RICE STRIPE NECROSIS VIRUSrepositorio.educacionsuperior.gob.ec/bitstream/28000/4694/2/Anexo 2... ·...
17
PROTOCOLOS MOLECULARES DE FITOPATOLOGÍA RICE STRIPE NECROSIS VIRUS INTRODUCCIÓN En el 2003 se detectó en Ecuador la presencia de la enfermedad entorchamiento del arroz, causada por el virus de la necrosis rayada del arroz (Rice Stripe Necrosis Virus) transmitido por el plasmodióforo Polymyxa graminis, ocurriendo pérdidas de hasta el 40% de la producción (Paz et al., 2009). La enfermedad del “entorchamiento” se inicia cuando las semillas y plántulas de arroz se siembran en un campo infestado con cistosoros que poseen el RSNV. El presente protocolo facilita la aplicación estándar de las técnicas moleculares en el laboratorio permitiendo la identificación del virus y el vector a través del uso de la reacción en cadena de la polimerasa o PCR con cebadores específicos.
Transcript of RICE STRIPE NECROSIS VIRUSrepositorio.educacionsuperior.gob.ec/bitstream/28000/4694/2/Anexo 2... ·...

PROTOCOLOS MOLECULARES DE FITOPATOLOGÍA
RICE STRIPE NECROSIS VIRUS
INTRODUCCIÓN
En el 2003 se detectó en Ecuador la presencia de la enfermedad entorchamiento del arroz,
causada por el virus de la necrosis rayada del arroz (Rice Stripe Necrosis Virus) transmitido
por el plasmodióforo Polymyxa graminis, ocurriendo pérdidas de hasta el 40% de la
producción (Paz et al., 2009).
La enfermedad del “entorchamiento” se inicia cuando las semillas y plántulas de arroz se
siembran en un campo infestado con cistosoros que poseen el RSNV.
El presente protocolo facilita la aplicación estándar de las técnicas moleculares en el
laboratorio permitiendo la identificación del virus y el vector a través del uso de la reacción en
cadena de la polimerasa o PCR con cebadores específicos.

I. Diseño de cebadores
Los cebadores (primers, iniciadores, oligonucleótidos) se diseñan con programas específicos
para ello, una vez se ha determinado el gen o secuencia de ácido desoxirribonucleico (ADN)
de interés. Se sugiere el uso del programa Primer3 (http://bioinfo.ut.ee/primer3/).
Para cada aplicación de la PCR es necesario ajustar las condiciones y el diseño de la reacción,
por ejemplo: la duración de las fases de hibridación y elongación; el tipo de cebadores o primer
empleados y sus temperaturas de alineamiento o annealing (Tm); el tipo de enzima ADN
polimerasa utilizada; la cantidad de los reactivos, sobretodo Mg+2
, dNTPs, ADN molde a
incluir; la longitud del amplicón o producto obtenido.
Las reglas fundamentales para el diseño de cebadores son las siguientes: 1) deben tener
longitudes de entre 19 y 25 nucleótidos; 2) el par de cebadores debe tener Tm muy cercana
entre ellos, para maximizar el rendimiento, lo cual suele traducirse en un contenido en GC
similar, entre 50% y 60%. Esta temperatura debe ser igual o superior a los 50°C para mantener
especificidad por la secuencia genética buscada, óptimo Tm= 58°C; 3) Los cebadores no deben
favorecer la formación de horquillas (hairpins) ni ser complementarios entre ellos; 4) Conviene
evitar secuencias que terminen en GGG, GGT, ATT, CGA, TAA o TTA.
En el caso del virus RSNV fueron diseñados un par de cebadores a partir de la secuencia
nucleotídica de la cápside proteica del virus RSNV (Genbank # XXX), en el Laboratorio de
Fitopatología de la Estación Experimental Litoral Sur, los cuales se identificaron como RSNV-
F (5’- GTGGGTTAGAACGTCTGAGGC- 3’) y RSNV-R (5’-GTCGTCAACCCACTTAAGC
3’). Estos cebadores amplifican un fragmento de ADN con tamaño esperado de 468
nucleótidos (468 pb).
Adicionalmente, a través de la colaboración con la Dra. Eugenie Hébrard (IRD, Montpellier),
se obtuvieron las secuencias RSNV_CP_F (5’ CATCTTGTCGAGATGAG 3’) y
RSNV_CP_R (5’ GCGTTGTCTTTATCAGTG 3’), las cuales identifican al virus, con un
amplicón esperado de 328 nucleótidos (328 pb) y se enviaron a sintetizar.
En el ARN 1 se sintetizaron los cebadores RSNV1-3827R (5’
TGTGGCGTTTCCAGACCTAAA 3’) y RSNV1-2901F (5’ TGAATTTGGTGCTCTCTTG
3’) (Hébrard, 2016, comunicación personal) amplicón esperado de 926 nucleótidos (926 pb).
Para realizar la amplificación por la reacción en cadena de la polimerasa (PCR) es necesario
contar con una secuencia molde de ADN, generada a partir del ácido ribonucleico (ARN) del
virus.
II. Extracción de ARN total del virus a partir de tejido vegetal de plantas
sintomáticas
Se utilizó el kit de SV RNA Total (Promega) siguiendo las instrucciones del fabricante
(Anexo 1).
UTILIZAR GUANTES LIMPIOS Y NUEVOS. USAR PIPETAS LIMPIAS DE ARNasas

1. Seleccionar plantas con los síntomas de la enfermedad en el invernadero de Fitopatología (al
menos dos plantas por cada genotipo), etiquetar y trasladar en sus respectivos maceteros al
laboratorio.
2. Obtener hojas frescas (aproximadamente 60 – 100 mg tejido fresco) y colocar en servilletas
humedecidas con agua destilada estéril para mantener el turgor hasta su procesamiento.
3. En el mesón del laboratorio de molecular etiquetar los microtubos de fondo redondeado con
un marcador indeleble. Llenar los microtubos con nitrógeno líquido (N2) previamente a la
colocación del tejido vegetal. La aplicación de N2 debe repetirse facilitando que el tejido
congelado sea macerado completamente con pestillos esterilizados tipo lápiz en el fondo de los
microtubos.
4. Transferir 175µL de RNA Lysis Buffer (con BME añadido) a un microtubo estéril
debidamente etiquetado. Añadir la muestra macerada a polvo homogéneo una vez que el N2 se
haya evaporado totalmente. Mezclar por inversión suave.
LA RELACIÓN TEJIDO Y BUFFER DEBE SER APROXIMADAMENTE 30MG/175µL
RNA LYSIS BUFFER. PARA PROCESAR 100 MG TEJIDO AÑADIR 250 µL.
5. Añadir 350µL de ARN buffer de dilución. Mezclar por inversión 3-4 veces. (Si se inicia con
100 mg, en este paso añadir 450 µL ARN buffer de dilución.
6. Colocar en baño María (o en calor seco en un Thermomixer) a 70°C por 3 min. MAYOR
TIEMPO COMPROMETE LA INTEGRIDAD DEL ARN.
7. Centrifugar a máxima velocidad (12 a 14 mil g) en una microcentrífuga por 10 min.
8. Ensamblar una columna y un tubo de colecta por cada muestra procesada etiquetando
debidamente y disponer en una gradilla de microtubos.
9. Transferir el lisado aclarado a un tubo de microcentrífuga fresco pipeteando, sin perturbar el
desecho precipitado. El volumen debe estar cercano a 500 µL.
10. Añadir 200µL etanol 95% al lisado aclarado y mezclar pipeteando 3 – 4 veces. Transferir a
la columna de spin. Centrifugar a 12,000–14,000 × g por 1 min.
11. Tomar la cesta de spin y descartar el líquido del tubo de colecta. Colocar nuevamente la
cesta sobre el tubo de colecta. Añadir 600 µL de solución de lavado ARN a la columna.
Centrifugar a 12,000–14,000 × g por 1 min.
12. Descartar el líquido del tubo de colecta y colocar en la gradilla. Para cada aislamiento
preparar la mezcla de incubación para ADNasa combinando en un tubo estéril 40µL Buffer
Núcleo amarillo, 5µL 0.09M MnCl2 y 5µL enzima ADNasa I por muestra, en este mismo
orden. Preparar sólo la cantidad de mezcla ADNasa requerida y pipeteando cuidadosamente.
NO USAR VÓRTEX.

13. Aplicar 50µL de la mezcla fresca de incubación con ADNasa directamente a la membrana
dentro de la cesta de spin. El color amarillo ayuda a visualizar si la solución cubre la
membrana.
14. Incubar a 20–25°C por 15 min. Después de la incubación añadir 200µL de la solución de
parada de ADNasa a la cesta de spin. Centrifugar a 12,000–14,000 × g por 1 min.
15. Añadir 600 µL Solución de lavado de ARN y centrifugar a 12,000–14,000 × g por 1 min.
16. Vaciar el tubo de colecta y añadir 250µL de la solución de lavado de ARN. Centrifugar a
alta velocidad por 2 min. Remover la tapa de la cesta de spin para las siguientes
centrifugaciones.
17. Preparar un tubo de elución de 1,5 mL con tapa por cada muestra. Transferir la cesta de
spin al tubo y añadir 100µL agua libre de nucleasas (NFW) a la membrana. Centrifugar a
12,000–14,000 × g por 1 min. Tapar el tubo de elución con el ARN purificado y guardar a –
70°C.
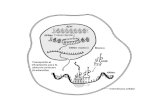
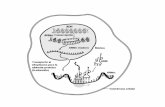
Figura 1. Protocolo rápido de extracción con el kit SV RNA extraction de Promega.

III. Determinación de rendimiento y calidad del ARN
Para determinar la concentración de ARN se puede medir la absorbancia a 260nm. Para ello se
usa un volumen de muestra entre 1 y 5μL. Como blanco se usa el NFW o ARNH2O-DEPC en
que hemos disuelto el ARN. Para calcular la cantidad de ARN hay que tener presente que
OD260 = 1 indica una concentración de ARN de 40μg/ml.
Puede usarse un espectrofotómetro. Se mide longitud de onda a 260nm, donde 1 unidad de
absorbancia es igual a 40µg de ARNss/ml. La pureza se estima relativa a 230, 260 y 280nm. Se
espera una relación de A260/A280 de 1.7–2.1 y A260/A230 de 1.8 – 2.2.
Para determinar el estado del ARN obtenido, si está degradado o no, conviene correr una
alícuota de aproximadamente 1μg en una electroforesis en agarosa en condiciones
desnaturalizantes. Se usan esas condiciones desnaturalizanes, y no las normales en geles de
agarosa para DNA, porque las moléculas de ARN tiene tendencia a formar estructuras
secundarias intracatenarias que si no se eliminan corren mal en la electroforesis y no forman
bandas nítidas. Esto supondría un problema ya que nuestro criterio para determinar que el
RNA obtenido no está degradado es observar bandas nítidas del ARN ribosomal en lugar de
manchas difusas.
RENDIMIENTO PROMEDIO DE ARN extraído de tejidos y células
(tomado del manual de Promega SV RNA TOTAL)
Muestras Cantidad tejido
máximo a
procesar
Rendimiento
promedio por
preparacion
(g)
Rendimiento
promedio por
mg de tejido
(g)
Relación
A260/A230
Relación
A260/A280
Bacteria
E. coli 1 x 10
9
células
36 na 1.6 2.0
Levadura
S. cerevisiae 4 x 10
7
células
19 na 1.7 2.1
Planta
S.
lycopersicum
30 mg 4.6 0,15 1,4 2,0
IV. Visualización del ARN
Si hay suficiente cantidad de ARN, se puede hacer una electroforesis en gel de agarosa
desnaturalizado (con formaldehido o glioxal). La relación de ARN ribosomal eucariótico 28S a
18S debe ser 2:1 aproximadamente, indicando baja degradación de ARN. Si ha ocurrido
degradación la relación se invierte, ya que el 28S ARNr se degrada a 18S.
Para la electroforesis se emplea un gel de agarosa al 3% que contiene formaldehído (eso es lo
que lo hace desnaturalizante): para 200mL de gel de agarosa pesar 3gr de agarosa,
Las muestras de ARN se calientan a 65ºC durante 10’ para que el ARN quede desnaturalizado
y se carga en los pocillos del gel.

V. Reacción de transcripción reversa (RT-PCR) usando la enzima MMLV- RT
La enzima Transcriptasa Reversa Moloney Murine Leukemia Virus (M-MLV RT) usa ARNss
o ADN en presencia de un primer o cebador para sintetizar una cadena de ADNc. Puede
sintetizar ADNc de hasta 7 kb.
Síntesis de la primera cadena de ADN complementario
1. Añadir todos los componentes a un tubo de microcentrífuga libre de nucleasas.
Volumen de reacción 20 µL
Total ARN o mRNA 1 ng - 5µg de ARN total ó 1 – 500 ng de
ARNm
Oligo (dT)12-18 (500 μg/mL), o 50–250 ng
random primers, o 2 pmole primer gen
específico
1 µL
10 mM dNTP Mix (10 mM cada dATP,
dGTP, dCTP y dTTP a pH neutro)
1 µL
Agua destilada estéril libre de nucleasas Hasta 12 µL
2. Calentar la mezcla a 65°C por 5 min para desarmar la estructura secundaria del ARN
total a usar como molde y enfriar rápido en hielo para prevenir que se forme la estructura
secundaria nuevamente.
3. Colectar el contenido del tubo por centrifugación breve para mantener la solución en el
fondo del microtubo.
4. Añadir los siguientes:
5X First-Strand Buffer 4 μL
0.1 M DTT 2 μL
RNaseOUT Recombinant Ribonuclease
Inhibitor (40 units/μL)
1 μL
Nota: Cuando se tienen menos de 50 ng de ARN muestra, es esencial la adición de RNaseOUT
5. Mezclar gentilmente e incubar la reacción a 37°C por 2 min.
6. Añadir 1 μL (200 unidades) de M-MLV RT, y mezcle por pipeteo gentilmente. Si usa
random primers, incube el microtubo a 25°C por 10 min.
Nota: Si usa menos de 1 ng de ARN, reduzca la cantidad de M-MLV RT en la reacción a 0.25
μL (50 unidades), y añada agua destilada estéril a 20 μL para completar el volumen.
7. Incubar durante 50 min a 37°C.
8. Inactivar la enzima transcriptasa reversa por choque térmico a 70°C por 15 min (puede
usar el bloque del termociclador).
El cDNA primera hebra sintetizado puede ser usado como molde para amplificación por
PCR. Sin embargo para algunos PCRs con productos de amplificación >1 kb se recomienda

remover el ARN complementario al cDNA utilizando 1 μL (2 unidades) de RNasa H e incubar
a 37°C por 20 min.
VI. Reacción de PCR
1. Se recomienda usar el 10% de la reacción de síntesis de la 1ra cadena ADNc (2 μL).
Este es el óptimo ya que añadir mayores volúmenes no aumenta la amplificación y puede
resultar en cantidades reducidas de productos PCR final.
2. Añadir los siguientes componentes en un tubo de reacción PCR:
Componente
Volumen (μL)
10X PCR Buffer 5
50 mM MgCl2 1.5
dNTPs (10 mM) 1
Cebador 1 (10 μM) 1
Cebador 2 (10 μM) 1
Taq DNA polymerase (5 U/μL) 0.2
cDNA template
(sintetizado con MMLV-RT)
2
NFW 38.3
Volumen final
50
Puede ser necesario optimizar la concentración de MgCl2 y dNTPs para cada par de cebadores.
Ciclo de PCR
Se recomienda utilizar una enzima polimerasa de alta fidelidad con capacidad de
lectura y corrección durante la polimerización de nucleótidos para enviar directamente a
secuenciar sin clonar.
94°C por 2 min para desnaturalizar.
94°C (15 a 40 ciclos de PCR).
La hibridación del primer y las condiciones de extensión dependen del primer per se y deben
determinarse empíricamente, generalmente se disminuyen o restan 2°C de la Tm (50mM Na+)
indicada por el fabricante.
Al finalizar la PCR se corre un gel de agarosa cuyo porcentaje varía de acuerdo al tamaño
esperado del fragmento. Como regla general hay que tener en cuenta que, a mayor
concentración menor es el tamaño de los fragmentos que se separarán. Según el objetivo
propuesto se elige el tampón, por ejemplo para clonaje o manipulación posterior del ADN se

recomienda el TAE, ya que el boro presente en el TBE buffer interacciona con los grupos
hidroxilos del polisacárido desoxiribosa formando complejos que dificultan la recuperación del
DNA. Para mejor resolución para fragmentos < 1kb se recomienda el TBE.
ANEXOS
ANEXO 1. RESUSPENSIÓN DE PRIMERS
FAVOR LEER LOS ENUNCIADOS ANTES DE INICIAR PROTOCOLO
Los primers son sintetizados de acuerdo a una secuencia determinada y entregados por los
fabricantes en estado liofilizado. Para comenzar a rehidratarlos o resuspenderlos se les aplica
un spin (en una microcentrífuga) para que todo el contenido se precipite al fondo del vial que
los contiene. Se debe calcular el volumen de resuspensión para que queden a una
concentración estándar de 100 µM.
A fin de garantizar su durabilidad, se aconseja que los primers sean resuspendidos utilizando
bufer Tris-EDTA (TE) 10:1, es decir 10 mM Tris:1 mM EDTA.
La resuspensión de los primers se realiza en una cámara de flujo laminar, utilizando puntas
(tips) con filtro estéril.
Preparación de Tris pH 8.0
El buffer (solución tampón) se puede preparar con Tris HCl o Tris Base. En este caso se
utilizará Tris HCl (PM: 157.56 g/mol). Tris HCl (Tris-Hydrochloride, Molecular Biology
Grade)
157.56 g 1000 mL
X 100 mL
R= 15.756 g
1g 1000 mg
15.756 g X
R= 15756 mg
Atención: Los 15756 mg aún están en concentración 1M requiriéndose 10 mM.
Por lo tanto, sabemos que 1M=1000 mM entonces:
1M 1000 mM
X 10 mM (lo deseado)
R= 0.01 M
A continuación se detallan los cálculos de la preparación:
Ah:
15756 mg 1 M

Seguidamente, se preparará el Tris HCl a una concentración de 10 mM pH 8.0 y 1 mM EDTA
Las cantidades de ambos componentes: Tris HCl y EDTA que se pesaron, se deberán diluir
inicialmente en 90 mL de agua ultrapura (Milli Q) autoclavada y ajustado el pH a 8.0 con
NaOH 1M (Base) y HCl 1M (Ácido).
IMPORTANTE:
El NaOH 1M y el HCl 1M se deben preparar con agua Milli Q autoclavada; estas dos
soluciones NaOH 1M y HCl 1M deben ser soluciones nuevas o de poco uso, no utilizar
soluciones viejas.
Además, es importante recordar que el EDTA sólo entrará en solución cuando el pH este
próximo a 8.0.
Seguidamente se complementará el volumen final a 100 mL con agua MilliQ autoclavada.
Posteriormente el tampón TE preparado y con pH ajustado a 8.0 se completa a volumen 100
mL se esterilizará en el autoclave.
Preparación del EDTA (Disodium salt, Molecular Biology Grade, Promega, PM: 372.20)
372.20 g 1000 mL
X 100 mL
R= 37.22 g
Los 37220 mg están calculados para 100 mL. Este peso (37220 mg) aún está a una
concentración de 1M por lo tanto se requiere que esté a 1mM.
1M 1000 mM
X 1 mM
R= 0.001 M
Seguidamente,
37220 mg 1 M
X 0.001 M
R= 37.22 mg de EDTA
1 g 1000 mg
37.22 g X
R= 37220 mg
EDTA 1mM pH 8.0 con volumen final (total) de 100 mL

RESUSPENSIÓN DE LOS PRIMERS
ATENCIÓN: Los primers deben quedar a una concentración stock de 100 µM.
Para llegar a 100µM se agregará un volumen de TE en µL equivalente a la multiplicación por
10 al valor de concentración en nanomoles (nmoles) detallado en el certificado de análisis de
cada primer sintetizado.
Ejemplo de Cálculo:
PRIMERS nmoles Volumen de TE a
adicionar a los Primers
liofilizados
RSNV F 45.8 45.8 x 10 = 458
µL
RSNV R 52.3 52.3 x 10 = 523
µL
Una vez adicionado el volumen del TE correspondiente se les dará agitación con el vortex, se
centrifuga a máxima velocidad por 30 s y se dejará a 4ºC hasta el día siguiente (overnight).
Los primers resuspendidos de esta manera quedarán a una concentración de 100 µM por lo
tanto, se les considerará como SOLUCION STOCK DE PRIMERS.
Para el STOCK DE TRABAJO DE LOS PRIMERS éstos deberán estar a una concentración
de 40 µM. La dilución se hará a partir del STOCK DE PRIMERS utilizando agua Milli Q
previamente esterilizada en autoclave.
Cálculos:
Se prepararán 100 µL de volumen final con una concentración final del primer a 10 mM:
Concentración 1 x Volumen 1 = Concentración 2 x Volumen 2
C1 x V1 = C2 x V2
100 mM x V1 = 10mM x 100 µL
V1 = 10mM x 100 µL
100 mM
V1 = 10 µL
De donde, de la solución stock del primer que se encuentra en 100 µM se tomarán 10 µL. A los
10 µL se le adicionarán 90 µL de agua Milli Q autoclavada para llegar al volumen final de 100
µL.
(IMPORTANTE: Este procedimiento de cálculo se deberá hacer para los primers en
Forward y Reverse).
A partir de ahora llamaremos PRIMERS DE TRABAJO estos primers diluidos con volumen
final de 100 µL y con concentración de 10 µM.
A partir del PRIMER DE TRABAJO se tomará el volumen adecuado para la reacción de
PCR.

ANEXO 2
Preparación e instrucciones del kit de SV RNA Total (Promega)
El Sistema SV RNA Total (Promega) contiene reactivos suficientes para 50 extracciones de
ARN total de tejidos, células o sangre. Incluye:
• 2 paquetes de tubos de recolección (25/paq)
• 2 paquetes de tubos de elución (25/paq)
• 2 paquetes columnas Spin (25/paq)
• 50 mL RNA Lysis Buffer (RLA)
• 20mL RNA Dilución Buffer (RDA) (azul)
• 2mL β-mercaptoetanol (BME) (48.7%)
• 1 vial DNasa I (liofilizada)
• 250µL MnCl 0.09M
• 2.5mL Buffer núcleo (YCB) (amarillo)
• 5.3mL Solución de parada de la DNasa (DSA) (concentrada)
• 58.8mL Solución de lavado de RNA (RWA) (concentrada)
• 13mL agua libre de nucleada (NFW)
NOTA 1: El RNA Dilución Buffer (RDA) y el Buffer núcleo (YCB) están coloreados en azul
y amarillo respectivamente para poder ser diferenciados fácilmente. El color amarillo del YCB
permite al usuario visualizar si la membrana ha sido completamente cubierta por la mezcla de
ADNasa en el paso de la ADNasa. Los tintes no tienen ningún efecto en la calidad o
comportamiento posterior del ARN.
Condiciones de almacenamiento: Una vez se le haya añadido β-Mercaptoetanol (BME) al
buffer de lisis (RLA) debe almacenarse a 4°C. Tapar muy bien entre usos.
Resto de componentes puede almacenarse a 22-25°C.
NOTA 2: No combine o reemplace componentes de SV Total RNA Isolation System con
componentes de otro kit.
Precaución: Tiocianato de Guanidina y β-mercaptoetanol son soluciones tóxicas. Use guantes
y siga procedimientos estándares de seguridad cuando trabaje con estas soluciones. Igualmente
para el manejo de desechos tóxicos.
Materiales a ser provistos por el usuario
• Homogenizador (Mortero y pestillo)
• 95% etanol, libre de ARNasa
• Microcentrífuga
• Baño de maría o bloque de calentamiento pre-calentado a 70°C

Preparación de reactivos del kit
ADNasa
ATENCION: Añadir la cantidad indicada de NFW incluida en el kit al vial de ADNasa I
liofilizada.
Mezclar suavemente el vial de la solución. La ADNasa rehidratada debe dispensarse en 5 – 10
alícuotas de trabajo usando tubos estériles libres de ARNasa. Por cada purificación de ARN se
requieren 5 µL de ADNasa I rehidratada. Almacenar a – 20°C.
PRECAUCION: NO se debe hacer vórtex a la solución de ADNasa I
NO congelar - descongelar más de tres veces la solución de ADNasa I
Buffer de lisis de ARN (RLA)
Añadir 1 mL de BME a 50 mL de RLA. Marcar la botella indicando que ya se ha añadido.
Almacenar a 4°C. Tapar bien entre usos.
Solución de lavado de ARN (RWA)
Añadir 100 mL de etanol 95% a la botella que contiene 58,8 mL de RWA concentrada.
Después de añadir el etanol, marcar la botella para indicar que ya se realizó este paso. Tapar
BIEN. Conservar a 22-25°C.
Solución de parada de la ADNasa
Añadir 8 mL de etanol al 95% a la botella con 5,3 mL de solución concentrada. Después de
añadir el etanol, marcar la botella para indicar que ya se realizó este paso. Tapar BIEN.
Conservar a 22-25°C.
AISLAMIENTO Y PURIFICACIÓN DE ARN
Para mejores resultados se recomienda usar muestras frescas cuando se procesa tejido.
Muestras viejas pueden rendir menos ARN total. Si es necesario se debe congelar las muestras
inmediatamente después de ser colectadas en nitrógeno líquido (N2) y almacenarlas a -70°C
para uso posterior.
Muestras ya homogeneizadas en el buffer de lisis RLA pueden almacenarse a -20°C o -70°C.
Muestras valiosas se sugiere que una parte sea reservada a -70°C en el caso de que se pierda la
muestra durante la purificación.
Debido a la toxicidad de los químicos usados en el proceso de purificación y la presencia de
ARNasas deben usarse guantes durante todo el proceso de lisis y purificación.

CONSIDERACIONES GENERALES
El ARN es por lo general más sensible que el DNA a la degradación por nucleasas. Las
nucleasas que actúan específicamente sobre el ARN, las ARNsas, son abundantes en las
muestras biológicas y también están presentes en las manos y en general por toda la superficie
de nuestros cuerpos. Además, muchas ARNsas son enzimas bastante estables y difíciles de
inactivar, incluso autoclavando las soluciones. Por ello, a la h de extraer ARN de un material
biológico hay que procurar eliminar rápidamente las que están presentes en el material de
partida con soluciones desnaturalizantes en el tampón de extracción y a partir de ese momento
evitar que se introduzcan nuevas ARNsas procedentes de nosotros mismos o presentes en las
soluciones que empleemos.
Las precauciones de orden general a tomar durante la extracción y manipulación del ARN son:
a) Usar siempre guantes y cambiarlos cada vez que se ensucien,
b) No hablar cuando se estén pipeteando soluciones con ARN o manipulando material o
reactivos que se vayan a emplear en su preparación. En la saliva hay ARNsas,
c) Emplear tubos eppendorf y puntas de micropipeta que no se hayan tocado con las manos
desnudas y que hayan sido convenientemente tratadas para eliminar restos de ARNsas,
d) Usar material de vidrio tratado con soluciones inactivadoras de ARNsas y manipularlo a
partir de ese momento siempre con guantes y cuidando de no contaminarlo con ARNsas,
e) Emplear soluciones preparadas con H2O-DEPC en vez de H2O normal. El DEPC (dietil
pirocarbonato) es un inhibidor de ARNsas. Se vende como una solución que tiene que añadirse
al H2O a una concentración final del 0.1%. Tras ello, se deja una noche agitando para que se
mezcle bien e inactive por completo las ARNsas y después, y antes de usarlo, se autoclava
(120ºC por 30 min) un par de veces el H2O-DEPC para eliminar el DEPC que ya ha cumplido
su misión (el DEPC produce modificaciones químicas en el ARN que lo degradan pero se
elimina con facilidad al autoclavar).
PREPARACIÓN DE UN AMBIENTE LIBRE DE RIBONUCLEASAS
Es sumamente difícil inactivar ribonucleasas, por ello debemos evitar introducirlas o activarlas
durante el procedimiento de aislamiento de ARN. Esto es muy importante ya que el material

inicial puede ser difícil de obtener o irreemplazable. Debemos prevenir la contaminación
accidental de las muestras. Para ello debemos seguir las siguientes pautas:
1. La primera fuente de ribonucleasas son las manos y bacterias o mohos que están
suspendidas en el aire. Para prevenir contaminación se deben usar técnicas asépticas o
estériles para el manejo de los reactivos presentes en el kit. Usar guantes todo el
tiempo.
2. Siempre que sea posible utilizar microtubos y otros insumos plásticos desechables
esterilizados para el manejo de ARN. Generalmente son materiales libres de AARNsas
y no requieren pretratamiento para inactivarlas. En el kit se proveen tubos de elución
autoclavados.
3. Recipientes de vidrio y plástico debe tratarse antes de usarla para que estén libres de
ARNasas. Para ello se recomienda el siguiente procedimiento: Colocar los recipientes
de vidrio a 200°C toda la noche. Los recipientes de plástico se enjuagan con NaOH 0.1
N y 1 mM EDTA; luego se enjuagan con agua esterilizada libre de ARNasa (NFW).
4. Tratar las soluciones a emplear añadiendo 0.1% dietilpirocarbonato (DEPC) e
incubando toda la noche a temperatura ambiente. Luego autoclavar por 30 min para
remover trazas de DEPC.
Nota: DEPC reacciona rápidamente con las aminas y NO PUEDE USARSE para tratar Tris
buffers.
Precaución: DEPC puede ser carcinogénico y debe usarse en cámara de extracción de gases.
La recomendación entonces es tratar el agua primero con DEPC y luego de que esta sustancia
es inactivada por autoclave, disolver el Tris para hacer el buffer apropiado.
5. Limpiar todas las superficies con 1% hipoclorito de sodio. Inmediatamente con alcohol
75%.
6. Para limpiar morteros y mazos usar NaOCl2 1% y EtOH 75%. Luego autoclavar y
conservar a -20°C.
7. Limpiar los extremos de las micropipetas con un algodón impregnado con agua
oxigenada comercial.
Purificación directa de ARN
El éxito en el aislamiento de ARN intacto requiere de 4 pasos esenciales: Rompimiento
efectivo de las células o tejido; desnaturalización de complejos de núcleo proteínas;
inactivación de ribonucleasas endógenas (se liberan inmediatamente de las membranas de los
organelos al romper las células); y remoción de ADN y proteínas contaminantes.

El kit combina las propiedades de guanidina tiocianato (GTC) y β-mercaptoetanol para
inactivar las ribonucleasas presentes en extractos celulares. GTC en asociación con SDS, actúa
para romper complejos de núcleo proteínas, permitiendo que el ARN se libere a la solución
libre de proteínas.
En altas concentraciones de GTC ocurre precipitación selectiva de proteínas celulares mientras
el ARN permanece en la solución. Después de la centrifugación el ARN se precipita con el
etanol uniéndose a la superficie de silica de las fibras de vidrio de la cesta de spin, mientras
que los desechos de proteínas y componentes celulares pasan. Las sales caotropicas favorecen
la adsorción de los ácidos nucleicos al silica. ADNasa I libre de ARNasa se aplica
directamente a la membrana de silica para digerir ADN genómico contaminante. El ARN
ligado al silica se purifica de sales, proteínas contaminantes e impurezas celulares en los pasos
de lavado simple. Finalmente el ARN se eluye de la membrana por adición de NFW.
El procedimiento rinde una fracción esencialmente pura de ARN total luego de una ronda de
purificación sin extracciones orgánicas o precipitaciones.
Capacidad de procesamiento: No se recomienda procesar más de 60 mg de tejido ya que se
tapan las membranas y la purificación es pobre. El volumen máximo de lisado que puede
procesarse en cada filtro (cesta de spin) es de 175µL.
AISLAMIENTO DE ARN TOTAL A PARTIR DE TEJIDO VEGETAL
1. Transferir 175µL de RNA Lysis Buffer (con BME añadido) a un microtubo estéril al cual se
añadirá la muestra. USAR PIPETAS LIBRES DE ARNASAS Y GUANTES para minimizar el
riesgo de contaminación con ARNasas.
2. Pesar el tubo que contiene el RNA Lysis Buffer y anote el peso.
3. Congelar el tejido vegetal en nitrógeno líquido y macerar con mortero y pestillo.
4. Transferir nitrógeno líquido y tejido macerado a un microtubo estéril. Permitir que el N2 se
evapore e inmediatamente transferir el tejido al tubo que contiene 175µL RNA Lysis Buffer.
Mezcle por inversión.
5. Pese el tubo que contiene tejido y buffer. Calcule el peso por diferencia. En general la
relación debe ser aproximadamente 30mg/175µL.
6. Añadir 350µL de ARN buffer de dilución. Mezclar por inversión 3-4 veces. Colocar en baño
María a 70°C por 3 min. Mayor tiempo compromete la integridad del ARN.
7. Centrifugar a máxima velocidad (12 a 14 mil g) en una microcentrífuga por 10 min.
Purificación de ARN por centrifugación
Use guantes y abra el paquete cuidadosamente. Tome una columna y un tubo de colecta para
cada muestra procesada. Decida si va a remover las tapas (con un movimiento de torsión se
pueden desprender). Si las deja, deben estar cerradas durante la centrifugación. Etiquete el tubo

y coloque este ensamble en una gradilla de microtubos. Es importante etiquetar los tubos para
mantener la identificación de las muestras. Use guantes para manejar los tubos.
1. Transfiera el lisado aclarado a un tubo de microcentrífuga fresco pipeteando. No perturbe el
desecho precipitado. El volumen debe estar cercano a 500 µL.
2. Añada 200µL etanol 95% al lisado aclarado y mezcle pipeteando 3 – 4 veces- Transfiera
esta mezcla a la columna de spin. Centrifugar a 12,000–14,000 × g por 1 min.
3. Tome la cesta de spin y descarte el líquido del tubo de colecta. Coloque nuevamente la cesta
sobre el tubo de colecta. Verifique que la solución de lavado de ARN ha sido diluida con
etanol como indicado. Añada 600 µL de solución de lavado ARN a la columna. Centrifugar a
12,000–14,000 × g por 1 min.
4. Descarte el líquido del tubo de colecta nuevamente y coloque en la gradilla. Para cada
aislamiento a realizar prepare la mezcla de incubación para ADNasa combinando en un tubo
estéril 40µL Buffer Núcleo amarillo, 5µL 0.09M MnCl2 y 5µL enzima ADNasa I por muestra
en un tubo estéril, en este mismo orden. Prepare sólo la cantidad de mezcla ADNasa requerida
y pipetee cuidadosamente. Mezcle pipeteando gentilmente: NO use vórtex. Mantenga la
ADNasa I en hielo mientras se descongela. Aplique 50µL de la mezcla fresca de incubación
con ADNasa directamente a la membrana dentro de la cesta de spin. Asegúrese que la solución
está en contacto y cubre toda la membrana. La solución es amarilla para ayudar a visualizarla.
NO mezcle el buffer núcleo amarillo y el MnCl2 antes de usarlos en paso 4. Deben conservarse
separados y mezclarse en fresco sólo cuando se van a usar.
5. Incubar por 15 min a 20–25°C. Después de la incubación añadir 200µL de la solución de
parada de ADNasa (verifique que contiene etanol)) a la cesta de spin; y centrifugue a 12,000–
14,000 × g por 1 min. No es necesario vaciar el tubo de colecta antes del siguiente paso.
6. Añada 600 µL Solución de lavado de ARN (con etanol añadido) y centrifugue a 12,000–
14,000 × g por 1 min.
7. Vacíe el tubo de colecta y añada 250µL Solución de lavado de ARN (con etanol añadido);
centrifugue a alta velocidad por 2 min.
8. Si no lo ha hecho aún, remueva la tapa de la cesta de spin torciéndola.
9. Para cada muestra, remueva un tubo de elución de 1,5 mL con tapa. Transfiera la cesta de
spin a ese tubo y añada 100 µL NFW a la membrana. Asegúrese de cubrir completamente la
membrana con el agua. Coloque los arreglos de cestas de spin en la centrífuga, con las tapas de
los tubos de elución hacia afuera. Centrifugue a 12,000–14,000 × g por 1 min. Remueva la
cesta de spin y descártela. Tape el tubo de elución que contiene el ARN purificado y almacene
a –70°C.
Nota: No se recomiendan volúmenes de elución menores que 100µL. Si se requiere concentrar
el RNA se puede usar secado en vacío y resuspenderlo en menor volumen de agua. Si es
esencial máxima recuperación de ARN se recomienda una nueva elución en un segundo tubo

estéril con 100µL adicionales de NFW seguido por centrifugación a 12,000–14,000 × g por 1
min. Se recobra un 10–20% más de ARN.
ANEXO 3.
MULTIPLOS
SUBMULTIPLOS
Factor Prefijo Símbolo Factor Prefijo Símbolo
1018
Exa E 10-1
deci d
1015
Peta P 10-2
centi c
1012
Tera T 10-3
mili m
109
Giga G 10-6
micro µ
106
Mega M 10-9
nano n
103
Kilo k 10-12
pico p
102
Hecto h 10-15
femto f
10 Deca da 10-18
atto a
REFERENCIAS BIBLIOGRÁFICAS
Koressaar T, Remm M (2007) Enhancements and modifications of primer design program
Primer3 Bioinformatics 23(10):1289-91
Untergasser A, Cutcutache I, Koressaar T, Ye J, Faircloth BC, Remm M, Rozen SG (2012)
Primer3 - new capabilities and interfaces. Nucleic Acids Research 40(15):115