
UNIVERSITAT DE VALÈNCIA FACULTAT DE MEDICINA · Tangier, déficit de apo A-I y C-III, D, déficit...
114
UNIVERSITAT DE VALÈNCIA FACULTAT DE MEDICINA APO B-100 DEFECTUOSA FAMILIAR: EFECTO FUNDADOR EN UNA POBLACIÓN DE LA COMUNITAT VALENCIANA Y COMPARACIÓN CON LA HIPERCOLESTEROLEMIA FAMILIAR. TESIS DOCTORAL Presentada por: Ismael Ejarque Doménech Valencia, 2006
Transcript of UNIVERSITAT DE VALÈNCIA FACULTAT DE MEDICINA · Tangier, déficit de apo A-I y C-III, D, déficit...

UNIVERSITAT DE VALÈNCIA FACULTAT DE MEDICINA
APO B-100 DEFECTUOSA FAMILIAR: EFECTO FUNDADOR EN UNA POBLACIÓN DE LA COMUNITAT VALENCIANA Y COMPARACIÓN CON LA HIPERCOLESTEROLEMIA FAMILIAR.
TESIS DOCTORAL Presentada por:
Ismael Ejarque Doménech Valencia, 2006


D. Rafael Carmena Rodríguez, Catedrático de Medicina de la Universitat de Valencia, D. Juan Francisco Ascaso Gimilio, Catedrático de Medicina de la Universitat de València y D. José T. Real Collado, Profesor asociado de Medicina de la Universitat de València certifican:
Que D. Ismael Ejarque Doménech, Licenciado en Medicina y Cirugía, ha realizado bajo nuestra dirección la tesis doctoral titulada: “Apo B-100 defectuosa: efecto fundador en una población de la Comunitat Valenciana y comparación con la hipercolesterolemia familiar”, y que está en condiciones de ser presentada para optar al grado de Doctor en Medicina y Cirugía.
Valencia, 6 de julio de 2006
Fdo. Prof. R. Carmena Fdo. Prof. JF. Ascaso Fdo. Prof. J.T. Real


AGRADECIMIENTOS
Con estas líneas quiero expresar mi más sincera gratitud a todos los que de alguna forma han hecho posible este trabajo.
A los Doctores Don Rafael Carmena y Don Juan Francisco Ascaso, por haberme dado ambos la oportunidad de llevar a cabo mi tesis doctoral dentro de su grupo de trabajo, ampliamente reconocido dentro del mundo de la medicina clínica y de la investigación.
Al Doctor Don José Tomás Real, por sus acertados consejos, apoyo y paciencia que siempre ha mostrado durante estos años de trabajo doctoral.
A los Doctores Doña Maria Eugenia Armengod y Don Felipe Javier Chaves, por sus orientaciones científicas y trabajo realizado en el laboratorio sobre genética molecular que entraña la presente tesis doctoral.
Al Doctor Don Enrique Milián, médico titular de la población de La Yesa, pues su ayuda ha sido fundamental a la hora de identificar las personas susceptibles de presentar la patología a estudio en esta tesis.
A la población de La Yesa y a su alcalde Don Alfredo Zaragozá, por las facilidades y colaboración prestadas en la presente investigación.
A los párrocos de la Iglesia Parroquial de Nuestra Señora de Los Ángeles de La Yesa, al facilitarme el proceso para dar con el árbol genealógico de las familias candidatas a estudio.
A mi padre, Don Ismael Ejarque Pardo, Ayudante Técnico Sanitario, por su colaboración desinteresada en la extracción de las muestras sanguíneas realizadas en La Yesa, así como a mi madre, Doña Carmen Doménech Pascual, pues ambos me han apoyado y animado durante todos estos años en la realización del presente estudio.


ABREVIATURAS
ADH: Hipercolesterolemia autosómica dominante.
ADN: Ácido desoxirribonucleico.
ADNc: Ácido desoxirribonucleico complementario.
Apo B: Apolipoproteína B-100.
Apo B-100: Apolipoproteína B-100.
Apo E: Apolipoproteína E.
APOB: Gen que codifica la apolipoproteína B (según la nomenclatura HUGO).
APOE: Gen que codifica la apolipoproteína E (según la nomenclatura HUGO).
ARH: Hipercolesterolemia autosómica recesiva.
ARNm: Ácido ribonucleico mensajero.
cHDL: Colesterol de lipoproteínas de alta densidad.
cIDL: Colesterol de lipoproteínas de densidad intermedia.
cLDL: Colesterol de lipoproteínas de baja densidad.
cVLDL: Colesterol de lipoproteínas de muy baja densidad.
CI: Cardiopatía isquémica.
CT: Colesterol total.
DFB: Defecto familiar de unión de apo B-100
FH: Hipercolesterolemia familiar.
HDL: Lipoproteínas de alta densidad.
HVR: Región hipervariable.
HF: Hipercolesterolemia familiar.
HUGO: Organización del genoma humano. IDL: Lipoproteínas de densidad intermedia.
IMC: Índice de masa corporal.

LDL: Lipoproteínas de baja densidad.
LDLR: Gen que codifica el receptor de las LDL (según la nomenclatura HUGO).
Lp(a): Lipoproteína (a).
LPA: Gen que codifica la lipoproteína (a) (según la nomenclatura HUGO).
Med-Ped: Make Early Diagnosis to Prevent Early Deaths; “hacer diagnóstico precoz para prevenir muertes precoces”.
OMIM: Herencia mendeliana humana en red (internet).
OMS: Organización Mundial de la Salud.
PCR: Reacción en cadena de la polimerasa.
PLG: Gen que codifica el plasminógeno (según la nomenclatura HUGO).
SNP: Polimorfismo de un único nucleótido.
SSCP: Polimorfismo conformacional de cadena sencilla.
TG: Triglicéridos.
VLDL: Lipoproteínas de muy baja densidad.

ÍNDICE


ÍNDICE
1. INTRODUCCIÓN (pág 1)
1. 1. Arteriosclerosis y enfermedad cardiovascular (pág 3).
1. 2. Dislipemias (pág 4).
1. 3. Hipercolesterolemia Familiar (pág 7)
- Recuerdo histórico (pág 7). - Clínica (pág 7). - Fisiopatología del receptor de LDL (pág 8). - Proteína y gen (pág 9). - Mutaciones del gen LDLR (pág 10). - Genética de poblaciones (pág 11). - Relación genotipo-fenotipo (pág 11). - Diagnóstico (pág 11).
1. 4. Defecto Familiar de unión de Apo B-100 (pág 12).
- Recuerdo histórico (pág 12). - Genética molecular (pág 13). - Clínica (pág 14). - Diagnóstico (pág 15).
1. 5. Efecto fundador (pág 15).
- Conceptos (pág 15). - Origen histórico de la mutación R3500Q (pág 19). - Datos históricos sobre la repoblación de la comarca de Los Serranos (p 20) - Mapa de localización de La Yesa (pág 23).
1. 6. Lipoproteína (a) (pág 23).
2. HIPÓTESIS Y OBJETIVOS (pág 25)
2. 1. Hipótesis (pág 27).
2. 2. Objetivos (pág 27).
3. RESULTADOS (pág 29)
3. 1. Resumen de los estudios (pág 31).

3. 2. Estudio 1 (pág 31).
3. 3. Estudio 2 (pág 32).
3. 4. Estudio 3 (pág 33).
3. 5. Estudio 4 (pág 33).
3. 6. Estudio 5 (pág 36).
4. DISCUSIÓN (pág 75)
5. CONCLUSIONES (pág 83)
6. BIBLIOGRAFÍA (pág 87)

1
INTRODUCCIÓN

2

3
1. INTRODUCCIÓN
1. 1. Arteriosclerosis y enfermedad cardiovascular
Las enfermedades cardiovasculares son la principal causa de morbi-mortalidad en España al igual que en el resto de los países occidentales. En la Comunidad Valenciana, según el Registro de Mortalidad de 1998, fueron la causa de muerte en el 45’2 % de las mujeres y el 34’2 % en hombres.
La base patogénica común de estas enfermedades es la arteriosclerosis, proceso inflamatorio crónico de la pared arterial, que en una primera etapa producirá una lesión proliferativa de las capas íntima y media, posteriormente afectará a la luz arterial y en combinación con procesos trombóticos podrá causar oclusión vascular e isquemia distal.
La aparición y evolución de la arteriosclerosis están determinadas por la interacción de factores genéticos y ambientales relacionados con el metabolismo lipídico e hidrocarbonado, la presión arterial, la coagulación, la respuesta inflamatoria y la función endotelial.
En la actualidad, no hay dudas sobre la implicación directa de los lípidos en la aparición y desarrollo de la placa ateromatosa. Esta afirmación se basa en diferentes tipos de pruebas y observaciones, algunas de ellas obtenidas a finales del siglo XIX y primeras décadas del siglo XX1, 2. Así, observaciones anatomopatológicas describieron la presencia de colesterol en las placas de ateroma; modelos animales demostraron que la hipercolesterolemia, generalmente inducida con manipulación dietética, provoca arteriosclerosis; de igual modo, los pacientes con hipercolesterolemias genéticas sufren con frecuencia arteriosclerosis coronaria a edades precoces. Además, estudios epidemiológicos y de intervención con fármacos hipolipemiantes, fundamentalmente estatinas, han comprobado y sustentado la teoría lipídica de la arteriosclerosis3. Dicha teoría propone que los lípidos que atraviesan el endotelio y se acumulan en la íntima, primer escalón de la reacción aterosclerótica4, proceden de las lipoproteínas circulantes, y por lo tanto, un alto nivel de lipoproteínas plasmáticas se asocia con concentraciones elevadas de éstas en la pared arterial.
Aparte de por factores genéticos, el metabolismo lipídico se ve influido por factores exógenos: hábitos dietéticos, ejercicio físico o por enfermedades intercurrentes, como disfunciones tiroideas, diabetes, síndrome nefrótico, colestasis, etc.
Habitualmente las alteraciones del metabolismo lipídico o dislipemias se clasifican en primarias (genéticas) y secundarias como las ya mencionadas. En el contexto de esta tesis nos centraremos exclusivamente en las dislipemias primarias o de origen genético, que afectan aproximadamente al 5 %5 de la población general.
Es importante recalcar que el fenotipo o expresión clínica de estas dislipemias primarias es el resultado de la interrelación del genotipo, en este caso la alteración genética causal, con los diversos factores ambientales. Dicho de otro modo, un mismo genotipo puede resultar en una cierta variabilidad fenotípica que va a depender de los hábitos de vida del sujeto, y otros factores exógenos. Muchas condiciones mendelianas son variables clínicamente incluso entre miembros afectos de una misma familia que portan la misma mutación. La variabilidad intrafamiliar puede ser causada por algún

4
tipo de combinación de los efectos de otros genes no-ligados (genes modificadores) y efectos del medio ambiente (incluidos los sucesos al azar)6.
Revisaremos a continuación una serie de conceptos importantes como paso previo para plantear nuestra hipótesis de trabajo y objetivos.
1. 2. Dislipemias
Las alteraciones del metabolismo lipídico o dislipemias se dividen en alteraciones por descenso de las lipoproteínas plasmáticas (hipolipoproteinemias o hipolipemias) y aquellas por aumento de las mismas (hiperlipoproteinemias o hiperlipemias).
En el grupo de las hipolipoproteinemias se incluyen diversos cuadros caracterizados por disminución de las HDL o por descenso de la concentración plasmática de lipoproteínas con apo B (VLDL y LDL). Es decir:
. Hipolipoproteinemias con niveles bajos de HDL: 1.a. Hipoalfalipoproteinemia familiar. 1.b. Hipoalfalipoproteinemia familiar con hipertrigliceridemia.
1.c. Síndromes genéticos que cursan con hipolipoproteinemia: enfermedad de Tangier, déficit de apo A-I y C-III, D, déficit de lecitin-colesterol aciltransferasa, enfermedad de los ojos de pez, y algunas variantes de Apo A-I (Milano, Munster, Marburg, etc).
. Hipolipoproteinemias con descenso de la concentración plasmática de las lipoproteínas que contienen Apo B (VLDL y LDL). 2.a. Hipobetalipoproteinemia familiar. 2.b. Abetalipoproteinemia familiar.
Se denomina hiperlipemia a los niveles lipídicos (colesterol y/o triglicéridos) séricos superiores a los valores plasmáticos ideales, compatibles con el mínimo riesgo de sufrir cardiopatía isquémica. Estos valores son muy variables dependiendo de las poblaciones estudiadas y han sido definidos por diversas asociaciones médicas mediante consenso internacional. Se aceptan como niveles deseables para la población general a los siguientes parámetros: colesterol total menor de 200 mg/dl, LDL colesterol inferior a 150 mg/dl, triglicéridos menores de 200 mg/dl y HDL colesterol superior a 35 mg/dl 7.En sujetos con otros factores de riesgo coronario los valores son, aproximadamente, 180, 130, 150 y 40 mg/dl respectivamente.
La hiperlipemia tiene una gran importancia dentro del riesgo cardiovascular global. Pero no hay que olvidar que se debe valorar al individuo en su conjunto y por ello junto con la hiperlipemia, debemos evaluar todos los factores de riesgo cardiovascular, mayores y menores como género, edad, antecedentes familiares de enfermedad cardiovascular precoz, hipertensión arterial, tabaquismo, diabetes, obesidad central, etc.
La clasificación de las hiperlipoproteinemias ha sufrido bastantes cambios, y aún hoy no existe ninguna que sea totalmente satisfactoria. La primera la llevó a cabo Fredrickson en 1967 8 y se basaba en el desplazamiento electroforético de las lipoproteínas; esta clasificación fue modificada en 1970 por un grupo de expertos de la

5
OMS 9, estableciendo 6 tipos. Más tarde, Havel simplificó el uso de esta clasificación pues la basó en parámetros más asequibles para el médico, como son el colesterol y los triglicé-ridos plasmáticos y además el aspecto del suero conservado a 4ºC durante doce horas (Tabla 1). El principal problema de esta clasificación es que al ser meramente descriptiva no diferenciaba entre hiperlipemias primarias y secundarias.
TABLA 1. CLASIFICACION FENOTIPICA DE LAS HIPERLIPOPROTEINEMIAS (OMS 1970), MODIFICADA POR HAVEL.
FENOTIPO LIPOPROTEINA ELEVADA LIPIDOS ELEVADOS
ASPEPTO DEL SUERO A 4ºC
Tipo I Quilomicrones TG Anillo cremoso sobrenadanteTipo IIA LDL CT Suero transparenteTipo IIB LDL y VLDL CT y TG Suero turbio Tipo III IDL CT y TG Suero turbio Tipo IV VLDL TG Suero turbio Tipo V Quilomicrones y VLDL TG Suero turbio con anillo cremoso sobrenadante
Como ya mencionamos, las hiperlipoproteinemias se dividen en la actualidad en primarias (o genéticas) y secundarias a otras alteraciones si nos basamos en los conocimientos etiopatogénicos de los que disponemos. Lo interesante de esta clasificación es que dentro de un mismo fenotipo existen diferentes entidades nosológicas con manifestaciones clínicas, riesgo cardiovascular, asesoramiento genético y tratamiento diferentes tanto en las primarias (Tabla 2) como en las secundarias (Tabla 3).
TABLA 2. CLASIFICACION DE LAS HIPERLIPOPROTEINEMIAS PRIMARIAS
NOMBRE FENOTIPO CT TG HERENCIA DEFECTO RESPONSABLE Hipercolesterolemia familiar IIA N Dominante Receptor LDL Defecto familiar de unión de apo B IIA N Dominante Apo B defectuosa Hipercolesterolemia poligénica IIA N Poligénica Desconocido Hipertrigliceridemia familiar IV N Dominante Desconocido Hiperlipemia familiar combinada IIA,IIB,IV oN oN Dominante Aumento Apo B Disbetalipoproteinemia familiar III Recesiva Alt. Apoproteina E Hiperlipemia exógena I N Recesiva Déficit de Lipoprotein Lipasa
Déficit de apo C-II Hiperlipemia mixta V No ? Apo E4, apo CIII ?
TABLA 3. HIPERLIPEMIAS SECUNDARIAS MÁS FRECUENTES (Fenotipo/Enfermedades responsables)
Tipo I Diabetes mellitus mal controlada Pancreatitis aguda Disgammaglobulinemia Lupus eritematosos diseminado
Tipo IIA Hipotiroidismo Síndrome nefrótico Colestasis Porfiria aguda intermitente Anorexia nerviosa Hepatoma Disgammaglobulinemias
Tipo IIB Síndrome nefrótico Contraceptivos Disgammaglobulinemia
Tipo III Hipotiroidismo Diabetes mellitus mal controlada Disgammaglobulinemia
Tipo IV Diabetes mellitus mal controlada Obesidad Hepatitis aguda Insuficiencia renal crónica Alcoholismo Disgammaglobulinemia Glucogenosis Trasplante renal Síndrome de Cushing Sepsis Estrés Acromegalia
Tipo V Diabetes mellitus mal controlada Alcoholismo Contraceptivos esteroideos Glucogenosis

6
En la tabla 4 presentamos el diagnóstico diferencial de las hipercolesterolemias primarias. Partimos de la base teórica que el Defecto Familiar de union de Apo B 100 tiene el mismo fenotipo lipoproteico que el de la Hipercolesterolemia Familiar.
TABLA 4. DIAGNÓSTICO DIFERENCIAL DE LAS HIPERCOLESTEROLEMIAS
HIPERCOLESTEROLEMIA FAMILIAR
HIPERLIPEMIA FAMILIAR COMBINADA
HIPERCOLESTEROLEMIA POLIGENICA
EDAD DE COMIENZO NACIMIENTO >16-20 AÑOS >20 AÑOS COLESTEROLEMIA HOMOCIGOTOS 600-1200
mg/dl HETEROCIGOTOS 250-300 mg/dl
250-400 mg/dl 260-350 mg/dl
LIPOPROTEINAS LDL -LDL, -VLDL o AMBAS LDL XANTOMAS TENDINOSOS SI NO SI MANIFESTACIONES DE ARTERIOSCLEROSIS CORONARIA
HOMOCIGOTO <20 AÑOS HETEROCIGOTO 30-50 AÑOS
<40 AÑOS >60 AÑOS
ANTECEDENTES DE HIPERLIPEMIA EN FAMILIARES DE PRIMER GRADO
50% 50% 10%
ASOCIACION CON DIABETES OBESIDAD E HIPERURICEMIA
NO SI NO
Más recientemente10 se ha propuesto una clasificación de las hipercolesterolemias primarias en formas dominantes y recesivas. Las hipercolesterolemias autosómicas dominantes (ADH) conocidas hasta ahora son la FH, DFB, PCSK9 y otras. Las hipercolesterolemias autosómicas recesivas (ARH) son muy poco frecuentes y se han descrito casi exclusivamente en la población sarda.
Creo conveniente recalcar que en atención primaria todo Médico de Familia deberá sospechar una hiperlipemia primaria en los siguientes casos, y remitirlos a la Unidad de Lípidos de Referencia:
Edad precoz de diagnóstico de la hiperlipemia (niños y jóvenes adultos). Presencia de xantomas tendinosos, arco corneal en menores de 45 años o presencia de xantomas eruptivos o palmares. Presencia de hiperlipemia o cardiopatía isquémica precoz en familiares de primer y segundo grado. Valores plasmáticos repetidos superiores al percentil 90 de colesterol y/o triglicéridos sin causas secundarias aparentes. Sujetos < 40 años con cardiopatia isquémica. Pancreatitis agudas de repetición no etílicas ni debidas a patología biliar.
La presente tesis doctoral versa sobre el Defecto Familiar de unión a Apo B-100 (DFB) en una población de la Comunitat Valenciana. En la misma se han comparado los pacientes DFB con pacientes con Hipercolesterolemia Familiar clásica (HF). Hemos analizado el efecto fundador de los pacientes DFB de nuestra población, así como las posibles alteraciones en sus valores de Lp(a). Por ello consideramos conveniente profundizar sobre dichos argumentos: HF, DFB, efecto fundador y Lp(a).

7
1. 3. Hipercolesterolemia familiar (HF)
La Hipercolesterolemia Familiar (HF) es una enfermedad hereditaria autosómica dominante debida a mutaciones en el gen del receptor de las LDL (gen LDLR), que codifica una glicoproteína de membrana fundamental para el aclaramiento de LDL colesterol cLDL del plasma. La enfermedad se caracteriza por la presencia de xantomas, arco corneal y placas ateromatosas coronarias que dan lugar a cardiopatía isquémica precoz. Estos signos traducen el depósito de cLDL en dichos tejidos11.
Los afectados manifiestan dos cuadros sindrómicos diferentes dependiendo de si existe un 50% de receptores funcionantes, como en el caso de los sujetos heterocigotos, o si no existe ningún receptor funcional como ocurre en los homocigotos.
Los heterocigotos tienen una frecuencia de 1 afectado por cada 500 sujetos sanos en la poblacion general, siendo la enfermedad monogénica más frecuente en humanos. Al tener una copia funcional del receptor LDL, presentan niveles de cLDL dos veces mayores que la de los sujetos sanos. Los homocigotos tienen una frecuencia en la población general de 1 en 1.000.000 de habitantes y presentan un síndrome más grave con elevaciones extremas de cLDL y cardiopatía isquémica en la infancia o en la juventud12.
Recuerdo histórico
Ya antes de 1900 fue descrita13 la aparición simultánea de xantomas tendinosos y aterosclerosis coronaria. En 1930 se describió la existencia de familias en las que aparecían xantomas, enfermedad isquémica coronaria precoz e hipercolesterolemia. Pero fue en 1938 cuando Müller describe por primera vez la enfermedad como un error hereditario del metabolismo que conduce a la presencia de xantomas, elevaciones del colesterol plasmático e infarto agudo de miocardio en pacientes jóvenes.
Los estudios de Wilkinson14 y Aldesberg15 de los años 40 y 50 del pasado siglo sugerían la presencia de una base genética que justificaba la agregación familiar de esta hipercolesterolemia. Khachadurian16 en los años 60 fue el autor que definió claramente las características clínicas y genéticas de la enfermedad al describir y diferenciar los heterocigotos de los homocigotos en familias libanesas afectas.
Goldstein y Brown, usando cultivos de fibroblastos humanos, descubrieron en la década de los 70 el receptor de las LDL y demostraron que la enfermedad se debía a mutaciones del gen del receptor para las partículas LDL17. Posteriormente, en 1982 el receptor fue purificado, en 1983 clonado su ADNc y en 1985 aislado y caracterizado su gen18, 19. Estos avances han permitido la caracterización molecular de la HF.
Clínica
Los signos y síntomas que presentan los heterocigotos dependen de su edad. Desde el nacimiento hasta la primera década de vida la mayor parte de los afectados sólo presenta elevaciones de cLDL. Los xantomas y el arco corneal suelen aparecer al final de la segunda década de la vida y aparecen entre un 30 y 50% de los afectados sobre la tercera década. Los síntomas de cardiopatía isquémica se manifiestan en hombres a partir de la cuarta década de vida y en mujeres a partir de la quinta década11.

8
En los homocigotos la clínica suele ser mas homogénea con desarrollo de xantomas tendinosos anaranjados y presencia de hipercolesterolemia grave desde el nacimiento. En la infancia o juventud la mayor parte de ellos mueren por cardiopatía isquémica20.
Los valores medios de colesterol total (CT) en los heterocigotos son de 350 mg/dl aunque existen considerables variaciones en individuos incluso dentro de la misma familia con cifras que oscilan entre 250 y 600 mg/dl. En los homocigotos los niveles de CT son muy elevados, desde 600 a 1200 mg/dl. Los niveles de triglicéridos son en la mayor parte de los afectados normales, aunque en algunos individuos pueden encontrarse niveles por encima de 250 mg/dl dado que el receptor de LDL también aclara las VLDL y las IDL11, 12.
La hipercolesterolemia se debe a un aumento de la fracción cLDL y cIDL. La media de cLDL en heterocigotos es 2-3 veces mayor que la media de la población general y la de los homocigotos 6 veces mayor. Estas partículas tiene un contenido lipídico, protéico, inmunoreactivo y de composición normales. Así, las partículas LDL de sujetos heterocigotos y homocigotos HF son adecuadamente metabolizadas por los receptores de sujetos normales11, 12. Los niveles de cHDL son discretamente menores que los de la población general.
El cLDL elevado se deposita en tendones y piel dando lugar a los xantomas. La aparición de estos depósitos dependen de la gravedad de la alteración y de la duración de la elevación del cLDL. Los depósitos se localizan fundamentalmente en tendones extensores de los dedos, codos y tendón de Aquiles. Los xantelasmas pueden aparecer en heterocigotos pero no son característicos de la enfermedad. Los xantomas cutáneos anaranjados y sobreelevados son típicos de los homocigotos y se localizan en muslos, manos y zona glútea.
El cLDL elevado también se deposita en las arterias coronarias en forma de placas de ateroma dando lugar a cardiopatía isquémica precoz (CI). En homocigotos la aterosclerosis afecta de forma muy precoz a las arterias coronarias y a la válvula aórtica, dando lugar a angor pectoris, infarto agudo de miocardio o muerte súbita antes de los 30 años21.
La CI en heterocigotos es heterogénea dependiendo de la edad del sujeto y del sexo. La CI en varones aparece 10 años antes que en mujeres HF. El riesgo de CI en varones es de aproximadamente un 5% a la edad de 30 años, de un 50% a la de 50 años y de un 80% a los 60 años22. Se especula con diferentes factores ambientales y genéticos que puedan contribuir a determinar el riesgo de CI en la HF. Entre estos factores destacan: el tabaquismo, los niveles de cHDL, el genotipo del gen apo E y los niveles de Lp(a)23, 24, 25.
Fisiología del receptor de LDL
La enfermedad se caracteriza por una elevación de las lipoproteínas de baja densidad o LDL, partículas que transportan la mayor parte del colesterol plasmático y lo transportan a las células de nuestro organismo para que sea utilizado en la formación de membranas, síntesis de hormonas esteroideas y precursores de los ácidos biliares. Esta

9
elevación de las lipoproteínas de baja densidad se debe a la existencia de alteraciones del receptor para LDL. El receptor podrá ser defectuoso funcionalmente o podrá no ser expresado a nivel de la membrana celular. Ambas alteraciones conducen a una disminución en el aclaramiento de las partículas LDL26.
Las alteraciones del receptor de LDL se deben a mutaciones que afectan al gen LDLR. El receptor de las LDL fue descubierto por Goldstein y Brown17 en estudios realizados sobre fibroblastos de pacientes con hipercolesterolemia familiar homocigota.
Se trata de una glicoproteína de membrana que contiene aproximadamente dos cadenas de oligosacáridos complejos unidos a asparragina (uniones N) y 18 cadenas de oligosacáridos unidos a serina/treoninas (uniones O)27. Esta glicoproteína se une a dos proteínas, una es apo B-100, que es el único componente proteico de las LDL y la otra es apo E, que se encuentra en diferentes copias en las HDL, IDL y VLDL.
El receptor es sintetizado en el retículo endoplásmico rugoso y sufre unos cambios a nivel de los azúcares, ganando en peso molecular por su paso a través del aparato de Golgi. A los 45 minutos de su síntesis aparece en la superficie celular. Una vez en la membrana celular, se una o no a las lipoproteínas LDL, sufre una invaginación a nivel de los hoyos revestidos de clatrina para formar endosomas. Los endosomas se unen a los lisosomas para que las partículas LDL captadas sean degradadas. Este ciclo se repite cada 10 minutos28.
Proteína y gen
El receptor de LDL es una glicoproteína de membrana en la que podemos diferenciar cinco dominios funcionales12.- El primer dominio está formado por 292 aminoácidos con una secuencia de 40 aminoácidos que se repiten de forma seriada 7 veces. Este dominio se denomina “ligando” o “de unión” puesto que reconoce a las apolipoproteínas B y E. - El segundo dominio se caracteriza por contener 400 aminoácidos con una secuencia que tiene un 30% de homología con el dominio extracelular del precursor del factor de crecimiento epidérmico. - El tercer dominio tiene 58 aminoácidos con 18 residuos de treonina o serina que se unen con azúcares (uniones O). - El cuarto dominio está constituido por 22 aminoácidos hidrofóbicos que se encuentran anclados en la membrana celular. - El quinto dominio, llamado “cola intracitoplásmica”, ancla el receptor en el interior celular y es fundamental para llevar acabo el proceso de endocitosis.
El ADNc del gen LDLR fue descrito y clonado por Russell y cols en 1983 18 y por Yamamoto y cols en 1984 19; tiene 5’3 kilobases y codifica una proteína de 860 aminoácidos. Aproximadamente la mitad del ARNm está constituido por una región no traducida en 3´que contiene tres copias de zonas repetitivas Alu.
El gen que codifica para esta proteína se encuentra en la zona distal del brazo corto del cromosoma 19 (19p13) (OMIM: 606945)12. Tiene 18 exones y 17 intrones, existiendo una gran correlación entre exones y partes funcionales de la glicoproteína de membrana.

10
El exón 1 codifica una corta región no traducida en 5´y para el péptido señal de 21 aminoácidos.
Los exones 2 a 6 codifican para la zona “ligando” o de unión, que tiene gran homología con proteínas del sistema de complemento. Esta zona está constituida por 7 repeticiones de 40 aminoácidos, conteniendo cada una de ellas seis residuos de cisteina que forman puentes disulfuro. La zona carboxiterminal de cada repetición contiene un triplete con cargas negativas (Ser-Asp-Glu) que se une a aminoácidos básicos de cargas positivas de las zonas de unión de apo B y apo E.
Los exones 7 a 14 codifican para la región con alta homología del factor de crecimiento epidérmico y es requerido para la disociación ácido dependiente de la lipoproteína LDL en los endosomas. EL exón 15 codifica para el dominio rico en residuos de treonina y serina que sirven de unión con azúcares.
Los exones 16 y primera porción del 17 codifican para la región transmembrana. El resto del exón 17 y el 18 codifican para la región intracitoplásmica que es clave para la localización del receptor en los hoyos revestidos.
Mutaciones del gen LDLR
Se calcula que existen más de 700 mutaciones que afectan al gen del receptor para LDL responsables de la hipercolesterolemia familiar29. Dependiendo de las poblaciones estudiadas del 5-15% de las mutaciones son grandes deleciones o inserciones y el restante son pequeñas mutaciones. Las mutaciones se clasifican según su expresión fenotípica en 5 clases:
- Clase 1 ó alelos nulos, puesto que no se detecta receptor inmunoprecipitable. Un grupo de ellas no producen ARNm como son la FH French Canadian 1 (deleción de 15 kb que incluye el promotor y el exón 1) y FH Denver 1 (deleción de 6 kb que incluye el exón 1 y la zona del promotor). La mayoría de ellas producen mensajero pero en concentraciones pequeñas. Han sido caracterizadas la FH Turkey y Nashville, que generan codones sin sentido en el exón 2 y 8 respectivamente. También se han descrito codones de terminación como en FH Italy 1 y Potenza.
- Clase 2 ó alelos defectivos para el transporte. Son mutaciones que impiden el paso del receptor del retículo endoplásmico al aparato de Golgi. Estas son las más frecuentes. Se han descrito diferentes mutaciones que afectan a los dominios de "binding" o de unión y al dominio de homología con el factor de crecimiento epidérmico. Las más importantes son la FH Lebanese, FH Genoa y FH Afrikaner 1.
- Clase 3 ó defectos en la unión. Afectan a los exones que codifican para el dominio ligando o de unión y a la zona adyacente de homología con el factor de crecimiento epidérmico. Un ejemplo de estas mutaciones es la FH Tomani 2 que produce una delección que elimina la primera y segunda de las repeticiones, disminuyendo la capacidad de unión del receptor a un 77%.
- Clase 4 ó alelos defectuosos para la internalización. Son mutaciones muy raras pero que permitieron conocer la importancia de los hoyos revestidos de clatrina en el proceso de endocitosis celular.

11
- Clase 5 ó defectos de reciclaje. Todas ellas afectan al dominio de homología con el factor de crecimiento epidérmico puesto que este dominio participa en el proceso de disociación ácido dependiente del receptor con la partícula. Son ejemplos de éstas la FH Afrikaner 2 y la FH Osaka 2.
Genética de poblaciones
La enfermedad se hereda de forma autosómica dominante. Tiene una prevalencia variable dependiendo de las poblaciones estudiadas. En poblaciones no endogámicas se estima en 1/500 en su forma heterocigota. En poblaciones endogámicas la frecuencia es mayor como ocurre con los Judíos Ashkenazi (1/67), los Cristianos del Líbano (1/170), los Afrikáners de Sudáfrica (1/100) y los Francocanadienses de Québec (1/270)30.
Relación genotipo-fenotipo
Existe una variabilidad en la expresión clínica de la enfermedad debida a la interacción con factores ambientales y con otros factores genéticos. Muchas mutaciones clase 2 no son capaces de aclarar el cLDL del plasma pero sí se unen con las partículas que contiene apo E. En estos sujetos la degradación de LDL esta alterada pero no su síntesis. Este tipo de mutantes tienen una forma de hipercolesterolemia menos grave31.Los sujetos con mutaciones clase 1, donde no existe receptor LDL, presentan niveles de cLDL más elevados, responden peor al tratamiento hipolipemiante y tienen mayor riesgo de cardiopatía isquémica32, 33.
Estudios en algunas familias han demostrado que la variabilidad clínica depende de un gen dominante que disminuye el cLDL 34.
Diagnóstico
El diagnóstico de la hipercolesterolemia familiar se basa en las características clínicas y bioquímicas de la enfermedad. Cuando éstas son evidentes es fácil de realizar, pero en la mayoría de los afectados estas características no se cumplen, sobre todo en niños y adultos jóvenes. Teniendo en cuenta los valores del colesterol total y de las LDL colesterol, existen tanto falsos positivos en niños y adultos jóvenes al medir sus cifras en una población general, como falsos negativos en niños de padres con hipercolesterolemia familiar35, 36.
Por ello, se está intentando desarrollar métodos fiables para la medición de la actividad de los receptores o la detección de los defectos moleculares, que permitan un diagnóstico seguro y precoz.
En homocigotos el diagnóstico suele ser fácil de realizar ante un niño con niveles extremadamente elevados de CT y cLDL que no presente ictericia y con de xantomas anaranjados. En heterocigotos dada la variabilidad clínica de los afectados en ocasiones no es fácil de realizar. No podemos basar nuestro diagnóstico atendiendo exclusivamente a los niveles de CT y cLDL puesto que sólo 1 de cada 20 hipercolesterolémicos será un HF37.
El diagnóstico diferencial se debe plantear con la hipercolesterolemia poligénica, el Defecto Familiar de Apo B-100, los sujetos con fenotipo IIa con hiperlipemia

12
familiar combinada y con las causas secundarias responsables de un fenotipo IIa (hipotiroidismo, síndrome nefrótico, hepatoma, anorrexia nerviosa, etc...). En la hipercolesterolemia poligénica no existe un patrón dominante de herencia de la dislipemia. Es raro encontrar xantomas en la HF y en el árbol genealógico es necesario que existan fenotipos lipoproteícos cambiantes en los familiares o en el probando. El DFB no es fácil de distinguir del fenotipo lipoprotéico y sólo descartando la presencia de las mutaciones del gen APOB se puede tener la certeza completa.
Para medir la actividad del receptor de LDL podemos utilizar cultivos de fibroblastos usando I125LDL o en linfocitos de pacientes evitando los cultivos estos cultivos con diferentes técnicas.
Finalmente, las técnicas de biología molecular permiten un diagnóstico directo que permite identificar las mutaciones responsables de la enfermedad. Esta aproximación es extremadamente útil en poblaciones endogámicas y para realizar un diagnóstico prenatal en caso de homocigotos38.
La extracción de ADN de nuestros pacientes se llevó a cabo a partir de linfocitos de sangre periférica mediante procedimiento estandar con fenol-cloforormo y el diagnóstico genético de HF se establecio a través de la identificacion de mutaciones en el gen LDLR por analisis de Southern Blot39 y secuenciación de ADN cuando se encontraba un patrón alterado de bandas mediante la técnica PCR-SSCP40.
Recientemente y de manera experimental algún grupo está realizando el cribado mutacional del gen LDLR mediante microchips de ADN41. No obstante, esta técnica no la consideramos adecuada para el cribado de nuestras muestras pues aun debe consolidarse dentro de los protocolos asistenciales y porque las técnicas habituales de biologia molecular tienen un óptimo rendimiento coste-efectivo.
1. 4. Defecto familiar de unión de apo B-100 (DFB)
El defecto familiar de unión de apo B-100 (DFB) (OMIM: 144010) es una enfermedad hereditaria autosómica dominante descrita recientemente que da lugar a un fenotipo de hipercolesterolemia42. Dicho fenotipo es similar al presentado por los sujetos afectados de HF. Esta alteración se debe a una serie de mutaciones puntuales que afectan a una región crítica del gen APOB (OMIM: 107730), localizada alrededor del codón 3500, que interviene en el reconocimiento y unión con su ligando, el receptor de las LDL. Por ello, las partículas apo B-100 son defectuosas y no se unen al receptor de LDL, por lo que se elevan estas partículas en el suero. La hipercolesterolemia dependiente de LDL va a generar depósitos de colesterol en piel y tendones (xantomas) y en las arterias va a dar lugar a cardiopatía isquémica.
Recuerdo histórico
En 1986 se describió que determinados individuos con hipercolesterolemia presentaban partículas LDL con una disminución en su aclaramiento o catabolismo al ser inyectadas en individuos normales43. En 1988 se encontró que estas lipoproteínas se unían de forma deficiente con el receptor LDL de fibroblastos normales. Posteriormente, se demostró que el anticuerpo monoclonal MB47 se unía con elevada

13
afinidad a dichas partículas defectuosas44. Estos datos sugirieron la existencia de una mutación que afectaría al gen APOB y que daría lugar a unas proteínas apo B-100 con disminución de su afinidad por el receptor LDL. En 1989 fue identificada dicha mutación, que da lugar a un cambio de un aminoácido (arginina por glutamina) en el codón 3500 (R3500Q)42. Posteriormente, se han localizado dos mutaciones más que dan lugar a formas de apo B defectuosas para la unión (R3500W, R3531C)45, 46.
Genética molecular
Las mutaciones localizadas alrededor del codón 3500 afectan a una zona de la proteína apo B 100 que es imprescindible para el reconocimiento y unión con el receptor LDL. Las proteínas defectuosas, como hemos descrito anteriormente, reducen de forma significativa el aclaramiento de las LDL, con lo que se produce una elevación plasmática de estas lipoproteínas.
La apolipoproteína B-100 es el único componente proteico de las lipoproteínas de baja densidad (LDL) y es el elemento que reconoce el receptor LDL para la unión y aclaramiento de la partícula. Se trata de una proteína de gran tamaño molecular, lo que hizo que su caracterización bioquímica fuera muy dificultosa47.
Recientemente, diferentes laboratorios de investigación han logrado establecer la estructura de la proteína al analizar, clonar y secuenciar el ADNc de la apo B 48. El ARNm tiene 14.5 kb de largo y codifica una proteína madura de 4536 aminoácidos con un peso molecular de aproximadamente 550.000 daltons, con un 10% de carbohidratos. El gen de la apo B tiene 43 kb con 29 exones y está localizado cerca del brazo corto del cromosoma 2 (2p24-p23)49. La distribución de los exones es característica, puesto que 24 de los 29 se encuentran en el tercio inicial de la porción 5´ del gen. La mitad de la proteína está codificada por el exón 26 que tiene 7572 pares de bases, siendo uno de los mayores exones descritos en el genoma humano.
Hasta ahora se han descrito 3 mutaciones en el gen de apo B (dos en el codón 3500 y una en el 3531) que alteran el aclaramiento de la partícula LDL 42, 45, 46,indicando la importancia funcional de esta región. La mutación apo B R3500Q fue la primera descrita45. Se debe a un cambio de adenina por guanina en el nucleótido 10708 del gen de apo B que da lugar a una sustitución del aminoácido arginina (R), el “fisiológico” para ese codón, por otro glutamina (Q) en el codón 3500. Recientemente, han sido descritas las mutaciones R3531C 45 que supone un cambio de arginina (R) por cisteina (C) en el codón 3531 y la mutación R3500W 46 que supone el cambio de la arginina por un triptófano (W). Estas mutaciones se heredan de forma autosómica dominante, afectando a la capacidad de reconocimiento y de unión de la apo B al receptor de las LDL, dando lugar al Defecto Familiar de unión de apo B 100 (DFB).
La mutación R3531C fue detectada en dos familias no relacionadas mediante técnica de PCR-SSCP. Se trata de un cambio de C a T en el nucleótido 10800 que produce una sustitución de cisteina por arginina en el resido 3531. El estudio permitió detectar 2 probandos no relacionados y 8 individuos afectados en los respectivos estudios familiares. El estudio de haplotipos indicó que el origen de la mutación es diferente en las dos familias. Utilizando un ensayo competitivo radioactivo se demostró que la unión de LDL de los sujetos afectados era del 70% con respecto a controles45.

14
Es de esperar que con la incorporación de nuevas técnicas más sensibles detectemos nuevas mutaciones que afecten a regiones clave para la unión con el receptor de LDL responsables de hipercolesterolemia.
Clínica
La mayoría de los afectados de DFB presentan niveles elevados de LDL colesterol, xantomas tendinosos, arco corneal y desarrollan de forma precoz cardiopatía isquémica. Todo ello impide que puedan ser clínicamente diferenciados de los individuos con el diagnóstico clínico-bioquímico de hipercolesterolemia familiar50, 51, 52.Hansen PS. y cols53 describieron 46 heterocigotos para la mutación R3500Q pertenecientes a 5 familias estudiadas, en los que las concentraciones plasmáticas de colesterol, de LDL colesterol y de apo B eran entre 50% y 70% más elevadas que en los familiares no afectados. Los niveles de triglicéridos a su vez también eran más elevados y los valores de HDL colesterol eran inferiores a los del grupo control. En mayores de 50 años, el 50% de los afectados presentaba cardiopatía isquémica frente a ninguno del grupo control. En este grupo de pacientes con manifestaciones de aterosclerosis coronaria existían niveles de colesterol total, LDL colesterol, triglicéridos, apo B y Lp(a) significativamente más elevados que en sus homólogos no afectados de cardiopatía. En dos de las cinco familias se encontraron xantomas53. Rauh G. y cols54
estudiaron las características clínico-bioquímicas de 54 heterocigotos para la mutación R3500Q, observando que el 22% presentaba arco corneal, el 25% tenía xantomas tendinosos y el 22% presentaba aterosclerosis coronaria. La media de colesterol total fue de 308 mg/dl y la de LDL colesterol de 242 mg/dl. Defesche JC y cols,55 realizaron un despistaje buscando la mutación R3500Q en 1248 pacientes con hiperlipidemia IIa encontrando a 18 afectados que estaban clasificados como heterocigotos de hipercolesterolemia familiar, 10 de los cuales tenían xantomas tendinosos y/o arco corneal, mientras que 8 de ellos ya presentaban clínica de cardiopatía isquémica.
Sin embargo, un reducido grupo de sujetos afectados presentan niveles normales de colesterol total y de LDL colesterol, no atribuibles a un alelo de apo B variante hipocolesterolemiante en las familias estudiadas. Tampoco se explica este hallazgo por polimorfismos de gen del receptor de LDL ni de apo E 56.
Se ha descrito el primer homocigoto para la enfermedad. Se trata de una paciente de 30 años que sorprendentemente no presenta niveles de colesterol tan elevados como era de esperar para un homocigoto y que no tiene xantomas tendinosos ni síntomas clínicos de cardipopatía isquémica.
La determinación de la frecuencia de la enfermedad se ha basado en los estudios realizados sobre pacientes con el diagnóstico clínico-biológico de hipercolesterolemia familiar ya que son indistingibles desde este punto de vista, y en sujetos hipercolesterolémicos, y se ha establecido en 1/300-1/700, dependiendo de la población estudiada57, 58.
En la actualidad todos los pacientes con el diagnóstico del defecto familiar de apo B 100, excepto un individuo, presentan el mismo haplotipo en el gen de la apo B lo que indica un antecesor común59.

15
Diagnóstico
En los últimos años se han desarrollado diferentes métodos, basados en la reacción en cadena de la polimerasa (PCR), que nos permiten detectar el defecto molecular de forma rápida y sencilla60.
Diferentes autores han demostrado con estas técnicas que entre un 3 y un 10 % de los sujetos diagnosticados de HF con los criterios clínico-bioquímicos padecen realmente DFB. Así, entre los sujetos HF de Gran Bretaña, un 4% padece realmente un DFB; parecida frecuencia se encuentra en población alemana, 5%57 y holandesa 2,8%62.En estudios similares analizando el fenotipo HF en Canadá hasta un 10% son portadores de la mutación R3500Q 57.
Por otra parte, estudios poblacionales realizados en diferentes países del Norte de Europa y América demuestran una alta frecuencia de la mutación R3500Q, que oscila entre 1/700 en población germánica a 1/300 en población suiza58.
Hay que destacar que en Finlandia no ha sido encontrada la mutación ni en pacientes con el diagnóstico clínico-biológico de hipercolesterolemia familiar ni en sujetos hiperlipémicos62.
En nuestro estudio el cribado para las mutaciones en el gen APOB se llevo a cabo mediante amplificación con PCR como se describe en Schuster et al63 y análisis de SSCP40. Se utilizaron muestras controles para las mutaciones R3480P, R3500Q, R3500W y R3531C responsables del Defecto Familiar de Apo B-100.
1. 5. Efector fundador
Conceptos
Entendemos el efecto fundador como las variaciones en las frecuencias genéticas debido al desarrollo de una población a partir de unos pocos individuos. Esto puede suceder en varias situaciones, como el establecimiento de una nueva población por parte de unos pocos individuos “fundadores” que proceden de una población originaria y donde dichos sujetos “fundadores” sólo portan una pequeña parte de la variabilidad genética de la población de la que proceden. Por ello, la nueva población fundadora podría tener una composición de alelos muy diferente al de la población originaria debido a que su información genética procede de una pequeña parte de dicha población64.
Cuando una pequeña porción de una población se traslada a un nuevo lugar, o cuando la población queda reducida a un pequeño tamaño por diversas causas, (por ejemplo cambios ambientales o enfermedades), los genes de los sujetos fundadores de la nueva población van a ser desproporcionadamente frecuentes en la población resultante.
Esta circunstancia suele ocurrir cuando se da un cierto grado de aislamiento poblacional a lo largo del tiempo, sobre todo debido a aislamiento geográfico, pero también por otras razones, como étnicas, religiosas, culturales, etc. En definitiva, todas aquellas circunstancias que conducen a endogamia y por lo tanto donde no se da la posibilidad de intercambiar sus alelos con los de otras poblaciones.

16
Un haplotipo es la combinación de diferentes alelos de distintas variantes genéticas (polimorfismos y/o mutaciones) presentes en una región de ADN 65.Normalmente, los haplotipos se consideran como tales cuando las variantes genéticas están vinculadas o en desequilibrio de ligamiento debido a su proximidad física y por lo tanto tienden a heredarse juntos, es decir, en bloques.
También podemos entender un haplotipo como el conjunto de SNPs (Single Nucleotide Polymorphism), u otras secuencias polimórficas, que se encuentran asociados estadísticamente a una única cromátida. La identificación de unos pocos alelos de un bloque de haplotipo identifica sin ambigüedades a todos los lugares polimórficos en esta región.
Los haplotipos se han producido en el genoma humano por los mecanismos moleculares de mutación y recombinación en las líneas germinales y a lo largo de toda la historia natural de nuestra especie. Los cromosomas de las células humanas se dan en parejas, es decir, en estado diploide, con excepción de las células sexuales. Cada miembro del par cromosómico será proporcionado por cada uno de sus progenitores. Pero los cromosomas no pasan de una generación a otra como copias idénticas.
En la gametogénesis, y más específicamente durante la meiosis, los pares cromosómicos se ven sometidos a un proceso conocido como recombinación génica o “crossing-over”. En dicho proceso los miembros de cada par cromosómico intercambian entre sí sus cromátidas. El resultado es un cromosoma híbrido, una especie de “mosaico cromosómico”, que posee segmentos de ADN que provienen de ambos progenitores. Y este cromosoma híbrido se transmite a la generación sucesiva.
Tras el paso de las generaciones, los segmentos de los cromosomas iniciales o ancestrales de una determinada población, se irán mezclando a través de fenómenos repetidos de recombinación génica.
Y se da la circunstancia que algunos de los segmentos de los cromosomas ancestrales se encuentran como secuencias de ADN que son compartidas por muchos individuos y que prácticamente no han sufrido modificaciones a lo largo del tiempo. Estos segmentos son regiones cromosómicas que no se han fragmentado mediante recombinación génica por lo que una mutación que se haya producido en un determinado haplotipo para estas regiones se heredará siempre con el mismo haplotipo a no ser que el evento mutacional se produzca “de novo” y, entonces, probablemente se produzca en otro alelo del mismo locus génico y por lo tanto presente un haplotipo diferente.
De la misma manera que cambia la frecuencia alélica en una población fundadora con respecto a la población originaria de la que deriva, también lo hará la frecuencia de los haplotipos de dicha población fundadora.
En la presente tesis doctoral hemos analizado el gen APOB con varias de sus secuencias polimórficas que constituyen su patrón de haplotipo.

17
El análisis del haplotipo en APOB es esencial para valorar si se ha dado efecto fundador en los sujetos DFB de nuestra población, lo cual es uno de los objetivos de la presente tesis doctoral.
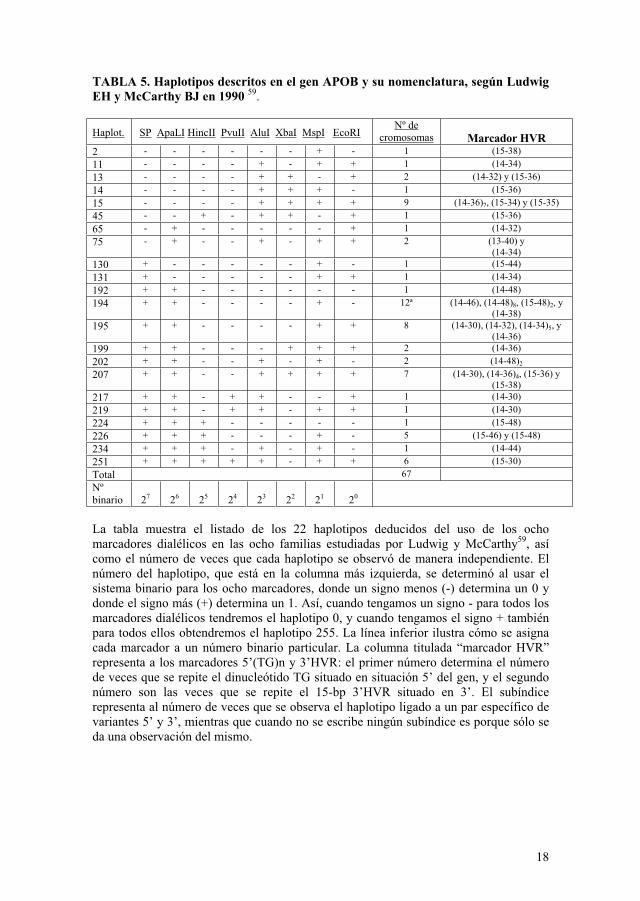
Los haplotipos descritos para el gen APOB son numerosos (Tabla 5) 59. Sólo para el polimorfismo 3’ HVR se han descrito muchos alelos posibles66.
El estudio de los haplotipos se realizó en todos los sujetos portadores de la mutación R3500Q para el gen APOB, pero no se llevó a cabo en los sujetos sanos dado que al existir muchos haplotipos de APOB en la población general, dicho estudio no hubiera aportado información de interés sobre el efecto fundador de nuestros pacientes DFB.
Esquema del gen APOB con los lugares polimórficos que constituyen su haplotipo.
R3500Q
5’
26
3’
18
TABLA 5. Haplotipos descritos en el gen APOB y su nomenclatura, según Ludwig EH y McCarthy BJ en 1990 59.
Haplot. SP ApaLI HincII PvuII AluI XbaI MspI EcoRINº de
cromosomas Marcador HVR 2 - - - - - - + - 1 (15-38)
11 - - - - + - + + 1 (14-34)
13 - - - - + + - + 2 (14-32) y (15-36)
14 - - - - + + + - 1 (15-36)
15 - - - - + + + + 9 (14-36)7, (15-34) y (15-35)
45 - - + - + + - + 1 (15-36)
65 - + - - - - - + 1 (14-32)
75 - + - - + - + + 2 (13-40) y (14-34)
130 + - - - - - + - 1 (15-44)
131 + - - - - - + + 1 (14-34)
192 + + - - - - - - 1 (14-48)
194 + + - - - - + - 12ª (14-46), (14-48)8, (15-48)2, y(14-38)
195 + + - - - - + + 8 (14-30), (14-32), (14-34)5, y (14-36)
199 + + - - - + + + 2 (14-36)
202 + + - - + - + - 2 (14-48)2
207 + + - - + + + + 7 (14-30), (14-36)4, (15-36) y (15-38)
217 + + - + + - - + 1 (14-30)
219 + + - + + - + + 1 (14-30)
224 + + + - - - - - 1 (15-48)
226 + + + - - - + - 5 (15-46) y (15-48)
234 + + + - + - + - 1 (14-44)
251 + + + + + - + + 6 (15-30)
Total 67
Nºbinario 27 26 25 24 23 22 21 20
La tabla muestra el listado de los 22 haplotipos deducidos del uso de los ocho marcadores dialélicos en las ocho familias estudiadas por Ludwig y McCarthy59, así como el número de veces que cada haplotipo se observó de manera independiente. El número del haplotipo, que está en la columna más izquierda, se determinó al usar el sistema binario para los ocho marcadores, donde un signo menos (-) determina un 0 y donde el signo más (+) determina un 1. Así, cuando tengamos un signo - para todos los marcadores dialélicos tendremos el haplotipo 0, y cuando tengamos el signo + también para todos ellos obtendremos el haplotipo 255. La línea inferior ilustra cómo se asigna cada marcador a un número binario particular. La columna titulada “marcador HVR” representa a los marcadores 5’(TG)n y 3’HVR: el primer número determina el número de veces que se repite el dinucleótido TG situado en situación 5’ del gen, y el segundo número son las veces que se repite el 15-bp 3’HVR situado en 3’. El subíndice representa al número de veces que se observa el haplotipo ligado a un par específico de variantes 5’ y 3’, mientras que cuando no se escribe ningún subíndice es porque sólo se da una observación del mismo.

19
Origen histórico de la mutación R3500Q
Se han detectado individuos con DFB en población caucásica de Alemania51, 67,
68, 69, Austria70, 66, 71, Dinamarca52, España72, 73, Holanda69, 74, 75, Hungría75, Italia76,Noruega74, Polonia76, Reino Unido52, República Checa77, 78, Suiza79, Canadá70, 59,Estados Unidos58, 70, 59, Méjico80, Australia74 y en no-caucásicos de China81. No se encontró en caucásicos de Finlandia62, Rusia (área de San Petersburgo)69 e Israel82, y tampoco en no-caucásicos de Japón83.
El hecho de que no se haya encontrado la mutación en población finlandesa62 ni en la rusa del área de San Petersburgo69 nos hace pensar que la mutación R3500Q se habría dado tras la divergencia entre la población finlandesa y las otras poblaciones caucásicas .
Dadas las distancias genéticas entre los finlandeses y las otras poblaciones europeas se estima que esta divergencia se habría dado hace aproximadamente entre 10.000 y 20.000 años. En cualquier caso, claramente tras la divergencia entre las poblaciones caucasianas y las mongoloides lo que se dio hace aproximadamente 41.000 años58, 84.
El hallazgo de un sujeto DFB de origen asiático (Szechuan, China) en San Francisco, Estados Unidos, pero sin el haplotipo 194 81 sugeriría que la mutación R3500Q no estaba presente todavía en la población cuando el hombre paleolítico emigró hacia el Oeste (hacia Europa) y hacia el Este (hacia Asia) hace 35.000 - 40.000 años85.
La segunda expansión en importancia dentro de Europa implicó la lenta extensión de agricultores neolíticos hacia el Oeste desde Oriente Próximo. La evidencia arqueológica indica85, 86 que esto tuvo lugar en un periodo que duraría hace unos 10.000-6.000 años, y donde una ruta migratoria importante era hacia Anatolia (Turquía asiática)87.
Mahley et al (1995)88 no detectó ninguna familia turca con DFB en el estudio en el que incluía a seis centros poblacionales de Turquía. Este hallazgo apoya el hecho de que la mutación R3500Q se originó en Europa, pues si el DFB hubiera estado presente en la población Neolítica que se extendió hacia Europa desde Oriente Próximo, se deberían haber observado vestigios de la mutación en la moderna Anatolia.
La mutación R3500Q ya estaba presente en los ancestros mesolíticos del pueblo celta (Helvetii) que vivían en una localización situada entre el río Rin y el Meno, lo que corresponde a la porción sur-occidental de la actual Alemania, y que inmigraron hacia el Noroeste de la Suiza moderna hace más de 2.000 años89.
Ello es congruente con el hecho de la baja prevalencia descrita al sur de los Alpes y de los Pirineos que actuarían como barrera migratoria, y también con la alta prevalencia descrita en Bélgica, área alemana del Rin-Meno y Suiza.
La presencia de la mutación R3500Q en poblaciones de Europa central, norte de los Alpes y de los Pirineos, pero también en poblaciones de Norte América y Australia colonizados por europeos, sugieren su origen centro-europeo.

20
Por lo tanto, todos los datos apuntan a que la mutación R3500Q emergió en los ancestros comunes del que vivieron durante el periodo Mesolítico (hace 6.000-10.000 años) en la región sur-occidental de Alemania.
En Europa central, las comunidades celtas aparecieron inicialmente en el primer milenio antes de cristo. La unidad céltica en Europa central estaba basada en una cultura y una lengua común, más que en un parentesco genético. La migración del pueblo celta desde partes del Sur-Oeste de Alemania hacia el Nor-Oeste de Suiza ocurrió cerca del año 200 antes de cristo. En aquel momento otras tribus celtas se fueron hacia el Este, Oeste y más lejos hacia el Sur (Italia y los Balcanes). Probablemente, fue justo el grupo suizo conquistador (Helvetii) el que portaba la mutación R3500Q.
De cualquier modo, cualquier intento de deducir la historia genética del DFB desde su distribución geográfica actual está obligado a tener en cuenta que existe un margen de incertezas. A parte de la dificultad de reconstruir los movimientos poblacionales en el pasado remoto, como distinto de la extensión cultural sin movimiento poblacional, hay vacíos en nuestro conocimiento de aspectos relevantes de DFB.
Datos históricos sobre la repoblación de la comarca de Los Serranos
Para ello voy a basarme en dos libros. En el primero, “Conquista y repoblación del Reino de Valencia”, Ramón Ferrer hace un estudio sobre la repoblación de las diversas comarcas del Reino de Valencia a partir de los datos del “Llibre del Repartiment”. En el segundo, “Els fundadors del regne de València: repoblament, antroponímia i llengua a la València medieval”, se estudia la repoblación de las diversas comarcas del Reino de Valencia tomando en cuenta la antroponimia, es decir, el estudio del origen y significado de los apellidos de la zona. Hay que decir que hace un estudio específico sobre la villa de Alpuente, localidad a la que perteneció La Yesa hasta 1587.
Ramón Ferrer Navarro en su obra “Conquista y repoblación del Reino de Valencia”90, refiere que en “El Llibre del Repartiment” se cuenta con concesiones de señorío referido a los lugares de Andilla, Gestalgar, Chulilla, Chelva y Pedralba durante los años 1237, 1238 y 1248. Se inicia en 1237 con la donación por parte del rey a P. Fernández de Albarracín del castro de Chelva, reteniendo la potestad, paz y guerra; ese mismo año se concede al caballero Berenguer de Entenza el poblado fortificado de Pedralba, al repostero real Jimeno Pérez le entrega el castro de Andilla. En 1238 el monarca dona Gestalgar a Rodrigo Ortiz; finalmente en 1248 don Jaime da a su escribano Pedro el castro de Chulilla.
En su obra “Els fundadors del regne de València: repoblament, antroponímia i llengua a la valència medieval”91, el historiador Enric Guinot nos muestra como fue la repoblación cristiana del municipio de Alpuente en los siglos XIII y XIV, es decir aquellos sucesivos a la conquista de la ciudad de Valencia y de la misma comarca de Los Serranos.
Enric Guinot establece seis mecanismos de diferenciación entre los modelos de antroponimia de Alpuente con respecto al de Castellón de la Plana, ya que describe a ambos modelos como paradigmas de repoblación predominantemente castellano-aragonesa y catalana respectivamente. Dichos mecanismos diferenciación son:

21
topónimos, nombres propios, apellidos geográfico-naturales, frecuencia de patronímicos, profesiones y lengua de los apodos.
No se nos cita directamente a la población de La Yesa, pues ésta era una aldea que pertenecía por aquel entonces al municipio de Alpuente. Por ello a partir de ahora hablaré de los datos de repoblación para la población de Alpuente.
Alpuente poseía un gran término municipal situado entre los límites del territorio aragonés del Consejo de Teruel (con las aldeas de Arcos de las Salinas, Torrijas y Ababuj), y los concejos orientales de la frontera castellana por la parte de Cuenca (Moya, Santa Cruz de Moya y Mira). Alpuente también ejercía su jurisdicción sobre sus aldeas: Aras de Alpuente, Titaguas y La Yesa. Por lo tanto nos encontramos en las tierras cercanas a la frontera aragonesa y castellana.
La historia de la conquista y de la primera repoblación cristiana de Alpuente es muy desconocida, dado que no se ha conservado documentación detallada local, y tampoco aparece en el “Llibre del Repartiment”.
Alpuente debió ser una de las villas bajo el control del último gobernador musulmán de Valencia, el Sayyid Abu Sayd, colaboracionista de Jaime I en los años de la conquista. Dado que las tierras controladas por este gobernador musulmán pasaron en tres cuartas partes a la corona en el año 1236, a partir del inicio de la campaña de conquista de la ciudad de Valencia y del nuevo pacto de vasallaje que hizo el rey Jaime I, parece razonable que fuera entonces cuando la Corona debió tomar posesión de Alpuente.
Sólo se conoce un documento de esta época: el otorgamiento por parte del rey de unas franquicias a sus primeros vecinos cristianos hacia el año 1240, pero por desgracia no constan los nombres de los beneficiarios.
Es por eso que tenemos que basarnos en información más tardía, como la lista del morabatí de 1396. Aquí consta el nombre de los cabezas de familia fiscales de Alpuente, y de sus aldeas que en conjunto sumaban 270 apellidos.
De estos, 106 llevan un topónimo identificado, es decir, poco más de un tercio del total (39 %). Pues bien, cerca de tres cuartas partes de los apellidos toponímicos de Alpuente hacen referencia a pueblos aragoneses, sobretodo de Teruel, seguido de otros de castellanos (10’5 %) y de navarros (8’6 %). Un total de pueblos de habla castellana absolutamente hegemónico (92 %).
Entre la gran mayoría aragonesa es muy evidente el predominio de gentes de las comarcas de Teruel, cerca de dos tercios del total, tal como se desprende de los apellidos formados de las aldeas de Teruel, Daroca y Albarracín.
Por lo que respecta a los apellidos basados en apodos y profesiones, las similitudes lingüísticas con los casos de Daroca y Teruel en los siglos XIII y XIV son evidentes. Además, pueden destacarse unos rasgos propios que diferencian este modelo antroponímico.

22
Las proporciones de los apellidos formados por apodos, nombres personales, profesiones y elementos geográficos o naturales son distintos.
En el año 1396 en la villa de Alpuente, después de los antroponímicos, los apellidos más habituales son los formados a partir de nombres propios de persona o de prenombres. Una vez más, el predominio de los nombres de origen aragonés resulta evidente, frente a una clara ausencia de catalanes.
En segundo término, hay que recalcar la exigüidad de los apellidos basados en la geografía, el pueblo y la naturaleza.
El otro bloque de apellidos significativo en proporción es el de patronímicos, es decir, aquellos formados a partir de un nombre propio –normalmente el del padre- con el sufijo -ez o –es –y con menor proporción –iz o –is (forma navarra), un modelo de apellido muy usual en el Aragón y la Navarra de los siglos XII y XIII, en cambio inexistente en Cataluña.
En Alpuente encontramos todos los habituales, hasta llegar a un 14 % del total de apellidos de la villa.
En cambio, los apellidos basados en profesiones son menos abundantes (6-7 %), quizá porque se trata de una comarca no tan desarrolladas económicamente en el siglo XIV. Lo que nos interesa es la forma ortográfica de estos apellidos, y hay que decir que pertenecen al léxico castellano, con la terminación –ero.
Finalmente, y además de la total ausencia de apellidos basados en nombres de santos –origen toponímico- los de apodos son también abundantes, si bien no llegan a quinta parte del total. Sin embargo, lo que es significativo es el hecho de que corresponden de manera casi general al léxico aragonés-castellano.
En un balance aproximativo de procedencias, podríamos considerar que cerca del 85 % de apellidos de Alpuente son aragoneses, y en menor medida, castellanos y navarros. La proporción de antropónimos procedentes de Cataluña es mínima (5 %), mientras que el resto podrían ser indistintos. Así la relación entre ambas procedencias es de 17 aragoneses por cada catalán.

23
Mapa de localización de La Yesa
1. 6. Lipoproteína Lp(a)
La lipoproteína(a), Lp(a), tiene gran importancia pues constituye un factor genético independiente de riesgo para enfermedad cardiovascular92-104.
Es una lipoproteína plasmática cuya composición química se parece bastante a la composición lipídica de las LDL. Además de la apolipoproteína B-100 [apo B], la Lp(a) contiene una segunda apolipoproteína, la apolipoproteína(a), [apo(a)], que puede ser liberada de la partícula por medio de agentes sulfidril-reductores. Apo B y apo(a) están unidas por medio de un puente disulfuro y se unen con una relación molar de 1:1 105-111.
La apo(a) es la apolipoproteína distintiva de la Lp(a). Es una glicoproteína polimórfica que contiene dominios repetidos de longitud variable que son homólogos al dominio Kringle-IV del plasminógeno, el cual es un componente clave del sistema

24
fibrinolítico que participa en la homeostasis de los procesos coagulativos. Además, la apo(a) posee una copia del dominio Kringle-V y una región proteasa. De este modo la Lp(a) combina físicamente una partícula rica en colesterol con una molécula relacionada con el sistema de la coagulación sanguínea112, 113.
Dicha homología estructural de la Lp(a) le dota de la capacidad de unirse a la fibrina y a las proteínas de membrana de las células endoteliales y de los monocitos, y por lo tanto de inhibir la unión de plasminógeno y la generación de plasmina. La inhibición de la producción de plasmina y el acúmulo de Lp(a) en la superficie de fibrina y membranas celulares favorecen el depósito de fibrina y de colesterol en lugares de daño vascular.
Se acepta actualmente que el efecto atero-trombótico de la Lp(a) depende de su concentración en plasma. Se ha citado que hay una relación inversa entre la concentración de Lp(a) y el tamaño de la isoforma de la apo(a) que está bajo control genético114-116.
Han sido descritas en humanos más de 30 isoformas diferentes de la Lp(a) con un rango de tamaños que van desde menos de 300 Kd hasta más de 800 Kd117-122.
La concentración plasmática de la Lp(a) está marcada genéticamente123-127.
Se ha demostrado que, al menos en caucásicos, el polimorfismo de tamaño de la apo(a) está inversamente relacionado con la concentración plasmática y que la Lp(a) se ha asociado con un mayor riesgo para la enfermedad coronaria, para el ictus y también para la reestenosis128.
El gen para la apolipoproteína(a), gen LPA, se sitúa en la citobanda 6p26-27, es decir, en una localización cercana al telómero del cromosoma 6 junto al gen del plasminógeno (gen PLG), que se sitúa inmediatamente junto al gen LPA en sentido telomérico129-134.
El gen LPA existe en varias isoformas de las que se conocen 34 isoformas135.
La apolipoproteína(a) es el determinante genético más importante de la concentración plasmática de lipoproteína(a). Esto es así porque las isoformas específicas determinan los diferentes tamaños de partículas apo(a), que varían de 300 a 850 kD119-122.
Por lo comentado, es interesante conocer los valores de Lp(a) en sujetos con DFB puesto que la la lipoproteína contiene apo B y esta apo B alterada podría estar modificando su catabolismo. Este objetivo es parte de la Tesis Doctoral presentada.

25
HIPÓTESIS Y OBJETIVOS

26

27
2. HIPÓTESIS Y OBJETIVOS
2. 1. Hipótesis
En el Sur de Europa apenas existen estudios sobre el fenotipo lipoproteico del DFB puesto que es una enfermedad muy poco frecuente. Sin embargo, nuestro grupo de trabajo ha detectado en los últimos años un número significativo de núcleos familiares y sujetos afectados por el DFB en la Comunitat Valenciana procedentes de la comarca de Los Serranos. Ello nos va a permitir analizar su fenotipo lipoproteico y estudiar si ha existido un efecto fundador que justifique su inesperadamente elevada prevalencia en una comunidad del Sur de Europa.
Por otro lado, los sujetos afectados por el DFB (mutación R3500Q) se deben comportar fenotípicamente de la misma forma que los sujetos con Hipercolesterolemia Familiar clásica (HF) que portan mutaciones en el dominio de unión al ligando del gen para el receptor de las LDL (gen LDLR).
2. 2. Objetivos
a) Conocer la prevalencia de la mutación R3500Q del gen APOB, responsable del DFB, en una determinada población de la Comunitat Valenciana, así como el posible efecto fundador mediante el estudio del haplotipo en dicho gen.
b) Describir el fenotipo lipoproteico, incluyendo el análisis de los valores plasmáticos de la Lp(a) de sujetos DFB con un mismo origen en una determinada población de la Comunitat Valenciana.
c) Comparar el fenotipo lipoproteico de dichos sujetos DFB con el de los pacientes con Hipercolesterolemia Familiar clásica ya estudiados molecularmente en nuestra Unidad de Lípidos y Arteriosclerosis.

28

29
RESULTADOS

30

31
3. RESUMEN DE LOS ESTUDIOS
El resultado de nuestro trabajo de investigación se expone en cinco partes presentados como artículos científicos. Cada uno de los artículos se presenta con el formato y en la lengua de la revista científica en la que ha sido publicado.
ESTUDIO 1. Real JT, Chaves FJ, Martin de Llano JJ, Ejarque I, Civera M, Ascaso JF, Knecht E, Armengod ME, Carmena R. “Identificación y caracterización del primer español con defecto homocigoto familiar de unión de la apolipoproteína B”. Med Clin (Barc). 2001 Feb 3;116(4):138-41.
ESTUDIO 2. Real JT, Chaves FJ, Ejarque I, Garcia-Garcia AB, Valldecabres C, Ascaso JF, Armengod ME, Carmena R. “Influence of LDR receptor gene mutations and the R3500Q mutation of the apoB gene on lipoprotein phenotype of familial hypercholesterolemic patients from a South European population. Eur J Hum Genet. 2003 Dec;11(12):959-65.
ESTUDIO 3. Ejarque I, Real JT, Ascaso JF, Chaves FJ, Milian E, Priego MA, Carmena R. “Estudio de los valores plasmáticos de Lp(a) en el defecto familiar de unión de la apo B 100 en una población mediterránea del sur de Europa”. An Med Interna. 2004 Jul;21(7):322-5.
ESTUDIO 4. Ejarque I, Real JT, Chaves FJ, Blesa S, González V, Milián E, Ascaso JF, Priego MA, Carmena R. “Estudio del defecto familiar de unión de la apolipoproteína B100 en una población mediterránea”. Med Clin (Barc). 2004 Oct 9;123(12):456-9.
ESTUDIO 5. Ejarque I, Real JT, Chaves FJ, García-García AB, Milián E, Ascaso JF, Carmena R. “A comparison of lipoprotein phenotype between LDL receptor gene mutations affecting ligand-binding domain and the R3500Q mutation of the apo B gene in patients from a South European population”. Enviado a la revista European Journal of Internal Medicine; pendiente de aceptación.
ESTUDIO 1
Hemos detectado y caracterizado el primer homocigoto español para el DFB (mutación R3500Q). Este paciente fue identificado tras cribar 95 pacientes españoles con fenotipo lipoprotéico de Hipercolesterolemia Familiar (HF) mediante técnicas de genética molecular. De este modo pudimos detectar el primer heterocigoto español DFB y al ampliar el estudio molecular a los familiares de primer y segundo grado, identificamos a un sujeto homocigoto y también a otros cuatro heterocigotos.
El paciente homocigoto DFB presenta valores moderadamente elevados de colesterol total y de cLDL a pesar de su situación de homocigosis. Padece una evidente enfermedad arteriosclerótica que ha dado manifestaciones clínicas en diversas localizaciones. No obstante, no presenta xantomas, ni xantelasmas, ni arco corneal.
A pesar de que los objetivos terapéuticos en prevención secundaria no se han conseguido con los fármacos suministrados, los descensos alcanzados en los niveles de CT y cLDL nos hacen considerar que se ha alcanzado una buena respuesta farmacológica. En

32
teoría, en la forma homocigota de HF, cabría esperar una falta de respuesta. En cambio en nuestro paciente se han conseguido reducciones importantes de CT y cLDL con dosis elevadas de estatinas y resinas. La sobrexpresión del receptor de LDL sano inducido por las estatinas, junto con una actividad residual de unión de la apo B defectuosa y un mayor aclaramiento de las VLDL (que contienen Apo E), precursoras de las LDL, podrían explicar la buena respuesta a las estatinas.
Por lo tanto, nuestro paciente homocigoto DFB presenta valores moderadamente elevados de colesterol total y de cLDL a pesar de su situación de homocigosis, lo que indica que el fenotipo lipoproteico de los homocigotos con DFB es diferente de la situación homocigota para HF.
Finalmente, subrayar que, dada la baja prevalencia para heterocigosis de sujetos DFB en nuestro medio, la existencia de un homocigoto DFB nos hace sospechar una situación de endogamia, pues todos los heterocigotos DFB proceden de una pequeña población aislada de la Comunidad Valenciana.
ESTUDIO 2
El objetivo de este estudio ha sido el de comparar el fenotipo lipoproteico de los pacientes HF con mutaciones en sus diversos dominios entre sí y con el fenotipo de los sujetos DFB con la mutación R3500Q. Queríamos saber si el fenotipo lipoproteico entre ellos era el mismo o no, sobre todo al comparar el fenotipo DFB con el fenotipo causado por mutaciones en el gen LDLR para su dominio de unión con la Apolipoproteína B-100.
Hemos comparado el fenotipo lipoprotéico de cierto número de pacientes con Hipercolesterolemia Familiar clásica, para varios de sus dominios, con pacientes con el Defecto Familiar de unión a apo B-100 (DFB) (mutación R3500Q).
Un total de 118 pacientes HF fueron clasificados en tres grupos según el tipo de mutación en el gen LDLR: mutaciones nulas (N = 45), mutaciones “missense” en el dominio de unión al ligando (N = 13), y mutaciones “missense” fuera de este dominio de unión al ligando (N = 60). Además, el fenotipo lipopoproteico de estos tres grupos HF fue comparado con el de 19 heterocigotos DFB.
Las mutaciones en el gen LDLR localizadas en el dominio de unión al ligando proporcionaron mayores niveles de colesterol total y de cLDL si las comparamos a las de otras mutaciones en LDLR o con heterocigotos DFB.
Las mutaciones nulas de LDLR proporcionan valores más bajos de HDL con respecto a las mutaciones LDLR “missense”.
Los pacientes DFB presentan valores más bajos de colesterol total y de cLDL si se comparan con los valores de los demás grupos HF.

33
ESTUDIO 3
El objetivo de este estudio ha sido proporcionar datos sobre los valores plasmáticos de Lp(a) en el DFB en el sur de Europa, dado que no existían datos al respecto.
Se estudiaron 19 heterocigotos DFB y 90 sujetos controles. Los sujetos DFB, portadores de la mutación R3500Q en el gen APOB, pertenecen a 12 familias relacionadas con la primera familia afectada por DFB descrita en nuestro país. Los controles proceden de una selección aleatoria de sujetos sanos que proceden del Banco de Sangre de nuestro centro.
Los portadores de la mutación R3500Q presentaron de forma estadísticamente significativa mayores concentraciones plasmáticas de CT, cLDL y apo B comparados con el grupo control. Estas diferencias se mantuvieron al realizar la comparación entre sexos.
Los valores plasmáticos de Lp(a) y su transformación logarítmica fueron significativamente mayores en el grupo DFB frente al grupo control. Además, la prevalencia de Lp(a) > 30 mg/dl, como punto de corte de alto riesgo para cardiopatía isquémica, fue significativamente mayor en el grupo portador de la mutación R3500Q.
Las dos mayores limitaciones de este estudio, son el bajo número de sujetos DFB y la relativa homogeneidad molecular que se da en ellos dado el vínculo familiar que les une. Dicha homogeneidad genética podría influir en la expresión del fenotipo lipoprotéico.
ESTUDIO 4
En estudio pretendemos: 1) comparar las características clínico-bioquímicas del DFB con las de la HF en una población mediterránea del sur de Europa, 2) conocer mejor las características clínico-bioquímicas del DFB de dicha población y 3) estudiar la prevalencia y el posible efecto fundador del DFB en una zona de la Comunidad Valenciana
Las concentraciones plasmáticas de colesterol total, cLDL, cHDL, apo B y apo A-I fueron significativamente mayores en el grupo con HF que en el DFB. También para la presencia de xantomas tendinosos y cardiopatía isquémica.
Cabe destacar que 12 sujetos DFB presentaban concentraciones de colesterol total menores del percentil 95 para nuestra población. Ningún heterocigoto DFB presentó xantomas tendinosos y un sujeto DFB, que además es diabético tipo 2, presentó cardiopatía isquémica. Estos resultados nos obligan a replantearnos los criterios diagnósticos de los sujetos con DFB de España.
No se hallaron diferencias estadísticamente significativas en IMC, concentración plasmática de triglicéridos o cVLDL ni en la distribución de genotipos de APOE entre sujetos DFB y HF.
Los 19 heterocigotos DFB fueron identificados a partir del diagnóstico molecular del primer heterocigoto DFB. El número de sujetos se fue ampliando través del cribado mutacional para R3500Q en todos los familiares de primer y segundo grado y también

34
ayudados con las partidas de bautismo de la población de La Yesa; ello nos permitió identificar más sujetos susceptibles de ser heterocigotos a los que se les ofreció el test genético. Las partidas de bautismo de La Yesa están disponibles desde el año 1898, lo cual nos ha permitido retrotraernos hasta finales del siglo XIX en la construcción de los árboles genealógicos.
La Yesa se sitúa en una zona rural bastante montañosa y aislada, dentro de la comarca de Los Serranos (norte de la provincia de Valencia, limítrofe con la de Teruel) en las estribaciones más meridionales del sistema ibérico. Esta población ha permanecido bastante aislada hasta mediados del siglo XX dado que se localiza en un área geográfica de difícil acceso, y también por diversas características histórico-culturales, todo lo cual ha favorecido una situación de endogamia. Ésta se puede comprobar en las partidas de bautismo pues los apellidos de los sujetos de La Yesa se repiten de manera constante.
De los actuales 350 habitantes censados, 4 son portadores de la mutación R3500Q, lo que nos da una prevalencia de 4/350, es decir, una ratio de 1 / 87’5. Esta prevalencia es incluso superior a la mayor descrita hasta ahora en la población suiza.
Todos los 19 heterocigotos DFB comparten el mismo haplotipo, denominado haplotipo 194, que posee estas características: ins / XbaI- / MspI+ / EcoRI- / 3HVR49. El estudio de los pedigríes y este resultado en el haplotipo confirman el efecto fundador en esta zona rural de la Comunidad Valenciana.
La valoración de los haplotipos del gen APOB de los portadores de la mutación R3500Q indica que posiblemente el origen mutacional es único y habría tenido lugar entre 6.000 y 7.000 años. Parece que la mutación se produjo en un único alelo que se ha transmitido hasta nuestros días. Ello permite concluir que el origen mutacional es el mismo en todas las poblaciones europeas estudiadas, entre las que se incluye nuestro grupo de 19 heterocigotos.
En otro estudio español se refiere una baja prevalencia en España para heterocigotos DFB, con una mayor parte de los afectados en Galicia. A diferencia de este hallazgo, en la Comunitat Valenciana nuestro grupo ha encontrado este núcleo poblacional con alta prevalencia de DFB por un efecto fundador.
La explicación de las diferencias encontradas en las concentraciones plasmáticas de colesterol total y cLDL entre sujetos DFB y HF podría residir en el diferente comportamiento metabólico de las partículas portadoras de apo B-100 (LDL, VLDL e IDL) observado en sujetos con DFB con respecto a aquellos HF.
En sujetos con DFB se ha demostrado “in vivo” un mayor aclaramiento de lipoproteínas IDL y VLDL, partículas precursoras de LDL. Además, la producción de LDL es menor en sujetos con DFB.
La mayor limitación para este estudio es el bajo número de sujetos DFB incluidos, así como la relativa homogeneidad molecular de los mismos, dado su vínculo familiar.
Dado que los pacientes con DFB analizados presentan el mismo haplotipo que los encontrados en los DFB de otros países se podría inferir que la mutación procede de un evento original ancestral y que llegó a la región estudiada como consecuencia de

35
fenómenos migratorios. Esta mutación se expandió en la población y no ha sido eliminada o disminuida a lo largo del tiempo por el escaso o nulo efecto que ha tenido sobre la población durante la mayor parte de la historia y al no actuar sobre la supervivencia de los individuos hasta edades posteriores a la de reproducción. La posibilidad de que la mutación R3500Q se hubiera producido “de novo” en un individuo de esta región y en el mismo haplotipo del gen APOB que se produjo en la población del norte de Europa es muy baja debido al numero de posibles haplotipos existentes.
Abajo se ve un árbol genealógico circular que muestra la parentela entre diversos núcleos familiares de La Yesa que portan la mutación R3500Q en el gen APOB. No incluye a todos los sujetos con la mutación R3500Q, pero sí aquellos donde se ha provado una parentela. Estos datos han sido obtenidos tras consultar las partidas de bautismo de la parroquia de La Yesa, que se inician en 1898 y se mantienen interrumpidamente hasta nuestros días, a excepción del periodo 1936-1939 donde los datos son escasos. Para su mejor visualización se ha remarcado con un círculo azul a los sujetos heterocigotos, y en rojo al único homocigoto encontrado.

36
ESTUDIO 5
Los objetivos de este estudio fueron 1) evaluar la sensibilidad de los criterios diagnósticos clínicos para defecto familiar de apo B y 2) comparar el fenotipo lipoproteico entre portadores de mutaciones en el dominio de unión a ligando del gen LDLR y los sujetos con la mutación R3500Q en el gen APOB, de una población del sur de Europa donde no había datos al respecto.
Se comparó el fenotipo lipoproteico de 30 sujetos FH portadores de una mutación “missense” en el gen LDLR que afecta al dominio de unión al ligando con 19 heterocigotos para la mutación R3500Q del gen APOB.
Los HF para el dominio de unión mostraron unos valores más elevados de colesterol y de cLDL que los pacientes DFB. Además tres de los diecinueve sujetos DFB presentaban niveles de CT y cLDL dentro del rango normal.
El uso de los criterios clínicos Med-Ped infradiagnostican a los sujetos DFB. Por ellos sugerimos que en la población del sur de Europa se revisen los criterios Med-Ped y se deberían generalizar el uso del diagnóstico genético para DFB.

138
El defecto familiar de unión de la apolipo-proteína B-100 (DFB) es una enferme-dad hereditaria autosómica dominante,debida a mutaciones localizadas en elgen de la apolipoproteína B-100, caracte-rizada por la presencia de xantomas ten-dinosos, arco corneal, concentracioneselevadas de colesterol total (CT) y coles-terol LDL (cLDL) y cardiopatía isquémicatemprana1-5. Se han descrito cuatro mu-taciones próximas al codón 3500 en elgen de la apo B (R3480P, R3500Q,R3500W y R3531C) que interfieren conel aclaramiento de las partículas LDL2-4 ydan lugar al DFB. La mutación R3500Q,que supone un cambio de Arg a Gln y re-duce en un 90% la capacidad de uniónde la apo B-100 con el receptor de LDL,fue la primera descrita y es la más fre-cuente, siendo el resto de mutacionespoco prevalentes2,5. Esta forma de hiper-colesterolemia primaria es clínicamenteindistinguible de la hipercolesterolemiafamiliar (HF), ocasionada por mutacionesheterogéneas localizadas en el gen delreceptor de LDL6. Por tanto, el diagnósti-co del DFB es genético, mediante la de-tección de las mutaciones causantes dela enfermedad.Al estudiar a 95 sujetos clínicamentediagnosticados de HF, nuestro grupoidentificó recientemente al primer sujetoafectado por esta enfermedad en nuestropaís7. En este grupo de sujetos con HF serealizó una detección de tres mutacionescausantes del DFB (R3500Q, R3500W yR3531C), utilizando la técnica de PCR-SSCP, encontrando un portador de lamutación R3500Q. Al ampliar el estudiode detección al resto de familiares de pri-mer y segundo grado hemos identificadoa cuatro sujetos más con la mutaciónR3500Q, y a un sujeto homocigoto paraesta enfermedad. En la actualidad, des-conocemos la prevalencia de la formahomocigota del DFB en nuestra pobla-ción, habiendo sido descritos pocos ca-sos de homocigotos DFB en diferentespaíses centroeuropeos8-12. En esta publi-cación describimos el primer homocigotoespañol para el DFB.
NOTA CLÍNICA
Identificación y caracterización del primerespañol con defecto homocigoto familiar de unión de la apolipoproteína B
José Tomás Real, Javier Francisco Chavesa, José Javier Martín de Llanoa,Ismael Ejarque, Miguel Civera, Juan Francisco Ascaso, Ervin Knechta,María Eugenia Armengoda y Rafael Carmena
Servicio de Endocrinología y Nutrición. Hospital Clínico Universitario. Universidad de Valencia.aInstituto de Investigaciones Citológicas de Valencia (FVIB).
Este trabajo ha sido financiado por el Fondo de Investigaciones Sanitarias (FIS 96/2063, 99/0008-01 y 03).
Correspondencia: Dr. J.T. Real.Servicio de Endocrinología y Nutrición. Hospital Clínico Universitario de Valencia.Avda. Blasco Ibáñez, 15. 46010 Valencia.
Recibido el 17-7-2000; aceptado para su publicación el 12-12-2000
FUNDAMENTO: El defecto familiar de unión de la apolipoproteína B-100 (DFB) es una enferme-dad hereditaria autosómica dominante debida a mutaciones localizadas en el gen de la apolipo-proteína B-100, clínicamente indistinguible de la hipercolesterolemia familiar. Describimos elprimer homocigoto español para el DFB.MÉTODOS: Estudiamos por técnica de PCR–SSCP la mutación R3500Q en los familiares de pri-mer y segundo grado de la familia con DFB previamente descrita por nuestro grupo. Además,analizamos la actividad del receptor de LDL en un ensayo con LDL conjugada con oro coloidal.RESULTADOS: El paciente presenta en ambos alelos la mutación R3500Q causante de DFB. Elestudio de la actividad del receptor de LDL es normal, lo que descarta que se trate de una hi-percolesterolemia familiar. El grado de hipercolesterolemia es menor del esperado tratándosede un homocigoto (colesterol total, 415, y cLDL, 352 mg/dl), y presenta una buena respuestaterapéutica a estatinas y resinas (descensos de hasta un 42% para el colesterol total y de un51% para el cLDL).CONCLUSIONES: Hemos detectado y caracterizado el primer homocigoto español para el DFB (mu-tación R3500Q), que presenta valores moderadamente elevados de colesterol total y cLDL apesar de su situación de homocigosis. Estos datos demuestran que el fenotipo lipoproteico delos homocigotos DFB es diferente de la situación de homocigosis para la hipercolesterolemiafamiliar.
Palabras clave: Defecto familiar de unión de apolipoproteína B. Homocigosis. Mutación R3500Q.Análisis por PCR-SSCP. Gen de la apo B. Actividad del receptor de LDL.
Identification and characterization of the first Spanish familial ligand-defectiveapolipoprotein B homozygote
BACKGROUND: Familial ligand-defective apolipoprotein B 100 (FDB) is an autosomal inherited di-sease due to mutations on apo B 100, clinically indistinguishable from familial hypercholeste-rolemia (FH). We described the first Spanish homozygote for FDB.METHODS: We have screened R3500Q mutation of apo B gene (PCR-SSCP analysis) in a largefamily with FDB and have identified the first Spanish homozygote for FDB.RESULTS: The homozygote is a 58 year-old man with coronary heart disease, no presence ofxanthomata and with total cholesterol and LDL cholesterol plasma levels of 415 and 352mg/dl. The response to statins and resins was up to 42% for total cholesterol and 51% forLDLc plasma values. The LDL receptor activity was normal in the FDB homozygote.CONCLUSIONS: We have identified and characterised the first Spanish homozygote for FDB(R3500Q mutation). Our data indicate a moderate lipoprotein phenotype in FDB homozygote,different as expected comparing to homozygous FH.
Key words: Homozygous familial ligand-defective apolipoprotein B. R3500Q mutation. SSCP analysis. Apo B gene. LDL receptor activity.
Med Clin (Barc) 2001; 116: 138-141


El estudio del gen del receptor de LDL del pacientehomocigoto con DFB, así como el de varios controlessanos y pacientes con HF, se realizó mediante ampli-ficación por PCR de todos los exones, regiones intró-nicas próximas y región promotora del gen del recep-tor de LDL y análisis de SSCP de todos ellosmediante el sistema anteriormente indicado y en di-ferentes condiciones de electroforesis (10-15% deacrilamida 29:1; 0-15% de glicerol; 2-25 ºC de tem-peratura, y bajo una diferencia de potencial entre350 y 500 W).La determinación del genotipo de apo E se realizó uti-lizando el método descrito por Hixon y Vernier15.
Discusión
Estudios epidemiológicos realizados endiferentes países del norte de Europa yAmérica demuestran una alta prevalenciade la mutación R3500Q en su forma he-terocigota, que oscila entre 1/700 habi-tantes en la población germánica y 1/300en la población suiza5. En el estudio deTybjaerg-Hansen et al16, la prevalencia deesta mutación en la población danesa ge-neral fue del 0,008, siendo significativa-mente superior en pacientes con isque-mia coronaria y en sujetos con HF.Además, se calcula que la prevalencia dela forma heterocigota entre sujetos conhipercolesterolemia oscila entre 0,01 y0,03, lo que indica que se trata de unaforma frecuente de hipercolesterolemiaprimaria5. En la actualidad, se desconocela prevalencia de la mutación R3500Q en la población española, aunque la de-tección de la mutación R3500Q en suje-tos con HF diagnosticados clínicamenteapunta que debe de ser muy baja7. Cu-riosamente, al ampliar el estudio familiarde un portador R3500Q detectado en taldetección, hemos sido capaces de identi-ficar cuatro nuevos casos de heterocigo-tos con DFB y un homocigoto con DFB(fig. 1). Esta aparente contradicción entrebaja prevalencia en nuestro país y la de-tección de una forma homocigota sedebe a la gran endogamia que existe enla zona de donde procede la familia estu-diada. El pueblo donde se detectó la pri-mera familia diagnosticada de DFB perte-nece a una comarca de montaña muyaislada por accidentes geográficos, don-de es frecuente encontrar una elevadaendogamia. De hecho, los padres del ho-mocigoto eran primos hermanos. Esta ca-racterística ha permitido detectar a dife-rentes heterocigotos y al homocigotodescrito.
La prevalencia de la forma homocigotano se conoce, y se han descrito pocoscasos. Gallagher et al8 describieron doscasos, un varón de 66 años con enferme-dad coronaria y valores de CT de 367mg/dl, y su hermana de 69 años, sin evi-dencia de enfermedad coronaria y conconcentraciones plasmáticas de CT de463 mg/dl. März et al9 han descrito otrohomocigoto con enfermedad coronaria ycifras moderadamente elevadas de CT ycLDL, al igual que el homocigoto descritopor Funke et al10.Como ocurre en el paciente que presen-tamos, los homocigotos con DFB hastaahora descritos presentan frecuentemen-te enfermedad coronaria y cifras modera-damente elevadas de CT y cLDL, diferen-tes de las esperadas. Así, en teoría, unhomocigoto con DFB que presente ensus dos alelos una mutación como laR3500Q, que reduce en un 90% la uniónde la partícula LDL a su receptor, deberíatener concentraciones de CT y cLDL delrango que presentan los homocigotoscon HF. Se han realizado diferentes estu-dios in vivo para tratar de explicar estacaracterística. Gallagher y Myant8 hanpropuesto que en estos homocigotos la
actividad residual de unión de apo B jun-to con un aclaramiento normal de losprecursores de LDL, las lipoproteínasVLDL, explicaría los valores moderada-mente elevados de CT y cLDL. Además,Schaefer et al17 han demostrado que lavía catabólica de las lipoproteínas VLDLque contienen apo E está elevada en elhomocigoto con DFB estudiado por sugrupo. Este mayor aclaramiento de laspartículas precursoras de LDL explicaríala moderada elevación de CT y cLDL quepresentan los homocigotos con DFB.También se han encontrado estas carac-terísticas en los sujetos con DFB hetero-cigotos. Igualmente Pietzsch et al18 de-mostraron in vivo un mayor aclaramientode los precursores de LDL en sujetos he-terocigotos con DFB.Estos hallazgos también podrían explicarla buena respuesta farmacológica a esta-tinas (descensos del 42% para el CT ydel 51% para cLDL) observada en el ho-mocigoto con DFB que presentamos,aunque no se hayan conseguido todavíalos objetivos terapéuticos de prevenciónsecundaria. En teoría, al igual que ocurrecon la forma homocigota para la HF, ca-bría esperar una falta de respuesta a es-
MEDICINA CLÍNICA. VOL. 116. NÚM. 4. 2001
140
Basal Febrero de 1998 Julio de 1998 Diciembre de 1998 Febrero de 1999 Septiembre de 1999 (atorvastatina, 30 mg/día) (atorvastatina, 40 mg/día) (atorvastatina, 60 mg/día) (simvastatina, 80 mg/día) (80 mg/día de simvastatina + 10 g/día de colestipol)
CT (mg/dl) 415 294 (29) 280 (33) 290 (30) 250 (40) 240 (42) cLDL (mg/dl) 352 229 (35) 225 (36) 240 (32) 189 (46) 173 (51) cHDL (mg/dl) 46 52 (13) 40 (13) 34 (26) 50 (9) 55 (20) TG (mg/dl) 84 64 (23) 74 (12) 87 (4) 58 (30) 62 (26) Apo B (mg/dl) 186 182 (2) 176 (5) 155 (17) 145 (20) 132 (29)
TABLA 1
Lípidos y apolipoproteína B plasmáticos durante el seguimiento clínico del paciente homocigoto con defecto familiar de unión de la apolipoproteína B-100
Entre paréntesis se indica el porcentaje de descenso o ascenso con respecto a la basal. CT: colesterol total; cLDL: colesterol LDL; cHDL: colesterol HDL; TG: triglicéridos; apo: apolipoproteína.
100
75
50
25
0Act
ivid
ad d
el r
ecep
tor
LDL
resp
ecto
a lo
s co
ntro
les
(%)
Controles HF(E256K)
Apo B 3500(homocigoto)
Apo B 3500(heterocigoto)
Fig. 2. Estudio de la acti-vidad del receptor de LDLpresentada como el por-centaje de actividad conrespecto a los controles.Los sujetos portadores dela mutación R3500Q tie-nen una actividad del re-ceptor de LDL normal. Encambio, el sujeto con hi-percolesterolemia familiar(HF) portador de la muta-ción E256K presenta unaactividad de aproximada-mente un 20% con res-pecto a la normalidad.

J.T. REAL ET AL.– IDENTIFICACIÓN Y CARACTERIZACIÓN DEL PRIMER ESPAÑOL CON DEFECTO HOMOCIGOTO FAMILIAR DE UNIÓN DE LA APOLIPOPROTEÍNA B
141
tatinas en el homocigoto con DFB, al te-ner completamente alterada la unión deapo B con el receptor de LDL. En cam-bio, como se observa en la tabla 1, seconsiguen importantes descensos de lasconcentraciones plasmáticas de CT ycLDL con dosis elevadas de diferentesestatinas y resinas. Por tanto, la sobrex-presión del receptor de LDL sano induci-do por las estatinas, junto con una activi-dad residual de unión de la apo Bdefectuosa y un mayor aclaramiento delos precursores de LDL, podría explicaresta buena respuesta a estatinas y resi-nas18.En conclusión, hemos detectado y carac-terizado el primer homocigoto españolpara el DFB que presenta valores mode-radamente elevados de CT y cLDL a pe-sar de su situación de homocigosis. Estehallazgo, junto con la buena respuesta alas estatinas, indica que el fenotipo lipo-proteico de los homocigotos con DFB esdiferente de la situación de homocigosispara la HF. Probablemente también en-contremos diferencias en la expresión fe-notípica de la enfermedad al compararlacon la HF en la situación de heterocigo-sis. En la actualidad, esta hipótesis estásiendo analizada por nuestro grupo de in-vestigación19.
AgradecimientoAgradecemos la inestimable colaboración dela familia afectada.
REFERENCIAS BIBLIOGRÁFICAS
1. Myant NB. Familial defective apolipoprotein B-100: a review, including some comparison withfamilial hypercholesterolemia. Atherosclerosis1993; 104: 1-18.
2. Soria LF, Ludwig EH, Clarke HR, Vega GL,Grundy SM, McCarthy BJ. Association betweena specific apoprotein B mutation and familial de-fective apo B-100. Proc Natl Acad Sci USA1989; 86: 587-591.
3. Pullinger CR, Hennessy LK, Chatterton JE, LiuW, Love JA, Mendel CM et al. Familial ligand-de-fective apolipoprotein B: identification of a newmutation that decreases LDL receptor bindingaffinity. J Clin Invest 1995; 95: 1225-1234.
4. Gaffney D, Reid JM, Cameron IM, Vass K, Casla-ke MJ, Sheperd J et al. Independent mutationsat codon 3500 of the apolipoprotein B gene areassociated with hyperlipidaemia. Arterioscler Thromb Vasc Biol 1995; 15: 1025-1029.
5. Rauh G, Keller C, Schuster H, Wolfgram G, Zöll-ner N. Familial defective apolipoprotein B-100: acommon cause of primary hyperlipemia. Clin In-vestig 1992; 70: 77-78.
6. Hobbs H, Brown MS, Goldstein JL. Moleculargenetics of the LDL receptor gene in familial hy-percholesterolemia. Hum Mutat 1992; 1: 445-466.
7. Real JT, Chaves FJ, Ascaso JF, Armengod ME,Carmena R. Estudio del defecto familiar de laapo B-100 en sujetos con el diagnóstico clínicode hipercolesterolemia primaria: identificaciónde la primera familia afectada en España. MedClin (Barc) 1999; 113: 15-17.
8. Gallagher JJ, Myant NB. The affinity of low den-sity lipoproteins and very low density lipoproteinremnants for the low density lipoprotein receptorin homozygous familial defective apolipoproteinB-100. Atherosclerosis 1995; 115: 263-272.
9. März W, Ruzicka V, Phol T, Usadel KH, GrossW. Familial defective apolipoprotein B-100: mildhypercholesterolemia without atherosclerosis ina homozygous patient. Lancet 1992; 340: 1362.
10. Funke H, Rust S, Seedorf U, Brennhausen B,Chirazi A, Motti C et al. Homozygosity for familialdefective apolipoprotein B 100 (FBD) is associa-
ted with lower plasma cholesterol concentrationsthan homozygosity for familial hypercholesterole-mia (FH). Circulation 1992; 86: 691 (resumen).
11. Horinek A, Ceska R, Sobra J, Vrablik M. Familialdefective apolipoprotein B-100 homozygote withpremature coronary atherosclerosis. A case re-port. J Intern Med 1999; 246: 2235-2236.
12. Gallagher JJ, Myant NB. Variable expression ofthe mutation in familial defective apolipoproteinB-100. Arterioscler Thromb 1993; 13: 973-976.
13. Martín de Llano JJ, Andreu EJ, Pastor A, De laGuardia M, Knecht E. Electrothermal atomic ab-sorption spectrometric diagnosis of familial hy-percholesterolemia. Analytical Chemistry 2000;72: 2406-2413.
14. Schuster H, Rauh G, Müller S, Keller C, Wol-gram G, Zöllner N. Allele-specific and asymme-tric PCR amplification in combination: a onestep protocol for rapid diagnosis of familial de-fective apo B-100. Anal Biochem 1992; 204:22-25.
15. Hixon JE, Vernier DT. Restriction isotyping of hu-man apolipoprotein E by gene amplification andcleavage with HhaI. J Lipid Res 1990; 31: 545-548.
16. Tybjaerg-Hansen A, Steffensen R, Meinertz H,Schnohr P, Nordestgaard BG. Association ofmutations in the apolipoprotein B gene with hy-percholesterolemia and risk of coronary heart di-sease. N Engl J Med 1998; 338: 1577-1583.
17. Schaefer JR, Scharngl H, Baumstark MW, Sch-weer H, Zech LA, Seyberth H et al. Homozygousfamilial defective apolipoprotein B 100. Enhacedremoval of apolipoprotein E containing VLDLsand decreased production of LDLs. ArteriosclerThromb Vasc Biol 1997; 17: 348-353.
18. Pietzsch J, Wiedemann B, Julius U, Nitzsche S,Gehrisch S, Bergmann S et al. Increased clea-rance of low density lipoprotein precursors in pa-tients with heterozygous familial defective apoli-poprotein B-100: a stable isotope approach. JLipid Res 1996; 37: 2074-2087.
19. Real JT, Ejarque I, Chaves FJ, Ascaso P, CiveraM, Ascaso JF et al. Fenotipo lipoproteico en eldefecto familiar de unión de apo B 100. Clin In-vest Arteriosclerosis 2000; 12: 12 (resumen).

ARTICLE
Influence of LDL receptor gene mutations and theR3500Q mutation of the apoB gene on lipoproteinphenotype of familial hypercholesterolemic patientsfrom a South European population
Jose T Real1, Felipe J Chaves2, Ismael Ejarque1, Ana B Garcıa-Garcıa2, Carmen Valldecabres1,Juan F Ascaso1, Marıa E Armengod2 and Rafael Carmena*,1
1Service of Endocrinology and Nutrition, Department of Medicine, Hospital Clınico Universitario, University ofValencia, Spain; 2Instituto de Investigaciones Citologicas, Fundacion Valenciana de Investigaciones Biomedicas,Valencia, Spain
Few data are available on genotype–phenotype interactions among familial hypercholesterolemia (FH)patients in South European populations and there are no data about the influence of R3500Q mutation onlipoprotein phenotype compared to low-density lipoprotein receptor (LDLR) mutations. The objective ofthe study is to analyze the influence of mutations in the LDLR and apolipoprotein B (apoB) genes onlipoprotein phenotype among subjects clinically diagnosed of FH living in East Spain. In all, 113 FH indexpatients and 100 affected relatives were studied. Genetic diagnosis was carried out following a protocolbased on Southern blot and PCR-SSCP analysis. A total of 118 FH subjects could be classified into threegroups according to the type of LDLR mutations (null mutations, missense mutations affecting the ligandbinding 3–5 repeat, and missense mutations outside this domain). In addition, the lipoprotein phenotypeof these FH groups was compared with 19 heterozygous subjects with familial ligand-defective apoB (FDB),due to R3500Q mutation. FH patients carrying missense mutations affecting the ligand binding repeat 3–5showed total and LDL cholesterol levels significantly higher than FH patients with missense mutations inother LDLR domains or FDB patients. FH subjects carrying null mutations showed lower high-densitylipoprotein cholesterol plasma values compared to FH carrying missense mutations. FDB subjects showedthe lowest total and LDL cholesterol plasma values. In conclusion, the type of LDLR gene mutation andR3500Q mutation influences the lipoprotein phenotype of FH population from East Spain.European Journal of Human Genetics (2003) 11, 959–965. doi:10.1038/sj.ejhg.5201079Published online 24 September 2003
Keywords: heterozygous familial hypercholesterolemia; genetic diagnosis; LDL receptor gene mutations;lipoprotein phenotype; familial ligand-defective apoB 100; R3500Q mutation
IntroductionFamilial hypercholesterolemia (FH) is an autosomal domi-
nant disease defined at the molecular level by the presence
of mutations in the low-density lipoprotein receptor
(LDLR) gene and characterized by markedly elevated low-
density lipoprotein cholesterol (LDLc) levels, tendon
xanthomata and increased risk of coronary heart disease
(CHD).1–4 FH shows great variability in phenotypicReceived 6 February 2003; revised 6 June 2003; accepted 4 July 2003
*Correspondence: Professor R Carmena, Department of Medicine,
Hospital Clınico Universitario, Avda. Blasco Ibanez 17, E-46010 Valencia,
Spain. Tel: þ 34 96 3862665; Fax: þ 34 96 3864767;
E-mail: [email protected]
European Journal of Human Genetics (2003) 11, 959–965& 2003 Nature Publishing Group All rights reserved 1018-4813/03 $25.00
www.nature.com/ejhg

expression, which may be influenced by factors such as
age, gender, diet, type of LDLR mutations or other genes.5–8
The type of LDLR gene mutation has been associated to
different phenotype expression,6,8,9 response to statins10,11
and risk of premature CHD.5 In Northern European FH
populations, several studies have demonstrated that the
type of mutation in the LDLR gene influences the FH
phenotype expression.12–15 Affected individuals with
LDLR mutations predicted to be severe, such as null
mutations or mutations affecting the ligand-binding repeat
3–5 of the receptor, presented higher plasma levels of total
and LDLc. In Southern European FH populations, only
Bertolini et al16 have studied the relationships between the
type of LDLR gene mutation and expression of the FH
phenotype, as far as we know. These authors have reported
that receptor-negative mutations are associated with high-
er plasma levels of total and LDLc compared to affected
subjects with receptor-defective mutations. Altogether,
these results suggest that DNA diagnosis could allow the
study of genotype–phenotype interactions in terms of
estimating the clinical severity and prognosis.
When compared with Northern Europeans, Spanish FH
subjects present lower levels of plasma LDLc and lower
prevalence of tendon xanthomata,17 making genetic
diagnosis even more important. We have previously
described large and point mutations in Spanish FH subjects
and analyzed the influence of LDLR gene mutation on
treatment response in our population.18–20 FH subjects
carrying large mutations affecting promotor region have
higher LDLc plasma values. FH subject carriers of null
mutations showed a poorer response to simvastatin
treatment than FH carriers of no null mutations.
Familial ligand-defective apolipoprotein B (apoB)-100
(FDB) is an autosomal dominant inherited disease due to
mutations in the apoB gene.21 It is characterized by
elevated plasma total and LDLc levels and premature
CHD. Three mutations (R3500Q, R3500W and R3531C)
are mostly responsible for the disease by reducing the
binding of LDL particles to the LDLR.21,22 The R3500Q
mutation was the first described and is the most prevalent,
while other point mutations (R3500W and R3531C) are
rare causes of FDB.23,24 The prevalence of FDB has been
estimated to be approximately 1/500 in North America,22
whereas in Europe seems to be high in the Northwestern
part of Switzerland (1/114), Eastern France and Southern
Germany22,25,26 and low in Italy and Spain. Miserez et al27
proposed that FDB emerged from mesolithic ancestors of
Celtic people.
FDB is clinically indistinguible from FH. However, the
plasma LDLc levels in age-matched FDB heterozygotes vary
over a wide range and, in some families, heterozygous
carriers of the R3500Qmutation show normal LDLc plasma
levels.21,25
In the Spanish population, we have recently described
the first FDB family and predicted a low prevalence of this
disease, since only one FDB out of 113 FH index patients
was detected.26 On the contrary, and due to a founder
effect in an isolated region of the Valencia province, we
have been able to identify seven affected pedigrees and 19
FDB heterozygotes and one homozygote.28 This has made
possible to compare the phenotype of this group of
patients with that of FH subjects.
The present study was undertaken to evaluate the
influence of the LDLR gene mutation type and the
R3500Q mutation of the apoB gene on the lipoprotein
phenotype in a South European population where few data
on this issue were available up to now.
Subjects and methodsSubjects
The study population consisted of a total of 213 clinically
diagnosed FH subjects: 113 index patients and 100 affected
relatives that had been referred to our Lipid Clinic (Table 1).
All subjects were Caucasian and lived in the Valencia
region. The institutional ethics committee from our
institution approved the protocol and all subjects gave
written informed consent to enter the study.
Diagnostic criteria for FH included: plasma levels of total
and LDLc above the 95th percentile corrected for both age
and sex,29 presence of tendon xanthomata, CHD in the
index patient or in a first-degree relative, and bimodal
distribution of total and LDLc levels in the family,
indicating an autosomal dominant pattern of phenotype
IIa.
A complete medical history and physical examination
were obtained in all participants. Body mass index (BMI)
was calculated as weight divided by height squared
(kg/m2). Blood pressure was measured with a von
Table 1 General characteristics and plasma lipids valuesin 213 FH subjects separated by gender
Men (n¼83) Women (n¼130)
Age (years) 36.5 (18.1) 39.9 (19.0)BMI (kg/m2) 25.6 (4.2) 24.9 (4.7)Xanthomata, n (%) 21 (25) 26 (20)Arcus cornealis, n (%) 40 (48) 56 (43)CHD, n (%) 20 (24) 23 (18)TC (mg/dl) 339.8 (63.6) 330.7 (67.6)TG (mg/dl) 129.9 (84.5) 106.2 (47.2)HDLc (mg/dl) 47.6 (14.4)** 53.6 (14.7)LDLc (mg/dl) 267.2 (63.8) 254.8 (67.6)VLDLc (mg/dl) 22.9 (12.1) 21.4 (10.9)Apo AI (mg/dl) 124.3 (30.4)* 135.5 (34.8)ApoB (mg/dl) 162.1 (43.1) 158.6 (39.7)
*Po0.05, **Po0.01, mean (SD).BMI, body mass index; CHD, coronary heart disease; TC, totalcholesterol; TG, triglycerides; HDLc, high-density lipoprotein choles-terol; LDLc, low-density lipoprotein cholesterol; VLDLc, very low-density lipoprotein cholesterol; apoB, apolipoprotein B; apoAI,apolipoprotein AI.
Genotype–phenotype in FHJT Real et al
960
European Journal of Human Genetics

Recklinghausen sphygmomanometer in the sitting posi-
tion and after a 5-min rest. The mean value of three
measurements was considered. CHD was diagnosed if there
was a documented history of previous myocardial infarc-
tion and/or coronary artery bypass surgery, or angioplasty,
or history of angina pectoris and alterations in the stress
ECG.
One of the clinically defined FH index patients was
found to be a carrier of the R3500Q mutation of the apoB
gene (1/113 FH index patients), responsible for FDB.26
Extending the study to seven affected pedigrees, we could
identify 19 heterozygotes for the R3500Q mutation and
one homozygote.26,28
Laboratory methodsMeasurement of lipids and lipoproteins After a 12–14h
fast and in basal situation (4 weeks without treatment with
drugs that affect lipid metabolism), blood samples were
drawn from an antecubital vein in tubes containing EDTA
(Vacutainer) and were centrifuged within 4h. Plasma was
stored at 41C for a maximum of 3 days. Cholesterol and
triglyceride levels were measured by enzymatic techni-
ques.30,31 High-density lipoprotein cholesterol (HDLc) was
measured after precipitation of apoB-containing lipopro-
teins with polyanions,32 and very-low-density lipoprotein
cholesterol (VLDLc) after separation of VLDL (do1.006 g/
ml) by ultracentrifugation.33 The LDLc was calculated by
subtraction of VLDL and HDL cholesterol from total
cholesterol (TC). Total plasma apoB was measured by
immunoturbimetry.34 Plasma Lp(a) concentrations were
determined by ELISA.35 The coefficients of variation for
lipids and lipoproteins were o5%.
Genetic methods DNA extraction was performed by a
standard procedure. Genetic diagnosis of FH was previously
established through identification of mutations of LDLR
gene by Southern blot analysis18 and DNA sequencing of
abnormal SSCP bands.20 Screening for the apoB mutations
was performed by PCR amplification, as described by
Schuster et al,36 and SSCP analysis;20 DNA samples carrying
the mutations R3480P, R3500Q, R3500W and R3531C,
responsible for FDB, were used as controls.
Genotyping for the ApoE polymorphisms was performed
following the method described by Hixon and Vernier37
with minor modifications.
Statistical analysis
Data were analyzed with the Statistical Package for the
Social Sciences (SPSS 6.1. 3 for Windows) and expressed as
mean7standard deviation (SD). The mean values of
quantitative variables were compared with one-way ANO-
VA. Proportions were compared with contingency tables
and the w2 test or the Fischer’s exact test (no5).
ANCOVA analysis was used to estimate the independent
contributions of the different LDLR gene mutations, age,
gender and BMI to the mean baseline lipid values.
ResultsGenetic diagnosis of FH and FDB
The genetic diagnosis of FH was previously established
using a three-step protocol that screened mutations at the
apoB gene, responsible for FDB, and major rearrangements
and minor or point mutations in the LDLR gene,
responsible for FH.18,20 Only one R3500Q mutation carrier
was identified among the 113 FH index patients.20 The
sample was screened for major rearrangements in the LDLR
gene by Southern blot analysis. Five rearrangements were
detected and characterized,18 two of them were due to
duplications of internal regions of the gene (FH Valencia-2
and FH Valencia-3), whereas the rest were caused by partial
deletions, which eliminate the promoter region in two
cases (FH Valencia-1 and -5). The mRNA analysis of the
alleles with internal rearrangements (FH Valencia-2, -3 and
-4) indicated that these resulted in in-frame alterations of
the RLDL gene.18 Thus, FH Valencia-1 and -5 may be
classified as null mutations and FH Valencia-2, -3 and -4 as
defective mutations.
SSCP analysis of the LDLR gene was used to screen for
point and minor mutations. A total of 42 minor mutations
causing FH were identified; a detailed description of them
can be found in a previous report.20
Effect of LDLR gene and R3500Q mutations on plasmalipids and lipoproteins
FH subjects were classified into four groups according to
the type of mutation. Group 1 included 45 subjects with
null mutations; group 2, 13 subjects with missense
mutations affecting ligand binding 3–5 repeat; group 3,
60 subjects with missense mutation not affecting this
domain; and group 4, 19 heterozygotes with the R3500Q
mutation in the apoB gene.
Mutations in the LDLR gene were considered null in the
case of: (a) major rearrangements deleting the promoter
region (FH Valencia-1 and -5), or (b) small mutations
producing stop codons (W-18X, E10X, Q133X) or frame-
shifts in the LDL open reading frame (112insA, 790delAT-
GA). Subjects with null mutations included: FH Valencia-1
(n¼4), FH Valencia-5 (n¼8), W-18X (n¼ 10), E10X (n¼2),
Q133X (n¼2), 112insA (n¼15) and 790delATGA (n¼4)
(see Figure 1).
Missense mutations in the exon 4 of the LDLR gene may
affect the role of the binding repeat 3–5 region of the
LDLR. The missense mutations used in this study were:
C95R (n¼4), E119K (n¼2), P160R (n¼3), D200G (n¼1)
and S205P (n¼3) (Figure 1). Carriers of S156L (n¼ 5) were
not included in this group because this mutation of exon 4
abolishes the binding of LDL, but not that of VLDL,
Genotype–phenotype in FHJT Real et al
961
European Journal of Human Genetics

causing only a mild reduction of the receptor function in
vitro.15
Missense mutations classified as no affecting the ligand
binding repeat 3–5 region were: C68W (n¼6), Q71E
(n¼2), E80K (n¼3), E246A (n¼3), E256K (n¼ 4), D280G
(n¼4), Y354C (n¼2), C358Y (n¼ 11), R395W (n¼8),
N407K (n¼1), T413R (n¼12), G528V (n¼ 2), D679G
(n¼2) and N804K (n¼2) (see Figure 1).
Finally, as previously mentioned, group 4 included 19
heterozygotes with the R3500Q mutation, which is
responsible of FDB.
Plasma lipid and apoB values at baseline of patients
classified according to their respective mutation type are
shown in Table 2. No differences were found between the
four groups with respect to age, BMI, gender distribution,
presence of CHD, and APOE genotype distribution.
Significant differences were observed with respect to TC
and LDLc depending on the type of LDLR gene mutation.
The mean plasma TC and LDLc levels were significantly
higher in subjects with missense mutations affecting the
binding repeat 3–5 domain, whereas the lowest values
were found in the FDB group. No statistical differences
were found between groups in plasma triglycerides (TG)
and VLDL values. HDLc values in the null mutation group
were significantly lower than in the missense mutation
group. In order to control covariables we have also used the
ANCOVA analysis. The intersubjects effect of the type of
LDLR mutation on HDLc plasma values was not signifi-
cantly different (P¼0.66) when BMI was included as
covariate and gender as independent variable.
Interestingly, in the whole group, the ANCOVA analysis
showed that the type of LDLR mutation (P¼0.006, 0.014),
but not age or gender, had an independent influence on TC
and LDLc plasma values.
DiscussionThis is the second study carried out in a South European FH
population aimed to analyze the relationships between the
type of mutation affecting genes involved in lipid meta-
bolism (the LDLR and ApoB genes) and phenotype. Our
results indicate that the type of LDLR gene mutation has an
impact on lipoprotein phenotype. The FH group with
missense mutations affecting the binding repeat 3–5
domain (group 2) presents the highest TC and LDLc
plasma concentrations, which determines a significant
difference with groups 3 and 4 (Po0.05). However, the TC
and LDLc values of the group with null mutations (group
1) were not statistically different than those of the other
groups. On the other hand, FDB subjects showed the
lowest TC and LDLc plasma values.
Figure 1 Description of LDLR gene mutations includedin the study.
Table 2 Characteristics of the FH subjects classified according to the type of mutation
Group 1 (n¼45) Group 2 (n¼13) Group 3 (n¼60) Group 4 (n¼ 19)
Age (years) 35.7 (18.4) 36.7 (18.8) 38.2 (17.6) 42.8 (15.9)Male/female 22/23 4/9 22/38 8/11CHD, n 7 (16%) 3 (20%) 9 (15%) 2 (10%)BMI, kg/m2 26.2 (5.2) 24.1 (3.4) 25.4 (3.2) 25.6 (4.2)TC, mg/dl* 343.7 (69.6) 394.4 (47.9)** 335.2 (60.2) 286.5 (57.9)TG (mg/dl) 102.9 (51.1) 102.8 (67.2) 105.3 (52.6) 114.1 (44.8)HDLc (mg/dl) 45.3 (14.1) *** 56.8 (18.5) 54.3 (12.6) 47.6 (11.1)LDLc (mg/dl)* 280.6 (65.9) 318.8 (49.7)** 256.1 (58.2) 220.1 (56.9)VLDLc (mg/dl) 19.9 (10.4) 16.9 (7.7) 20.6 (11.6) 20.7 (12.9)Apo B (mg/dl) 151.7 (30.8) 170.6 (19.3) 153.1 (38.6) 149.1 (32.7)Lp(a) (mg/dl) 19.6 (15.9) 15.9 (5.8) 19.6 (19.3) 26.4 (19.8)Apo E genotypeE3/E3 32 (71%) 7 (57%) 41 (69%) 13 (70%)E3/E4 8 (18%) 11 (18%) 3 (13%)E3/E2 4 (9%) 4 (28%) 5 (8%) 2 (11%)E2/E4 1 (3%) 2 (5%) 3 (5%) 1 (6%)
*Po0.05 ANOVA, **Po0.05 group 2 vs groups 3 and 4, ***Po0.05 group1 vs group2, mean (SD).Group 1: null mutation. Group 2: missense mutations affecting the binding repeat 3–5 region. Group 3: missense mutations not affecting the bindingrepeat 3–5. Group 4: R3500Q mutation of the apoB gene. For abbreviations, see Table 1.
Genotype–phenotype in FHJT Real et al
962
European Journal of Human Genetics

Our results are partially in agreement with previous data
from Northern European FH populations. Thus, Gudnas-
son and Humphries15 reported that FH subjects with null
mutations and mutations affecting repeat 5 had the
highest mean LDLc plasma values. Sun et al12 showed that
carriers of LDLR mutations predicted to be ‘severe’ (like
null mutations or mutations that affect exon 4 repeat 3–5)
have higher plasma TC and LDLc values and will not
decrease lipid levels with treatment to the same extent
than carriers of ‘mild mutations’. Finally, a study per-
formed by Graham et al14 indicated that families with
LDLR defects had higher total and LDLc plasma levels and
higher incidence of xanthomas, than those with FDB.
Our study expands the few data available from South
European FH populations about genotype–phenotype
relationships. As far as we know, only Bertolini et al16 has
addressed this issue. They studied 282 FH subjects from
Italy, and identified 71 LDLR gene mutations responsible
for the disease. Separating the molecularly defined FH
subjects into two groups (receptor negative and receptor
defective), they showed that FH subjects with receptor-
negative mutations had higher TC plasma values (18%)
and lower HDLc (�5%) than subjects in the receptor-
defective group. The percentage of xanthomas and CHD in
the receptor-negative group was two-fold higher than in
the receptor-defective group.
All these observations suggest that different mutations of
the LDLR gene are associated with differences in plasma
lipid levels, although the basis for these differences are not
clear. One possible explanation could be that upregulation
of the wild-type LDLR allele was affected by the nature of
the mutant allele, or that some defective mutations had
residual LDLR activities13,14 On the other hand, genetic
variability due to DNA polymorphisms of the wild type
allele could quantitatively influence LDLc concentrations,
as has been reported by Betard et al38 in French Canadian
FH women carrying the French Canadian 10 kb deletion,
which may further complicate the interpretation of data.
One could expect that null mutations where nonrecep-
tor is expressed and missense mutations affecting the
binding domain will correlate with a severe phenotype,
compared to other mutations where residual activity or
expression of the receptor will be predicted. In contrast to
this hypothesis, the results of previous studies14,15,39,40
indicate that FH patients carrying null mutation not
always have the highest TC and LDLc plasma values. In
most studies, FH subject carriers of a mutation predicted to
be severe (not only null mutations) or that affects five
repeat of the binding domain region have the most
atherogenic lipid profile (higher plasma values of TC and
LDLc). This is consistent with our results. In our study, we
have found that FH patients carrying mutations predicted
to be severe (null plus missense mutations affecting the
binding repeat 3–5 region) have the worst lipid phenotype
(higher plasma values of TC and LDLc and lower HDLc).
The differences found in the revised studies could be
explained by methodological differences, selection vias
and the classification used for LDLR gene mutations.
We have not found statistically significant differences in
TC and LDLc plasma values between the null mutation
group (group 1) and the group with missense mutations
affecting 3–5 binding repeat (group 2). Similar results have
been previously demonstrated.14,15,40 One possible expla-
nation is that FH patient carriers of null mutations
(predicted to be severe) only express the normal receptor
in the membrane of their cells. In contrast, FH patient
carriers of the missense mutations express both the normal
receptor and the altered receptors. This situation could
facilitate a local competition between both receptors so
that the internalization process could be affected. In
addition, in the case of missense mutations affecting the
3–5 repeat, the situation would worsen, because the
affected protein could not bind the apoB.
Another possibility is that, in group 3 (missense muta-
tions not affecting the binding repeat 3–5 region), we
could have included some mutations affecting the binding
domain (missense mutations localized in exon 3 and 6),
which could increase the average values of TC and LDLc of
the entire group.
Finally, we cannot exclude differences in lipid profiles
due to the influence of environmental or other genetic
factors in our FH population. It should be noted that
interaction with other genes41 and environmental factors42
could also influence the phenotype in FH patients.
Pimstone et al42 have demonstrated that Chinese FH
heterozygotes living in Canada exhibit a similar phenotype
to that of other FH patients from Western societies, and
different to those living in China. Leitersdorf et al.43 have
shown the important effect of environmental factors like
diet, physical activity, weight and others on the lipid
phenotype in FH patients. Finally, the contribution of the
ApoE genotype has been studied by Bertolini et al.16 These
authors have reported a lowering effect of the e2 allele, and
a raising effect of the e4 allele on the LDL cholesterol levels
in both receptor-negative and -defective groups. In our
study, the effects of environment (age, gender, BMI) and
genetic factors (apoE genotype) that might modulate the
lipid phenotype of FH or FDB subjects appear similar in the
four groups.
On the other hand, and in agreement with Sijbrands
et al44 and Bertolini et al,16 our FH subjects with null
mutations showed plasma HDLc values significantly lower
than those carrying missense mutations. Our observation
on HDLc values suggests that the LDLR could be important
in HDL metabolism of FH patients. The role of the LDLR on
the clearance of remnant chylomicrons and VLDL particles
could explain the HDLc plasma levels found in our group
of FH subjects. In recent human studies, there was a
markedly delayed clearance of retinol-labelled triglyceride-
rich lipoproteins following an oral fat load in six Japanese
Genotype–phenotype in FHJT Real et al
963
European Journal of Human Genetics

homozygous FH subjects. In addition, the binding and
clearance of chylomicron remnants by fibroblasts was
substantially decreased in the homozygous FH group.45
Along the same lines, Castro-Cabezas et al46 reported, in
heterozygous FH subjects, a two-fold delay in the area
under the curve for clearance of remnant particles. Thus, it
is possible that FH subjects with null mutations would have
a lower clearance of remnant particles (chylomicron and
IDL) and, as a consequence, lower HDLc concentrations. In
addition, this subgroup of FH presented the highest risk of
CHD in the study of Vohl et al.5
Different studies in North European populations47,48
have demonstrated that FDB subjects have lower total and
LDLc plasma values than FH subjects. This is also the case
in our study done in a South European population. The
explanation for these differences has been attributed to the
increased clearance of LDL precursors that has been
observed in heterozygous and homozygous FDB patients.49
There are different possible classifications of FH accord-
ing to the type of LDLR mutations.12–16 An alternative is to
classify them into two groups (receptor negative and
receptor defective). The problem is that the defective
mutations group can include severe or mild mutations,
limiting the study of genotype–phenotype relationship in
FH subjects.
Our study points out, in a South European outbreed FH
population, that the type of LDLR mutation and the
R3500Q mutation of the apoB gene have an impact on the
lipid phenotype expression. In the general population, a
direct relationship exists between the magnitude of
hypercholesterolemia and prevalence and incidence of
CHD, as well as onset of symptoms. Therefore, the
differences in TC and LDLc, found between molecularly
defined FH subjects, could explain the different risk for
CHD observed in these patients. Moreover, Vohl et al5 and
Bertolini et al16 have demonstrated that FH subjects
carrying a receptor-negative mutation have an increased
risk for CHD compared to receptor-defective subjects. In
addition, Jansen et al50 have demonstrated that LDLc
plasma values alone could not account for the difference in
CHD risk in these FH subjects.
We have not found significant differences in CHD
prevalence between the groups. This can be attributed to
the low number of subjects studied that limited the power
of the study to investigate this issue.
Our results and those of others5,8,9,11,12,15,16 suggest that
identification of mutations in the LDLR gene may allow a
better assessment of clinical expression in these subjects
with high CHD risk. In this sense, prospective studies
should be performed to evaluate further the impact of
genotype on severity of FH. In addition, the genetic
diagnosis and molecular characterisation of LDLR gene
mutations will allow a better assessment of response to
treatment, prevention and family screening in these
subjects.
AcknowledgementsWe thank the patients and their relatives for their cooperation. Thiswork was supported by grants from Fondo de InvestigacionesSanitarias and Generalitat Valenciana to JT Real (FIS 02/1875 andGV01-46) and Red de Centros Metabolismo y Nutricion del InstitutoCarlos III (C 03/08).
References1 Brown MS, Goldstein JL: A receptor-mediated pathway for
cholesterol homeostasis. Science 1986; 232: 34–47.2 Hobbs H, Brown MS, Goldstein JL: Molecular genetics of the LDL
receptor gene in familial hypercholesterolemia. Hum Mutat 1992;1: 445–466.
3 Varret M, Rabes JP, Thiart R et al: LDLR database (second edition):new additions to the database and the software, and results of thefirst molecular analysis. Nucl Acid Res 1998; 26: 248–252.
4 Wilson DJ, Gahan M, Haddad L et al: A world wide website forLDLR gene mutations in FH: sequence-based, tabular, and directsubmission data handling. Am J Cardiol 1998; 81: 1509–1511.
5 Volh MC, Gaudet D, Moorjani S et al: Comparison of the effect oftwo low-density lipoprotein receptor class mutations on coronaryheart disease among French–Canadian patients heterozygousfor familial hypercholesterolemia. Eur J Clin Invest 1997; 27:366–373.
6 Hoeg JM: Homozygous familial hypercholesterolemia: aparadigm for phenotypic variation. Am J Cardiol 1993; 72:11D–14D.
7 Ferrieres J, Lambert J, Lussier-Cacan S, Davignon J: Coronaryartery disease in heterozygous familial hypercholesterolemiapatients with the same LDL receptor gene mutation. Circulation1995; 92: 290–295.
8 Kotze MJ, de Villiers WJS, Stein K et al: Phenotypic variationamong familial hypercholesterolemic heterozygous for either oneof two Afrikaner founder LDL receptor mutations. ArterioscleThromb 1993; 13: 1460–1468.
9 Gudnason V, Day INM, Humphries SE: Effect on plasma lipidlevels of different classes of mutations in the low-densitylipoprotein receptor gene in patients with familialhypercholesterolemia. Arterioscle Thromb 1994; 17: 1717–1722.
10 Vuorio AF, Ojala JP, Sarna S, Turtola H, Tikkanen MJ, Kontula K:Heterozygous familial hypercholesterolaemia: the influence ofthe mutation type of the low-density-lipoprotein receptor geneand PvuII polymorphism of the normal allele on serum lipidlevels and response to lovastatin treatment. J Intern Med 1995;237: 43–48.
11 Leren TP, Hjermann I: Is responsiveness to lovastatin in familialhypercholesterolaemia influenced by the specific mutation in thelow density lipoprotein receptor gene? Eur J Clin Invest 1995; 25:967–973.
12 Sun XM, Neuwirth C, Patel DD, Knight BL, Soutar AK, with thefamilial hypercholesterolemia regression study group: Influenceof genotype at the low density lipoprotein receptor gene locus ofthe clinical phenotype and response to lipid lowering drugtherapy in heterozygous familial hypercholesterolemia.Atherosclerosis 1998; 136: 175–185.
13 Moorjani S, Roy M, Torres A et al: Mutations of low densitylipoprotein receptor gene, variation in plasma cholesterol, andexpression of coronary heart disease in homozygous familialhypercholesterolemia. Lancet 1993; 341: 1303–1306.
14 Graham CA, McClean E, Ward AJM et al: Mutation screening andgenotype: phenotype correlation in familial hypercholesterol-emia. Atherosclerosis 1999; 147: 309–316.
15 Gudnasson V, Humphries SE: Effect on plasma lipid levels ofdifferent classes of mutations in the low density lipoproteinreceptor gene in patients with familial hypercholesterolemia.Arterioscle Thromb 1994; 14: 1717–1722.
Genotype–phenotype in FHJT Real et al
964
European Journal of Human Genetics

16 Bertolini A, Cantafora A, Averna M, Cortese C, Motti C, MartiniC: Clinical expression of familial hypercholesterolemia in clusterof mutations of the LDL receptor gene that cause a receptordefective or receptor negative phenotype. Arterioscle Thromb VascBiol 2000; 20: e41–e52.
17 Villaverde CA, Sarda P, Vallbe JC et al: Manifestaciones clınicas dela hipercolesterolemia familiar en una poblacion mediterranea.Med Clin (Barna) 1999; 113: 521–525.
18 Chaves FJ, Real JT, Garcıa-Garcıa AB et al: Large rearrangements ofthe LDL receptor gene and lipid profile in a Spanish population.Eur J Clin Invest 2001; 31: 309–317.
19 Chaves FJ, Real JT, Garcıa-Garcıa AB et al: Genetic diagnosis offamilial hypercholesterolemia in a south European outbreedpopulation: influence of low density lipoprotein (LDL) receptorgene mutations on treatment response to simvastatin in total,LDL and high density lipoprotein cholesterol. J Clin EndocrinolMetab 2001; 86: 4926–4932.
20 Garcıa-Garcıa AB, Real JT, Puig O et al: Molecular genetics offamilial hypercholesterolemia in Spain: ten novel LDLRmutations and population analysis. Hum Mutat 2001; 454: 1–9.
21 Myant NB: Familial defective apolipoprotein B-100: a review,including some comparison with familial hypercholesterolemia.Atherosclerosis 1993; 104: 1–18.
22 Soria LF, Ludwig EH, Clarke HR, Vega GL, Grundy SM, McCarthyBJ: Association between a specific apoprotein B mutation andfamilial defective apo B-100. Proc Nat Acad Sci USA 1989; 86:587–591.
23 Pullinger CR, Hennessy LK, Chatterton JE et al: Familial ligand-defective apolipoprotein B: identification of a new mutation thatdecreases LDL receptor binding affinity. J Clin Invest 1995; 95:1225–1234.
24 Gaffney D, Reid JM, Cameron IM et al: Independent mutations atcodon 3500 of the apolipoprotein B gene are associated withhyperlipidaemia. Arterioscle Thromb Vasc Biol 1995; 15:1025–1029.
25 Rauh G, Seller C, Schuster H, Wolfgram G, Zollnner N: Familialdefective apolipoprotien B-100: a common cause of primaryhyperlipemia. Clin Investigator 1995; 95: 1225–1234.
26 Real JT, Chaves FJ, Ascaso JF, Armengod ME, Carmena R: Estudiodel defecto familiar de la apo B-100 en sujetos con el diagnosticoclınico de hipercolesterolemia primaria: identificacion de laprimera familia afectada en Espana. Med Clin (Barna) 1999; 113:15–17.
27 Miserez AR, Muller PY: Familial defective apolipoprotein B-100: amutation emerged in the Mesolithic ancestor of Celtic people?Atherosclerosis 2000; 148: 433–436.
28 Real JT, Chaves JF, Martın de Llano JJ et al: Identificacion ycaracterizacion del primer homocigoto espanol con defectofamiliar de union de la apolipoproteina B. Med Clin (Barna)2001; 116: 138–141.
29 Gutierrez Fuentes JA, Gomez Gerique JA: Estudio DRECE. Dieta yriesgo de enfermedades cardiovasculares en Espana.; Ministeriode Sanidad y Consumo, Direccion General de Salud Publica,Madrid, 1993.
30 Allain CC, Poon LS, Chan CSG, Richmond W, Fu PC: Enzymaticdetermination of total serum cholesterol. Clin Chem 1974; 20:470–475.
31 Ter Welle HF, Baartscheer T, Fiolet JWT: Influence of free glycerolon enzymatic evaluation of triglycerides. Clin Chem 1984; 30:1102–1103.
32 Burstein M, Scholnick HR, Morfin R: Rapid method for theisolation of lipoproteins from human serum by precipitationwith polyanions. J Lipid Res 1970; 11: 583–595.
33 Havel RJ, Eder HJ, Bragdon JH: The distribution and chemicalcomposition of centrifugally separated lipoproteins in humanserum. Eur J Clin Invest 1995; 34: 1345–1354.
34 Rosseneu M, Vercaemst R, Steinberg KK, Cooper GR: Someconsiderations of methodology and standarization of apolipo-protein B immunoassays. Clin Chem 1983; 29: 427–433.
35 Real JT, Ascaso JF, Chaves FJ et al: Plasma Lp(a) values in familialhypercholesterolemia and its relation to coronary heart disease.Nutr Metab Cardiovas Dis 1999; 9: 41–44.
36 Schuster H, Rauh G, Muller S, Keller C, Wolfram G, Zollner N:Allele specific and asymetric polymerase chain reactionamplification in combination: a step polymerase chain reactionprotocol for rapid diagnosis of familial defective apolipoproteinB-100. Ann Bioch 1992; 204: 22–25.
37 Hixon JE, Vernier DT: Restriction isotyping of humanapolipoprotein E by gene amplification and cleavage with HhaI.J Lipid Res 1990; 31: 545–548.
38 Betard C, Kessling AM, Roy M, Davignon J: Influence of geneticvariability in the non deletion LDL-receptor allele on phenotypicvariation in French Canadian familial hypercholesterolemiaheterozygotes sharing a ‘null’ LDL receptor gene defect.Atherosclerosis 1996; 119: 43–55.
39 Ward AJ, OKane M, Nicholls DP, Young IS, Nevin NC, GrahamCA: A novel single base deletion in the LDLR gene (211delG):effect on serum lipid profiles and the influence of other geneticpolymorphism in the ACE gene, apoE and apoB genes.Atherosclerosis 1996; 120: 83–91.
40 Jensen HK, Jensen LG, Hansen PS, Faergeman O, Gregersen N:The Trp23Stop and Trp66Gly mutations in the LDL receptor gene:common causes of familial hypercholesterolemia in Denmark.Atherosclerosis 1996; 120: 57–65.
41 Carmena-Ramon R, Ascaso JF, Real JT, Ordovas JM, Carmena R:Genetic variation at the apo A IV gene locus and response to dietin familial hypercolesterolemia. Arterioscler Thromb Vasc Biol1998; 18: 1266–1274.
42 Pimstone SN, Sun XM, du Souich C, Frohlich JJ, Hayden MR,Soutar AK: Phenotypic variation in hetrozygous familialhypercholesterolemia. A comparison of Chinese patientswith the same or similar mutations in the LDL receptor genein China or Canada. Arterioscler Thromb Vasc Biol 1998; 18:309–315.
43 Leitersdorf E, Eisenberg E, Eliav O et al: Genetic determinants ofresponsiveness to HMG-CoA reductase inhibitor fluvastatin inpatients with molecularly defined heterozygous familialhypercholesterolemia. Circulation 1993; 87: 35–44.
44 Sijbrands EJG, Lombardi MP, Westendorp RGJ et al: Similarresponse to simvastatin in patients heterozigous for familialhypercholesterolemia with mRNA negative and mRNA positivemutations. Atherosclerosis 1998; 136: 247–254.
45 Mamo JCL, Smith D, Yu KC et al: Accumulation of chylomicronremnants in homozygous subjects with familialhypercholesterolemia. Eur J Clin Invest 1998; 28: 379–384.
46 Castro MC, De Bruin TWA, Westerveld HE, Meijer E, ErkelensDW: Delayed chylomicron remnants clearance in subjects withheterozygous familial hypercholesterolemia. J Internal Med 1998;244: 299–307.
47 Marz W, Ruzicka V, Phol T, Usadel KH, Gross W: Familial defectiveapolipoprotein B-100: mild hypercholesterolemia withoutatherosclerosis in a homozygous patient. Lancet 1992; 340: 1362.
48 Gallagher JJ, Myant NB: Variable expression of the mutation infamilial defective apolipoprotein B-100. Arterioscler Thromb 1993;13: 973–976.
49 Schaefer JR, Scharngl H, Baumstark MW et al: Homozygousfamilial defective apolipoprotein B 100. Enhaced removal ofapolipoprotein E containing VLDLs and decreased production ofLDLs. Arterioscler Thromb Vasc Biol 1997; 17: 348–353.
50 Jansen ACM, van Wissen S, Defesche JC, Kastelein JJP:Phenotypic variability in familial hypercholesterolemia: anupdate. Curr Opin Lipidol 2002; 13: 165–171.
Genotype–phenotype in FHJT Real et al
965
European Journal of Human Genetics


14
INTRODUCCIÓN
El defecto familiar de unión de apo B 100 (DFB) se heredade forma autosómica dominante y se caracteriza por la presen-cia de xantomas tendinosos, concentraciones plasmáticas ele-vadas de colesterol total (CT) y colesterol de la lipoproteínade baja densidad (LDL), y cardiopatía isquémica (CI) precoz(1,2). Cuatro mutaciones localizadas en el gen de la apo B
(R3500Q, R3500W, R3531C y R3480P) son las responsablesdel DFB. La más prevalente de ellas es la mutación R3500Q(2-5).
La prevalencia estimada del DFB en nuestro país es muybaja, 2,8 x 10-5 para población general y aproximadamentedel 1% en sujetos con diagnóstico clínico de HF, siendo máselevada en poblaciones centroeuropeas y norteamericanas(2,5-8). Nuestro grupo encontró y caracterizó en 1999 la pri-
[0212-7199 (2004) 21: 7; pp 322-325]ANALES DE MEDICINA INTERNA
Copyright © 2004 ARAN EDICIONES, S.L.
AN. MED. INTERNA (Madrid)Vol. 21, N.º 7, pp. 322-325, 2004
Trabajo aceptado: 11 de marzo de 2004
Correspondencia: Rafael Carmena, Departamento de Medicina. Hospital Clínico Universitario. Avda. Blasco Ibáñez, 17, E. 46010 Valencia. e-mail: [email protected]
Este trabajo ha sido financiado por el Fondo de Investigaciones Sanitarias proyecto FIS 01/0056-02, Generalitat Valenciana proyecto GV01-46 y Red de Centrosde Metabolismo y Nutrición del Instituto Carlos III (C 03/08).
Ejarque I, Real JT, Ascaso JF, Chaves FJ, Milian E, Priego MA, Carmena R. Estudio de los valores plasmáticos de Lp(a) en el defecto fami-liar de unión de la apo B 100 en una población mediterránea del sur de Europa. An Med Interna (Madrid) 2004; 21: 322-325.
RESUMENObjetivo: Estudiar los valores plasmáticos de Lp(a) en el defecto
familiar de unión de apo B 100 (DFB) en poblaciones sur Europea dondeno existen datos al respecto.
Métodos: Hemos estudiado a 19 heterocigotos DFB (8 varones), por-tadores de la mutación R3500Q del gen de la apo B y a 90 controles (34varones). El diagnóstico genético se realizó por técnica de PCR-SSCP ysecuenciación automática. En todos los sujetos se determinó de formaestandarizada las concentraciones plasmáticas de lípidos, apolipoproteí-na B y Lp(a).
Resultados: Los valores plasmáticos de Lp(a) y su transformaciónlogarítmica fueron significativamente mayores en el grupo con DFBfrente al grupo control. Además, la prevalencia de Lp(a) > de 30 mg/dl,como punto de corte de alto riesgo para cardiopatía isquemia fue signifi-cativamente mayor en el grupo portador de la mutación R3500Q.
Conclusión: Los sujetos portadores de la mutación R3500Q del gende apo B mostraron valores plasmáticos superiores de Lp(a) que los con-troles, sin que conozcamos por el momento el mecanismo y sus implica-ciones clínicas.
PALABRAS CLAVE: Defecto familiar de unión de apo B 100. Lipopro-teína A. Fenotipo lipoproteico.
ABSTRACTAims: 1) to study lipoprotein (a) (Lp(a)) plasma values in subjects
with familial ligand-defective apo B 100 (FDB) .Methods: We studied 19 heterozygous FDB subjects (8 males) from
12 families, carriers of R3500Q mutation on apo B gene and 90 controls(34 males). The genetic diagnosis was established with PCR-SSCPanalysis and automatic sequencing. In all subjects plasma lipids, apoli-poprotein B and Lp(a) levels were determined with standard procedures.
Results: Subjects carriers of R3500Q mutation on apo B gene havesignificantly higher plasma Lp(a) and log transformed Lp(a) values andprevalence of Lp(a) > 30 cut point for coronary heart disease than con-trols.
Conclusions: Subjects with FDB showed higher Lp(a) plasma valuesthan controls, although the mechanism and the clinical consequences ofthese result are not known.
KEY WORDS: Familial ligand defective apo B. Lipoprotein A,Lipid andlipoproteins.
Estudio de los valores plasmáticos de Lp(a) en eldefecto familiar de unión de la apo B 100 en unapoblación mediterránea del sur de Europa
I. EJARQUE, J. T. REAL, J. F. ASCASO, F. J. CHAVES, E. MILIAN, M. A. PRIEGO,R, CARMENA
Servicio de Endocrinología y Nutrición. Hospital Clínico Universitario. Departamento deMedicina. Universidad de Valencia
PLASMA LIPOPROTEIN A VALUES IN FAMILIAL LIGAND DEFEC-TIVE APO B 100 IN A SOUTH EUROPEAN POPULATION

17
mera familia española afectada de DFB (6) y el primer homo-cigoto (8). El despistaje de portadores de la mutación R3500Qen la zona donde se encontró la primera familia ha permitidodetectar un mayor número de casos y conocer las característi-cas clínico – biológicas de la enfermedad en una zona del surde Europa donde existen pocos datos sobre la misma.
La lipoproteína a (Lp(a)) es una lipoproteína LDL modifi-cada, al unirse una apoproteína (a) por puentes disulfuro conla apoproteína B de la LDL (9). En múltiples estudios epide-miológicos, se ha demostrado que las elevaciones plasmáticasde la Lp(a) son un factor de riesgo cardiovascular (10). LaLp(a) es en parte aclarada del plasma por el receptor LDL (9).En sujetos con DFB se ha demostrado que la Lp(a) contieneapo B defectuosa, lo que podría contribuir a la elevación plas-mática de esta partícula (11), al disminuir su aclaramientoplasmático por la falta de unión de la apo B defectuosa con elreceptor de LDL.
Nuestro objetivo ha sido estudiar los valores plasmáticosde Lp(a) en el DFB en una población del sur de Europea don-de no existen datos al respecto.
MATERIAL Y MÉTODO
SUJETOS
Hemos estudiado 19 heterocigotos DFB (8 varones) porta-dores de la mutación R3500Q del gen de la apo B 100 y 90controles (34 varones). Los 19 heterocigotos DFB pertenecena 12 familias relacionadas con la primera familia afectada porDFB descrita en nuestro país (7). Los 90 controles procedende una selección aleatoria de sujetos sanos, sin antecedentespersonales ni familiares de enfermedad coronaria, ni dislipe-mia. Estos sujetos sanos proceden de trabajadores, estudian-tes, donantes de plasma y sangre del Banco de Sangre denuestro Centro previamente seleccionados y reclutados pornuestro grupo de investigación.
Todos los participantes son caucasianos y residen en laComunidad Autónoma de Valencia. El comité de ética denuestro Centro aprobó el protocolo de estudio y todos lossujetos dieron su consentimiento por escrito, para participaren el mismo.
En todos ellos se realizó una historia clínica y exploraciónfísica completas. El índice de masa corporal (IMC) se calculódividiendo el peso entre la talla al cuadrado (kg/m2). La car-diopatía isquémica se definió por una historia clínica docu-mentada de infarto agudo de miocardio (clínica, elevación deCPK-MB y ECG patológico) o presencia de angor pectoriscon test de esfuerzo y coronariografía patológicas.
MÉTODOS DE LABORATORIO
Medición de lípidos y apo B
Tras 12-14 horas de ayuno y en situación basal se recogióuna muestra de sangre de la vena antecubital en tubos conEDTA (sistema Vacutainer) siendo centrifugadas en menos de4 horas. Con situación basal nos referimos a la suspensión defármacos hipolipemiantes 6 semanas previas a la toma demuestras para el estudio.
El colesterol y los triglicéridos (TG) fueron medidos por
técnicas enzimáticas (12). El colesterol HDL (cHDL) sedeterminó tras precipitación de lipoproteínas con apo B conpolianiones (13) el colesterol VLDL (cVLDL) tras separaciónde VLDL (d < 1.006 g/mL) por ultracentrifugación (14). Elcolesterol LDL (cLDL) se calculó por diferencia. Los nivelesplasmáticos totales de apo B se midieron por inmunoturbime-tría (15). La Lp(a) se midió por ELISA con un kit comercial(Macra Lp(a) SDI strategic diagnosis).
Los coeficientes de variación para los lípidos y lipoproteí-nas de nuestro laboratorio son < 5%.
MÉTODOS GENÉTICOS
La extracción de ADN se realizó con un método estandari-zado (16). El diagnóstico genético de DFB se basó en análisisde PCR-SSCP para despistaje de la presencia de mutacioneslocalizadas en el gen de apo B responsables del DFB(R3480P, R3500Q, R3500W y R3531C). Posteriormente sellevó a cabo la secuenciación. El protocolo escalonado dediagnóstico molecular para hipercolesterolemias primariasque se realiza en nuestro centro se detalla en las referencias 17y 18.
ANÁLISIS ESTADÍSTICO
Los datos fueron analizados utilizando el “Statistical Pac-kage for the Social Sciences (SPSS versión 9 para Windows)”y se expresan como medias, mediana y desviación estándar.Las medias de las variables cuantitativas se compararon utili-zando el test de Mann Whitney. Las proporciones se compara-ron utilizando tablas de contingencia y Chi cuadrado (χ2) o eltest de Fischer (n < 5).
Utilizamos el análisis de correlación de Pearson para ana-lizar las asociaciones entre edad, IMC, lípidos y Lp(a) plas-máticos.
RESULTADOS
Despistaje de la mutación R3500Q en una zona de laComunidad Valenciana
Partiendo de la primera familia diagnosticada de DFB ennuestro país y de la familia del homocigoto (6,8) se realizó undespistaje de la mutación R3500Q en todos los familiaresaccesibles relacionados.
Logramos contactar y estudiar la presencia o no de lamutación R3500Q en 51 sujetos, pertenecientes a 12 familiasrelacionadas. 19 de los 51 sujetos resultaron ser portadores dela mutación R3500Q.
CARACTERÍSTICAS BIOQUÍMICAS DE LOS SUJETOS DFB
Las características clínicas y concentraciones de lípidos yapoproteínas estudiados se presentan en la tabla I. No encon-tramos diferencias estadísticamente significativas en los pará-metros referidos entre varones y mujeres portadores de lamutación R3500Q (datos no mostrados). En el grupo comple-to, no hallamos correlaciones entre la edad o el IMC y los lípi-dos y apoproteínas.
Vol. 21, N.º 7, 2004 VALORES DE Lp(A) EN EL DEFECTO FAMILIAR DE UNIÓN DE APO B 100 EN POBLACION MEDITERRANEA 323

Los portadores de la mutación R3500Q presentaron deforma estadísticamente significativa mayores concentracionesplasmáticas de CT, cLDL y apo B comparados con el grupocontrol (Tabla I). Estas diferencias se mantuvieron al realizarla comparación entre géneros.
No hallamos diferencias estadísticamente significativas enel IMC, concentraciones plasmáticas de TG o cVLDL entrecontroles y portadores de la mutación R3500Q.
Al compararlos con el grupo control, los valores plasmáti-cos de Lp(a) y su transformación logarítmica fueron significa-tivamente mayores en el grupo con DFB (Tabla II). Además,la prevalencia de Lp(a) > de 30 mg/dl, como punto de corte dealto riesgo para cardiopatía isquemia fue significativamentemayor en el grupo portador de la mutación R3500Q (28%frente a 8% respectivamente).
DISCUSIÓN
En nuestro estudio queda demostrado que los sujetos por-tadores de la mutación R3500Q del gen de la apo B presentan
una elevación significativa de los valores plasmáticos de laLp(a). Estudios anteriores, realizados en población holandesay del Reino Unido, también observaron en los sujetos DFBuna elevación plasmática de la Lp(a), comparado con losvalores presentados por sus familiares no afectados. En elestudio de Perombelon y cols. (11) los sujetos portadores de lamutación R3500Q presentaban mayores valores de Lp(a)plasmática y menor tamaño de apo a, aunque existía una granvariabilidad en el fenotipo de apo a. En 5 sujetos DFB aparea-dos por tamaño de apo a con sus familiares de primer grado noafectados también se demostró una mayor elevación plasmáti-ca de la Lp(a).
En el estudio de van der Hoek y cols. (19), la mediana dela Lp(a) plasmática no fue diferente entre los 85 sujetos DFBy sus 106 familiares DFB no afectados, sin embargo, al anali-zar parejas de hermanos sí se encontraron diferencias signifi-cativas, siendo los valores plasmáticos de la Lp(a) mayores enlos portadores de la mutación R3500Q.
La elevación plasmática de la Lp(a) en sujetos DFB hasido relacionada con la disminución del aclaramiento de laspartículas de Lp(a) portadoras de la apoB defectuosa.
Por otra parte, diferentes estudios han demostrado que laLp(a) plasmática también se encuentra elevada en sujetos conhipercolesterolemia familiar (HF) (20,21). Además, esta ele-vación ha sido relacionada con el papel que juega el receptorde LDL en el aclaramiento de la Lp(a) plasmática (9). Así, lossujetos HF con diferentes defectos en el receptor de LDL,deberían presentar un aclaramiento alterado de la Lp(a) plas-mática, lo que justificaría la consiguiente elevación plasmáti-ca de la partícula. De forma contraria a esta hipótesis, un estu-dio en homocigotos HF con nula actividad del receptor deLDL mostraba que los valores plasmáticos de Lp(a) no depen-dían del aclaramiento de dicha partícula por el receptor deLDL (22). El mismo estudio mostraba también que los valoresplasmáticos de Lp(a) dependían más de la síntesis de Lp(a)que de su catabolismo (22). Por otro lado, si bien el aclara-miento de la Lp(a) es posible por vía del receptor de LDL,este aclaramiento es mínimo por la gran afinidad de la partícu-la LDL por el mismo, que compite con la partícula de Lp(a)(23). Además, estudios de intervención con estatinas queinducen la sobreexpresión del receptor de LDL no demuestrandisminuciones de la Lp(a) (24). Finalmente, en estudios deheterocigotos HF genéticamente caracterizados y de sus fami-liares de primer grado las elevaciones plasmáticas de la Lp(a)no se relacionaban con el tipo de mutación del receptor deLDL ni con la condición de HF (25,26).
Por tanto, la hipótesis de que la interacción de la apo Bdefectuosa que portan las partículas de Lp(a) de los sujetoscon DFB es la causa de las elevaciones plasmáticas de Lp(a)encontradas en diferentes poblaciones estudiadas, al igual queen la nuestra, es probablemente falsa. De hecho, en el estudiode Perombelon y cols. (11), se demostró que las partículas deLp(a) contenían una menor proporción de apo B defectuosaque las LDL de los sujetos DFB, indicando que la contribu-ción de la interacción de la apoB defectuosa con el receptor deLDL en la elevación plasmática de la Lp(a) era pequeña y nojustificaba la elevación plasmática de la Lp(a) en estos suje-tos. Por otro lado y en este sentido, estudios in vivo del meta-bolismo de la Lp(a) demuestran que la fracción catabólica dela apo B y apo a contenidas en la Lp(a) son similares pero un50% menores a la fracción catabólica observada para la LDL(9).
324 I. EJARQUE ET AL. AN. MED. INTERNA (Madrid)
18
CARACTERÍSTICAS GENERALES, LÍPIDOS Y APOPROTEÍNA BPLASMÁTICOS DE LOS SUJETOS ESTUDIADOS
DFB Controles(n=19) (n=90)
Edad (años) 45,36 (15,93) 42,31 (14,25)Varón/mujer 8/11 34/56IMC (kg/m2) 27,47 (4,65) 26,08 (4,01)CT (mmol/l) 7,4 (1,49) 5,12 (0,82)*TG (mmol/l) 1,28 (0,51) 1,22 (0,51)cLDL (mmol/l) 5,69 (1,47) 3,30 (0,72*)Apo B (g/l) 1,49 (0,32) 0,92 (0,25)** p < 0,01 DFB frente a controlesMedia (desviación estándar). IMC: índice de masa corporal. CT: coleste-rol total. TG: triglicéridos. cLDL: colesterol LDL. apo B: apolipoproteínaB.
TABLA I
VALORES PLASMÁTICOS DE LP(A) EN CONTROLES YSUJETOS CON DEFECTO FAMILIAR DE UNIÓN DE APO B 100
DFB ControlesN = 19 N = 90
Lp(a) mg/dlMedia 32,94* 12,8DE 27,67 14,22Mediana 21,25* 7,25
Log Lp(a)Media 1,32* 0,85DE 0,48 0,51*p < 0,05DFB: defecto familiar de unión de apo B. Lp(a): lipoproteína A. DE: des-viación estándar.
TABLA II

19
Vol. 21, N.º 7, 2004 VALORES DE Lp(A) EN EL DEFECTO FAMILIAR DE UNIÓN DE APO B 100 EN POBLACION MEDITERRANEA 325
Es pues necesario realizar un mayor número de estudios invivo del metabolismo de la Lp(a) en sujetos DFB para poderconocer la causa de la elevación de esta partícula en estossujetos.
Si bien la Lp(a) plasmática se encuentra elevada enpacientes afectados del DFB, no se han realizado estudiospara conocer el impacto de esta elevación en el riesgo cardio-vascular de estos sujetos, teniendo en cuenta que en otraspoblaciones la Lp(a) es un reconocido factor de riesgo cardio-vascular (10).
La mayor limitación de nuestro estudio, es el bajo númerode sujetos con DFB incluidos, lo que implica un elevado error
beta y la relativa homogeneidad molecular por el vínculofamiliar existente entre los sujetos con DFB. Esta relativahomogeneidad podría influir en la expresión del fenotipo lipo-proteico.
En conclusión, la Lp(a) plasmática se encuentra elevadaen sujetos con DFB. Es necesario realizar estudios del meta-bolismo de la Lp(a) en sujetos portadores de la mutaciónR3500Q para aclarar el papel que juega la apoB defectuosa yel receptor de LDL en el aclaramiento de la partícula Lp(a).Además, también es necesario aclarar si esta elevación de laLp(a) plasmática es un factor de riesgo cardiovascular adicio-nal en los sujetos con DFB.
1. Myant NB. Familial defective apolipoprotein B-100: a review, inclu-ding some comparison with familial hypercholesterolemia. Atheroscle-rosis 1993; 104: 1-18.
2. Rauh G, Séller C, Schuster H, Wolfgram G, Zöllnner N. Familial defec-tive apolipoprotien B-100: a common cause of primary hyperlipemia.Clin Investigator 1995; 95: 1225-1234.
3. Pullinger CR, Hennessy LK, Chatterton JE, et al. Familial ligand-defec-tive apolipoprotein B: identification of a new mutation that decreasesLDL receptor binding affinity. J Clin Invest 1995; 95: 1225-1234.
4. Gaffney D, Reid JM, Cameron IM, et al. Independent mutations atcodon 3500 of the apolipoprotein B gene are associated with hyperlipi-daemia. Arterioscle Thromb Vasc Biol 1995; 15: 1025-1029.
5. Miserez AR, Keller U. Differences in the phenotypic characteristics ofsubjects with familial defective apolipoprotein B-100 and familialhypercholesterolemia. ATVB 1995; 15: 1719-1729.
6. Real JT, Chaves FJ, Ascaso JF, Armengod ME, Carmena R. Estudio deldefecto familiar de la apo B-100 en sujetos con el diagnóstico clínico dehipercolesterolemia primaria: identificación de la primera familia afec-tada en España. Med Clin (Barc) 1999; 113: 15-17.
7. Castillo S, Tejedor D, Mozas P, Reyes G, Civeira F, Alonso R, Ros E,Pocovi M, Mata P. Atherosclerosis 2002; 165: 127-135.
8. Real JT, Chaves JF, Martín de Llano JJ, et al. Identificación y caracteri-zación del primer homocigoto español con defecto familiar de unión dela apolipoproteína B. Med Clin (Barc) 2001; 116: 138-141.
9. Demant T, Seeberg K, Bedynek A, Seidel D. The metabolism of lipo-protein(a) and other apolipoprotein B-containing lipoproteins: a kineticstudy in humans. Atherosclerosis 2001; 157: 325-39.
10. Pedro- Botet J, Rubies-Prat J. Lipoproteína (a) y aterotrombosis. ClinInvest Arterioscler 2003; 15: 258-260.
11. Perombelon YF, Gallagher JJ, Myant NB, Soutar AK, Knight BL. Lipo-protein(a) in subjects with familial defective apolipoprotein B100.Atherosclerosis 1992; 92: 203-12.
12. Allain CC, Poon LS, Chan CSG, Richmond W, Fu PC. Enzymaticdetermination of total serum cholesterol. Clin Chem 1974; 20: 470-5.
13. Burstein M, Scholnick HR, Morfin R. Rapid method for the isolation oflipoproteins from human serum by precipitation with polyanions. JLipid Res 1970; 11: 583-95.
14. Havel RJ, Eder HJ, Bragdon JH. The distribution and chemical compo-sition of centrifugally separated lipoproteins in human serum. Eur J ClinInvest 1995; 34: 1345-54.
15. Rosseneu M, Vercaemst R, Steinberg KK, Cooper GR. Some considera-
tions of methodology and standarization of apolipoprotein B immunoas-says. Clin Chem 1983; 29: 427-33.
16. Tilzer L, Thomas S, Moreno RF. Use of silica gel polymer for DNAextraction with organic solvents. Anal Biochem 1989; 183: 13-15.
17. García-García AB, Real JT, Puig O, et al. Molecular genetics of familialhipercolesterolemia in Spain: ten novel LDLR mutations and populationanalysis. Hum Mutat 2001; 454: 1-9.
18. Chaves FJ, Real JT, García-García AB, et al. Genetic diagnosis of fami-lial hypercholesterolemia in a south European outbreed population:influence of low density lipoprotein (LDL) receptor gene mutations ontreatment response to simvastatin in total, LDL and high density lipo-protein cholesterol. J Clin Endocrinol Metab 2001; 86: 4926-4932.
19. van der Hoek YY, Lingenhel A, Kraft HG, Defesche JC, Kastelein JJ,Utermann G. Sib-pair analysis detects elevated Lp(a) levels and largevariation of Lp(a) concentration in subjects with familial defectiveApoB. J Clin Invest 1997; 99: 2269-73
20. Seed M, Hopplicher F, Reaveley D, Mc Carthy S, Thompson G, Boer-winkle E, Uterman G. relation of serum lipoprotein (a) concentrationand apolipoprotein a phenotype to coronary heart disease in patientswith familial hypercholesterolemia. N Engl J Med 1990; 322: 1494-1499.
21. Kraft HG, Lingenhel A, Raal FJ, Hohenegger M, Utermann G. Lipopro-tein(a) in homozygous familial hypercholesterolemia. ArteriosclerThromb Vasc Biol 2000; 20: 522-8.
22. Rader DJ, Mann WA, Cain W, Kraft HG, Usher D, Zech La, et al. Thelow density lipoprotein receptor is not required for normal catabolism ofLp(a) in humans. J Clin Invest 1995; 95: 1403-1408.
23. Armstrong VW, Walli AK, Seidel D. Isolation characterization anduptake in human fibroblasts of an apo(a) free lipoprotein obtained onreduction of lipoprotein(a). J Lipid Res 1985; 26: 1314-1318.
24. Thiery J, Armstrong VW, Schleef J, Creutzfeld C, Creutzfeld W, SeidelD. Serum lipoprotein Lp(a) concentrations are not influence by anHMG CoA reductase inhibitor. Klin Wochenschr 1988; 66: 462-466.
25. Carmena R, Lussier-Cacan S, Roy M, Minnich A, Lingenhel A, Kro-nenberg F, Davignon J. Lp(a) levels and atherosclerotic vascular diseasein a sample of patients with familial hypercholesterolemia sharing thesame gene defect. Arterioscler Thromb Vasc Biol 1996; 16: 129-36.
26. Real JT, Ascaso JF, Chaves FJ, Tenes S, Priego MA, Puig O, ArmengodME, Carmena R. Plasma Lp(a) values in familial hypercholesterolemiaand its relation to coronary heart disease. Nutr Metab Cardiovasc Dis1999; 941-4.
Bibliografía




57
A comparison of lipoprotein phenotype between LDL receptor gene mutations
affecting ligand-binding domain and the R3500Q mutation of the apo B gene in
patients from a South European population.
Ismael Ejarque, José T. Real, Felipe J. Chaves, Ana B. García-García, Enrique Millan*,
Juan F. Ascaso, Rafael Carmena.
Service of Endocrinology and Nutrition, Hospital Clínico Universitario, Department of
Medicine, University of Valencia, Spain.
* Physician La Yesa, Valencia, Spain.
Address for reprints:
Prof. Rafael Carmena, Department of Medicine, University of Valencia, Hospital Clínico
Universitario, Avda. Blasco Ibáñez 17, E-46010 Valencia, Spain (telephone number
+34-96-3862665; fax +34-96-3864767; E-mail: [email protected]).
Running title: Genetic hypercholesterolemias

58
Abstract
Aims: (1) to examine the sensitivity of clinical diagnosis criteria for familial
defective apo B 100 (FDB); (2) to compare the lipoprotein phenotype between carriers
of LDL receptor (LDLR) gene mutations affecting ligand-binding domain and subjects
with the R3500Q mutation in apo B gene, from a South European population where no
data are still available.
Subjects and Methods: 213 subjects (113 probands) with familial
hypercholesterolemia (FH) and 19 heterozygous FDB subjects were studied. Genetic
diagnosis was carried out following a protocol based on Southern blot and PCR-SSCP
analysis. 42 FH subjects carried LDLR gene missense mutations affecting ligand-
binding domain. Thirty of them were matched by age, gender, and body mass index to
19 heterozygous FDB subjects carriers of the R3500Q mutation. Lipoprotein phenotype
comparison was carried out between the two groups.
Results: FH patients carrying missense mutations affecting the ligand-binding
domain showed plasma total and LDL cholesterol levels significantly higher than FDB
patients. Three out of the 19 heterozygous FDB subjects showed plasma total and LDL
cholesterol values in the normal range. Using the 1999 clinical Med-Ped criteria for
diagnosis of genetic hypercholesterolemia, none of the FDB subjects had a confirmed
diagnosis; it was probable in 36%, possible in 32% and could be excluded in the
remaining 32%.
Conclusions: The type IIa lipoprotein phenotype in FDB subjects was
significantly less severe than that observed in FH carriers of LDLR gene missense
ligand-binding domain mutations. Clinical Med-Ped diagnosis criteria for genetic
hypercholesterolemia under-diagnose FDB subjects.
Key words: heterozygous familial hypercholesterolemia, genetic diagnosis, LDL
receptor gene ligand-binding mutations, lipoprotein phenotype, familial defective apo
B100, R3500Q mutation.

59

60
Introduction
Familial hypercholesterolemia (FH) is an autosomal dominant disease defined
at the molecular level by the presence of mutations in the LDL receptor (LDLR) gene
and characterized by markedly elevated plasma low density lipoprotein cholesterol
(LDLc) levels, tendon xanthomata and increased risk of coronary heart disease (CHD)
(1).
The type of LDLR gene mutation has been associated to different phenotype
expressions (2,3). The ligand-binding domain of LDLR gene is included in exons 2-6
and is an important functional domain that interacts with the apo B ligand binding
domain (1).
Familial defective apolipoprotein B-100 (FDB) is an autosomal dominant
inherited disease due to mutations in the apo B gene affecting the ligand binding
domain of apo B to LDLR (4). It is characterised by elevated plasma total and LDL
cholesterol levels and premature CHD. Four mutations, R3480P, R3500Q, R3500W
and R3531C are responsible for the disease by reducing the binding of LDL particles to
the LDL receptor (4-6). The R3500Q mutation was the first described and is the most
prevalent. The prevalence of FDB has been estimated to be approximately 1/500 in
North America, whereas in Europe it seems to be higher in the North-western part of
Switzerland (1/114), Eastern France, and Southern Germany (7) and lower in Italy and
Spain (8).
FDB is clinically indistinguible from FH. Therefore, Med-Ped criteria (9) for
diagnosing FH have been used to clinically diagnose FDB. However, in North
European populations, the plasma LDLc levels in age-matched FDB heterozygotes
vary over a wide range and, in some families, heterozygous carriers of the R3500Q
mutation show normal plasma LDLc levels (7).
We have recently described the first affected FDB family in Spain carrying the
R3500Q mutation and predicted a low prevalence of this disease, since only one FDB
out of 113 FH probands was detected (8). On the contrary, and due to a founder effect

61
in an isolated region of the Valencia province (Eastern Spain), we have now identified a
total of 19 FDB heterozygotes and 1 homozygote (10). This has allowed us to compare
the lipoprotein phenotype of this group of patients with that of FH subjects in a South
European population, where no study on this issue has yet been done.
Our working hypothesis was that FDB carriers of R3500Q mutation in apo B
gene, which affects the ligand binding domain region of apo B, and FH subjects
carriers of missense mutations in LDLR gene, that affect the ligand-binding domain
region, should express similar lipoprotein phenotype.
The aims of the study were: to analyze and compare the lipoprotein phenotype
between FH carriers of LDLR gene missense mutations affecting ligand-binding
domain and FDB subjects carriers of the R3500Q mutation on apo B gene in a South
European population. In addition, the sensitivity of the MED-Ped clinical criteria (9) for
diagnosis of genetic hypercholesterolemia has been evaluated in the FDB group.
Subjects and Methods
Subjects
The study population consisted of 213 FH subjects (113 probands and 100
affected relatives) that had been referred to our Lipid Clinic. All subjects were
Caucasian and lived in the Valencia region. The ethics committee from our institution
approved the protocol and all subjects gave written informed consent to enter the
study.
Clinical diagnostic criteria for FH or genetic hypercholesterolemia, according to
Med-Ped, 1999 (9), included: plasma levels of total and LDL cholesterol above the 95th
percentile corrected for both age and sex, presence of tendon xanthomata or arcus
cornealis in subjects aged below 45 years, CHD in the proband or in a first-degree
relative, and bimodal distribution of plasma total and LDL cholesterol levels in the
family, indicating an autosomal dominant pattern of lipoprotein phenotype IIa.

62
A complete medical history and physical examination were obtained in all
participants. Body mass index (BMI) was calculated as weight divided by height
squared (kg/m2). CHD was diagnosed if there was a documented history of previous
myocardial infarction and/or coronary artery bypass surgery or angioplasty.
Laboratory Methods
Measurement of Lipids and lipoproteins
After a 12 to 14 hours fast, blood samples were drawn in tubes containing
EDTA and were centrifuged within 4 hours. Plasma was stored at 4 C for a maximum
of 3 days. Plasma cholesterol and triglyceride levels were measured by standard
enzymatic techniques (11, 12). High density lipoprotein cholesterol (HDLc) was
measured after precipitation of apoB-containing lipoproteins with polyanions (13) and
very low density lipoprotein cholesterol (VLDLc) after separation of VLDL (d < 1.006
g/mL) by ultracentrifugation (14). The LDLc was calculated by subtraction of VLDL and
HDL cholesterol from total cholesterol. Total plasma apoB was measured by
immunoturbimetry (15).
Genetic methods
DNA extraction was performed by a standard procedure (16). Genetic diagnosis
of FH was previously established through identification of mutations of LDL receptor
gene by Southern blot analysis (17) and DNA sequencing of abnormal SSCP bands
(18).
Screening for the apoB mutations was performed by PCR amplification, as
described by Schuster et al (19), and SSCP analysis. DNA samples carrying the
mutations R3480P, R3500Q, R3500W and R3531C, responsible for FDB, were used
as controls.
Genotyping for the ApoE polymorphisms was performed following the method
described by Hixon and Vernier (20) with minor modifications.

63
Statistical Analysis
Data were analyzed with the Statistical Package for the Social Sciences (SPSS
6.1.3 for Windows) and expressed as mean standard deviation (SD). Mean values of
quantitative variables were compared with t-Student. Proportions were compared with
contingency tables and the chi-square ( 2) test or the Fischer’s exact test (n< 5).
Results
Genetic diagnosis of FDB and FH.
The genetic diagnosis of FDB and FH was previously established using a three-
step protocol that screened mutations at the apoB gene, responsible for FDB, and
major rearrangements and minor or point mutations in the LDLR gene, responsible for
FH (17,18) . Only one R3500Q mutation carrier was identified among 113 FH probands
(8). The screening study of R3500Q mutation was then extended to 51 subjects form
seven pedigrees related to the affected FDB family. A total of 19 heterozygotes for the
R3500Q mutation and 1 homozygote subject were identified. The general
characteristics, lipid and lipoprotein plasma values and the 95 and 85 percentiles of
plasma total and LDL cholesterol levels for our population (21) are shown in table 1.
The percentiles were obtained from a random sample of 1063 subjects (475 men and
587 women, age 18-68 years) from our population (21).
SSCP analysis of the LDLR gene was used to screen for point and minor
mutations of the LDLR gene. The methods and procedures have been previously
reported (18).
75 FH subjects from a total of 213 FH genetically screened were carriers of
point and minor mutations affecting exon 2-6 (ligand-binding domain of LDLR gene). 42
FH subjects out of the 75, carried missense mutations in exons 2-6 from the LDLR
gene, which predictably affect the ligand-binding domain region. The mutations
included in the study are: C68W n= 5, Q71E n= 1, C74G n= 3, E80K n=3, C95R n=8,
E158K n= 4, S156L n=2, P160R n=5, D200G n=1, S205P n=1, E246A n= 4, E256K

64
n=4 and I289T n= 1. The remaining 33 FH subjects carried point and minor mutations
causing stop codons in exons 2-6 (111insA n=18, Q133X n=3, 790delATG n=4, W-18X
n=1, 884delT n=4) or splicing mutations (313+1G, n=3), and were excluded from the
study.
Lipoprotein phenotype in FDB and FH carriers of ligand-binding mutations in LDLR
gene
No significant statistical differences were found between non affected and
affected FDB subjects from the seven studied pedigrees in age, BMI, and gender
distribution (table 2). As expected, we found significant differences in total, LDLc and
apo B plasma values and prevalence of CHD between carriers and non carriers of
R3500Q mutation. 3 out of the 19 heterozygous FDB patients presented
normolipidemic values (table 1). These 3 subjects are members of different pedigrees.
The 95 and 85 percentiles of plasma total and LDL cholesterol values from a
randomly selected sample representing the general population in our Community are
shown in table 1 (20). 3 FDB subjects (15%) have total cholesterol and LDLc plasma
values below the 85 percentile. 15 FDB subjects presented a total cholesterol plasma
value above the 95 th and in 14 FDB subjects the plasma LDLc value was above the
95th. None of the 19 heterozygous FDB subjects had tendon xanthomata and 1
showed CHD.
Therefore, using the Med-Ped clinical criteria (9) for diagnosing FH or genetic
hypercholesterolemia, none of the genetically diagnosed FDB subjects had a confirmed
genetic hypercholesterolemia; 7 (36%) FBD were “probable”, 6 (32%) were “possible”,
while 6 (32%) did not meet such diagnostic criteria.
30 FH subjects carriers of missense mutation affecting the ligand-binding
domain of LDLR gene were matched by age, gender distribution and BMI to the 19
heterozygous FBD, in order to compare their lipoprotein phenotype (table 3). When the
lipoprotein phenotype of FH carriers of missense mutations was compared with that of

65
FDB subjects, significant differences were found in total cholesterol and LDLc plasma
values, with no differences in triglycerides, VLDLc, apoB and HDLc plasma levels
(table 3). In addition, the prevalence of tendon xanthomata was statistical significantly
higher in the FH group.
No significant differences were found in apoE genotype distribution between the
two groups.
Discussion
This is the first study carried out in a South European population with the aim of
comparing the lipoprotein phenotype of heterozygous FDB carriers of mutation
R3500Q with that of FH subjects carriers of ligand-binding domain LDLR gene
mutations. We have observed that FDB subjects have significantly lower total and LDL
plasma cholesterol values than FH of the same age, BMI and gender distribution.
Our results are in agreement with a previous study from a Northern European
FDB and molecularly characterized FH population. The study conducted by Graham et
al (22) indicated that families with LDLR gene defects frameshift (n=12); nonsense
(n=8); and missense (n=21) LDLR gene mutations had higher total and LDL plasma
cholesterol levels and higher incidence of xanthomas, than those with FDB.
Previous studies in Northern European populations (4,7) have also
demonstrated that FDB subjects have lower plasma total and LDL cholesterol values
than non molecularly characterized FH subjects. In addition, some carriers of the
R3500Q mutation did not show elevations in their plasma values of total and LDL
cholesterol.
The explanation for these findings could be attributed to the different
metabolism of apoB-100-containing particles in FDB compared with that in FH. In vivo
kinetic studies, conducted in homozygous FDB, have revealed an increase removal of
VLDL apoE, a reduction in LDL apoB-100 production and an increased LDL residence
time (23, 24). Taken together, these observations (elevated clearance of LDL

66
precursors and reduced LDL production) help to explain why homozygous FDB
subjects have plasma values of LDLc lower than predicted when compared to
homozygous FH patients.
Moreover, FDB homozygotes have later onset and less severe CHD than FH
homozygotes, and respond more effectively to treatment with statins (10, 23, 25). It is
of interest that the production rate of apoA-I in FDB is significantly higher than in
control subjects, (26) suggesting that defective apoB-100 may influence HDL kinetics.
The increase in total HDL turnover, by enhancing reverse cholesterol transport, could
contribute to the seemingly benign clinical course of FDB compared with that of FH.
Another possibilities, i.e. mutations in the normal allele at the apo B locus that
could protect from LDLc elevations were rejected by Gallagher’s findings (27) who
failed to observe association of normocholesterolemia in FDB with inheritance of a
variant normal allele at the apoB-100 locus, apoE locus or polymorphic variations on
the LDLR locus.
In our population, if the established Med-Ped criteria for diagnosing FH
are used for FDB diagnosis, we will misdiagnosed 32 % of our FDB patients. These
results possible indicate the need to screen mutations causing FDB in all subjects
suspected to have a primary hypercholesterolemia. In addition, regardless of the
lipoprotein phenotype, in all first and second degrees relatives form affected carriers. In
our study, 3 out of the 19 heterozygous carriers of the R3500Q mutation showed
normolipidemic plasma values.
Due to the relative lower number of FDB patients included in the study and the
relative genetic homogeneity of these patients, we need more studies in South
European population to confirm our results.
In conclusion, FDB subjects showed a less severe hypercholesterolemia
than FH carriers of LDLR gene missense ligand-binding domain mutations, indicating
that the metabolism of apoB-100-containing particles is different in FDB compared with
that in FH. Moreover, in our experience, the clinical Med-Ped diagnosis criteria for

67
genetic hypercholesterolemia under-diagnose FDB subjects. Our results indicate that in
South European population the clinical diagnostic criteria for FDB should be reviewed
and the need to generalise the use of genetic diagnosis of FDB.
Acknowledgements
We thank the patients and their relatives for their cooperation. This work was supported
by grants from Fondo de Investigaciones Sanitarias (FIS 01/0056-02) and Generalitat
Valenciana (GV01-46) to Jose T. Real, and Red de Centros de Metabolismo y
Nutrición from Instituto Carlos III (C 03/08).
68
Table 1. General characteristics and lipid plasma values of the 19 heterozygous
FDB and the total and LDL cholesterol 95 and 85 percentiles of our population.
Gender R3500Q
Age
(years)
TC
(mmol·l-1)
TC
95th TC 85th
LDLc
(mmol·l-1)
LDLc
95th
LDLc
85th
Male Heterozygote 60 4.65
7.02 6.38 3.15
4.96 4.39
Male Heterozygote 25 4.96
7.02 6.38 3.82
4.96 4.39
Female Heterozygote 23 5.27
6.61 5.94 3.77
4.55 3.98
Female Heterozygote 24 6.72
6.61 5.94 4.88
4.55 3.98
Female Heterozygote 27 6.85
6.61 5.94 5.06
4.55 3.98
Female Heterozygote 49 7.03
6.61 5.94 5.14
4.55 3.98
Male Heterozygote 60 7.08
7.02 6.38 4.68
4.96 4.39
Female Heterozygote 52 7.11
6.61 5.94 5.12
4.55 3.98
Male Heterozygote 18 7.29
7.02 6.38 5.53
4.96 4.39
Male Heterozygote 64 7.52
7.02 6.38 5.84
4.96 4.39
Female Heterozygote 47 8.04
6.61 5.94 6.28
4.55 3.98
Female Heterozygote 53 8.24
6.61 5.94 6.51
4.55 3.98
Male Heterozygote 56 8.66
7.02 6.38 6.69
4.96 4.39
Female Heterozygote 53 8.89
6.61 5.94 7.29
4.55 3.98
Female Heterozygote 54 9.77
6.61 5.94 7.93
4.55 3.98
Male Heterozygote 56 7.78
7.02 6.38 7.70
4.96 4.39
Male Heterozygote 57 7.49
7.02 6.38 4.75
4.96 4.39
Female Heterozygote 54 8.40
6.61 5.94 5.99
4.55 3.98
Female Heterozygote 23 7.60
6.61 5.94 5.06
4.55 3.98
Abbreviations: TC: total cholesterol, LDLc: low density lipoprotein cholesterol.

69
Table 2: General characteristics and plasma lipids and lipoprotein values in 19
carriers and 32 non carriers of R3500Q mutation from seven pedigrees.
R3500Q + (n=19) R3500- (n=32)
Age (years) 45.4 (15.9) 41.3 (17.0)
Male/female (n) 8/11 8/24
BMI (kg/m2) 25.6 (4.2) 24.9 (4.7)
CHD (n) 1 0
TC (mmol·l-1) 7.4 (1.49)** 5.1 (0.92)
TG (mmol·l-1) 1.28 (0.5) 1.08 (0.52)
HDLc (mmol·l-1) 1.23 (0.28) 1.37 (0.38)
LDLc (mmol·l-1) 5.69 (1.47)** 3.27 (0.76)
VLDLc (mmol·l-1) 0.53 (0.33) 0.53 (0.24)
ApoB (g·l-1) 1.49 (0.32)** 0.92 (0.28)
*P < 0.05
**P < 0.01
Mean (SD)
Abbreviations: BMI: body mass index, CHD: coronary heart disease, TC: total
cholesterol, TG: triglycerides, HDLc: high-density lipoprotein cholesterol, LDLc: low
density lipoprotein cholesterol, VLDLc: very low-density lipoprotein cholesterol, apoB:
apolipoprotein B.
70
Table 3: Comparison of lipoprotein phenotype between FDB and FH subjects
carrying ligand-binding domain mutations in LDLR gene with similar age, gender
distribution and BMI.
FDB
(n=19)
FH carriers of ligand-binding
domain mutations (n=30)
Age (years) 45.4 (15.9) 44.7 (19.6)
Male / female (n) 8/11 10/20
CHD (n) 1 3
BMI, kg/m2 25.6 (4.2) 25.5 (4.2)
Xanthomas (n) 0 12*
TC (mmol·l-1) 7.4 (1.49) 9.04 (1.89)*
TG (mmol·l-1) 1.28 (0.5) 1.23 (0.68)
HDLc (mmol·l-1) 1.23 (0.28) 1.40 (0.39)
LDLc (mmol·l-1) 5.69 (1.47) 7.02 (1.88)*
VLDLc (mmol·l-1) 0.53 (0.33) 0.52 (0.27)
Apo B (g·l-1) 1.49 (0.32) 1.57 (0.30)
Apo E genotype
(%)
E3 / E3
E3 /E4
E3 / E2
E2 /E4
70
12
12
6
76
15
5
4
*p < 0.05
Mean (SD)
Abbreviations: LDLR: low density lipoprotein receptor, BMI: body mass index, CHD:
coronary heart disease, TC: total cholesterol, TG: triglycerides, HDLc: high-density
lipoprotein cholesterol, LDLc: low density lipoprotein cholesterol, VLDLc: very low-
density lipoprotein cholesterol, apoB: apolipoprotein B.

71
References
1) Goldstein JL, Hobbs HH, Brown MS. Familial hypercholesterolemia. In: Scriver CR
et al (eds.) The Metabolic and Molecular Bases of Inherited Disease, 8th edition.
McGraw Hill New York; 2863-2913, 2001.
2) Hoeg JM. Homozygous familial hypercholesterolemia: a paradigm for phenotypic
variation. Am J Cardiol; 72: 11D-14D, 1993.
3) Gudnason V, Day INM, Humphries SE. Effect on plasma lipid levels of different
classes of mutations in the low-density lipoprotein receptor gene in patients with
familial hypercholesterolemia. Arterioscle Thromb; 17: 1717-1722, 1994.
4) Myant NB. Familial defective apolipoprotein B-100: a review, including some
comparison with familial hypercholesterolemia. Atherosclerosis; 104: 1-18, 1993.
5) Pullinger CR, Hennessy LK, Chatterton JE, Liu W, Love JA, Mendel CM, et al.
Familial ligand-defective apolipoprotein B: identification of a new mutation that
decreases LDL receptor binding affinity. J Clin Invest; 95: 1225-1234, 1995.
6) Gaffney D, Reid JM, Cameron IM, Vass K, Caslake MJ, Shepherd J, Packard CJ.
Independent mutations at codon 3500 of the apolipoprotein B gene are associated with
hyperlipidaemia. Arterioscle Thromb Vasc Biol; 15: 1025-1029, 1995.
7) Rauh G, Séller C, Schuster H, Wolfgram G, Zöllnner N. Familial defective
apolipoprotien B-100: a common cause of primary hyperlipemia. Clin Investigator; 95:
1225-1234, 1995.
8) Real JT, Chaves FJ, Ascaso JF, Armengod ME, Carmena R. Estudio del defecto
familiar de la apo B-100 en sujetos con el diagnóstico clínico de hipercolesterolemia
primaria: identificación de la primera familia afectada en España. Med Clin (Barna);
113: 15-17, 1999.
9) Human Genetic Program. Division of Noncomunicable Diseases. Familial
hypercholesterolemia (FH). Report of a WHO consultation. World Health Organization,
1997; 1-45.

72
10) Real JT, Chaves JF, Martín de Llano JJ, Ejarque I, García-García AB, Knecht E, et
al. Identificación y caracterización del primer homocigoto español con defecto familiar
de unión de la apolipoproteina B. Med Clin (Barna); 116: 138-141, 2001.
11) Allain CC, Poon LS, Chan CSG, Richmond W, Fu PC. Enzymatic determination of
total serum cholesterol. Clin Chem; 20: 470-475, 1974.
12) Ter Welle HF, Baartscheer T, Fiolet JWT. Influence of free glycerol on enzymatic
evaluation of triglycerides. Clin Chem; 30: 1102-1103, 1984.
13) Burstein M, Scholnick HR, Morfin R. Rapid method for the isolation of lipoproteins
from human serum by precipitation with polyanions. J Lipid Res; 11: 583-595, 1970.
14) Havel RJ, Eder HJ, Bragdon JH. The distribution and chemical composition of
centrifugally separated lipoproteins in human serum. Eur J Clin Invest; 34: 1345-1354,
1995.
15) Rosseneu M, Vercaemst R, Steinberg KK, Cooper GR. Some considerations of
methodology and standarization of apolipoprotein B immunoassays. Clin Chem;29:
427-433, 1983.
16) Tilzer L, Thomas S, Moreno RF. Use of silica gel polymer for DNA extraction with
organic solvents. Anal Biochem; 183: 13-15, 1989.
17) Chaves FJ, Real JT, García-García AB, Puig O, Ordovas JM, Ascaso JF, et al. Large
rearrangements of the LDL receptor gene and lipid profile in a Spanish population. Eur J
Clin Invest; 31: 309-317, 2001.
18) García-García AB, Real JT, Puig O, Cebolla E, Marín-García P, Martínez Ferrandis
JI, et al. Molecular genetics of familial hipercolesterolemia in Spain: ten novel LDLR
mutations and population analysis. Hum Mutat; 454: 1-9, 2001.
19) Schuster H, Rauh G, Müller S, Keller C, Wolfram G, Zöllner N. Allele specific and
asymetric Polymerase chain reaction amplification in combination: a step polymerase
chain reaction protocol for rapid diagnosis of familial defective apolipoprotein B-100.
Ann Bioch; 204: 22-25, 1992.

73
20) Hixon JE, Vernier DT. Restriction isotyping of human apolipoprotein E by gene
amplification and cleavage with HhaI. J Lipid Res; 31: 545-548, 1990.
21) Corella D, Guillen M, Portoles O, Sorli JV, Alonso V, Folch J, Saiz C. Gender
specific association of the Trp64Arg mutation in the beta3-adrenargic receptor gene
with obesesity related phenotypes in a Mediterranean population: interaction with a
common lipoprotein gene variation. J Intern Med; 250: 348-360, 2001.
22) Graham CA, McClean E, Ward AJ, Beattie ED, Martin S, O'Kane M, et al. Mutation
screening and genotype: phenotype correlation in familial hypercholesterolemia.
Atherosclerosis; 147: 309-316, 1999.
23) Schaefer JR, Scharnagl H, Baumstark MW, Schweer H, Zech LA, Seyberth H, et
al. Homozygous familial defective apolipoprotein B 100. Enhaced removal of
apolipoprotein E containing VLDLs and decreased production of LDLs. Arteriosclerosis
Thrombosis Vascular Biology; 17: 348-353, 1997.
24) Pietzsch J, Wiedemann B, Julius U, Nitzsche S, Gehrisch S, Bergmann S et al.
Increased clearance of low density lipoprotein precursors in patients with heterozygous
familial defective apolipoprotein B-100: a stable isotope approach. J Lipid Res; 37:
2074-2087, 1996.
25) März W, Ruzicka V, Phol T, Usadel KH, Gross W. Familial defective apolipoprotein
B-100: mild hypercholesterolemia without atherosclerosis in a homozygous patient.
Lancet; 340: 1362, 1992.
26) Schaefer JR, Winkler K, Schweer H, Hoffmann MM, Soufi M, Scharnagl H, Maisch
B, Wieland H, Steinmetz A, März W: Increased production of HDL ApoA-I in
homozygous familial defective apoB-100. Arterioscler Thromb Vasc Biol; 20: 1796-
1799, 2000.
27) Gallagher JJ, Myant NB. Variable expression of the mutation in familial defective
apolipoprotein B-100. Arteriosclerosis Thrombosis; 13: 973-976, 1993.

74

75
DISCUSIÓN

76

77
4. DISCUSIÓN
Los cinco trabajos que componen la presente tesis doctoral como compendio de publicaciones comparten una misma línea de investigación centrada en el estudio del Defecto Familiar de unión de Apo B-100. Inicialmente se identificaron 4 heterocigotos y un homocigoto DFB72, 136, pertenecientes a una familia con un origen geográfico común, lo que nos hizo pensar en un posible efecto fundador y en la posibilidad de detectar un mayor número de sujetos afectados. Recordemos que esta enfermedad es poco prevalente en países del sur de Europa como la costa mediterránea de Francia o en Italia donde sólo se había descrito una familia afectada. Por tanto, existían pocos datos sobre las características clínico-biológicas de la enfermedad en esta zona. La ampliación del estudio a familiares de segundo y tercer grado nos permitió detectar y aumentar los heterocigotos DFB, estudiar la prevalencia y el efecto fundador137 de la enfermedad en una zona concreta de la Comunitat Valenciana origen geográfico de todas las familias estudiadas. Al incrementar el tamaño muestral de los heterocigotos DFB pudimos conocer y definir su fenotipo lipoproteico. Posteriormente, comparamos este fenotipo137,
138 con el de una muestra de heterocigotos HF que también procedían de la Comunitat Valenciana139, y ello lo hicimos clasificando a los heterocigotos HF según los tipos de mutación del gen LDLR.
1. Estudio del homocigoto DFB.
En el primer artículo hemos identificado al primer homocigoto DFB español136.Actualmente, se desconoce la prevalencia precisa de la mutación R3500Q en homocigosis en la población española, aunque el bajo índice de detección de heterocigotos R3500Q en sujetos con diagnóstico clínico de HF apunta a que debe de ser muy baja. La aparente contradicción entre la baja prevalencia de DFB en España y la existencia de una forma homocigota DFB se debe a la gran endogamia que existe en la zona de donde procede la familia estudiada. La Yesa, el pueblo donde se detectó la primera familia diagnosticada de DFB, pertenece a la comarca de Los Serranos, comarca montañosa aislada por diversos accidentes geográficos, donde es frecuente encontrar una elevada endogamia. De hecho, los padres del homocigoto son primos hermanos.
Hemos estudiado el fenotipo lipoproteico de nuestro homocigoto y su respuesta a fármacos hipolipemiantes. Sus valores de CT y cLDL son moderados a pesar de su situación de homocigosis, lo cual no era de esperar. En teoría, cabría esperar una falta de respuesta a estatinas, al igual que ocurre en los homocigotos HF, dado que en ambas situaciones se da una alteración en la interacción entre la apolipoproteína B-100 con el receptor hepático de las LDL.
Realizando una búsqueda bibliográfica sistemática se han descrito otros 6 homocigotos DFB en diversas poblaciones. Gallagher et al140 describieron dos homocigotos, un varón de 66 años, y su hermana de 69 años, März et al141 detectó un homocigoto varón de 54 años, Funke et al142 identificaron una homocigota de 31 años, Horinek et al143
detectaron otro varón homocigoto de 43 años, y por último Gaffney et al144 también identificó otro homocigoto. Sus valores de CT fueron de 367 mg/dl, 463 mg/dl, 331 mg/dl, 299-331 mg/dl, 394 mg/dl y 398 mg/dl respectivamente. Además, en tres de los

78
seis homocigotos no se detectaron cardiopatía isquémica ni xantomas tendinosos. En todos ellos los valores de CT y LDL colesterol descendieron con estatinas.
Estos datos nos muestran que a diferencia de lo que ocurre en el caso de homocigosis por mutaciones en el receptor de LDL, los homocigotos para mutaciones de su ligando, la apo B-100, presentan un fenotipo lipoproteico moderado y pocas manifestaciones clínicas de la enfermedad (xantomas, cardiopatía isquémica). Como ocurre en el paciente que presentamos, los homocigotos DFB hasta ahora descritos presentan frecuentemente enfermedad coronaria y cifras elevadas de CT y cLDL pero sólo de forma moderada, diferentes de las esperadas. En teoría, un homocigoto con DFB que presente en sus dos alelos la mutación R3500Q, que reduce en un 90 % la unión de la partícula LDL a su receptor, debería tener concentraciones de CT y cLDL del rango que presentan los homocigotos con HF.
Se han realizado diferentes estudios “in vivo” para tratar de explicar este moderado fenotipo lipoproteico. Gallagher y Myant140 han propuesto que en los homocigotos DFB la actividad residual de unión de apo B junto con un aclaramiento normal de las VLDL (precursoras de las LDL), explicarían los valores moderadamente elevados de CT y cLDL. Además, Schaefer et al145 han demostrado que la vía catabólica de las lipoproteínas VLDL, que contienen apo E, está elevada en el homocigoto DFB estudiado por su grupo. Este mayor aclaramiento probablemente compensatorio de las partículas VLDL explicaría la moderada elevación de CT y cLDL que presentan los homocigotos DFB. También, se han encontrado este mayor aclaramiento en los heterocigotos DFB146.
Estos hallazgos también podrían explicar la buena respuesta farmacológica a estatinas (descensos del 42 % para CT y del 51 % para cLDL) observada en nuestro homocigoto DFB, aunque no se hayan conseguido totalmente los objetivos terapéuticos de la prevención secundaria. En teoría, al igual que ocurre con la forma homicogota para la HF, cabría esperar una falta de respuesta a estatinas en el homocigoto con DFB, al tener completamente alterada la unión de la apo B con el receptor de LDL. En cambio, se consiguen importantes descensos de las concentraciones plasmáticas de CT y cLDL con dosis elevadas de diferentes estatinas y resinas. Por lo tanto, la buena respuesta a estatinas y resinas podría explicarse por la sobrexpresión del receptor sano de las LDL inducido por estatinas, junto con una actividad residual de unión de la apo B defectuosa y un mayor aclaramiento de los precursores de LDL146.
En conclusión, hemos detectado y caracterizado el primer homocigoto español DFB que presenta valores moderadamente elevados de CT y cLDL a pesar de su situación de homocigosis. Este hallazgo, junto con la buena respuesta a las estatinas, indica que el fenotipo lipoproteico de los homocigotos DFB es diferente de la situación de homocigosis para la HF.
2. Estudio del fenotipo lipoproteico de los heterocigotos DFB y comparación con la HF.
- CT y cLDL
En los artículos nº 2, 3, 4 y 5 mostramos las diversas características clínico-bioquímicas de los sujetos DFB estudiados, así como, las diferencias fenotípicas con respecto a los

79
heterocigotos con HF de nuestra población. Los signos clínicos (xantomas, cardiopatía isquémica temprana) no son tan prevalentes en sujetos con el DFB. En el artículo nº 2 hemos estudiado una población de heterocigotos con HF y de heterocigotos con DFB pertenecientes al sur de Europa con el objetivo de comparar el fenotipo lipoproteico según se afecte el gen LDLR o bien el gen APOB. Nuestros resultados indican que el diverso dominio en que mute el gen LDLR tiene un impacto en el fenotipo lipoproteico. El grupo HF con mutaciones “missense” que afectan al dominio de unión presenta las concentraciones plasmáticas más elevadas de CT y cLDL, con una diferencia significativa respecto al dominio que no afecta a la unión al ligando y al grupo de heterocigotos DFB. Sin embargo, los valores de CT y cLDL del grupo con mutaciones nulas no eran estadísticamente diferentes que el de los otros grupos. Por otro lado, los heterocigotos DFB para la mutación R3500Q mostraron los valores plasmáticos más bajos de CT y cLDL. Incluso, como hemos mostrado nosotros y también otros investigadores56, 72, no es infrecuente encontrar portadores de la mutación R3500Q normocolesterolémicos.
Estos datos han sido encontrados parcialmente al compararlos con datos previos de otras poblaciones HF del norte y del sur de Europa. Así, en estudios realizados en el Norte de Europa, Gudnason y Humphries147, Sun et al148, así como Graham et al149
demostraron que los sujetos con mutaciones nulas y con aquellas que afectan al dominio de unión poseen niveles lipídicos más elevados. En una población del Sur de Europa, Bertolini et al150 demostró también las diferencias entre el grupo de pacientes con receptor LDL negativo y aquellos con receptor defectivo, siendo el grupo del receptor negativo el que mostró los niveles de CT más elevados y de cHDL más bajos. Los xantomas y la enfermedad coronaria fue también más frecuente en el grupo del receptor negativo. Todas estas observaciones sugieren que las diferentes mutaciones del gen LDLR están asociadas con diferentes niveles lipídicos en plasma, aunque la base molecular para explicar estas diferencias aun no está del todo clara.
Una posible explicación podría ser que la sobrerregulación del alelo salvaje del gen LDLR esté afectado por la naturaleza del alelo mutante, o que algunas mutaciones defectivas del gen LDLR tengan actividad residual149, 151. Por otro lado, la variabilidad genética debida a los polimorfismos de ADN del alelo salvaje podrían influenciar las concentraciones de cLDL, como ya ha sido citado por Betard et al152.
Podría esperarse que las mutaciones nulas y las mutaciones “missense” que afectan al dominio de unión correlacionen con el fenotipo más grave al compararlo con otras mutaciones donde se espere una actividad residual o la expresión del receptor. En contraste con esta hipótesis, los resultados previos147, 149, 153, 154 indican que los pacientes HF con mutaciones nulas no siempre poseen los valores plasmáticos más elevados de CT y de cLDL.
En la mayoría de los estudios, los sujetos HF portadores de una mutación en la que se espera un fenotipo grave (no sólo mutaciones nulas) o que afectan al dominio de unión al ligando, tienen el perfil lipídico más aterogénico (valores plasmáticos más altos de CT y cLDL). Esto es congruente con nuestros resultados. En nuestro estudio hemos encontrado que los pacientes HF portadores de mutaciones que se esperan graves (las nulas y las “missenses” que afectan al dominio de unión) tienen el fenotipo lipídico mas desfavorable (valores plasmáticos más altos de CT y cLDL, y cHDL más bajo).

80
Las diferencias encontradas en los estudios revisados podrían ser explicados por diferencias metodológicas, por sesgo de selección y también por la clasificación que se haya utilizado para clasificar a las mutaciones en LDLR. No hemos encontrado diferencias estadísticamente significativas en los valores plasmáticos de CT y cLDL entre el grupo de mutaciones nulas y el grupo con mutaciones “missense” que afectan al dominio de unión. Se han encontrado resultados similares con anterioridad147, 149, 154.Una posible explicación es que los pacientes HF con mutaciones nulas sólo expresan el receptor normal en la membrana de sus células. En cambio, el paciente HF con mutaciones “missense“ para el dominio de unión expresan tanto el receptor normal como el alterado. Esta situación podría facilitar una competición local entre ambos receptores afectándose el proceso de internalización. Además, en el caso de mutaciones “missense” que afectan al dominio de unión la situación empeoraría porque la proteína afectada no podría unirse a la apo B. Otra posibilidad es que, en el grupo de mutaciones “missense” que no afectan al dominio de unión pudiéramos haber incluido algunas mutaciones que sí que afectan al dominio de unión (mutaciones “missense” localizadas entre los exones 3 y 6), lo que podría aumentar los valores de CT y cLDL de todo el grupo.
La explicación de las diferencias encontradas en las concentraciones plasmáticas de colesterol total y cLDL entre sujetos con DFB y HF, tanto en países del norte de Europa como en nuestro estudio, puede residir en el diferente comportamiento metabólico de las partículas portadoras de apo B-100 (LDL, VLDL y lipoproteínas de densidad intermedia) observado en sujetos con DFB respecto a aquellos con HF145, 146.
Nuestros resultados apoyan la necesidad de modificar los criterios diagnósticos Med-Ped basados en presencia de xantomas y cardiopatía isquémica temprana junto con valores superiores al percentil 95 de cLDL para identificar a sujetos con DFB.
- cHDL
Hemos encontrado diferencias estadísticamente significativas en las concentraciones plasmáticas de cHDL y apo A-I al comparar sujetos con DFB y sujetos con HF. Este hallazgo va en contra de lo demostrado por Schaefer et al155 que encontraron una mayor producción de HDL y apo A-I en un homocigoto con DFB. Si analizamos las diferencias observadas en nuestro trabajo, los varones con DFB presentan de forma significativa un mayor IMC, lo que podría explicar esta diferencia, dada la conocida asociación inversa entre la obesidad y las concentraciones plasmáticas de cHDL156.
Por otro lado, y de acuerdo con Sijbrands et al157 y Bertolini et al150 nuestros sujetos HF con mutaciones nulas mostraban valores plasmáticos de cHDL significativamente más bajos que aquellos que portan mutaciones “missense”. Nuestra observación de los valores cHDL sugiere que el LDLR podría ser importante en el metabolismo de las HDL de pacientes HF. El papel del LDLR en el aclaramiento de quilomicrones remanentes y partículas VLDL podría explicar los niveles plasmáticos de cHDL encontrados en nuestro grupo de sujetos HF.
Tras una carga grasa oral en seis sujetos homocigotos HF japoneses había un aclaramiento marcadamente retrasado de triglicéridos ricos en retinol marcado. Además, la unión y el aclaramiento de los quilomicrones remanentes por parte de los fibroblastos

81
estaban disminuidos sustancialmente en el grupo HF homocigoto158. En esta misma línea, Castro-Cabezas et al159 informaron sobre un retraso de dos veces en el área bajo la curva para el aclaramiento de las partículas remanentes en sujetos heterocigotos HF. Así, es posible que los sujetos HF con mutaciones nulas tengan un aclaramiento más bajo de quilomicrones e IDL y, como consecuencia, concentraciones más bajas de cHDL. Además, este subgrupo de HF presentó el riesgo más elevado de CHD en el estudio de Vohl et al33.
Finalmente, no podemos excluir diferencias en los perfiles lipídicos debido a la influencia de factores ambientales u otros genéticos en nuestra población HF. Debería recalcarse que la interacción con otros genes160 y con factores ambientales también podría influenciar en el fenotipo de pacientes HF. Pimstone et al161 mostraron que los heterocigotos chinos HF que viven en Canadá presentan un fenotipo lipoproteico similar al de otros pacientes HF de sociedades occidentales, y diferente de aquellos que viven en China. Leitersdorf et al162 han mostrado el efecto que tienen los factores ambientales como la dieta, la actividad física, el peso y otros factores sobre el fenotipo lipídico en pacientes HF. Finalmente, la contribución del genotipo del gen APOE ha sido estudiada por Bertolini et al150. Estos autores han encontrado un efecto hipolipemiante por parte del alelo e2, y un efecto hiperlipemiante por parte del alelo e4, siempre sobre los niveles de cLDL tanto en el grupo receptor-negativo como en el defectivo. En nuestro estudio, la distribución de los factores ambientales (edad, sexo, IMC) y el del genotipo APOE que podrían modular el fenotipo lipídico de sujetos HF y DFB es similar en los cuatro grupos.
3. Efecto fundador y prevalencia de la mutación R3500Q.
En el cuarto trabajo el estudio del haplotipo del gen APOB en los sujetos con la mutación R3500Q nos llevó a evidenciar que el origen de dicho haplotipo era el mismo, lo que nos confirma la endogamia. Además, este haplotipo lo portan la mayor parte de los sujetos con DFB lo que indica el origen común de todos ellos. Se ha estudiado el origen cronológico de dicha mutación localizándola hace 6.000-7.000 años en una región geográfica comprendida entre los ríos Rin y Meno (suroeste de Alemania)87.
En poblaciones no endogámicas como la española, factores culturales, históricos y geográficos pueden facilitar la endogamia en zonas aisladas, generalmente rurales. Este ha sido el caso del DFB en la Comunitat Valenciana. Otro estudio español73, que mostró una baja prevalencia del DFB en España, localizó la mayor parte de los afectados en Galicia, lo que apoya una supuesta hipótesis del origen celta de la mutación R3500Q. A diferencia de este hallazgo, y como demuestra nuestro estudio, hemos encontrado un núcleo de alta prevalencia del DFB en una población de la Comunitat Valenciana debido a un efecto fundador. Esta prevalencia es incluso superior a la mayor citada hasta ahora, descrita en población suiza58, 163.
La comparación del haplotipo del gen APOB de los diversos portadores de la mutación R3500Q con el haplotipo del resto de sujetos DFB de otras poblaciones europeas indica que lo más probable es que existió un único episodio mutacional que originó la mutación R3500Q hace unos 6.000-7.000 años87, como hemos comentado previamente. Todo parece indicar que la mutación R3500Q se produjo en un alelo que se ha transmitido desde su origen hasta nuestros días. Esto permite suponer que el origen de la

82
mutación es común en todas las poblaciones estudiadas87. Diferentes fenómenos migratorios habrían extendido dicha mutación desde su núcleo original -suroeste de Alemania- hasta las diversas localizaciones en que se ha descrito.
El análisis de los diversos datos sobre la conquista y la repoblación de la comarca de Los Serranos en el siglo XIII90, 91, así como el análisis de los apellidos encontrados en las partidas de bautismo de la parroquia de La Yesa no nos permite establecer un claro vínculo con los sujetos DFB del suroeste de Alemania. No obstante, el análisis de los datos nos hace suponer que las migraciones sufridas por los descendientes de los DFB de origen céltico-alemán repoblarían la comarca de Los Serranos tras emigrar desde su localización inicial, pasando la barrera alpina, luego la pirenaica y situándose en la comarca de Los Serranos en el periodo histórico de la reconquista para posteriormente producir un efecto fundador.

83
CONCLUSIONES

84

85
4. CONCLUSIONES
1. En contra de los datos publicados sobre prevalencia en el Sur de Europa, la prevalencia de la mutación R3500Q en el gen APOB es muy elevada en la población que hemos estudiado, La Yesa (comarca de los Serranos, provincia de Valencia). Es mayor que la existente para otras poblaciones con gran prevalencia.
2. Hemos demostrado un claro efecto fundador y evidente endogamia, conclusión derivada del estudio de los árboles genealógicos de la población de La Yesa y de los haplotipos en el locus APOB de los 19 heterocigotos DFB. Todos ellos tienen el haplotipo ins / XbaI- / MspI+ / EcoRI- / 3HVR49.
3. El fenotipo lipoproteico de los heterocigotos DFB estudiados consiste en niveles moderadamente elevados de colesterol total, cLDL, apo B y Lp(a). Estos resultados están en sintonía con los de otros estudios realizados con heterocigotos DFB del norte de Europa.
4. Al comparar el fenotipo lipoproteico de los heterocigotos DFB con el de los heterocigotos HF, tanto en su conjunto como divididos según el tipo de mutación (nulas, dominio de unión al ligando y fuera del dominio de unión al ligando), el fenotipo de los DFB es el más leve. Por ello, se descarta la hipótesis inicial de trabajo que hipotetizaba que heterocigotos DFB y HF expresarían un fenotipo lipoproteico similar.
5. Las diferencias encontradas en las concentraciones plasmáticas de colesterol total y cLDL entre sujetos DFB y HF podría explicarse por el diferente comportamiento metabólico de las partículas portadoras de apo B-100 (LDL, VLDL e IDL) en los sujetos DFB con respecto a aquellos HF. Es fundamental el papel de las apo E que llevarían al aumento del catabolismo en las partículas VLDL, lo cual haría disminuir los valores de cLDL.
6. Los valores plasmáticos de Lp(a) y su transformación logarítmica fueron significativamente mayores en el grupo DFB frente al grupo control.
7. A tenor de nuestros resultados, los criterios diagnósticos MedPed para hipercolesterolemias genéticas primarias tienen escaso valor para el diagnóstico de DFB. El 64 % de nuestros sujetos tenían colesterolemias inferiores al percentil 95 y sólo en el 36 % del grupo se consideró un diagnóstico “probable”. Usando los criterios MedPed, para el diagnóstico de HF o de hipercolesterolemia genética, ninguno de los sujetos DFB diagnosticados genéticamente tendría una hipercolesterolemia genética confirmada.

86

87
BIBLIOGRAFÍA

88

89
5. BIBLIOGRAFÍA
1 Virchow R: Gesammelte Adhandlunger zur Wissenschaftlichen Medicin Meidingir Sohn and Company, Frankfurt am main.1856:pag.458
2 Anistschkow.N.:Uber die Veranderungen der kaninchen aorta bei experimenteller Cholesterinsteatose. Beit Path Anat.1913:56-79
3 Álvarez-Sala LA, Millán J. Evidencias de la eficacia del tratamiento hipocolesterolemiante en la prevención primaria y secundaria de la cardiopatía isquémica. Med Clin (Barc) 2000; 114(Supl 2):1-10.
4 Berliner JA, Navab M, Fogelman AM, Frank JS, Demer LL, Edwards PA, Watson AD, Lusis AJ. Atherosclerosis: basic mechanisms. Oxidation, inflammation, and genetics. Circulation. 1995 May 1;91(9):2488-96.
5 Lewis B, Myant NB. Studies in the metabolism of cholesterol in subjects with normal plasma cholesterol levels and in patients with essential hypercholesterolaemia. Clin Sci. 1967 Apr;32(2):201-13.
6 Human Molecular Genetics. Strachan T, Read AP. 3ª edición. BIOS Scientific Publisher.
7 Report of the National Cholesterol Education Program Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults. The Expert Panel. Arch Intern Med. 1988 Jan;148(1):36-69.
8 Fredrickson DS, Levy RI, Lees RS. Fat transport in lipoproteins--an integrated approach to mechanisms and disorders. N Engl J Med. 1967 Jan 5;276(1):34-42.
9 Beaumont JL, Carlson LA, Cooper GR, Fejfar Z, Fredrickson DS, Strasser T. Classification of hyperlipidaemias and hyperlipoproteinaemias. Bull World Health Organ. 1970;43(6):891-915.
10 Rader DJ, Cohen J, Hobbs HH. Monogenic hypercholesterolemia: new insights in pathogenesis and treatment. J Clin Invest. 2003 Jun;111(12):1795-803.
11 Brown MS, Goldstein JL. Expression of the familial hypercholesterolemia gene in heterozygotes: mechanism for a dominant disorder in man. Science. 1974 Jul 5;185(4145):61-3.
12 Südhof TC, Goldstein JL, Brown MS, Russell DW. The LDL receptor gene: a mosaic of exons shared with different proteins. Science. 1985 May 17;228(4701):815-22.
13 Fox TC. A case of xanthelasma multiplex. Lancet 1879; 2: 688.
14 Wilkinson CF, Hand EA, Fliebelman MT. Essential familial hypercholesterolemia. Ann Intern Med 1948; 29: 671-686.

90
15 Adlesberg D. Hypercholesterolemia with predisposition to atherosclerosis: An inborn error of lipid metabolism. Am J Med 1951; 11: 600.
16 Khachadurian AK. The inheritance of essential familial hypercholesterolemia. Am J Med. 1964 Sep;37:402-7.
17 Brown MS, Goldstein JL. Analysis of a mutant strain of human fibroblasts with a defect in the internalization of receptor-bound low density lipoprotein. Cell. 1976 Dec;9(4 PT 2):663-74.
18 Russell DW, Yamamoto T, Schneider WJ, Slaughter CJ, Brown MS, Goldstein JL. cDNA cloning of the bovine low density lipoprotein receptor: feedback regulation of a receptor mRNA. Proc Natl Acad Sci U S A. 1983 Dec;80(24):7501-5.
19 Yamamoto T, Davis CG, Brown MS, Schneider WJ, Casey ML, Goldstein JL, Russell DW. The human LDL receptor: a cysteine-rich protein with multiple Alu sequences in its mRNA. Cell. 1984 Nov;39(1):27-38.
20 Hobbs HH, Russell DW, Brown MS, Goldstein JL. The LDL receptor locus in familial hypercholesterolemia: mutational analysis of a membrane protein. Annu Rev Genet. 1990;24:133-70.
21 Uauy R, Zwiener RJ, Phillips MJ, Petruska ML, Bilheimer DW. Treatment of children with homozygous familial hypercholesterolemia: safety and efficacy of low-density lipoprotein apheresis. J Pediatr. 1992 Jun;120(6):892-8.
22 Sugrue DD, Thompson GR, Oakley CM, Trayner IM, Steiner RE. Contrasting patterns of coronary atherosclerosis in normocholesterolaemic smokers and patients with familial hypercholesterolaemia. Br Med J (Clin Res Ed). 1981 Nov 21;283(6303):1358-60.
23 Slack J. Risks of ischaemic heart-disease in familial hyperlipoproteinaemic states. Lancet. 1969 Dec 27;2(7635):1380-2.
24 Seed M, Hoppichler F, Reaveley D, McCarthy S, Thompson GR, Boerwinkle E, Utermann G. Relation of serum lipoprotein(a) concentration and apolipoprotein(a) phenotype to coronary heart disease in patients with familial hypercholesterolemia. N Engl J Med. 1990 May 24;322(21):1494-9.
25 Ferrieres J, Lambert J, Lussier-Cacan S, Davignon J. Coronary artery disease in heterozygous familial hypercholesterolemia patients with the same LDL receptor gene mutation. Circulation. 1995 Aug 1;92(3):290-5.
26 Havel JR, Goldstein JL, Brown MS. Lipoprotein and lipid transport. En: Bondey PK, Rosemberg LE, ed. Metabolic Control and Disease. Filadelfia: WB Saunders 1980; 393-498.
27 Davis CG, Lehrman MA, Russell DW, Anderson RG, Brown MS, Goldstein JL. The J.D. mutation in familial hypercholesterolemia: amino acid substitution in cytoplasmic domain impedes internalization of LDL receptors. Cell. 1986 Apr 11;45(1):15-24.

91
28 Mahley RW, Innerarity TL, Weisgraber KH, Rall SC Jr, Hui DY, Lalazar A, Boyles JK, Taylor JM, Levy-Wilson B. Cellular and molecular biology of lipoprotein metabolism: characterization of lipoprotein receptor-ligand interactions. Cold Spring Harb Symp Quant Biol. 1986;51 Pt 2:821-8.
29 Hobbs HH, Brown MS, Goldstein JL. Molecular genetics of the LDL receptor gene in familial hypercholesterolemia. Hum Mutat. 1992;1(6):445-66.
30 Varret M, Rabes JP, Collod-Beroud G, Junien C, Boileau C, Beroud C. Software and database for the analysis of mutations in the human LDL receptor gene. Nucleic Acids Res. 1997 Jan 1;25(1):172-80.
31 Fass D, Blacklow S, Kim PS, Berger JM. Molecular basis of familial hypercholesterolaemia from structure of LDL receptor module. Nature. 1997 Aug 14;388(6643):691-3.
32 Jeenah M, September W, Graadt van Roggen F, de Villiers W, Seftel H, Marais D. Influence of specific mutations at the LDL-receptor gene locus on the response to simvastatin therapy in Afrikaner patients with heterozygous familial hypercholesterolaemia. Atherosclerosis. 1993 Jan 4;98(1):51-8.
33 Vohl MC, Gaudet D, Moorjani S, Tremblay G, Perron P, Gagne C, Lesiege D, Bergeron J, Lupien PJ, Despres JP. Comparison of the effect of two low-density lipoprotein receptor class mutations on coronary heart disease among French-Canadian patients heterozygous for familial hypercholesterolaemia. Eur J Clin Invest. 1997 May;27(5):366-73.
34 Brown MS, Goldstein JL. The SREBP pathway: regulation of cholesterol metabolism by proteolysis of a membrane-bound transcription factor. Cell. 1997 May 2;89(3):331-40.
35 Williams RR, Hunt SC, Schumacher MC, Hegele RA, Leppert MF, Ludwig EH, Hopkins PN. Diagnosing heterozygous familial hypercholesterolemia using new practical criteria validated by molecular genetics. Am J Cardiol. 1993 Jul 15;72(2):171-6.
36 Koivisto PV, Koivisto UM, Miettinen TA, Kontula K. Diagnosis of heterozygous familial hypercholesterolemia. DNA analysis complements clinical examination and analysis of serum lipid levels. Arterioscler Thromb. 1992 May;12(5):584-92.
37 Williams RR, Schumacher MC, Barlow GK, Hunt SC, Ware JL, Pratt M, Latham BD. Documented need for more effective diagnosis and treatment of familial hypercholesterolemia according to data from 502 heterozygotes in Utah. Am J Cardiol. 1993 Sep 30;72(10):18D-24D.
38 Kotze MJ, Langenhoven E, Kriek JA, Oosthuizen CJ, Retief AE. DNA screening of hyperlipidemic Afrikaners for familial hypercholesterolemia. Clin Genet. 1992 Jul;42(1):43-6.

92
39 Chaves FJ, Real JT, Garcia-Garcia AB et al: Large rearrangements of the LDL receptor gene and lipid profile in a Spanish population. Eur J Clin Invest 2001; 31: 309–317.
40 Garcia-Garcia AB, Real JT, Puig O et al: Molecular genetics of familial hypercholesterolemia in Spain: ten novel LDLR mutations and population analysis. Hum Mutat 2001; 454: 1–9.
41 Tejedor D, Castillo S, Mozas P, Jimenez E, Lopez M, Tejedor MT, Artieda M, Alonso R, Mata P, Simon L, Martinez A, Pocovi M; Spanish FH Group. Reliable low-density DNA array based on allele-specific probes for detection of 118 mutations causing familial hypercholesterolemia. Clin Chem. 2005 Jul;51(7):1137-44.
42 Soria LF, Ludwig EH, Clarke HR, Vega GL, Grundy SM, McCarthy BJ. Association between a specific apolipoprotein B mutation and familial defective apolipoprotein B-100. Proc Natl Acad Sci U S A. 1989 Jan;86(2):587-91.
43 Vega GL, Grundy SM. In vivo evidence for reduced binding of low density lipoproteins to receptors as a cause of primary moderate hypercholesterolemia. J Clin Invest. 1986 Nov;78(5):1410-4.
44 Weisgraber KH, Innerarity TL, Newhouse YM, Young SG, Arnold KS, Krauss RM, Vega GL, Grundy SM, Mahley RW. Familial defective apolipoprotein B-100: enhanced binding of monoclonal antibody MB47 to abnormal low density lipoproteins. Proc Natl Acad Sci U S A. 1988 Dec;85(24):9758-62.
45 Pullinger CR, Hennessy LK, Chatterton JE, Liu W, Love JA, Mendel CM, Frost PH, Malloy MJ, Schumaker VN, Kane JP. Familial ligand-defective apolipoprotein B. Identification of a new mutation that decreases LDL receptor binding affinity. J Clin Invest. 1995 Mar;95(3):1225-34.
46 Gaffney D, Reid JM, Cameron IM, Vass K, Caslake MJ, Shepherd J, Packard CJ. Independent mutations at codon 3500 of the apolipoprotein B gene are associated with hyperlipidemia. Arterioscler Thromb Vasc Biol. 1995 Aug;15(8):1025-9.
47 Blackhart BD, Ludwig EM, Pierotti VR, Caiati L, Onasch MA, Wallis SC, Powell L, Pease R, Knott TJ, Chu ML, Mahley RW, Scott J, McCarthy BJ, Levy-Wilson B. Structure of the human apolipoprotein B gene. J Biol Chem. 1986 Nov 25;261(33):15364-7.
48 Chen SH, Yang CY, Chen PF, Setzer D, Tanimura M, Li WH, Gotto AM Jr, Chan L. The complete cDNA and amino acid sequence of human apolipoprotein B-100. J Biol Chem. 1986 Oct 5;261(28):12918-21.
49 Entrez Nucleotide para APOB:http://www.ncbi.nlm.nih.gov/entrez/viewer.fcgi?val=M14162

93
50 Myant NB, Gallagher JJ, Knight BL, McCarthy SN, Frostegard J, Nilsson J, Hamsten A, Talmud P, Humphries SE. Clinical signs of familial hypercholesterolemia in patients with familial defective apolipoprotein B-100 and normal low density lipoprotein receptor function. Arterioscler Thromb. 1991 May-Jun;11(3):691-703.
51 Schuster H, Rauh G, Kormann B, Hepp T, Humphries S, Keller C, Wolfram G, Zollner N. Familial defective apolipoprotein B-100. Comparison with familial hypercholesterolemia in 18 cases detected in Munich. Arteriosclerosis. 1990 Jul-Aug;10(4):577-81.
52 Tybjaerg-Hansen A, Gallagher J, Vincent J, Houlston R, Talmud P, Dunning AM, Seed M, Hamsten A, Humphries SE, Myant NB. Familial defective apolipoprotein B-100: detection in the United Kingdom and Scandinavia, and clinical characteristics of ten cases. Atherosclerosis. 1990 Jan;80(3):235-42.
53 Hansen PS, Meinertz H, Jensen HK, Fruergaard P, Launbjerg J, Klausen IC, Lemming L, Gerdes U, Gregersen N, Faergeman O. Characteristics of 46 heterozygous carriers and 57 unaffected relatives in five Danish families with familial defective apolipoprotein B-100. Arterioscler Thromb. 1994 Feb;14(2):207-13.
54 Rauh G, Keller C, Kormann B, Spengel F, Schuster H, Wolfram G, Zollner N. Familial defective apolipoprotein B100: clinical characteristics of 54 cases. Atherosclerosis. 1992 Feb;92(2-3):233-41.
55 Defesche JC, Pricker KL, Hayden MR, van der Ende BE, Kastelein JJ. Familial defective apolipoprotein B-100 is clinically indistinguishable from familial hypercholesterolemia. Arch Intern Med. 1993 Oct 25;153(20):2349-56.
56 Gallagher JJ, Myant NB. Variable expression of the mutation in familial defective apolipoprotein B-100. Arterioscler Thromb. 1993 Jul;13(7):973-6.
57 Rauh G, Keller C, Schuster H, Wolfram G, Zollner N. Familial defective apolipoprotein B-100: a common cause of primary hypercholesterolemia. Clin Investig. 1992 Jan;70(1):77-84.
58 Miserez AR, Laager R, Chiodetti N, Keller U. High prevalence of familial defective apolipoprotein B-100 in Switzerland. J Lipid Res. 1994 Apr;35(4):574-83.
59 Ludwig EH, McCarthy BJ. Haplotype analysis of the human apolipoprotein B mutation associated with familial defective apolipoprotein B100. Am J Hum Genet. 1990 Oct;47(4):712-20.
60 Chaves FJ, Real JT, Garcia-Garcia AB, Civera M, Armengod ME, Ascaso JF, Carmena R. Genetic diagnosis of familial hypercholesterolemia in a South European outbreed population: influence of low-density lipoprotein (LDL) receptor gene mutations on treatment response to simvastatin in total, LDL, and high-density lipoprotein cholesterol. J Clin Endocrinol Metab. 2001 Oct;86(10):4926-32.

94
61 Koivisto PV, Koivisto UM, Miettinen TA, Kontula K. Diagnosis of heterozygous familial hypercholesterolemia. DNA analysis complements clinical examination and analysis of serum lipid levels. Arterioscler Thromb. 1992 May;12(5):584-92.
62 Hämäläinen T, Palotie A, Aalto-Setala K, Kontula K, Tikkanen MJ. Absence of familial defective apolipoprotein B-100 in Finnish patients with elevated serum cholesterol. Atherosclerosis. 1990 Jun;82(3):177-83.
63 Schuster H, Rauh G, Mu ller S, Keller C, Wolfram G, Zollner N: Allele specific and asymetric polymerase chain reaction amplification in combination: a step polymerase chain reaction protocol for rapid diagnosis of familial defective apolipoprotein B-100. Ann Bioch 1992; 204: 22– 25.
64 Mueller RF, Young ID. Emery’s elements of medical genetics. 10 edición. Churchill Livingstone.
65 Lewin B. Genes VI. Oxford University Press. 1997.
66 Friedl W, Ludwig EH, Paulweber B, Sandhofer F, McCarthy BJ. Hypervariability in a minisatellite 3' of the apolipoprotein B gene in patients with coronary heart disease compared with normal controls. J Lipid Res. 1990 Apr;31(4):659-65.
67 Rauh G, Schuster H, Fischer J, Keller C, Wolfram G, Zollner N. Familial defective apolipoprotein B-100: haplotype analysis of the arginine(3500)----glutamine mutation. Atherosclerosis. 1991 Jun;88(2-3):219-26.
68 Motti C, Funke H, Rust S, Dergunov A, Assmann G. Using mutagenic polymerase chain reaction primers to detect carriers of familial defective apolipoprotein B-100. Clin Chem. 1991 Oct;37(10 Pt 1):1762-6.
69 Schuster H, Humphries S, Rauh G, Keller C. First International Workshop on Familial Defective apo B-100, Munich, November 1991. Clin Investig. 1992 Oct;70(10):961-4.
70 Innerarity TL, Mahley RW, Weisgraber KH, Bersot TP, Krauss RM, Vega GL, Grundy SM, Friedl W, Davignon J, McCarthy BJ. Familial defective apolipoprotein B-100: a mutation of apolipoprotein B that causes hypercholesterolemia. J Lipid Res. 1990 Aug;31(8):1337-49.
71 Friedl W, Ludwig EH, Balestra ME, Arnold KS, Paulweber B, Sandhofer F, McCarthy BJ, Innerarity TL. Apolipoprotein B gene mutations in Austrian subjects with heart disease and their kindred. Arterioscler Thromb. 1991 Mar-Apr;11(2):371-8.
72 Real JT, Chaves JF, Ascaso JF, Armengod ME, Carmena R. Estudio del defecto familiar de la apo B-100 en sujetos con el diagnóstico clínico de hipercolesterolemia primaria: identificación de la primera familia afectada en España. Med Clin (Barc). 1999 Jun 12;113(1):15-7.

95
73 Castillo S, Tejedor D, Mozas P, Reyes G, Civeira F, Alonso R, Ros E, Pocovi M, Mata P. The apolipoprotein B R3500Q gene mutation in Spanish subjects with a clinical diagnosis of familial hypercholesterolemia. Atherosclerosis. 2002 Nov;165(1):127-35.
74 Tybjaerg-Hansen A, Humphries SE. Familial defective apolipoprotein B-100: a single mutation that causes hypercholesterolemia and premature coronary artery disease. Atherosclerosis. 1992 Oct;96(2-3):91-107.
75 Kalina A, Csaszar A, Czeizel AE, Romics L, Szaboki F, Szalai C, Reiber I, Nemeth A, Stephenson S, Williams RR. Frequency of the R3500Q mutation of the apolipoprotein B-100 gene in a sample screened clinically for familial hypercholesterolemia in Hungary. Atherosclerosis. 2001 Jan;154(1):247-51.
76 Gorski B, Kubalska J, Naruszewicz M, Lubinski J. LDL-R and Apo-B-100 gene mutations in Polish familial hypercholesterolemias. Hum Genet. 1998 May;102(5):562-5.
77 Freiberger T, Kuhrova V, Kozak L, Soska V, Pekarik V, Merinska L, Fajkusova L, Francova H. Direct detection of mutations in the LDL receptor gene in patients with familial hypercholesterolemia. Cas Lek Cesk. 1998 Dec 14;137(24):750-2.
78 Ceska R, Vrablik M, Horinek A. Familial defective apolipoprotein B-100: a lesson from homozygous and heterozygous patients. Physiol Res. 2000;49 Suppl 1:S125-30.
79 Corsini A, McCarthy BJ, Granata A, Soria LF, Fantappie S, Bernini, Romano C, Romano L, Fumagalli R, Catapano AL. Familial defective apo B-100, characterization of an Italian family. Eur J Clin Invest. 1991 Aug;21(4):389-97.
80 Robles-Osorio L, Ordonez ML, Aguilar-Salinas CA, Auron-Gomez M, Tusie-Luna MT, Gomez-Perez FJ, Rull-Rodrigo JA. Familial hypercholesterolemia due to ligand-defective apolipoprotein B100: first case report in a Mexican family. Arch Med Res. 2003 Jan-Feb;34(1):70-5.
81 Bersot TP, Russell SJ, Thatcher SR, Pomernacki NK, Mahley RW, Weisgraber KH, Innerarity TL, Fox CS. A unique haplotype of the apolipoprotein B-100 allele associated with familial defective apolipoprotein B-100 in a Chinese man discovered during a study of the prevalence of this disorder. J Lipid Res. 1993 Jul;34(7):1149-54.
82 Friedlander Y, Dann EJ, Leitersdorf E. Absence of familial defective apolipoprotein B-100 in Israeli patients with dominantly inherited hypercholesterolemia and in offspring with parental history of myocardial infarction. Hum Genet. 1993 Apr;91(3):299-300.
83 Yu W, Nohara A, Higashikata T, Lu H, Inazu A, Mabuchi H. Molecular genetic analysis of familial hypercholesterolemia: spectrum and regional difference of LDL receptor gene mutations in Japanese population. Atherosclerosis. 2002 Dec;165(2):335-42.

96
84 Nei M, Roychoudhury. 1967. Genetic relationship and evolution of human races. En: Evolutionary Biology. Hecht MK, Wallace B, Prance GT editores. Plenum Press. Nueva York, NY 3-15.
85 Cavalli-Sforza LL y Bodmer WF. 1991. The genetics of human populations. pp. 483-488. Freeman. San Francisco. 86 Renfrew C (1990). Before civilization: the radiocarbon revolution and prehistoric Europe. pp. 69-71. Penguin books, London.
87 Myant NB, Forbes SA, Day IN, Gallagher J. Estimation of the age of the ancestral arginine3500-->glutamine mutation in human apoB-100. Genomics. 1997 Oct 1;45(1):78-87.
88 Mahley RW, Palaoglu KE, Atak Z, Dawson-Pepin J, Langlois AM, Cheung V, Onat H, Fulks P, Mahley LL, Vakar F, et al. Turkish Heart Study: lipids, lipoproteins, and apolipoproteins. J Lipid Res. 1995 Apr;36(4):839-59.
89 Miserez AR, Muller PY. Familial defective apolipoprotein B-100: a mutation emerged in the mesolithic ancestors of Celtic peoples? Atherosclerosis. 2000 Feb;148(2):433-6.
90 Conquista y repoblación del Reino de Valencia. Ramón Ferrer Navarro. Valencia. Del Senia al Segura, 1999.
91 Els fundadors del Regne de València: repoblament, antroponímia i llengua a la València medieval. Enric Guinot Rodríguez. València. Eliseu Climent, 1999.
92 Berg K, Dahlen G, Frick MH. Lp(a) lipoprotein and pre-beta1-lipoprotein in patients with coronary heart disease. Clin Genet. 1974;6(3):230-5.
93 Kostner GM, Avogaro P, Cazzolato G, Marth E, Bittolo-Bon G, Qunici GB. Lipoprotein Lp(a) and the risk for myocardial infarction. Atherosclerosis. 1981 Jan-Feb;38(1-2):51-61.
94 Rhoads GG, Dahlen G, Berg K, Morton NE, Dannenberg AL. Lp(a) lipoprotein as a risk factor for myocardial infarction. JAMA. 1986 Nov 14;256(18):2540-4.
95 Dahlen GH, Guyton JR, Attar M, Farmer JA, Kautz JA, Gotto AM Jr. Association of levels of lipoprotein Lp(a), plasma lipids, and other lipoproteins with coronary artery disease documented by angiography. Circulation. 1986 Oct;74(4):758-65.
96 Ariyo AA, Thach C, Tracy R; Cardiovascular Health Study Investigators. Lp(a) lipoprotein, vascular disease, and mortality in the elderly. N Engl J Med. 2003 Nov 27;349(22):2108-15.
97 Perombelon YF, Gallagher JJ, Myant NB, Soutar AK, Knight BL. Lipoprotein(a) in subjects with familial defective apolipoprotein B100. Atherosclerosis. 1992 Feb;92(2-3):203-12.

97
98 Van der Hoek YY, Lingenhel A, Kraft HG, Defesche JC, Kastelein JJ, Utermann G. Sib-pair analysis detects elevated Lp(a) levels and large variation of Lp(a) concentration in subjects with familial defective ApoB. J Clin Invest. 1997 May 1;99(9):2269-73.
99 Rader DJ, Mann WA, Cain W, Kraft HG, Usher D, Zech LA, Hoeg JM, Davignon J, Lupien P, Grossman M, et al. The low density lipoprotein receptor is not required for normal catabolism of Lp(a) in humans. J Clin Invest. 1995 Mar;95(3):1403-8.
100 Utermann G. 1994. Lipoprotein(a). In The Metabolic and Molecular Bases of Inherited Disease. C. R. Scriver, A. L. Beaudet, W. S. Sly, J. B. Stanbury, J. B. Wyngaarden, and D. S. Frederickson, editors. McGraw-Hill Inc., New York. 1887-1912.
101 Luc G, Bard JM, Arveiler D, Ferrieres J, Evans A, Amouyel P, Fruchart JC, Ducimetiere P; PRIME Study Group. Lipoprotein (a) as a predictor of coronary heart disease: the PRIME Study. Atherosclerosis. 2002 Aug;163(2):377-84.
102 Watts GF, Kearney EM, Taub NA, Slavin BM. Lipoprotein (a) as an independent risk factor for myocardial infarction in patients with common hypercholesterolaemia. J Clin Pathol. 1993 Mar;46(3):267-70.
103 Fujino A, Watanabe T, Kunii H, Yamaguchi N, Yoshinari K, Watanabe Y, Mutou M, Ishikawa S, Ogyuu A, Ashikawa K, Maruyama Y. Lipoprotein(a) is a potential coronary risk factor. Jpn Circ J. 2000 Jan;64(1):51-6.
104 Peltier M, Iannetta Peltier MC, Sarano ME, Lesbre JP, Colas JL, Tribouilloy CM. Elevated serum lipoprotein(a) level is an independent marker of severity of thoracic aortic atherosclerosis. Chest. 2002 May;121(5):1589-94.
105 McCormick SP, Ng JK, Taylor S, Flynn LM, Hammer RE, Young SG. Mutagenesis of the human apolipoprotein B gene in a yeast artificial chromosome reveals the site of attachment for apolipoprotein(a). Proc Natl Acad Sci U S A. 1995 Oct 24;92(22):10147-51.
106 Brunner C, Kraft HG, Utermann G, Muller HJ. Cys4057 of apolipoprotein(a) is essential for lipoprotein(a) assembly. Proc Natl Acad Sci U S A. 1993 Dec 15;90(24):11643-7.
107 Callow MJ, Rubin EM. Site-specific mutagenesis demonstrates that cysteine 4326 of apolipoprotein B is required for covalent linkage with apolipoprotein (a) in vivo. J Biol Chem. 1995 Oct 13;270(41):23914-7.
108 Guevara J Jr, Spurlino J, Jan AY, Yang CY, Tulinsky A, Prasad BV, Gaubatz JW, Morrisett JD. Proposed mechanisms for binding of apo[a] kringle type 9 to apo B-100 in human lipoprotein[a]. Biophys J. 1993 Mar;64(3):686-700.
109 Gaubatz JW, Heideman C, Gotto AM Jr, Morrisett JD, Dahlen GH. Human plasma lipoprotein (a). Structural properties. J Biol Chem. 1983 Apr 10;258(7):4582-9.

98
110 Koschinsky ML, Cote GP, Gabel B, van der Hoek YY. Identification of the cysteine residue in apolipoprotein(a) that mediates extracellular coupling with apolipoprotein B-100. J Biol Chem. 1993 Sep 15;268(26):19819-25.
111 Koschinsky ML, Marcovina SM, May LF, Gabel BR. Analysis of the mechanism of lipoprotein(a) assembly. Clin Genet. 1997 Nov;52(5):338-46.
112 McLean JW, Tomlinson JE, Kuang WJ, Eaton DL, Chen EY, Fless GM, Scanu AM, Lawn RM. cDNA sequence of human apolipoprotein(a) is homologous to plasminogen. Nature. 1987 Nov 12-18;330(6144):132-7.
113 Eaton DL, Fless GM, Kohr WJ, McLean JW, Xu QT, Miller CG, Lawn RM, Scanu AM. Partial amino acid sequence of apolipoprotein(a) shows that it is homologous to plasminogen. Proc Natl Acad Sci U S A. 1987 May;84(10):3224-8.
114 Marcovina SM, Zhang ZH, Gaur VP, Albers JJ. Identification of 34 apolipoprotein(a) isoforms: differential expression of apolipoprotein(a) alleles between American blacks and whites. Biochem Biophys Res Commun. 1993 Mar 31;191(3):1192-6.
115 Perombelon YF, Soutar AK, Knight BL. Variation in lipoprotein(a) concentration associated with different apolipoprotein(a) alleles. J Clin Invest. 1994 Apr;93(4):1481-92.
116 Gavish D, Azrolan N, Breslow JL. Plasma Ip(a) concentration is inversely correlated with the ratio of Kringle IV/Kringle V encoding domains in the apo(a) gene. J Clin Invest. 1989 Dec;84(6):2021-7.
117 Trommsdorff M, Kochl S, Lingenhel A, Kronenberg F, Delport R, Vermaak H, Lemming L, Klausen IC, Faergeman O, Utermann G, et al. A pentanucleotide repeat polymorphism in the 5’ control region of the apolipoprotein(a) gene is associated with lipoprotein(a) plasma concaentrations in Caucasians. J Clin Invest. 1995; 96: 150-57.
118 Lackner C, Boerwinkle E, Leffert CC, Rahmig T, Hobbs HH. Molecular basis of apolipoprotein (a) isoform size heterogeneity as revealed by pulsed-field gel electrophoresis. J Clin Invest. 1991 Jun;87(6):2153-61.
119 Gaubatz JW, Chari MV, Nava ML, Guyton JR, Morrisett JD. Isolation and characterization of the two major apoproteins in human lipoprotein [a]. J Lipid Res. 1987 Jan;28(1):69-79.
120 Gaubatz JW, Ghanem KI, Guevara J Jr, Nava ML, Patsch W, Morrisett JD. Polymorphic forms of human apolipoprotein[a]: inheritance and relationship of their molecular weights to plasma levels of lipoprotein[a]. J Lipid Res. 1990 Apr;31(4):603-13.
121 Utermann G, Menzel HJ, Kraft HG, Duba HC, Kemmler HG, Seitz C. Lp(a) glycoprotein phenotypes. Inheritance and relation to Lp(a)-lipoprotein concentrations in plasma. J Clin Invest. 1987 Aug;80(2):458-65.

99
122 Fless GM, Rolih CA, Scanu AM. Heterogeneity of human plasma lipoprotein (a). Isolation and characterization of the lipoprotein subspecies and their apoproteins. J Biol Chem. 1984 Sep 25;259(18):11470-8.
123 Utermann G, Kraft HG, Menzel HJ, Hopferwieser T, Seitz C. Genetics of the quantitative Lp(a) lipoprotein trait. I. Relation of LP(a) glycoprotein phenotypes to Lp(a) lipoprotein concentrations in plasma. Hum Genet. 1988 Jan;78(1):41-6.
124 Boerwinkle E, Leffert CC, Lin J, Lackner C, Chiesa G, Hobbs HH. Apolipoprotein(a) gene accounts for greater than 90% of the variation in plasma lipoprotein(a) concentrations. J Clin Invest. 1992 Jul;90(1):52-60.
125 Boerwinkle E, Menzel HJ, Kraft HG, Utermann G. Genetics of the quantitative Lp(a) lipoprotein trait. III. Contribution of Lp(a) glycoprotein phenotypes to normal lipid variation. Hum Genet. 1989 Apr;82(1):73-8.
126 DeMeester CA, Bu X, Gray RJ, Lusis AJ, Rotter JI. Genetic variation in lipoprotein (a) levels in families enriched for coronary artery disease is determined almost entirely by the apolipoprotein (a) gene locus. Am J Hum Genet. 1995 Jan;56(1):287-93.
127 Kraft HG, Kochl S, Menzel HJ, Sandholzer C, Utermann G. The apolipoprotein (a) gene: a transcribed hypervariable locus controlling plasma lipoprotein (a) concentration. Hum Genet. 1992 Nov;90(3):220-30.
128 Frank SL, Klisak I, Sparkes RS, Mohandas T, Tomlinson JE, McLean JW, Lawn RM, Lusis AJ. The apolipoprotein(a) gene resides on human chromosome 6q26-27, in close proximity to the homologous gene for plasminogen. Hum Genet. 1988 Aug;79(4):352-6.
129 Malgaretti N, Acquati F, Magnaghi P, Bruno L, Pontoglio M, Rocchi M, Saccone S, Della Valle G, D'Urso M, LePaslier D, et al. Characterization by yeast artificial chromosome cloning of the linked apolipoprotein(a) and plasminogen genes and identification of the apolipoprotein(a) 5' flanking region. Proc Natl Acad Sci U S A. 1992 Dec 1;89(23):11584-8.
130 Weitkamp LR, Guttormsen SA, Schultz JS. Linkage between the loci for the Lp(a) lipoprotein (LP) and plasminogen (PLG). Hum Genet. 1988 May;79(1):80-2.
131 Murray JC, Buetow KH, Donovan M, Hornung S, Motulsky AG, Disteche C, Dyer K, Swisshelm K, Anderson J, Giblett E, et al. Linkage disequilibrium of plasminogen polymorphisms and assignment of the gene to human chromosome 6q26-6q27. Am J Hum Genet. 1987 Apr;40(4):338-50.
132 Drayna DT, Hegele RA, Hass PE, Emi M, Wu LL, Eaton DL, Lawn RM, Williams RR, White RL, Lalouel JM. Genetic linkage between lipoprotein(a) phenotype and a DNA polymorphism in the plasminogen gene. Genomics. 1988 Oct;3(3):230-6.
133 Lindahl G, Gersdorf E, Menzel HJ, Duba C, Cleve H, Humphries S, Utermann G. The gene for the Lp(a)-specific glycoprotein is closely linked to the gene for plasminogen on chromosome 6. Hum Genet. 1989 Jan;81(2):149-52.

100
134 Frank SL, Klisak I, Sparkes RS, Mohandas T, Tomlinson JE, McLean JW, Lawn RM, Lusis AJ. The apolipoprotein(a) gene resides on human chromosome 6q26-27, in close proximity to the homologous gene for plasminogen. Hum Genet. 1988 Aug;79(4):352-6.
135 Lackner C, Cohen JC, Hobbs HH. Molecular definition of the extreme size polymorphism in apolipoprotein(a). Hum Mol Genet. 1993 Jul;2(7):933-40.
136 Real JT, Chaves FJ, Martín de Llano JJ, Ejarque I, Civera M, Ascaso JF, Knecht E, Armengod ME, Carmena R. Identificación y caracterización del primer español con defecto homocigoto familiar de unión de apolipoproteína B. Med Clin (Barc). 2001 Feb 3;116(4):138-41.
137 Ejarque I, Real JT, Chaves FJ, Blesa S, González V, Milián E, Ascaso JF, Priego MA, Carmena R. Estudio del defecto familiar de unión de la apolipoproteína B100 en una población mediterránea. An Med Interna. 2004 Jul;21(7):322-5.
138 Ejarque I, Real JT, Ascaso JF, Chaves FJ, Milian E, Priego MA, Carmena R. Estudio de los valores plasmáticos de Lp(a) en el defecto familiar de unión de la apo B 100 en una población mediterránea del sur de Europa. An Med Interna. 2004 Jul;21(7):322-5.
139 Real JT, Chaves FJ, Ejarque I, Garcia-Garcia AB, Valldecabres C, Ascaso JF, Armengod ME, Carmena R. Influence of LDL receptor gene mutations and the R3500Q mutation of the apoB gene on lipoprotein phenotype of familial hypercholesterolemic patients from a South European population. Eur J Hum Genet. 2003 Dec;11(12):959-65.
140 Gallagher JJ, Myant NB. The affinity of low-density lipoproteins and of very-low-density lipoprotein remnants for the low-density lipoprotein receptor in homozygous familial defective apolipoprotein B-100. Atherosclerosis. 1995 Jun;115(2):263-72.
141 Marz W, Ruzicka C, Pohl T, Usadel KH, Gross W. Familial defective apolipoprotein B-100: mild hypercholesterolaemia without atherosclerosis in a homozygous patient. Lancet. 1992 Nov 28;340(8831):1362.
142 Funke H, Rust S, Seedorf U, Brennhausen B, Chirazi A, Motti C, Assmann G. Homozygosity for familial defective apolipoprotein B-100 (FDB) is associated with lower plasma cholesterol concentrations than homozygosity for familial hypercholesterolemia (FH). Circulation 86 (Suppl I): 691, 1992.
143 Horinek A, Ceska R, Sobra J, Vrablik M. Familial defective apolipoprotein B-100 homozygote with premature coronary atherosclerosis. A case report. J Intern Med. 1999 Aug;246(2):235-6.
144 Gaffney D, Forster L, Caslake MJ, Bedford D, Stewart JP, Stewart G, Wieringa G, Dominiczak M, Miller JP, Packard CJ. Comparison of apolipoprotein B metabolism in familial defective apolipoprotein B and heterogeneous familial hypercholesterolemia. Atherosclerosis. 2002 May;162(1):33-43.

101
145 Schaefer JR, Scharnagl H, Baumstark MW, Schweer H, Zech LA, Seyberth H, Winkler K, Steinmetz A, Marz W. Homozygous familial defective apolipoprotein B-100. Enhanced removal of apolipoprotein E-containing VLDLs and decreased production of LDLs. Arterioscler Thromb Vasc Biol. 1997 Feb;17(2):348-53.
146 Pietzsch J, Wiedemann B, Julius U, Nitzsche S, Gehrisch S, Bergmann S, Leonhardt W, Jaross W, Hanefeld M. Increased clearance of low density lipoprotein precursors in patients with heterozygous familial defective apolipoprotein B-100: a stable isotope approach. J Lipid Res. 1996 Oct;37(10):2074-87.
147 Gudnason V, Day IN, Humphries SE. Effect on plasma lipid levels of different classes of mutations in the low-density lipoprotein receptor gene in patients with familial hypercholesterolemia. Arterioscler Thromb. 1994 Nov;14(11):1717-22.
148 Sun XM, Patel DD, Knight BL, Soutar AK, with the familial hypercholesterolaemia regression study group. Influence of genotype at the low density lipoprotein (LDL) receptor gene locus on the clinical phenotype and response to lipid-lowering drug therapy in heterozygous familial hypercholesterolaemia. Atherosclerosis. 1998 Jan;136(1):175-85.
149 Graham CA, McClean E, Ward AJ, Beattie ED, Martin S, O'Kane M, Young IS, Nicholls DP. Mutation screening and genotype:phenotype correlation in familial hypercholesterolaemia. Atherosclerosis. 1999 Dec;147(2):309-16.
150 Bertolini S, Cantafora A, Averna M, Cortese C, Motti C, Martini S, Pes G, Postiglione A, Stefanutti C, Blotta I, Pisciotta L, Rolleri M, Langheim S, Ghisellini M, Rabbone I, Calandra S. Clinical expression of familial hypercholesterolemia in clusters of mutations of the LDL receptor gene that cause a receptor-defective or receptor-negative phenotype. Arterioscler Thromb Vasc Biol. 2000 Sep;20(9):E41-52.
151 Moorjani S, Roy M, Torres A, Betard C, Gagne C, Lambert M, Brun D, Davignon J, Lupien P. Mutations of low-density-lipoprotein-receptor gene, variation in plasma cholesterol, and expression of coronary heart disease in homozygous familial hypercholesterolaemia. Lancet. 1993 May 22;341(8856):1303-6.
152 Betard C, Kessling AM, Roy M, Davignon J. Influence of genetic variability in the nondeletion LDL-receptor allele on phenotypic variation in French-Canadian familial hypercholesterolemia heterozygotes sharing a 'null' LDL-receptor gene defect. Atherosclerosis. 1996 Jan 5;119(1):43-55.
153 Ward AJ, O'Kane M, Nicholls DP, Young IS, Nevin NC, Graham CA. A novel single base deletion in the LDLR gene (211delG): Effect on serum lipid profiles and the influence of other genetic polymorphisms in the ACE, APOE and APOB genes. Atherosclerosis. 1996 Feb;120(1-2):83-91.
154 Jensen HK, Jensen LG, Hansen PS, Faergeman O, Gregersen N. The Trp23-Stop and Trp66-Gly mutations in the LDL receptor gene: common causes of familial hypercholesterolemia in Denmark. Atherosclerosis. 1996 Feb;120(1-2):57-65.

102
155 Schaefer JR, Winkler K, Schweer H, Hoffmann MM, Soufi M, Scharnagl H, Maisch B, Wieland H, Steinmetz A, Marz W. Increased production of HDL ApoA-I in homozygous familial defective ApoB-100. Arterioscler Thromb Vasc Biol. 2000 Jul;20(7):1796-9.
156 Abbasi F, Brown BW Jr, Lamendola C, McLaughlin T, Reaven GM. Relationship between obesity, insulin resistance, and coronary heart disease risk. J Am Coll Cardiol 2002;40:937-43.
157 Sijbrands EJ, Lombardi MP, Westendorp RG, Leuven JA, Meinders AE, Van der Laarse A, Frants RR, Havekes LM, Smelt AH. Similar response to simvastatin in patients heterozygous for familial hypercholesterolemia with mRNA negative and mRNA positive mutations. Atherosclerosis. 1998 Feb;136(2):247-54.
158 Mamo JC, Smith D, Yu KC, Kawaguchi A, Harada-Shiba M, Yamamura T, Yamamoto A. Accumulation of chylomicron remnants in homozygous subjects with familial hypercholesterolaemia. Eur J Clin Invest. 1998 May;28(5):379-84.
159 Castro-Cabezas M, De Bruin TWA, Westerveld HE, Meijer E, Erkelens DW. Delayed chylomicron remnant clearance in subjects with heterozygous familial hypercholesterolaemia. J Intern Med. 1998 Oct;244(4):299-307.
160 Carmena-Ramon R, Ascaso JF, Real JT, Ordovas JM, Carmena R. Genetic variation at the apoA-IV gene locus and response to diet in familial hypercholesterolemia. Arterioscler Thromb Vasc Biol. 1998 Aug;18(8):1266-74.
161 Pimstone SN, Sun XM, du Souich C, Frohlich JJ, Hayden MR, Soutar AK. Phenotypic variation in heterozygous familial hypercholesterolemia: a comparison of Chinese patients with the same or similar mutations in the LDL receptor gene in China or Canada. Arterioscler Thromb Vasc Biol. 1998 Feb;18(2):309-15.
162 Leitersdorf E, Eisenberg S, Eliav O, Friedlander Y, Berkman N, Dann EJ, Landsberger D, Sehayek E, Meiner V, Wurm M, et al. Genetic determinants of responsiveness to the HMG-CoA reductase inhibitor fluvastatin in patients with molecularly defined heterozygous familial hypercholesterolemia. Circulation. 1993 Apr;87(4 Suppl):III35-44.
163 Miserez AR, Keller U. Differences in the phenotypic characteristics of subjects with familial defective apolipoprotein B-100 and familial hypercholesterolemia. Arterioscler Thromb Vasc Biol 1995;15:1719-29.


















