
TRASTORNOS MOTORES ESOFÁGICOS PRIMARIOS · do ondas terciarias simultáneas de predominio dis-tal...
28
TRASTORNOS MOTORES ESOFÁGICOS PRIMARIOS J. F. Suárez Crespo, T. Jordán Madrid, J. Esteban Carretero Servicio de Aparato Digestivo. Complejo Hospitalario Torrecárdenas. Almería E n los últimos años la patología funcional di- gestiva ha mostrado un interés creciente. La creación en nuestro país de nuevas unidades de exploraciones funcionales digestivas y laborato- rios de motilidad, con personal claramente motiva- do por el estudio y tratamiento de esta patología, da fe de ello. No obstante son múltiples las patolo- gías funcionales que aún precisan una profunda in- vestigación. Los trastornos motores esofágicos, aunque apa- rentemente bien conocidos por el médico general, gastroenterólogo, cardiólogo, etcétera, son un gru- po de enfermedades que aún plantean múltiples confusiones, dudas y controversias. Así, sigue sien- do indeseablemente frecuente encontrar aún al pa- ciente que pasa largos años sin diagnóstico y trata- miento concreto, ya que en su momento el médico que lo atendió sólo se preocupó de que su disfagia o dolor torácico no fueran síntomas secundarios a un problema orgánico. También es frecuente que los dolores torácicos recurrentes de presumible ori- gen no coronario sean con frecuencia catalogados sin más de espasmo difuso esofágica, no sabiendo el médico que lo trata que el reflujo gastroesofági- co ácido es una causa sin duda mucho más común, o que la peristalsis esofágico sintomática existe co- mo trastorno motor y es aún más frecuente que el primero. Pero también para los que nos dedica- mos más de lleno a su estudio siguen planteándo- nos múltiples controversias en su etiopatogenia, fi- siopatología y tratamiento. Éstas y otras muchas cosas son la que nos mueven a profundizar en su estudio y difusión. P odemos definir como trastornos motores esofágicos primarios a un grupo de enfer- medades que afectan única y exclusivamen- te al esófago, y que se caracterizan por la existen- cia de anomalías en el control de la peristalsis del cuerpo esofágico y/o función del esfínter esofágico inferior, siendo su etiología actualmente de origen desconocido. Existen otros trastornos motores esofágicos, a veces indistinguibles de los primarios en sus mani- festaciones clínicas, radiológicas y manométricas, que aparecen en el contexto de una enfermedad conocida de origen neoplásico, neuromuscular, in- feccioso o sistémico, y que se denominan trastor- nos motores esofágicos secundarios. U na clasificación es siempre necesaria, ya que aunque muchos de estos trastornos comparten manifestaciones clínicas pareci- das, enfoques terapéuticos similares y posiblemen- te un mecanismo fisiopatológico común, poseen también rasgos clínicos, diagnósticos y terapéuti- cos propios. INTRODUCCIÓN Revis Gastroenterol 2002; 1: 1-28 DEFINICIÓN CLASIFICACIÓN
Transcript of TRASTORNOS MOTORES ESOFÁGICOS PRIMARIOS · do ondas terciarias simultáneas de predominio dis-tal...

TRASTORNOS MOTORES ESOFÁGICOS PRIMARIOS
J. F. Suárez Crespo, T. Jordán Madrid, J. Esteban CarreteroServicio de Aparato Digestivo. ComplejoHospitalario Torrecárdenas. Almería
En los últimos años la patología funcional di-gestiva ha mostrado un interés creciente. Lacreación en nuestro país de nuevas unidades
de exploraciones funcionales digestivas y laborato-rios de motilidad, con personal claramente motiva-do por el estudio y tratamiento de esta patología,da fe de ello. No obstante son múltiples las patolo-gías funcionales que aún precisan una profunda in-vestigación.
Los trastornos motores esofágicos, aunque apa-rentemente bien conocidos por el médico general,gastroenterólogo, cardiólogo, etcétera, son un gru-po de enfermedades que aún plantean múltiplesconfusiones, dudas y controversias. Así, sigue sien-do indeseablemente frecuente encontrar aún al pa-ciente que pasa largos años sin diagnóstico y trata-miento concreto, ya que en su momento el médicoque lo atendió sólo se preocupó de que su disfagiao dolor torácico no fueran síntomas secundarios aun problema orgánico. También es frecuente quelos dolores torácicos recurrentes de presumible ori-gen no coronario sean con frecuencia catalogadossin más de espasmo difuso esofágica, no sabiendoel médico que lo trata que el reflujo gastroesofági-co ácido es una causa sin duda mucho más común,o que la peristalsis esofágico sintomática existe co-mo trastorno motor y es aún más frecuente que elprimero. Pero también para los que nos dedica-mos más de lleno a su estudio siguen planteándo-nos múltiples controversias en su etiopatogenia, fi-siopatología y tratamiento. Éstas y otras muchas
cosas son la que nos mueven a profundizar en suestudio y difusión.
Podemos definir como trastornos motoresesofágicos primarios a un grupo de enfer-medades que afectan única y exclusivamen-
te al esófago, y que se caracterizan por la existen-cia de anomalías en el control de la peristalsis delcuerpo esofágico y/o función del esfínter esofágicoinferior, siendo su etiología actualmente de origend e s c o n o c i d o .
Existen otros trastornos motores esofágicos, aveces indistinguibles de los primarios en sus mani-festaciones clínicas, radiológicas y manométricas,que aparecen en el contexto de una enfermedadconocida de origen neoplásico, neuromuscular, in-feccioso o sistémico, y que se denominan trastor-nos motores esofágicos secundarios.
Una clasificación es siempre necesaria, yaque aunque muchos de estos trastornoscomparten manifestaciones clínicas pareci-
das, enfoques terapéuticos similares y posiblemen-te un mecanismo fisiopatológico común, poseentambién rasgos clínicos, diagnósticos y terapéuti-cos propios.
INTRODUCCIÓN
Revis Gastroenterol 2002; 1: 1-28
DEFINICIÓN
CLASIFICACIÓN

Generalmente encontramos un buen consensode todos los autores cuando decidimos clasificareste tipo de trastornos, habiéndose descrito al me-nos dos clasificaciones. Una clásica, conocida y se-guida por la inmensa mayoría de los gastroenteró-logos; otra práctica o terapéutica, propuesta poralgunos autores. (Ambas clasificaciones se mues-tran en la T a b l a 1 . )
TEORÍA FISIOPATOLÓGICA COMÚN
A pesar de que estos trastornos son conocidosdesde hace muchos años y que son muchos lostrabajos publicados hasta la fecha, siguen existien-do muchos puntos oscuros en la fisiopatología ehistopatología de los trastornos motores esofágicosprimarios. La dificultad radica en la imposibilidadde crear un modelo experimental fiable para uncorrecto estudio in vivo y la escasez de muestrasde necropsia.
Tan sólo en la acalasia existe una fisio-histopa-tología más o menos clara y aceptada por la ma-yoría de los autores. Así, se sospecha que esta en-fermedad está provocada por un mecanismo dedenervación de origen desconocido que afecta fun-
damentalmente a las neuronas del esfínter esofági-co inferior (EEI) y en menor medida a las del cuer-po esofágico.
De forma resumida podríamos decir que la mo-tilidad esofágica está regulada por una correcta in-teracción entre dos tipos fundamentales de neuro-nas del plexo mientérico. Cuando el mediadorquímico es la acetilcolina (ACH), la neurona actúacomo activadora. Cuando el mediador químico esel óxido nítrico (ON), y en menor medida el pépti-do intestinal vasoactivo (VIP), la neurona actúa co-mo inhibidora. En la acalasia, como veremos másadelante, existe una pérdida selectiva y progresivade neuronas inhibidoras, lo que provoca una pérdi-da del equilibrio neurológico y por tanto del meca-nismo de control, existiendo excitación neuromus-cular permanente sin oposición inhibitoria. Elporqué se produce este daño neuronal selectivo esaún desconocido, existiendo como en todas las en-fermedades idiopáticas una alta sospecha de quepueda tratarse de un mecanismo de origen viraly/o autoinmune desconocido.
En el resto de los trastornos motores esofágicosprimarios el mecanismo fisiopatológico es desco-nocido. No obstante, existe la atractiva teoría deque posiblemente estos trastornos motores puedanser una misma enfermedad en distintos estadiosevolutivos. La base de esta afirmación no es muyfirme y se basa fundamentalmente en observacio-nes clínicas evolutivas de pacientes aislados, a lasque todos hemos asistido en alguna ocasión, y quehan quedado reflejadas en la literatura generalmen-te con la secuencia Peristalsis Esofágica Sintomáti-ca (PES)→ Espasmo Difuso Esofágico (EDE)→acalasia (1,2,3). Tampoco es infrecuente encontrarretrospectivamente en la anamnesis de pacientescon acalasia como existe una evolución en susintomatología a lo largo del tiempo, con síntomasiniciales de dolor torácico que posteriormente seconvierten en disfagia y finalmente en regurgita-ción no ácida y pérdida de peso, lo que nos puedehacer sospechar en la existencia de una PES oEDE en las fases iniciales. Experimentalmente seha demostrado que estimulando la motilidad diges-tiva se puede pasar de una PES a un EDE (4).También algunos autores encontraron alteracioneshistopatológicas en el EDE similares, aunque demenor grado, a las encontradas en la acalasia, su-
REVIS GASTROENTEROL Vol. 4. Núm.1. 2002
2
Clasificación de los trastornos motores esofágicos primarios
Clasificación clásica:
Esfínter esofágico inferior hipertónico (EEIH)
Peristalsis esofágica sintomática (PES)
Espasmo difuso esofágico (EDE)
Acalasia
Formas intermedias
Trastornos motores inespecíficos
Clasificación práctica o terapéutica:
Acalasia
Otros trastornos motores primarios que cursan con
hiperactividad contráctil
Otros trastornos motores primarios sin hiperactivi-
dad contráctil
Tabla 1

TRASTORNOS MOTORES ESOFÁGICOS PRIMARIOS J. F. SUÁREZ CRESPO ET AL.
giriendo que ambos trastornos pueden ser una mis-ma enfermedad en distintos estadios evolutivos (5).
El mecanismo inicial podría estar en fallo de larelajación del EEI. Esta obstrucción funcional de-sencadenaría un incremento de la potencia de con-tracción en el cuerpo esofágico, apareciendo on-das peristálticas de gran amplitud que intentaríanvencer el obstáculo distal de un EEI con relajacióninadecuada (PES). Posteriormente se produciríauna claudicación parcial de la contractilidad esofá-gica con fenómenos de cavidad común, aparecien-do ondas terciarias simultáneas de predominio dis-tal que alternarían con ondas peristálticas (EDE).Más tarde existiría un agotamiento progresivo de laactividad contráctil esofágica, apareciendo primerouna acalasia vigorosa y posteriormente, cuando eldaño es máximo, una acalasia clásica. Existenotros hechos que pueden apoyar de forma indirec-ta esta teoría y que pasaremos a describir a conti-nuación de una forma resumida.
Quién no ha visto en alguna ocasión como al-gunos de sus pacientes con PES y EDE, en los queteóricamente existe un EEI normal, se beneficiande una dilatación o miotomía; cabe pensar por su-puesto que el EEI no era tan normofuncionante co-mo parecía en la manometría. También en algunaocasión hemos visto como algún paciente con aca-
lasia no muy evolucionada recuperaba parte de superistaltismo tras una dilatación o miotomía delEEI. Tampoco es infrecuente encontrar pacientescon criterios diagnósticos de acalasia esofágica queen la manometría tienen relajaciones completas delEEI. Mearin y Malagelada (6) demuestran con resis-tometría que este hallazgo sólo se trata de un arte-facto técnico intrínseco de la propia manometría yque el EEI no es normofuncionante en estos pa-cientes. ¿Podría ocurrir también esto en la PES y elEDE?; serían de agradecer nuevos trabajos en estes e n t i d o .
(En la f i gura 1 se resume de forma gráfica lateoría fisiopatológica común).
No es infrecuente encontrar un EEIH en laPES, y sobre todo en el EDE y la acalasia.Incluso hay algunos autores que lo encon-
traron, aunque parezca paradójico, asociado a en-fermedad por reflujo gastroesofágico (ERGE) (7).No obstante, como trastorno motor esofágico pri-
Figura 1
Figura 1: Teoría fisiopatológicacomún: una atractiva teoríapara explicar el posibleorigen común de lostrastornos motoresesofágicos primarios.
ESFÍNTER ESOFÁGICO INFERIOR HIPERTÓNICO O
HIPERTENSO (EEIH)

mario aislado, en nuestra experiencia y en la de lamayoría de los autores, tal vez sea el menos co-mún del grupo; es más, podríamos considerarlocomo casi excepcional.
El EEIH se define como la existencia de un tonoo presión media máxima basal del EEI igual o su-perior a 2 desviaciones estándar a la del grupocontrol; es decir, más de 40-45 mm Hg. Siendoademás condiciones indispensables que existan re-lajaciones completas del EEI y que el peristaltismoesofágico sea normal (8,9,10).
Pero existen algunas consideraciones que hayque tener en cuenta. Así, la relajación completadel EEI viene definida por un número relativo, esdecir relajación igual o superior al 75-80% de lapresión basal inicial (9,11). Lógicamente cabríaesperar que en los pacientes con EEIH exista unapresión residual absoluta en reposo mayor a laobservada en pacientes normales. ¿Estamos con-siderando erróneamente como normal algo queno lo es? Efectivamente, en los pacientes conEEIH no sólo existe una presión residual de rela-jación más elevada que en los sujetos control, si-no que habitualmente el porcentaje de relajacióndel EEI también suele ser inferior (9). No obstanteestudios funcionales de vaciado de líquidos conesofagograma baritado, y de vaciado de sólidoscon radionúclidos, suelen ser siempre normalesen estos pacientes, por lo que la funcionalidad delEEI a pesar de todas estas consideraciones pare-ce integra (9).
Los síntomas con mayor frecuencia descritosson dolor torácico y/o disfagia. Mejorando gene-ralmente el dolor tras el tratamiento, siendo la dis-fagia más difícil de tratar (9).
Un gran número de estos pacientes muestranen estudios psicológicos la existencia de episodiosde ansiedad, somatización y trastornos de índoleafectiva (9), por lo que a igual que en otros trastor-nos motores esofágicos la sintomatología puedemejorar tratando el trasfondo psicopatológico.
El tratamiento médico con relajantes de lamusculatura lisa (antagonistas de los canales delcalcio y nitritos) puede mejorar la sintomatologíaen muchos pacientes. Las inyecciones intraesfin-terianas de toxina botulínica puede ser una alter-nativa terapéutica en los pacientes que no res-ponden al tratamiento médico convencional (12).
Queda por definir el papel de la dilatación y lamiotomía en los enfermos no respondedores.
DEFINICIÓN
Es fundamentalmente manométrica (13). Podría-mos definirlo como el trastorno de la motilidadesofágica que se caracteriza por la presencia decontracciones de gran amplitud, sin alteracionesen el peristaltismo del cuerpo esofágico y la relaja-ción del esfínter esofágico inferior, y que se acom-paña generalmente de dolor torácico recurrente yen ocasiones también de disfagia.
FRECUENCIA
Se trata del un trastorno motor muy común,siendo con frecuencia un hallazgo manométricocasual. Se piensa que puede aparecer en algo másdel 2% de la población general (14) y en cerca deun 15% de los pacientes estudiados por dolor torá-cico recurrente no coronario (14,15).
ETIOLOGÍA, FISIOPATOLOGÍA E HISTOPATOLOGÍA
Su etiología y fisiopatología es desconocida, noobstante con bastante frecuencia aparece asociadoa ERGE (15), siendo el motivo de dicha asociacióndesconocido. También se ha descrito con mayorfrecuencia en pacientes con angina microvascularo síndrome X (16,17), que se caracteriza por doloranginoso y cambios isquémicos electrocardiográfi-cos durante la ergometría con coronariografía nor-mal. Algunos autores encontraron mayor inciden-c ia en pacientes con diabetes m e l l i t u sinsulinodependiente (18).
No existe ninguna alteración histológica o mor-fológica conocida en el esófago de estos pacientes,
REVIS GASTROENTEROL Vol. 4. Núm.1. 2002
4
PERISTALSIS ESOFÁGICA SIN-TOMÁTICA O ESÓFAGO EN
"CASCANUECES" (PES)

TRASTORNOS MOTORES ESOFÁGICOS PRIMARIOS J. F. SUÁREZ CRESPO ET AL.
destacando la escasez de trabajos publicados en es-te sentido. Algunos autores han realizado estudiosde ultrasonografía perendoscópica, demostrandoque en algunos de estos pacientes existe un au-mento del grosor de la muscularis mucosae ( 1 9 ) .No obstante cuando se compara el grupo de pa-cientes con esta alteración anatómica y los que nola poseen, no se aprecian diferencias significativasen las características o gravedad de la sintomatolo-gía, ni en los parámetros manométricos (19), porlo que la significación fisiopatológica de dicho ha-llazgo podría ser simplemente anecdótica.
MANIFESTACIONES CLÍNICAS
Aunque este trastorno de la motilidad esofágicapuede cursar como asintomático, los pacientesdescriben con frecuencia dolor torácico recurrentey con menor frecuencia disfagia.
Este trastorno motor, asociado o no ERGE y/oangina microvascular, es tal vez el hallazgo mano-métrico más común en los pacientes con dolor to-rácico recurrente (15).
Damos por hecho que un esófago que se con-trae con tanta violencia y espectacularidad en lamanometría, alcanzando las ondas en muchasocasiones presiones superiores a los 300 mm Hg
(F i g. 2), debe producir dolor. No obstante nos se-guimos planteando muchas dudas al respecto:
—Nuestros pacientes se muestran casi invaria-blemente asintomáticos durante la realización de lamanometría estacionaria mientras se suceden unay otra vez las violentas contracciones de la PES, sinembargo, ¿cómo es posible que en ese momentono exista dolor?
—Se ha demostrado en estudios serios y biendiseñados, que cuando el PES se asocia a ERGE yse trata esta última, muchos pacientes quedan asin-tomáticos a pesar de que persiste el trastorno mo-tor desde el punto de vista manométrico (20). ¿Sipersiste una actividad peristáltica tan intensa, por-que ya no existe dolor?
—Todos hemos visto PES asintomáticos, comohallazgo casual en el paciente al que se le realizauna manometría esofágica por otro motivo, gene-ralmente ERGE, y que en la anamnesis nuncacuentan dolores torácicos y/o disfagia. ¿Cómo esp o s i b l e ?
Para producir el PES dolor por si mismo, sin laayuda de una ERGE o una angina microvascular,debe existir seguramente algo más que una meraactividad peristáltica intensa; ¿tal vez fenómenosde hiperalgesia visceral? Efectivamente, nadie du-da hoy en día que existen factores emocionalesque pueden modular la percepción de dolor en los
Figura 2
Figura 2: Peristalsis esofágicasintomática (manometría):paciente remitida paraestudio de dolor torácicorecurrente no coronario.Obsérvese la presencia deondas peristálticas de granamplitud (> 180 mm Hg) a 3cm (P5) y 8 cm (P4) porencima del borde superiordel EEI.

pacientes con PES, haciendo no sólo que aparez-can los síntomas, sino que éstos se presenten deforma más frecuente e intensa (21,22). Existenotros datos que apoyan un estado de hiperalgesiavisceral en estos pacientes; así cuando se realiza ladilatación esofágica con balón, hasta un 60% delos pacientes con dolor torácico recurrente no co-ronario experimentan dolor frente al 20% de con-trol, sin embargo con volúmenes de 8 ml hasta un50% experimentaban dolor frente al 0% del con-trol (23).
DIAGNÓSTICO
Se establece por la manometría y se caracterizapor la aparición de ondas de gran amplitud (≥180mm Hg), que ocasionalmente también son de largaduración (≥6 segundos), siendo condición indispen-
sable que el peristaltismo esofágico sea normal(10) (T a b l a 2 y F i g. 2).
Hay que tener en cuenta que muchas veces eltrastorno motor ocurre exclusivamente en un cortosegmento del tercio distal esofágico, por lo que noes infrecuente que el trastorno sólo se manifiesteen un registro. Es obligado, más que nunca, que latécnica de registro se realice de forma cuidadosa,con un canal de registro colocado 3 cm por enci-ma del borde superior del EEI.
La radiología baritada suele ser normal, habién-dose descrito en algunos pacientes alteraciones ra-diológicas inespecíficas, fundamentalmente tercia-rismo esofágico distal (13,24).
Las pruebas de provocación, fundamentalmen-te dilatación esofágica con balón, son necesariaspara establecer la relación entre dolor torácico ytrastorno motor. El estándar es un balón de polivi-
REVIS GASTROENTEROL Vol. 4. Núm.1. 2002
6
Criterios manométricos de diagnóstico en los trastornos motores esofágicos primarios
EEI hipertónico Tono del EEI ≥ 40 mm Hg Relajación del EEI normal
Peristalsis esofágica normal
Peristalsis sintomática Amplitud ondas ≥ 180 mm Hg Duración ondas ≥ 6 segundos
Peristalsis esofágica normal
Espasmo difuso esofágico Ondas simultáneas ≥ 30% Ondas repetitivas
Peristalsis esofágica presente Ondas espontáneas
Amplitud ondas ≥ 180 mm Hg
Duración ondas ≥ 6 segundos
Hipertonía del EEI
Relajación incompleta del EEI
Acalasia clásica Aperistalsis esofágica Relajación incompleta del EEI
Amplitud ondas < 60 mm Hg Hipertonía del EEI
Aumento presión basal cuerpo
Acalasia vigorosa Aperistalsis esofágica Relajación incompleta del EEI
Amplitud ondas ≥ 60 mm Hg Hipertonía del EEI
Aumento presión basal cuerpo
Trastornos motores Cualquier alteración motoraesofágicos Inespecíficos que por sí misma o unida a
otras, no permite su clasificaciónen los grupos específicos
TRASTORNO MOTOR ESOFÁGICO CRITERIOS OBLIGADOS CRITERIOS OPCIONALES
Tabla 2

TRASTORNOS MOTORES ESOFÁGICOS PRIMARIOS J. F. SUÁREZ CRESPO ET AL.
nilo de 30 mm de longitud y que alcanza 25 mmde diámetro con una insuflación de 10 ml. Se colo-ca a 10 cm por encima del EEI y se rellena con in-flados progresivos de 1 ml, considerándose positi-vo cuando aparece dolor con menos de 8 ml devolumen (23).
Por último decir que en estos pacientes siemprees obligado descartar una ERGE, por lo que es ne-cesario realizar una endoscopia digestiva alta y pH-metría esofágica de 24 horas.
TRATAMIENTO
S i existe ERGE, puede ser razonable el trata-miento inicial de ésta con inhibidores de la bombade protones (IBPs), evitando el uso de miorrelajan-tes que podrían empeorar el reflujo gastroesofági-co y por tanto, la sintomatología (20).
Cuando no existe ERGE o cuando existiendo és-ta no se consiguen controlar los síntomas, se pue-den usar miorrelajantes de la musculatura lisa, fun-damentalmente nifedipino, diltiazen y nitritos. Elnifedipino a dosis de 10-30 mgr por toma, disminu-ye significativamente la amplitud de las contraccio-nes esofágicas, siendo ésta reducción dosis depen-diente y de hasta un 50% a dosis máximas,mejorando en muchos pacientes el dolor torácico yla disfagia (25). El diltiazen aunque se muestra me-nos eficaz para disminuir la contractilidad esofágica,mejora también los síntomas en muchos pacientescuando se usa a largo plazo (26). Los nitritos de ac-ción rápida y prolongada pueden ser iguales de efi-caces. No obstante la mejoría manométrica nosiempre se acompaña de mejoría clínica (27).
Hoy en día se da mucha importancia a los trata-mientos que pueden actuar como modulares de lapercepción visceral. Las benzodiazepinas y sobretodo los antidepresivos tricíclicos, parecen espe-cialmente útiles en estos pacientes. Faltan ensayoscontrolados con nuevos fármacos como antago-nistas de serotonina, fedotiazina, octeotride, etcé-tera (15).
La dilatación del EEI puede ser una alternativa te-rapéutica en casos no respondedores. No obstante,hay que tener en cuenta que existen pocos estudiosque evalúen su eficacia. Algunos autores encontra-ron una mejoría significativa en estos pacientes tras
la dilatación, que no se correlacionó en sí con la ver-dadera eficacia manométrica de ésta (28).
A igual que ocurre en otros trastornos motoresesofágico primarios, excluyendo la acalasia, los re-sultados de la inyección intraesfinteriana de toxinabotulínica son generalmente muy pobres (27).
La esofagomiotomía ampliada hasta el cayadoaórtico puede ser una alternativa en casos gravesintratables (29), ofreciendo por vía toracoscópicaaceptables resultados hasta en un 70-80% de lospacientes (30,31).
DEFINICIÓN
Descrito por primera vez en 1889 por Osgood(32) y manométricamente definido en 1958 porCreamer y cols. (33), se trata de un trastorno mo-tor esofágico que afecta fundamentalmente a lamusculatura lisa y que se caracteriza por la apari-ción de actividad contráctil no propulsiva que alter-na con episodios de peristalsis normal, siendo susmanifestaciones clínicas más frecuentes episodiosde dolor torácico y disfagia intermitente.
ETIOLOGÍA, FISIOPATOLOGÍA E HISTOPATOLOGÍA
Aunque con frecuencia se presenta de formaidiopática, se ha relacionado también con ERGE,obstrucción mecánica del cardias, hipomagnese-mia, amiloidosis y neuropatía visceral secundaria adiabetes m e l l i t u s o en el contexto de un síndromeparaneoplásico (10,34-36).
Las alteraciones histológicas y ultraestructuralescelulares encontradas en estos pacientes no son es-pecíficas y ni mucho menos constantes, aunquecon cierta frecuencia nos evocan la posibilidad deuna acalasia en una fase muy inicial. Consisten fun-damentalmente en hiperplasia de las células muscu-lares y ocasionalmente mínimas alteraciones a nivelde las fibras nerviosas terminales del plexo de Aure-bach, no existiendo alteraciones significativas en lascélulas ganglionares y nervios (37) o en todo casoestas son mínimas y en menor grado que en la aca-
ESPASMO DIFUSO ESOFÁGICO

lasia (5). Estudios recientes sugieren que en estospacientes puede existir un déficit de síntesis o unaumento de la degradación del óxido nítrico (38),tal y como ya veremos ocurre en la acalasia.
MANIFESTACIONES CLÍNICAS
Como en el PES, no son infrecuentes las formassubclínicas. Nosotros lo hemos encontrado concierta frecuencia de forma casual en pacientes es-tudiados generalmente por ERGE cuando se lesrealizaba una seriada EGD y sobre todo una mano-metría esofágica.
El dolor torácico es la manifestación clínica máscaracterística (10). Tiene una frecuencia e intensi-dad variable. Su localización es fundamentalmenteretroesternal, aunque a veces se irradia al cuello yhombros, simulando una enfermedad coronaria.Puede aparecer durante la ingesta, aunque a vecesno tiene relación con ésta, siendo entonces concierta frecuencia de aparición nocturna provocan-do el despertar del paciente (11).
La disfagia es tan frecuente o más que el dolortorácico (10). También es intermitente y de intensi-dad variable, desencadenándose con frecuenciacon la toma de bolos sólidos y bebidas frías carbó-nicas (11). Como ocurre con el dolor, es habitualque los episodios sean más frecuentes e intensoscuando el paciente está sometido a situaciones deestrés psicológico o emocional (22). Algunos estu-dios han demostrado la existencia de trastornospsiquiátricos mayores en un porcentaje importantede pacientes (39).
La regurgitación también es un síntoma comúny aparece en relación con la alimentación o bienes provocada (10). Nuestros pacientes nos refierencon cierta frecuencia regurgitaciones de líquido osaliva no relacionadas con la ingesta.
La pérdida de peso o las complicaciones pul-monares son propias de la acalasia y excepciona-les en el EDE.
DIAGNÓSTICO
Los criterios diagnósticos del EDE han ido va-riando con el tiempo. Actualmente sólo son nece-
sarias tres condiciones: síntomas clínicos compati-bles, ausencia de lesión orgánica y trastorno motorespecífico en la manometría estacionaria (40). Po-siblemente en un futuro y en el contexto del estu-dio del dolor torácico recurrente no coronario, conel empleo tal vez más frecuente y generalizado dela manometría ambulatoria de 24 horas, haya quemodificar sensiblemente este concepto, sobre tododesde el punto de vista manométrico (41).
Radiología baritada (seriada esofágica,e s o f a g o g r a m a )
Puede ser normal (en nuestra experiencia conbastante frecuencia).
Los hallazgos característicos son la aparición defrecuentes ondas no propulsivas en músculo lisoesofágico que identan la columna de bario y retra-san su evacuación, habiéndose descrito como esó-fago en sacacorchos, esófago arrosariado o esófa-go en tirabuzón (42). El esófago superior puedeaparecer dilatado y con nivel hidroaéreo de la pa-pilla de bario (10). En todos los casos, leves o gra-ves, las contracciones simultáneas se entremezclancon contracciones peristálticas (42).
Como también ocurre en la manometría, la in-tensidad de los hallazgos radiológicos no se corre-lacionan bien con las características o gravedad dela sintomatología (40,42). En cuanto a la correla-ción entre los hallazgos manométricos y los hallaz-gos radiológicos existe en la literatura gran dispa-ridad, para algunos autores es buena (43,44),para otros muy mala (45) y para otros buena perosin que exista en la radiología signos específicos( 4 6 ) .
Manometría esofágica
Las alteraciones manométricas suelen ser seg-mentarias, apareciendo generalmente en los dostercios inferiores del cuerpo esofágico (musculaturalisa). No obstante, también pueden existir alteracio-nes en el tercio superior (musculatura estriada) yE E I .
Generalmente existe unanimidad en los crite-rios diagnósticos (T a b l a 2), aunque antes de expo-
REVIS GASTROENTEROL Vol. 4. Núm.1. 2002
8

TRASTORNOS MOTORES ESOFÁGICOS PRIMARIOS J. F. SUÁREZ CRESPO ET AL.
nerlos vamos a realizar una serie de aclaraciones.El EDE se caracteriza fundamentalmente por fe-nómenos peristálticos que se mezclan con otrosno peristálticos simultáneos, siendo la gravedad deesta alteración variable (F i gs. 3 y 4). El porcentajede ondas simultáneas necesarias para el diagnósti-co varía según los diferentes autores de >10%(47, 48) a ≥30% (49,50). Bajo nuestro punto devista, y creemos que el de la mayoría de los auto-
res, parece razonable al menos un 30% de simul-taneidad. No obstante pacientes con un 20% deestas ondas, podrían ser subsidiarios de una pe-queña prolongación de la manometría en buscade una muestra más amplia para valorar el por-centaje final, o de una valoración más cuidadosade otros parámetros diagnósticos opcionales. Así,las salvas de ondas, ondas espontáneas, ondas re-petitivas y ondas de tres o más picos presivos,
Figura 3
Figura 4
Figura 3: Espasmo difuso esofágico(manometría): obsérvesetras la primera deglución lapresencia de ondassimultáneas repetivias.Posteriormente existeperistaltismo esofágiconormal (degluciones 2 y 3)que en ocasiones alternacon ondas simultáneas entercio medio y distal(degluciones 4 y 5).
Figura 4:Espasmo difuso esofágico(manometría): ¿EDE enevolución a acalasia?Presencia en tercio medio(P4) y distal (P5) de ondassimultáneas de granamplitud en tercio distal(degluciones 1,2 y 4) quealternan muyocasionalmente con ondasperistálticas normales(deglución 3). EI EEI (P6) esnormal.

aunque no son obligatorias ni exclusivas, suelenser muy características del EDE y relativamentefrecuentes. La amplitud de las ondas aunque pue-de ser elevada, con mucha mayor frecuencia esnormal, sobre todo cuando son simultáneas (48).Hipertonía del EEI y alteraciones en su relajacióndurante la deglución son relativamente infrecuen-tes (48,49); éstas últimas se manifiestan general-mente por ser incompletas y en menor medida decorta duración (51).
Endoscopia digestiva alta
Obligatoria en todos los casos antes de la ma-nometría. Si por cualquier circunstancia no se rea-lizó es obligatoria su realización posterior. Hay quedescartar obstrucción orgánica y lesiones secunda-rias a ERGE.
En ocasiones se pueden ver ondas terciarias.Cuando el trastorno es grave, en similitud con laradiología, el esófago adopta forma de sacacor-chos o escalera de caracol (F i g. 5).
pHmetría esofágica ambulatoria de 24 horas
Prácticamente es obligada para descartar ERGEcomo posible causa del EDE.
Manometría esofágica ambulatoria de24 horas
Parece razonable su realización en pacientescon dolor torácico y disfagia, en los que se sospe-cha EDE u otra alteración motora esofágica y pre-sentan manometría estacionaria normal, ya que sepuede tratar de un fenómeno temporal. Parecemás razonable aún para correlacionar síntomas yalteraciones manométricas. Además se puede y sedebe combinar con registro pHmétrico esofágico.Puede ser una alternativa futura, dada su escasadisponibilidad actual, fundamentalmente por altocoste y escasa experiencia. Posiblemente haya quereconsiderar los parámetros diagnósticos. Así, unadefinición aceptable del EDE en manometría am-bulatoria sería la presencia de ondas multipico enlos últimos diez centímetros del cuerpo esofágico,con una duración mínima de 15 segundos y unaamplitud máxima superior a 200 mm Hg, a lasque se debe asociar dolor torácico y/o disfagia( 4 1 ) .
TRATAMIENTO
Básicamente lo ya descrito para el tratamientodel PES es válido en los pacientes con EDE.
Los miorrelajantes, fundamentalmente los anta-gonistas de los canales del calcio, pueden ser bene-ficiosos en algunos pacientes (27).
Los antidepresivos y en ocasiones también losansiolíticos, pueden ser especialmente útiles en pa-cientes con EDE (27,39).
La inyección intraesfinteriana de toxina botulíni-ca, aunque generalmente presenta malos resulta-dos (27), ha sido bastante eficaz en algunos estu-dios publicados (12), por lo que podría plantearsecomo iniciativa terapéutica al ser inicialmente pocoi n v a s i v a .
Es bien conocido que los resultados de la dilata-ción neumática son peores en el EDE que en laacalasia. Para algunos autores sólo algo más del50% de los pacientes dilatados obtienen una mejo-ría sintomática significativa (50). Sin embargootros autores seleccionando sólo a pacientes conpatología grave refractaria al tratamiento médico ydisfunción demostrada del EEI, obtienen muy bue-
REVIS GASTROENTEROL Vol. 4. Núm.1. 2002
10
Figura 5
Figura 5: Espasmo difuso esofágico(endoscopia): una imagen
poco habitual de esófago en“sacacorchos” o “escalera
de caracol”.

TRASTORNOS MOTORES ESOFÁGICOS PRIMARIOS J. F. SUÁREZ CRESPO ET AL.
nos resultados en algo mas de un 80% de pacien-tes dilatados (52).
La esofagomiotomía ampliada hasta el cayadoaórtico puede ser una alternativa muy acertada encasos graves refractarios a cualquier otro trata-miento. Por vía toracoscópica se obtienen resulta-dos generalmente buenos y muy similares a la ciru-gía abierta (30,31,53).
DEFINICIÓN
Descrita por primera vez en 1672 por Sir Tho-mas Willis (54), fue definida en 1904 como cardio-espasmo por Von Mikulizci (55), recibiendo sunombre definitivo de acalasia en 1930 por Hust yRake (56).
La podríamos definir como aquella enfermedadmotora esofágica que se caracteriza por la ausen-cia completa de peristaltismo esofágico asociada adisfunción de la relajación del esfínter esofágico in-ferior, provocando dilatación esofágica progresivacon retención de alimentos y disfagia grave, apare-ciendo en estadios más avanzados una pérdidaprogresiva de peso y regurgitación alimentaria concomplicaciones respiratorias por aspiración.
EPIDEMIOLOGÍA
En Estados Unidos y Europa se estima que laacalasia primaria tiene una incidencia de 0,4–1,2casos/100.000 habitantes/año y una prevalenciade 7–13 pacientes/100.000 habitantes. Afectapor igual a ambos sexos, apareciendo sobre todoentre los 25-60 años. Puede afectar también a ni-ños, representando hasta un 5% de los pacientestotales diagnosticados (57).
ETIOPATOGENIA
La acalasia hoy en día sigue siendo consideradacomo una enfermedad de origen desconocido. Noobstante tenemos bastantes conocimientos en
cuanto a su fisiopatología e histopatología, exis-tiendo casi una absoluta certeza de que su desarro-llo se debe finalmente a un fallo en la inervacióndel músculo liso esofágico.
A lo largo de los años y sobre la base de obser-vaciones clínicas, inmunológicas e histopatológi-cas, se han construido muchas teorías sobre cuálpodría ser su verdadero origen:
—Teoría genética: aunque con mucha mayorfrecuencia no existen antecedentes familiares, laobservación de aparición de dos o más casos den-tro de una misma familia y con una frecuencia su-perior a lo que realmente puede ser previsible enuna enfermedad esporádica, ha hecho que se pien-se en el posible origen genético de la acalasia. Laherencia podría ser autosómica recesiva (58-60),habiéndose descrito casos en padre-hijo (61), ma-dre-hijo (62), hermano-hermano (63). También sehan descrito casos únicos de 3 hermanos (59) y ge-melos monocigotos (64). No obstante algunos au-tores sólo encontraron la enfermedad en uno delos dos gemelos idénticos (65). Además tambiénhan sido descritos casos familiares de acalasia yotros trastornos motores esofágicos primarios, fun-damentalmente EDE (60,66,67), lo que nos vuelvea evocar la teoría fisiopatológica común descrita alprincipio del artículo.
—Teoría infecciosa: el tryponosoma cruzi,agente causal de la enfermedad de Chagas, cursacon unas manifestaciones esofágicas similares a lasde la acalasia primaria. Desde hace muchos añosse han intentado relacionar con acalasia otrosagentes infecciosos bacterianos (C o r y n e b a c t e r i u md i p h t e r i a e, Mycobacterium tuberculosis, T r e p o -nema pallidum y algunos Clostridium) o virales(Herpes virus varicella-zoster, virus de la polio yvirus del sarampión) (57). No obstante el agenteque más interés ha despertado ha sido el H e r p e svirus varicella-zoster. Este virus, con una clarapredilección por el epitelio escamoso y tejido ner-vioso, ha sido descubierto en el esófago de algunospacientes con acalasia por técnicas de hibridacióndel DNA (68); posteriores trabajos con técnicas dereacción de la cadena de la polimerasa han des-mentido este hecho (69).
—Teoría autoinmune: estudios prospectivoscon inmunofluorescencia indirecta han encontrado laexistencia de anticuerpos Ig G contra las neuronas
11
ACALASIA ESOFÁGICA

del plexo mientérico en pacientes con acalasia(70,71). Estudios de inmunohistoquímica han de-mostrado la presencia de linfocitos T activados en elinfiltrado inflamatorio de estos pacientes. Éstos, me-diados por un antígeno desconocido, podrían des-truir progresivamente las estructura nerviosas(72,73). Algunos trabajos han intentando relacionarla acalasia con la expresión de algunas clases deHLA, encontrando una clara correlación general-mente raza dependiente. Así parece existir una aso-ciación significativamente positiva entre acalasia y laexpresión de HLA de tipo II DQw1 (74) yDQA1*0101 (75), así como con los HLADQB1*0620 y DRB1*15 sólo en caucásicos (76).Los HLA DRw53 en caucásicos (74) y los HLADQB1*02 (75) pueden tener una función protectora.
—Teoría degenerativa: la acalasia podría tra-tarse de una enfermedad neurológica degenerativacon predilección por las neuronas del plexo mien-térico. Esta sospecha se basa fundamentalmenteen la observación de la aparición de acalasia en al-gunas enfermedades neurológicas degenerativascomo la enfermedad de Parkinson, la ataxia cere-belar hereditaria y enfermedad de von Reckling-hausen (57).
Sobre la base de todo lo expuesto, posiblemen-te la acalasia primaria podría estar provocada poruna infección viral que actúa sobre un organismopredispuesto genéticamente, provocando una re-acción autoinmune cuya consecuencia última seríala destrucción selectiva de las neuronas inhibidorasdel plexo mientérico esofágico (77).
HISTOPATOLOGÍA
La mayoría de los cambios histológicos descritosproceden de biopsias obtenidas durante la cirugíao la necropsia, es decir, de enfermedades sufi-cientemente evolucionadas, existiendo escasez demuestras de los estadios iniciales (57). Por tantopoco sabemos hasta la fecha de la evolución histo-lógica de la acalasia.
Recientemente Goldblum y cols. (78) han publi-cado estudios histológicos de paciente en una faseinicial de la enfermedad; la principal alteración des-crita se localiza en la zona del EEI, aunque tambiénen el cuerpo esofágico, y se caracteriza por la apa-
rición de un infiltrado inflamatorio crónico que seacompaña de una progresiva pérdida de las célulasganglionares y fibrosis neural. Sin embargo, en es-tadios avanzados domina un daño más generaliza-do, con vaciamiento ganglionar extenso, gravedestrucción nerviosa, fibrosis intensa e infiltrado in-flamatorio inconstante.
El papel que juega este infiltrado inflamatorio,claramente dominante en las fases iniciales de laenfermedad, no es del todo conocido. Estudios re-alizados demostraron la existencia predominantede linfocitos T, eosinófilos, mastocitos y célulasplasmáticas, provocando fenómenos de perineuri-tis, neuritis y periganglionitis (79). Aún no se cono-ce bien si lo que primero que aparece es el infiltra-do inflamatorio que actúa como citotóxico o sirealmente éste sólo aparece de forma reactiva se-c u n d a r i a .
Otras alteraciones descritas son hipertrofia mus-cular, fibrosis de la muscularis mucosae, hiperpla-sia del epitelio escamoso, pérdida de glándulassubmucosas, etcétera. Estos hallazgos podrían sersecundarios a la inflamación, denervación y estasisalimentario en la luz esofágica (79).
No obstante la degeneración nerviosa no quedaconfinada al esófago, habiéndose descrito tambiénen algunos pacientes cambios degenerativos en losnervios vagos y en sus núcleos motores dorsales si-tuados a nivel troncoencefálico, abriendo por tantola posibilidad de que la acalasia se trate en realidadde la manifestación esofágica de una enfermedadgeneralizada del sistema autonómico (80). Así, di-ferentes autores han encontrado en pacientes conacalasia alteraciones vagales en otras localizacionesdel tubo digestivo, fundamentalmente en estómagoproximal (81,82), intestino delgado proximal (83),vesícula (84,85) y esfínter de Oddi (86). Sin embar-go, aunque existe una alta sospecha de que efecti-vamente también existe un daño en el sistema ner-vioso autonómico extradigestivo, algunos de losestudios realizados hasta la fecha solo han mostra-do hallazgos contradictorios (87-89).
FISIOPATOLOGÍA
Hasta hace poco tiempo se pensaba que la re-gulación neuronal del tracto gastrointestinal era bá-
REVIS GASTROENTEROL Vol. 4. Núm.1. 2002
12

TRASTORNOS MOTORES ESOFÁGICOS PRIMARIOS J. F. SUÁREZ CRESPO ET AL.
sicamente central y dependía del sistema simpáti-co-parasimpático. Hoy en día se sabe que existeademás un sistema neuronal postganglionar entéri-co denominado no adrenérgico no colinérgico yque usa mediadores químicos diferentes. Hasta ha-ce pocos años se pensaba que el principal neuro-transmisor inhibidor en este sistema era el VIP; sinembargo estudios realizados en los últimos añoshan puesto de manifiesto que aunque éste puedeseguir jugando un importante papel, el óxido nítri-co (ON) es el principal neurotransmisor inhibitoriodel sistema no adrenérgico no colinérgico (57,90).
El ON es una pequeña molécula que se sintetizade la L-arginina a través de la ON-sintetasa. A niveldel sistema nervioso central y periférico actúa co-mo neurotransmisor-neuromodulador. En el tubodigestivo se fabrica a demanda, sin sufrir almace-namiento, y se libera de las terminaciones nervio-sas nitrinérgicas, no colinérgicas no adrenérgicas,induciendo relajación de la fibra muscular lisa (91).La actividad de la ON-sintetasa puede ser medidamediante la transformación de L-arginina a L-citru-llina marcadas con C14. Estudios realizados porMearin y cols. (92) con tejido de la unión esófago-gástrica de pacientes con acalasia han demostradouna ausencia completa de la actividad de esta enzi-ma, a diferencia del grupo de pacientes sin acalasiausados como control. Este hecho también fue con-firmado en estos mismos pacientes con estudios in-munohistoquímicos de anticuerpos monoclonalesdirigidos contra la ON-sintetasa. La diaforasa deNADPH puede ser también utilizada como marca-dor del ON neuronal, habiéndose demostrado porLefebvre (91) deficiente no sólo en el tejido de pa-cientes con acalasia, sino también en el de pacien-tes con enfermedad de Hirschsprung y estenosispilórica hipertrófica.
También se han intentado involucrar en el desa-rrollo de la acalasia la deficiencia de otros neuro-péptidos hoy en día considerados como menos im-portantes, como son el VIP (93-96) y elneuropéptido Y (96), siendo ya muy improbable laasociación con déficit de encefalina y sustancia P( 9 3 , 9 6 ) .
Por tanto, podemos concluir que la acalasia fi-nalmente se desarrolla por la pérdida selectiva yprogresiva de células ganglionares nitrinérgicas enla zona del esfínter esofágico inferior, lo que pro-
voca estimulación muscular constante sin fenóme-nos de relajación.
MANIFESTACIONES CLÍNICAS
La principal manifestación clínica de la acalasiaes la disfagia, apareciendo hasta en el 99% de lospacientes. Su localización es retroesternal, general-mente referida al esófago medio y distal, aunqueen ocasiones también la refieren en el área cervi-cal. Suele presentarse desde el inicio tanto para só-lidos como para líquidos en un 70-97% de los pa-c ientes. Aunque existe variabil idad en suinstauración y progresión, generalmente al princi-pio es ocasional y fluctuante, convirtiéndose des-pués de un tiempo variable en prácticamente con-tinua. Como en otros trastornos motoresesofágicos, algunos pacientes experimentarán tam-bién episodios de empeoramiento ante situacionesde ansiedad. También es típico que los pacientespara aliviar sus síntomas adopten determinadasconductas o posturas durante la comida, tales co-mo levantar los brazos por encima de la cabeza yadoptar una postura bien erguida, tragar agua obebidas carbónicas para evitar la detención del bo-lo sólido, hacer degluciones con pequeñas porcio-nes de alimento, comer de forma lenta con deglu-ciones deliberadas, etcétera (10,42,77,97).
El segundo síntoma más común es la regurgita-ción, apareciendo hasta en el 75% de los pacien-tes. Es característicamente no ácida y está com-puesta por alimentos sin digerir y saliva. En lasfases iniciales suele ser activa, es decir, provocadapor el propio paciente para aliviar la disfagia, pu-diéndose confundir con trastornos de la conductaalimentaria (anorexia nerviosa y bulimia). En fasesavanzadas existe una regurgitación pasiva, es decirno provocada por el paciente, y que representa elestasi s alimentario en un esófago dilatado( 1 0 , 4 2 , 7 7 , 9 7 ) .
El dolor torácico aparece en un 40% de los pa-cientes, siendo más característico de las fases ini-ciales de la enfermedad y de los individuos más jó-venes, desapareciendo de forma espontánea enmuchos pacientes con el paso del tiempo. Sueleser retroesternal, pudiéndose irradiar a los hom-bros, el cuello y la espalda. Aunque puede apare-
13

cer de forma espontánea, suele desencadenarsecon mayor frecuencia con la ingesta y en situacio-nes de ansiedad o estrés. El dolor a veces puedeser tan intenso y provocar tanto miedo en el pa-ciente, que puede ser el principal síntoma referidoy en ocasiones tan responsable en la pérdida depeso como la disfagia (10,42,52,97,98).
Hasta un 40% de los pacientes experimentanpirosis. Ésta se ha relacionado con las propias alte-raciones de la motilidad, ingesta de bebidas carbó-nicas, formación de ácido láctico por fermentaciónbacteriana en la comida retenida e incluso con po-sibles relajaciones transitorias del EEI que favore-cen una ERGE (77,97,99).
COMPLICACIONES
Las complicaciones fundamentales descritas enla acalasia son la pérdida progresiva de peso y lapresencia de aspiraciones respiratorias, con acce-sos de tos nocturna hasta en un 30% de los pa-cientes e infecciones broncopulmonares hasta enun 10% (42).
También en estos pacientes existe una mayorprobabilidad de desarrollar un carcinoma de célulasescamosas; sin embargo, no existe mayor riesgode adenocarcinoma o tumores extraesofágicos(10). La prevalencia puede ser de un 2-7% (77),aunque para algunos autores ésta puede ser inclu-so más alta (100). Se consideran factores de riesgode cáncer en la acalasia: enfermedad con más de20 años de evolución, megaesófago con retenciónde importante cantidad de alimentos, edad mayorde 60 años, tabaquismo y consumo excesivo de al-cohol (100). En estos pacientes estaría justificadala realización periódica de endoscopia con mues-tras para estudio histológico (100). La tinción concolorante vital como el lugol, es de gran utilidad ala hora de dirigir la biopsia, ya que en fases tem-pranas suele ser difícil la distinción macroscópicaentre mucosa con cambios inflamatorios crónicosy carcinoma precoz (100,101).
La esofagitis por estasis y candidiásica es unacomplicación relativamente frecuente (57), ha-biéndose descrito otras complicaciones excepcio-nales como la formación de bezoar esofágico( 1 0 2 ) .
DIAGNÓSTICO
La acalasia se sospecha por la aparición clínicade disfagia a sólidos y líquidos, con regurgitaciónde alimentos y saliva. Esta sospecha debe confir-marse con la realización de un esofagograma bari-tado. Posteriormente se realizará siempre una ma-nometría esofágica, que será la que finalmentehaga el diagnóstico. Además todos los pacientestienen que ser sometidos a una endoscopia digesti-va alta para descartar pseudoacalasia por tumor enla zona del cardias (97).
Radiología baritada (esofagograma ba -r i t a d o )
La radiología simple sólo es útil en casos avan-zados, apareciendo en las placas anteroposterio-res un ensanchamiento mediastínico y en las late-rales un ensanchamiento del espacioretrocardiaco, apreciándo en algunos pacientesun claro nivel hidroaéreo mediastínico o cervical( 1 1 ) .
El estudio baritado es el que realmente nos va aaportar datos para el diagnóstico. Se inicia con elpaciente en decúbito, viendo que el bolo de barioingresa en el esófago de forma normal, pero ésteno suele progresar, moviéndose hacia arriba y ha-cia abajo con la aparición de contracciones tercia-rias, o simplemente se queda quieto en un órganoatónico; el EEI puede relajarse intermitentementesin relación con la deglución. Cuando se da unamayor cantidad de bario y se coloca al pacientebipedestado, se observan las características radio-lógicas típicas de la acalasia: esófago dilatado so-bre todo en su porción distal, nivel hidroaéreo yterminación con afilamiento distal a nivel del EEIen forma de pico de pájaro. La dilatación del esó-fago es variable y en enfermos con enfermedadde corta evolución puede ser mínima o incluso es-tar ausente. En fases avanzadas se encuentra muydilatado, adoptando forma sigmoidea. También sepueden observar divertículos epifrénicos en posi-ción inmediatamente proximal al EEI, siendo enocasiones muy abultados, obstaculizando otraspruebas diagnósticas y maniobras terapéuticas( 4 2 ) .
REVIS GASTROENTEROL Vol. 4. Núm.1. 2002
14

TRASTORNOS MOTORES ESOFÁGICOS PRIMARIOS J. F. SUÁREZ CRESPO ET AL.
Hasta en un 8% de los pacientes con acalasia sepuede identificar radiológicamente una hernia hia-tal, siendo no obstante mucho más infrecuente queen la población general. Estos pacientes no difie-ren significativamente del resto de pacientes conacalasia en cuanto a su edad, sexo, manifestacio-nes clínicas, hallazgos manométricos en el EEI ycomplicaciones terapéuticas (103).
La radiología digital puede aportar ciertas mejo-ras a la radiología convencional, fundamentalmen-te una mayor comodidad para el paciente, mayorrapidez, posibilidad de reproducibilidad y menor ín-dice de radiación (104).
Aunque la radiología generalmente no puedesuperar a la endoscopia en el diagnóstico de pseu-doacalasia, en muchas ocasiones es muy útil.Cuando los hallazgos no son concluyentes, unahistoria de disfagia de corta duración y un segmen-to esofágico distal estenosado de más de 3,5 cm,con pequeña o ninguna dilatación proximal, essiempre sugerente de enfermedad neoplásica( 1 0 5 ) .
Manometría esofágica
La manometría esofágica se puede definir comola exploración “llave” (97) o “gold standar” (57) enel diagnóstico de la acalasia, ya que no sólo lo con-firma, sino que también lo establece (42). Ademáshay que tener en cuenta que es la única explora-ción que nos puede ofrecer el diagnóstico en las fa-ses iniciales de la enfermedad, cuando las radiogra-f ías y endoscopias son normales o pococoncluyentes (10,42), siendo además prácticamen-te imprescindible en el diagnóstico diferencial conotros trastornos motores esofágicos (11). Podría-mos definirla como inexcusablemente obligada.
La aperistalsis esofágica es el único criteriodiagnóstico obligado en la acalasia. Se caracterizapor la aparición invariable de ondas simultáneas debaja amplitud en todos los segmentos esofágicos yque se acompaña con frecuencia de repetitividadtambién simultánea (97). No obstante, en las fasesiniciales pueden existir ondas peristálticas en el ter-cio proximal esofágico (97), o al menos en sus 4primeros cm (11), ya que en esta zona existe unclaro predominio de musculatura estriada. De for-
ma característica estas contracciones estriadas, encomparación con los grupos de control, son siem-pre de baja amplitud, aunque su duración y morfo-logía son normales (106).
El EEI puede ser hipertenso, considerándose es-te hallazgo sólo un criterio diagnóstico opcional ono obligatorio, ya que en algo más del 40% de lospacientes el EEI será nomotónico (57).
Con mayor frecuencia las relajaciones del EEIcon la deglución estarán ausentes o en todo casoserán incompletas. Pero la existencia de relajacio-nes aparentemente normales, aunque generalmen-te breves, no excluye el diagnóstico manométricode acalasia (107). Aunque parezca un contrasenti-do, este hallazgo es relativamente frecuente, ha-biéndose calculado que puede aparecer hasta enun 30% de todos los pacientes (107). Se piensaque se debe a un artefacto manométrico, posible-mente secundario al pequeño calibre de las sondasde manometría, ya que estudios de vaciamientocon radionúclidos (107) y más recientemente estu-dios de resistometría del EEI (6), han demostradoque en estos pacientes el EEI no es funcional, yaque ofrece una importante resistencia al vacia-miento esofágico.
El cuerpo esofágico con mucha frecuenciamostrará una presión basal positiva; es decir, su-perior o igual a la de referencia fúndica. Este ha-llazgo se debe a la retención de alimentos y se-crec iones en el esófago (11,57) . Así, no esinfrecuente que durante el desarrollo de la mano-metría observemos cómo con cada deglución au-menta la presión basal de forma progresiva, exis-tiendo en algunos momentos caídas bruscas convuelta a las presiones de inicio, interpretándoseeste hecho como la retención progresiva de líqui-do y saliva que en un momento determinado con-sigue sobrepasar el EEI.
El esfínter esofágico superior también puedemostrar con cierta frecuencia alteraciones como hi-pertonía, relajaciones de corta duración, ligeros au-mentos presivos coincidiendo con la aparición deondas simultáneas en el cuerpo esofágico y altera-ciones en el reflejo del eructo. Estas alteracionestienen posiblemente como único objetivo la pre-vención de regurgitaciones con aspiración respira-toria y no son consideradas como criterios diag-nósticos (57).
15

Por último decir que la manometría no sólo tie-ne un incalculable valor diagnóstico, sino que co-mo veremos más tarde, también puede ofrecernosdatos predictivos de la eficacia del tratamiento( 1 0 ) .
(En la t a b l a 2 se resumen los criterios diagnósti-cos y en las f i guras 6 a 9 se muestran distintos re-gistros manométricos de acalasia.)
Endoscopia digestiva alta
Es obligada en todos los pacientes, siendo suprincipal objetivo descartar la existencia de una ne-oplasia que afecte a la unión esófago-gástrica (57).
La pseudoacalasia es relativamente frecuente,siendo en muchas ocasiones indistinguible clínica ymanométricamente de la acalasia primaria. Nor-malmente ocurre en pacientes mayores (>60años), con disfagia rápidamente progresiva (<1año) e importante pérdida de peso (>7 kgr); peroel valor predictivo de los síntomas desgraciada-mente es muy bajo (108). Las radiografías, aunquecomo vimos anteriormente pueden ser útiles en eldiagnóstico diferencial, a veces muestran un grannúmero de falsos negativos. La endoscopia esinexcusable y no le podemos negar su gran valoren el diagnóstico diferencial, pero hay que tener
en cuenta que sólo identifica el 65-75% de los pa-cientes con pseudoacalasia (109,110).
En la endoscopia los hallazgos más frecuentesde la acalasia son la presencia de un cuerpo esofá-gico dilatado y atónico que retiene saliva y alimen-tos, siendo frecuente la existencia de esofagitis porestasis, cándidas o medicamentos. El EEI aparececerrado de forma permanente y no se abre con ladistensión, sin embargo no suele existir gran difi-cultad de paso al estómago tras ejercer una míni-ma presión, siendo este hallazgo cuando falta máscaracterístico de la pseudoacalasia. Posteriormentehay que evaluar el cardias y región subcardial des-de el estómago por retroflexión. La toma de biop-sias de la unión esófago-gástrica y cardias es siem-pre obligada (57).
E c o e n d o s c o p i a
En un principio parecía que la ecoendoscopiaiba a jugar un gran papel en el estudio de la acala-sia (111). Sin embargo sus hallazgos siguen siendocontrovertidos, posiblemente debido a problemasde tipo anatómico y técnico, y aunque podría tenerespecial utilidad en el diagnóstico diferencial depseudoacalasia, sobre todo en los pacientes conendoscopia normal, los datos existentes hasta la fe-
REVIS GASTROENTEROL Vol. 4. Núm.1. 2002
16
Figura 6
Figura 6: Acalasia esofágica
(manometría): registroclásico. Ausencia completade peristaltismo en cuerpo
esofágico (P2, P3 y P4), conausencia de relajación delEEI (P5). La presión basaldel cuerpo esofágico está
elevada (con respecto a lareferencia fúndica que es 0).
TRASTORNOS MOTORES ESOFÁGICOS PRIMARIOS J. F. SUÁREZ CRESPO ET AL.
cha hablan también de la posibilidad de frecuentesfalsos positivos por los mismos motivos (112-114).
Tomografía computerizada (TC)
No debe ser un estudio rutinario, pero puedeser útil en el diagnóstico diferencial de la pseudoa-calasia (115). Para algunos autores, cuando no hay
masa evidente, el hallazgo de una pared del EEIasimétrica y con más de 1 cm de espesor podríaser sugerente de enfermedad maligna (116).
DIAGNÓSTICO DIFERENCIAL
En la t a b l a 3 se exponen las causas reconocidashasta la fecha de acalasia secundaria, y que debe-
17
Figura 7
Figura 8
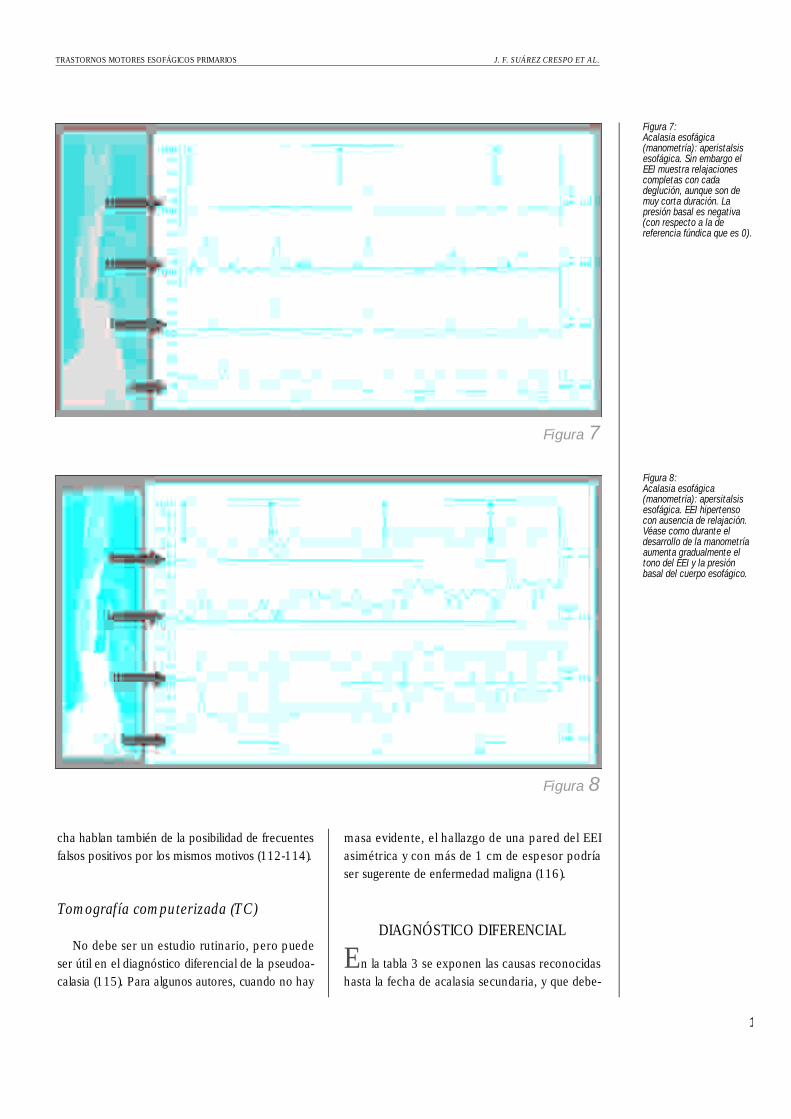
Figura 7: Acalasia esofágica(manometría): aperistalsisesofágica. Sin embargo elEEI muestra relajacionescompletas con cadadeglución, aunque son demuy corta duración. Lapresión basal es negativa(con respecto a la dereferencia fúndica que es 0).
Figura 8:Acalasia esofágica(manometría): apersitalsisesofágica. EEI hipertensocon ausencia de relajación.Véase como durante eldesarrollo de la manometríaaumenta gradualmente eltono del EEI y la presiónbasal del cuerpo esofágico.

mos incluir siempre en el diagnóstico diferencial.Especial interés ofrece el diagnóstico diferencial
de la pseudoacalasia, es decir, de la acalasia secun-daria a lesión neoplásica, y que por motivos didác-ticos hemos desarrollado ampliamente en el apar-tado anterior. Solamente resumir que representa el3% de todos los casos de acalasia, aumentandosignificativamente este porcentaje con la edad, re-presentando según diversos estudios hasta el 9%de acalasia en pacientes de más de 60 años (110).La principal causa son tumores que afectan direc-tamente a la unión esófago-gástrica, fundamen-talmente adenocarcinoma gástrico del cardias, se-guido en frecuencia por el carcinoma esofágico(110). Otros tumores más raros, bien por infiltra-ción de la zona del EEI o bien a distancia en elcontexto de un síndrome paraneoplásico, puedenser causa de pseudoacalasia. Aunque por la clínicacon frecuencia se sospecha, puede ser indistingui-ble por sus síntomas de la acalasia. Las alteracio-nes manométricas también son similares en ambosgrupos de pacientes y por tanto no tiene valor enel diagnóstico diferencial. El esofagograma puedeser útil, pero será la endoscopia con biopsias la ex-ploración obligada por su mayor rentabilidad. Noestá definido el papel de la ecoendoscopia y TC,aunque podría resultar de utilidad en algunos pa-cientes. No obstante, cuando existe una alta sospe-cha de neoplasia y no se ha confirmado por las
técnicas diagnósticas disponibles, la cirugía conbiopsias intraoperatorias es la que debe aclarar las i t u a c i ó n .
TRATAMIENTO
Ninguno de los tratamientos disponibles hasta lafecha cura la acalasia, siendo tan sólo su objetivoaliviar los síntomas mejorando el vaciamiento eso-fágico. Actualmente se consideran como más efi-caces la dilatación neumática y la miotomía quirúr-gica. En los pacientes con elevado riesgo sonaceptables alternativas las inyecciones de toxinabotulínica en el EEI y el tratamiento farmacológicocon nitritos o bloqueantes de los canales del calcio( 9 7 ) .
A continuación desarrollaremos de forma ex-tensa cada uno de los tratamientos disponibles. (Enla f i gura 10 se expone un algoritmo de estrategiadel tratamiento).
Tratamiento farmacológico clásico
Los tratamientos farmacológicos actuales dismi-nuyen notablemente el tono del EEI, pero semuestran incapaces para mejorar sus relajacionesy la apersitalsis del cuerpo esofágico. En la actuali-
REVIS GASTROENTEROL Vol. 4. Núm.1. 2002
18
Figura 9
Figura 9: Acalasia esofágica vigorosa(manometría): paciente de
38 años estudiado porepisodios de dolor torácicocon disfagia ocasional. Su
madre había sidodiagnosticada unos meses
antes de acalasia esofágica(figura 8). Existe
aperistalsis esofágica conondas multipico de gran
amplitud (>60 mm Hg), EEIcon frecuentes relajaciones
completas y presión basaldel cuerpo esofágico
negativa.

TRASTORNOS MOTORES ESOFÁGICOS PRIMARIOS J. F. SUÁREZ CRESPO ET AL.
dad se usan fundamentalmente tan sólo dos gruposde fármacos: agentes bloqueadores de los canalesdel calcio (sobre todo nifedipino) y nitritos de ac-ción prolongada (sobre todo dinitrato de isosorbi-de). Con ellos se consigue disminuir el tono del EEIhasta en un 50% (97).
El Nifedipino se usa a dosis de 10 a 30 mgs porvía sublingual, 30 a 45 minutos antes de cada co-mida, y en los casos con regurgitación y tos noc-turna se debe dar una dosis más antes de acostarse(57). Su efecto dura unos 30 a 120 minutos, mejo-rando los síntomas hasta en un 75% de los pacien-tes (97).
El dinitrato de isosorbide se usa a dosis de 5 a20 mgs por vía sublingual, 10 a 15 minutos antes
de las comidas (57). Su efecto máximo dura de 3 a27 minutos, mejorando los síntomas hasta en un87% de los pacientes (97).
A pesar de que los resultados expuestos pare-cen muy satisfactorios, diversas circunstancias hancondicionado su escasa utilidad clínica. Funda-mentalmente su eficacia limitada e impredecible,su efecto transitorio y de muy corta duración, laaparición de tolerancia en poco tiempo y la fre-cuente existencia de efectos secundarios casi siem-pre mal tolerados por los pacientes (10). Hoy endía podrían estar sólo indicados en pacientes conenfermedad temprana, escasamente sintomática ycon poca dilatación esofágica, pacientes con ele-vado riesgo para la dilatación o la cirugía, y pa-
19
Causas de acalasia secundaria
Enfermedades neoplásicas (pseudoacalasia):—Por infiltración del EEI:
Adenocarcinoma de cardiasCarcinoma esofágicoAdenocarcinoma de mamaAdenocarcinoma prostáticoAdenocarcinoma pulmonarLinfoma gástricoLinfoma esofágicoLinfangioma esofágico
—Por mecanismo a distancia (por afectación vagal y/o sd. paraneoplásico):Metástasis en el tronco del encéfaloEnfermedad de HodgkinHepatocarcinomaAdenocarcinoma gástricoCarcinoma pulmonar pobremente diferenciadoSarcoma de células reticularesMesotelioma peritoneal
Enfemedades infiltrativas no neoplásicas:—Amiloidois—Leiomiomatosis—Esofagitis eosinofílica—Sarcoidosis—Enfermedad de Fabri
Miscelánea—Enfermedad de Chagas—Cirugía de la unión esofagogástrica y vagotomía—Diabetes mellitus—Síndrome de Allgrove (insuficiencia adrenocortical familiar, acalasia y alacrima)—MEN tipo IIb—Pseudoquiste pancreático—Síndrome de Sicca con hiposecreción gástrica—Pseudoobstrucción intestinal crónica—Acalasia y piloroespasmo
Tabla 3

cientes que rechazan tratamientos invasivos ytampoco quieren someterse a inyecciones con to-xina botulínica, o cuando lo hicieron ésta fracasó( 9 7 , 1 1 7 ) .
Nuevos fárm a c o s
Recientes estudios (118) han demostrado que elSildenafil (Viagra®), un nuevo fármaco que ha sidodiseñado para el tratamiento de la impotencia fun-cional, puede ser útil en el tratamiento de la acala-sia. El Sildenafil es un potente inhibidor específicode la enzima fosfodiesterasa tipo 5 de los cuerposcavernosos humanos. Esta enzima destruye e inhi-be la formación de ON, que es el mediador necesa-rio para la actuación de la guanilatociclasa forma-dora de guanosin monofosfato cíclico (GMPc),responsable final de la relajación de la musculaturalisa. El Sildenafil a nivel del esófago se ha mostradoeficaz no sólo disminuyendo el tono del EEI, sinotambién la presión residual durante la deglución yla amplitud de las ondas esofágicas. Pero no todoson ventajas, así se ha mostrado incapaz de resta-blecer el peristaltismo, su vía de administración noparece muy adecuada (precisa llegar al estómago),tiene un efecto muy corto (< 1 hora), su potenciatiene una gran variabilidad paciente dependiente(aceptable integridad del sistema nitrinérgico) y tie-ne demostrados efectos indeseables y secundarios.Por lo tanto su uso sólo puede considerarse experi-
mental, pero abre una puerta al desarrollo de nue-vos fármacos formadores o liberadores de ON.
Inyección intraesfinteriana de toxina bo -t u l í n i c a
Este novedoso tratamiento fue descrito por pri-mera vez como posible tratamiento de la acalasiaen 1993 por Pasricha y cols. (119), tras demostraren animales de experimentación su eficacia pararelajar de forma mantenida el EEI sin que aparecie-ran signos de toxicidad (120). Una vez descritacompletamente la técnica de inyección y los pri-meros resultados en humanos (121,122), se nosabrían muchas expectativas y todos comenzamos ausar con cierto entusiasmo aquel remedio mágicoen nuestros pacientes. Posteriormente "las aguasvolvieron a su cauce" y comprendimos que comoel resto de los tratamientos también tiene sus limi-taciones. Actualmente solo es considerado comoun tratamiento de primera línea en unas circuns-tancias muy concretas que más adelante describire-m o s .
La toxina botulínica tipo A es un inhibidor cal-cio dependiente de la acetilcolina, comercializadaen viales liofilizados de 100 UI, disolviéndose en 5-10 ml de suero salino antes de su uso. Ésta unavez preparada se inyecta en el EEI con una agujade escleroterapia en dosis de 20-25 unidades encuatro cuadrantes (97). En los estudios más opti-
REVIS GASTROENTEROL Vol. 4. Núm.1. 2002
20
Figura 10
Figura 10: Algoritmo terapéutico: en
consonancia con la mayoríade los autores y en base a lo
expuesto en el texto,mostramos gráficamente
nuestra propuesta detratamiento en la aclasia.

TRASTORNOS MOTORES ESOFÁGICOS PRIMARIOS J. F. SUÁREZ CRESPO ET AL.
mistas hasta un 90% de los pacientes mejoran suclínica en la primera semana tras una primera yúnica inyección (123). Pero esta respuesta es fugazy las recaídas muy frecuentes, ya que sólo un 70%de los inicialmente respondedores mantendrán res-puesta a los 3 meses (123), un 60% a los 6 meses(124), un 50% a los 9 meses (125) y un 30% alaño (126). Tan sólo hemos encontrado un trabajoen niños, siendo los resultados muy parecidos a losde los pacientes adultos, con respuesta inicial favo-rable en un 82% y mantenimiento de ésta en un50% después de los 6 meses (127).
Con la finalidad de mejorar estos resultados, enlos últimos tiempos se han buscado factores pre-dictivos para la respuesta y también se ha intenta-do optimizar la técnica y dosis de la inyección.Así, algunos autores encontraron mejor respuestaen pacientes mayores de 50 años (123) y sobretodo en los que t ienen acalasia v igorosa(123,128). El sexo, el tiempo transcurrido desdeel inicio de los síntomas, las características de és-tos o su gravedad, la presión en reposo del EEI,las características radiológicas y la existencia o node dilatación previa, no se consideran factorespredictivos de la respuesta a la inyección con toxi-na botulínica (123,124). En cuanto a la técnica deinyección, estudios iniciales parecen mostrar quesi ésta es guiada por ecoendoscopia los resultadospodrían ser mejores (129,130). En cuanto a lasdosis podría ofrecer mejores resultados la inyec-ción de 100 U en dos sesiones separadas 30 días,ya que hasta un 68% de los pacientes tratados deesta forma pueden mantener remisión durante 24meses (128).
Los parámetros manométricos y radiológicostras la inyección de toxina botulínica, a diferenciade lo que ocurre con la dilatación, generalmenteno se correlacionan bien con la mejoría sintomáti-ca de los pacientes y por tanto no parecen útilespara predecir el fracaso o éxito de ésta (131). Auna segunda inyección responden el 76% de losque respondieron inicialmente a la primera y tansólo el 20% de los que no lo hicieron (123,126).Se piensa que estas respuestas inicialmente bue-nas, pero pasajeras o simplemente malas desde unprincipio, se producen por un fenómeno de taquifi-laxia secundaria al desarrollo de anticuerpos contrala toxina botulínica ya vistos anteriormente en el
tratamiento de otras patologías (97). Nos pregun-tamos si en la acalasia el uso de toxina botulínica Bpodría ser eficaz en estas situaciones, al igual queya ocurre en otras enfermedades neuromusculares( 1 3 2 ) .
La inyección de toxina botulínica es más costo-sa comparada con la dilatación neumática. El gastoagregado al retratamiento frecuente con la toxinabotulínica pesa más que los beneficios económicospotenciales de la seguridad del procedimiento dila-tador a menos que la esperanza de vida sea de 2años o menos (133).
La inyección de toxina botulínica aumentan elriesgo de la cirugía, ya que se incrementan por 7las dificultades técnicas y también por 7 el riesgode perforación, pero cuando se ha usado previa aésta no disminuye su eficacia final (134).
Por tanto, la inyección intraesfinteriana con to-xina botulínica sólo se puede considerar como tra-tamiento de primera línea en pacientes con eleva-do riesgo quirúrgico y aquéllos que rechazan ladilatación o la cirugía.
Inyección intraesfinteriana de sustanciase s c l e ro s a n t e s
Moretó y cols. (135-137) nos proponen la in-yección en el EEI de oleato de etanolamina al 5%como alternativa posible en el tratamiento de laacalasia.
La técnica inicialmente diseñada consistía en lainyección de 2 cc de lidocaína al 2% con 5 cc deoleato de etanolamina al 5% en cada uno de loscuadrantes, con sesiones quincenales de reinyec-ción en sólo 2 cuadrantes hasta cumplir los objeti-vos de respuesta. Al mes de seguimiento el 80%de los pacientes habían tenido una respuesta exce-lente y sólo en un 10% se consideró la respuestamala; al final de un seguimiento, 3 años como me-dia, no sólo los pacientes con respuesta excelentemantenían su situación, sino que el 10% no res-pondedor presentaba en ese momento una res-puesta buena. El único inconveniente era el desa-rrollo de estenosis significativas en el 20% de lospacientes, que no obstante se solucionó bien conbalón dilatador, y el desarrollo de esofagitis grado Ien el 25% de los pacientes.
21

Tras una modificación de la técnica inicial, usan-do tan sólo en la sesión inicial la inyección en dospuntos, posponiendo nuevas sesiones si aparecíansignos inflamatorios o úlceras, y usando omeprazoloral, los resultados no sólo eran tan buenos a cortoy largo plazo, sino que además la esofagitis sóloaparecía en un 8% y la estenosis no apareció enningún paciente. Con un mayor tiempo de segui-miento hasta el 14% de los pacientes presentabanRGE ácido patológico en la pHmetría esofágica de24 horas (sólo se sometieron a esta exploración lamitad del total), pero la esofagitis sólo persistía enel 2% de los pacientes, presentando estenosis sóloel 7% del total de pacientes que fue sometido a lasegunda técnica ya descrita, no obstante la respues-ta a las dilataciones fue posteriormente buena.
Por tanto este tipo de tratamiento podría seruna alternativa válida al tratamiento clásico, aun-que indudablemente se precisan más estudios parasacar conclusiones más sólidas.
DILATACIÓN FORZADA DEL CARDIAS
El primer tratamiento diseñado para la acalasiafue la dilatación del EEI. El primer dilatador cono-cido, fabricado de una barba de una ballena conuna esponja en uno de sus extremos, fue diseñadopor Sir Thomas Willis en 1672 (54). Posterior-mente se diseñaron dilatadores más sofisticadoscomo bujías de mercurio y dilatadores metálicos.En nuestros días los dilatadores utilizados son debalón, usando para su expansión aire.
Los dos modelos más usados actualmente sonlos dilatadores de Rigiflex y Witzel . El balón de Ri-giflex está fabricado en polietileno, tiene 10 cm delargo y se fabrica en tres diámetros: 3, 3,5 y 4 cm.Se trata de un balón no deformable, con nula c o m -p l i a n c e, lo que le permite no sobrepasar diámetromáximo aunque lo sometamos a sobreinsuflación.Se coloca con guía previamente pasada al estóma-go con el endoscopio, insuflándose a presionesque oscilan entre 7-10 psi durante unos 3 minutosde forma continuada o en 3 periodos de 1 minutocada uno. El balón de Witzel es también de polieti-leno y viene montado sobre un tubo de poliviniloque permite sujetarlo al endoscopio. La dilataciónse realiza con las mismas presiones y durante el
mismo tiempo que el balón de Rigiflex, pero el ba-lón de Witzel no precisa guía y se controla con elendoscopio desde el estómago por retroflexión(57, 97,138, 139).
Los resultados para la mayoría de los autoresson muy buenos y dependen básicamente del diá-metro del balón. Después de analizar la tasa acu-mulativa de 13 trabajos con 359 pacientes, sepuede concluir que usando balones de 3, 3,5 y 4cms mejorarán sus síntomas el 74, 86 y 90% delos pacientes tratados, respectivamente (97). Sinembargo para obtener una buena respuesta, hastaen el 26 y 16% de los pacientes hay que realizaruna segunda o tercera dilatación, respectivamente;éstas se realizarán con una periodo de descanso deunas 4 semanas (57). La mejoría, a diferencia de lainyección de toxina botulínica, se suele mantenerpor periodos largos de tiempo en muchos pacien-tes; así hasta un 72% de los pacientes finalmenterespondedores seguirán asintomáticos después dealgo más de 4 años de seguimiento (140) y hastaun 51% a los 8 años (141).
Algunos factores pueden tener valor predictivoa la hora de iniciar una dilatación. Así, parece queresponden peor a una dilatación: los pacientes va-rones, los menores de 20 años, los que tienen undiámetro esofágico medido por esofagograma in-ferior o igual a 3 cm, y los que durante la mano-metría tienen una presión basal del cuerpo esofági-co superior a 15 mm Hg y/o un tono del EEImayor de 30 mm Hg (141).
Después de la dilatación cabría esperar una me-joría sintomática directamente proporcional al gra-do de efectividad de esta medida por manometría,esofagograma baritado o estudio de vaciamientocon radionúclidos. Birgisson y Ritchter (57) hacenuna revisión de la literatura y encuentran los si-guientes hallazgos: la dilatación consigue disminuirel tono del EEI entre un 43-70%; algunos autoresencuentran una significativa mejoría con la dismi-nución del tono del EEI por debajo de 10-15 mmHg, o al menos un 50% del valor basal; otros auto-res no encuentran correlación entre la mejoría sin-tomática y los valores finales del tono del EEI; aun-que hasta un 20% de los pacientes recuperan encierto grado su peristalsis, ésta no se correlacionacon mejoría de los síntomas o el vaciamiento eso-fágico; tampoco la mejoría sintomática se asocia
REVIS GASTROENTEROL Vol. 4. Núm.1. 2002
22

TRASTORNOS MOTORES ESOFÁGICOS PRIMARIOS J. F. SUÁREZ CRESPO ET AL.
bien a una mejoría del vaciamiento esofágico conradionúclidos o disminución del calibre esofágicoen el esofagograma.
Las principales complicaciones descritas son laperforación y la aparición de ERGE (10). Otrascomplicaciones posibles son: neumonía por aspira-ción, hemorragia digestiva, rotura de la mucosa sinperforación y la formación de hematoma esofágico(57).
En una revisión de 6 estudios con 265 pacien-tes y 325 dilataciones, la tasa acumulativa de per-foraciones usando balones de Rigiflex es del 1,8%(57). Con el balón de Witzel el índice de perfora-ción parece mayor (142,143). Se sabe que el ries-go de perforación, a igual que ocurría con la pro-babilidad de mejoría sintomática, aumenta con eldiámetro de balón usado (57). Hoy en día se consi-dera que la técnica que entraña un menor riesgode perforación, es la que usa los balones de 3, 3,5y 4 cm de forma gradual y según la respuesta delpaciente (144,145). La perforación se suele identi-ficar en las primeras 5 horas que siguen a la dilata-ción, siendo más frecuente en los pacientes quepresentan sangre en el balón, taquicardia, dolor demás de 4 horas de duración y fiebre (146). El eso-fagograma con gastrografín es la técnica más ade-cuada para identificar la perforación. El tratamien-to de elección de la perforación libre es la cirugía;sin embargo, cuando la perforación es incompleta,puede intentarse un tratamiento médico con sondanasogástrica, nutrición parenteral total y antibióti-cos de amplio espectro (57). La identificación pre-coz de ésta, con cierre quirúrgico y esofagomioto-mía en los casos que sea posible, se acompañaráde un buen pronóstico, siendo los resultados fina-les similares a la de los pacientes con esofagomito-mía quirúrgica electiva (146).
(En la t a b l a 4 se exponen cuáles son los factoresque aumentan el riesgo real de perforación, y lascontraindicaciones de la dilatación o situaciones enlas que es preferible usar la cirugía.)
TRATAMIENTO QUIRÚRGICO
El primer tratamiento quirúrgico para la acalasiafue diseñado en 1913 por Heller (147) y consistíaen una cardiomitomía extramucosa anterior y pos-
terior. En 1923 Zaaijer (148) modifica la técnicarealizando una, sólo una cardiomitomía extramu-cosa anterior, que es la que se usa en la actualidad.En la última década del siglo XX se desarrolla la ci-rugía mínimamente invasiva de la acalasia; en1991 Shimi y cols. (149) describen por primeravez la cadiomiotomía por vía laparoscópica y en1992 Pellegrini y cols. (150) publican su experien-cia con 17 casos de cardiomiotomia por vía tora-coscópica y 2 casos más por vía laparoscópica.
Los resultados de la cardiomiotomía extramuco-sa suelen ser inicialmente excelentes y posterior-mente duraderos en la mayoría de los pacientes.Con cirugía abierta se considera efectiva en hastaun 83% de los pacientes y con cirugía mínimamen-te invasiva estos resultados llegan hasta un 94% delos pacientes. La mortalidad intraoperatoria con ci-rugía abierta es inferior al 1%, siendo mayor si lacirugía abierta se realizó por vía transtorácica; en la
23
—Factores locales que aumentan el riesgo de perfo-ración:Hernia hiatalDivertículo epifrénicoHistoria previa de perforación o desgarroDilatación neumática previaDilatación con balones de gran calibreDilatación con presión > 11 psi
—Contraindicaciones o situaciones en la que es pre-ferible realizar cirugía:Paciente no colaborador (niños, pacientes psicóticos)Imposibilidad de descartar enfermedad neoplasiaPresencia de divertículo epifrénicoPresencia de hernia hiatalEsófago sigmoide con esofagitis grave por retenciónFracaso de dilataciones previasFracaso de cirugía previaCirugía previa sobre la unión esófago-gástrica
FACTORES QUE AUMENTAN EL RIESGOEN LA DILATACIÓN FORZADA DEL CAR-DIAS, Y SUS CONTRAINDICACIONES O
SITUACIONES EN LA QUE ES PREFERIBLEREALIZAR CIRUGÍA
Tabla 4

cardiomiotomía laparoscópica la mortalidad esprácticamente nula. La estancia hospitalaria es de7-10 días para la cirugía abierta y de 2-4 días parala mínimamente invasiva. El riesgo de RGE es ma-yor en la cirugía abierta por laparotomía (hasta un22%), siendo menor por cirugía abierta transtorá-cica (10%) y cirugía mínimamente invasiva (11%);por esto la mayoría de los autores realizan una ci-rugía antirreflujo (Nissen incompleto, Toupet in-completo o preferiblemente una fundusplicatura deDor), siendo los resultados generalmente muy bue-nos. Otras complicaciones de la cirugía como sonatelectasias, neumonía, perforación, mediastinitis,empiema pleural y tromboflebitis, aparecen gene-ralmente en menos del 1% de los pacientes( 5 7 , 9 7 ) .
En los pacientes en los que han fallado reitera-damente todos los tratamientos descritos y sobretodo en aquéllos que presenta megaesófago (sig-moide de >8 cm de diámetro), hay que considerarla posibilidad de esofagectomía con gastroplastia(“estiramiento” gástrico) o coloplastia (interposi-ción colónica) (57).
ALGUNAS CONSIDERACIONES SOBRELA ACALASIA VIGOROSA
Clásicamente se define como acalasia vigorosa aaquella forma de acalasia, posiblemente inicial, quedesde el punto de vista manométrico se presentacon ondas de amplitud ≥ 60 mm Hg, que clínica-mente se manifiesta fundamentalmente por dolortorácico y que radiológicamente cursa con escasadilatación esofágica y abundante terciarismo.
Esta definición actualmente es cuestionada poralgunos autores, ya que suele existir una importan-te disociación entre los hallazgos manométricos,
clínicos y radiológicos (151). Es decir, la presenciade ondas simultáneas de gran amplitud no siemprese acompaña de una mayor repetitividad o un ma-yor tono del EEI, tampoco siempre se acompañade una mayor presencia de dolor torácico, ni tam-poco de una mayor terciarismo o menor dilataciónen el esofagograma.
Pero también se cuestiona su verdadera utilidad,ya que la presencia de acalasia vigorosa no sueleimplicar grandes variaciones en cuanto a la actitudterapéutica a seguir o sus resultados finales. A con-tinuación servimos algunos ejemplos:
—Aunque para algunos autores es incuestiona-ble una mejor respuesta a la inyección de toxina bo-tulínica en los pacientes con acalasia vigorosa(123,128), para otros la respuesta fue similar a lade los pacientes que padecían acalasia clásica (124).
—La respuesta a la dilatación neumática estambién similar para ambos grupos de pacientes( 1 5 2 , 1 5 3 ) .
—Aunque tras la miotomía algunos autores en-contraron una mayor tendencia a la vuelta de laperistalsis en la acalasia vigorosa (153), como yavimos antes este hallazgo no significa necesaria-mente una mejoría de los síntomas o del vacia-miento esofágico (57).
—La respuesta a la miotomía corta en los pa-cientes con acalasia vigorosa, es tan buena comoen los que padecen una acalasia clásica (154), porlo que puede estar muy cuestionada la miotomíaampliada.
REVIS GASTROENTEROL Vol. 4. Núm.1. 2002
24
1. Traube M, Aaronson RM, McCallum RW. Transition fromperistaltic esophageal contractions to diffuse esophage-al spasm. Arch Intern Med 1986; 146: 1844-6.
2. Kramer P, Harris LD, Donaldson RM Jr. Transitionfrom symptomatic diffuse spasm to cardiospasm.Gut 1967; 8: 115-9.
3. Millan MS, Bourdages R, Beck IT, DaCosta LR Tran-sition from diffuse esophageal spasm to achalasia. JClin Gastroenterol 1979; 1: 107-17.
4. Cole MJ, Paterson WG, Beck IT, DaCosta LR. The ef-fect of acid and bethanechol stimulation in patientswith symptomatic hypertensive peristaltic (nutcrac-
BIBLIOGRAFÍA
CORRESPONDENCIA: J. F. Suárez CrespoServicio Aparato DigestivoHospital TorrecárdenasPasaje de Torrecárdenas s/n04009 Almería

TRASTORNOS MOTORES ESOFÁGICOS PRIMARIOS J. F. SUÁREZ CRESPO ET AL.
ker) esophagus. Evidence that this disorder may bea precursor of diffuse esophageal spasm. J Clin Gas-troenterol 1986 ; 8 : 223-9
5. Adams CW, Brain RH, Trounce JR. Ganglion cells inachalasia of the cardia. Virchows Arch A PatholAnat Histol 1976; 372: 75-9.
6. Mearin F, Malagelada JR. Complete lower esopha-geal sphincter relaxation observed in some achala-sia patients is functionally inadequate. Am J Phy-siol Gastrointest L iver Physiol 2000; 278:G 3 7 6 - 8 3 .
7. Katzka DA, Sidhu M, Castell DO. Hypertensive loweresophageal sphincter pressures and gastroesopha-geal reflux: an apparent paradox that is not unusual.Am J Gastroenterol 1995; 90: 280-4.
8. Bassotti G, Alunni G, Cocchieri M, Pelli MA, MorelliA. Isolated hypertensive lower esophageal sphinc-ter. Clinical and manometric aspects of an uncom-mon esophageal motor abnormality. J Clin Gastroen-terol 1992; 14: 285-7.
9. Waterman DC, Dalton CB, Ott DJ, Castell JA, Brad-ley LA, Castell DO, et al. Hypertensive lower esopha-geal sphincter: what does it mean? J Clin Gastroen-terol 1989; 11: 139-46.
10. Garriges V, Ponce J. Trastornos motores del esófa-go. En: Motilidad Digestiva. Patología. Diagnóstico ytratamiento. De: Ponce J. Barcelona: J R Prous,1996; 213-38.
11. Ruiz de León A, Sevilla-Mantilla C, Pérez de la SernaJ. Trastornos motores esofágicos primarios. En: Tras-tornos motores del Aparato Digestivo. De: Díaz-RubioM. Madrid: Médica Panamerican, 1996; 69-79.
12. Miller LS, Parkman HP, Schiano TD, Cassidy MJ, TerRB, Dabezies MA, et al. Treatment of symptomaticnonachalasia esophageal motor disorders with botuli-num toxin injection at the lower esophageal sphinc-ter. Dig Dis Sci 1996; 41: 2025-31.
13. Ott DJ, Richter JE, Wu WC, Chen YM, Gelfand DW,Castell DO. Radiologic and manometric correlationin "nutcracker esophagus". AJR Am J Roentgenol1986; 147: 692-5.
14. Castell JA, Castell DO. Manometría esofágica. En:Exploraciones funcionales en la enfermedad esofági-ca. De: Scarpignato C y Galmiche JP. Barcelona: Edi-ka Med, 1994; 109-29.
15. Achem SR, Devault K. Dolor torácico no cardiaco.Revis Gastroenterol 1998; 1: 67-84.
16. Cannon RO 3d, Cattau EL Jr, Yakshe PN, Maher K,Schenke WH, Benjamin SB, et al. Coronary flow re-serve, eshofageal motility, and chest pain in pa-tients with angiograhically normal coronary arteries.Am J Med 1990; 88: 217-22.
17. Borjesson M, Albertsson P, Dellborg M, Eliasson T,Pilhall M, Rolny P, et al. Esophageal dysfunction insyndrome X. Am J Cardiol 1998; 82: 1187-91.
18. Solzi GF, Di Lorenzo C. Nutcracker esophagus in achild with insulin-dependent diabetes mellitus. JPediatr Gastroenterol Nutr 1999; 29: 482-4.
19. Melzer E, Ron Y, Tiomni E, Avni Y, Bar-Meir S. As-sessment of the esophageal wall by endoscopic ul-trasonography in patients with nutcracker esopha-gus. Gastrointest Endosc 1997; 46: 223-5.
20. Achem SR, Kolts BE, Wears R, Burton L, Richter JE.Chest pain associated with nutcracker esophagus: apreliminary study of the role of gastroesophageal re-flux. Am J Gastroenterol 1993; 88: 187-92.
21. Richter JE, Obrecht WF, Bradley LA, Young LD, An-derson KO. Psychological comparison of patients
with nutcracker esophagus and irritable bowel syn-drome. Dig Dis Sci 1986; 31: 131-8.
22. Clouse RE. Psychiatric disorders in patients withesophageal disease. Med Clin North Am 1991; 75:1081-96.
23. Richter JE, Barish CF, Castell DO. Abnormal sensoryperception in patients with esophageal chest pain.Gastroenterology 1986 ; 91: 845-52.
24. Drane WE, Johnson DA, Hagan DP, Cattau EL Jr."Nutcracker" esophagus: diagnosis with radionucli-de esophageal scintigraphy versus manometry. Ra-diology 1987; 163: 33-7.
25. Richter JE, Dalton CB, Buice RG, Castell DO. Nifedi-pine: a potent inhibitor of contractions in the body ofthe human esophagus. Studies in healthy volunteersand patients with the nutcracker esophagus. Gastro-enterology 1985; 89: 549-54.
26. Richter JE, Spurling TJ, Cordova CM, Castell DO. Ef-fects of oral calcium blocker, diltiazem, on esopha-geal contractions. Studies in volunteers and pa-tients with nutcracker esophagus. Dig Dis Sci 1984;29: 649-56.
27. Storr M, Allescher HD. Esophageal pharmacologyand treatment of primary moti lity disorders. DisEsophagus 1999; 12: 241-57.
28. Winters C, Artnak EJ, Benjamin SB, Castell DO.Esophageal bougienage in symptomatic patientswith the nutcracker esophagus. A primary esophage-al motility disorder. JAMA 1984; 252: 363-6.
29. Horton ML, Goff JS. Surgical treatment of nutcrackeresophagus. Dig Dis Sci 1986; 31: 878-83.
30. Feussner H, Kauer W, Siewert JR. The surgical ma-nagement of motility disorders. Dysphagia 1993; 8:135-45.
31. Patti MG, Pellegrini CA, Arcerito M, Tong J, MulvihillSJ, Way LW. Comparison of medical and minimallyinvasive surgical therapy for primary esophageal mo-tility disorders. Arch Surg 1995; 130: 609-15.
32. Osgood H. A peculiar form of oesophagismus. Bos-ton Medical Sciences Journal 1889; 120: 401-405.
33. Creamer B, Donoghue FE, Code CF. Pattern of esop-hageal motility in diffuse spasm. Gastroenterology1958; 34: 782-96.
34. Iannello S, Spina M, Leotta P, Prestipino M, Spina S,Ricciardi N, Belfiore F. Hypomagnesemia and smo-oth muscle contractility: diffuse esophageal spasmin an old female patient. Miner Electrolyte Metab1998; 24: 348-56.
35. Crozier RE, Glick ME, Gibb SP, Ellis FH Jr, VeermanJM. Acid-provoked esophageal spasm as a cause ofnoncardiac chest pain. Am J Gastroenterol 1991;86: 1576-80.
36 Di Baise JK, Quigley EM. Tumor-related dysmotility:gastrointestinal dysmotility syndromes associatedwith tumors. Dig Dis Sci 1998; 43: 1369-401.
37. Friesen DL, Henderson RD, Hanna W. Ultrastructure ofthe esophageal muscle in achalasia and diffuse esop-hageal spasm. Am J Clin Pathol 1983; 79: 319-25.
38. Konturek JW, Gillessen A, Domschke W. Diffuseesophageal spasm: a malfunction that involves nitricoxide? Scand J Gastroenterol 1995; 30: 1041-5.
39. Handa M, Mine K, Yamamoto H, Hayashi H, Tsuchi-da O, Kanazawa F, et al. Antidepressant treatmentof patients with diffuse esophageal spasm: a psy-chosomatic approach. J Clin Gastroenterol 1999;28: 228-32.
40. Ruiz de León A. Diagnóstico de los trastornos moto-res del esófago. Revis Gastroenterol 1998; 1: 85-93.
25

41. Barham CP, Gotley DC, Fowler A, Mills A, AldersonD. Diffuse oesophageal spasm: diagnosis by ambula-tory 24 hour manometry. Gut 1997; 41: 151-5.
42. Clouse RE. Trastornos motores. (En: Enfermedadesgastrointestinales. Fisiopatología, diagnóstico y tra-tamiento). De: Sleisenger MH, Fordtran JS. Madrid:Medica Panamericana, 1994; 349-85.
43. Hewson EG, Ott DJ, Dalton CB, Chen YM, Wu WC,Richter JE. Manometry and radiology. Complemen-tary studies in the assessment of esophageal moti-lity disorders. Gastroenterology 1990; 98: 626-32.
44. Ott DJ, Chen YM, Hewson EG, Richter JE, DaltonCB, Gelfand DW, et al. Esophageal motility: assess-ment with synchronous video tape fluoroscopy andmanometry. Radiology 1989; 173: 419-22.
45. Bassotti G, Pelli MA, Morelli A. Clinical and mano-metric aspects of diffuse esophageal spasm in a co-hort of subjects evaluated for dysphagia and/orchest pain. Am J Med Sci 1990; 300: 148-51.
46. Chen YM, Ott DJ, Hewson EG, Richter JE, Wu WC,Gelfand DW, Castell DO. Diffuse esophageal spasm:radiographic and manometric correlation. Radiology1989; 170: 807-10.
47. Richter JE, Castell DO. Diffuse esophageal spasm: areappraisal. Ann Intern Med 1984; 100: 242-5.
48. Dalton CB, Castell DO, Hewson EG, Wu WC, RichterJE. Diffuse esophageal spasm. A rare motility disor-der not characterized by high-amplitude contrac-tions. Dig Dis Sci 1991; 36: 1025-8.
49. Bassotti G, Pelli MA, Morelli A. Clinical and mano-metric aspects of diffuse esophageal spasm in a co-hort of subjects evaluated for dysphagia and/orchest pain. Am J Med Sci 1990; 300: 148-51.
50. Irving JD, Owen WJ, Linsell J, McCullagh M, Keigh-tley A, Anggiansah A. Management of diffuse esop-hageal spasm with balloon dilatation. GastrointestRadiol 1992; 17: 189-92.
51. Campo S, Traube M. Lower esophageal sphincterdysfunction in diffuse esophageal spasm. Am J Gas-troenterol 1989; 84: 928-32.
52. Ebert EC, Ouyang A, Wright SH, Cohen S, LipshutzWH. Pneumatic dilatation in patients with sympto-matic diffuse esophageal spasm and lower esopha-geal sphincter dysfunction. Dig Dis Sci 1983; 28:481-5.
53. Filipi CJ, Hinder RA. Thoracoscopic esophageal myo-tomy a surgical technique for achalasia diffuse esop-hageal spasm and "nutcracker esophagus". Surg En-dosc 1994; 8: 921-5.
54. Willis T. Pharmaceutice rationalis sive diatriba domedicamentorum operationibus in humano corpore.London, Hagae Comitis, 1674.
55. Von Mikuliczi J. Pathologie und Therapie des Car-diospasmus. Dtsch Med Wochenschr 1904; 30: 17.
56. Hurst AF, Rake GW. Achalasia of the cardia. Q JMed 1930; 23: 491-507.
57. Birgisson S, Richter JE. Achalasia: what's new indiagnosis and treatment? Dig Dis 1997; 15 (Suppl1): 1-27.
58. Westley CR, Herbst JJ, Goldman S, Wiser WC. Infan-tile achalasia; inherited as an autosomal recessivedisorder. J Pediatr 1975; 87: 243-6.
59. London FA, Raab DE, Fuller J. Achalasia in three si-blings: a rare occurrence. Mayo Clin Proc 1977; 52:97-100.
60. O'Brien CJ, Smart HL. Familial coexistence of acha-lasia and non-achalasic oesophageal dysmotility:
evidence for a common pathogenesis. Gut 1992;33: 1421-3.
61. Mackler D, Schneider R. Achalasia in father andson. Am J Dig Dis 1978; 23: 1042-5.
62. Chawla K, Chawla SK, Alexander LL. Familial achala-sia of the esophagus in mother and son: a possiblepathogenetic relationship. J Am Geriatr Soc 1979;27: 519-21.
63. Stoddard CJ, Johnson AG. Achalasia in siblings. Br JSurg 1982; 69: 84-5.
64. Stein DT, Knauer CM. Achalasia in monozygotictwins. Dig Dis Sci 1982; 27: 636-40.
65. Eckrich JD, Winans CS. Discordance for achalasia inidentical twins. Dig Dis Sci 1979; 24: 221-4.
66. Kaye MD, Demeules JE. Achalasia and diffuse oe-sophageal spasm in siblings. Gut 1979; 20: 811-4.
67. Frieling T, Berges W, Borchard F, Lubke HJ, Enck P,Wienbeck M. Family occurrence of achalasia anddiffuse spasm of the oesophagus. Gut 1988; 29:1595-602.
68. Robertson CS, Martin BA, Atkinson M. Varicella-zos-ter virus DNA in the oesophageal myenteric plexusin achalasia. Gut 1993; 34: 299-302.
69. Niwamoto H, Okamoto E, Fujimoto J, Takeuchi M,Furuyama J, Yamamoto Y. Are human herpes virusesor measles virus associated with esophageal achala-sia? Dig Dis Sci 1995; 40: 859-64.
70. Verne GN, Sallustio JE, Eaker EY. Anti-myentericneuronal antibodies in patients with achalasia. Aprospective study. Dig Dis Sci 1997; 42: 307-13.
71. Storch WB, Eckardt VF, Wienbeck M, Eberl T, AuerPG, Hecker A, et al. Autoantibodies to Auerbach'splexus in achalasia. Cell Mol Biol (Noyse-le-grand)1995; 41: 1033-8.
72. Clark SB, Rice TW, Tubbs RR, Richter JE, GoldblumJR. The nature of the myenteric infiltrate in achala-sia: an immunohistochemical analysis. Am J SurgPathol 200; 24: 1153-8.
73. Raymond L, Lach B, Shamji FM. Inflammatory ae-tiology of primary oesophageal achalasia: an im-munohistochemical and ultrastructural study ofAuerbach's plexus. Histopathology 1999; 35:4 4 5 - 5 3 .
74. Wong RK, Maydonovitch CL, Metz SJ, Baker JR Jr.Significant DQw1 association in achalasia. Dig DisSci 1989; 34: 349-52.
75. De la Concha EG, Fernández-Arquero M, Mendoza JL,Conejero L, Figueredo MA, Perez de la Serna J, Diaz-Rubio M, Ruiz de Leon. A Contribution of HLA classII genes to susceptibility in achalasia. Tissue Anti-gens 1998; 52: 381-4.
76. Verne GN, Hahn AB, Pineau BC, Hoffman BJ, Wojcie-chowski BW, Wu WC. Association of HLA-DR and -DQ alleles with idiopathic achalasia. Gastroentero-logy 1999; 117: 26-31.
77. Ponce J, Bustamante M, Garriges V, Urquijo J. Tras-tornos motores esofágicos primarios y secundarios.Etiopatogenia y manifestaciones clínicas. Rev EspEnferm Dig 1997; 98: 10-8.
78. Goldblum JR, Rice TW, Richter JE. Histopathologicfeatures in esophagomyotomy specimens from pa-tients with achalasia. Gastroenterology 1996; 111:648-54.
79. Goldblum JR, Whyte RI, Orringer MB, Appelman HD.Achalasia. A morphologic study of 42 resected spe-cimens. Am J Surg Pathol 1994; 18: 327-37.
REVIS GASTROENTEROL Vol. 4. Núm.1. 2002
26

TRASTORNOS MOTORES ESOFÁGICOS PRIMARIOS J. F. SUÁREZ CRESPO ET AL.
80. Trudgill NJ, Hussain FN, Smith LF, Riley SA. Studiesof autonomic function in patients with achalasia andnutcracker oesophagus. Eur J Gastroenterol Hepatol1999; 11: 1349-54.
81. Bittinger M, Barnert J, Eberl T, Wienbeck M. Post-prandial gastric relaxation in achalasia. Eur J Gastro-enterol Hepatol 1998; 10: 741-4.
82. Mearin F, Papo M, Malagelada JR. Impaired gastricrelaxation in patients with achalasia. Gut 1995; 36:363-8.
83. Schmidt T, Pfeiffer A, Hackelsberger N, Widmer R,Pehl C, Kaess H. Dysmotility of the small intestinein achalasia. Neurogastroenterol Motil 1999; 11:11-7.
84. Caturelli E, Squillante MM, Fusilli S, Aliotta A, Celle-rino C, Mangia A, et al. Gallbladder emptying in pa-tients with primary achalasia. Digestion 1992; 52:152-6.
85. Annese V, Caruso N, Accadia L, Gabbrielli A, ModoniS, Frusciante V, et al. Gallbladder function and gas-tric liquid emptying in achalasia. Dig Dis Sci 1991;36: 1116-20.
86. Hagenmuller F, Classen M. Motility of Oddi's sphinc-ter in Parkinson's disease, progressive systemicsclerosis, and achalasia. Endoscopy 1988; 20(Suppl 1): 189-92.
87. Rinaldi R, Cortelli P, Di Simone MP, Pierangeli G,D'Alessandro R, Mattioli S. Cardiovascular autono-mic function in patients with primary achalasia. DigDis Sci 2000; 45: 825-9.
88. von Herbay A, Heyer T, Olk W, Kiesewalter B, AuerP, Enck P, et al. Autonomic dysfunction in patientswith achalasia of the oesophagus. Neurogastroen-terol Motil 1998; 10: 387-93.
89. Eckardt VF, Stenner F, Liewen H, Roder R, Koop H,Bernhard G. Autonomic dysfunction in patients withachalasia. Neurogastroenterol Motil 1995; 7: 55-61.
90. Piqué JM. Regulación de la motilidad digestiva. (En:Motilidad Digestiva. Patología. Diagnóstico y trata-miento). De: Ponce J. Barcelona: J R Prous, 1996:1 - 1 7 .
91. Lefebvre RA. Nitric oxide in the peripheral nervoussystem. Ann Med 1995; 27: 379-88.
92. Mearin F, Mourelle M, Guarner F, Salas A, Riveros-Moreno V, Moncada S, et al. Patients with achalasialack nitric oxide synthase in the gastro-oesophagealjunction. Eur J Clin Invest 1993; 23: 724-8.
93. Aggestrup S, Uddman R, Jensen SL, Sundler F,Schaffalitzky de Muckadell O, Holst JJ, et al. Regula-tory peptides in the lower esophageal sphincter ofman. Regul Pept 1985; 10: 167-78.
94. Guelrud M, Rossiter A, Souney PF, Sulbaran M.Transcutaneous electrical nerve stimulation decrea-ses lower esophageal sphincter pressure in patientswith achalasia. Dig Dis Sci 1991; 36: 1029-33.
95. Guelrud M, Rossiter A, Souney PF, Rossiter G, Fani-kos J, Mujica V. The effect of vasoactive intestinalpolypeptide on the lower esophageal sphincter inachalasia. Gastroenterology 1992; 103: 377-82.
96. Wattchow DA, Costa M. Distribution of peptide-con-taining nerve fibres in achalasia of the oesophagus.J Gastroenterol Hepatol 1996; 11: 478-85.
97. Vaezi MF, Richter JE. Diagnosis and management ofachalasia. American College of GastroenterologyPractice Parameter Committee. Am J Gastroenterol1999; 94: 3406-12.
98. Eckardt VF, Stauf B, Bernhard G. Chest pain in acha-lasia: patient characteristics and clinical course.Gastroenterology 1999; 116: 1300-4.
99. Crookes PF, Corkill S, DeMeester TR. Gastroesopha-geal reflux in achalasia. When is reflux really reflux?Dig Dis Sci 1997; 42: 1354-61.
100. Loviscek LF, Cenoz MC, Badaloni AE, AgarinakazatoO. Early cancer in achalasia. Dis Esophagus 1998;11: 239-47.
101. Yamamuro EM, Cecconello I, Iriya K, Tomishigue T,Oliveira MA, Pinotti HW. Lugol dye endoscopy foranalysis of esophageal mucosa in achalasia. Hepato-gastroenterology 1999; 46: 1687-91.
102. Shah SW, Khan AA, Alam A, Butt AK, Shafqat F.Esophageal bezoar in achalasia: a rare condition. JClin Gastroenterol 1997; 25: 395-6.
103. Ott DJ, Hodge RG, Chen MY, Wu WC, Gelfand DW.Achalasia associated with hiatal hernia: prevalenceand potential implications. Abdom Imaging 1993;18: 7-9.
104. Aly YA. Digital radiography in the evaluation of oe-sophageal motility disorders. Clin Radiol 2000; 55:561-8.
105. Woodfield CA, Levine MS, Rubesin SE, Langlotz CP,Laufer I. Diagnosis of primary versus secondaryachalasia: reassessment of clinical and radiographiccriteria. AJR Am J Roentgenol 2000; 175: 727-31.
106. Dunaway PM, Maydonovitch CL, Wong RK. Charac-terization of esophageal striated muscle in patientswith achalasia. Dig Dis Sci 2000; 45: 285-8.
107. Katz PO, Richter JE, Cowan R, Castell DO. Apparentcomplete lower esophageal sphincter relaxation inachalasia. Gastroenterology 1986; 90: 978-83.
108. Sandler RS, Bozymski EM, Orlando RC. Failure of cli-nical criteria to distinguish between primary achala-sia and achalasia secondary to tumor. Dig Dis Sci1982; 27: 209-13.
109. Rozman RW Jr, Achkar E. Features distinguishing se-condary achalasia from primary achalasia. Am J Gas-troenterol 1990; 85: 1327-30.
110. Kahrilas PJ, Kishk SM, Helm JF, Dodds WJ, HarigJM, Hogan WJ. Comparison of pseudoachalasia andachalasia. Am J Med 1987; 82: 439-46.
111. Deviere J, Dunham F, Rickaert F, Bourgeois N, Cre-mer M. Endoscopic ultrasonography in achalasia.Gastroenterology 1989 Apr; 96 (4): 1210-3.
112. Barthet M, Mambrini P, Audibert P, Boustiere C, Hel-bert T, Bertolino JG, et al. Relationships between en-dosonographic appearan e and clinical or manome-t ric features in patients with acha lasia. Eur JGastroenterol Hepatol 1998; 10: 559-64.
113. Hatlebakk JG, Odegaard S. Endoscopic ultrasound: anew look at achalasia? Eur J Gastroenterol Hepatol1998; 10: 543-5.
114. Van Dam J. Endosonographic evaluation of the pa-tient with achalasia. Endoscopy 1998; 30 (Suppl 1):A48-50.
115. Rabushka LS, Fishman EK, Kuhlman JE. CT evalua-tion of achalasia. J Comput Assist Tomogr 1991; 15:434-9.
116. Carter M, Deckmann RC, Smith RC, Burrell MI, Trau-be M. Differentiation of achalasia from pseudoacha-lasia by computed tomography. Am J Gastroenterol1997; 92: 624-8.
117. Vaezi MF, Richter JE. Current therapies for achala-sia: comparison and efficacy. J Clin Gastroenterol1998; 27: 21-35.
27

118. Bortolotti M, Mari C, Lopilato C, Porrazzo G, MiglioliM. Effects of sildenafil on esophageal motility of pa-tients with idiopathic achalasia. Gastroenterology2000; 118: 253-7.
119. Pasricha PJ, Ravich WJ, Kalloo AN. Botulinum toxinfor achalasia. Lnacet 1993; 341: 244-5.
120. Pasricha PJ, Ravich WJ, Kalloo AN. Effects of in-trasphincteric botulinum toxin on the lower esopha-geal sphincter in piglets. Gastroenterology 1993;105: 1045-9.
121. Pasricha PJ, Ravich WJ, Hendrix TR, Sostre S, JonesB, Kallo AN. Treatment of achalasia with intrasp-hincteric injection of botulinum toxin. A pilot trial.Ann Intern Med 1994; 121: 590-1.
122. Pasricha PJ, Ravich WJ, Hendrix TR, Sostre S, Jones B,Kallo AN. Intrasphincteric botulinum toxin for the treat-ment of achalasia. N Engl J Med 1995; 332: 774-8.
123. Pasricha PJ, Rai R, Ravich WJ, Hendrix TR, KallooAN. Botulinum toxin for achalasia: long-term outco-me and predictors of response. Gastroenterology1996; 110: 1410-5.
124. Cuilliere C, Ducrotte P, Zerbib F, Metman EH, de Lo-oze D, Guillemot F, et al. Achalasia: outcome of pa-tients treated with intrasphincteric injection of botu-linum toxin. Gut 1997; 41: 87-92.
125. Prakash C, Freedland KE, Chan MF, Clouse RE. Bo-tulinum toxin injections for achalasia symptoms canapproximate the short term efficacy of a singlepneumatic dilation: a survival analysis approach. AmJ Gastroenterol 1999; 94: 328-33.
126. Kolbasnik J, Waterfall WE, Fachnie B, Chen Y, Tou-gas G. Long-term efficacy of Botulinum toxin in clas-sical achalasia: a prospective study. Am J Gastroen-terol 1999; 94: 3434-9.
127. Hurwitz M, Bahar RJ, Ament ME, Tolia V, MollestonJ, Reinstein et al. Evaluation of the use of botulinumtoxin in children with achalasia. J Pediatr Gastroen-terol Nutr 2000; 30: 509-14.
128. Annese V, Bassotti G, Coccia G, Dinelli M, D'OnofrioV, Gatto G, et al. A multicentre randomised study ofintrasphincteric botulinum toxin in patients with oe-sophageal achalasia. GISMAD Achalasia StudyGroup. Gut 2000; 46: 597-600.
129. Hoffman BJ, Knapple WL, Bhutani MS, Verne GN, Ha-wes RH. Treatment of achalasia by injection of botu-linum toxin under endoscopic ultrasound guidance.Gastrointest Endosc 1997; 45: 77-9.
130. Maiorana A, Fiorentino E, Genova EG, Murata Y, Su-zuki S. Echo-guided injection of botulinum toxin inpatients with achalasia: initial experience. Endos-copy 1999; 31: S3-4.
131. Vaezi MF, Richter JE, Wilcox CM, Schroeder PL, Bir-gisson S, Slaughter RL, et al. Botulinum toxin ver-sus pneumatic dilatation in the treatment of achala-sia: a randomised trial. Gut 1999; 44: 231-9.
132. Brin MF, Lew MF, Adler CH, Comella CL, Factor SA,Jankovic J, et al. Safety and efficacy of NeuroBloc(botulinum toxin type B) in type A-resistant cervicaldystonia. Neurology 1999; 53: 1431-8.
133. Panaccione R, Gregor JC, Reynolds RP, PreiksaitisHG. Intrasphincteric botulinum toxin versus pneuma-tic dilatation for achalasia: a cost minimizationanalysis. Gastrointest Endosc 1999; 50: 492-8.
134. Horgan S, Hudda K, Eubanks T, McAllister J, Pelle-grini CA. Does botulinum toxin injection make esop-hagomyotomy a more difficult operation?. Surg En-dosc 1999; 13: 576-9.
135. Moreto M, Ojembarrena E, Rodríguez ML. Endoscopicinjection of ethanolamine as a treatment for achala-sia: a first report. Endoscopy 1996; 28: 539-45.
136. Moreto M. Barturen A, Ojembarrena E, Casado I.Treatment of achalasia by injection of sclerosants: a
long-term experience. Gastrointest Endosc 1999;99: AB 128.
137. Moreto M, Ojembarrena E. Treatment of achalasiaby injection of botulinum toxin or sclerosants?. En-doscopy 2000; 32: 361-2.
138. Alonso P, Castro ML, Vázquez-Iglesias JL. Tratamien-to endoscópico de las estenosis esofágicas benig-nas. (En: Endoscopia Digestiva alta II. Terapéutica).De: Vázquez Iglesias JL. A Coruña: Galicia Editorial,1995; 41-62.
139. Vazquez-Iglesias JL, Alonso P. Tratamiento de las es-tenosis esofágicas benignas. (En: Gastroenterolo-gía. Endoscopia diagnóstica y terapéutica). Madrid:Abreu L. Eurobook, 1998: 133-138.
140. Vaezi MF, Richter JE. Current therapies for achala-sia: comparison and efficacy. J Clin Gastroenterol1998; 27: 21-35.
141. Ponce J, Garrigues V, Pertejo V, Sala T, Berenguer J.Individual prediction of response to pneumatic dila-tion in patients with achalasia. Dig Dis Sci 1996;41: 2135-41.
142. Borotto E, Gaudric M, Danel B, Samama J, QuartierG, Chaussade S, Couturier D. Risk factors of oesop-hageal perforation during pneumatic dilatation forachalasia. Gut 1996; 39: 9-12.
143. Barnett JL, Eisenman R, Nostrant TT, Elta GH. Wit-zel pneumatic dilation for achalasia: safety and long-term efficacy. Gastrointest Endosc 1990; 36: 482-5.
144. Metman EH, Lagasse JP, d'Alteroche L, Picon L, Scot-to B, Barbieux JP. Risk factors for immediate compli-cations after progressive pneumatic dilation for acha-lasia. Am J Gastroenterol 1999 ; 94: 1179-85.
145. Kadakia SC, Wong RK. Graded pneumatic dilationusing Rigiflex achalasia dilators in patients with pri-mary esophageal achalasia. Am J Gastroenterol1993; 88: 34-8.
146. Nair LA, Reynolds JC, Parkman HP, Ouyang A, StromBL, Rosato EF, et al. Complications during pneumaticdilation for achalasia or diffuse esophageal spasm.Analysis of risk factors, early clinical characteristics,and outcome. Dig Dis Sci 1993; 38: 1893-904.
147. Heller E. Extramuköse Kardioplastick beim chronis-chen Kardiospasmus mit Dilatation des Oesophagus.Mitt Grenzgeb Med Chir 1914; 27: 141-9.
148. Zaaijer JH. Cardiospasm in the aged. Ann Surg1923; 77: 615-7.
149. Shimi S, Nathanson LK, Cuschieri A. Laparoscopiccardiomyotomy for achalasia. J R Coll Surg Edinb1991; 36: 152-4 (abstract).
150. Pellegrini C, Wetter LA, Patti M, Leichter R, MussanG, Mori T, et al. Thoracoscopic esophagomyotomy.Initial experience with a new approach for the treat-ment of achalasia. Ann Surg 1992; 216: 291-6.
151. Goldenberg SP, Burrell M, Fette GG, Vos C, TraubeM. Classic and vigorous achalasia: a comparison ofmanometric, radiographic, and clinical findings. Gas-troenterology 1991; 101: 743-8.
152. Ponce García J, Garrigues Gil V, Pertejo Pastor V,Valverde de la Osa J, Galvez Castillo C, BerenguerLapuerta J. Existen diferencias clínicas y en la res-puesta a la dilatación neumática entre la acalasia tí-pica y vigorosa. Gastroenterol Hepatol 1995; 18:315-8.
153. Parrilla P, Martínez de Haro LF, Ortiz A, Morales G,Garay V, Aguilar J. Factors involved in the return ofperistalsis in patients with achalasia of the cardiaafter Heller's myotomy. Am J Gastroenterol 1995 ;90: 713-7.
154. Parrilla Paricio P, Martinez de Haro LF, Ortiz Escan-dell A, Morales Cuenca G, Molina Martinez J. Shortmyotomy for vigorous achalasia. Br J Surg 1993; 80:1540-2.
REVIS GASTROENTEROL Vol. 4. Núm.1. 2002
28


















