tratamiento con Crestor. RazaLos estudios farmacocinéticos muestran un aumento de la exposición en pacientes de origen asiático en comparación con los pacientes caucásicos (ver sección “Posología y forma de administración”, sección “Contraindicaciones” y sección “Propiedades farmacocinéticas”). Inhibidores de la proteasa No se recomienda el uso concomitante con inhibidores de la proteasa (ver sección “Interacción con otros medicamentos y otras formas de interacción”). Intolerancia a lactosa Los pacientes con intolerancia hereditaria a galactosa, insuficiencia de lactasa de Lapp (insuficiencia observada en ciertas poblaciones de Laponia) o malabsorción de glucosa o galactosa
no deben tomar este medicamento. Enfermedad pulmonar intersticial Se han registrado casos excepcionales de enfermedad pulmonar intersticial con algunas estatinas, especialmen-te con tratamientos a largo plazo (ver sección “Reacciones adversas”). Los principales signos que se presentan pueden incluir disnea, tos no productiva y deterioro del estado general de salud (fatiga, pérdida de peso y fiebre). Si se sospecha que un paciente ha desarrollado enfermedad pulmonar intersticial, debe interrumpirse el tratamiento con estatinas.Diabetes Mellitus Algunas evidencias sugieren que las estatinas como clase, elevan la glucosa en sangre y en algunos pacientes, con alto riesgo de diabetes en un futuro, pueden producir un
nivel de hiperglicemia para el cual un cuidado convencional de la diabetes es apropiado. Este riesgo, sin embargo, está compensado con la reducción del riesgo vascular con las estatinas y por tanto no debería ser una razón para abandonar el tratamiento con estatinas. Los pacientes con riesgo (glucosa en ayunas de 5,6 a 6,9 mmol/L, IMC>30kg/m2, triglicéridos elevados, hipertensión) deberían ser controlados clínica y bioquímicamente de acuerdo con las directrices nacionales. En el estudio JUPITER, la frecuencia global notificada de la diabetes mellitus fue un 2,8% en rosuvastatina y un 2,3% en placebo, principalmente en pacientes con un nivel de glucosa en ayunas de 5,6 a 6,9 mmol/L. Población
pediátrica El estudio del crecimiento lineal (altura), peso, IMC (índice de masa corporal) y las características secundarias de la madurez sexual, según los estadios de Tanner en población pediátrica de 10 a 17 años de edad tratados con rosuvastatina está limitado a un periodo de un año. En un estudio de 52 semanas de duración, no se detectó ningún efecto sobre el crecimiento, peso, IMC ni madurez sexual (ver sección “Propiedades farmacodinámicas”). La experiencia en ensayos clínicos en niños y adolescentes es limitada y se desconocen los efectos de la rosuvastatina a largo plazo (> 1 año) sobre la pubertad. En un ensayo clínico aleatorizado, de niños y adolescentes a los que se les administró
rosuvastatina durante 52 semanas, se observó un incremento de CK > 10x LSN y aumento de los síntomas musculares tras el ejercicio o actividad física, más frecuentemente en comparación con los datos observados en los ensayos clínicos realizados en adultos (ver sección “Reacciones adversas”). Interacción con otros medicamentos y otras formas de interacción Ciclosporina: durante el tratamiento concomitante con Crestor y ciclosporina, los valores del AUC de rosuvastatina fueron, como media, 7 veces superiores a los observados en individuos sanos (ver sección “Contraindicaciones”). La administración concomitante de Crestor y ciclosporina no afectó a los niveles plasmáticos de la ciclosporina.
Antagonistas de la vitamina K: como con otros inhibidores de la HMG-CoA reductasa, el inicio del tratamiento o la escalada de la dosis con Crestor en pacientes tratados de forma concomitante con antagonistas de la vitamina K (p.ej. warfarina u otros anticoagulantes cumarínicos) puede dar lugar a incrementos del Indice Normalizado Internacional (INR). La interrupción del tratamiento o la disminución de la dosis de Crestor pueden resultar en una disminución del INR. En tales casos, es recomendable llevar a cabo una monitorización adecuada del INR. Gemfibrozilo y otros medicamentos reductores del colesterol: la administración concomitante de Crestor y gemfibrozilo duplicó la Cmax y el AUC de la
rosuvastatina (ver sección “Advertencias y precauciones especiales de empleo”). De acuerdo con los resultados de los estudios de interacción específica no se espera ninguna interacción farmacocinética significativa con el fenofibrato, sin embargo, sí podría darse una interacción farmacodinámica. El gemfibrozilo, fenofibrato, otros fibratos y dosis hipolipemiantes (mayores o iguales a 1g/día) de niacina (ácido nicotínico), aumentan el riesgo de miopatía cuando se administran de forma concomitante con inhibidores de la HMG-CoA reductasa, probablemente debido a que pueden provocar miopatía cuando se administran solos. La dosis de 40 mg está contraindicada con el uso concomitante con fibratos (ver
sección “Contraindicaciones” y sección “Advertencias y precauciones especiales de empleo”). Estos pacientes deberán iniciar también el tratamiento con una dosis de 5 mg. Ezetimiba: el uso concomitante de Crestor con ezetimiba no provocó cambios en el AUC ni en la Cmax de ninguno de los dos fármacos. Sin embargo, no se puede descartar una interacción farmacodinámica, en términos de reacciones adversas, entre Crestor y ezetimiba (ver sección “Advertencias y precauciones especiales de empleo”). Inhibidores de la proteasa: aunque se desconoce el mecanismo exacto de interacción, el uso concomitante de los inhibidores de la proteasa puede aumentar de manera importante la exposición a la
rosuvastatina. En un estudio farmacocinético, la administración concomitante de 20 mg de rosuvastatina y un medicamento compuesto por la combinación de dos inhibidores de la proteasa (400 mg de lopinavir/ 100 mg de ritonavir) en individuos sanos se asoció con un aumento de aproximadamente dos y cinco veces respectivamente en el AUC(0-24) y la Cmax en el estado de equilibrio. Por lo tanto, no se recomienda el uso concomitante de rosuvastatina en pacientes con VIH tratados con inhibidores de la proteasa (ver sección “Advertencias y precauciones especiales de empleo”). Antiácidos: la administración concomitante de Crestor con una suspensión antiácida a base de hidróxido de aluminio y
magnesio, originó una disminución de la concentración plasmática de la rosuvastatina de 50% aproximadamente. Este efecto se vio mitigado cuando se administró el antiácido 2 horas después de la administración de Crestor. No se ha establecido la importancia clínica de esta interacción. Eritromicina: el uso concomitante de Crestor y eritromicina originó una disminución del 20% del AUC(0-t) y una disminución del 30% de la Cmax de la rosuvastatina. Esta interacción puede estar causada por un incremento en la motilidad intestinal provocada por la eritromicina. Anticonceptivos orales/terapia hormonal sustitutiva (THS): la administración conjunta de Crestor y un anticonceptivo oral originó un incremento del
AUC de etinilestradiol y norgestrel del 26% y 34%, respectivamente. Deben tenerse en cuenta estos aumentos de los niveles plasmáticos a la hora de establecer la dosis del anticonceptivo oral. No hay datos farmacocinéticos disponibles de pacientes con tratamiento concomitante de Crestor y THS y, por lo tanto, no se puede descartar un efecto similar. Sin embargo, durante los ensayos clínicos, esta combinación fue empleada ampliamente por mujeres y fue bien tolerada. Otros medicamentos: de acuerdo a los resultados de estudios específicos de interacción no se esperan interacciones importantes con la digoxina. Enzimas del citocromo P450: los resultados de los estudios in vitro e in vivo muestran
que la rosuvastatina no es ni un inhibidor ni un inductor de las isoenzimas del citocromo P450. Además, la rosuvastatina es un sustrato con poca afinidad para estas isoenzimas. No se han observado interacciones clínicamente importantes entre la rosuvastatina y el fluconazol (un inhibidor CYP2C9 y CYP3A4) ni el ketoconazol (un inhibidor de CYP2A6 y CYP3A4). La administración concomitante de itraconazol (un inhibidor de la CYP3A4) y rosuvastatina originó un incremento del 28% del AUC de la rosuvastatina. Este pequeño aumento no se considera clínicamente significativo. Por lo tanto, no se esperan interacciones medicamentosas debidas al metabolismo mediado por el citocromo P450. Embarazo y
lactancia Crestor está contraindicado durante el embarazo y la lactancia.Las mujeres en edad fértil deben emplear medidas anticonceptivas adecuadas. Debido a que el colesterol y otros productos de la biosíntesis del colesterol son esenciales para el desarrollo del feto, el riesgo potencial de la inhibición de la HMG-CoA reductasa sobrepasa las ventajas del tratamiento durante el embarazo. Los estudios en animales proporcionan una evidencia limitada de la toxicidad reproductiva (ver sección “Datos preclínicos sobre seguridad”). Si una paciente queda embarazada durante el tratamiento con este medicamento, deberá interrumpirse el tratamiento inmediatamente. La rosuvastatina se excreta en la leche de ratas. No
existen datos respecto a la excreción en la leche humana (ver sección “Contraindicaciones”). Efectos sobre la capacidad para conducir y utilizar máquinas No se han llevado a cabo estudios para determinar el efecto de Crestor sobre la capacidad de conducir o utilizar máquinas. Sin embargo, de acuerdo a sus propiedades armacodinámicas, no es probable que Crestor afecte esta capacidad. Cuando se conduzcan vehículos o se utilice maquinaria, debe tenerse en cuenta la posibilidad de mareos durante el tratamiento. Reacciones adversas Las reacciones adversas observadas con Crestor son generalmente de carácter leve y
transitorio. En ensayos clínicos controlados menos del 4% de los pacientes tratados con Crestor abandonaron el estudio debido a las reacciones adversas. Los acontecimientos adversos se han clasificado en función de su frecuencia en: Frecuentes (>1/100, <1/10); Poco frecuentes (>1/1.000, <1/100); Raros (>1/10.000, <1/1.000); Muy raros (<1/10.000); Frecuencia no conocida (no puede estimarse a partir de los datos disponibles).Trastornos del sistema inmunológico.Raros: reacciones de hipersensibilidad incluyendo angioedema. Trastornos endocrino.Frecuentes: diabetes1 .Trastornos del sistema nervioso. Frecuentes: cefalea, mareos. Trastornos gastrointestinales. Frecuentes: estreñimiento, náuseas, dolor
abdominal. Raros: pancreatitis. Trastornos de la piel y del tejido subcutáneo. Raros: prurito, rash y urticaria. Trastornos musculoesqueléticos y del tejido conjuntivo Frecuente: mialgia. Raros: miopatía (incluyendo miositis) y rabdomiolisis. Trastornos generale. Frecuentes: astenia. 1 La frecuencia dependerá de la presencia o ausencia de factores de riesgo (glucosa en sangre en ayunas ≥ 5.6 mmol/L, BMI>30kg/m2, triglicéridos elevados, historia de hipertensión). Como con otros inhibidores de la HMG-CoA reductasa, la incidencia de reacciones adversas al medicamento tiende a ser dosis-dependiente. Efectos renales: se ha observado proteinuria, detectada mediante tira reactiva y principalmente de origen tubular, en
pacientes tratados con Crestor. Se observaron cambios en la proteinuria desde nada o trazas hasta un resultado ++ o superior en <1% de los pacientes en algún momento del tratamiento con 10 y 20 mg y aproximadamente en el 3% de los pacientes tratados con 40 mg. Con la dosis de 20 mg se observó un pequeño incremento en el cambio desde nada o
trazas a +. En la mayoría de los casos, la proteinuria disminuye o desaparece de forma espontánea al continuar con el tratamiento, y no se ha demostrado que sea indicativa de enfermedad renal aguda o progresiva. Se ha observado hematuria en pacientes tratados con Crestor y los datos clínicos muestran que la frecuencia de aparición es baja.Efectos sobre el músculo esquelético: se han registrado efectos sobre el músculo esquelético, por ej. mialgia, miopatía (incluyendo miositis) y, muy raramente, rabdomiolisis con o sin fallo renal agudo con todas las dosis, en pacientes tratados con todas las dosis de Crestor y especialmente con dosis superiores a 20 mg. Se ha observado un incremento dosis-
dependiente de los niveles de CK en pacientes tratados con rosuvastatina, siendo la mayoría de los casos leves, asintomáticos y transitorios. Si los niveles de CK son elevados (>5xLSN), se deberá interrumpir el tratamiento (ver sección “Advertencias y precauciones especiales de empleo”). Efectos hepáticos: como con otros inhibidores de la HMG-CoA reductasa, se ha observado un incremento dosis-dependiente de las transaminasas en un reducido número de pacientes tratados con rosuvastatina; la mayoría de los casos fueron leves, asintomáticos y transitorios. Experiencia post-comercialización: Además de lo mencionado anteriormente, se han registrado las siguientes reacciones adversas durante la
experiencia post-comercialización con Crestor: Trastornos del sistema nervioso: Muy raros: polinefropatía, pérdida de memoria. Trastornos respiratorios, torácicos y mediastínicos: Frecuencia no conocida: tos, disnea. Trastornos gastrointestinales: Frecuencia no conocida: diarrea. Trastornos hepatobiliares: Muy raros: ictericia, hepatitis; Raros: aumento de las transaminasas hepáticas. Trastornos de la piel y del tejido subcutáneo: Frecuencia no conocida: síndrome de Stevens-Johnson. Trastornos musculoesqueléticos: Muy raros: artralgia. Frecuencia no conocida: miopatía necrotizante mediada por el sistema inmune. Trastornos renales: Muy raros: hematuria. Trastornos del aparato reproductor y de la mama: Muy
raros: ginecomastia. Trastornos generales y alteraciones en el lugar de administración: Frecuencia no conocida: edema. Las siguientes reacciones adversas han sido registradas con algunas estatinas: Depresión. Alteraciones del sueño, incluyendo insomnio y pesadillas. Disfunción sexual. Casos excepcionales de enfermedad pulmonar intersticial, especialmente en tratamientos a largo plazo (ver sección “Advertencias y precauciones especiales de empleo”). Alteraciones en los tendones, a veces agravadas por rotura. La frecuencia de notificaciones de rabdomiolisis, acontecimientos renales graves y acontecimientos hepáticos graves (que consisten principalmente en el aumento de las transaminasas hepáticas) es
mayor con la dosis de 40 mg. Población pediátrica: En un ensayo clínico de 52 semanas de duración de tratamiento, realizado en niños y adolescentes, se observó un incremento en los niveles de creatina cinasa >10xLSN y aumento de los síntomas musculares después del ejercicio o actividad física, con mayor frecuencia en comparación con los datos de seguridad observados en los ensayos clínicos en adultos (ver sección “Advertencias y precauciones especiales de empleo”). En otros aspectos, el perfil de seguridad de la rosuvastatina fue similar en niños y adolescentes en comparación con adultos.Sobredosis
No existe un tratamiento específico en caso de sobredosis. Si se produce una sobredosis, debe tratarse al paciente sintomáticamente e instaurar medidas de soporte, según sea necesario. Deben monitorizarse la función hepática y los niveles de CK. No es probable que la hemodiálisis proporcione algún beneficio. PROPIEDADES FARMACOLÓGICAS Propiedades farmacodinámicas. Grupo farmacoterapéutico: Inhibidores de la HMG-CoA reductasa Código ATC: C10A A07. Mecanismo de acciónLa rosuvastatina es un inhibidor competitivo y selectivo de la HMG-CoA reductasa, la enzima limitante que convierte la 3-hidroxi-3-metilglutaril coenzima A en mevalonato, un precursor del colesterol. El
principal lugar de acción de la rosuvastatina es el hígado, el órgano diana para la disminución de los niveles de colesterol.La rosuvastatina aumenta el número de receptores LDL
hepáticos en la superficie celular, aumentando la absorción y el catabolismo de LDL e inhibe la síntesis hepática de VLDL, reduciendo así el número total de partículas VLDL y LDL.Efectos farmacodinámicos
Crestor reduce los niveles elevados de colesterol-LDL, colesterol total y triglicéridos e incrementa el colesterol-HDL. También disminuye los valores de ApoB, C-noHDL, C-VLDL, TG-VLDL e incrementa los valores de ApoA1 (ver Tabla 1). Crestor también disminuye los cocientes de C-LDL/C-HDL, C-total/C-HDL, C-noHDL/C-HDL y ApoB/ApoA1.Tabla 1Dosis-respuesta en pacientes con hipercolesterolemia primaria (tipo IIa y IIb) (porcentaje medio de cambio ajustado por el valor basal) El efecto terapéutico se obtiene 1 semana después del inicio del tratamiento y el 90% de la respuesta máxima se alcanza a las 2 semanas. La respuesta máxima se alcanza
generalmente a las 4 semanas de tratamiento y se mantiene a partir de ese momento.Eficacia clínicaCrestor es eficaz en pacientes adultos con hipercolesterolemia, con o sin hipertrigliceridemia, independientemente de la raza, sexo o edad, y en poblaciones especiales de pacientes tales como diabéticos o pacientes con hipercolesterolemia familiar.Los datos combinados de la fase III han mostrado que el tratamiento con Crestor es eficaz en alcanzar los objetivos determinados por la guía de la Sociedad Europea de Aterosclerosis (EAS; 1998) en la mayoría de los pacientes con hipercolesterolemia tipo IIa y IIb (valor inicial medio de C-LDL aproximadamente 4,8 mmol/l); aproximadamente un 80% de los
pacientes tratados con 10 mg alcanzaron los niveles objetivo de la EAS de C-LDL (< 3 mmol/l).En un amplio estudio, 435 pacientes con hipercolesterolemia familiar heterocigótica recibieron desde 20 mg a 80 mg de Crestor según un diseño de escalada de dosis. Todas las dosis mostraron un efecto beneficioso sobre los parámetros lipídicos y en la obtención de los objetivos de tratamiento. Tras la escalada de dosis hasta 40 mg al día (12 semanas de tratamiento), los niveles de C-LDL disminuyeron en un 53%. Un 33% de los pacientes alcanzó los niveles de C-LDL (<3mmol/l) establecidos por la guía de la EAS. En un ensayo abierto de escalada de dosis, se evaluó la respuesta de 42 pacientes con hipercolesterole-
mia familiar homocigótica a Crestor 20 y 40 mg. En la población global del estudio, la reducción media de C-LDL fue del 22%. En estudios clínicos realizados con un número limitado de pacientes se ha demostrado que Crestor tiene una eficacia aditiva en la disminución de los triglicéridos cuando se emplea en combinación con fenofibrato, y en el aumento de los niveles de C-HDL cuando se emplea en combinación con niacina (ver sección “Advertencias y precauciones especiales de empleo”). En un estudio clínico multicéntrico, doble-ciego, controlado con placebo (METEOR), 984 pacientes entre 45 y 70 años de edad y con bajo riesgo de enfermedad coronaria (definido como riesgo Framingham <10% en 10 años), con
un C-LDL medio de 4,0 mmol/l (154,5 mg/dl), pero con aterosclerosis subclínica medida por el grosor de la capa íntima-media carotídea (CIMT: “Carotid Intima Media Thickness”) se aleatorizaron a 40 mg de rosuvastatina una vez al día o placebo durante 2 años. La rosuvastatina disminuyó significativamente la velocidad de progresión del CIMT medido en 12 localizaciones carotídeas en comparación con placebo en -0,0145 mm/año [intervalo de confianza al 95%: -0,0196, -0,0093; p<0,0001]. El cambio desde el valor basal fue de -0,0014 mm/año (-0,12%/año; p no significativa) para rosuvastatina en comparación con el aumento de +0,0131 mm/año (+1,12%/año; p<0.0001) en el grupo placebo. Aún no se ha
demostrado ninguna correlación directa entre la disminución del CIMT y la reducción del riesgo de acontecimientos cardiovasculares. La población estudiada en el estudio METEOR presentaba bajo riesgo de enfermedad cardiaca coronaria y no representa la población diana de Crestor 40 mg. La dosis de 40 mg sólo debe ser prescrita en pacientes con hipercolesterolemia grave con elevado riesgo cardiovascular (ver sección “Posología y forma de administración”). En el estudio denominado “Justification for the Use of Statins in Primary Prevention: An Intervention Trial Evaluation Rosuvastatin (JUPITER)” (Justificación del Uso de Estatinas en la Prevención Primaria: Un Ensayo de Intervención para Evaluar
Rosuvastatina), se evaluó el efecto de la rosuvastatina sobre la aparición de eventos cardiovasculares ateroescleróticos mayores en 17.802 hombres (≥50 años de edad) y mujeres (≥60 años de edad). Los participantes del estudio fueron asignados de forma aleatoria a placebo (n=8.901) o rosuvastatina 20 mg una vez al día (n=8.901) y se les realizó un seguimiento de duración media de 2 años. La concentración de colesterol LDL disminuyó un 45% (p<0,001) en el grupo con rosuvastatina en comparación con el grupo placebo. En un análisis a posteriori de un subgrupo de sujetos de alto riesgo con un riesgo inicial > 20% en la escala de Framingham (1.558 sujetos), hubo una reducción significativa en la
variable combinada de muerte cardiovascular, ictus e infarto de miocardio (p=0,028) en el tratamiento con rosuvastatina frente a placebo. La reducción absoluta del riesgo en la tasa de eventos fue de 8,8 por cada 1.000 pacientes-año. La mortalidad total no se vio afectada en este grupo de alto riesgo (p=0,193). En un análisis a posteriori del subgrupo de sujetos de alto riesgo (un total de 9.302 pacientes) con un riesgo inicial en la escala SCORE de ≥5% (extrapolado para incluir pacientes mayores de 65 años de edad) hubo una reducción significativa en la variable combinada de muerte cardiovascular, ictus e infarto de miocardio (p=0,0003) en el tratamiento con rosuvastatina frente a placebo. La reducción absoluta del
riesgo en la tasa de acontecimientos fue de 5,1 por cada 1.000 pacientes-año. La mortalidad total no se vio afectada en este grupo de alto riesgo (p=0,076). En el ensayo JUPITER un 6,6% de los pacientes con rosuvastatina y un 6,2% de los pacientes con placebo interrumpieron el uso de la medicación del estudio debido a una reacción adversa. Las reacciones adversas más comunes que llevaron a una interrupción del tratamiento fueron: mialgia (0,3% rosuvastatina, 0,2% placebo), dolor abdominal (0,03% rosuvastatina, 0,02% placebo) y erupción cutánea (0,02% rosuvastatina, 0,03% placebo). Las reacciones adversas más comunes con una frecuencia mayor o igual a placebo fueron infección del tracto urinario (8,7%
rosuvastatina, 8,6% placebo), nasofaringitis (7,6% rosuvastatina, 7,2% placebo), dolor de espalda (7,6% rosuvastatina, 6,9% placebo) y mialgia (7,6% rosuvastatina, 6,6% placebo). Población pediátrica En un estudio, doble ciego, aleatorizado, multicéntrico y controlado con placebo de 12 semanas (n=176, 97 niños y 79 niñas), seguido de una fase abierta de 40 semanas de titulación de dosis de rosuvastatina (n=173, 96 niños y 77 niñas), los pacientes de entre 10-17 años de edad (estadio de Tanner II-V y adolescentes del sexo femenino al menos 1 año después de la menarquia) con hipercolesterolemia familiar heterocigótica, recibieron 5, 10 ó 20 mg de rosuvastatina o placebo diariamente, durante 12 semanas y
posteriormente, todos recibieron rosuvastatina diariamente durante 40 semanas. En el estudio, aproximadamente el 30% de los pacientes tenían entre 10-13 años de edad y aproximadamente el 17%, 18%, 40% y 25% estaban en los estadios de Tanner II, III, IV y V respectivamente. El C-LDL disminuyó un 38,3%, un 44,6% y un 50,0% con rosuvastatina 5, 10, y 20 mg, respectivamente, en comparación con un 0,7% con placebo. Al final de la fase abierta de 40 semanas de titulación de dosis hasta un máximo de 20 mg una vez al día, 70 de los 173 pacientes (40,5%) habían alcanzado el objetivo de C-LDL < 2,8 mmol/l. Tras 52 semanas de tratamiento de estudio, no se detectó ningún efecto sobre el crecimiento,
peso, IMC ni madurez sexual (sección “Advertencias y precauciones especiales de empleo”). La experiencia en ensayos clínicos con niños y adolescentes es limitada y se desconocen los efectos a largo plazo de la rosuvastatina (> 1 año) sobre la pubertad. Este ensayo (n=176) no era adecuado para realizar una comparación de acontecimientos adversos del medicamento. Propiedades farmacocinéticas Absorción: las concentraciones plasmáticas máximas de rosuvastatina se alcanzan aproximadamente 5 horas después de la administración ora l. La
biodisponibilidad absoluta es de aproximadamente un 20%. Distribución: la rosuvastatina es extensamente absorbida por el hígado, principal lugar de síntesis del colesterol y de aclaramiento del C-LDL. El volumen de distribución de la rosuvastatina es de aproximadamente 134 l. La rosuvastatina se une a proteínas plasmáticas aproximadamente en un 90%, principalmente a la albúmina. Metabolismo: la rosuvastatina se metaboliza de forma limitada (aproximadamente un 10%). Estudios in vitro de metabolismo realizados en hepatocitos humanos indican que la rosuvastatina no es un
buen sustrato del metabolismo mediado por el citocromo P450. La principal isoenzima implicada es la CYP2C9, y en menor medida la 2C19, 3A4 y la 2D6. Los principales metabolitos identificados son el N-desmetilado y el lactónico. El metabolito N-desmetilado es aproximadamente un 50% menos activo que la rosuvastatina, mientras que el lactónico se considera clínicamente inactivo. Más de un 90% de la actividad de inhibición de la HMG-Co A reductasa circulante se atribuye a la rosuvastatina.Excreción: aproximadamente un 90% de la rosuvastatina se excreta inalterada en las heces (incluyendo el principio activo absorbido y no absorbido) y el resto se excreta en orina. Aproximadamente el 5% se excreta inalterado
en la orina. La semivida de eliminación plasmática es de aproximadamente 19 horas. La semivida de eliminación no aumenta al incrementar la dosis. La media geométrica del aclaramiento plasmático es aproximadamente 50 litros/hora (coeficiente de variación 21,7%). Como con otros inhibidores de la HMG-CoA reductasa, el transportador de membrana OATP-C está implicado en la absorción hepática de la rosuvastatina. Este transportador es importante en la eliminación hepática de la rosuvastatina.Linealidad: la exposición sistémica a la rosuvastatina aumenta de forma proporcional a la dosis. No hay cambios en los parámetros farmacocinéticos después de la administración de dosis diarias repeti-
das.Poblaciones especiales:Edad y sexo: la edad y el sexo no afectan de forma clínicamente significativa a la farmacocinética de la rosuvastatina en adultos. Los datos de farmacocinética de rosuvastatina en niños y adolescentes con hipercolesterolemia familiar heterocigótica fueron similares a los de los voluntarios adultos (ver “Población pediátrica” más adelante).Raza: los estudios farmacocinéticos muestran un aumento de aproximadamente el doble en el AUC medio y en la Cmax en pacientes de origen asiático (japoneses, chinos, vietnamitas y coreanos), en comparación con los pacientes de origen caucásicos. Los pacientes indo-asiáticos presentan un aumento de 1,3 veces en el AUC medio y la Cmax.
Un análisis farmacocinético de la población no mostró ninguna diferencia clínicamente significativa en la farmacocinética entre pacientes de raza blanca y de raza negra.Insuficiencia renal: en un estudio llevado a cabo en pacientes con distintos grados de insuficiencia renal, la enfermedad renal leve a moderada no afectó a las concentraciones plasmáticas de rosuvastatina ni de su metabolito N-desmetilado. Los pacientes con insuficiencia renal grave (CrCl < 30 ml/min) presentaron un incremento de las concentraciones plasmáticas tres veces mayor y un incremento de la concentración de metabolito N-desmetilado nueve veces mayor que el de los voluntarios sanos. Las concentraciones plasmáticas de rosuvastatina
en el estado de equilibrio en pacientes sometidos a hemodiálisis fueron aproximadamente un 50% más elevadas en comparación con voluntarios sanos.Insuficiencia hepática: en un estudio llevado a cabo con pacientes con diversos grados de insuficiencia hepática no existió evidencia de un aumento de la exposición a la rosuvastatina, en pacientes con puntuación Child-Pugh de 7 o menos. Sin embargo, dos pacientes con puntuaciones Child-Pugh de 8 y 9 presentaron un aumento de la exposición sistémica de casi dos veces la de los pacientes con valores más bajos de Child-Pugh. No existe experiencia con pacientes con puntuaciones Child-Pugh superiores a 9.Población pediátrica: Los parámetros
farmacocinéticos en pacientes pediátricos con hipercolesterolemia familiar heterocigótica de edades comprendidas entre los 10 y 17 años no han sido descritos en su totalidad. Un estudio farmacocinético pequeño con rosuvastatina (administrada en forma de comprimidos) en 18 pacientes pediátricos, demostró que la exposición en pacientes pediátricos parece ser comparable a la exposición en pacientes adultos. Además, los resultados indican que no cabe esperar una gran desviación de la proporcionalidad de dosis. Datos preclínicos sobre seguridad. Los datos preclínicos muestran que de acuerdo con los estudios convencionales de seguridad farmacológica, genotoxicidad y potencial carcinogénico no existe un
riesgo especial en humanos. No se han evaluado ensayos específicos sobre los efectos en canales ERGh. Las reacciones adversas no descritas en estudios clínicos, pero observadas en animales a niveles de exposición similares a los niveles de exposición clínica fueron las siguientes: en los estudios de toxicidad de dosis repetidas se observaron cambios histopatológicos hepáticos en ratón y rata, probablemente debidos a la acción farmacológica de la rosuvastatina y, en menor medida, con efectos sobre la vesícula en perros, pero no en monos. Además, se observó toxicidad testicular en monos y perros a dosis más altas. La toxicidad reproductiva fue evidente en ratas y quedó demostrada por la
disminución de los tamaños de las camadas, del peso de la camada y de la supervivencia de las crías observados a dosis tóxicas para la madre, en las que los niveles de exposición sistémica fueron muy superiores a los niveles de exposición terapéutica. DATOS FARMACÉUTICOS Lista de excipientes Núcleo del comprimidoLactosa monohidrato.Celulosa microcristalina. Fosfato de calcio. Crospovidona. Estearato de magnesio. RecubrimientoLactosa monohidrato. Hipromelosa. Triacetato de glicerol. Dióxido de titanio (E171). Oxido de hierro, amarillo (E172) (Crestor 5 mg). Oxido de hierro, rojo (E172) (Crestor 10 mg y 20 mg). Incompatibilidades. No procede. Periodo de validez 3 años. Precauciones especiales
de conservación Blister: No conservar a temperatura superior a 30ºC. Conservar en el envase original. Frasco de HDPE: No conservar a temperatura superior a 30°C. Mantener el frasco perfectamente cerrado. Naturaleza y contenido del recipiente Blister de aluminio/aluminio de 7, 14, 15, 20, 28, 30, 42, 50, 56, 60, 84, 90, 98 y 100 comprimidos y frascos de HDPE de 30 y 100 comprimidos. Puede que no se comercialicen todos los tamaños de envases. Instrucciones de uso y manipulación
Ninguna especialTITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN AstraZeneca Farmacéutica Spain, S.A. C/ Serrano Galvache, 56 – Edificio Roble.Madrid 28033 NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN Crestor 5 mg comprimidos recubiertos con película: 70.334 Crestor 10 mg comprimidos recubiertos con película: 70.243 Crestor 20 mg comprimidos recubiertos con película: 70.244FECHA DE LA PRIMERA AUTORIZACIÓN/RENOVACIÓN DE LA AUTORIZACIÓNCrestor 5 mg comprimidos recubiertos con película: Enero 2009Crestor 10 y 20 mg comprimidos recubiertos con película: Noviembre 2008FECHA DE LA REVISIÓN DEL TEXTO
Julio 2012REGIMEN DE PRESCRIPCION Y DISPENSACIONSujeto a prescripción médica. Medicamento de dispensación renovable.PRESENTACIONES Y PRECIOSCrestor 5 mg comprimidos recubiertos con película: envase de 28 comprimidos, PVPiva: 18,90 €.Crestor 10 mg comprimidos recubiertos con película: envase de 28 comprimidos, PVPiva: 25,95 €; envase clínico de 100 comprimidos, PVPiva: 72,20 €.Crestor 20 mg comprimidos recubiertos con película: envase de 28 comprimidos, PVPiva: 38,92 €; envase clínico de 100 comprimidos, PVPiva: 108,31 €.Para mayor información, consultar la Ficha Técnica completa y/o dirigirse a AstraZeneca Farmacéutica Spain, S.A., Tfno: 900 200 444.FA
COMBI- completa 5, 10, 20/FT 12.07.2012 (Var II-050 Miopatía)/SmPC 09 July 2012
Dosis N C-LDL C-Total c-HDL TG C-noHDL ApoB ApoA1
Placebo 13 -7 -5 3 -3 -7 -3 0
5 17 -45 -33 13 -35 -44 -38 4
10 17 -52 -36 14 -10 -48 -42 4
20 17 -55 -40 8 -23 -51 -46 5
40 18 -63 -46 10 -28 -60 -54 0

NOMBRE DEL MEDICAMENTOBrilique 90 mg comprimidos recubiertos con película. COMPOSICIÓN CUALITATIVA Y CUANTITATIVACada comprimido contiene 90 mg de ticagrelor.Para consultar la lista completa de excipientes, ver sección Lista de excipientes. FORMA FARMACÉUTICAComprimido
recubierto con película (comprimido). Comprimidos redondos, biconvexos, de color amarillo, marcados con ‘90’ sobre una ‘T’ en una cara y lisos por la otra. DATOS CLÍNICOS. Indicaciones terapéuticasBrilique, administrado conjuntamente con ácido acetilsalicílico (AAS), está indicado para la prevención de acontecimientos aterotrombóticos en pacientes adultos con Síndromes Coronarios Agudos (angina inestable, infarto de miocardio sin elevación del segmento ST [IMSEST] o infarto de miocardio con elevación del segmento ST [IMCEST]), incluidos los pacientes controlados con tratamiento médico y los sometidos a
una intervención coronaria percutánea (ICP) o a un injerto de derivación de arteria coronaria (IDAC). Para más información ver sección Propiedades farmacodinámicas. Posología y forma de administraciónPosología.El tratamiento con Brilique debe iniciarse con una única dosis de carga de 180 mg (dos comprimidos de 90 mg), para continuar con 90 mg dos veces al día. Los pacientes tratados con Brilique deben tomar también AAS diariamente, a menos que esté expresamente contraindicado. Tras una dosis inicial de AAS, Brilique debe utilizarse con una dosis de mantenimiento de 75‑150 mg de AAS (ver sección
Propiedades farmacodinámicas ).Se recomienda continuar con el tratamiento hasta 12 meses a menos que la interrupción de Brilique esté clínicamente indicada (ver sección Propiedades farmacodinámicas ). La experiencia durante más de 12 meses es limitada.En pacientes con Síndrome Coronario Agudo (SCA), la interrupción prematura de cualquier antiagregante plaquetario, incluyendo Brilique, puede aumentar el riesgo de muerte cardiovascular o infarto de miocardio debido a la enfermedad subyacente del paciente. Por lo tanto, debe evitarse la interrupción prematura del tratamiento.Deben evitarse también faltas
en el tratamiento. El paciente que se olvide de tomar una dosis de Brilique, debe tomar sólo un comprimido de 90 mg (su siguiente dosis) a su hora habitual. Se puede cambiar a los pacientes tratados con clopidogrel directamente a Brilique si fuera necesario (ver sección Propiedades farmacodinámicas). No se ha investigado el cambio de prasugrel a Brilique. Poblaciones Especiales. Pacientes de edad avanzada.No es necesario ajustar la dosis en los pacientes de edad avanzada (ver sección Propiedades farmacocinéticas). Pacientes con insuficiencia renal. No es necesario ajustar la dosis en pacientes con insuficiencia renal
(ver sección Propiedades farmacocinéticas). No hay datos disponibles sobre el tratamiento de los pacientes en diálisis renal y por lo tanto, Brilique no está recomendado en estos pacientes. Pacientes con insuficiencia hepática. No es necesario ajustar la dosis en los pacientes con insuficiencia hepática leve. Brilique no se ha estudiado en pacientes con insuficiencia hepática moderada o grave. Su uso en pacientes con insuficiencia hepática moderada a grave está por lo tanto contraindicada (ver sección Contraindicaciones, Advertencias y precauciones especiales de empleo y Propiedades farmacocinéticas).Pacientes
pediátricos. No se ha establecido la seguridad y eficacia de Brilique en niños menores de 18 años de edad en la indicación aprobada para adultos. No hay datos disponibles (ver sección Propiedades farmacodinámicas y Propiedades farmacocinéticas). Forma de administración . Vía oral. Brilique puede administrarse con o sin alimentos.Contraindicaciones. -Hipersensibilidad al principio activo o a alguno de los excipientes incluidos en la sección Lista de excipientes (ver sección Reacciones adversas).Hemorragia patológica activa. Historial de hemorragia intracraneal (ver sección Reacciones adver-
sas).Insuficiencia hepática moderada a grave (ver sección Posología y forma de administración Advertencias y precauciones especiales de empleo y Propiedades farmacocinéticas).La administración concomitante de ticagrelor con inhibidores potentes de CYP3A4 (por ejemplo, ketoconazol, claritromicina, nefazodona, ritonavir y atazanavir) está contraindicada, debido a que la co‑administración puede llevar a un aumento considerable en la exposición a ticagrelor (ver sección Advertencias y precauciones especiales de empleo y Interacción con otros medicamentos y otras formas de interacción). Advertencias
y precauciones especiales de empleo. Riesgo de hemorragia. En el estudio pivotal de fase 3 (PLATO [PLATelet Inhibition And Patient Outcomes], 18.624 pacientes) los principales criterios de exclusión incluyeron el aumento del riesgo de hemorragia, trombocitopenia o anemia clínicamente importante, hemorragia intracraneal previa, hemorragia gastrointestinal en los últimos 6 meses o cirugía mayor en los últimos 30 días. Los pacientes con síndromes coronarios agudos tratados con Brilique y AAS mostraron un aumento del riesgo de hemorragia grave no relacionada con un IDAC y también de forma más
general, en hemorragias que requirieron atención médica, es decir, hemorragias PLATO Mayor + Menor, pero no hemorragias Mortales ni Potencialmente Mortales (ver sección Reacciones adversas). Por lo tanto, el uso de Brilique en pacientes con alto riesgo conocido de hemorragia debe sopesarse frente a su beneficio en la prevención de acontecimientos aterotrombóticos. Si está clínicamente indicado, Brilique debe emplearse con precaución en los siguientes grupos de pacientes: -Pacientes con propensión a las hemorragias (por ejemplo, debido a un traumatismo reciente, cirugía reciente, trastornos de coagula-
ción, hemorragia digestiva activa o reciente). El uso de Brilique está contraindicado en pacientes con hemorragia patológica activa, en aquellos con un historial de hemorragia intracraneal y en pacientes con insuficiencia hepática moderada a grave (ver sección Contraindicaciones). -Pacientes con administración concomitante de medicamentos que pueden aumentar el riesgo de hemorragia (por ejemplo, fármacos antiinflamatorios no esteroideos (AINE), anticoagulan-tes orales y/o fibrinolíticos) en las 24 horas siguientes a la administración de Brilique. No existen datos sobre Brilique en relación al beneficio hemostático de
las transfusiones de plaquetas; la presencia de Brilique circulante puede inhibir a las plaquetas transfundidas. Dado que la administración concomitante de Brilique con desmopresina no redujo el tiempo de sangrado, es improbable que la desmopresina sea eficaz para el manejo de acontecimientos hemorrágicos clínicos (ver sección Interacción con otros medicamentos y otras formas de interacción). El tratamiento antifibrinolítico (ácido aminocaproico o ácido tranexámico) y/o el factor recombinante VIIa pueden aumentar la hemostasia. El tratamiento con Brilique puede reanudarse después de haber identificado y
controlado la causa de la hemorragia. Cirugía. Se debe advertir a los pacientes que informen a su médico y dentista de que están tomando Brilique antes de programar cualquier cirugía y antes de tomar cualquier medicamento nuevo. En los pacientes del estudio PLATO sometidos a un injerto de derivación de arteria coronaria (IDAC), Brilique presentó más hemorragias que clopidogrel cuando se interrumpió 1 día antes de la cirugía pero obtuvo una tasa de hemorragias graves similar a la de clopidogrel después de la interrupción del tratamiento 2 ó más días antes de la cirugía (ver sección Reacciones adversas).
Si un paciente va a someterse a una intervención quirúrgica programada y no se requiere un efecto antiagregante plaquetario, debe suspenderse el tratamiento con Brilique 7 días antes de la intervención (ver sección Propiedades farmacodinámicas). Pacientes con riesgo de acontecimientos de bradicardia. Al haberse observado pausas ventriculares, en su mayoría asintomáticas, en un estudio clínico previo, los pacientes con mayor riesgo de acontecimientos de bradicardia (es decir, pacientes sin marcapasos que presenten síndrome de disfunción de nódulo sinusal, bloqueo AV de 2º o 3º grado, o síncope relacionado
con bradicardia) fueron excluidos del estudio principal PLATO en el que se evaluó la seguridad y la eficacia de Brilique. Por consiguiente, debido a la experiencia clínica limitada, se recomienda administrar Brilique con precaución en esos pacientes (ver sección Propiedades farmacodinámicas). Además, se debe tener precaución cuando se administre Brilique de forma concomitante con medicamentos conocidos por inducir bradicardia. Sin embargo, no se observó
ninguna evidencia de reacciones adversas clínicamente significativas en el estudio PLATO tras la administración concomitante de uno o más medicamentos conocidos por inducir bradicardia (por ejemplo, 96% betabloqueantes, 33% antagonistas de los canales de calcio, diltiazem y verapamilo y 4% digoxina) (ver sección Interacción con otros medicamentos y otras formas de interacción).Durante el subestudio Holter en PLATO, más pacientes presentaron pausas ventriculares ≥3 segundos con ticagrelor que con clopidogrel durante la fase aguda de su SCA. El aumento de pausas ventriculares detectadas mediante
Holter con ticagrelor fue mayor en pacientes con insuficiencia cardíaca crónica (ICC) que en la población general del estudio durante la fase aguda de SCA,
pero no después de un mes con ticagrelor, ni en comparación con clopidogrel. No se produjeron consecuencias clínicas adversas asociadas a dicho desequilibrio (incluyendo síncope o la implantación de un marcapasos) en esta población de pacientes (ver sección Propiedades farmacodinámicas). DisneaLa disnea se registró en un 13,8% de los pacientes tratados con Brilique y en un 7,8% de los pacientes tratados con clopidogrel. En 2,2% de los pacientes, los investigadores consideraron la disnea como causalmente relacionada con el tratamiento con Brilique. Normalmente es de intensidad leve o
moderada y a menudo desaparece sin necesidad de interrumpir el tratamiento. Los pacientes con asma/EPOC pueden presentar un aumento del riesgo absoluto de padecer disnea con Brilique (ver sección Reacciones adversas). Ticagrelor debe emplearse con precaución en pacientes con historial de asma y/o EPOC. No se ha determinado el mecanismo. Si un paciente informa sobre la aparición, prolongación o empeoramiento de la disnea, debe realizarse una investigación exhaustiva y, si no es tolerada, debe interrumpirse el tratamiento con Brilique. Aumentos de creatinina. Los niveles de creatinina pueden
aumentar durante el tratamiento con Brilique (ver sección Reacciones adversas). No se ha determinado el mecanismo. Debe controlarse la función renal después de un mes y a partir de entonces, de acuerdo con la práctica médica habitual, prestando especial atención a los pacientes ≥75 años, pacientes con insuficiencia renal moderada/grave y aquellos que estén recibiendo tratamiento concomitante con un ARA. Aumento del ácido úrico. En el estudio PLATO, los pacientes con ticagrelor presentaron mayor riesgo de hiperuricemia que aquellos pacientes que recibían clopidogrel (ver sección Reacciones adversas). Debe
tenerse precaución al administrar ticagrelor a pacientes con un historial de hiperuricemia o artritis gotosa. Como medida de precaución, no se recomienda el uso de ticagrelor en pacientes con nefropatía por ácido úrico. OtrosEn función de la relación observada en PLATO entre la dosis de mantenimiento de AAS y la eficacia relativa de ticagrelor en comparación con clopidogrel, no se recomienda la administración concomitante de Brilique con dosis altas de mantenimien-to de AAS (>300 mg) (ver sección Propiedades farmacodinámicas). La administración concomitante de Brilique con inhibidores potentes del CYP3A4 (por
ejemplo, ketoconazol, claritromicina, nefazodona, ritonavir y atazanavir) está contraindicada (ver sección Contraindicaciones y Interacción con otros medicamentos y otras formas de interacción). La administración concomitante puede ocasionar un aumento considerable de la exposición a Brilique (ver sección Interacción con otros medicamentos y otras formas de interacción).No se recomienda la administración concomitante de ticagrelor con inductores potentes del CYP3A4 (por ejemplo, rifampicina, dexametasona, fenitoína, carbamazepina, fenobarbital), debido a que la administración concomitante puede
llevar a una disminución en la exposición y eficacia del ticagrelor (ver sección Interacción con otros medicamentos y otras formas de interacción).No se recomienda la administración concomitante de Brilique con sustratos del CYP3A4 con índices terapéuticos estrechos (por ejemplo, cisaprida y alcaloides del cornezuelo del centeno) ya que ticagrelor puede aumentar la exposición a estos medicamentos (ver sección Interacción con otros medicamentos y otras formas de interacción). No se recomienda el uso concomitante de Brilique con dosis de simvastatina o lovastatina mayores de 40 mg (ver sección Interacción
con otros medicamentos y otras formas de interacción). Se recomienda llevar a cabo una estrecha monitorización clínica y analítica cuando se administre digoxina de forma concomitante con Brilique (ver sección Interacción con otros medicamentos y otras formas de interacción). No hay datos disponibles sobre el uso concomitante de Brilique con inhibidores potentes de la P‑glucoproteína (P‑gp) (por ejemplo verapamilo, quinidina, ciclosporina) que podrían aumentar la exposición al ticagrelor. Si no puede evitarse la asociación, debe realizarse la administración concomitante con precaución (ver sección Interacción con
otros medicamentos y otras formas de interacción). Interacción con otros medicamentos y otras formas de interacción. Ticagrelor es principalmente un substrato del CYP3A4 y un inhibidor leve del CYP3A4. Ticagrelor también es un substrato de la P‑gp y un inhibidor débil de la P‑gp y puede aumentar la exposición a substratos de la P‑gp. Efectos de otros medicamentos sobre Brilique. Medicamentos metabolizados por CYP3A4.Inhibidores del CYP3A4. +Inhibidores potentes del CYP3A4 - La administración concomitante de ketoconazol y ticagrelor aumentó la Cmáx y el AUC del ticagrelor 2,4 y 7,3 veces,
respectivamente. La Cmáx y el AUC del metabolito activo se redujeron en un 89% y un 56%, respectivamente. Cabe esperar que otros inhibidores potentes del CYP3A4 (claritromicina, nefazodona, ritonavir y atazanavir) tengan efectos similares y la administración concomitante con Brilique está contraindicada (ver sección Contraindicaciones y Advertencias y precauciones especiales de empleo). +Inhibidores moderados del CYP3A4 - La administración concomitante de
diltiazem y ticagrelor aumentó la Cmáx en un 69% y el AUC 2,7 veces, y redujo la Cmáx del metabolito activo en un 38%, sin cambios en el AUC. El ticagrelor no afectó a las concentraciones plasmáticas del diltiazem. Cabe esperar que otros inhibidores moderados del CYP3A4 (por ejemplo, amprenavir, aprepitant,
eritromicina y fluconazol) tengan un efecto similar y pueden administrarse también conjuntamente con Brilique.Inductores del CYP3A. La administración concomitante de rifampicina y ticagrelor redujo la Cmáx y el AUC del ticagrelor en un 73% y un 86%, respectivamente. La Cmáx del metabolito activo no varió y el AUC se redujo en un 46%, respectivamente. Cabe esperar que otros inductores del CYP3A (por ejemplo, dexametasona, fenitoína, carbamazepina y fenobarbital) reduzcan también la exposición a Brilique. La administración concomitante de ticagrelor e inductores potentes del CYP3A puede disminuir la
exposición y eficacia de ticagrelor (ver sección Advertencias y precauciones especiales de empleo). Otros. Los estudios clínicos de interacciones farmacológi-cas han revelado que la administración concomitante de ticagrelor y heparina, enoxaparina y AAS o desmopresina no tuvo ningún efecto sobre la farmacoci-nética de ticagrelor o su metabolito activo, ni sobre la agregación plaquetaria inducida por ADP en comparación con el ticagrelor solo. Si están clínicamente indicados, los medicamentos que alteran la hemostásis deberán ser usados con precaución en combinación con Brilique (ver sección Advertencias y
precauciones especiales de empleo). No hay datos disponibles sobre el uso concomitante de Brilique con inhibidores potentes de P‑gp (por ejemplo, verapamilo, quinidina y ciclosporina) que pudieran aumentar la exposición de ticagrelor. Si están clínicamente indicados, la co‑administración debe realizarse con precaución (ver sección Advertencias y precauciones especiales de empleo ). Efectos de Brilique sobre otros medicamentos. Medicamentos metaboliza-dos por CYP3A4. -Simvastatina - La administración concomitante de ticagrelor y simvastatina aumentó la Cmáx de la simvastatina en un 81% y el AUC en un
56%, y también aumentó la Cmáx del ácido de simvastatina en un 64% y el AUC en un 52%, con algunos casos individuales en los que aumentó 2 ó 3 veces. La administración concomitante de ticagrelor con dosis de simvastatina superiores a 40 mg diarios podría provocar efectos adversos de la simvastatina y debe sopesarse frente a los beneficios potenciales. La simvastatina no afectó a las concentraciones plasmáticas del ticagrelor. Brilique puede tener un efecto similar sobre la lovastatina. No se recomienda el uso concomitante de Brilique con dosis de simvastatina o lovastatina mayores de 40 mg (ver sección Advertencias y
precauciones especiales de empleo). -Atorvastatina - La administración concomitante de atorvastatina y ticagrelor aumentó la Cmáx del ácido de atorvastatina en un 23% y el AUC en un 36%. Se observaron incrementos similares en el AUC y la Cmáx de todos los metabolitos del ácido de atorvastatina. Esos incrementos no se consideran clínicamente significativos. - No se puede excluir un efecto similar sobre otras estatinas metabolizadas por CYP3A4. Los pacientes en PLATO que recibieron ticagrelor tomaron diversas estatinas, sin problemas asociados con la seguridad de la estatina en el 93% de la cohorte de
PLATO que tomaban estos medicamentos. Ticagrelor es un inhibidor leve del CYP3A4. No se recomienda la administración concomitante de Brilique con sustratos del CYP3A4 con índices terapéuticos estrechos (por ejemplo, cisaprida y alcaloides del cornezuelo del centeno) ya que puede aumentar la exposición a estos medicamentos (ver sección Advertencias y precauciones especiales de empleo). Medicamentos metabolizados por CYP2C9. La administración concomitante de Brilique y tolbutamida no alteró las concentraciones plasmáticas de ninguno de los dos medicamentos, lo que indica que el
ticagrelor no es un inhibidor del CYP2C9 y, por tanto, es improbable que altere el metabolismo mediado por CYP2C9 de medicamentos como la warfarina y la tolbutamida. Anticonceptivos orales. La administración concomitante de Brilique y levonorgestrel y etinilestradiol aumentó la exposición a etinilestradiol aproximadamente en un 20%, pero no alteró la farmacocinética del levonorgestrel. No se prevé que la administración concomitante de Brilique con levonorgestrel y etinilestradiol tenga un efecto clínicamente relevante en la eficacia de los anticonceptivos orales. Substratos de la P‑glucoproteína (P‑gp)
(incluyendo la digoxina y ciclosporina). La administración concomitante de Brilique aumentó la Cmáx de la digoxina en un 75% y el AUC en un 28%. Los niveles mínimos medios de digoxina aumentaron aproximadamente un 30% con la administración concomitante de ticagrelor con algunos aumentos máximos individuales de 2 veces. En presencia de digoxina, la Cmáx y el AUC de ticagrelor y su metabolito activo no se vieron afectados. Por consiguiente, se recomienda realizar los controles médicos y análisis clínicos pertinentes cuando se administren medicamentos dependientes de la P‑gp con un índice
terapéutico estrecho, como digoxina y ciclosporina, de forma concomitante con Brilique (ver sección Advertencias y precauciones especiales de empleo). Otros tratamientos concomitantes. Medicamentos conocidos por inducir bradicardia. Debido a las observaciones de pausas ventriculares, en su mayoría asintomáticas, y bradicardia, se debe tener precaución cuando se administre Brilique de forma concomitante con medicamentos conocidos por inducir bradicardia (ver sección Advertencias y precauciones especiales de empleo). Sin embargo, no se observó ninguna evidencia de reacciones adversas
clínicamente significativas en el estudio PLATO tras la administración concomitante de uno o más medicamentos conocidos por inducir bradicardia (por ejemplo, 96% betabloqueantes, 33% antagonistas de los canales de calcio, diltiazem y verapamilo y 4% digoxina). En el estudio PLATO, Brilique se administró en muchos casos junto con AAS, inhibidores de la bomba de protones, estatinas, betabloqueantes, inhibidores de la enzima convertidora de la angiotensina y antagonistas de los receptores de la angiotensina, según fuera necesario para el tratamiento de enfermedades concomitantes a largo plazo, así como
heparina, heparina de bajo peso molecular, inhibidores de GpIIb/IIIa por vía intravenosa, en tratamientos de corta duración (ver sección Propiedades farmacodinámicas). No se observó ninguna evidencia de interacciones adversas clínicamente significativas con estos medicamentos. La administración concomitante de Brilique con heparina, enoxaparina o desmopresina no tuvo efecto en el tiempo de tromboplastina parcial activado (TTPa) y el tiempo de coagulación activado (TCA) ni la determinación del factor Xa. Sin embargo, debido a las interacciones farmacodinámicas potenciales, debe tenerse precaución
con la administración concomitante de Brilique con medicamentos conocidos por alterar la hemostasis (ver sección Advertencias y precauciones especiales de empleo). Debido a las notificaciones de anomalías hemorrágicas cutáneas con ISRS (por ejemplo, paroxetina, sertralina y citalopram) se recomienda precaución al administrar ISRS con Brilique, debido a que esto puede aumentar el riesgo de hemorragia. Fertilidad, embarazo y lactancia: Mujeres en edad fértil. Las mujeres en edad fértil deben tomar métodos anticonceptivos adecuados para evitar el embarazo durante el tratamiento con Brilique. Embarazo. No
existen datos sobre el uso de ticagrelor en mujeres embarazadas o son limitados. Los estudios realizados en animales han mostrado toxicidad para la reproducción (ver sección Datos preclínicos sobre seguridad). Brilique no está recomendado durante el embarazo. Lactancia. Los datos farmacodinámicos/toxicológicos disponibles en animales han demostrado la excreción de ticagrelor y sus metabolitos activos en la leche (ver sección Datos preclínicos sobre seguridad). No se puede excluir el riesgo para neonatos/lactantes. Debe decidirse si interrumpir la lactancia o interrumpir/abstenerse del tratamiento con
Brilique, teniendo en consideración el beneficio de la lactancia para el niño y el beneficio del tratamiento para la mujer. Fertilidad. El ticagrelor no tuvo ningún efecto en la fertilidad masculina ni femenina en animales (ver sección Datos preclínicos sobre seguridad). Efectos sobre la capacidad para conducir y utilizar máquinas. No se han realizado estudios de los efectos de Brilique sobre la capacidad para conducir y utilizar máquinas. Cabe esperar que la influencia de Brilique sobre la capacidad para conducir y utilizar máquinas sea nula o insignificante. Reacciones adversas: Resumen del perfil de seguridad.
La seguridad de Brilique en pacientes con síndrome coronario agudo (AI, IMSEST e IMCEST) se ha evaluado en un único estudio pivotal de fase 3 PLATO (estudio [PLATelet Inhibition And Patient Outcomes], 18.624 pacientes), en el que se comparó a pacientes tratados con Brilique (dosis de carga de 180 mg seguida de 90 mg dos veces al día como dosis de mantenimiento) frente a pacientes tratados con clopidogrel (dosis de carga de 300‑600 mg seguida de 75 mg una vez al día como dosis de mantenimiento) en combinación con ácido acetilsalicílico (AAS) y con otros tratamientos convencionales. Las reacciones
adversas notificadas con más frecuencia durante el tratamiento con ticagrelor fueron disnea, contusión y epistaxis, y estos acontecimientos ocurrieron con unas tasas más altas que en el grupo que recibió tratamiento con clopidogrel. Resumen tabulado de reacciones adversas. Se han identificado las siguientes reacciones adversas tras los estudios con Brilique (Tabla 1). Las reacciones adversas se ordenan según frecuencia y Clasificación de Órganos y Sistemas. La frecuencia de las reacciones adversas se define mediante la siguiente convención: muy frecuentes (≥1/10), frecuentes (≥1/100 a <1/10), poco frecuentes
(≥1/1.000 a <1/100), raras (≥1/10.000 a <1/1.000), muy raras (<1/10.000), frecuencia no conocida (no puede estimarse a partir de los datos disponibles). En la tabla se han agrupado múltiples reacciones adversas relacionadas e incluyen términos médicos como los descritos a continuación: A. Hiperuricemia, Aumento del ácido úrico en sangre. B. Hemorragia cerebral, Hemorragia intracraneal, Ictus hemorrágico. C. Disnea, Disnea de esfuerzo,
Disnea en reposo, Disnea nocturna. D. Hemorragia gastrointestinal, Hemorragia rectal, Hemorragia intestinal, Melena, Hemorragia digestiva oculta. E. Hemorragia por úlcera gastrointestinal, Hemorragia por úlcera gástrica, Hemorragia por úlcera duodenal, Hemorragia por úlcera péptica. F. Hematoma subcutáneo, Hemorragia cutánea, Hemorragia subcutánea, Petequias. G. Contusión, Hematoma, Equimosis, Aumento de la tendencia de hematomas, Hematoma traumático. H. Hematuria, Presencia de sangre en orina, Hemorragia del tracto urinario. I. Hemorragia en el lugar de perforación del vaso,
Hematoma en el lugar de punción del vaso, Hemorragia en el lugar de inyección, Hemorragia en el lugar de punción, Hemorragia en el lugar de inserción del catéter # No hubo casos de AA de hemartros registrados en la rama de ticagrelor (n=9.235) del estudio PLATO, la frecuencia se ha calculado utilizando el límite superior del intervalo de confianza del 95% para el punto de estimación (basado en 3/X, en donde X representa el tamaño total de la muestra, por ejemplo 9.235). Esto se calcula como 3/9.235 lo que equivale a una categoría de frecuencia de “raro”. Descripción de algunas reacciones adversas.
Hemorragia. En la tabla 2 se muestran los resultados globales de la frecuencia de hemorragia ocurridas en el estudio PLATO. Tabla 2 - Estimación Kaplan-Meier de la frecuencia de hemorragias por tratamiento Definiciones de las categorías de hemorragias: Mayor Mortal/ Potencialmente Mortal: Clínicamente aparente con una disminución de >50 g/L de
hemoglobina o transfusión de ≥4 unidades de hematíes; o mortal; o intracraneal; o intrapericardial con taponamiento cardíaco; o con shock hipovolémico o hipotensión grave requiriendo vasopresores o cirugía. Otros Mayores: Clínicamente aparente con una disminución de 30‑50 g/L de hemoglobina o transfusión de 2‑3 unidades de hematíes; o significativamente discapacitante. Hemorragia Menor: Requiere intervención médica para parar o tratar la hemorragia. Mayor según la escala TIMI: Clínicamente aparente con una disminución de >50 g/L en la hemoglobina o hemorragia intracraneal. Menor
según la escala TIMI: Clínicamente aparente con una disminución de 30‑50 g/L de hemoglobina. Brilique y clopidogrel no se diferenciaron en cuanto a las tasas de hemorragia PLATO Mayor Mortal/Potencialmente Mortal, hemorragia PLATO Mayor Total, Hemorragia Mayor según la escala de TIMI o Hemorragia Menor según la escala de TIMI (Tabla 2). Sin embargo, hubo más hemorragia PLATO Mayor + Menor combinada con ticagrelor en comparación con clopidogrel. Pocos pacientes incluidos en PLATO presentaron hemorragias mortales: 20 (0,2%) con ticagrelor y 23 (0,3%) con clopidogrel (ver sección
Advertencias y precauciones especiales de empleo). Ni la edad, sexo, peso, raza, región geográfica, enfermedades coexistentes y tratamientos concomitantes e historial clínico, incluyendo ictus previo o accidente isquémico transitorio, pronosticaron ni la hemorragia total ni la PLATO Mayor no relacionada con un procedimiento. Por lo tanto, no se identificó ningún grupo de riesgo concreto para ningún tipo de hemorragia.Hemorragia relacionada con un IDAC: En PLATO, 42% de 1.584 pacientes (12% de la cohorte) que fueron sometidos a un injerto de derivación de arteria coronaria (IDAC) presentaron una hemorragia PLATO
Mayor Mortal/Potencialmente Mortal sin diferencia entre los grupos de tratamiento. La Hemorragia Mortal relacionada con un IDAC ocurrió en 6 pacientes de cada grupo de tratamiento (ver sección Advertencias y precauciones especiales de empleo). Hemorragias no relacionadas con un IDAC y hemorragias no relacionadas con un procedimiento: Brilique y clopidogrel no se diferenciaron en cuanto a las hemorragias Mayor Mortal/Potencialmente Mortal no relaciona-das con un IDAC según definición de PLATO, pero las hemorragias Mayor Total, Mayor según la escala TIMI y Mayor + Menor según la escala TIMI, según
definición de PLATO, fueron más comunes con ticagrelor. De forma similar, al eliminar todas las hemorragias relacionadas con procedimientos, hubo más hemorragias con ticagrelor que con clopidogrel (Tabla 2). La interrupción del tratamiento debido a hemorragia no relacionada con un procedimiento fue más habitual con ticagrelor (2,9%) que con clopidogrel (1,2%; p<0,001). Hemorragias intracraneales: Se produjeron más hemorragias intracraneales no relacionadas con un procedimiento con ticagrelor (n=27 hemorragias en 26 pacientes, 0,3%) que con clopidogrel (n=14 hemorragias, 0,2%), de las cuales, 11
de las hemorragias con ticagrelor y 1 con clopidogrel fueron mortales. No hubo diferencia en las hemorragias mortales totales. Disnea. La disnea, una sensación de falta de aire, se ha notificado en pacientes tratados con Brilique. Los acontecimientos adversos (AA) relacionados con la disnea (disnea, disnea en reposo, disnea de esfuerzo, disnea paroxística nocturna y disnea nocturna), en conjunto, se registró en un 13,8% de los pacientes tratados con ticagrelor y en 7,8% de los pacientes tratados con clopidogrel. Los investigadores consideraron la disnea causalmente relacionada con el tratamiento en el 2,2% de los
pacientes tratados con ticagrelor y en el 0,6% de los tratados con clopidogrel en el estudio PLATO, y pocos casos fueron graves (0,14% ticagrelor; 0,02% clopidogrel), (ver sección Advertencias y precauciones especiales de empleo). La mayoría de los síntomas de disnea fueron de intensidad leve o moderada y la mayoría fueron registrados como un episodio aislado poco después del inicio del tratamiento. En comparación con clopidogrel, los pacientes con asma/EPOC tratados con ticagrelor pueden presentar un aumento del riesgo de experimentar disnea no grave (3,29% con ticagrelor frente a 0,53% con clopidogrel)
y disnea grave (0,38% con ticagrelor frente a 0,00% con clopidogrel). En términos absolutos, este riesgo fue mayor en la población global de PLATO. Ticagrelor debe emplearse con precaución en pacientes con historial de asma y/o EPOC (ver sección Advertencias y precauciones especiales de empleo). Aproximadamente el 30% los casos remitieron en el plazo de 7 días. El estudio PLATO incluía pacientes con insuficiencia cardíaca congestiva inicial, enfermedad pulmonar obstructiva crónica o asma; era más probable que estos pacientes, así como los pacientes de edad avanzada, registraran disnea. Para
Brilique, 0,9% de los pacientes interrumpieron la medicación de estudio debido a la disnea en comparación con un 0,1% que tomaban clopidogrel. La mayor incidencia de disnea con Brilique no está asociada a la aparición o empeoramiento de una enfermedad cardíaca o pulmonar (ver sección Advertencias y precauciones especiales de empleo). Brilique no afecta las pruebas de la función respiratoria. Exploraciones complementarias. Aumentos de creatinina: En PLATO, la concentración sérica de creatinina aumentó de forma significativa en >30% en el 25,5% de los pacientes que recibían ticagrelor en comparación
con el 21,3% de los pacientes que recibían clopidogrel y en >50% en 8,3% de los pacientes que recibían ticagrelor en comparación con el 6,7% de pacientes que recibían clopidogrel. Las elevaciones de creatinina en >50% fueron más pronunciadas en pacientes de >75 años de edad (ticagrelor 13,6% frente a clopidogrel 8,8%) en pacientes con insuficiencia renal grave en los valores iniciales (ticagrelor 17,8% frente a clopidogrel 12,5%) y en pacientes que recibían tratamiento concomitante con ARA (ticagrelor 11,2% frente a clopidogrel 7,1%). Dentro de estos subgrupos, los acontecimientos adversos graves relacionados
con el riñón y los acontecimientos adversos que conducen a la interrupción de la medicación de estudio fueron similares entre los grupos de tratamiento. La totalidad de los AAs renales notificados fue de 4,9% para ticagrelor frente a 3,8% para clopidogrel, sin embargo un porcentaje similar de pacientes registraron acontecimientos que fueron considerados por los investigadores como causalmente relacionados con el tratamiento; 54 (0,6%) para ticagrelor y 43 (0,5%) con clopidogrel. Aumentos de ácido úrico: En el estudio PLATO, la concentración plasmática de ácido úrico aumentó hasta por encima del límite superior de
normalidad en el 22% de los pacientes tratados con ticagrelor, frente al 13% de los pacientes tratados con clopidogrel. La concentración plasmática media de ácido úrico aumentó en aproximadamente un 15% con ticagrelor en comparación con casi un 7,5% con clopidogrel y una vez interrumpido el tratamiento, disminuyó a aproximadamente un 7% con ticagrelor pero no se observaron disminuciones con clopidogrel. Los AAs notificados de hiperuricemia fueron 0,5% para ticagrelor frente a 0,2% para clopidogrel. De estos AAs, 0,05% para ticagrelor frente a 0,02% para clopidogrel fueron considerados por los investigadores
como causalmente relacionados. Para la artritis gotosa, los AAs notificados fueron de 0,2% para ticagrelor frente a 0,1% para clopidogrel; ninguno de estos acontecimientos adversos fue considerado por los investigadores como causalmente relacionado. Sobredosis: Ticagrelor es bien tolerado en dosis únicas de hasta 900 mg. La toxicidad gastrointestinal fue dosis limitante en un único estudio de aumentos de dosis. Otras reacciones adversas clínicamente significati-vas que pueden aparecer con la sobredosis incluyen disnea y pausas ventriculares (ver sección Reacciones adversas). En caso de sobredosis, vigilar la
aparición de estas posibles reacciones adversas y considerar la monitorización ECG. No existe actualmente ningún antídoto conocido para neutralizar los efectos de Brilique, y no cabe esperar que el medicamento sea dializable (ver sección Advertencias y precauciones especiales de empleo). El tratamiento de la sobredosis debe realizarse conforme a la práctica médica habitual local. El efecto esperado de una sobredosis de Brilique es la duración prolongada del riesgo de hemorragias asociada a la inhibición plaquetaria. Si apareciera hemorragia, deben tomarse las medidas de apoyo oportunas. PROPIEDADES
FARMACOLÓGICAS. Propiedades farmacodinámicas: Grupo farmacoterapéutico: Inhibidores de la agregación plaquetaria, excluyendo heparina, Código ATC: B01AC24. Mecanismo de acción. Brilique contiene ticagrelor, un medicamento que pertenece a la clase química de la ciclopentiltriazolopirimidinas (CPTP), que es un antagonista selectivo de los receptores del adenosín difosfato (ADP) que actúa sobre el receptor P2Y12 del ADP que puede prevenir la activación y agregación de las plaquetas mediada por ADP. El ticagrelor es activo por vía oral y se une de forma reversible al receptor P2Y12 del ADP en las
plaquetas. El ticagrelor no se une al mismo lugar de unión del ADP, pero interacciona con el receptor P2Y12 de ADP en las plaquetas para impedir la transmisión de señales. Efectos farmacodinámicos. Inicio de la acción. En pacientes con enfermedad arterial coronaria estable que toman AAS, ticagrelor presenta un inicio rápido del efecto farmacológico, como refleja la Inhibición de la Agregación Plaquetaria (IAP) media para ticagrelor a 0,5 horas tras una
Tabla 1 - Reacciones adversas según frecuencia y clasificación por Órganos y Sistemas (SOC) Frecuentes Poco frecuentes Raras Clasificación por Órganos y Sistemas Trastornos del metabolismo y de la nutrición
Hiperuricemiaa
Trastornos psiquiátricos Confusión Trastornos del sistema nervioso Hemorragia intracranealb, Mareo, Cefalea Parestesia Trastornos oculares Hemorragia ocular (intraocular, conjuntival, retinal) Trastornos del oído y del laberinto Hemorragia del oído, Vértigo Trastornos de la piel y del tejido subcutáneo Disneac, Epistaxis Hemoptisis Trastornos gastrointestinales Hemorragia gastrointestinald Hematemesis, Hemorragia por úlcera gastrointestinale,
Hemorragia hemorroidal, Gastritis, Hemorragia bucal (incluyendo sangrado gingival), Vómitos, Diarrea, Dolor abdominal, Náuseas, Dispepsia
Hemorragia retroperitonealb, Estreñimiento
Skin and subcutaneous tissue disorders Hemorragia subcutánea o dérmicaf, Hematomasg Exantema, Prurito Trastornos musculoesqueléticos y del tejido conjuntivo Hemartros# Trastornos renales y urinarios Hemorragias del tracto urinarioh Trastornos del aparato reproductor y de la mama Hemorragia vaginal (incluyendo metrorragia) Exploraciones complementarias Aumento de la creatinina sérica Lesiones traumáticas, intoxicaciones y complicaciones de procedimientos terapéuticos Hemorragia en el lugar de intervención i Hemorragia después del procedimiento, Hemorragia Hemorragia de heridas, Hemorragia
traumática Trastornos del sistema inmunológico Hipersensibilidad incluyendo angioedema
Brilique/ (%/año)/N=9.235 Clopidogrel/(%/año)/N=9.186 P PLATO Mayor Total 11,6 11,2 0,4336 PLATO Mayor Mortal/ Potencialmente Mortal 5,8 5,8 0,6988 PLATO Mayor no relacionada con un IDAC 4,5 3,8 0,0264 PLATO Mayor no relacionadas con un procedimiento 3,1 2,3 0,0058 PLATO Mayor Total + Menor 16,1 14,6 0,0084 PLATO Mayor + Menor no relacionadas con un procedimiento 5,9 4,3 <0,0001 Mayor según la escala TIMI 7,9 7,7 0,5669 Mayor + Menor según la escala TIMI 11,4 10,9 0,3272
dosis de carga de 180 mg de aproximadamente el 41%, con un efecto máximo de IAP del 89% 2‑4 horas después de la administración, manteniéndose entre 2‑8 horas. El 90% de los pacientes presentó una IAP prolongada final >70% 2 horas después de administrar la dosis. Fin de la acción. Si se programa un
procedimiento de IDAC, el riesgo de hemorragia con ticagrelor está aumentado en comparación con clopidogrel cuando se interrumpe menos de 96 horas antes del procedimiento. Datos sobre el cambio de tratamiento. El cambio de clopidogrel a ticagrelor produce un aumento absoluto de la IAP del 26,4% y el cambio de ticagrelor a clopidogrel produce una disminución absoluta de la IAP del 24,5%. Los pacientes pueden pasar de clopidogrel a ticagrelor sin interrumpir el efecto de inhibición de la agregación plaquetaria (ver sección Posología y forma de administración). Eficacia y seguridad clínica. El estudio PLATO incluyó 18.624
pacientes que presentaban síntomas de angina inestable (AI), infarto de miocardio sin elevación del segmento ST (IMSEST) o infarto de miocardio con elevación del segmento ST (IMCEST) en las 24 horas después del inicio y, en un principio, se controlaron médicamente o con intervención coronaria percutánea (ICP), o con un injerto de derivación de arteria coronaria (IDAC) (ver sección Indicaciones terapéuticas). Con un tratamiento de base con AAS diario, ticagrelor 90 mg dos veces al día mostró superioridad a clopidogrel 75 mg al día en la prevención de los criterios de valoración de la eficacia combinados de muerte cardiovascular
[CV], infarto de miocardio [IM] e ictus, con una diferencia debida a la mortalidad CV y el IM. Los pacientes recibieron una dosis de carga de 300 mg de clopidogrel (600 mg posible en presencia de ICP) o de 180 mg de ticagrelor. El resultado apareció pronto (reducción absoluta del riesgo [RAR] 0,6% y Reducción Relativa del Riesgo [RRR] del 12% a los 30 días), con un efecto del tratamiento constante durante todo el intervalo de 12 meses, consiguiendo una RAR del 1,9% por año con una RRR del 16%. Esto sugiere que es apropiado tratar a pacientes con ticagrelor hasta 12 meses (ver sección Posología y forma de administración). El
tratamiento de 54 pacientes con SCA con ticagrelor en lugar de clopidogrel evitará 1 acontecimiento aterotrombótico; el tratamiento de 91 evitará 1 muerte CV (ver Figura 1 y la Tabla 3). El efecto del tratamiento con ticagrelor frente a clopidogrel parece ser el mismo en todos los subgrupos de pacientes, incluyendo peso, género, historial clínico de diabetes mellitus, accidente isquémico transitorio o ictus no hemorrágico o revascularización, tratamientos concomitantes incluyendo heparinas, inhibidores GpIIb/IIIa e inhibidores de la bomba de protones (ver sección Interacción con otros medicamentos y otras formas de interacción),
diagnóstico final del episodio (IMCEST, IMSEST y AI) y el protocolo de tratamiento propuesto en la aleatorización (invasiva o médica). Se observó una interacción de los tratamientos débilmente significativa en relación con la región, según la cual la RRI para el criterio de valoración principal favorece a ticagrelor en el resto del mundo, pero favorece a clopidogrel en Norteamérica, que representa aproximadamente el 10% de la población total estudiada (valor p de la interacción = 0,045). Los análisis exploratorios sugieren una posible asociación con la dosis de AAS, tal, que se observó una eficacia reducida de ticagrelor al
aumentar las dosis de AAS. Las dosis diarias crónicas de AAS para acompañar a Brilique deben ser de 75‑150 mg (ver sección Posología y forma de administración y Advertencias y precauciones especiales de empleo). En la Figura 1 se indica el valor estimado del riesgo hasta un primer episodio del criterio de valoración combinado de la eficacia.
Figura 1 - Tiempo transcurrido hasta un primer acontecimiento de muerte CV, IM e Ictus (PLATO). Brilique redujo la incidencia de l criterio principal de valoración combinado en comparación con clopidogrel, tanto en los pacientes con AI/IMSEST como en los pacientes con IMCEST (Tabla 3).
Tabla 3 - Resultados de los acontecimientos en PLATO
a RAR = reducción absoluta del riesgo; RRR= reducción relativa del riesgo (1- razón de riesgos instantáneos) x 100%. Los valores con una reducción relativa del
riesgo negativa indican un aumento relativo del riesgo. b excluyendo el infarto de miocardio silente. c IGR = isquemia grave recurrente; IR = isquemia recurrente; AIT = accidente isquémico transitorio; AAT = acontecimiento arterio trombótico. IM total incluye el IM silente con fecha de inicio del acontecimiento fijada en la
fecha de diagnostico. d valor nominal de significación; todos los demás son formalmente de significación estadística según un análisis jerárquico predefinido. Subestudio Holter: Para evaluar la aparición de pausas ventriculares y otros episodios arrítmicos durante el estudio PLATO, los investigadores realizaron una monitorización con Holter a un subgrupo de casi 3.000 pacientes, obteniéndose registros en casi 2.000 pacientes tanto en la fase aguda de su SCA como un mes después. La variable principal de interés fue la aparición de pausas ventriculares ≥3 segundos. El número de pacientes que presentaron pausas ventriculares fue
mayor con ticagrelor (6,0%) que con clopidogrel (3,5%) en la fase aguda; el 2,2% y el 1,6%, respectivamente, después de un mes (ver sección Advertencias y precauciones especiales de empleo). El aumento en las pausas ventriculares en la fase aguda del SCA fue más pronunciado en pacientes con ticagrelor con un historial de ICC (9,2% frente a 5,4% en pacientes sin ICC previa; para los pacientes con clopidogrel, 4,0% en los que sí tenían frente a 3,6% en los que no tenían ICC previa). Este desequilibrio no ocurrió en el primer mes: 2,0% frente a 2,1% para pacientes con ticagrelor con y sin historial de ICC, respectivamente; y 3,8%
frente a 1,4% con clopidogrel. No se produjeron consecuencias clínicas adversas asociadas a dicho desequilibrio (como la implantación de un marcapasos) en esta población de pacientes. Subestudio genético de PLATO: La determinación del genotipo del CYP2C19 y el ABCB1 en 10.285 pacientes en PLATO proporcionó asociaciones de los grupos de genotipos con los resultados de PLATO. La superioridad de ticagrelor sobre clopidogrel en la reducción de los acontecimientos CV mayores no se vio
significativamente afectada por el genotipo del ABCB1 o del CYP2C19 del paciente. Al igual que en el estudio global de PLATO, la hemorragia PLATO ‘Mayor Total’ no se diferenció entre ticagrelor y clopidogrel, con independencia del genotipo del CYP2C19 o del ABCB1. La hemorragia PLATO ‘Mayor’ no relacionada con un IDAC según PLATO aumentó con ticagrelor en comparación con clopidogrel en pacientes uno o más alelos con pérdida de función del CYP2C19, pero fue similar al clopidogrel en pacientes sin alelos con pérdida de función.
Criterio de valoración combinado de eficacia y seguridad : El criterio de valoración combinado de eficacia y seguridad (muerte CV, IM, ictus, o hemorragia ‘Mayor Total’ según la definición del estudio PLATO) indica que el beneficio en la eficacia de Brilique en comparación con el clopidogrel no finaliza por los acontecimien-tos hemorrágicos graves (RAR 1,4%, RRR 8%, RRI 0,92; p = 0,0257) en los 12 meses siguientes al SCA. Población pediátrica : La Agencia Europea de Medicamentos ha eximido al titular de la obligación de presentar los resultados de los ensayos realizados con Brilique en todos los grupos de la población
pediátrica establecida en la indicación autorizada (ver sección Posología y forma de administración y Propiedades farmacocinéticas). Propiedades farmacociné-ticas: El ticagrelor presenta una farmacocinética lineal y la exposición a ticagrelor y al metabolito activo (AR‑C124910XX) son aproximadamente proporcionales a la dosis hasta 1.260 mg.Absorción. El ticagrelor se absorbe rápidamente, con una mediana del tmáx de 1,5 horas aproximadamente. La formación del principal metabolito circulante AR‑C124910XX (también activo) del ticagrelor es rápida, con una mediana del tmáx de 2,5 horas aproximadamente. Tras la administración
oral de ticagrelor 90 mg en condiciones de ayuno, la Cmáx es de 529 ng/ml y el AUC es de 3.451 ng*h/ml. Las relaciones para el metabolito original son de 0,28 para la Cmáx y 0,42 para el AUC. La biodisponibilidad absoluta media del ticagrelor se estimó en un 36%. La ingestión de una comida rica en grasas aumentó en un 21% el AUC del ticagrelor y redujo en un 22% la Cmáx del metabolito activo pero no tuvo efecto alguno en la Cmáx del ticagrelor ni en el AUC del metabolito activo. Estos pequeños cambios se consideraron de importancia clínica mínima; por consiguiente, el ticagrelor se puede administrar con o sin alimentos.
Ticagrelor así como su metabolito activo son substratos de la P‑gp. Distribución. El volumen de distribución en estado de equilibrio del ticagrelor es de 87,5 l. El ticagrelor y el metabolito activo se unen en un gran porcentaje a las proteínas plasmáticas humanas (>99,0%). Biotransformación. CYP3A4 es el principal enzima responsable del metabolismo del ticagrelor y la formación del metabolito activo, y sus interacciones con otros sustratos del CYP3A van desde la activación hasta la inhibición. El principal metabolito del ticagrelor es AR‑C124910XX, también es activo según demuestra su unión in vitro al receptor P2Y12 de ADP en las
plaquetas. La exposición sistémica al metabolito activo es aproximadamente un 30‑40% de la obtenida con ticagrelor. Eliminación. La principal vía de eliminación del ticagrelor es por metabolismo hepático. Cuando se administra ticagrelor con marcaje radiactivo, la recuperación media de la radiactividad es de aproximada-mente el 84% (57,8% en las heces, 26,5% en orina). La recuperación de ticagrelor y del metabolito activo en la orina fue inferior al 1% de la dosis. La principal vía de eliminación del metabolito activo probablemente sea por secreción biliar. La media de la t1/2 fue de aproximadamente 7 horas para ticagrelor y de 8,5 horas
para el metabolito activo. Poblaciones especiales. Pacientes de edad avanzada. Se observaron exposiciones más altas a ticagrelor (aproximadamente 25% tanto para la Cmáx como para el AUC) y al metabolito activo en pacientes de edad avanzada (≥75 años) con SCA que en pacientes jóvenes según el análisis farmacocinético de la población. Esas diferencias no se consideran clínicamente significativas. (ver sección Posología y forma de administración).Población pediátrica. El ticagrelor no se ha evaluado en población pediátrica (ver sección Posología y forma de administración y Propiedades farmacodinámicas). Sexo. Se
observaron exposiciones más altas a ticagrelor y al metabolito activo en las mujeres que en los hombres. Las diferencias no se consideran clínicamente significativas. Insuficiencia renal. La exposición a ticagrelor y al metabolito activo fue casi un 20% menor en los pacientes con insuficiencia renal grave (aclaramiento de creatinina <30 ml/min) en comparación con sujetos con una función renal normal (ver sección Posología y forma de administración). Insuficiencia hepática. La Cmáx y el AUC del ticagrelor fueron un 12% y un 23% mayores en los pacientes con insuficiencia hepática leve que en sujetos sanos
correspondientes, respectivamente (ver sección Posología y forma de administración). No se ha estudiado el ticagrelor en pacientes con insuficiencia hepática moderada o grave y su uso en estos pacientes está contraindicado (ver sección Contraindicaciones y Advertencias y precauciones especiales de empleo). Origen étnico. Los pacientes de ascendencia asiática tienen una biodisponibilidad media que es un 39% mayor que la observada en pacientes de raza blanca. Los pacientes que se consideran de raza negra tuvieron una biodisponibilidad de ticagrelor que fue un 18% menor que los pacientes de raza blanca. En los estudios
de farmacología clínica, la exposición (Cmáx y AUC) a ticagrelor en pacientes japoneses fue aproximadamente un 40% mayor (un 20% tras realizar los ajustes necesarios para tener en cuenta el peso corporal) que en los pacientes de raza blanca. Datos preclínicos sobre seguridad. Los datos preclínicos correspon-dientes al ticagrelor y a su metabolito principal no han demostrado un riesgo inaceptable de efectos adversos para el ser humano tras realizar los estudios convencionales de farmacología de seguridad, toxicidad a dosis únicas y repetidas y potencial genotóxico. Se observó irritación gastrointestinal en varias
especies animales a niveles de exposición clínicamente relevantes (ver sección Reacciones adversas).En ratas hembra, el ticagrelor a dosis altas presentó un aumento de la incidencia de tumores uterinos (adenocarcinomas) y un aumento de la incidencia de adenomas hepáticos. El mecanismo de los tumores uterinos se debe probablemente a un desequilibrio hormonal cuyo efecto en ratas conduce al desarrollo de tumores. El mecanismo para los adenomas hepáticos se debe probablemente a una inducción enzimática en el hígado específica en roedores. Por lo tanto, no se considera que los hallazgos carcinogenéticos tengan
importancia en humanos. En ratas se observaron anomalías menores en el desarrollo con la dosis de toxicidad materna (margen de seguridad de 5,1). En conejos se observó un ligero retraso en la madurez hepática y el desarrollo esquelético en fetos de madres con la dosis alta, sin que mostrasen toxicidad materna (margen de seguridad de 4,5).Los estudios en ratas y conejos han mostrado toxicidad reproductiva, con un ligero descenso del aumento de peso materno y una reducción de la viabilidad neonatal y de peso al nacer, y retraso en el crecimiento. El ticagrelor produce ciclos irregulares (principalmente alargamiento del ciclo)
en ratas hembra, pero no afectó a la fertilidad global en ratas macho y hembra. Los estudios farmacocinéticos llevados a cabo con ticagrelor radio‑marcado han demostrado que el compuesto original y sus metabolitos se excretan en la leche de las ratas (ver sección Embarazo y lactancia). DATOS FARMACÉUTICOS. Lista de excipientes. Núcleo/ Manitol (E421)/ Fosfato dibásico de calcio / Estearato de magnesio (E470b) / Carboximetilalmidón sódico / Hidroxipropilcelulosa (E463) / Recubrimiento / Talco / Dióxido de titanio (E171) / Óxido de hierro amarillo (E172) / Polietilenglicol 400 / Hipromelosa (E464). Incompatibilidades. No
procede. Periodo de validez. 3 años. Precauciones especiales de conservación. Este medicamento no requiere condiciones especiales de conserva-ción.Naturaleza y contenido del envase. - Blister transparente de PVC‑PVDC/Al (con símbolos de sol/luna) de 10 comprimidos en cajas de 60 comprimidos (6 blisters) y 180 comprimidos (18 blisters). - Blister transparente de PVC‑PVDC/Al calendario (con símbolos de sol/luna) de 14 comprimidos en cajas de 14 comprimidos (1 blister), 56 comprimidos (4 blisters) y 168 comprimidos (12 blisters). - Blister transparente de dosis unitaria con lámina perforada de PVC‑PVDC/
Al de 10 comprimidos en cajas de 100x1 comprimidos (10 blisters). Puede que solamente estén comercializados algunos tamaños de envase. Precauciones especiales de eliminación. Ninguna especial. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN. AstraZeneca AB. S‑151 85. Södertälje. Suecia. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN. EU/1/10/655/001-006. FECHA DE LA PRIMERA AUTORIZACIÓN/RENOVACIÓN DE LA AUTORIZACIÓN. Fecha de la primera autorización: 03 diciembre 2010. FECHA DE LA REVISIÓN DEL TEXTO. Octubre 2012. REGIMEN DE PRESCRIPCION Y
DISPENSACION. Con receta médica. CONDICIONES DE LA PRESTACION DEL SISTEMA NACIONAL DE SALUD. Brilique 90mg, 56 comprimidos recubiertos con película: CN 665929.8 - incluido en la oferta del SNS. Brilique 90mg, 100 comprimidos recubiertos con película: CN 603681.5 – no incluido en la oferta del SNS. PRESENTACION Y PRECIOS. Brilique 90 mg comprimidos con película: envase de 56 comrpimidos, CN 665929.8; PVL: 57,40 €; PVP: 86,16 €; PVP IVA: 89,61 €.. Brilique 90 mg comprimidos con película: envase de 100 comrpimidos, CN 603681.5 PVL: 102,50 €; PVP: 119,88; PVP IVA:
124,68 €. La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos http://www.ema.europa.eu. FAcompleta/FT 24-Oct-2012(Hipersensibilidad)/QRD 24-Oct-2012 (IB-01).
Brilique (% de pacientes con acontecimientos) N=9.333
Clopidogrel (% pacientes con acontecimientos) N=9.291
RARa (%/año)
RRRa (%) (IC del 95%) P
Muerte CV, IM (excl. IM silente) o ictus. 9,3 10,9 1,9 16 (8, 23) 0,0003
Intención invasiva 8,5 10,0 1,7 16 (6, 25) 0,0025
Intención médica 11,3 13,2 2,3 15 (0,3, 27) 0,0444d
Muerte CV 3,8 4,8 1,1 21 (9, 31) 0,0013
IM (excl. IM silente)b 5,4 6,4 1,1 16 (5, 25) 0,0045 Ictus 1,3 1,1 -
0,2 -17 (-52, 9) 0,2249
Mortalidad por cualquier causa, IM (excl. IM silente) o ictus 9,7 11,5 2,1 16 (8, 23) 0,0001
Muerte CV, IM total, ictus, IGR, IR, AIT, u otros AATc 13,8 15,7 2,1 12 (5,19) 0,0006
Mortalidad por cualquier causa
4,3 5,4 1,4 22 (11, 31) 0,0003d Trombosis confirmada del stent
1,2 1,7 0,6 32 (8, 49) 0,0123d



















