
Trastornos de la hemostasia
63
TRASTORNOS DE LA HEMOSTASIA EN PEDIATRIA Dra. Pamela Bolaños Melara Dr. Edgardo Ortiz Castaneda RESIDENTES DE TERCER AÑO HNNBB
-
Upload
edgardomdneo -
Category
Documents
-
view
6.295 -
download
13
Transcript of Trastornos de la hemostasia

TRASTORNOS DE LA HEMOSTASIA EN PEDIATRIA
Dra. Pamela Bolaños MelaraDr. Edgardo Ortiz CastanedaRESIDENTES DE TERCER AÑOHNNBB

OBJETIVOS
Enunciar los conceptos básicos de la hemostasia
Identificar los principales métodos diagnósticos de los trastornos hemorragíparos
Establecer pautas de aproximación ante un paciente con sospecha de trastorno de la coagulación
Descibir los trastornos hemorragíparos más frecuentes en edad pediátrica (PTI, hemofilias, Enfermedad de VonWilembrant)

HEMOSTASIA
Proceso activo que coagula la sangre en las zonas donde se haya producido una lesión de los vasos sanguíneos.
Limita la extensión del coágulo
Determina la desaparición del coágulo

Numerosas enzimas participan en su regulación
El ión calcio es escencial para su funcionamiento
Existen dos vías de activación

Secuencia…
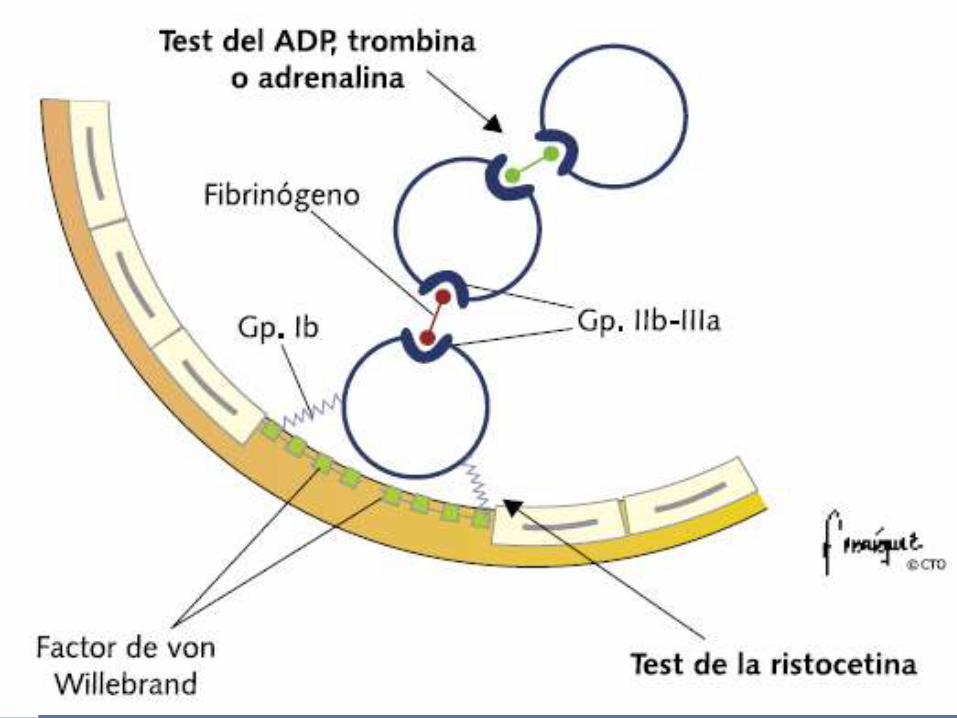
HEMOSTASIA PRIMARIA
1. Vasoconstricción refleja
2. Adhesión plaquetaria (FVW)
3. Activación plaquetaria
4. Agregación plaquetaria

HEMOSTASIA SECUNDARIA
1. Tiene como objetivo formar un coágulo estable de fibrina
2. Se realiza principalmente por los factores de la coagulación
3. Intervienen además los fosfolípidosplaquetarios y de los tejidos y el calcio

Sistema de fibrinolisis
Destrucción de la fibrina
Se realiza por medio de la plasmina

Aproximación diagnóstica
El tipo de sangramiento orienta hacia la causa
Sangramiento de mucosas (defectos plaquetarios, VW, disfibrinogenemias)
Hemorragia de tejido blando o hemartrosis (hemofilias)
Hemorragia umbilical o retraso en la curación de heridas (FXIII)

Aproximación diagnóstica
Antecedentes familiares de sangramientoexcesivo
Hasta 30% de los diagnósticos nuevos de hemofilia no tienen historia familiar


Evaluación de laboratorio
Debe recogerse una buena muestra
No usar heparina
Se debe recoger 4,5 ml en tubo de ensayo con citrato de sodio 0,5 ml (relación 1:10)
Se debe procesar en las primeras 2 horas (4 si esta refrigerada)

Pruebas de cribado
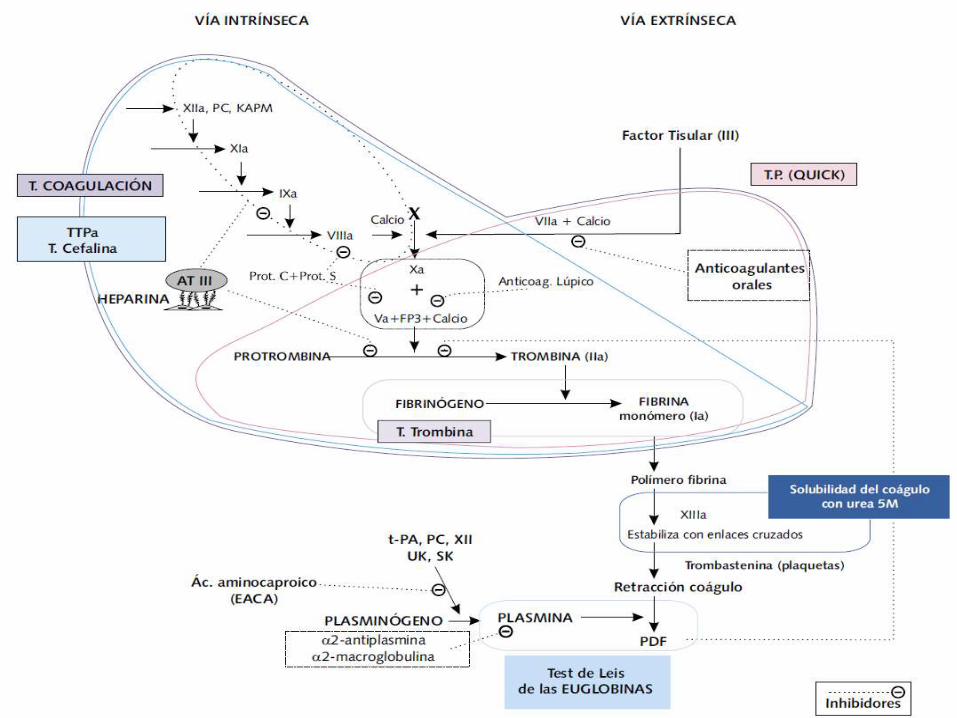
Tiempo de protrombina
Tiempo de tromboplastina tisular activado
Recuento de plaquetas
Frotis de sangre periférica
Dosificación de factores específicos de la coagulación

Tiempo de protrombina TP
Se realiza añadiendo un “reagente” de tromboplastina que contiene Ca
Se mide el tiempo de formación del coágulo por un medio automatizado
Es por lo general de 10 a 11 seg
Mide la actividad de los factores:
a) Fibrinógeno (I)
b) Protrombina (II)
c) V, VII y X

Se prolonga el tiempo hasta que uno de los factores involucrados esta por debajo del 30%
O que el fibrinógeno es menor a 100 mg/dl

Tiempo de tromboplastina parcial activada
Se añaden fosfolípidos sin factor tisular
Además introduciendo activación prolongada de los factores de contacto (F. XI, XII, prekalikreina, cininógeno de alto peso molecular)
Por medio de preincubación con factores de superficie (celita, kaolin, silica)
Posteriormente se agrega cloruro de calcio y se mide el tiempo en formar el coagulo.

TTPA
Mide los factores:
a) Fibrinógeno (I)
b) Protrombina (II)
c) V, VIII, IX, X, XI, XII
d) Calicreina
e) Cininógeno de bajo peso molecular

Un paciente con sangramiento clínico con prolongación del TTPA, se debe a deficiencia de F. VIII, IX y XI.
En la mayoría de casos se prolonga el TTPA hasta que el F. VIII ha descendido a menos del 35%.
Valores de referencia son por lo genera de 26 a 35 seg. Pero varía según el método.

Tiempo de Trombina
Mide el tiempo de conversión del fibrinógeno en fibrina inducida por la protrombina.
Se agrega trombina bovina a la muestra citratada
Valor medio entre 11 y 15 seg.
Se prolonga si el fibrinógeno esta disminuido en cantidad o calidad.

Conteo de plaquetas
Rango normal de 150 a 450 mil /cc.
Puede se determinado por medios opticos o electrónicos

Tiempo de sangrado
Se realiza por dos incisiones cutáneas en el antebrazo tras colocar un esfingomanómetroa 40 mmHg
Tiempo normal entre 4 y 8 min.
Se prolonga con plaquetopenia por debajo de 100 mil o por defectos en la función plaquetaria.
Se ha sustituido por el PFA

Disificación de factores de coagulación
Cuantifica de forma específica la concentración de los factores.
Se realiza por medio de espectroscopía

Alteraciones Plaquetarias
Disminución en el número de plaquetas
a) Trombocitopernias centrales
b) Trombocitopenias periféricas
Alteración en la función plaquetaria

Trombocitopenia
Reducción del volumen plaquetario por debajo de 150 mil/mm3
1. Por disminución en la trombopoyesis
2. Por secuestro esplénico o en otros órganos
3. Por aumento en su destrucción por causas inmunitarias o no inmunitarias

Disminución de la trombopoyesis Enfermedades hereditarias
Enfermedades adquiridas
a) Anemia aplásica
b) Sindrome mielodisplásico
c) Enfermedad infiltrativa
d) Osteopetrosis
e) Deficiencia nutricional
f) Inducida por fármacos
g) Hipoxia neonatal o insuficiencia placentaria

Secuestro
Hiperesplenismo
Hipotermia
Quemaduras

Aumento en la destrucción plaquetaria Síndromes de consumo primario
a) Inmunitario
b) No inmunitario
Síndromes de consumo combinado de plaquetas y fibrinógeno
a) CID
b) Kasabach – Merrit
c) Síndrome hemofagocítico de origen viral

Purpura TrombocitopenicaIdiopática Es la causa más frecuente
Anticuerpos dirigidos contra la superficie plaquetaria
Inducida por infección viral (1-4 sem)
Se asocia a infecciones por VEB y VIH

Manifestaciones
Presencia de petequias y púrpura en niño de uno a cuatro años
Los hallazgos clínicos se relacionan con el grado de plaquetopenia (menos de 20 mil)
En el hemograma y FSP las demás líneas se encuentran respetadas

Pruebas de Laboratorio
MO: Megacariocitos normal o aumentado, las demás líneas están conservadas
Indicaciones de AMO:
1. Formula leucocitaria anormal
2. Anemia no explicada
3. Indicios de enfermedad medular.

Diagnostico diferencial
Inducida por medicamentos
Secuestro esplénico por Htportal inadvertida
Fanconi en sus fases iniciales
Puede se manifestación de otras enfermedades sistemicas (LED, VIH)

Tratamiento
Su utilidad es en el ascenso más rápido de plaquetas
Se reserva la transfusión plaquetaria para casos graves

Tratamiento.
Asesoramiento familiar
Ig IV, 0,8 a 1 g/kg/día por 1-2 días
Tratamiento AntiD IV. 50-75 mg/kg en pacientes Rh positivo
Prednisona: 1-4 mg/kg/día. Se debe descartar otras causas por medio de Mos.
Se cumple por dos o tres semanas y luego se retira paulatinamente.

Esplenectomía
PTI crónica mayores de cuatro años
PTI complicada por hemorragia severa y recuento plaquetario que no sube a pesar de tratamiento médico.

PTI crónica.
Persistencia por más de 6 meses
La esplenectomía induce remisión en 64 –48%

Deficiencia de los Factores de la Coagulación
Hemofilias (A, B y C)
Deficiencia de Factor VII
Deficiencia de Factor X
Deficiencia de Protrombina
Deficiencia de Factor V
Deficiencia Combinada de Factor V y VIII
Deficiencia de Fibrinógeno
Deficiencia de Factor XIII
Deficiencia de Antiplasmina

Hemofilia A y B
GENERALIDADES:
Enfermedades hemorrágicas hereditarias graves mas frecuentes.
Ligadas al cromosoma X
Gen Factor VIII: brazo largo cromosoma Xq28
Gen Factor IX: cromosoma Xq27
Vida media mas larga del F IX

Hemofilias A y B
Complejo activador del factor X (Tenasa: Ca, fosfolÍpido, factores VIII y IX).
Factor VIIa + Factor Tisular activan el F IX para iniciar la coagulación.
TP mide la activación del FX por el FVII, siendo normal el TP en deficiencia de FVIII y IX.

Hemofilias A y B
FISIOPATOLOGIA:
Retraso en la formación del coagulo
Coagulo blando y friable (inadecuada generación de trombina).
Hemorragia intraarticular cesa por taponamiento
Hemorragia abierta: coágulo friable con tendencia a producir nuevas hemorragias.

Hemofilias A y B
MANIFESTACIONES CLINICAS:
Manifestaciones fetales o desde el nacimiento
2% con hemorragias intracraneales
30% sangran con la circuncisión
Síntomas evidentes: equimosis, hematomas intramusculares y hemartrosis.

Hemofilias A y B
MANIFESTACIONES CLINICAS:
Hemartrosis: tobillos, rodillas y codos. Tumefacción, acumulo de liquido, sensación de calor y hormigueo articular.
Articulación DIANA
Hemorragias musculares: M. psoas-iliaco
Hemorragias del SNC, vía respiratoria alta, gastrointestinal.

Hemofilias A y B
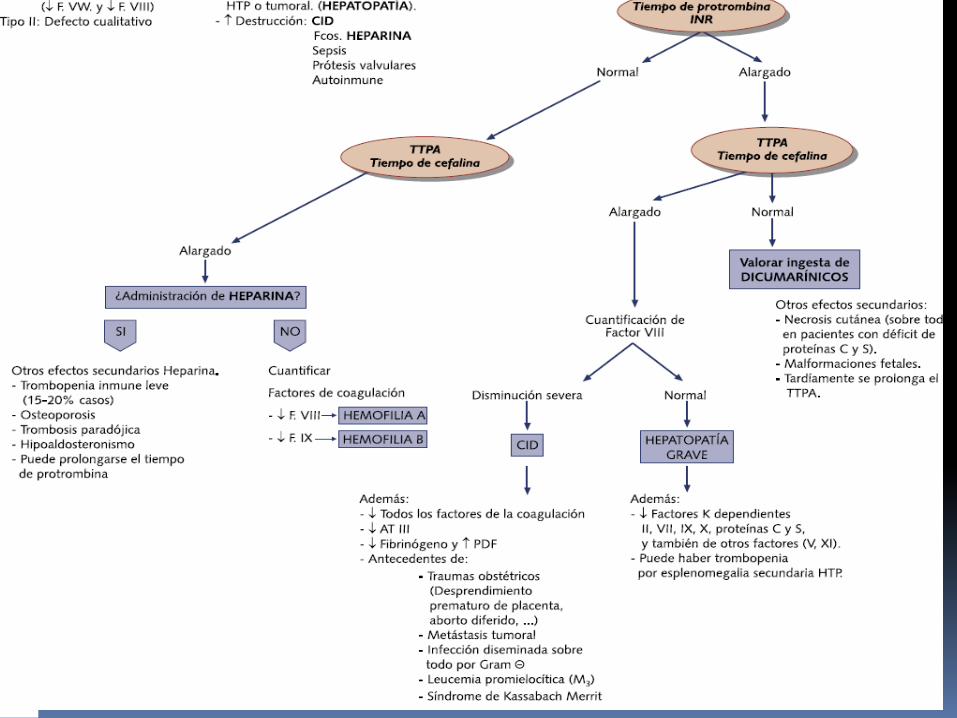
PRUEBAS DE LABORATORIO:
TTPA prueba de detección analítica
Hemofilia grave: TTPA 2-3 veces mayor
Inhibidor (Anticuerpo) de F VIII o IX: mezcla de plasma normal con la del paciente no corrige el TTP.
Análisis especifico de FVIII Y F IX confirman el Dx.
Inhibidores: dirigidos contra el lugar de coagulación activa.

EVALUACIÓN
TTPA TP normal
TP TTPA normal
TP TTPA
Hemofilia?Dosif FVII y IX
Dosif. FVII Dosif. FV
HemofiliaA
HemofiliaB
Deficitcongentito de
Fact VII
Deficitcongenitode
Fact V

Hemofilias A y B
DIAGNOSTICO DIFERENCIAL:
Manifestaciones hemorrágicas graves: trombocitopenia grave.
Trastornos graves de la Fx plaquetaria:
Sd. Bernard-Soulier
Tromboastenia de Glanzmann.
Enfermedad de von Willebrand tipo 3
Deficiencia de vitamina K.

Hemofilias A y B
GENETICA Y CLASIFICACION:
Afecta a 1:5000 varones, de estos el 85% con déficit de FVIII y 10-15% de FIX.
Afecta a todos los grupos étnicos.
Gravedad se clasifica en función de la concentración basal del paciente de F VIII o FIX (1ml de plasma normal= 1UI de factor).
Tienen disminuida la proteína factor de coagulación o esta no es funcional

Hemofilias A y B
CLASIFICACION:
Hemofilia grave: actividad <1%, hemorragias espontáneas.
Hemofilia moderada: 1-5%, hemorragias por traumas leves.
Hemofilia leve: >5%, por traumatismos importantes.
Concentración hemostática de F VIII: >30-40% y de F IX >25-30%.

DEFICIENCIA DE FACTOR VIII Y IX
TRATAMIENTO:
Dosis de FVIII (UI)=% deseado (↑de FVIII)x peso (kg)x 0.5
Dosis de FIX (UI)=% deseado (↑ de FIX)x peso (kg)x 1.4

Hemofilias A y B
TRATAMIENTO:
Hemorragia leve a moderada: incrementar la concentración del factor hasta niveles hemostáticos del 35-50%.
Hemorragias graves: se debe intentar alcanzar niveles de actividad del 100%
Tratamiento profilactico

TIPO DE HEMORRAGIA HEMOFILIA A HEMOFILIA B
HEMARTROSIS 40UI/Kg 1º día, 20UI/Kg días 2,3,y 5; con fx articular normal. Días alternos.
60-80UI/Kg 1º día, luego 40UI/Kg días 2,4. Valorar días alternos (7-10)
HEMATOMA MUSCULAR O SUBCUTANEO
20UI/Kg, valorar días alternos hasta su resolución
40UI/Kg, valorar usar tx cada 2-3 días hasta su resolución.
EXTRACCION DENTAL 20UI/Kg, tx antifibrinolitico, extraer dientes de leche sueltos.
40UI/Kg, , tx antifibrinolitico, extraer dientes de leche sueltos.
EPISTAXIS Presión por 15-20 min., taponar con gasa con vaselina, tx antibibrinolitico. 20UI/Kg .
Presión por 15-20 min., taponar con gasa con vaselina, tx antibibrinolitico. 30UI/Kg .
CIRUGIA MAYOR, HEMORRAGIA CON RIESGO VITAL
50-75UI/Kg en bolo, infusión 2-4 UI/Kg /h 1º día, luego 2-3 UI 5-7 días
120UI/Kg en bolo, luego 50-60UI/Kg 5-7 días
HEMORRAGIA PSOAS-ILIACO 50UI/Kg, luego 25UI/Kg cada 12 hrs hasta q este asintomático. 20UI/Kg días alternos hasta 14 días
120UI/Kg, luego 50-60UI/Kg cada 12-24 hrs, hasta q este asintomático. 40-50 UI/Kg días alternos por 14 días.
HEMATURIA Reposo en cama, líquidos; si no hay mejoría 20UI/Kg , no mejoría prednisona (no VIH).
Reposo en cama, líquidos; si no hay mejoría 40UI/Kg , no mejoría prednisona (no VIH).
PROFILAXIS 20-40UI/Kg días alternos hasta conseguir concentraciones > 1%.
30-50UI/Kg cada 2-3 días hasta conseguir concentraciones >1%.

DEFICIENCIA DE FACTOR XI (HEMOFILIA C)
Enfermedad autosómica, con síntomas hemorrágicos leves a moderados.
La gravedad de la hemorragia no es proporcional a la cantidad de factor existente.
No existe ningún concentrado de FXI aprobado, por lo que es necesario usar PFC.
La deficiencia de FXI se confirma mediante análisis específico.

DEFICIENCIA DE FACTOR XI (HEMOFILIA C)
La infusión de plasma con 1UI/Kg suele incrementar la concentración plasmática a 2%.
La vida media del FXI es ≥48 hrs.

ENFERMEDAD DE VON WILLEBRAND
Trastorno hemorrágico hereditario mas frecuente.
Prevalencia del 1-2%.
Herencia autosómica dominante, predomina en el sexo femenino.
Menorragia es uno de los síntomas principales
Se clasifica en tres tipos, se deben a mutaciones de los loci que codifican dominios funcionales distintos de la proteina del FvW.

ENFERMEDAD DE VON WILLEBRAND
FISIOPATOLOGIA:
FvW glucoproteína multimérica sintetizada por los megacariocitos y cels. Endoteliales
Deficiencia cualitativa y cuantitativa de FvW.
FvW actúa como proteína transportadora de F VIII.
Su deficiencia produce deficiencia de F VIII.

ENFERMEDAD DE VON WILLEBRAND
MANIFESTACIONES CLINICAS:
Hemorragia mucocutánea: equimosis, epistaxis, menorragia y hemorragias postoperatorias (amigdalectomías, exodoncias).
FvW es una proteína de fase aguda, el estrés aumenta su concentración.
Durante el embarazo tiende a duplicarse o triplicarse.
Telangiectasias gastrointestinales

ENFERMEDAD DE VON WILLEBRAND
VARIANTES:
TIPO 1: 85%, epistaxis, equimosis y menorragia, tratamiento con desmopresina.
TIPO 2A: proteólisis anormal de FvW.
TIPO2B: FvW hiperactivo, tratamiento IV con FvW.
TIPO 3: concentración inadecuada de FvW y baja de F VIII.

ENFERMEDAD DE VON WILLEBRAND
TRATAMIENTO:
Desmopresina: 0.3 µg/kg diluido en 100 ml SSN pasar en 30 min. Aumenta niveles de F VIIIC y FvW y acorta el tiempo de sangría.
Productos derivados del plasma, concentrados de F VIIIC que contengan FvW.

ENFERMEDAD DE VON WILLEBRAND
TRATAMIENTO:
Antifibrinoliticos: Tx coadyuvante hemorragia de mucosas, hematomas leves y menorragia.
Ac. Epsilonaminocaproico: 100 mg/kg c/6 h
Ac. Tranexamenico: 20 mg/kg c/8 h

ENFERMEDAD DE VON WILLEBRAND
COMPLICACIONES:
Anemia grave
Aguda (hipovolemia)
Crónica (ferropenia)
Hemorragias musculares o articulares (tipo 3)

BIBLIOGRAFIA
Tratado de Pediatría Nelson, 18° Edición, Volumen 2, capitulo20, Págs.. 2060 – 2074.
Tratado de Pediatría M. Cruz, Nueva Edición 2007, Volumen 2, capitulo 18.10, Pág. 1526 –1537.
Hematology of Infancy and Chilhood Nathan and Oski´s, 6° Edición, Volumen 2, 2003. Capitulo 39, Págs.. 1515 – 1522.













![TEMAS& DIAGNÓSTICO& TRATAMIENTO& SEGUIMIENTO&tipbook.iapp.cl/empresa/2/pdf/207/2-trastornos-de-coagulacion.pdf · 2.]!TRASTORNOS!DE!LA!HEMOSTASIA!SECUNDARIA!! EXÁMENES.DE.LABORATORIO.BÁSICOS!](https://static.fdocuments.co/doc/165x107/60157cc3364a2a321526b777/temas-diagnstico-tratamiento-seguimiento-2trastornosdelahemostasiasecundaria.jpg)




