







Idiomas
Páginas
Jurídico


UNIVERSIDAD NACIONAL DE SAN LUIS
MAESTRIA EN CIENCIAS DE SUPERFICIE Y MEDIOS POROSOS
ADSORCIÓN CON MÚLTIPLE OCUPACIÓN DE SITIOS
SOBRE SUPERFICIES HEXAGONALES Y TRIANGULARES
Maestrando: Ing. José Eduardo González
Director: Dr. Antonio José Ramirez Pastor
Co-Director: Dr. Víctor Daniel Pereyra
AÑO 2002

A mi familia.
A la memoria de mi padre y mi hermana.

AGRADECIMIENTOS
• A Antonio José Ramirez Pastor, por el respaldo profesional y humano
brindado permanentemente durante la realización de esta tesis.
• Al Director y Cuerpo de Profesores de la Maestría en Ciencias de Superficie
y Medios Porosos.
• A Víctor Pereyra y José Riccardo, por su generoso aporte a mi formación
profesional.
• A la Universidad Nacional de San Luis, por permitirme realizar
gratuitamente mis estudios de posgrado.
• A Federico José Romá, por la colaboración brindada.
• A mis compañeros de estudio: Rosa, Ana María, Dolly y Hugo, por todos los
momentos gratos compartidos.
* * * * * * * * *

INDICE
Capítulo 1: Introducción Pág. 1
1.1 Adsorción con múltiple ocupación de sitios: breve reseña de antecedentes. Pág. 31.2 Objetivos de la tesis. Pág. 41.3 Organización y secuencia de contenidos. Pág. 4
Capítulo 2: Adsorción de dímeros sobre redes homogéneas Pág. 7
2.1 Extensión a n dimensiones de los resultados exactos en una dimensión. Pág. 72.2 Teoría de Flory-Huggins de polímeros en solución. Pág. 142.3 Resumen. Pág. 17
Capítulo 3: Técnicas de simulación y diseño de redes Pág. 18
3.1 Simulación de Monte Carlo. Pág. 183.2 Cálculo de la entropía. Pág. 233.3 Diseño de redes. Pág. 253.4 Resumen. Pág. 31
Capítulo 4: Adsorción de dímeros sobre redes hexagonales homogéneas Pág. 32
4.1 Interacciones laterales atractivas. Pág. 324.2 Interacciones laterales repulsivas. Pág. 434.3 Resumen. Pág. 56

Capítulo 5: Adsorción de dímeros sobre redes triangulares homogéneas Pág. 58
5.1 Interacciones laterales atractivas. Pág. 585.2 Interacciones laterales repulsivas. Pág. 695.3 Resumen. Pág. 87
Capítulo 6: Adsorción de dímeros sobre redes heterogéneas hexagonales y triangulares Pág. 89
6.1 Modelos de adsorción sobre superficies heterogéneas sin interacciones laterales. Pág. 896.2 Modelos de adsorción sobre superficies heterogéneas con interacciones laterales. Pág. 946.3 Simulación de Monte Carlo: adsorción sobre superficies heterogéneas sin interacciones laterales. Pág. 986.4 Simulación de Monte Carlo: adsorción sobre superficies heterogéneas con interacciones laterales. Pág. 1136.5 Resumen . Pág. 133
Capítulo 7: Conclusiones y perspectivas Pág. 134
7.1 Conclusiones . Pág. 1347.2 Perspectivas. Pág. 136
* * * * * * * * * *

CAPITULO 1
INTRODUCCION
Los fenómenos de superficie juegan un importante papel en diferentes
aplicaciones que van desde la microelectrónica hasta a la producción química
de alto volumen. El estudio de estos fenómenos es también de interés en el
mundo académico ya que se trata de un campo multidisciplinario en el cual hay
muchos problemas interesantes aún no resueltos [1].
Los fenómenos superficiales son usualmente difíciles de observar y de
medir, y la moderna físico-química de superficies no habría sido posible sin el
desarrollo de las técnicas UHV (Ultra high vacuum) que permitieron preparar
sustratos limpios y bien definidos, sobre los cuales generalmente se realizan los
estudios de superficie [2]. Además, los fenómenos superficiales no habrían
alcanzado el presente nivel de entendimiento teórico sin la posibilidad de realizar
grandes y complejos cálculos computacionales [2].
La adsorción de moléculas sobre superficies sólidas constituye un tema
central dentro de la física de superficies, y es un fenómeno muy importante en
una gran variedad de procesos de mucho interés para la ciencia moderna, tales
como catálisis heterogénea, oxidación, lubricación, procesos separativos en
membranas, almacenamiento de gases en medios porosos, lo mismo que en
numerosas aplicaciones en la tecnología actual [3-8].
La situación anterior ha hecho necesario comprender los mecanismos
elementales involucrados en el proceso superficial mencionado. Con esta
finalidad se han desarrollado técnicas experimentales [9-23] tales como STEM
(scanning-transmission electron microscopy), STM (scanning tunneling

2
microscopy) o FIM (field ion microscopy), lo mismo que simulaciones
computacionales [24-35] las que, especialmente en las últimas décadas, han
cobrado importancia como una rama intermedia entre el experimento y la teoría,
ya que facilitan la interpretación de los datos experimentales, por un lado, y
permiten verificar la validez de las hipótesis y predicciones teóricas por otro.
La adsorción sobre superficies sólidas es un fenómeno complejo, ya que,
en general, las superficies no son uniformes sino que consisten de sitios con una
amplia distribución de energías de adsorción. Además, la densidad de sitios
puede ser muy alta o muy baja como para formar un continuo, y aquéllos
pueden ser independientes unos de otros, o bien interactuar con sus vecinos
próximos, y aún con los más lejanos. Asimismo, las fuerzas que unen las
moléculas de adsorbato a una superficie pueden ser de origen físico, o bien
tratarse de fuerzas equivalentes a las existentes en los enlaces químicos, las
cuales involucran el solapamiento de orbitales [36].
Teóricamente la adsorción se ha descripto principalmente en función de la
adsorción localizada [36-39], ya que ésta presenta menores dificultades de
modelado analítico. Los trabajos comprendidos en este grupo se basan en el
modelo de adsorción de Langmuir, o en generalizaciones del mismo que tienen
en cuenta las interacciones entre moléculas adsorbidas [40-41], la heterogeneidad
superficial [37, 42-49] o los efectos de adsorción en multicapas [37, 50-52].
Estas teorías conservan una característica fundamental del modelo de
Langmuir, la de considerar que cada molécula adsorbida ocupa un sitio sobre la
red de sitios de adsorción, considerando generalmente que un sitio se
corresponde en dimensiones con el tamaño de la molécula adsorbida.
Si bien el concepto de adsorción unisitio puede aceptarse en el caso de
moléculas compactas, falla totalmente en casos como el de la adsorción de
cadenas de hidrocarburos, que son altamente flexibles y cuyos grupos CHx
tienden a adsorberse como segmentos individuales. De ahí que sea necesario
incorporar la denominada adsorción con múltiple ocupación de sitios que, según

3
varios autores, constituye uno de los temas principales en la moderna
físicoquímica de superficies, y en los que el modelado teórico y de simulación
numérica están aún en etapas primarias de desarrollo [42, 53-58].
Es por esta razón, que un objetivo de la presente Tesis, es el de contribuir
al entendimiento de la termodinámica de adsorción de sistemas con múltiple
ocupación de sitios.
1.1 ADSORCIÓN CON MÚLTIPLE OCUPACIÓN DE SITIOS: BREVERESEÑA DE ANTECEDENTES
Los primeros antecedentes sobre estudios de adsorción con múltiple
ocupación de sitios sobre superficies homogéneas se remontan a la década del
30, y se relacionan a problemas de soluciones binarias compuestas por molécula
de polímeros disueltas en un solvente monomérico, las que constituyen un
sistema isomorfo con el de adsorción de cadenas lineales de k segmentos iguales
(k-meros) sobre una red regular de sitios.
La teoría fue desarrollada independientemente por Flory [53] y por
Huggins[54-56], como una generalización de la aproximación de Bragg-
Williams en el modelo de red de soluciones binarias compuestas por especies de
igual tamaño [38].
Posteriormente, diversos motivos llevaron a estudiar el problema, entre
ellos cabe mencionar: la posibilidad de modelar pares de Cooper [59]; la difusión
de dímeros sobre metales [60-61]; la adsorción de H2O / Ni (110) [62]; la
migración de Pt2, Pt3, Pt4 sobre tungsteno [63]; y la adsorción de cadenas de
hidrocarburos. Mediante la técnica de STM se han podido observar algunos de
estos sistemas, lo que ha permitido corroborar las hipótesis sobre múltiple
ocupación de sitios.
Además de la metodología de gas de red, empleada por Flory y por
Huggins, existen otros métodos para estudiar el problema, los que se basan en

4
relaciones recursivas para la energía libre [64] y en cálculos de matriz de
transferencia [65].
En los últimos años, Marczewski [57] y Nitta [58] realizaron una
contribución importante al tema al incluir la heterogeneidad superficial en los
estudios de adsorción con múltiple ocupación de sitio, agregando así mayor
complejidad y realismo a los modelos existentes hasta entonces.
Por último, cabe destacar las aproximaciones teóricas desarrolladas por
Ramírez Pastor [66] para describir la adsorción de moléculas poliatómicas sobre
superficies homogéneas y heterogéneas.
1.2 OBJETIVOS DE LA TESIS
En base a lo expresado anteriormente, los objetivos de la presente tesis
pueden resumirse así:
a) Estudiar la termodinámica de adsorción de sistemas formados por dímeros
interactuantes adsorbidos sobre redes homogéneas hexagonales y
triangulares. Para ello, extender la aplicación de modelos teóricos de
adsorción existentes al caso de las redes mencionadas.
b) Diseñar las redes triangular y hexagonal que representarán al sustrato en los
procesos de adsorción. Asimismo diseñar algoritmos y metodologías de
simulación de Monte Carlo, y comparar los resultados obtenidos
computacionalmente con los que resultan de la aplicación de los modelos
existentes.
c) Estudiar el efecto de las heterogeneidades sobre la adsorción de dímeros en
redes bidimensionales, hexagonales y triangulares.
d) Desarrollar un método de caracterización de la topografía energética de
sustratos que pueden aproximarse por medio de superficies bivariadas.

5
1.3 ORGANIZACIÓN Y SECUENCIA DE CONTENIDOS
La secuencia de contenidos de la tesis es la siguiente:
En el capítulo 2 se detallan las teorías sobre adsorción con múltiple
ocupación de sitios sobre superficies homogéneas, entre ellas, la extensión a n-
dimensiones de los resultados exactos en una dimensión, desarrollada por
Ramírez Pastor, y la aproximación de Flory y Huggins.
En el capítulo 3 se describen las técnicas de simulación de Monte Carlo
[24,25] que se utilizan luego en dos formas diferentes: 1) Para analizar el alcance
de diferentes modelos teóricos referidos a multiple ocupación de sitios y 2) Para
acceder a condiciones en las cuales las teorías existentes no dan una descripción
acabada del sistema en estudio. También se presentan las distintas cantidades
termodinámicas a medir por simulación y se describe la forma de determinar la
entropía del sistema en estudio. Por último, también se efectúa el diseño de las
redes triangular y hexagonal que luego se utilizan para representar el sustrato en
los procesos de adsorción y se indican los aspectos básicos de los programas
computacionales diseñados para trabajar con las redes hexagonal y triangular.
En el capítulo 4 se estudia la adsorción de dímeros sobre superficies
homogéneas hexagonales, utilizándose las técnicas de simulación de Monte
Carlo para analizar el comportamiento de diversas cantidades termodinámicas
asociadas a la capa adsorbida. Asimismo, se comparan los resultados que se
obtienen por aplicación de los modelos teóricos mencionados, con los obtenidos
a partir de las técnicas de simulación.

6
En el capítulo 5 se estudia la adsorción de dímeros sobre superficies
homogéneas triangulares, empleándose las técnicas de simulación de Monte
Carlo para analizar el comportamiento de distintas cantidades termodinámicas
asociadas a la capa adsorbida. Luego se comparan los resultados obtenidos por
aplicación de los modelos teóricos mencionados con los que se obtienen a partir
de las técnicas de simulación.
En el capítulo 6 se presentan distintas teorías de adsorción con múltiple
ocupación de sitios sobre superficies heterogéneas. Luego se comparan los
resultados que resultan de la aplicación de dichas teorías al caso de redes
hexagonales y triangulares con los que se obtienen mediante simulación de
Monte Carlo. Finalmente se desarrolla un método de caracterización de la
topografía energética de sustratos que pueden aproximarse por medio de
superficies bivariadas.
Por último, en el capítulo 7 se resumen las principales conclusiones
obtenidas, indicándose también las perspectivas que cabe esperar de los estudios
realizados en la presente tesis.
* * * * * * * * * *

7
CAPITULO 2
ADSORCION DE DIMEROS SOBRE REDES HOMOGENEAS
Desde hace varias décadas se viene estudiando la adsorción de moléculas
poliatómicas, asociada a sistemas de mezclas binarias de polímeros (k-meros) en
solventes monoméricos, problema que es isomorfo con el de adsorción con
múltiple ocupación de sitios. Una de las características más importantes de este
tipo de sistemas es que las propiedades termodinámicas de la capa adsorbida son
muy afectadas por la estructura de las moléculas que forman el adsorbato,
incluso cuando las moléculas adsorbidas no interactúan entre sí.
En este capítulo se detallan algunas teorías sobre adsorción con múltiple
ocupación de sitios sobre superficies homogéneas, entre ellas, la extensión a n-
dimensiones de los resultados exactos obtenidos para una dimensión,
desarrollada en REF[66], y la aproximación de Flory y Huggins [53-56].
2.1 EXTENSIÓN A N DIMENSIONES DE LOS RESULTADOSEXACTOS OBTENIDOS EN UNA DIMENSION
2.1.1 Adsorción sin interacciones laterales
Esta aproximación, que fue desarrollada por Ramirez Pastor y col.[66], y a
la que en adelante llamaremos aproximación (RP), considera inicialmente una
red unidimensional de M sitios (distinguibles e independientes), de constante de

8
red a y con condiciones periódicas de borde. Bajo éstas hipótesis todos los sitios
son equivalentes, evitándose así problemas de contorno.
También se considera que N moléculas lineales, constituidas por k
segmentos iguales (k-meros), están adsorbidas sobre la red, de modo que cada
sitio puede estar vacío u ocupado por un segmento de adsorbato. Se prohibe la
doble ocupación de un mismo sitio, y se restringe el estudio al régimen de
monocapa. Las moléculas de adsorbato proceden de una fase gaseosa en
equilibrio con la fase adsorbida (o gas de red).
Se supone que las fuerzas que ligan al sólido (adsorbente) son mucho
mayores que las fuerzas de adsorción de modo que el sólido no es perturbado
por la presencia de las moléculas de gas sobre su superficie, y es el responsable
del campo de potencial para las moléculas de adsorbato, o sea que el sistema
termodinámico que se estudia es un gas de moléculas ligadas al campo de
potencial mencionado. Se propone también un potencial del tipo Lennard-Jones
para la energía de interacción entre un segmento k-mero y cada átomo del sólido,
y se describen las propiedades considerando que la adsorción es localizada, y que
las moléculas de adsorbato se adsorben y desorben como un todo, sin tener en
cuenta efectos de disociación o religadura de aquéllas [67].
Se considera, además, que la superficie de la red es homogénea, o sea que
todos los sitios, con energía U0, son equivalentes para la adsorción. Luego, un k-
mero adsorbido sobre la superficie tiene una energía de adsorción Ek dada por:
Ek = k U0 (2.1), donde:
k = número de segmentos que forman la molécula de adsorbato.
U0 = Energía de adsorción para cada segmento.
También se supone que las moléculas de adsorbato no interactúan
(adsorción sin interacciones laterales), por lo tanto, todas las configuraciones
posibles de N k-meros sobre M sitios son igualmente probables. Así, la función
de partición canónica es:

9
Q(M,N,T) = Ω(M,N)exp(–βN Ek) (2.2), donde:
Ω(M,N) = Número total de configuraciones posibles de N k-meros sobre M
sitios.
Ek = Energía de un k-mero adsorbido.
β =1/kBT
Ω(M,N) se puede calcular como el número total de permutaciones de N k-
meros indistinguibles sobre ne entidades, donde:
ne = n° de k-meros + n° de sitios vacíos
ne = N + M – kN = M – (k – 1)N (2.3)
Luego:
ne
Ω(M,N) = N = [M – (k–1).N]!/N![M – kN]! (2.4)
En el conjunto canónico, la energía libre de Helmholtz, F(M,N,T),
resulta:
β F(M,N,T) = – lnQ (M,N,T) = – ln Ω(M,N) + βN Ek (2.5)
Las principales funciones termodinámicas se obtienen de la siguiente
forma diferencial general [38]:
dF = – SdT – πdM + µdN (2.6)
donde S, π, y µ representan la entropía, la presión (spreading pressure1) y
el potencial químico, respectivamente, los cuales resultan:
S = – (∂F/∂T)M,N; π = – (∂F/∂M)T,N; µ = – (∂F/∂N)T,M (2.7)
De (2.4) y (2.5), se tiene: 1 π representa, salvo por una constante, la llamada “spreading pressure’’, definida así: φ = (∂F/∂A)T,N,donde A es el área del adsorbente. Luego: π = constante.φ.

10
β F(M,N,T) = – ln [M – (k–1).N]! – lnN! – ln[M – kN! + βN Ek (2.8)
Usando la aproximación de Stirling, lnx! ≅ xlnx – x, la (2.8) se puede escribir:
β F(M,N,T) = – [M – (k–1).N ln [M – (k–1).N] + NlnN
+ [M – kN]ln[M – kN] + βN Ek (2.9)
Derivando la energía libre de Helmholtz con respecto a la temperatura,
manteniendo N y M constantes, se obtiene una expresión para la entropía:
S(M,N)/kB = [M – (k–1).N] ln [M – (k–1).N] – NlnN
– (M – kN)ln(M – kN) (2.10)
Derivando la energía libre de Helmholtz con respecto al número de sitios,
manteniendo N y T constantes, se obtiene una expresión para la presión:
βπ(M,N,T) = ln [M – (k–1).N] – ln(M – kN) (2.11)
Derivando la energía libre de Helmholtz con respecto al número de
partículas, manteniendo M y T constantes, se obtiene una expresión para el
potencial químico:
βµ(M,N,T) = ln(kN/M) + (k – 1) ln 1 – [(k–1).N/M]
– kln[1 – (kN/M)] + β Ek (2.12)
Definiendo el cubrimiento, θ = kN/M, la energía libre por sitio f = S/M, y
la entropía por sitio s = S/M, las ecuaciones (2.9) a (2.12) se pueden escribir en
función de las variables intensivas θ y T.
βf(θ,T) = – [1 – (k – 1)(θ/k)]ln[1 – (k – 1)(θ/k)] + (θ/k)ln(θ/k)
+ (1 – θ)ln(1 – θ) + βθUo (2.13)

11
s(θ)/kB = [1 – (k – 1)(θ/k)]ln[1 – (k – 1)(θ/k)] – (θ/k)ln(θ/k)
– (1 – θ)ln(1 – θ) (2.14)
exp(βπ) = [1 – (k – 1)(θ/k)]/ (1 – θ) (2.15)
y(θ) = kexp[β(µ–k.Uo)] = [θ/(1–θ)k][1–(k–1)(θ/k)](k-1) (2.16)
La (2.16) es la ecuación de la isoterma de adsorción para un gas de red de k–
meros, sin interacciones laterales en 1-D.
A partir de la función de partición calculada en 1-D, se obtiene una
expresión aproximada para la función de partición de un gas de k-meros no
interactuantes, adsorbidos sobre un sustrato en n dimensiones.
La dimensionalidad se introduce a través de un número de coordinación o
conectividad de la red, c (donde c es el número de primeros vecinos de un sitio
cualquiera, por ejemplo, c vale 3 para red hexagonal, y 6 para triangular). Esta es
una forma efectiva de considerar la conectividad del sistema ya que redes de
igual conectividad pueden corresponder a problemas de dimensionalidad
diferente (por ejemplo, c es igual a 6 para una red triangular en dos dimensiones
y para una red cúbica simple en 3 dimensiones). Así, se supone que la cantidad
Ω expresada en la ecuación (2.4) para M y N dados, también es función de la
conectividad c, o sea que Ω ≡ Ω(M,N,c).
Ω(M,N,c) se calcula con un criterio usado por diferentes autores [53,
68, 69], el cual permite relacionar Ω(M,N,c) con la misma cantidad obtenida en
1-D, Ω(M,N,c =2), a través de la siguiente expresión:
Ω(M,N,c)/ Ω(M,N,2) = K(c,k)N (2.17)
donde K(c,k) depende de la conectividad de la red, y del tamaño de las moléculas
de adsorbato. En el caso particular de k-meros lineales, K(c,k) = c/2.

12
Bajo esta hipótesis, la ecuación (2.5) se reescribe así:
β F(M,N,c,T) = – lnQ(M,N,c,T) = – ln Ω(M,N,c) + βNEk (2.18)
donde, por la ecuación (2.17):
lnΩ(M,N,c) = lnΩ(M,N,2) + NlnK(c,k) (2.19)
Las principales funciones termodinámicas se recalculan a partir de las
ecuaciones (2.13) a (2.16), generalizándose ahora para una conectividad dada c:
β f(θ,c,T) = – [1 – (k – 1)(θ/k)]ln[1 – (k – 1)(θ/k)] + (θ/k)ln(θ/k)
+ (1 - θ)ln(1 - θ) + βθUo – (θ/k)lnK(c,k) (2.20)
s(θ,c)/kB = [1 – (k – 1)(θ/k)]ln[1 – (k – 1)(θ/k)] – (θ/k)ln(θ/k)
– (1 – θ)ln(1 – θ) + (θ/k)lnK(c,k) (2.21)
exp[βπ(θ,c)] = [1 – (k – 1)(θ/k)]/ (1 – θ) (2.22)
y(θ,c) = k K(c,k)exp[β(µ–k.Uo)] = [θ/(1 – θ)k][1 – (k–1)(θ/k)](k - 1) (2.23)
Las ecuaciones (2.20) a (2.23) dan las funciones termodinámicas básicas
para estudiar la adsorción de k-meros no interactuantes sobre una red de
conectividad c.
2.1.2 Adsorción con interacciones laterales
Las interacciones laterales se introducen mediante la aproximación de
Bragg-Williams o campo medio, que es la forma más sencilla de obtener
algunas características cualitativas importantes del efecto de dichas interacciones
sobre la adsorción de k-meros.

13
En la aproximación de campo medio para un gas de red con interacciones
laterales de magnitud w entre primeros vecinos ocupados, el factor
configuracional se calcula suponiendo que las moléculas de adsorbato están
distribuidas al azar (o sea, suponiendo w = 0), por lo tanto esta aproximación es
correcta en el límite termodinámico de T Õ ∞. Luego, la función de partición del
sistema, en el conjunto canónico. se puede escribir así:
Q(M,N,c,T) = K(c,k)N[M– (k–1).N]!/N![M – kN]!exp(–βNEk)exp(–βN11mw)
(2.24)
donde:
N11m = número medio de pares de sitios ocupados, cada uno de los cuales
contribuye con una energía w, y donde cada sitio del par corresponde a un k-
mero distinto.
N11mw = energía de interacción promedio.
exp(– β N11mw) = término de campo medio
Luego se calcula N11m considerando que un k-mero lineal, adsorbido
sobre una red de conectividad c, tiene (c – 1) vecinos en sus extremos, y (c – 2)
vecinos en los restantes (k – 2) componentes, o sea que el número total de
primeros vecinos de un k-mero, 2λ , es:
2λ = [2(c–1) + (k–2)(c–2)]
La probabilidad de que estos vecinos estén ocupados es: θ = kN/M
Luego el número de pares de sitios ocupados, para N k-meros adsorbidos es:
N11m = (1/2) (n° de k-meros).(n° de vecinos).(prob. de ocupación de cada vecino)
N11m = (1/2)N.[2(c – 1) + (k – 2).(c – 2)].[kN/M]
N11m = λkN2/M (2.25)
donde el (1/2) se introduce para evitar el doble conteo de cada par.
Por otro lado, Ekm = kNUo + λN(kN/M)w (2.26)
Usando la ecuación (2.24) se escribe:

14
ln[Q(M,N,c,T)] = ln[Ω(M,N,c)] – βNEk – β N11m w
ln[Q(M,N,c,T)] = ln[M – (k–1).N]! – lnN! – ln [M – kN]!
+ NlnK(c,k) – βkNUo – βwλkN2/M (2.27)
La energía libre de Helmholtz por sitio, en función de θ = kN/M, resulta:
β f(θ,c,T) = – [1 – (k – 1)(θ/k)]ln[1 – (k – 1)(θ/k)] + (θ/k)ln(θ/k)
+ (1 - θ)ln(1 - θ) + βθUo + βwλ θ2/k – (θ/k)lnK(c,k) (2.28)
A partir de la ecuación anterior se pueden deducir las funciones
termodinámicas de interés:
s(θ,c)/kB = [1 – (k – 1)(θ/k)]ln[1 – (k – 1)(θ/k)] – (θ/k)ln(θ/k)
– (1 – θ)ln(1 – θ) + (θ/k)lnK(c,k) (2.29)
Esta ecuación, que da la entropía por sitio, es igual que la calculada para el gas
de red ideal (2.21), debido a que en la aproximación de Bragg-Williams se
supone que las moléculas se distribuyen al azar sobre la superficie.
Además, se tienen:
La presión bidimensional:
βπ(θ,c,T) = ln[1 – (k – 1)(θ/k)] – ln(1 – θ) + βwλkθ2/k (2.30)
La isoterma de adsorción:
kK(c,k)exp[β(µ – k.Uo)] = [θ/(1 – θ)k][1 – (k – 1)(θ/k)](k - 1)exp(2βλwθ) (2.31)
El factor termodinámico:
th = [∂(βµ)/∂lnθ]T = (1 - θ)[1 - θ (k -1)/k) ]-1 + 2βλwθ (2.32)
El calor diferencial de adsorción:
qd = - 2λwθ - kUo (2.33)

15
La energía de adsorción total por sitio:
ε(θ) = θUo + λwθ2/k (2.34)
2.2 TEORIA DE FLORY-HUGGINS (F-H) DE POLIMEROS ENSOLUCION
La adsorción con múltiple ocupación de sitios sobre superficies
homogéneas comenzó a estudiarse en la década del treinta, en relación con
problemas de soluciones binarias compuestas por moléculas de k segmentos
(polímeros) disueltas en un solvente monomérico. Este sistema es isomorfo con
la adsorción de k-meros sobre una red regular, que es el tema que estamos
tratando.
2.2.1 Adsorción sin interacciones laterales
La teoría que se presenta aquí fue desarrollada en forma independiente
por Flory [53,68] y Huggins [54-56].
El sistema que se estudia es una solución condensada, incompresible, y a
temperatura T, compuesta por N1 moléculas de solvente, cada una de las cuales
ocupa un sitio sobre una red rígida, y N2 moléculas de polímero, cada una de las
cuales ocupa k sitios. Cada sitio tiene c primeros vecinos, y no hay sitios vacíos.
Por lo tanto, el tamaño M de la red es: M = N1 + k N2.
Se supone que las moléculas presentes en la red no interactúan entre sí, o
sea w = 0, y que no hay energía relacionada a la existencia de polímero o
solvente sobre la red (en adsorción equivale a trabajar con Uo = 0).
Aunque no aparecen restricciones sobre la forma de las cadenas del polímero,
suposiciones relacionadas con la forma de colocar las cadenas, indican que son
estrictamente válidas para polímeros lineales.
Las fracciones ocupadas por solvente y polímero se definen así:

16
θ1 = N1/(N1 + k N2) (2.35)
θ2 = k N2/(N1 + k N2) (2.36)
siendo θ1 + θ2 = 1 (2.37)
El sistema descripto es isomorfo al de adsorción con múltiple ocupación
de sitios, ya que la fracción de sitios ocupados por los k-meros, θ, equivale a la
fracción ocupada por el polímero, θ2, y la fracción de sitios vacíos, 1 - θ, es
equivalente a la fracción ocupada por el solvente, θ1.
Por un procedimiento semejante al visto en la sección 3.1.1, se obtiene la
siguiente ecuación para la función de partición Q del sistema en estudio:
lnQ(N1, N2) = – N2lnN2 + N2 – N1lnN1 + N1 + MlnM
– M + N2(k – 1)ln[(c – 1)/e] (2.38)
A partir de la ecuación anterior, la energía molar libre de Helmholtz en
función de θ1 y θ2 , se escribe:
βf(θ1,θ2) = (θ2/k)ln(θ2/k) + θ1lnθ1 - [(k-1)/k] θ2ln[(c – 1)/e] (2.39)
La ecuación anterior se puede expresar en función de las variables del
problema de múltiple ocupación de sitios, así:
βf(θ) = (θ/k)ln(θ/k) + (1 – θ)ln(1 – θ) – [(k – 1)/k]θln[(c – 1)/e] (2.40)
Análogamente para las otras funciones termodinámicas vistas:
s(θ,)/kB = – (θ/k)ln(θ/k) – (1 – θ)ln(1 – θ) + [(k – 1)(θ/k)] ln[(c – 1)/e] (2.41)
exp(βπ) = exp[1 – (k – 1)(θ/k)]/(1 – θ) (2.42)

17
y(θ) = k K(c,k)exp(βµ)] = [θ/(1 – θ)k] (2.43)
2.2.2 Adsorción con interacciones laterales
Flory y Huggins introducen las interacciones laterales, entre moléculas,
w, con una técnica semejante a la vista en la sección 2.1.2.
A partir de las ecuaciones (2.24) y (2.25) se escriben nuevas expresiones
para las ecuaciones (2.38) y (2.39):
ln[Q(N1, N2)] = – N2lnN2 + N2 – N1lnN1 + N1 + MlnM – M
+ N2(k – 1)ln[(c – 1)/M] – βwλkN2/M (2.44)
βf(θ1,θ2) = (θ2/k)ln(θ2/k) + θ1lnθ1 - [(k-1)/k] θ2ln[(c – 1)/e]
+ βwλ θ22/ k (2.45)
Expresando la ecuación anterior en función de θ, se tiene:
βf(θ) = (θ/k)ln(θ/k) + (1 – θ)ln(1 – θ) – [(k-1)/k] θln[(c – 1)/e]
+ βwλ θ2/ k (2.46)
Usando la ecuación (2.46) y en base a la teoría de Flory-Huggins, se
deducen las cantidades termodinámicas ya vistas en las ecuaciones (2.29) a
(2.34).
s(θ,)/kB = – (θ/k)ln(θ/k) – (1 – θ)ln(1 – θ) + [(k – 1)(θ/k)] ln[(c – 1)/e] (2.47)
exp(βπ) = exp[1 – (k – 1)(θ/k)]exp[βwλ θ22/ k]/(1 – θ) (2.48)
(θ) = kK(c,k)exp(βµ)] = [θ/(1 – θ)k] exp(2 βwλ θ) (2.49)
th = [∂(βµ)/∂lnθ]T = [1 + (k-1)θ]/(1 - θ)] + 2βλwθ (2.50)

18
ε = λwθ2/k (2.51)
qd = - 2λwθ (2.52)
2.3 RESUMEN
En este capítulo se describieron algunas teorías sobre adsorción con
múltiple ocupación de sitios sobre superficies homogéneas, entre ellas, la
extensión a n-dimensiones de los resultados exactos en 1 – D, y la aproximación
de Flory y Huggins.
* * * * * * * * *

19
CAPITULO 3
TECNICAS DE SIMULACION Y DISEÑO DE REDES
En el presente capítulo se describen las técnicas de simulación de Monte
Carlo [24] que se utilizarán luego en dos formas diferentes: 1) Para analizar el
alcance de diferentes modelos teóricos referidos a multiple ocupación de sitios y
2) Para acceder a condiciones en las cuales las teorías existentes no dan una
descripción acabada del sistema en estudio. También se presentan las distintas
cantidades termodinámicas a medir por simulación, y se describe la forma de
determinar la entropía del sistema en estudio, ya que esta propiedad requiere de
técnicas especiales para su obtención. Por último, se diseñan las redes triangular
y hexagonal que posteriormente se usarán para representar al sustrato en las
simulaciones computacionales, y se indican los aspectos básicos de los
programas computacionales diseñados para trabajar con las redes hexagonal y
triangular.
3.1 SIMULACION DE MONTE CARLO
El método de Monte Carlo fue desarrollado a fines de la segunda guerra
mundial por von Neumann, Ulam y Metrópolis con el objeto de estudiar la
difusión de neutrones en materiales fisionables. El nombre de Monte Carlo se lo
asignó Metrópolis debido al empleo de números aleatorios en los cálculos [24].
El sistema termodinámico a estudiar consiste en una red regular
homogénea (hexagonal o triangular), cuyos sitios de adsorción se ubican en
posiciones fijas sobre la misma. Esta red juega el papel de sustrato, en contacto

20
con el cual se encuentra una fase gaseosa a temperatura T, constituida por
dímeros homonucleares, modelados como dos centros de interacción separados
una distancia fija igual al parámetro de red a. En todos los casos, y para evitar
efectos de borde, se usan condiciones periódicas de contorno. Se supone que las
energías de adsorción son lo suficientemente pequeñas como para no romper los
enlaces del dímero, de modo que las moléculas de adsorbato se adsorben y
desorben como un todo. Además, se considera que no existen posibilidades de
migración sobre la superficie (modelo de adsorción localizada).
Con el objeto de definir el Hamiltoniano H, para un conjunto de N dímeros
adsorbidos sobre M sitios, a temperatura T, se introduce la variable ci,j, la cual
puede tomar los siguientes valores: ci,j = 0, si el correspondiente sitio (i,j) está
vacío, y ci,j = 1, si el correspondiente sitio (i,j) está ocupado por uno de los
elementos del dímero. Con estas consideraciones, H viene dado por:
H = wΣ cij.ci’j’ − Nw + ΣUi,j.ci,j (3.1) <(i,j),(i’,j’)> (i,j)
donde w es la constante de interacción a primeros vecinos, la cual puede ser
positiva (caso repulsivo) o negativa (caso atractivo) y <(i,j),(i’,j’)> representa
todos los pares de primeros vecinos. El primer término de la ecuación (3.1)
representa el aporte de energía de todos los pares de sitios ocupados de la red,
mientras que el segundo término resta las interacciones entre los sitios ocupados
que pertenecen a un mismo dímero. En el tercer término de (3.1), Ui,j, representa
la interacción entre una unidad de cualquier dímero y un sitio(i,j); por
simplicidad, se considerará Ui,j = Uo = 0 para una superficie homogénea.

21
3.1.1 Simulación de Monte Carlo en el Conjunto Gran Canónico
En el conjunto Gran Canónico se trabaja con la temperatura T, el potencial
químico µ y el volumen V del sistema como parámetros termodinámicos fijos,
siendo variable el número N de moléculas de adsorbato.
En el conjunto Gran Canónico, la probabilidad P(N,X) [27,28], de tener N
moléculas adsorbidas en un estado dado de ocupación X, definido por el
conjunto de valores ci,j de las N moléculas, puede escribirse así:
P(N,X) = [1/Q(µ,T,V)]exp[ − 1/kBT][H(X) − µ Σcij] (3.2) (i,j)
donde Q(µ,T,V) es la gran función de partición y H(X) viene dado por la
ecuación (3.1).
Continuando con el esquema de Metrópolis [24,25] la probabilidad de
transición W (Xi → Xf) desde un estado inicial Xi, a uno final Xf, viene dado
por:
W (Xi → Xf) = mín1, [P(Xf)/P(Xi)] (3.3)
la expresión anterior satisface el principio de reversibilidad microscópica, y es
condición suficiente para obtener una cadena con una distribución estacionaria
de estados dada por la ecuación (3.2).
Una vez definida la probabilidad de transición, el equilibrio de adsorción-
desorción se alcanza mediante un algoritmo tipo “spin flip”(dinámica de
Glauber), aplicado a moléculas biatómicas, y que se puede describir de la forma
que se indica a continuación para un paso elemental de Monte Carlo (MCs) [41].
Dado el substrato con las energías de adsorción ya asignadas:
1) Se fijan un valor de µ y uno de T.
2) Se fija un estado inicial de cubrimiento XN sobre la red, colocando
N moléculas sobre 2N sitios de adsorción.

22
3) Se elige al azar una dupla lineal de sitios vecinos próximos en la
red, y se genera un número aleatorio ξ ∈ [0,1].
- Si la dupla elegida está vacía, y ξ ≤ W (XN → XN+1)
se adsorbe un dímero.
- Si la dupla elegida está ocupada por un dímero,
éste se desorbe si ξ ≤ W(XN → XN-1).
- En cualquier otro caso se vuelve a (3).
4) Se repite desde (3) M veces.
En todas las medidas se descartan al comienzo una cantidad m’ de MCs
hasta alcanzar el régimen de equilibrio e independizarse de las condiciones
iniciales, y luego se promedia sobre un número m de MCs. Los valores de m’ y
m dependen de la propiedad que quiera calcularse, y de las condiciones
termodinámicas o región del diagrama de fases en la que se está trabajando. En
ningún caso se usan valores de m’ y m que conduzcan a errores mayores que un
5%.
A continuación se presentan las cantidades termodinámicas medidas en
este esquema de simulación, recordando que ellas se calculan para distintos
valores de µ y T.
a) Isoterma de adsorción o cubrimiento medio (θθ).
θ(µ) = (1/M)<N>T = (1/M) Σ<cij>T (3.4) (i,j)
b) Factor termodinámico o inversa de las fluctuaciones cuadráticas medias normalizadas del cubrimiento (th).
th = [(<(δN)2>T)/<N>T]−1 = [(<N2>T − <N>T2)/<N>T]−1 (3.5)

23
c) Energía media de adsorción por sitio (εε).
ε = (1/M)<H>T (3.6)
d)Calor diferencial de adsorción (qd).
El calor diferencial de adsorción se puede obtener de la siguiente manera.
A partir de la gran función de partición Q(µ,T,M), se puede deducir una
conocida relación [70]:
(∂lnz/∂β)<N> = ∂<E>/∂<N> (3.7)
donde <E> = <H>, β = 1/kBT y z = exp(µ/kBT)/[h3(2πmkBT)3/2]. Para un gas
ideal, z → kBTP. Finalmente [71],
qd = RT2 (∂lnP/∂T)<N> - RT = - (∂<E>/∂<N>)
qd = (- <EN> - <E><N>)/(<N2> - <N>2) (3.8)
R es la constante de los gases (R/kB = Número de Avogadro); <EN>, <E>, <N>,
<N2> y <N>2 se pueden evaluar por simulación de Monte Carlo a µ y T
constantes.
En todos los casos, con <--->T se representa el llamado promedio
térmico sobre los m pasos de Montecarlo antes mencionados, después de haber
descartado m’ pasos de Montecarlo.
3.1.2 Simulación de Monte Carlo en el Conjunto Canónico
Se considera el sistema a estudiar a temperatura T y volumen V
constantes, y con un número fijo N de moléculas de adsorbato.
La probabilidad P(N,X) [27,28] de tener N moléculas adsorbidas en un
estado dado de ocupación X, definido por el conjunto de valores cij de las N
moléculas en el conjunto canónico es:
P(N,X) = [1/Z(N,T,V)]exp[− H(X)/kBT] (3.9)

24
donde H(X) viene dado por la ecuación (3.1) y Z(N,T,V) es la función de
partición canónica.
Siguiendo el esquema de Metrópolis [24,25], la probabilidad de transición
W (Xi → Xf) desde un estado inicial Xi, a uno final Xf, viene dado por:
W (Xi → Xf) = min[1,exp(−δH/kBT)] = min1,exp[(−)(H(Xf) − H(Xi))/kBT]
(3.10)
donde δH es la diferencia de energías entre los estados finales e iniciales del
sistema. La expresión (3.10) satisface el principio de reversibilidad
microscópica.
Definida la probabilidad de transición, el equilibrio se alcanza mediante un
algoritmo del tipo “intercambio de spin”(dinámica de Kawasaki), generalizado a
moléculas poliatómicas, y que se puede describir como se indica a continuación
para un paso elemental de Monte Carlo (MCs) [41]:
Dado el sustrato con las energías de adsorción ya asignadas:
1) Se fija un valor de T y se distribuyen N dímeros sobre la red.
2) Se selecciona al azar un dímero y una dupla lineal de sitios vecinos
próximos vacíos cuyas ocupaciones se intenta intercambiar.
3) Se calcula δδH y con ella W(δδH) para el posible cambio, y se genera
un número aleatorio ξ ∈ [0,1].
- el cambio se realiza si ξ ≤ W(δδH).
- en cualquier caso se vuelve a 2
4) Se repite M veces desde (2).
En todas las medidas se descartan al comienzo una cantidad m’ de MCs
hasta alcanzar el régimen de equilibrio, y luego se promedia sobre un número m
de MCs. Los valores de m’ y m dependen de la propiedad que quiera calcularse

25
Mediante este esquema de simulación se determina la energía media de
adsorción por sitio, la cual se emplea posteriormente para la determinación de la
entropía.
ε = (1/M)<H>T (3.11)
La diferencia entre las ecuaciones (3.6) y (3.11) es que la (3.6) se calcula a
potencial químico constante, mientras que la (3.11) se determina a cubrimiento
constante.
3.2 CALCULO DE LA ENTROPIA
El cálculo de entropía mediante simulación numérica requiere el uso de
métodos especiales ya que, como no se puede asociar una entropía a cada uno de
los estados accesibles al sistema (la entropía depende del volumen total del
espacio de fase accesible al sistema), surgen dificultades cuando se quieren
realizar cálculos mediante promedios efectuados en el equilibrio [72]. Es por ello
que se han desarrollado diferentes métodos para salvar este inconveniente, entre
ellos: el método de Salsburg, el método de integración termodinámica, el método
de coincidencias o de Ma, y el método de los estados locales o de Meirovitvch
[72].
3.2.1 Método de integración termodinámica
Este método es de los más usados debido a su eficiencia, a que es posible
implementarlo fácilmente, y a que se puede aplicar a sistemas de cualquier
tamaño.
Sean algunas derivadas de la entropía S y de la energía libre de Helmholtz
F: 1/T = – (∂S/∂E)V,N; (3.12)

26
µ = – (∂F/∂N)V,T; (3.13)
CV/T = – (∂S/∂T)V,N; (3.14)
donde V es el volumen, CV la capacidad calorífica a volumen constante, N el
número de partículas y µ el potencial químico. A partir de las ecuaciones
anteriores, mediante integración se puede calcular la entropía en un estado de
equilibrio.
E(T1)
S(V,N,T1) – S(V,N,To) = dE’/T’ (3.15)
E(To)
N1
S(V,N1,T) – S(V,No,T) = (1/T)[E(V,N1,T) – E(V,No,T)] – µ’dN’ (3.16)
No
T1
S(V,N,T1) – S(V,N,To) = (C’V/T’)dT’ (3.17)
To
Para evaluar la integral de la (3.15) en el conjunto canónico, se calculan
por simulación los valores que toma E en un número finito de estados de
equilibrio comprendidos entre To y T1, y luego se integra mediante algún método
numérico adecuado. Conociendo la entropía en un estado de referencia S(V, N,
To), se puede calcular la entropía en cualquier otro estado S(V, N, T1).
El procedimiento para calcular la entropía a partir de la (3.16) es el mismo
que el descripto anteriormente, sólo que en este caso la integral es distinta.
Para aplicar la (3.17) hay que trabajar en el conjunto gran canónico para
calcular el número de partículas en los estados intermedios, y también hay que
conocer la energía media en los estados extremos.
En general, cualquiera sea la ecuación que se utilice, para calcular la
entropía en un estado determinado hay que conocer:

27
- la entropía en un estado de referencia que sea accesible a la simulación.
- la cantidad a integrar en un número considerable de estados de
intermedios entre el estado de referencia y el estado en cuestión.
De estos dos requisitos el segundo es fácil de cumplir, no así el primero ya
que en el conjunto canónico es difícil calcular estados de referencia de entropía
que puedan emplearse en la integración termodinámica. Sin embargo, Romá [69]
ha desarrollado recientemente un método para estimar numéricamente estados de
referencia de entropía que se pueden usar para la integración termodinámica en
el conjunto canónico.
3.3 DISEÑO DE REDES
A fin de realizar los cálculos computaciones mediante simulación de
Monte Carlo es necesario diseñar las redes hexagonal y triangular, lo cual se
lleva a cabo a partir de una red cuadrada de L x L sitios, (figura 3.1), que de aquí
en adelante denominaremos red cuadrada básica (r.c.b.).
Figura 3.1. Red cuadrada básica (r.c.b.).

28
3.3.1 Red hexagonal
Tomando como base la r.c.b., en la cual cada sitio está conectado a otros
cuatro (c = 4), la red hexagonal, cuya conectividad es 3, se obtiene manteniendo
las uniones verticales de cada sitio, pero eliminando las horizontales, una por
medio, como se muestra en la figura 3.2.
Figura 3.2. Red hexagonal.
Llamando i a las filas de la red cuadrada básica, y j a las columnas, en la
figura 3.3 se puede apreciar que no todos los sitios tienen los mismos vecinos
próximos, como ocurre en el caso de red cuadrada, sino que existen dos tipos de
sitio, que hemos designado por A y B.
A B AB B A B A A B A B B AB A
Figura 3.3

29
Los sitios A (i,j) tienen por vecinos próximos a los sitios (i,j +1), (i - 1, j)
e (i + 1, j) y los sitios B (i, j) tiene por vecinos a ( i, j - 1), ( i - 1, j) e (i + 1, j).
Numerando filas y columnas a partir de 0, se cumple que para los sitios A la
suma (i + j) es cero o par, mientras que para los sitios B es impar. Además,
como se puede apreciar en la figura, todo sitio A tiene por vecino próximo un
sitio B y viceversa. También se puede ver en la figura que la r.c.b. más pequeña
con la que se puede trabajar es de 4 x 4 y que, a fin de establecer condiciones de
borde periódicas, en general la r.c.b. debe ser de L x L, donde L es un múltiplo
entero de 4.
Los aspectos básicos del programa computacional diseñado para trabajar
con la red hexagonal son los que se indican a continuación.
La r.c.b se representa con un arreglo (matriz) bidimensional. Se usan dos
matrices bidimensionales, una, a[L][L], para representar a los sitios vacíos y
ocupados, con ceros y unos, respectivamente, y la otra, b[L][L], para ubicar a
los dímeros con un número de orden identificatorio, el mismo en los dos sitios
ocupados por el dímero. En esta matriz también los sitios vacíos se llenan con
ceros. Por ejemplo, si los sitios vecinos próximos, (i,j) y (p,q) están ocupados por
el dímero 8, se tiene: a[i][j] = 1, a[p][q] = 1, b[i][j] = 8, y b[p][q] = 8.
Se usan además cuatro matrices unidimensionales c[H], d[H], e[H], f[H],
para almacenar las cuatro coordenadas del dímero (i,j,p,q), y una quinta matriz,
g[H], para almacenar su número identificatorio.
Inicialmente los sitios de todas las matrices mencionadas se llenan con
ceros.
Para distribuir los dímeros en la red, primero se elige al azar un sitio (i,j) y
se determina si es A, o B. Luego se elige al azar un vecino próximo del anterior,
(p,q), que será B, o A, respectivamente. Se determina si el par de vecinos
próximos está vacío y, en caso afirmativo, se coloca el dímero y se almacenan
sus coordenadas y su número identificatorio. Todo el proceso anterior se repite
hasta adsorber un número predeterminado de dímeros.

30
Si se quiere mover un dímero a una posición vecina próxima, hay que
tener en cuenta que esto se puede lograr sólo mediante reptado por rotación
alrededor de un extremo de aquél. Además, hay que considerar que el dímero
puede ocupar en la red tres posiciones diferentes, lo cual fija las coordenadas de
sus cuatro vecinos próximos: a) Horizontal. b) Vertical, con el extremo superior
en un sitio A. c) Vertical, con el extremo superior en un sitio B, como se aprecia
en la figura 3.4.
A B AB A B AB A B AB B A B A B A B A B A B A A B A B A B AB A B AB B AB A B A B A B A B A (a) (b) (c)Figura 3.4
Para mover el dímero a una posición vecina próxima, se elige un dímero al
azar, eligiendo aleatoriamente un número identificatorio, obteniéndose así sus
cuatro coordenadas (i,j,p,q). Estas determinan cual de las posiciones posibles
mencionadas ocupa el dímero. Luego se elige uno de los cuatro vecinos
próximos del dímero, se averigua si está vacío y se determina si se mueve o no
uno de los monómeros a esa posición; en caso de hacerlo, se actualizan sus
coordenadas.
Cuando se quiere adsorber o desorber un dímero, se elige al azar un par de
sitios vecinos próximos, (i,j) y (p,q), y se averigua si están vacíos u ocupados.
Si ambos sitios están vacíos, o sea si a[i][j] = 0, y a[p][q] = 0, se
determina si se adsorbe o no un dímero. En caso afirmativo, se hace a[i][j] = 1, y
a[p][q] = 1, b[i][j] = dim, b[p][q] = dim, donde dim es el número identificatorio

31
del nuevo dímero. Además, se almacenan sus coordenadas y su número
identificatorio en las matrices correspondientes.
Si ambos sitios están ocupados, y ambos monómeros pertenecen al mismo
dímero, o sea si a[i][j] = 1, a[p][q] = 1, y b[i][j] = b[p][q], se determina si se
desorbe o no el dímero. En caso de desorber, se hace a[i][j] = 0, a[p][q] = 0,
b[i][j] = 0, b[p][q] = 0, y en las matrices correspondientes se hacen cero las
coordenadas y el número identificatorio que tenía el dímero.
3.3.2 Red triangular
Para obtener la red triangular a partir de la r.c.b., a las cuatro uniones de
cada sitio de aquélla se agregan dos uniones diagonales a 45 grados, como se
observa en la figura 3.5.
Figura 3.5. Red triangular.
En este caso, al igual que en el caso de red cuadrada, todos los sitios
tienen los mismos vecinos próximos, 6 en este caso. Para un sitio (i,j) dado, ellos
son: (i, j - 1), (i, j + 1), (i - 1, j), (i + 1, j), (i - 1, j + 1), (i + 1, j – 1).
El número total de vecinos próximos de un dímero es diez, como se puede
apreciar en la figura 3.6, donde los sitios marcados con x se cuentan dos veces ya
que son vecinos de lo dos monómeros que forman el dímero.

32
x
x
Figura 3.6. Red triangular. Vecinos próximos de un dímero.
Los diez vecinos próximos de un dímero no son los mismos en todos los
casos, sino que dependen de la ubicación de aquél en la red.
Considerando una r.c.b. de L x L, y llamando 1(i,j) y 2(p,q) a los
monómeros que forman el dímero, a efectos de diferenciarlos, pese a que son
iguales, las seis ubicaciones distintas y las coordenadas de los vecinos próximos
del dímero son:
1) Dímero [1-2] horizontal, 1 a la derecha de 2, pero 1 no se encuentra en
la columna L –1, ni 2 en la 0, o dímero[1-2] horizontal, 1 en la columna cero y 2
en la L-1.
(i, j + 1), (i - 1, j), (i + 1, j), (i - 1, j +1), (i + 1, j – 1),
(p, q - 1), (p - 1, q), (p + 1, q –1).
2) Dímero [1-2] horizontal, 1 a la izquierda de 2, pero 1 no está en la
columna 0, ni 2 en la L-1, o dímero [1-2] horizontal, 1 en la columna L -1 y 2
en la 0.
(i, j - 1), ( i - 1, j), (i + 1, j), (i - 1, j +1), (i + 1, j – 1),
(p, q + 1), (p + 1,q), (p - 1, q +1).

33
3) Dímero [1-2] vertical, 1 debajo de 2, pero1 no está en la fila L –1, ni 2
en la fila 0, o dímero [1-2] vertical, 1 en la fila 0 y 2 en la L-1.
(i, j - 1), (i, j + 1), (i + 1, j), (i - 1, j + 1), (i + 1, j – 1),
(p, q - 1), (p - 1, q), (p - 1, q +1).
4) Dímero [1-2] vertical, 1 encima de 2, pero1 no está en la fila 0, ni 2 en
la fila L-1, o dímero [1-2] vertical, 1 en la fila L -1 y 2 en la fila 0.
(i, j - 1), (i, j + 1), (i - 1, j), (i –1,j + 1 ), (i + 1, j – 1),
(p, q + 1), (p + 1, q), (p +1, q - 1).
5) Dímero [1-2] diagonal, 1 a la derecha y arriba de 2, pero 1 no está en (0,
L-1) , ni 2 en (L-1, 0),o dímero [1-2] diagonal, 1 en (L-1, 0) y 2 en (0, L-1).
(i, j - 1), (i, j + 1), (i - 1, j), (i + 1, j), (i - 1, j + 1),
(p, q - 1), (p +1, q ), (p +1, q - 1).
6) Dímero [1-2] diagonal, 1 a la izquierda y abajo de 2, pero 1 no está en
(L-1, 0), ni 2 en (0, L-1), o dímero[1-2] diagonal, 1 en (0, L-1) y 2 en (L-1, 0).
(i, j - 1), (i, j + 1), (i - 1, j), (i + 1, j), (i + 1, j – 1),
(p, q + 1), (p - 1, q), (p - 1, q +1).
Los aspectos básicos del programa computacional diseñado para trabajar
con la red triangular son en general los mismos que se vieron para red hexagonal.
Cabe destacar que en el caso de red triangular, el movimiento de un
dímero a una posición vecina próxima se puede realizar de dos formas: por salto
a lo largo del eje del dímero, o mediante reptado por rotación alrededor de un
extremo de aquél.

34
3.4 RESUMEN
En este capítulo se describieron las técnicas de simulación de Monte Carlo
cuyos resultados serán empleados más adelante en dos formas diferentes: por un
lado, permitirán analizar el alcance de los modelos teóricos presentados en el
capítulo anterior y, por otro, posibilitarán el acceso a condiciones en las cuales la
teoría no brinda una descripción adecuada del sistema en estudio. También se
presentaron las distintas cantidades termodinámicas a medir por simulación, y se
describió la forma de determinar la entropía del sistema en estudio. Por último,
se diseñaron las redes triangular y hexagonal que posteriormente se usarán para
representar al sustrato en las simulaciones computacionales, y se indicaron los
aspectos básicos de los programas computacionales diseñados para trabajar con
las redes hexagonal y triangular.
* * * * * * * * * *

CAPITULO 4
ADSORCION DE DIMEROS SOBRE REDESHEXAGONALES HOMOGENEAS
En éste capítulo se estudia la adsorción de dímeros sobre
superficies homogéneas hexagonales, empleándose las técnicas de
simulación de Monte Carlo, vistas en el capítulo 3. Además, se analiza el
comportamiento de diversas cantidades termodinámicas asociadas a la
capa adsorbida (isoterma de adsorción, factor termodinámico, energía,
calor diferencial de adsorción y entropía), para interacciones laterales
tanto atractivas como repulsivas.
Asimismo se comparan los resultados obtenidos por aplicación de
los modelos teóricos vistos en el capítulo 2, con los que se obtienen a
partir de las técnicas de simulación.
La simulación computacional se realiza para redes hexagonales de
M = L x L sitios, con L = 144 y condiciones periódicas de borde. Con este
tamaño de red se verifica que los efectos de tamaño finito son
despreciables.
4.1 INTERACCIONES LATERALES ATRACTIVAS
4.1.1 Simulación de Monte Carlo: isoterma de adsorción, calordiferencial de adsorción, energía de adsorción, factor termodinámicoy entropía configuracional.
En la simulación realizada se descartaron los primeros 105 MCs
para permitir que se alcanzara el equilibrio, mientras que se usaron los
siguientes 105 MCs para obtener promedios.
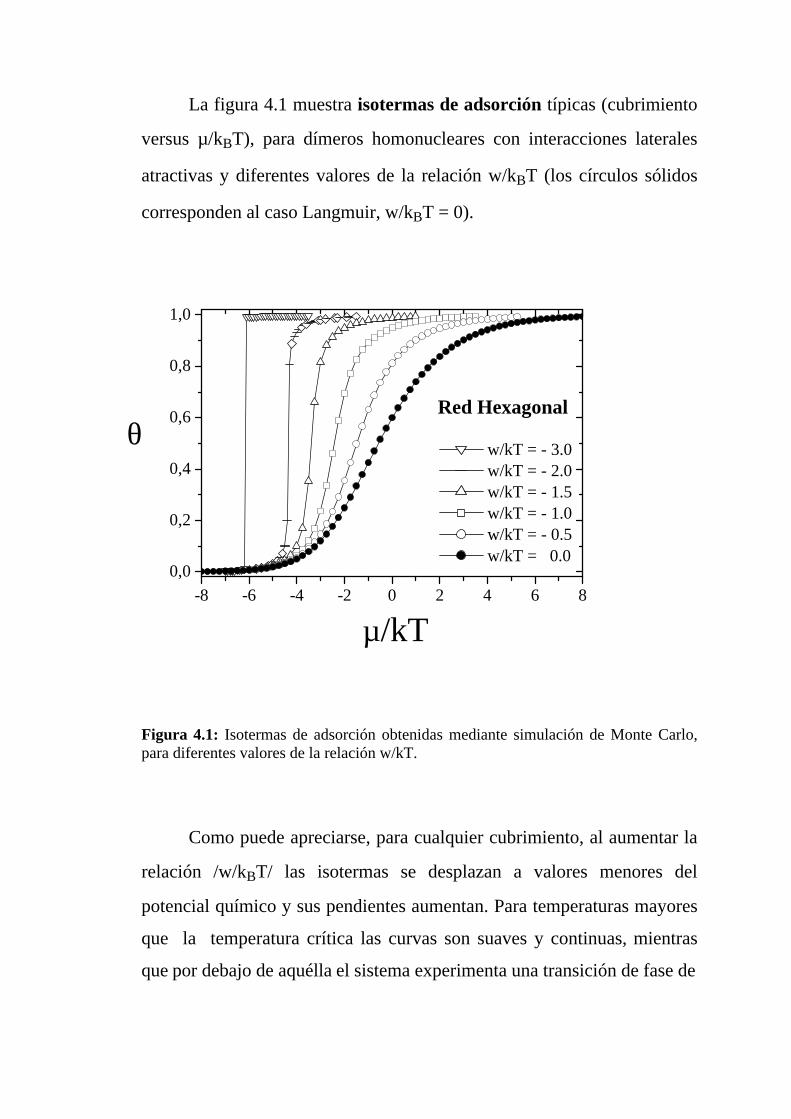
La figura 4.1 muestra isotermas de adsorción típicas (cubrimiento
versus µ/kBT), para dímeros homonucleares con interacciones laterales
atractivas y diferentes valores de la relación w/kBT (los círculos sólidos
corresponden al caso Langmuir, w/kBT = 0).
Figura 4.1: Isotermas de adsorción obtenidas mediante simulación de Monte Carlo,para diferentes valores de la relación w/kT.
Como puede apreciarse, para cualquier cubrimiento, al aumentar la
relación /w/kBT/ las isotermas se desplazan a valores menores del
potencial químico y sus pendientes aumentan. Para temperaturas mayores
que la temperatura crítica las curvas son suaves y continuas, mientras
que por debajo de aquélla el sistema experimenta una transición de fase de
-8 -6 -4 -2 0 2 4 6 80,0
0,2
0,4
0,6
0,8
1,0
Red Hexagonal
θ
µ/kT
w/kT = - 3.0 w/kT = - 2.0 w/kT = - 1.5 w/kT = - 1.0 w/kT = - 0.5 w/kT = 0.0

primer orden (con un agrupamiento de los dímeros adsorbidos), lo cual se
pone de manifiesto en la clara discontinuidad o salto que muestran
las isotermas de adsorción. El valor crítico (kBTc/w) depende de la
estructura de la red, siendo de aproximadamente 0,33 para red hexagonal,
0,68 para red cuadrada y 1 para red triangular. Una explicación simple
para este comportamiento es la siguiente: a fin de sacar un dímero de una
estructura l x l, y de destruir la isla en el adsorbato, se necesita una energía
equivalente a 2(c-1)w, donde 2(c-1) es el número de vecinos próximos del
dímero. O sea que a mayor conectividad se necesita mayor energía para
destruir el agrupamiento de dímeros. Por esta razón la estructura es
energéticamente más estable a medida que aumenta c, lo cual explica los
valores obtenidos para (kBTc/w), dado que c vale 3, 4 y 6 para redes
hexagonal, cuadrada y triangular respectivamente.
En la figura 4.2 se representa el calor diferencial de adsorción en
función del cubrimiento, para diferentes valores de la relación w/kBT. Al
aumentar la relación w/kBT se observan dos características importantes:
a) Aparece una meseta, siendo qd ≅ (c –1)w. Este comportamiento
demuestra que la isla de dímeros adsorbidos crece a través del
perímetro, siendo (c–1)w la energía involucrada en la adsorción
(desorción) de un dímero en el perímetro de la isla de adsorbato.
b) La meseta se extiende sobre un rango amplio de cubrimiento ya
que las islas de adsorbato son más compactas para valores
mayores de la relación w/kBT.

Figura 4.2: Calor diferencial de adsorción en función del cubrimiento, para diferentesvalores de la relación w/kT.
La figura 4.3 muestra las gráficas del factor termodinámico en
función del cubrimiento, para diferentes valores de la relación w/kBT.
Figura 4.3: Factor termodinámico en función del cubrimiento, para diferentesvalores de la relación w/kT.
0,0 0,2 0,4 0,6 0,8 1,0
0
2
4
6
8
10
12
14
16
Red Hexagonal
w/kT = 0.0 w/kT = - 0.5 w/kT = - 1.0 w/kT = - 1.5 w/kT = - 2.0 w/kT = - 3.0
qd
θ
0,0 0,2 0,4 0,6 0,8 1,0
0,1
1
10
Red Hexagonal
th
θ
w/kT = 0,0 w/kT = - 0,5 w/kT = - 1,0 w/kT = - 1,5 w/kT = - 2,0

En la figura 4.4 se representa la energía de adsorción en función delcubrimiento, para diferentes valores de la relación w/kBT.
Figura 4.4: Energía de adsorción en función del cubrimiento, para diferentesvalores de la relación w/kT.
Figura 4.5: Entropía configuracional en función del cubrimiento, para diferentesvalores de la relación w/kT.
0,0 0,2 0,4 0,6 0,8 1,0
0,0
0,5
1,0
1,5
2,0
2,5
3,0 Red Hexagonal
- ε
/ kT
θ
w/kT = - 3.0 w/kT = - 2.0 w/kT = - 1.5 w/kT = - 1.0 w/kT = - 0.5
0,0 0,2 0,4 0,6 0,8 1,00,0
0,2
0,4
0,6Red Hexagonal
w/kT = 0.0 w/kT = - 0.5 w/kT = - 1.0 w/kT = - 1.5 w/kT = - 2.0
s/kB
θ

Las curvas de entropía configuracional s(θ,T), figura 4.5, se han
obtenido de la ecuación (3.15), donde las dependencias µ (θ,T) y ε(θ,T) se
han tomado de las isotermas de adsorción y de la gráfica de energía de
adsorción, obtenidas por simulación, respectivamente.
En todos los casos las curvas son asimétricas con respecto a un
cubrimiento de θ = 0.5, y la entropía disminuye para todo θ a medida
que aumenta el valor de las interacciones laterales.
4.1.2 Comparación de valores obtenidos por simulación y teóricos
Las expresiones teóricas que se dan a continuación, y que expresan
la ley de variación de diferentes variables termodinámicas para la
adsorción de dímeros sobre red hexagonal, se obtienen a partir de las
expresiones generales vistas en el capítulo 2.
Las isotermas de adsorción para la adsorción de dímeros sobre
red hexagonal, se obtienen a partir de las siguientes ecuaciones:
Para la aproximación R.P.:
(µ/kT) = lnθ – 2ln(1 – θ) + ln[1 – (θ/2)] + 4(w/kT)θ – ln3 (4.1)
Para la teoría de F-H:
(µ/kT) = lnθ – 2ln(1 – θ) + 4(w/kT)θ – ln3 (4.2)
En la figura 4.6 se muestran las isotermas de adsorción obtenidas
por simulación de Monte Carlo y a partir de las aproximaciones teóricas
mencionadas, para w/kT = 0, -1.0 y -2.0, de derecha a izquierda. En ellas
se puede observar que para w/kT = 0 (caso Langmuir) ambas
aproximaciones muestran un muy buen acuerdo con la simulación,
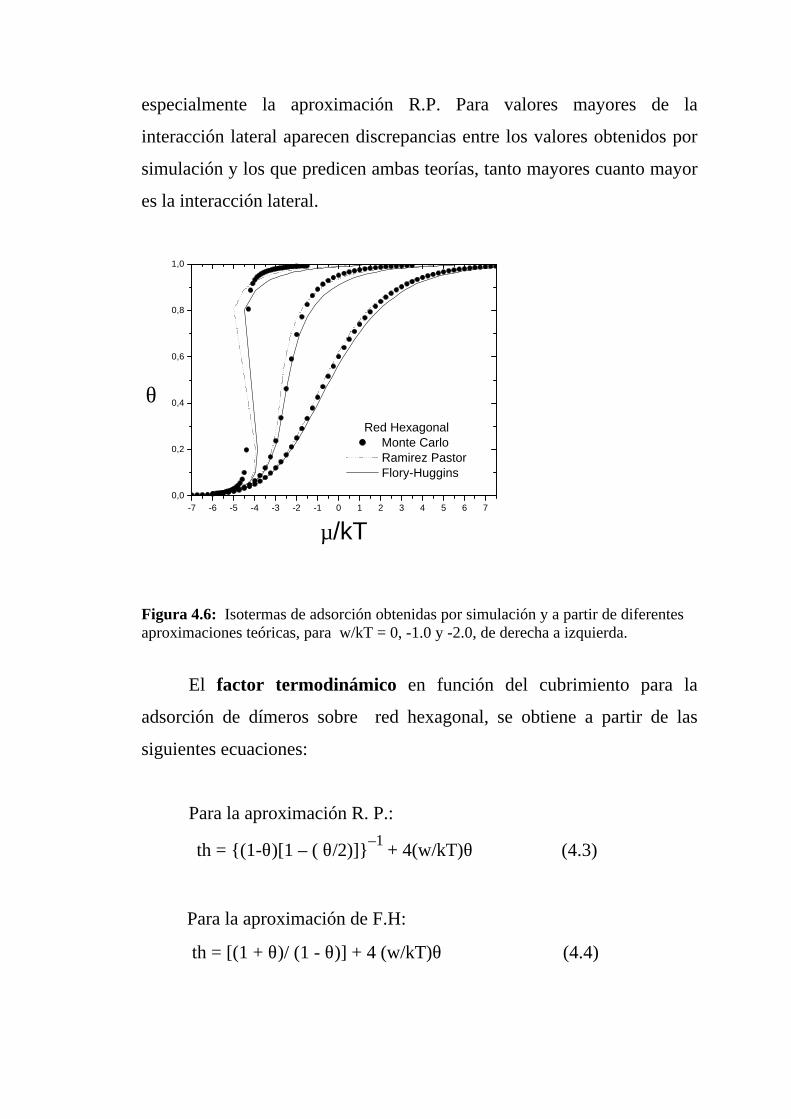
especialmente la aproximación R.P. Para valores mayores de la
interacción lateral aparecen discrepancias entre los valores obtenidos por
simulación y los que predicen ambas teorías, tanto mayores cuanto mayor
es la interacción lateral.
Figura 4.6: Isotermas de adsorción obtenidas por simulación y a partir de diferentesaproximaciones teóricas, para w/kT = 0, -1.0 y -2.0, de derecha a izquierda.
El factor termodinámico en función del cubrimiento para la
adsorción de dímeros sobre red hexagonal, se obtiene a partir de las
siguientes ecuaciones:
Para la aproximación R. P.:
th = (1-θ)[1 – ( θ/2)]–1
+ 4(w/kT)θ (4.3)
Para la aproximación de F.H:
th = [(1 + θ)/ (1 - θ)] + 4 (w/kT)θ (4.4)
0,0
0,2
0,4
0,6
0,8
1,0
-7 -6 -5 -4 -3 -2 -1 0 1 2 3 4 5 6 7
Red Hexagonal Monte Carlo Ramirez Pastor Flory-Huggins
µ/kT
θ

En la figura 4.7 se muestran las gráficas del factor termodinámico
en función del cubrimiento, para distintos valores de la relación (w/kT),
obtenidas por simulación de Monte Carlo y a partir de las aproximaciones
teóricas mencionadas.
Figura 4.7: Curvas del factor termodinámico en función del cubrimiento, obtenidaspor simulación y a partir de diferentes aproximaciones teóricas, para diferentes valoresde la relación w/kT (-0,5, -1 y –1,5, de arriba hacia abajo).
En la figura se observa que el mejor ajuste entre los modelos
teóricos y la simulación se produce para valores elevados de la
temperatura.
La ecuación que expresa la energía de adsorción por sitio
(normalizada a su valor de máximo cubrimiento) en función del
cubrimiento, para la adsorción de dímeros sobre red hexagonal, según las
aproximaciones de F-H, y de R-P es:
ε(θ)/ε(1) = θ2 (4.5)
0,0 0,2 0,4 0,6 0,8 1,00,1
1
10
100
Red Hexagonal
th
θ
Simulación Ramirez Pastor Flory-Huggins

A continuación se muestran las gráficas de la energía de adsorción
normalizada, en función del cubrimiento, para distintos valores de la
relación (w/kT).
Figura 4.8: Energía de adsorción por sitio (normalizada a su valor de máximocubrimiento), en función del cubrimiento, obtenidas por simulación y a partir deaproximaciones teóricas, para diferentes valores de la relación w/kT (-0,5, -1 y –1,5,de abajo hacia arriba).
Como indica la ecuación (4.5), y se puede observar en la figura,
ambas aproximaciones predicen un comportamiento de la energía
normalizada que es independiente del valor de la interacción lateral w,
produciéndose un mejor ajuste para valores más bajos de la interacción
lateral.
Según las aproximaciones de F.H y R. P., el calor diferencial de
adsorción en función del cubrimiento, para la adsorción de dímeros
sobre red hexagonal, se obtiene a partir de la siguiente ecuación:
0,0 0,2 0,4 0,6 0,8 1,00,0
0,2
0,4
0,6
0,8
1,0
Simulación Ramírez Pastor
y Flory-Huggins
ε(θ)
/ε(1
)
θ

qd = - 4wθ (4.6)
Figura 4.9: Curvas de calor diferencial de adsorción, en función del cubrimiento,obtenidas por simulación, y a partir de aproximaciones teóricas, para diferentesvalores de la relación w/kT (-0,5, -1 y –1,5, de abajo hacia arriba).
En la figura 4.9 se muestran las gráficas del calor diferencial de
adsorción en función del cubrimiento, para distintos valores de la relación
(w/kT), obtenidas por simulación de Monte Carlo y a partir de las
aproximaciones teóricas mencionadas. Como se desprende de la ecuación
(4.7), ambos modelos predicen un crecimiento lineal del calor diferencial
de adsorción con el cubrimiento.
El acuerdo entre ambos modelos y la simulación es bueno en el
régimen de altas temperaturas, y el ajuste es total cuando la temperatura
tiende a infinito.
La entropía por sitio en función del cubrimiento, para la adsorción
de dímeros sobre red hexagonal, se obtiene a partir de las siguientes
ecuaciones:
0,0 0,2 0,4 0,6 0,8 1,00
1
2
3
4
5
6
7
Simulación Ramírez Pastor
y Flory-Huggins
qd
θ
Para la aproximación R.P.:
s(θ,c)/kB = [1 – (θ/2)]ln[1 – (θ/2)] – (θ/2)ln((θ/2) – (1 – θ)ln(1 – θ) + (θ/2)0,693; (4.7)
Para el modelo de F. H.:
s(θ,c)/kB = [1 – (θ/2)]ln[1 – (θ/2)] – (θ/2)ln((θ/2) – (1 – θ)ln(1 – θ) + (θ/2)(c – 1); (4.8)
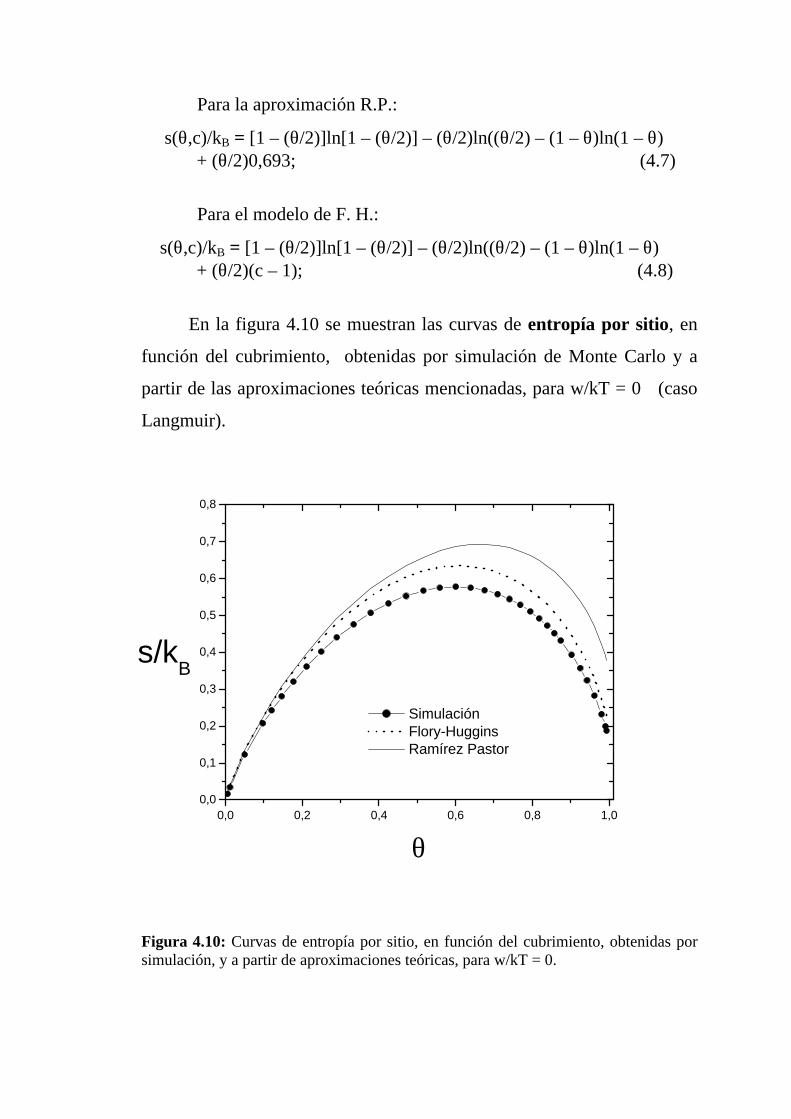
En la figura 4.10 se muestran las curvas de entropía por sitio, en
función del cubrimiento, obtenidas por simulación de Monte Carlo y a
partir de las aproximaciones teóricas mencionadas, para w/kT = 0 (caso
Langmuir).
Figura 4.10: Curvas de entropía por sitio, en función del cubrimiento, obtenidas porsimulación, y a partir de aproximaciones teóricas, para w/kT = 0.
0,0 0,2 0,4 0,6 0,8 1,00,0
0,1
0,2
0,3
0,4
0,5
0,6
0,7
0,8
Simulación Flory-Huggins Ramírez Pastor
s/kB
θ

Como se puede observar en la figura, para cubrimientos bajos las
aproximaciones de F-H y de R.P. muestran un buen acuerdo con la
simulación. Para cubrimientos superiores a 0,25 se advierten diferencias
apreciables entre aquéllas y la simulación.
4.2 INTERACCIONES LATERALES REPULSIVAS
4.2.1 Simulación de Montecarlo: isoterma de adsorción, calordiferencial de adsorción, energía de adsorción, factor termodinámicoy entropía diferencial de adsorción.
En este caso, a temperaturas alejadas del punto crítico, el estado de
equilibrio se alcanzó luego de descartar los primeros 105 MCs, mientras
que se usaron los siguientes 105 MCs para la obtención de promedios. En
la proximidad de los puntos críticos debieron usarse hasta 106 MCs a fin
de permitir que el sistema se relajara a partir de estados metaestables. Hay
que señalar que, a fin de alcanzar el equilibrio en un tiempo razonable,
debe permitirse el desplazamiento (relajación difusiva) de las moléculas
adsorbidas a posiciones vecinas próximas, mediante reptado por rotación
alrededor de un extremo de aquél.
En la figura 4.11 se muestran isotermas de adsorción para el caso
de interacciones laterales atractivas obtenidas: a)Con relajación difusiva.
b) Sin relajación difusiva.
Puede observarse que en el segundo caso no se observa la
formación de la meseta para θ = 2/3, que se aprecia en la primer isoterma.
Además, en el segundo caso, una vez que el cubrimiento alcanza un valor
de 0.76 no sigue aumentando, aún con valores mayores del potencial
químico.
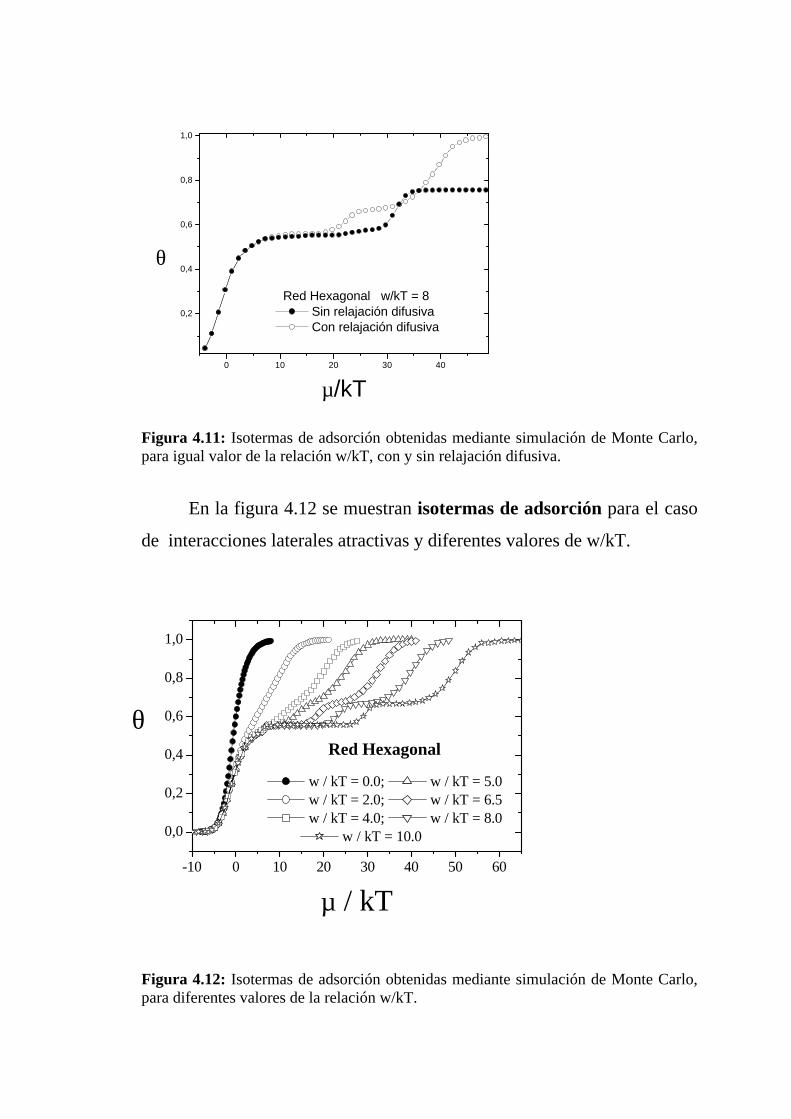
Figura 4.11: Isotermas de adsorción obtenidas mediante simulación de Monte Carlo,para igual valor de la relación w/kT, con y sin relajación difusiva.
En la figura 4.12 se muestran isotermas de adsorción para el caso
de interacciones laterales atractivas y diferentes valores de w/kT.
Figura 4.12: Isotermas de adsorción obtenidas mediante simulación de Monte Carlo,para diferentes valores de la relación w/kT.
-10 0 10 20 30 40 50 60
0,0
0,2
0,4
0,6
0,8
1,0
Red Hexagonal
θ
µ / kT
w / kT = 0.0; w / kT = 5.0 w / kT = 2.0; w / kT = 6.5 w / kT = 4.0; w / kT = 8.0
w / kT = 10.0
0 10 20 30 40
0,2
0,4
0,6
0,8
1,0
θ
µ/kT
Red Hexagonal w/kT = 8 Sin relajación difusiva Con relajación difusiva

Como puede observarse, al aumentar la interacción lateral aparecen
dos escalones pronunciados y bien definidos para θ1 = 5/9 y θ2 = 2/3.
Estas estructuras, que denominamos FOBC (fase ordenada de bajo
cubrimiento) y FOAC (fase ordenada de alto cubrimiento), las
reportamos recientemente en la literatura [73], y son una clara evidencia
de un comportamiento subcrítico. En efecto, el sistema experimenta una
transición de fase, de estructuras desordenadas a estructuras ordenadas.
En la figura 4.13 se muestra una instantánea de las configuraciones
del estado fundamental correspondiente a las estructuras FOBC y FOAC.
(a) (b)
Figura 4.13: Configuraciones del estado fundamental correspondiente a lasestructuras FOBC (a) y FOAC (b).
La figura 4.14 muestra la variación de la energía de adsorción con
el cubrimiento para diferentes valores de la relación w/kT.

Figura 4.14: Curvas de energía de adsorción obtenidas mediante simulación de Monte
Carlo, para diferentes valores de la relación w/kT.
Se puede observar que para la mayor temperatura, T = ∞, la energía
promedio no depende del valor del cubrimiento. A medida que la
temperatura disminuye, aumenta la influencia del ordenamiento
estructural. La pendiente de ε/w (siendo ε la energía por sitio, ε = E/M) en
función de θ cambia en forma discontinua para los cubrimientos críticos,
y se pueden apreciar claramente los tres regímenes siguientes:
I) Para 0 ≤ θ ≤ θ1, los dímeros no interactúan, siendo por lo tanto ε/w =
0. Para cubrimientos bajos los dímeros adsorbidos están aislados; a
medida que el cubrimiento aumenta, aquéllos forman islas ordenadas y
finalmente se alcanza una fase perfectamente ordenada para θ = θ1.
II) Para θ1 ≤ θ ≤ θ2, el agregado de un nuevo dímero aumenta el
cubrimiento en un factor de 2/M, mientras que ε/w aumenta linealmente,
desde ε/w = 0, hasta ε/w = 1/6.
0,0 0,2 0,4 0,6 0,8 1,0
-8
-6
-4
-2
0
Red Hexagonal
-ε/k
T
θ
w / kT = 0.0 w / kT = 2.0 w / kT = 4.0 w / kT = 5.0 w / kT = 6.5 w / kT = 8.0

III) Para θ2 ≤ θ ≤ 1, la energía aumenta linealmente con el cubrimiento
hasta ε/w = (c – 1)/2 = 1 para cubrimiento total.
La dependencia del calor diferencial de adsorción (qd) con el
cubrimiento se muestra en la figura 4.15.
Figura 4.15: Calor diferencial de adsorción en función del cubrimiento, para diferentes valores de la relación w/kT.
A medida que la temperatura disminuye, qd refleja claramente los
tres regímenes de adsorción antes descriptos. El comportamiento puede
analizarse teniendo en cuenta tres características de las curvas: mesetas,
escalones y hombros.
0,0 0,2 0,4 0,6 0,8 1,0
-40
-30
-20
-10
0
w/kT = 0.0 w/kT = 2.0 w/kT = 4.0 w/kT = 5.0 w/kT = 6.5 w/kT = 8.0
qd
θ

Las mesetas indican la existencia de diferentes regímenes de
adsorción:
• I) Para 0 ≤ θ ≤ θ1, los dímeros no interactúan y la adsorción-
desorción de un dímero involucra un calor diferencial nulo.
• II) Para θ1 ≤ θ ≤ θ2, el sistema pasa de una fase ordenada de
bajo cubrimiento (FOBC) a una fase ordenada de alto
cubrimiento (FOAC). El valor de qd en este régimen se puede
obtener a partir de la pendiente de ε vs θ, resultando, en general,
qd = [(c – 2)w]/[3(θ2 − θ1)]. Luego, para redes hexagonales es
qd = 3w.
• III) Para θ2 ≤ θ ≤ 1, la energía necesaria para adsorber un
nuevo dímero se puede estimar considerando su adsorción sobre
una estructura zig-zag perfecta. Para ello es necesario crear
previamente un par de sitios vacíos moviendo un dímero ya
adsorbido. Este proceso involucra una variación de energía igual
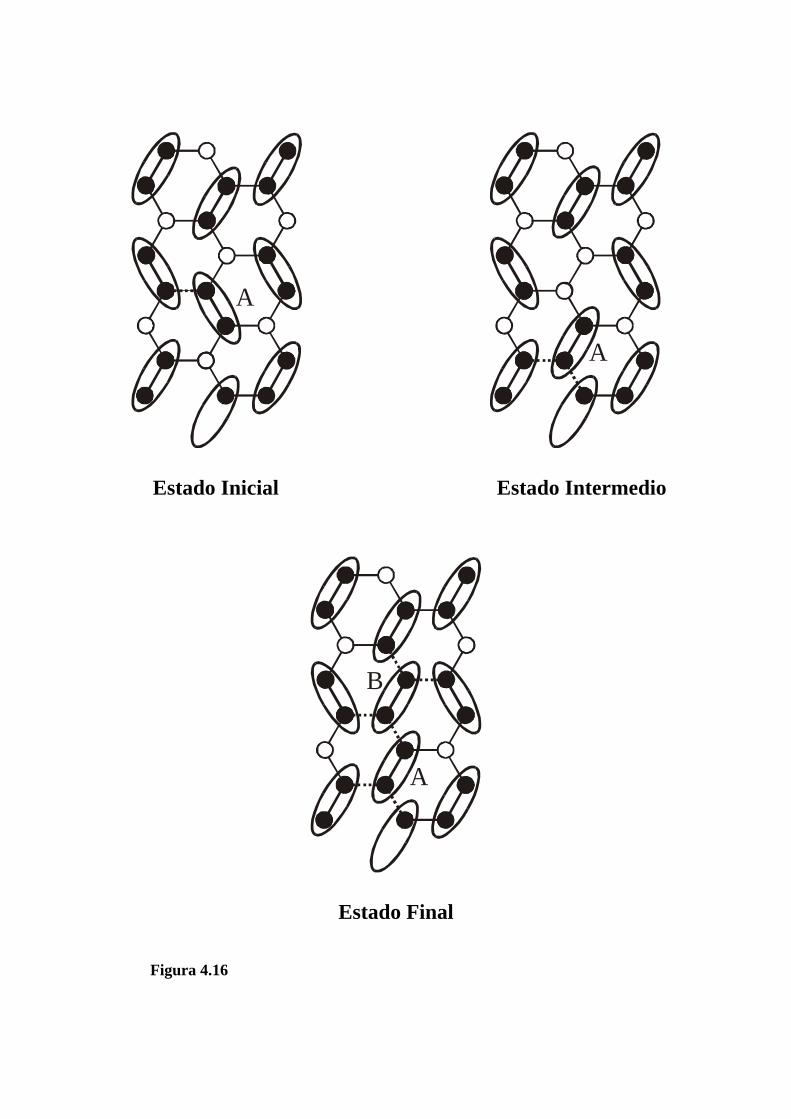
a w, como se muestra en la figura 4.16, donde el dímero A
cambia de una posición en la que interactúa con un vecino
próximo(NN), a otra en la que interactúa con dos NN (estado
intermedio: las interacciones de los NN se indican con líneas de
trazo). Así, el dímero entrante (indicado con B), se puede alojar
en la red a un costo adicional de 4w (ver figura 4.16).
Finalmente, la variación total de energía es de 5w,
correspondiendo a la meseta de qd para θ2 ≤ θ ≤ 1 en la figura
4.15.
A
A
Estado Inicial Estado Intermedio
A
B
Estado Final
Figura 4.16

Los escalones corresponden a cubrimientos críticos (θ1 y θ2) y
separan diferentes regímenes de adsorción.
Los hombros aparecen alrededor de los cubrimientos críticos. La
posición de un hombro corresponde a un cubrimiento crítico en el cual
ocurre un cambio drástico de orden en el sistema. Los hombros
suministran información muy importante acerca del comportamiento
crítico del sistema.
En la figura 4.17 se representa la entropía configuracional como
una función del cubrimiento.
Figura 4.17: Entropía configuracional en función del cubrimiento, para diferentes valores de la relación w/kT.
Para dímeros que no interactúan (w/kT = 0), s(θ,T) tiene un
máximo para θ > 0,5. El efecto global de las interacciones es la
disminución de la entropía para todos los cubrimientos. A medida que la
0,0 0,2 0,4 0,6 0,8 1,00,0
0,1
0,2
0,3
0,4
0,5
0,6
w/kT = 0.0 w/kT = 2.0 w/kT = 4.0 w/kT = 5.0 w/kT = 6.5 w/kT = 8.0 w/kT = 10.0
Red Hexagonal
s/kB
θ

temperatura disminuye, aparecen dos mínimos locales de s(θ,T) para θ =
θ1 y θ = θ2, lo cual confirma la existencia de estructuras ordenadas para
estos valores del cubrimiento.
Las temperaturas críticas correspondientes a los cubrimientos
críticos θ = θ1 y θ = θ2, se estimaron graficando los valores de s(θ1) y
s(θ2) (y sus derivadas) en función de la temperatura.
Figura 4.18: s(θ1), y su derivada, en función de la temperatura.
0,0 0,2 0,4 0,6 0,8 1,00,0
0,2
0,4
0,6
s(θ
= 5/
9)
kT/w
0,0 0,2 0,4 0,6 0,8 1,00,0
0,5
1,0
1,5
2,0
kTc/w ≈ 0.22
ds(θ
= 5
/9)/
dT
kT/w
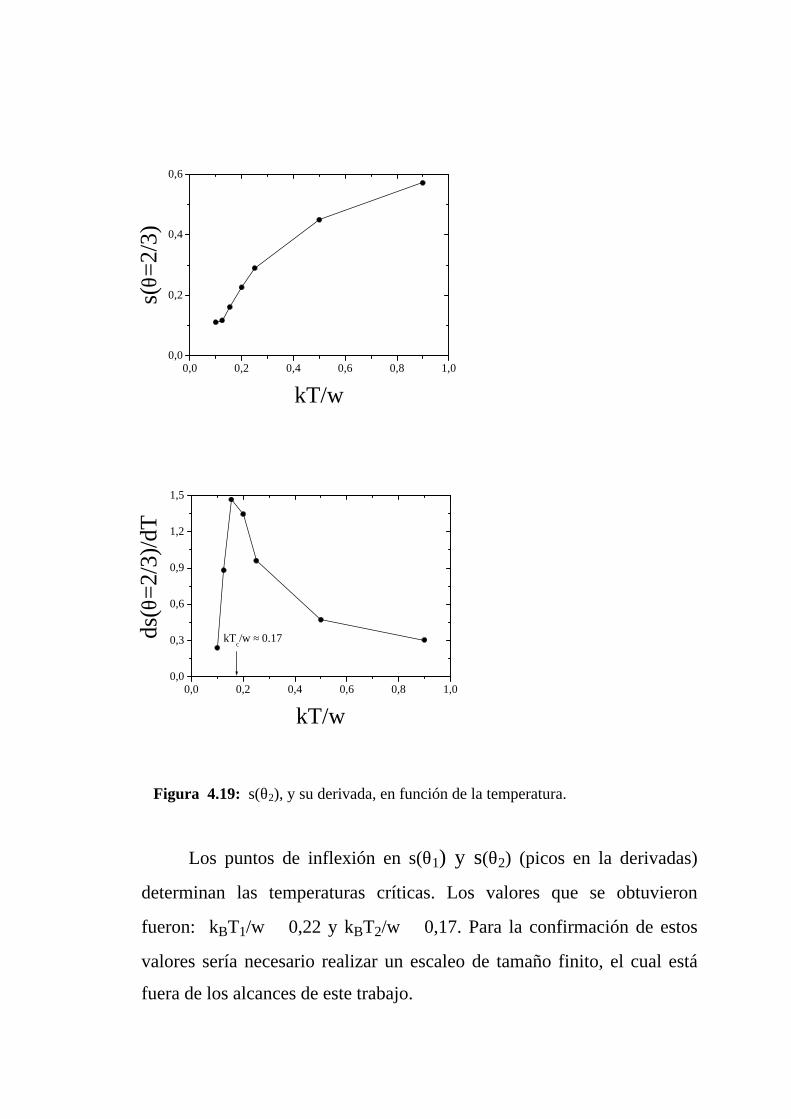
Figura 4.19: s(θ2), y su derivada, en función de la temperatura.
Los puntos de inflexión en s(θ1) y s(θ2) (picos en la derivadas)
determinan las temperaturas críticas. Los valores que se obtuvieron
fueron: kBT1/w ≅ 0,22 y kBT2/w ≅ 0,17. Para la confirmación de estos
valores sería necesario realizar un escaleo de tamaño finito, el cual está
fuera de los alcances de este trabajo.
0,0 0,2 0,4 0,6 0,8 1,00,0
0,2
0,4
0,6
s(
θ =2/
3)
kT/w
0,0 0,2 0,4 0,6 0,8 1,00,0
0,3
0,6
0,9
1,2
1,5
kTc/w ≈ 0.17
ds(θ
=2/3
)/dT
kT/w
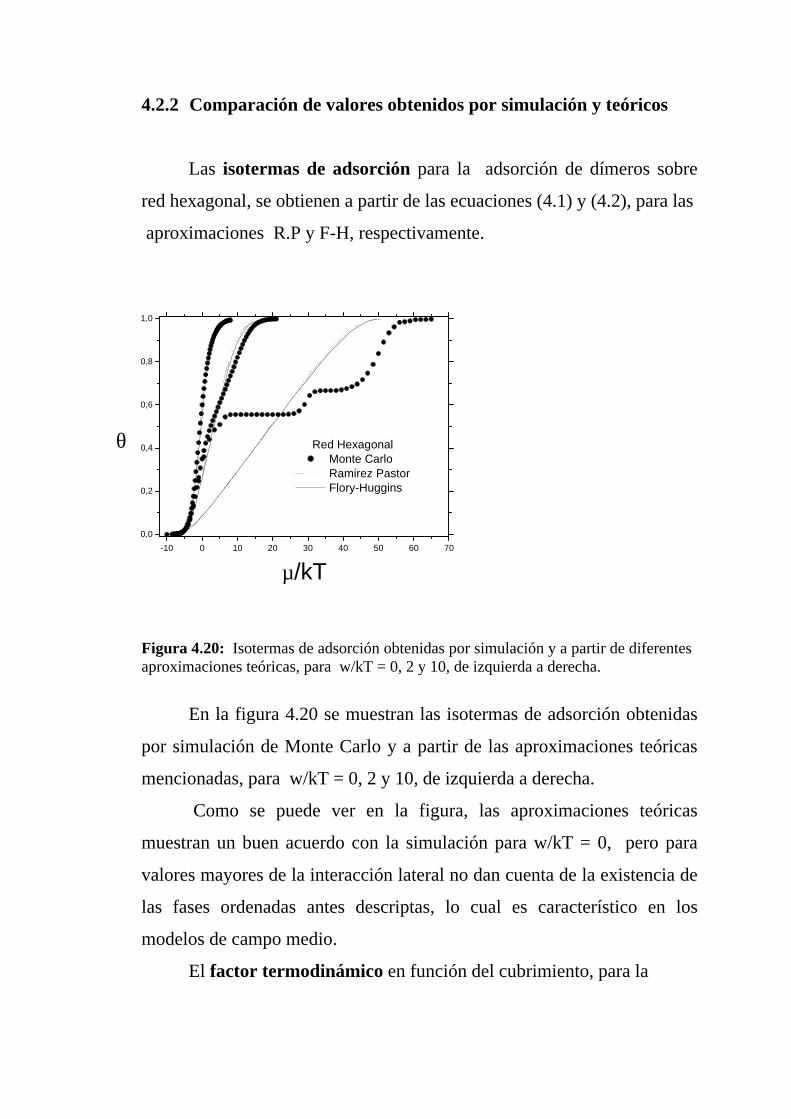
4.2.2 Comparación de valores obtenidos por simulación y teóricos
Las isotermas de adsorción para la adsorción de dímeros sobre
red hexagonal, se obtienen a partir de las ecuaciones (4.1) y (4.2), para las
aproximaciones R.P y F-H, respectivamente.
Figura 4.20: Isotermas de adsorción obtenidas por simulación y a partir de diferentesaproximaciones teóricas, para w/kT = 0, 2 y 10, de izquierda a derecha.
En la figura 4.20 se muestran las isotermas de adsorción obtenidas
por simulación de Monte Carlo y a partir de las aproximaciones teóricas
mencionadas, para w/kT = 0, 2 y 10, de izquierda a derecha.
Como se puede ver en la figura, las aproximaciones teóricas
muestran un buen acuerdo con la simulación para w/kT = 0, pero para
valores mayores de la interacción lateral no dan cuenta de la existencia de
las fases ordenadas antes descriptas, lo cual es característico en los
modelos de campo medio.
El factor termodinámico en función del cubrimiento, para la
0,0
0,2
0,4
0,6
0,8
1,0
-10 0 10 20 30 40 50 60 70
µ/kT
θ Red Hexagonal Monte Carlo Ramirez Pastor Flory-Huggins
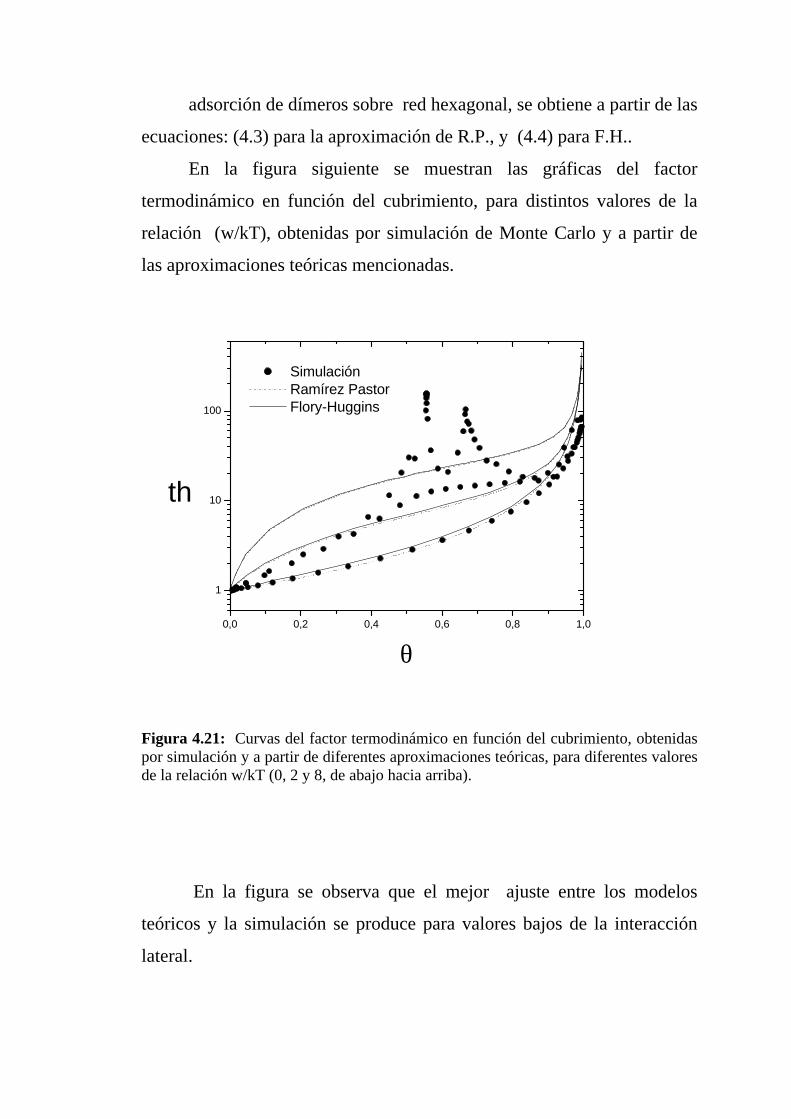
adsorción de dímeros sobre red hexagonal, se obtiene a partir de las
ecuaciones: (4.3) para la aproximación de R.P., y (4.4) para F.H..
En la figura siguiente se muestran las gráficas del factor
termodinámico en función del cubrimiento, para distintos valores de la
relación (w/kT), obtenidas por simulación de Monte Carlo y a partir de
las aproximaciones teóricas mencionadas.
Figura 4.21: Curvas del factor termodinámico en función del cubrimiento, obtenidaspor simulación y a partir de diferentes aproximaciones teóricas, para diferentes valoresde la relación w/kT (0, 2 y 8, de abajo hacia arriba).
En la figura se observa que el mejor ajuste entre los modelos
teóricos y la simulación se produce para valores bajos de la interacción
lateral.
0,0 0,2 0,4 0,6 0,8 1,0
1
10
100
Simulación Ramírez Pastor Flory-Huggins
th
θ
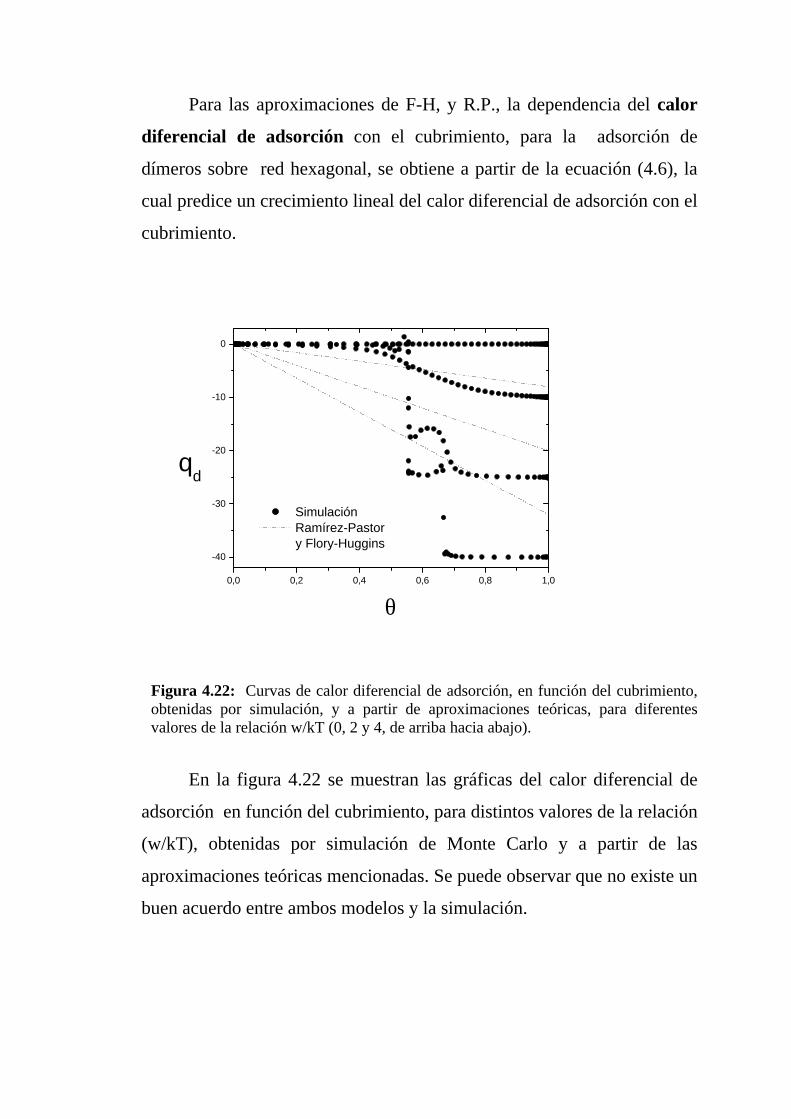
Para las aproximaciones de F-H, y R.P., la dependencia del calor
diferencial de adsorción con el cubrimiento, para la adsorción de
dímeros sobre red hexagonal, se obtiene a partir de la ecuación (4.6), la
cual predice un crecimiento lineal del calor diferencial de adsorción con el
cubrimiento.
Figura 4.22: Curvas de calor diferencial de adsorción, en función del cubrimiento,obtenidas por simulación, y a partir de aproximaciones teóricas, para diferentesvalores de la relación w/kT (0, 2 y 4, de arriba hacia abajo).
En la figura 4.22 se muestran las gráficas del calor diferencial de
adsorción en función del cubrimiento, para distintos valores de la relación
(w/kT), obtenidas por simulación de Monte Carlo y a partir de las
aproximaciones teóricas mencionadas. Se puede observar que no existe un
buen acuerdo entre ambos modelos y la simulación.
0,0 0,2 0,4 0,6 0,8 1,0
-40
-30
-20
-10
0
Simulación Ramírez-Pastor
y Flory-Huggins
qd
θ

La ecuación (4.7) expresa la energía de adsorción por sitio
(normalizada a su valor de máximo cubrimiento) en función del
cubrimiento, para la adsorción de dímeros sobre red hexagonal, según las
aproximaciones de F-H, y R.P.
Figura 4.23: Energía de adsorción por sitio (normalizada a su valor de máximocubrimiento), en función del cubrimiento, obtenidas por simulación y a partir deaproximaciones teóricas, para diferentes valores de la relación w/kT (2, 4 y 5, dearriba hacia abajo).
Como se observa en la figura, ambas aproximaciones predicen un
comportamiento de la energía normalizada que es independiente del valor
de la interacción lateral w, produciéndose un mejor ajuste para los valores
más bajos de la interacción lateral.
4.3 RESUMEN
En el presente capítulo se estudió la adsorción de dímeros
interactuantes (en forma atractiva y repulsiva) sobre superficies
homogéneas hexagonales. Se utilizaron técnicas de simulación de Monte
0,0 0,2 0,4 0,6 0,8 1,0
0,0
0,2
0,4
0,6
0,8
1,0
Simulación Ramírez Pastor
y Flory-Huggins
ε(θ)
/ε(1
)
θ

Carlo para analizar el comportamiento de diversas cantidades
termodinámicas asociadas a la capa adsorbida. En el caso particular de
interacciones laterales repulsivas y valores altos de la relación w/kBT, se
encontraron dos nuevas fases ordenadas en el adsorbato, para θ1 = 5/9 y
θ2 = 2/3. Esto indica que el sistema experimenta una transición de fase,
de estructuras desordenadas a estructuras ordenadas, que denominamos
FOBC (fase ordenada de bajo cubrimiento) y FOAC (fase ordenada de
alto cubrimiento), las cuales reportamos por primera vez recientemente
en la literatura [73]. Asimismo, en la figura 4.13 se mostraron
instantáneas de las configuraciones del estado fundamental de las
estructuras mencionadas.
También se estimaron las temperatura críticas correspondientes a
los cubrimientos críticos. Para confirmar los valores obtenidos es
necesario realizar un escaleo de tamaño finito, el cual está fuera de los
alcances de este trabajo.
En las secciones 4.1.2 y 4.2.2. se compararon los resultados
obtenidos aplicando los modelos teóricos vistos en el capítulo 2, con
aquellos provenientes desde las técnicas de simulación vistas en el
capítulo 3, para interacciones laterales atractivas y repulsivas,
respectivamente. Los alcances de la teoría fueron analizados y discutidos.
* * * * * * * * *

CAPITULO 5
ADSORCION DE DIMEROS SOBRE REDESTRIANGULARES HOMOGENEAS
En el presente capítulo se estudia la adsorción de dímeros sobre
superficies triangulares homogéneas. Se analiza en primer término el
comportamiento de diversas cantidades termodinámicas asociadas a la
capa adsorbida (isoterma de adsorción, factor termodinámico, energía,
calor diferencial de adsorción y entropía), tanto para interacciones
laterales atractivas como repulsivas, usándose para ello las técnicas de
simulación de Montecarlo vistas en el capítulo 3.
Posteriormente se comparan los resultados obtenidos al aplicar los
modelos teóricos vistos en el capítulo 2, con los obtenidos por medio de
las técnicas de simulación mencionadas.
La simulación de Monte Carlo se lleva a cabo para redes
triangulares de M = L x L sitios, con L = 150, empleándose condiciones
periódicas de borde. El tamaño de red mencionado permite despreciar los
efectos de tamaño finito.
5.1 INTERACCIONES LATERALES ATRACTIVAS
5.1.1 Simulación de Montecarlo: isoterma de adsorción, calordiferencial de adsorción, energía de adsorción, factor termodinámicoy entropía configuracional.
La simulación se llevó a cabo descartando los primeros 105 pasos
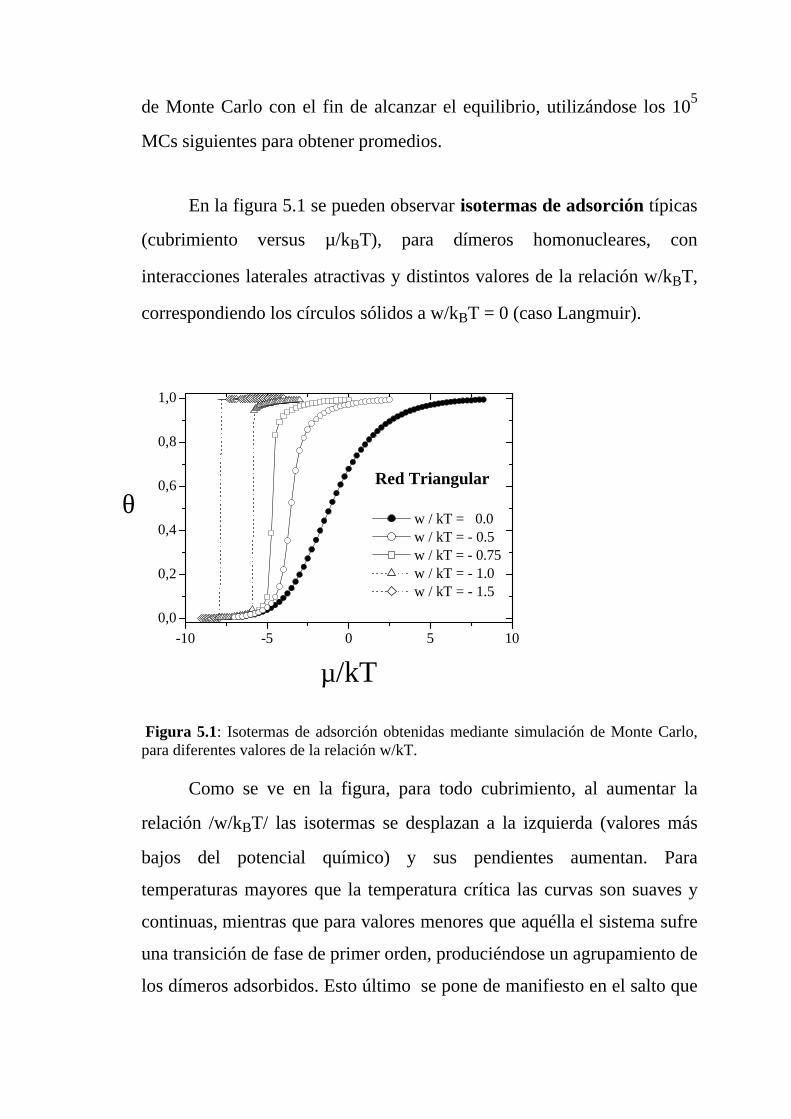
de Monte Carlo con el fin de alcanzar el equilibrio, utilizándose los 105
MCs siguientes para obtener promedios.
En la figura 5.1 se pueden observar isotermas de adsorción típicas
(cubrimiento versus µ/kBT), para dímeros homonucleares, con
interacciones laterales atractivas y distintos valores de la relación w/kBT,
correspondiendo los círculos sólidos a w/kBT = 0 (caso Langmuir).
Figura 5.1: Isotermas de adsorción obtenidas mediante simulación de Monte Carlo,para diferentes valores de la relación w/kT.
Como se ve en la figura, para todo cubrimiento, al aumentar la
relación /w/kBT/ las isotermas se desplazan a la izquierda (valores más
bajos del potencial químico) y sus pendientes aumentan. Para
temperaturas mayores que la temperatura crítica las curvas son suaves y
continuas, mientras que para valores menores que aquélla el sistema sufre
una transición de fase de primer orden, produciéndose un agrupamiento de
los dímeros adsorbidos. Esto último se pone de manifiesto en el salto que
-10 -5 0 5 100,0
0,2
0,4
0,6
0,8
1,0
Red Triangular
θ
µ/kT
w / kT = 0.0 w / kT = - 0.5 w / kT = - 0.75 w / kT = - 1.0 w / kT = - 1.5
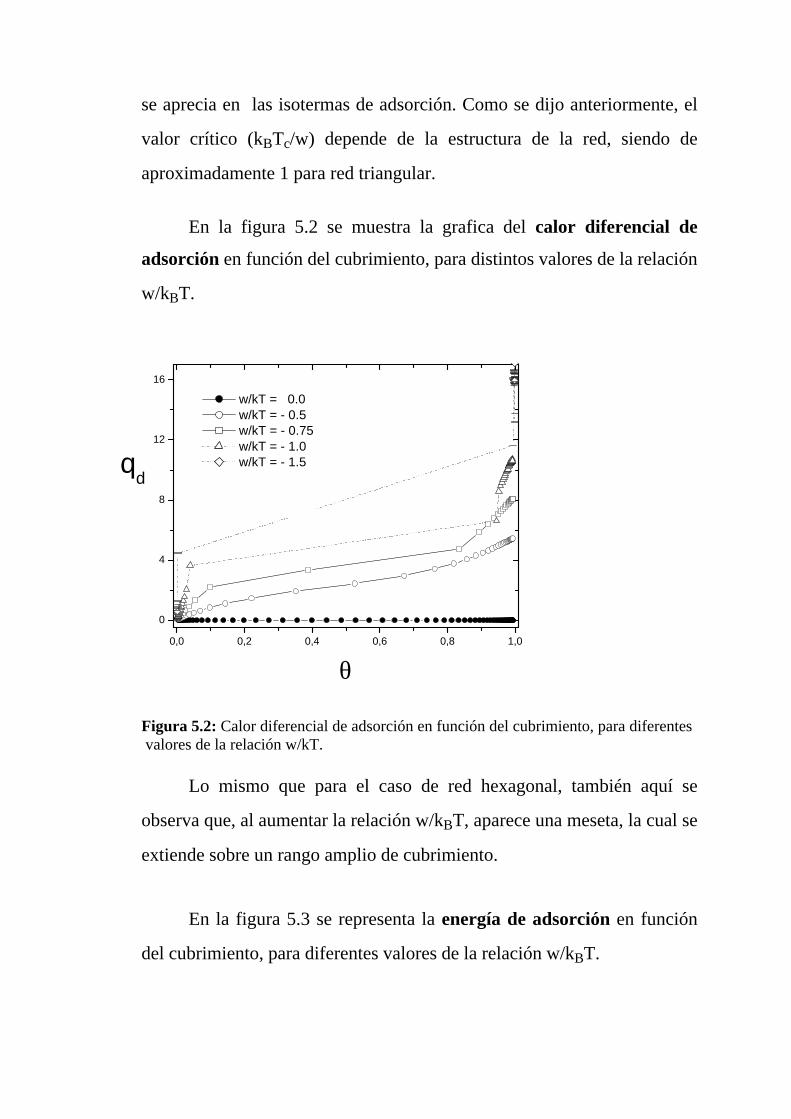
se aprecia en las isotermas de adsorción. Como se dijo anteriormente, el
valor crítico (kBTc/w) depende de la estructura de la red, siendo de
aproximadamente 1 para red triangular.
En la figura 5.2 se muestra la grafica del calor diferencial de
adsorción en función del cubrimiento, para distintos valores de la relación
w/kBT.
Figura 5.2: Calor diferencial de adsorción en función del cubrimiento, para diferentes valores de la relación w/kT.
Lo mismo que para el caso de red hexagonal, también aquí se
observa que, al aumentar la relación w/kBT, aparece una meseta, la cual se
extiende sobre un rango amplio de cubrimiento.
En la figura 5.3 se representa la energía de adsorción en función
del cubrimiento, para diferentes valores de la relación w/kBT.
0,0 0,2 0,4 0,6 0,8 1,0
0
4
8
12
16
qd
θ
w/kT = 0.0 w/kT = - 0.5 w/kT = - 0.75 w/kT = - 1.0 w/kT = - 1.5

La figura 5.4 muestra las gráficas del factor termodinámico en
función del cubrimiento, para diferentes valores de la relación w/kBT.
Figura 5.3: Energía de adsorción en función del cubrimiento, para diferentesvalores de la relación w/kT.
Figura 5.4: Factor termodinámico en función del cubrimiento, para diferentes valores de la relación w/kT.
0,0 0,2 0,4 0,6 0,8 1,0
0
1
2
3
4Red Triangular
-ε/k
T
θ
w / kT = 0.00 w / kT = - 0.5 w / kT = - 0.75 w / kT = - 1.0 w / kT = - 1.5
0,0 0,2 0,4 0,6 0,8 1,0
10-1
100
101
102
103
Red Triangular
th
θ
w / kT = 0.00 w / kT = - 0.5 w / kT = - 0.75 w / kT = - 1.0 w / kT = - 1.5

En la figura 5.5 se describe la entropía configuracional s(θ,T). Las
curvas se han obtenido a partir de la ecuación (3.15), y las dependencias µ
(θ,T) y ε (θ,T) se han tomado de las isotermas de adsorción y de la gráfica
de energía de adsorción respectivamente, ambas obtenidas por simulación.
Figura 5.5: Entropía configuracional en función del cubrimiento, para diferentes
valores de la relación w/kT.
Como puede observarse, para todos los valores de wkB/T las curvas
son asimétricas con respecto a un cubrimiento de θ = 0.5, y la entropía
disminuye para todo θ a medida que aumenta el valor de las interacciones
laterales.
5.1.2 Comparación de valores obtenidos por simulación y teóricos
Las siguientes ecuaciones expresan la ley de variación de diferentes
variables termodinámicas para la adsorción de dímeros sobre red
0,0 0,2 0,4 0,6 0,8 1,00,0
0,2
0,4
0,6
0,8
Red Triangular
w / kT
0.00 0.50 0.75 1.00
s/kB
θ
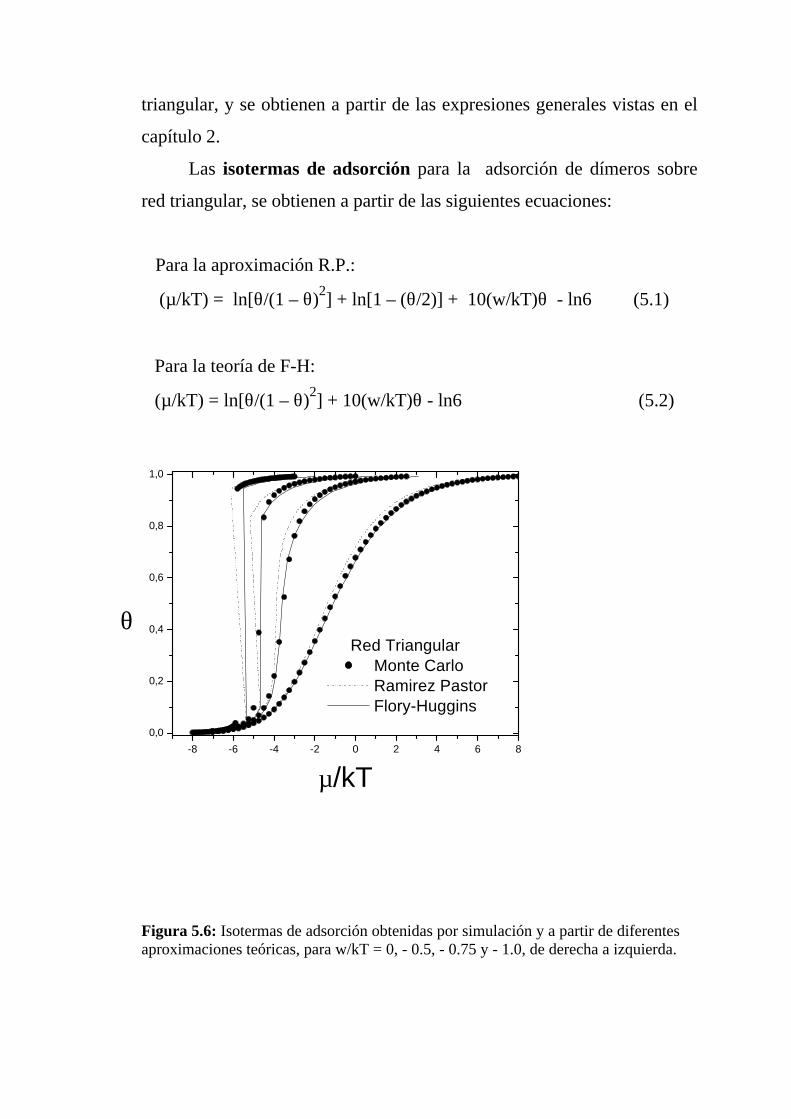
triangular, y se obtienen a partir de las expresiones generales vistas en el
capítulo 2.
Las isotermas de adsorción para la adsorción de dímeros sobre
red triangular, se obtienen a partir de las siguientes ecuaciones:
Para la aproximación R.P.:
(µ/kT) = ln[θ/(1 – θ)2] + ln[1 – (θ/2)] + 10(w/kT)θ - ln6 (5.1)
Para la teoría de F-H:
(µ/kT) = ln[θ/(1 – θ)2] + 10(w/kT)θ - ln6 (5.2)
Figura 5.6: Isotermas de adsorción obtenidas por simulación y a partir de diferentesaproximaciones teóricas, para w/kT = 0, - 0.5, - 0.75 y - 1.0, de derecha a izquierda.
0,0
0,2
0,4
0,6
0,8
1,0
-8 -6 -4 -2 0 2 4 6 8
µ/kT
θ Red Triangular
Monte Carlo Ramirez Pastor Flory-Huggins

En la figura 5.6 se pueden ver las isotermas de adsorción obtenidas
por simulación de Monte Carlo y a partir de las aproximaciones teóricas
mencionadas, para valores de w/kT iguales a 0, - 0.5, - 0.75 y - 1.0, de
derecha a izquierda. Para w/kT = 0 ambas aproximaciones muestran un
muy buen acuerdo con la simulación. Para valores mayores de la
interacción lateral se pueden apreciar discrepancias entre los valores
teóricos y simulados, siendo mejor el acuerdo que muestra la
aproximación de F-H..
El factor termodinámico en función del cubrimiento para la
adsorción de dímeros sobre red triangular, se obtiene a partir de las
siguientes ecuaciones:
Para la aproximación R.P.:
th = (1-θ)[1 – ( θ/2)]–1
+ 10 (w/kT)θ (5.3)
Para la aproximación F-H:
th = [(1 + θ)/ (1 - θ)] + 10 (w/kT)θ (5.4)
En la figura 5.7 se observa que el mejor ajuste entre los modelos
teóricos y la simulación se verifica para w/k/T = 0, produciéndose
discrepancias entre aquéllos para valores mayores de las interacciones
laterales, siendo mejor el acuerdo que muestra la aproximación R.P..

Figura 5.7: Curvas del factor termodinámico en función del cubrimiento, obtenidaspor simulación y a partir de diferentes aproximaciones teóricas, para diferentes valoresde la relación w/kT (0, -0,5, –0,75 de arriba hacia abajo).
La energía de adsorción por sitio (normalizada a su valor de
máximo cubrimiento) en función del cubrimiento, para la adsorción de
dímeros sobre red triangular, según las aproximaciones de F-H, y de R.P.
viene dada por:
ε(θ)/ε(1) = θ2 (5.5)
Como se ve en la figura 5.8, ambas aproximaciones predicen un
comportamiento de la energía normalizada que es independiente del valor
de la interacción lateral w, produciéndose un mejor ajuste para valores
más bajos de la interacción lateral.
0,0 0,2 0,4 0,6 0,8 1,0
0,1
1
10
100
1000
th
θ
Simulación Ramírez Pastor Flory-Huggins

Figura 5.8: Energía de adsorción por sitio (normalizada a su valor de máximocubrimiento), en función del cubrimiento, obtenidas por simulación y a partir deaproximaciones teóricas, para diferentes valores de la relación w/kT (-0,5, -,75 y –1,de abajo hacia arriba).
De acuerdo con las aproximaciones de F-H y R.P., el calor
diferencial de adsorción en función del cubrimiento, para la adsorción
de dímeros sobre red triangular, se obtiene a partir de la siguiente
ecuación:
qd = - 10wθ (5.6)
En la figura 5.9 se muestran las gráficas del calor diferencial
de adsorción en función del cubrimiento, para distintos valores de la
0,0 0,2 0,4 0,6 0,8 1,00,0
0,2
0,4
0,6
0,8
1,0
ε(
θ)/ε
(1)
θ
Simulación Ramirez Pastor y
Flory-Huggins

relación (w/kT), obtenidas por simulación de Monte Carlo y a partir de
las aproximaciones teóricas mencionadas.
Figura 5.9: Curvas de calor diferencial de adsorción, en función del cubrimiento,obtenidas por simulación, y a partir de aproximaciones teóricas, para diferentesvalores de la relación w/kT (-0.5, -0.75 y –1, de abajo hacia arriba).
De la ecuación (5.6) resulta que ambos modelos predicen un
aumento lineal del calor diferencial de adsorción con el cubrimiento. El
acuerdo entre aquéllos y la simulación es bueno para altas temperaturas, y
el ajuste es perfecto cuando kw/T = 0.
La entropía por sitio en función del cubrimiento, para la adsorción
de dímeros sobre red triangular, se obtiene a partir de las siguientes
ecuaciones:
Para la aproximación R.P.:
s(θ,c)/kB = [1 – (θ/2)]ln[1 – (θ/2)] – (θ/2)ln((θ/2) – (1 – θ)ln(1 – θ)
+ (θ/2)0,693; (5.7)
0,0 0,2 0,4 0,6 0,8 1,00
2
4
6
8
10
qd
θ
Simulación Ramírez Pastor y
Flory-Huggins

Para el modelo de F-H:
s(θ,c)/kB = [1 – (θ/2)]ln[1 – (θ/2)] – (θ/2)ln((θ/2) – (1 – θ)ln(1 – θ)
+ (θ/2)(c – 1); (5.8)
En la figura 5.10 se muestran las curvas de entropía por sitio, en
función del cubrimiento, obtenidas por simulación de Monte Carlo y a
partir de las aproximaciones teóricas mencionadas, para w/kT = 0 (caso
Langmuir).
Figura 5.10: Curvas de entropía por sitio, en función del cubrimiento, obtenidas porsimulación, y a partir de aproximaciones teóricas.
Como se puede observar en la figura, se advierten diferencias
apreciables entre ambas aproximaciones teóricas y la simulación en todo
el rango del cubrimiento.
0,0 0,2 0,4 0,6 0,8 1,0
0,0
0,1
0,2
0,3
0,4
0,5
0,6
0,7
0,8
0,9
1,0
1,1
s/kB
θ
Simulación Flory-Huggins Ramirez Pastor

5.2 INTERACCIONES LATERALES REPULSIVAS
5.2.1 Simulación de Montecarlo: isoterma de adsorción, calordiferencial de adsorción, energía de adsorción, factor termodinámicoy entropía diferencial de adsorción.
Para temperaturas alejadas del punto crítico el estado de equilibrio
se alcanzó luego de descartar los primeros 105 pasos de Monte Carlo,
mientras que los siguientes 105 MCs se emplearon para obtener
promedios. En la cercanía de los puntos críticos debieron usarse hasta 106
MCs para permitir que el sistema se relajara a partir de estados
metaestables. A fin de alcanzar el equilibrio en un tiempo razonable, se
permite el desplazamiento de las partículas adsorbidas a posiciones
vecinas próximas (relajación difusiva), por salto a lo largo del eje del
dímero, o mediante reptado por rotación alrededor de un extremo de
aquél.
En la figura 5.11 se muestran isotermas de adsorción para el caso
de interacciones laterales repulsivas y diferentes valores de la relación
w/kBT. Los círculos sólidos representan el estado de referencia (isoterma
de Langmuir, w/kBT = 0).
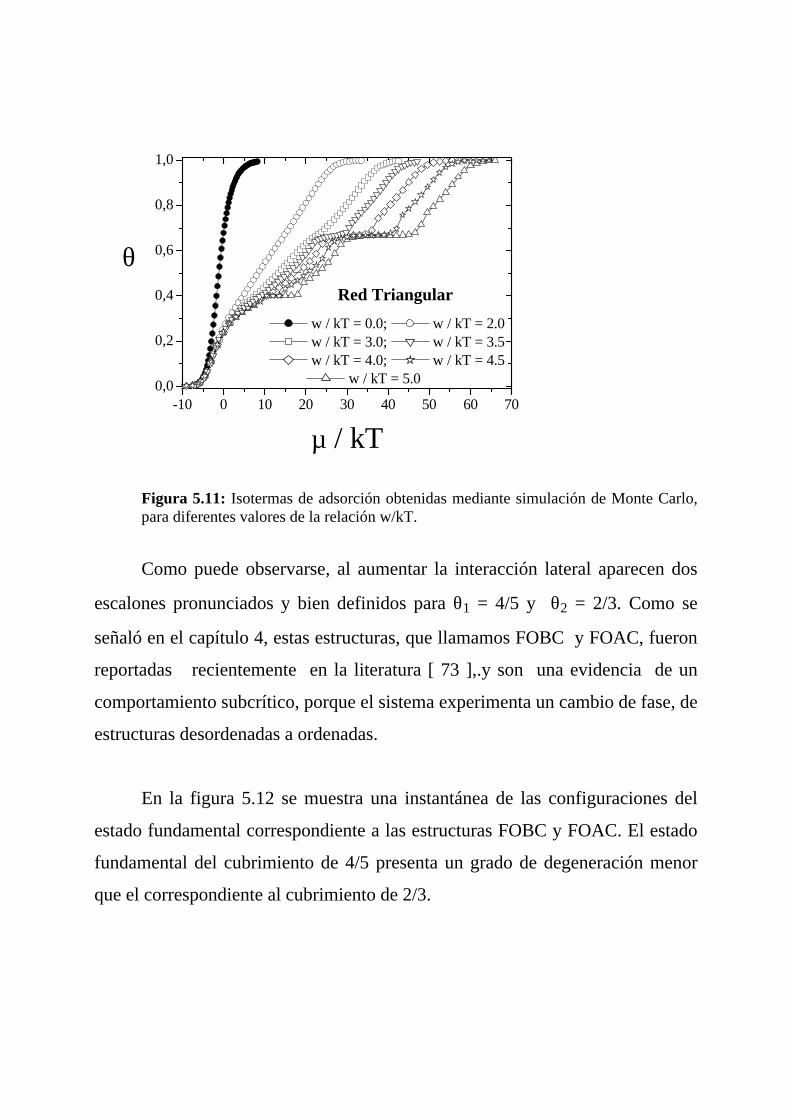
Figura 5.11: Isotermas de adsorción obtenidas mediante simulación de Monte Carlo,para diferentes valores de la relación w/kT.
Como puede observarse, al aumentar la interacción lateral aparecen dos
escalones pronunciados y bien definidos para θ1 = 4/5 y θ2 = 2/3. Como se
señaló en el capítulo 4, estas estructuras, que llamamos FOBC y FOAC, fueron
reportadas recientemente en la literatura [ 73 ],.y son una evidencia de un
comportamiento subcrítico, porque el sistema experimenta un cambio de fase, de
estructuras desordenadas a ordenadas.
En la figura 5.12 se muestra una instantánea de las configuraciones del
estado fundamental correspondiente a las estructuras FOBC y FOAC. El estado
fundamental del cubrimiento de 4/5 presenta un grado de degeneración menor
que el correspondiente al cubrimiento de 2/3.
-10 0 10 20 30 40 50 60 700,0
0,2
0,4
0,6
0,8
1,0
Red Triangular
θ
µ / kT
w / kT = 0.0; w / kT = 2.0 w / kT = 3.0; w / kT = 3.5 w / kT = 4.0; w / kT = 4.5
w / kT = 5.0

(a)
(b)
Figura 5.12: Configuraciones del estado fundamental correspondiente a las estructuras FOBC(a) y FOAC (b).
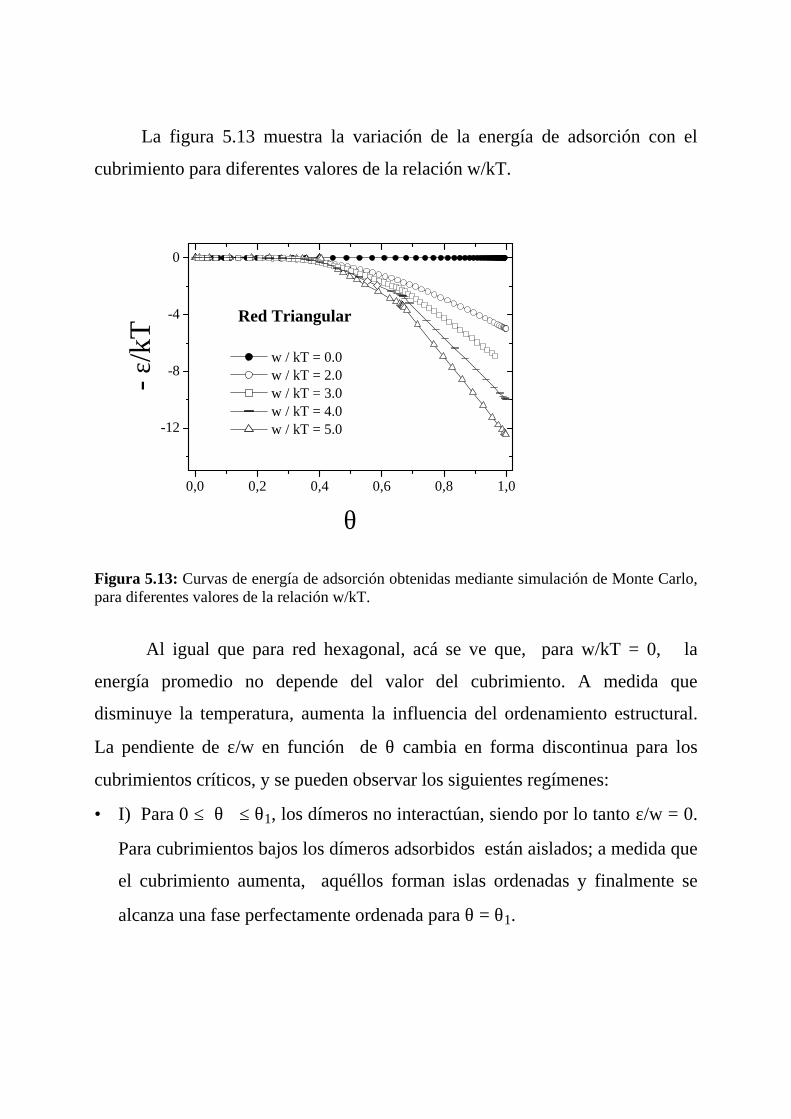
La figura 5.13 muestra la variación de la energía de adsorción con el
cubrimiento para diferentes valores de la relación w/kT.
Figura 5.13: Curvas de energía de adsorción obtenidas mediante simulación de Monte Carlo,para diferentes valores de la relación w/kT.
Al igual que para red hexagonal, acá se ve que, para w/kT = 0, la
energía promedio no depende del valor del cubrimiento. A medida que
disminuye la temperatura, aumenta la influencia del ordenamiento estructural.
La pendiente de ε/w en función de θ cambia en forma discontinua para los
cubrimientos críticos, y se pueden observar los siguientes regímenes:
• I) Para 0 ≤ θ ≤ θ1, los dímeros no interactúan, siendo por lo tanto ε/w = 0.
Para cubrimientos bajos los dímeros adsorbidos están aislados; a medida que
el cubrimiento aumenta, aquéllos forman islas ordenadas y finalmente se
alcanza una fase perfectamente ordenada para θ = θ1.
0,0 0,2 0,4 0,6 0,8 1,0
-12
-8
-4
0
Red Triangular
- ε/k
T
θ
w / kT = 0.0 w / kT = 2.0 w / kT = 3.0 w / kT = 4.0 w / kT = 5.0
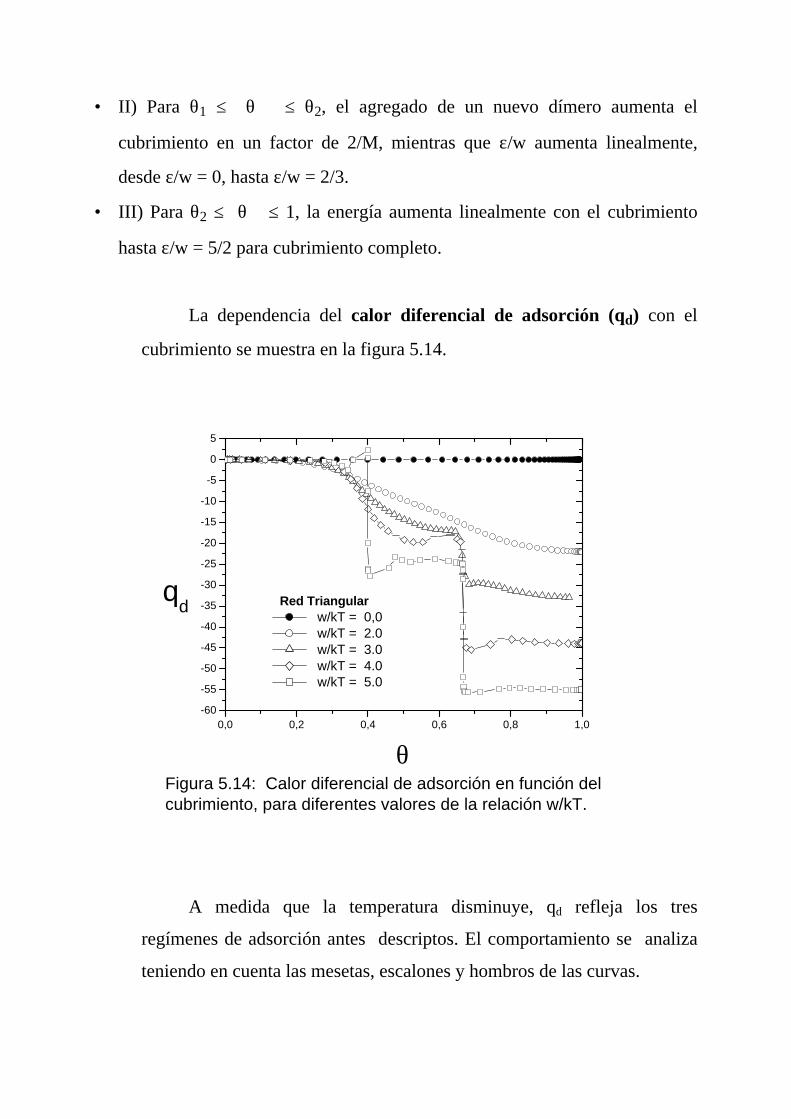
• II) Para θ1 ≤ θ ≤ θ2, el agregado de un nuevo dímero aumenta el
cubrimiento en un factor de 2/M, mientras que ε/w aumenta linealmente,
desde ε/w = 0, hasta ε/w = 2/3.
• III) Para θ2 ≤ θ ≤ 1, la energía aumenta linealmente con el cubrimiento
hasta ε/w = 5/2 para cubrimiento completo.
La dependencia del calor diferencial de adsorción (qd) con el
cubrimiento se muestra en la figura 5.14.
A medida que la temperatura disminuye, qd refleja los tres
regímenes de adsorción antes descriptos. El comportamiento se analiza
teniendo en cuenta las mesetas, escalones y hombros de las curvas.
0,0 0,2 0,4 0,6 0,8 1,0-60
-55
-50
-45
-40
-35
-30
-25
-20
-15
-10
-5
0
5
Figura 5.14: Calor diferencial de adsorción en función del cubrimiento, para diferentes valores de la relación w/kT.
qd
θ
Red Triangular w/kT = 0,0 w/kT = 2.0 w/kT = 3.0 w/kT = 4.0 w/kT = 5.0

Se tienen tres mesetas:
• I) Para 0 ≤ θ ≤ θ1, los dímeros no interactúan, y la adsorción o desorción de
un dímero involucra un calor diferencial igual a cero.
• II) Para θ1 ≤ θ ≤ θ2, el sistema pasa de una fase ordenada de bajo
cubrimiento (FOBC) a una fase ordenada de alto cubrimiento (FOAC). El
valor de qd en este régimen se puede obtener a partir de la pendiente de ε vs
θ, resultando, en general, qd = [(c – 2)w]/[3(θ2 − θ1)]. Luego, para redes
triangulares es qd = 5w.
• III) Para θ2 ≤ θ ≤ 1, la energía necesaria para adsorber un nuevo dímero se
puede estimar considerando su adsorción sobre una estructura zig-zag
perfecta. Para ello es necesario crear previamente un par de sitios vacíos
moviendo un dímero ya adsorbido. Este proceso involucra una variación de
energía igual a 3w, como se muestra en la figura 5.15, donde el dímero A
cambia de una posición en la que interactúa con cuatro vecinos próximos
(NN), a otra en la que interactúa con siete NN (estado intermedio: las
interacciones de los NN se indican con líneas de trazo). Así, el dímero
entrante (indicado con B), se puede alojar en la red con un costo adicional de
8w (ver estado final en figura 5.15). Finalmente, la variación total de energía
es de 11w, correspondiendo a la meseta de qd para θ2 ≤ θ ≤ 1 en la figura
5.14.

Red Triangular
A
Estado Inicial
A
Estado Intermedio
A
B
Estado Final
Figura 5.15

En la figura 5.16 se representa la entropía configuracional como
una función del cubrimiento.
Figura 5.16: Entropía configuracional en función del cubrimiento, paradiferentes valores de la relación w/kT.
Para dímeros que no interactúan, s(θ,T) tiene un máximo para θ >
0,5. El efecto global de las interacciones es la disminución de la entropía
para todos los cubrimientos. A medida que la temperatura disminuye,
aparecen dos mínimos locales de s(θ,T) para θ = θ1 y θ = θ2, lo cual
confirma la existencia de estructuras ordenadas para estos valores del
cubrimiento.
El hecho de que el segundo mínimo corresponda a un estado
fundamental altamente degenerado se pone de manifiesto en su
profundidad, que es menor que la del primer mínimo.
0,0 0,2 0,4 0,6 0,8 1,00,0
0,1
0,2
0,3
0,4
0,5
0,6
0,7
0,8
Red Triangular
s/kB
θ
w/kT 0,0 2,0 3,0 3,5 4,0 4,5 5,0
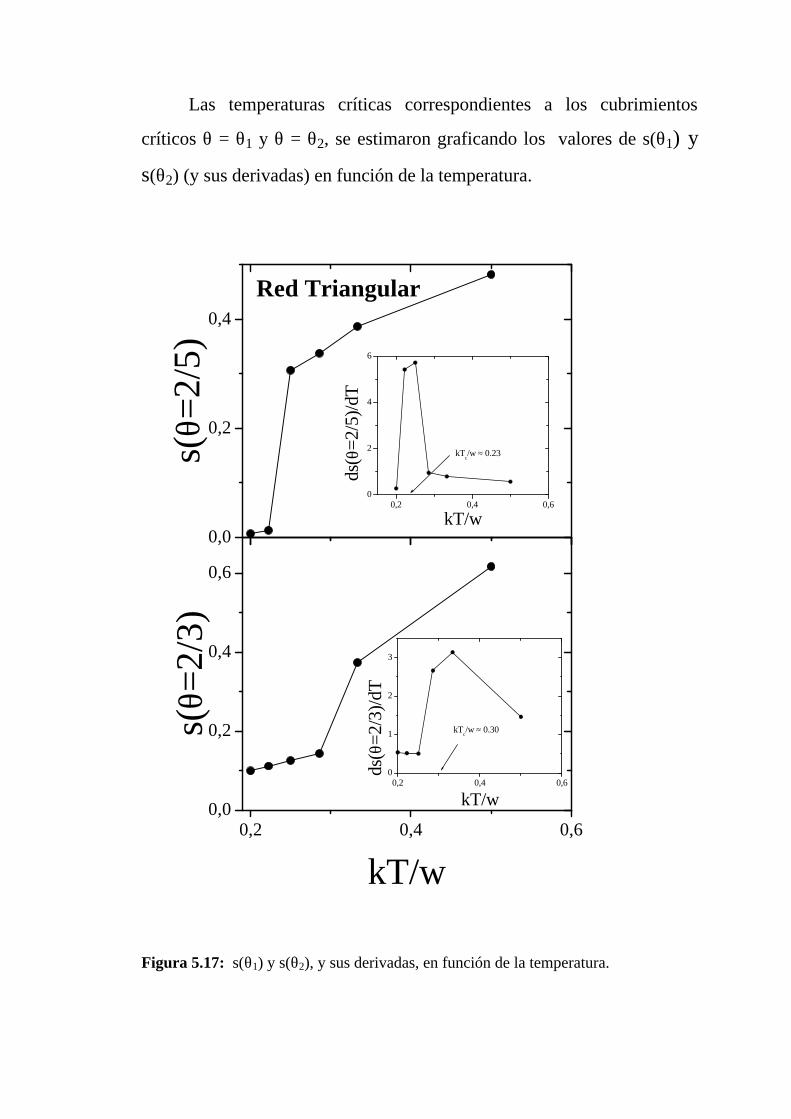
Las temperaturas críticas correspondientes a los cubrimientos
críticos θ = θ1 y θ = θ2, se estimaron graficando los valores de s(θ1) y
s(θ2) (y sus derivadas) en función de la temperatura.
Figura 5.17: s(θ1) y s(θ2), y sus derivadas, en función de la temperatura.
0,0
0,2
0,4
0,2 0,4 0,60,0
0,2
0,4
0,6
0,2 0,4 0,60
2
4
6
kTc/w ≈ 0.23
ds
( θ=2
/5)/
dT
kT/w
Red Triangular
s(θ=
2/5)
s(θ=
2/3)
kT/w
0,2 0,4 0,60
1
2
3
kTc/w ≈ 0.30
ds(θ
=2/3
)/dT
kT/w

Los puntos de inflexión en s(θ1) y s(θ2) (picos en la derivadas)
determinan las temperaturas críticas. Los valores que se obtuvieron
fueron: kBT1/w ≅ 0,23 y kBT2/w ≅ 0,30. La confirmación de estos valores
debería realizarse mediante un escaleo de tamaño finito, lo cual, como se
indicó en el capítulo 4, está fuera de los alcances de este trabajo.
5.2.2 Comparación de valores obtenidos por simulación y teóricos.
Las isotermas de adsorción para la adsorción de dímeros sobre
una red triangular, se obtienen a partir de las ecuaciones (5.1) y (5.2), para
las aproximaciones R.P y F-H, respectivamente.
Figura 5.18: Isotermas de adsorción obtenidas por simulación y a partir de diferentesaproximaciones teóricas, para w/kT = 0, 2 y 5, de izquierda a derecha.
Como se aprecia en la figura 5.18, las aproximaciones teóricas
mencionadas muestran un buen acuerdo con la simulación para w/kT = 0,
0,0
0,2
0,4
0,6
0,8
1,0
0 10 20 30 40 50 60
µ/kT
θ Red Triangular
Monte Carlo Ramirez Pastor Flory-Huggins

pero para valores mayores de la interacción lateral no dan cuenta de la
existencia de fases ordenadas, como es característico de los modelos de
campo medio.
El factor termodinámico en función del cubrimiento, para la
adsorción de dímeros sobre red triangular, se obtiene a partir de las
ecuaciones (5.3) para la aproximación R.P., y (5.4) para la teoría de F.H..
Figura 5.19: Curvas del factor termodinámico en función del cubrimiento, obtenidaspor simulación y a partir de diferentes aproximaciones teóricas, para diferentes valoresde la relación w/kT (0, 2 y 4, de abajo hacia arriba).
En la figura anterior se muestran las gráficas del factor
termodinámico en función del cubrimiento, para distintos valores de la
relación (w/kT), obtenidas por simulación de Monte Carlo y a partir de
las aproximaciones teóricas mencionadas. Puede observarse que el mejor
ajuste entre los modelos teóricos y la simulación se produce para valores
bajos de la interacción lateral.
Para las aproximaciones de F.H. y de R.P., la dependencia del calor
0,0 0,2 0,4 0,6 0,8 1,0
1
10
100
1000
th
θ
Red triangular Simulación Ramirez Pastor Flory Huggins

diferencial de adsorción con el cubrimiento, para la adsorción de
dímeros sobre red triangular, se obtiene a partir de la ecuación (5.6), la
cual predice un crecimiento lineal del calor diferencial de adsorción con el
cubrimiento.
Figura 5.20: Curvas de calor diferencial de adsorción, en función del cubrimiento,obtenidas por simulación, y a partir de aproximaciones teóricas, para diferentesvalores de la relación w/kT (0, 2 y 4, de arriba hacia abajo).
En la figura anterior se muestran las gráficas del calor diferencial de
adsorción en función del cubrimiento, para distintos valores de la relación
(w/kT), obtenidas por simulación de Monte Carlo y a partir de las
aproximaciones teóricas mencionadas. Se puede observar que no existe un
buen acuerdo entre ambos modelos y la simulación.
La ecuación (5.5) expresa la energía de adsorción por sitio
normalizada a su valor de máximo cubrimiento) en función del
cubrimiento, para la adsorción de dímeros sobre red triangular, según las
0,0 0,2 0,4 0,6 0,8 1,0
-35
-30
-25
-20
-15
-10
-5
0
qd
θ
Simulación Ramírez Pastor y
Flory-Huggins

aproximaciones de F.H. y R.P..
Figura 5.21: Energía de adsorción por sitio (normalizada a su valor de máximocubrimiento), en función del cubrimiento, obtenidas por simulación y a partir deaproximaciones teóricas, para diferentes valores de la relación w/kT (2, 4 y 5, dearriba hacia abajo).
Como se observa en la figura anterior, ambas aproximaciones
predicen un comportamiento de la energía normalizada que es
independiente del valor de la interacción lateral w, produciéndose un
mejor ajuste para los valores más bajos de la interacción lateral.
5.2.3 Comparación de valores obtenidos por simulación para redeshexagonal y triangular.
En esta sección se comparan los resultados obtenidos por
simulación para la isoterma de adsorción, el calor diferencial de
adsorción, y la entropía configuracional, para redes hexagonales y
triangulares.
0,0 0,2 0,4 0,6 0,8 1,0
0,0
0,2
0,4
0,6
0,8
1,0
ε(
θ)/ε
(1)
θ
Red Triangular Simulación Ramírez Pastor
y Flory-Huggins

(a)
(b)
Figura 5.22: Isotermas de adsorción obtenidas mediante simulación de Monte Carlo,para diferentes valores de la relación w/kT: a) Red Hexagonal, b) Red Triangular.
-10 0 10 20 30 40 50 600,0
0,2
0,4
0,6
0,8
1,0
θ
µ/kT
Red Triangular
w/kT = 4 w/kT = 5
-10 0 10 20 30 40 500,0
0,2
0,4
0,6
0,8
1,0
θ
µ/kT
Red Hexagonal
w/kT = 5 w/kT = 8

(a)
(b)
Figura 5.23: Calor diferencial de adsorción en función del cubrimiento, paradiferentes valores de la relación w/kT: a) Red Hexagonal, b) Red Triangular.
0,0 0,2 0,4 0,6 0,8 1,0
-40
-35
-30
-25
-20
-15
-10
-5
0
qd
θ
Red Hexagonal
w/kT = 5 w/kT = 8
0,0 0,2 0,4 0,6 0,8 1,0
-60
-55
-50
-45
-40
-35
-30
-25
-20
-15
-10
-5
0
5
qd
θ
Red Triangular
w/kT = 3 w/kT = 5

(a)
(b)
Figura 5.24: Entropía configuracional en función del cubrimiento, para diferentesvalores de la relación w/kT.: a) Red Hexagonal, b) Red Triangular.
0,0 0,2 0,4 0,6 0,8 1,00,0
0,1
0,2
0,3
0,4
0,5
0,6
s/kB
θ
Red Triangular
w/kT= 3,0 w/kT= 4,5
0,0 0,2 0,4 0,6 0,8 1,00,00
0,05
0,10
0,15
0,20
0,25
0,30
0,35
0,40
s/kB
θ
Red Hexagonal
w/kT = 5.0 w/kT = 10.0
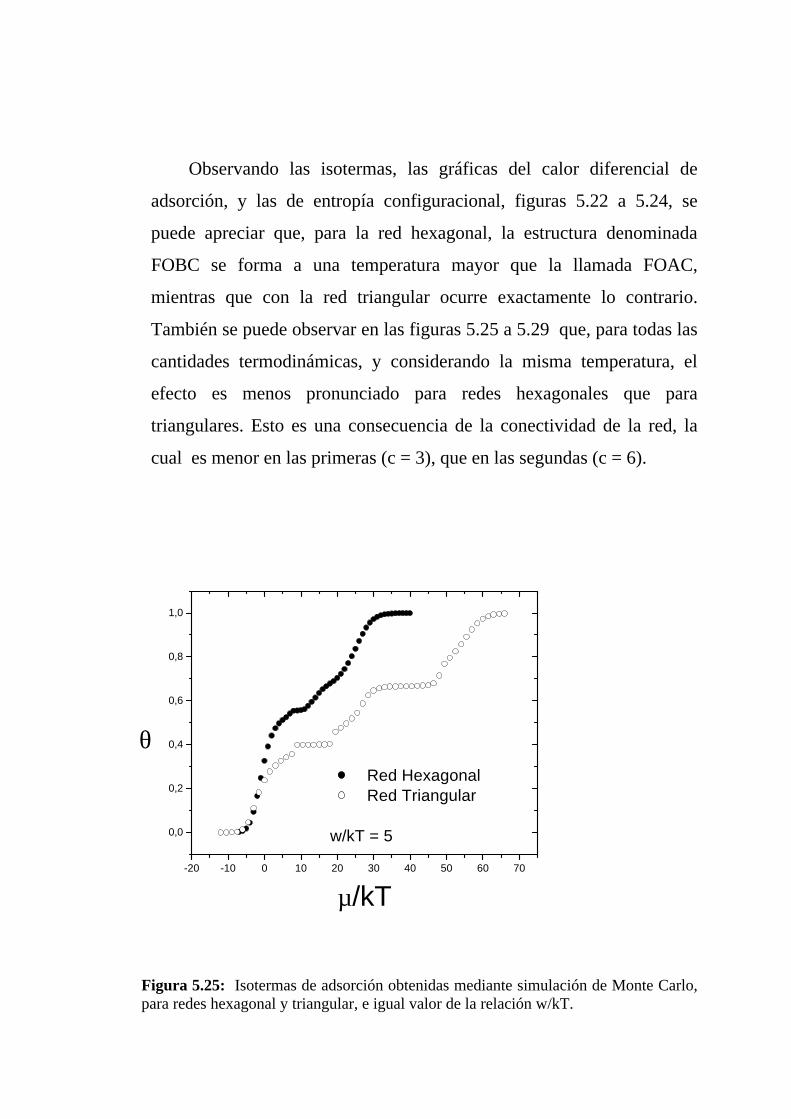
Observando las isotermas, las gráficas del calor diferencial de
adsorción, y las de entropía configuracional, figuras 5.22 a 5.24, se
puede apreciar que, para la red hexagonal, la estructura denominada
FOBC se forma a una temperatura mayor que la llamada FOAC,
mientras que con la red triangular ocurre exactamente lo contrario.
También se puede observar en las figuras 5.25 a 5.29 que, para todas las
cantidades termodinámicas, y considerando la misma temperatura, el
efecto es menos pronunciado para redes hexagonales que para
triangulares. Esto es una consecuencia de la conectividad de la red, la
cual es menor en las primeras (c = 3), que en las segundas (c = 6).
Figura 5.25: Isotermas de adsorción obtenidas mediante simulación de Monte Carlo,para redes hexagonal y triangular, e igual valor de la relación w/kT.
-20 -10 0 10 20 30 40 50 60 70
0,0
0,2
0,4
0,6
0,8
1,0
θ
µ/kT
Red Hexagonal Red Triangular
w/kT = 5

Figura 5.26: Calor diferencial de adsorción en función del cubrimiento, para redeshexagonal y triangular, e igual valor de la relación w/kT.
Figura 5.27: Energía en función del cubrimiento, para redes hexagonal y triangular, eigual valor de la relación w/kT.
0,0 0,2 0,4 0,6 0,8 1,0
-10
-8
-6
-4
-2
0
-ε/k
T
θ
Red Hexagonal Red Triangular
w/kT = 4
0,0 0,2 0,4 0,6 0,8 1,0-60
-55
-50
-45
-40
-35
-30
-25
-20
-15
-10
-5
0
5
qd
θ
Red Hexagonal Red Triangular
w/kT = 5
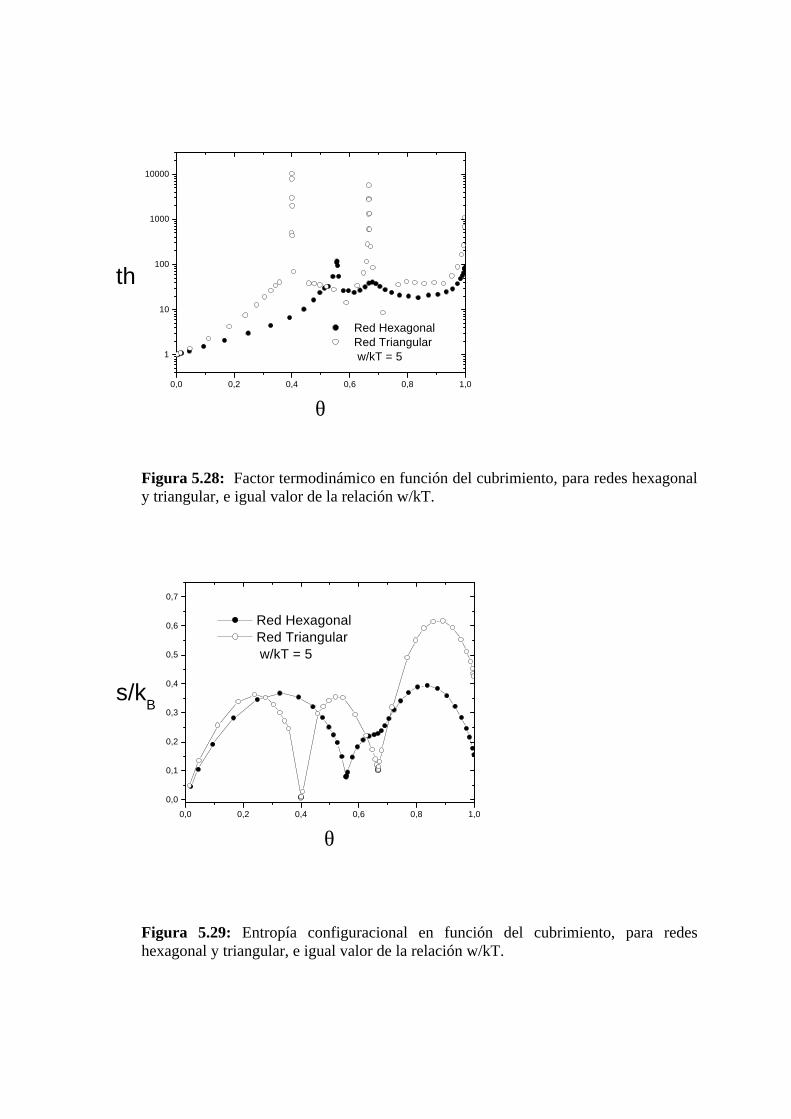
Figura 5.28: Factor termodinámico en función del cubrimiento, para redes hexagonaly triangular, e igual valor de la relación w/kT.
Figura 5.29: Entropía configuracional en función del cubrimiento, para redeshexagonal y triangular, e igual valor de la relación w/kT.
0,0 0,2 0,4 0,6 0,8 1,0
1
10
100
1000
10000
th
θ
Red Hexagonal Red Triangular
w/kT = 5
0,0 0,2 0,4 0,6 0,8 1,0
0,0
0,1
0,2
0,3
0,4
0,5
0,6
0,7
s/kB
θ
Red Hexagonal Red Triangular
w/kT = 5

5.3 RESUMEN
En este capítulo se estudió la adsorción de dímeros sobre
superficies triangulares homogéneas. En primer lugar se analizó el
comportamiento de diversas cantidades termodinámicas asociadas a la
capa adsorbida, tanto para interacciones laterales atractivas como
repulsivas (secciones 5.1.1 y 5.1.2), usándose para ello las técnicas de
simulación de Monte Carlo vistas en el capítulo 3. En el caso particular de
interacciones laterales repulsivas y valores altos de la relación
temperaturas, se encontraron dos muevas fases ordenadas en el adsorbato,
para θ1 = 4/5 y θ2 = 2/3. Lo anterior indica que el sistema experimenta
una transición de fase, de estructuras desordenadas a estructuras
ordenadas, que llamamos FOBC (fase ordenada de bajo cubrimiento) y
FOAC (fase ordenada de alto cubrimiento), las cuales reportamos
recientemente en la literatura [ 73 ], y que constituyen una clara evidencia
de un comportamiento subcrítico. Asimismo, en la figura 5.12 se
mostraron instantáneas de las configuraciones del estado fundamental de
las estructuras mencionadas. También se estimaron las temperatura
críticas correspondientes a los cubrimientos críticos. Para confirmar los
valores obtenidos es necesario realizar un escaleo de tamaño finito, el
cual está fuera de los alcances de este trabajo.
Luego se compararon los resultados obtenidos al aplicar los
modelos teóricos vistos en el capítulo 2, con los obtenidos por medio de
las técnicas de simulación de Monte Carlo.
Por último, en la sección 5.2.3 se compararon los resultados
obtenidos por simulación para ciertas cantidades termodinámicas
(isotermas de adsorción, calor diferencial de adsorción, y entropía
configuracional), para redes hexagonales y triangulares, y valores iguales
de interacción lateral repulsiva.
* * * * * * * *

CAPITULO 6
ADSORCION DE DIMEROS SOBRE REDESHETEROGENEAS HEXAGONALES Y TRIANGULARES
En éste capítulo se estudia la adsorción de dímeros sobre
superficies heterogéneas hexagonales y triangulares, por medio de
modelos teóricos, entre ellos los de Nitta [69], y los de Ramirez Pastor,
recientemente presentados en Ref. [66], comparándose luego los valores
que surgen de la aplicación de estas teorías, con los resultados obtenidos
mediante simulación de Monte Carlo. Asimismo se propone un método
para caracterizar la topografía energética de sustratos que pueden
aproximarse por medio de superficies bivariadas, a través de medidas de
adsorción de dímeros no interactuantes.
6.1 MODELOS DE ADSORCIÓN DE K-MEROS NOINTERACTUANTES SOBRE SUPERFICIES HETEROGÉNEAS
6.1.1 Estudio de Marczewski [57]
Marczewski estudió el efecto de la múltiple ocupación de sitios
sobre la distribución de las energías de adsorción y también intentó
encontrar una expresión para la isoterma total de adsorción, usando la
ecuación integral de la isoterma:
θ (p,T) = θ(Ek,p,T) χk(Ek) dEk (6.1) ∆Ek
donde:
Ek: Energía de adsorción de una molécula de adsorbato, consistente en un
número k de átomos o segmentos iguales.
θ(Ek,p,T): isoterma de adsorción local para cierto tipo de topografía
superficial.

χk(Ek): Función de distribución de energías de adsorción.
Marczewski no pudo obtener una expresión rigurosa para la
isoterma de adsorción local, y si bien para el caso de grandes parches
puede usarse como isoterma local a la isoterma homogénea, para otras
topografías hubo que esperar a que Nitta desarrollara una primer solución
para superficies aleatorias.
6.1.2 Modelo de Nitta
En este modelo, desarrollado por Nitta [69] para superficies
constituidas por sitios distribuidos al azar, aquél considera una superficie
con W tipos diferentes de sitios, siendo M el número total de éstos.
Además supone que una molécula adsorbida consta de s tipos de
segmentos, siendo k el número de sitios ocupados por aquélla.
Cuando N moléculas se adsorben sobre una superficie heterogénea,
la función de partición canónica del sistema Q (N,M,T) se puede escribir:
Q = (qse)NΣ g(N,M,Nij)exp[ΣΣ(Nijεij/kBT)] (6.2) Nij i j
Nij: número de pares de adsorción constituidos por sitios tipo i y
segmentos tipo j.
Nij: configuración de los anteriores pares de adsorción.
εij : energía de adsorción de un segmento tipo j sobre un sitio tipo i.
g(N,M,Nij): número de formas de distribuir N moléculas sobre M sitios
bajo la condición de una configuración particular Nij.
qse: función de partición interna y vibracional de la molécula.
Suponiendo que los pares ij son independientes unos de otros,
con la única condición de respetar una cierta configuración Nij, y

siguiendo un procedimiento similar al presentado por Guggenheim [38]
para la interacción adsorbato-adsorbato, se llega a la siguiente expresión:
W sln[g(N,M,Nij)] = ln [go (N,M)] − [Σ Σ(Nij! / Nij*!)] (6.3) i=1 j=0
donde el supraíndice “*” indica pertenencia a una distribución aleatoria
Nij* correspondiente a una superficie homogénea, y go es el factor
combinatorio que caracteriza a esa superficie, siendo:
go = [M!/N!(M – kN)!][ξN/M(k – 1)N] (6.4)
donde ξ es una constante relacionada con la flexibilidad y simetría de las
moléculas de adsorbato.
La expresión de la isoterma de adsorción para superficies
heterogéneas, escrita en función del cubrimiento total, que se obtiene para
este modelo, es:
sln(kKp) = lnθ − kln(1 − θ) + Σ kj ln[Yj/Yj*] (6.5) j =1donde:
Yj* = f1/(1 − θ) (6.6)
sYj es una variable positiva definida así: Yj = θij/θj(1− Σθij) (6.7) j=1
θ = kN/m (fracción de sitios ocupados) (6.8)
θj = kjN/m (j = 1,2...s) (fracción de sitios ocupados por segmentos
tipo j) (6.9)

θij = Nij/Mi (j = 1,2...s, i = 1,2,..,W) (fracción de sitios tipo “i” ocupados
por segmentos tipo “j”) (6.10)
Además, K es la constante de equilibrio de adsorción definida por:
s K = (qseξΛ3/qgkBT)exp[Σ (kjε1j/kBT)] (6.11) j=1
siendo Λ la longitud de onda térmica de De Broglie y qg la función de
partición molecular interna para la fase gas.
6.1.3 Modelo de Nitta modificado
En el segundo miembro de la ecuación (6.3) se pueden distinguir
dos partes: el primer término, característico de una superficie homogénea,
y el segundo, en el cual se incorpora la heterogeneidad al problema.
Ramirez Pastor [66] modificó el modelo de Nitta reemplazando go de la
ecuación (6.3) por Ω(N,M) de la ecuación (2.4), el cual es exacto para una
superficie homogénea unidimensional, y obtuvo así una nueva expresión
para la isoterma de adsorción heterogénea:
sln(kKp) = lnθ − kln(1 − θ) + (k – 1)ln1 – [(k – 1)/k]θ + Σ kj ln[Yj/Yj*] j=1 (6.12)
La ecuación anterior difiere de la originalmente propuesta por Nitta en el
término (k – 1)ln1 – [(k – 1)/k]θ.
Se realizaron estudios [66] en los que se compararon los valores
obtenidos por aplicación de los modelos de Nitta y de Nitta modificado,
con los que resultan de la simulación de Monte Carlo en el conjunto Gran
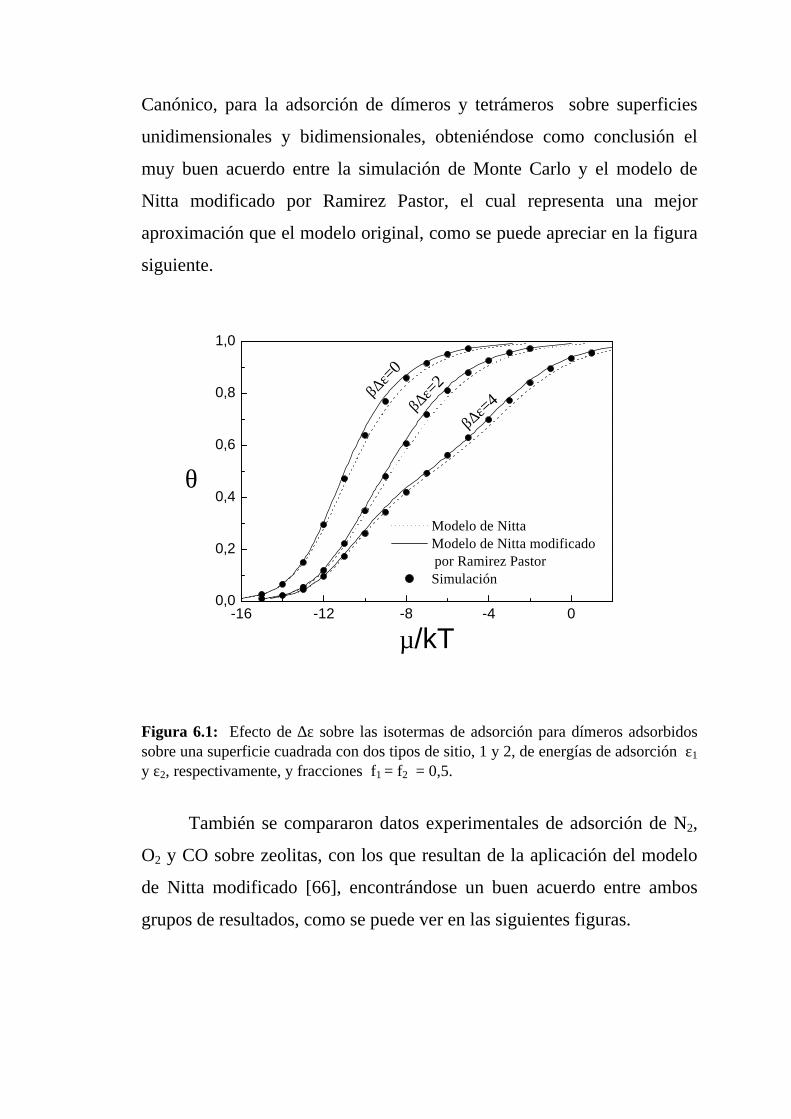
Canónico, para la adsorción de dímeros y tetrámeros sobre superficies
unidimensionales y bidimensionales, obteniéndose como conclusión el
muy buen acuerdo entre la simulación de Monte Carlo y el modelo de
Nitta modificado por Ramirez Pastor, el cual representa una mejor
aproximación que el modelo original, como se puede apreciar en la figura
siguiente.
Figura 6.1: Efecto de ∆ε sobre las isotermas de adsorción para dímeros adsorbidossobre una superficie cuadrada con dos tipos de sitio, 1 y 2, de energías de adsorción ε1
y ε2, respectivamente, y fracciones f1 = f2 = 0,5.
También se compararon datos experimentales de adsorción de N2,
O2 y CO sobre zeolitas, con los que resultan de la aplicación del modelo
de Nitta modificado [66], encontrándose un buen acuerdo entre ambos
grupos de resultados, como se puede ver en las siguientes figuras.
-16 -12 -8 -4 00,0
0,2
0,4
0,6
0,8
1,0
β∆ε=4β∆ε=
2β∆ε=
0
Modelo de Nitta Modelo de Nitta modificado
por Ramirez Pastor Simulación
θ
µ/kT

-2 -1 0 1 2 30.0
0.2
0.4
0.6
0.8
1.0
O2
N2
CO
CO, N2, O
2/5A
144 K
θ
log(p/po)
-2 -1 0 1 2 30.0
0.2
0.4
0.6
0.8
1.0
O2N
2
CO
CO, N2, O
2/10X
144 K
θ
log(p/po)
Figura 6.2: Comparación entre isotermas experimentales (círculos abiertos) y
teóricas (líneas) para los sistemas que se indican en las gráficas.
6.2 MODELOS DE ADSORCIÓN DE K-MEROSINTERACTUANTES SOBRE SUPERFICIES HETEROGÉNEAS
En los dos modelos anteriores, desarrollados sólo para las
topografías límites de sitios al azar y de dos grandes parches, se
consideraba que la adsorción con múltiple ocupación de sitios tenía lugar
sin interacción entre las moléculas adsorbidas. Ramirez Pastor [66]
desarrolló dos nuevos modelos introduciendo la heterogeneidad, y las
interacciones laterales, mediante un formalismo basado en la ecuación

integral de la isoterma, y en modelos simples para las isotermas locales.
Para ello Ramirez Pastor considera que cada molécula de adsorbato está
compuesta de k monómeros iguales, y supone una distribución discreta
de W energías de adsorción. El cubrimiento medio total, θm, se obtiene
mediante la expresión:
kW
θm = Σχk(Eki)θ(Eki) (6.13) i=1
donde Eki es el i-ésimo nivel de energía de los kW niveles posibles.
La ecuación anterior resulta de la ecuación integral de la isoterma
de adsorción (ecuación 6.1) cuando la distribución de energía de los sitios
es discreta.
La función χk(Eki) se puede escribir en función de las frecuencias
f1,...,fW, con que aparecen los W sitios, y puede calcularse en forma
analítica suponiendo una topografía particular para la superficie.
Una vez obtenida la expresión analítica de χk se calcula la isoterma
local, y mediante la ecuación (6.13) se obtiene la isoterma total de
adsorción. Ramirez Pastor [66] desarrolló dos modelos para obtener la
isoterma local, uno basado en una estadística tipo Fermi-Dirac, y el otro
basado en la ecuación homogénea exacta en una dimensión. En los dos
casos las interacciones laterales se introdujeron por medio de un término
de campo promedio.
6.2.1 Modelo tipo Fermi-Dirac para la isoterma local de adsorción
En este modelo Ramirez Pastor supone que los kW niveles posibles,
de energías Eki, son diferentes entre sí, y mediante la estadística de

Fermi-Dirac obtiene la ecuación para la isoterma local sobre un conjunto
de k sitios, sin interacción lateral entre las moléculas adsorbidas:
θFD(Eki) = exp[−β(Eki −µ)]/1 + exp[−β(Eki −µ)] (6.14)
donde µ es el potencial químico, e i es uno de los kW niveles posibles de
energía.
Las interacciones laterales se introducen en la ecuación (6.14)
agregando una contribución energética de campo medio, con lo cual
resulta:
θFD(Eki*) = exp[−β(Eki + 2λwθ*−µ)]/1 + exp[−β(Eki + 2λwθ*−µ)]
(6.15)
donde:
Eki* = Eki + 2λwθ*
θ* = θFD(Eki*), para una topografía de grandes parches, y θ* = θm para
una topografía al azar.
2λwθ* = máxima interacción lateral para un k-mero con todos sus
primeros vecinos ocupados.
A partir de las ecuaciones (6.14) y (6.15) se puede calcular la
isoterma total de adsorción heterogénea, con múltiple ocupación de sitios
e interacciones laterales. Para el caso de dímeros (k = 2), Ramirez Pastor
obtuvo las siguientes expresiones para las topografías límites de
distribución al azar y en grandes parches.
a) Distribución aleatoria
θm(µ,T) = (f12)θFD(E21
*) + 2 f1f2θFD(E22*) + (f2
2)θFD(E23*) (6.16)
donde:
θFD(E21*) = exp[−β(2ε1+2λwθm−µ)]/1 + exp[−β(2ε1+2λwθm−µ)] (6.17)

θFD(E22*) = exp[−β(ε1+ε2 +2λwθm −µ)]/1 + exp[−β(ε1+ε2 +2λwθm −µ)]
(6.18)
θFD(E23*) = exp[−β(2ε2+2λwθm−µ)]/1+exp[−β(2ε2+ 2λwθm−µ)] (6.19)
b) Distribución en grandes parches
θm (µ,T) = f1θFD(E21*) + f2θFD(E22
*) (6.20)
donde:
θFD(E21*) = exp[−β(2ε1 + 2λwθFD(E21
*) − µ)]/1 + exp[−β(2ε1 +
2λwθFD(E21*) − µ)] (6.21)
θFD(E22*) = exp[−β(2ε2 + 2λwθFD(E22
*) − µ)]/1 + exp[−β(2ε2 +
2λwθFD(E22*) − µ)] (6.22)
6.2.2 Modelo para la isoterma local basado en la isoterma homogéneaexacta en una dimensión
En este caso Ramirez Pastor representa el sustrato como un
conjunto de dominios homogéneos, cada uno con energía Eki, o sea que
considera a cada uno de los kW niveles antes mencionados como una
superficie homogénea, y resuelve θ a partir de la ecuación (3.16) para la
isoterma unidimensional exacta.
Lo mismo que en el modelo anterior, las interacciones laterales se
introducen mediante una aproximación de campo promedio.
La isoterma local que resulta en el caso de dímeros es:
θ1-D(E2i*) = 1 − 1 +4exp[−β(E2i
*−µ)]½/1 +4exp[−β(E2i*−µ)]
(6.23)
donde:

E2i* = E2i + 2λwθ*
θ* = θ1-D(E2i*), para una topografía de grandes parches, y θ* = θm para
una topografía al azar.
Las isoterma totales en los dos casos límite de topografías
energéticas son:
a) Distribución aleatoria
θm (µ,T) = (f12)θ1-D(E21
*) + 2 f1f2θ1-D(E22*) + (f2
2)θ1-D(E23*) (6.24)
donde:
θ1-D(E21*) = 1 − 1 + 4exp[−β(2ε1 + 2λwθm −µ)]½/1 + 4exp[−β(2ε1
+ 2λwθm −µ)] (6.25)
θ1-D(E22*) = 1 − 1 + 4exp[−β(ε1+ε2 + 2λwθm −µ)]½/1 + 4exp[−β(ε1+ε2
+ 2λwθm −µ)] (6.26)
θ1-D(E23*) = 1 − 1 + 4exp[−β(2ε2 + 2λwθm − µ)]½/1 + 4exp[−β(2ε2
+ 2λwθm − µ)] (6.27)
b) Distribución en grandes parches
θm(µ,T) = f1θ1-D(E21*) + f2θ1-D(E22
*) (6.28)
donde:
θ1-D(E21*) = 1 − 1 + 4exp[−β(2ε1 + 2λwθ1-D(E21
*) − µ)]½/1 +
4exp[−β(2ε1 + 2λwθ1-D(E21*) − µ)] (6.29)
θ1-D(E22*) = 1 − 1 + 4exp[−β(2ε2 + 2λwθ1-D(E22
*) − µ)]½/1 +
4exp[−β(2ε2 + 2λwθ1-D(E22*) − µ)] (6.30)

6.3 SIMULACION DE MONTECARLO: ADSORCIÓN DE DIMEROSNO INTERACTUANTES SOBRE SUPERFICIES HETEROGÉNEAS
Se supone que el sustrato está representado por una red
bidimensional, hexagonal o triangular, de L x L sitios de adsorción, con
condiciones periódicas de borde. La heterogeneidad se introduce
considerando dos tipos de sitio de adsorción: débiles y fuertes, con
energías de adsorción ε1 y ε2, respectivamente, siendo ε1 < ε2. La
superficie heterogénea se modela como un conjunto de parches, que son
dominios de igual tamaño MP = l x l, distribuidos al azar, o en forma de
tablero de ajedrez, como se ven en las figuras 6.3 y 6.4. Los símbolos
negros representan a los sitios de fuertes, y los blancos, a los débiles.
Figura 6.3
(a) Tablero de ajedrez
RED HEXAGONAL
(b) Parches al azar

(a) Tablero de ajedrez
(b) Parches al azar
Figura 6.4
El proceso de adsorción se simula a través del método de Monte
Carlo en el Conjunto Gran Canónico.
.No se pierde generalidad si se supone que ε1 = 0, y ε2 = ε1 + ∆ε, de
modo que la energía adsortiva se caracteriza por el parámetro ∆ε.
También se considera que todas las energías se miden en unidades de kT
(∆ε/kT). Además, se considera que ambos tipos de sitios están en igual
concentración (f1 = f2 = 0,5), y de acuerdo a una distribución bimodal de
energía de sitios, como se aprecia en la siguiente figura.
F(ε)
0,5
ε2 ε1 ε
Figura 6.5

La simulación computacional se llevó a cabo para redes
hexagonales de M = L x L sitios, con L = 144, empleándose condiciones
periódicas de borde. El tamaño de red mencionado permite despreciar los
efectos de tamaño finito. Se descartaron los primeros 105 pasos de Monte
Carlo con el fin de alcanzar el equilibrio, utilizándose los 105 MCs
siguientes para obtener promedios. Los resultados obtenidos se analizan a
continuación.
6.3.1 Adsorción sobre redes hexagonales
La figura 6.6 muestra un conjunto de isotermas de adsorción de
dímeros sobre superficies heterogéneas bivariadas, con una distribucion
de sitios tipo tablero de ajedrez, con ∆ε/kT = 12, f1 = f2 = 0,5 y diferentes
tamaños de los parches (l = 1, 2, 3,). También se ha representado la
isoterma de adsorción correspondiente a una superficie formada por dos
grandes parches y el mismo valor de ∆ε/kT.
-10 -5 0 5 10 15 20 25 30 35
0,0
0,2
0,4
0,6
0,8
1,0
Simulación de MonteCarlo Red Hexagonal ∆ε/kT = 12
Figura 6.6
Grandes Parches Ajedrez 1 x 1 Ajedrez 2 x 2 Ajedrez 3 x 3
θ
µ/kT

Dependiendo del valor de l, y de la topografía, las isotermas
presentan uno, dos o tres regímenes de cubrimiento diferentes, asociados
con las tres energías posibles de adsorción de un dímero:
1) ε11, correspondiente a un dímero adsorbido sobre dos sitios
débiles.
2) ε22, correspondiente a un dímero adsorbido sobre dos sitios
fuertes.
3) ε12, correspondiente a un dímeros adsorbido sobre un par de
sitios vecinos próximos débil-fuerte.
En la figura 6.6 se pueden distinguir tres tipos de isotermas:
1) Para l = 1, los dímeros están siempre adsorbidos sobre pares de
sitios débil-fuerte, y un régimen de adsorción único caracteriza a
todo el proceso de adsorción.
2) Para l par, y dos grandes parches, aparecen dos regímenes
marcados de adsorción: a) Para 0 < θ < 0,5: las moléculas se
adsorben sobre pares fuerte-fuerte ocupando totalmente los
parches fuertes; b) Para 0,5 < θ < 1: los sitios débiles (pares
débil-débil) son ocupados hasta que se alcanza el cubrimiento
total.
3) Para l impar y distinto de uno, el número de sitios en los parches
fuertes y débiles es también impar. Por esta razón, cada parche
no puede ser llenado totalmente por dímeros, quedando sitios
vacíos en los parches. Entonces, los pares vacíos fuerte-débil que
aparecen en las fronteras entre parches se involucran en el
proceso de adsorción. Así, el proceso de llenado de sitios es el
siguiente: a valores bajos del potencial químico, los dímeros
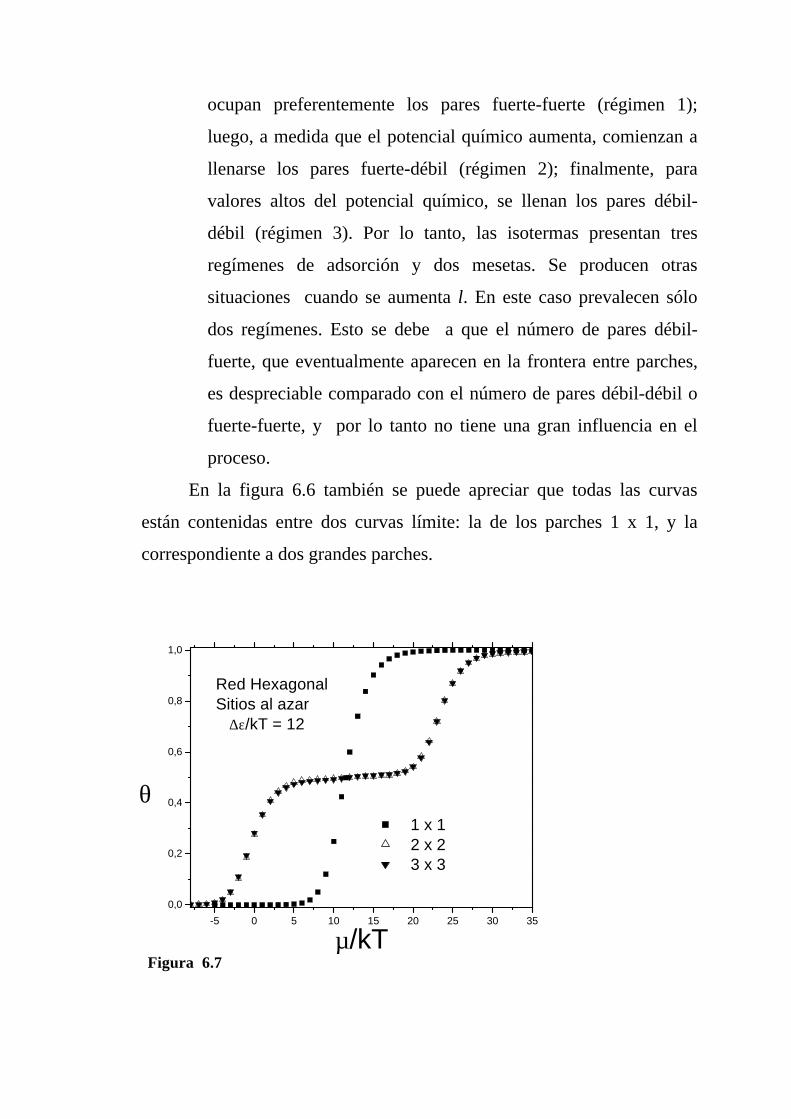
ocupan preferentemente los pares fuerte-fuerte (régimen 1);
luego, a medida que el potencial químico aumenta, comienzan a
llenarse los pares fuerte-débil (régimen 2); finalmente, para
valores altos del potencial químico, se llenan los pares débil-
débil (régimen 3). Por lo tanto, las isotermas presentan tres
regímenes de adsorción y dos mesetas. Se producen otras
situaciones cuando se aumenta l. En este caso prevalecen sólo
dos regímenes. Esto se debe a que el número de pares débil-
fuerte, que eventualmente aparecen en la frontera entre parches,
es despreciable comparado con el número de pares débil-débil o
fuerte-fuerte, y por lo tanto no tiene una gran influencia en el
proceso.
En la figura 6.6 también se puede apreciar que todas las curvas
están contenidas entre dos curvas límite: la de los parches 1 x 1, y la
correspondiente a dos grandes parches.
-5 0 5 10 15 20 25 30 35
0,0
0,2
0,4
0,6
0,8
1,0
Red Hexagonal Sitios al azar ∆ε/kT = 12
Figura 6.7
1 x 1 2 x 2 3 x 3
θ
µ/kT
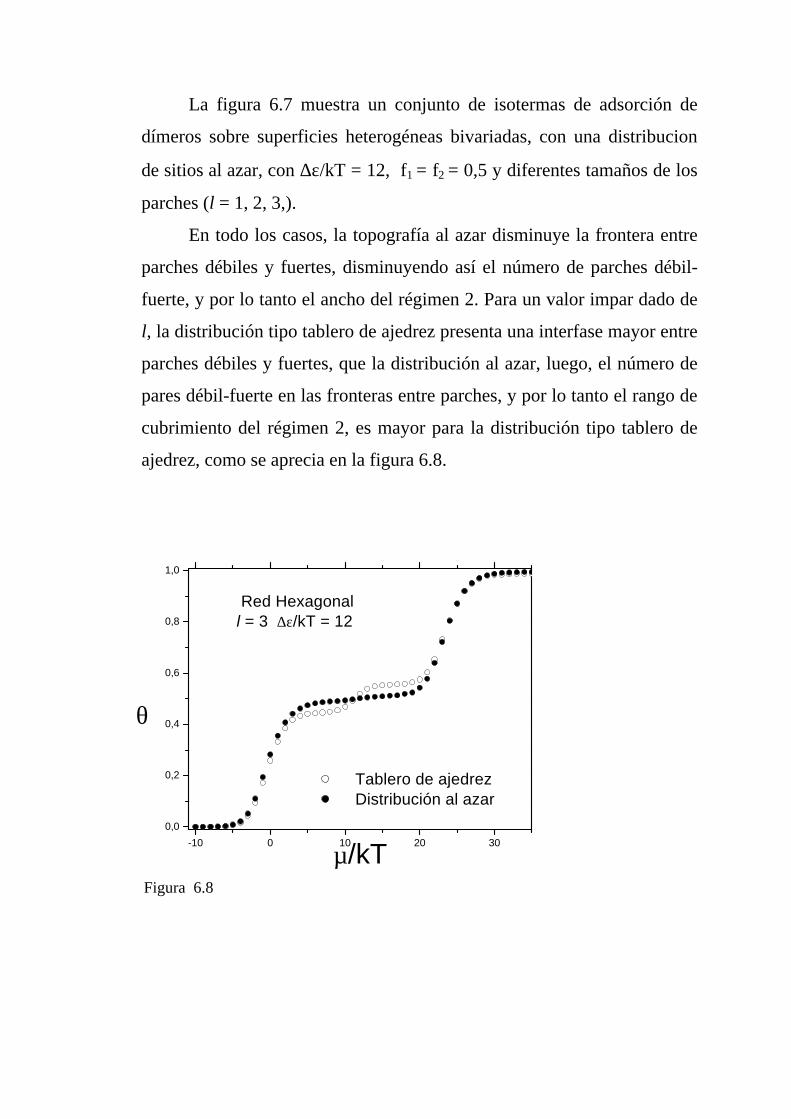
La figura 6.7 muestra un conjunto de isotermas de adsorción de
dímeros sobre superficies heterogéneas bivariadas, con una distribucion
de sitios al azar, con ∆ε/kT = 12, f1 = f2 = 0,5 y diferentes tamaños de los
parches (l = 1, 2, 3,).
En todo los casos, la topografía al azar disminuye la frontera entre
parches débiles y fuertes, disminuyendo así el número de parches débil-
fuerte, y por lo tanto el ancho del régimen 2. Para un valor impar dado de
l, la distribución tipo tablero de ajedrez presenta una interfase mayor entre
parches débiles y fuertes, que la distribución al azar, luego, el número de
pares débil-fuerte en las fronteras entre parches, y por lo tanto el rango de
cubrimiento del régimen 2, es mayor para la distribución tipo tablero de
ajedrez, como se aprecia en la figura 6.8.
-10 0 10 20 30
0,0
0,2
0,4
0,6
0,8
1,0
Red Hexagonal l = 3 ∆ε/kT = 12
Figura 6.8
θ
µ/kT
Tablero de ajedrez Distribución al azar

6.3.2 Adsorción sobre redes triangulares
La figura 6.9 muestra las isotermas de adsorción de dímeros sobre
superficies heterogéneas bivariadas, con una distribucion de sitios tipo
tablero de ajedrez, con ∆ε/kT = 12, f1 = f2 = 0,5 y diferentes tamaños de
los parches (l = 1, 2, 3,). También se ha representado la isoterma de
adsorción para una distribución en dos grandes parches e igual ∆ε/kT.
En este caso el mecanismo de llenado de la red se puede explicar
usando argumentos similares a los empleados para el caso de red
hexagonal, con la diferencia que en este caso la alta conectividad de la
red permite que se cubran totalmente los pares de sitios débil-débil para
todos los valores de l, por lo tanto, las isotermas que resultan presentan
-10 0 10 20 30
0,0
0,2
0,4
0,6
0,8
1,0
Grandes Parches Ajedrez 1 x 1 Ajedrez 2 x 2 Ajedrez 3 x 3
Simulación de MonteCarlo Red Triangular ∆ε/kT = 12
Figura 6.9
θ
µ/kT
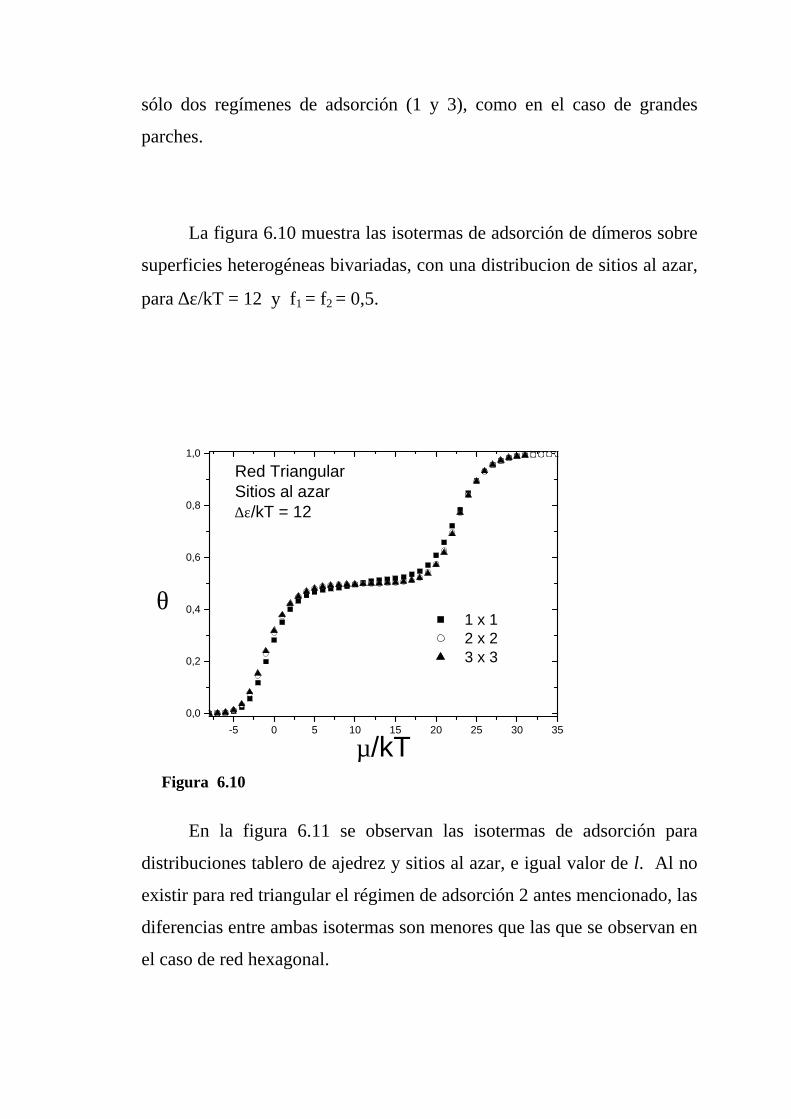
sólo dos regímenes de adsorción (1 y 3), como en el caso de grandes
parches.
La figura 6.10 muestra las isotermas de adsorción de dímeros sobre
superficies heterogéneas bivariadas, con una distribucion de sitios al azar,
para ∆ε/kT = 12 y f1 = f2 = 0,5.
En la figura 6.11 se observan las isotermas de adsorción para
distribuciones tablero de ajedrez y sitios al azar, e igual valor de l. Al no
existir para red triangular el régimen de adsorción 2 antes mencionado, las
diferencias entre ambas isotermas son menores que las que se observan en
el caso de red hexagonal.
-5 0 5 10 15 20 25 30 35
0,0
0,2
0,4
0,6
0,8
1,0
Figura 6.10
1 x 1 2 x 2 3 x 3
Red Triangular Sitios al azar ∆ε/kT = 12
θ
µ/kT
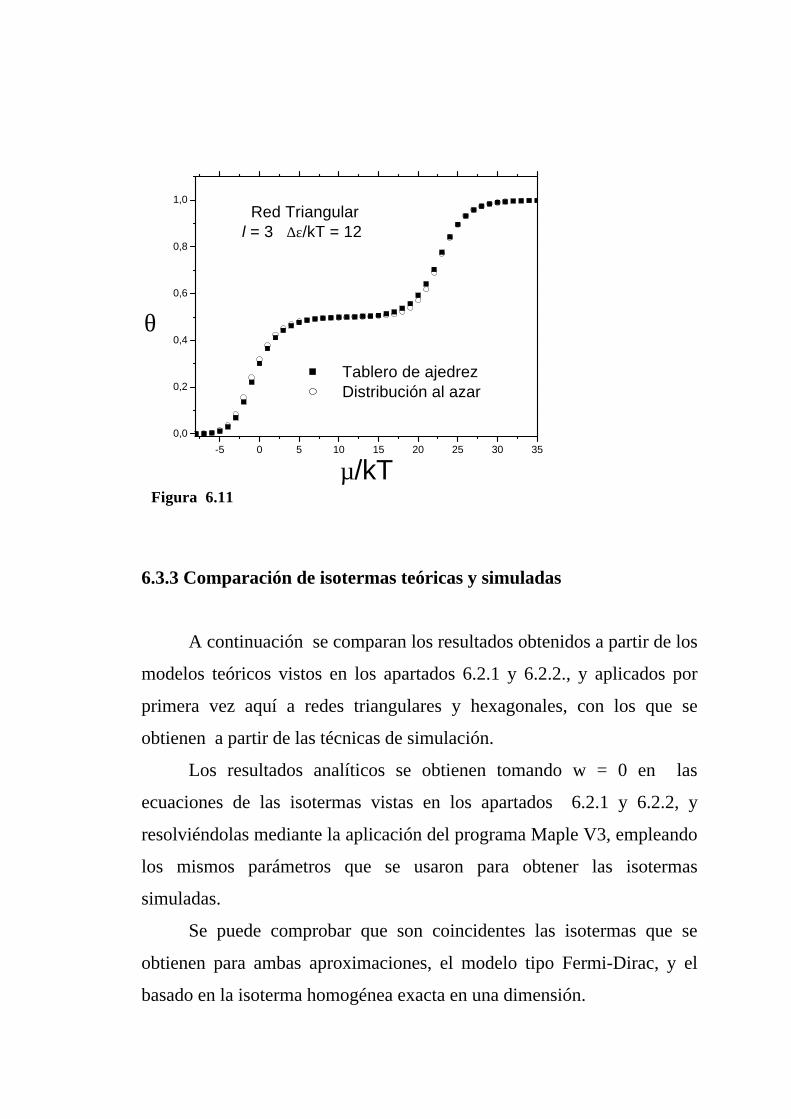
6.3.3 Comparación de isotermas teóricas y simuladas
A continuación se comparan los resultados obtenidos a partir de los
modelos teóricos vistos en los apartados 6.2.1 y 6.2.2., y aplicados por
primera vez aquí a redes triangulares y hexagonales, con los que se
obtienen a partir de las técnicas de simulación.
Los resultados analíticos se obtienen tomando w = 0 en las
ecuaciones de las isotermas vistas en los apartados 6.2.1 y 6.2.2, y
resolviéndolas mediante la aplicación del programa Maple V3, empleando
los mismos parámetros que se usaron para obtener las isotermas
simuladas.
Se puede comprobar que son coincidentes las isotermas que se
obtienen para ambas aproximaciones, el modelo tipo Fermi-Dirac, y el
basado en la isoterma homogénea exacta en una dimensión.
-5 0 5 10 15 20 25 30 35
0,0
0,2
0,4
0,6
0,8
1,0 Red Triangular l = 3 ∆ε/kT = 12
Figura 6.11
θ
µ/kT
Tablero de ajedrez Distribución al azar

-10 0 10 20 30
0,0
0,2
0,4
0,6
0,8
1,0
Figura 6.12
Aproximación de Fermi-Dirac Red Hexagonal ∆ε/kT = 12
θ
µ/kT
Grandes Parches Ajedrez 1 x 1 Ajedrez 2 x 2 Ajedrez 3 x 3
-5 0 5 10 15 20 25 30
0,0
0,2
0,4
0,6
0,8
1,0
Figura 6.13
Red Hexagonal Tablero de Ajedrez l = 3 ∆ε/kT = 12
θ
µ/kT
Simulación de Monte Carlo
Aproximación de Fermi-Dirac
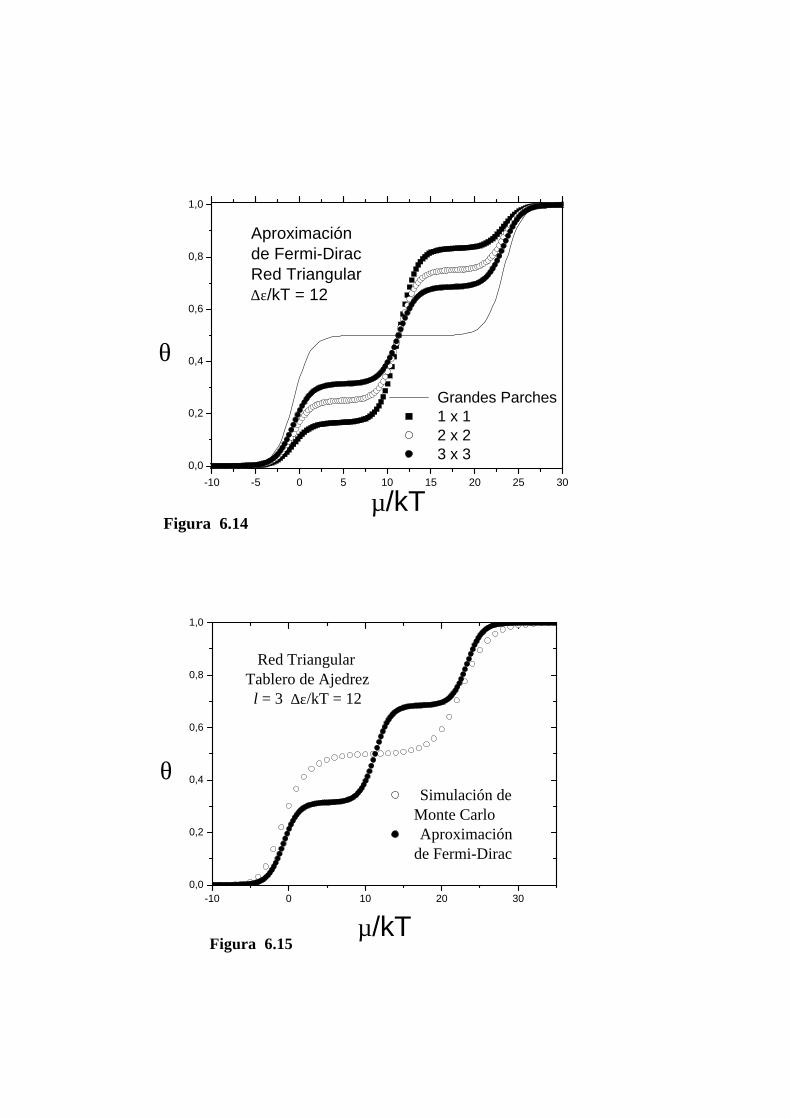
-10 -5 0 5 10 15 20 25 30
0,0
0,2
0,4
0,6
0,8
1,0
Figura 6.14
Aproximación de Fermi-Dirac Red Triangular ∆ε/kT = 12
Grandes Parches 1 x 1 2 x 2 3 x 3
θ
µ/kT
-10 0 10 20 300,0
0,2
0,4
0,6
0,8
1,0
Figura 6.15
Red Triangular Tablero de Ajedrez l = 3 ∆ε/kT = 12
Simulación de Monte Carlo
Aproximación de Fermi-Dirac
θ
µ/kT

Si bien existe una diferencia cuantitativa entre las curvas analíticas y
simuladas, el comportamiento cuantitativo es el mismo ya que:
1) Las isotermas teóricas y simuladas presentan diferentes regímenes de
cubrimiento, separados por mesetas bien definidas (Figuras 6.13 y
6.15).
2) 2) Tanto las isotermas teóricas como las simuladas están
comprendidas entre dos curvas límite: tablero de ajedrez, con parches
1 x 1, y grandes parches (Figuras 6.6, 6.9, 6.12 y 6.13).
6.3.4 Escaleo y comportamiento exponencial
El hecho de que para diferentes valores de la longitud de escala (l), las
isotermas de adsorción estén comprendidas entre dos curvas límite, permite
definir la siguiente cantidad:
∞χ = [ |θ(µ) − θR(µ)| dµ]2 (6.31) -∞
donde θR(µ) es la isoterma de adsorción de referencia.
La cantidad χ, que representa el cuadrado del área comprendida entre una
curva dada y la curva de referencia, es apropiada para medir la desviación entre
las isotermas y para estudiar su comportamiento cuando se varía la longitud de
escala. Tomando de referencia la curva correspondiente a la topografía de
grandes parches, se obtiene la figura 6.16 en la cual se puede ver que χ se
comporta como una exponencial en l, con un exponente α = - 2.
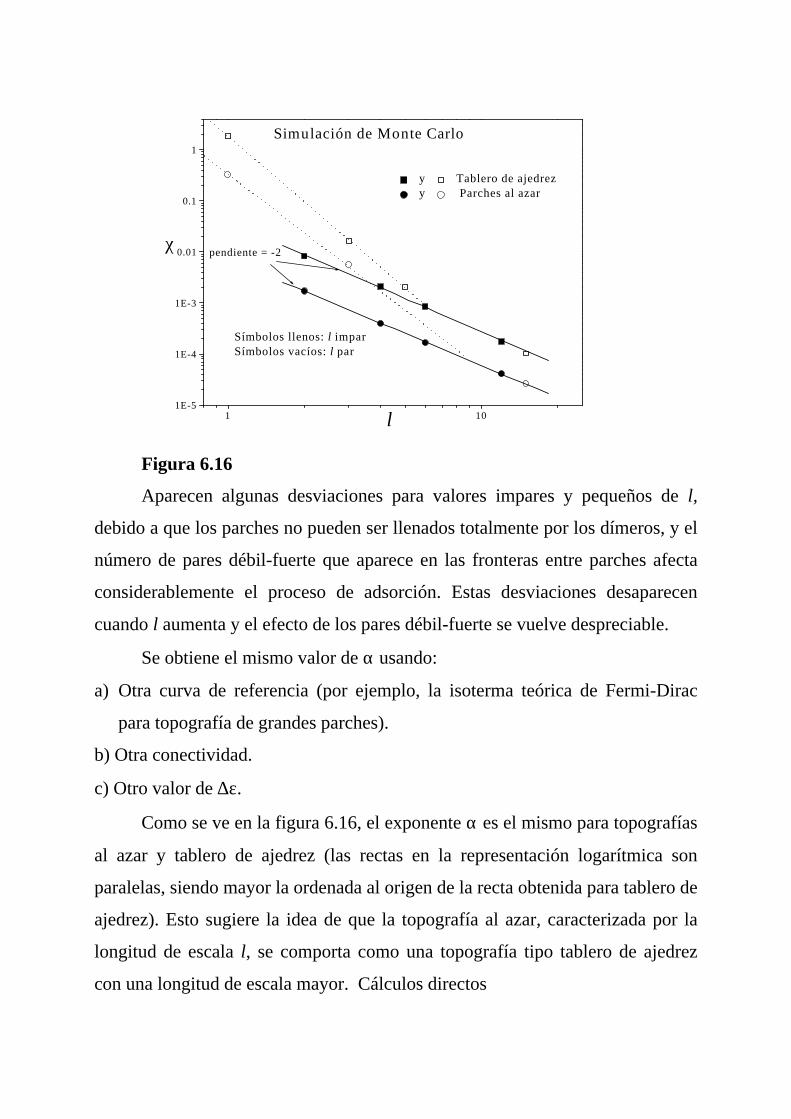
1 101E-5
1E-4
1E-3
0.01
0.1
1
y Tablero de ajedrezy Parches al azar
Símbolos llenos: l imparSímbolos vacíos: l par
Simulación de Monte Carlo
pendiente = -2
χ
l
Figura 6.16
Aparecen algunas desviaciones para valores impares y pequeños de l,
debido a que los parches no pueden ser llenados totalmente por los dímeros, y el
número de pares débil-fuerte que aparece en las fronteras entre parches afecta
considerablemente el proceso de adsorción. Estas desviaciones desaparecen
cuando l aumenta y el efecto de los pares débil-fuerte se vuelve despreciable.
Se obtiene el mismo valor de α usando:
a) Otra curva de referencia (por ejemplo, la isoterma teórica de Fermi-Dirac
para topografía de grandes parches).
b) Otra conectividad.
c) Otro valor de ∆ε.
Como se ve en la figura 6.16, el exponente α es el mismo para topografías
al azar y tablero de ajedrez (las rectas en la representación logarítmica son
paralelas, siendo mayor la ordenada al origen de la recta obtenida para tablero de
ajedrez). Esto sugiere la idea de que la topografía al azar, caracterizada por la
longitud de escala l, se comporta como una topografía tipo tablero de ajedrez
con una longitud de escala mayor. Cálculos directos
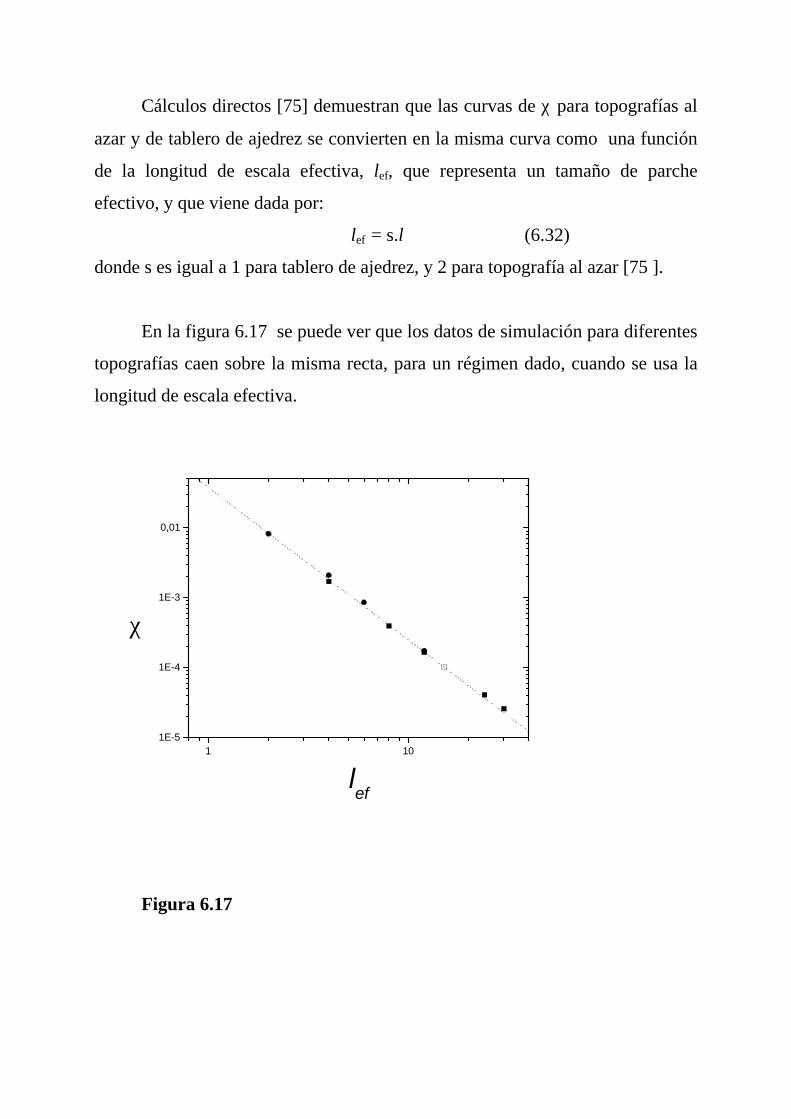
Cálculos directos [75] demuestran que las curvas de χ para topografías al
azar y de tablero de ajedrez se convierten en la misma curva como una función
de la longitud de escala efectiva, lef, que representa un tamaño de parche
efectivo, y que viene dada por:
lef = s.l (6.32)
donde s es igual a 1 para tablero de ajedrez, y 2 para topografía al azar [75 ].
En la figura 6.17 se puede ver que los datos de simulación para diferentes
topografías caen sobre la misma recta, para un régimen dado, cuando se usa la
longitud de escala efectiva.
Figura 6.17
1 101E-5
1E-4
1E-3
0,01
χ
lef

2
La figura 6.18 muestra los resultados obtenidos por aplicación de la
aproximación tipo F-D.
Figura 6.18
Como se puede apreciar, en todos los casos se sigue la misma ley
exponencial (con exponente α = -2) que se observa en la figura 6.16. La muy
buena concordancia entre el comportamiento de χ para la aproximación
analítica y para la simulación de Monte Carlo, refuerza la solidez de la ley
exponencial introducida anteriormente.
Como conclusión de todo lo anterior, se puede establecer que la cantidad
χ definida en la ecuación (6.31), y calculada mediante una curva de referencia
teórica o simulada, se comporta como una ley exponencial de la longitud de
escala efectiva.
lnχ = constante + αln lef (6.33)
donde el exponente α tiene un comportamiento universal dado por α = -2.
Este resultado suministra un método para caracterizar la topografía energética
1 10
0,01
0,1
1
10 Aproximación Fermi-Dirac Red hexagonal Red cuadrada Red triangular
pendiente (α) = -2
χ
l

3
de sustratos heterogéneos que pueden aproximarse por medio de superficies
bivariadas, a través de medidas de adsorción de dímeros no interactuantes. En
efecto, eligiendo una aproximación teórica adecuada como curva de referencia,
se puede calcular el valor de χ, permitiendo que se obtenga lef de la ecuación
(6.33).
6.4 SIMULACION DE MONTE CARLO: ADSORCIÓN DE DIMEROSINTERACTUANTES SOBRE SUPERFICIES HETEROGÉNEAS
A continuación se analizan las isotermas de adsorción obtenidas mediante
simulación de Monte Carlo, las que luego se comparan con las que resultan de
la aplicación de los modelos teóricos ya vistos.
También acá el sustrato se representa por una red bidimensional,
hexagonal o triangular, de L x L sitios de adsorción, con condiciones periódicas
de borde. Existen dos tipos de sitio de adsorción: débiles, con energías de
adsorción ε1, y fuertes, con energías ε2, los cuales forman dominios de igual
tamaño, MP = l x l, que están distribuidos al azar, o formando dos grandes
parches.
La simulación computacional del proceso adsortivo se llevó a cabo sobre
redes bidimensionales de 200 x 200 sitios. Se descartaron los primeros 106 pasos
de Monte Carlo (MCs) para permitir que se alcanzara el equilibrio, mientras que
se usaron los siguientes 106 MCs para obtener promedios.

4
6.4.1 Adsorción sobre redes triangulares
En la figura 6.19 se muestran las isotermas obtenidas para un
sistema constituido por dímeros interactuantes adsorbidos sobre una
superficie triangular de grandes parches. Cada curva corresponde a un
valor diferente de la energía de interacción lateral, siendo la diferencia de
energía entre los parches (∆ε/kT) constante e igual a 48.
En el caso de w/kT = 0 (círculos vacíos) la isoterma presenta una
meseta para θ = 0,5, la cual separa dos regímenes de adsorción diferente:
1) Llenado del parche fuerte. 2) Llenado del parche débil.
Cuando w/kT < 0 (interacciones atractivas), en el adsorbato tiende a
producirse una transición de primer orden a medida que decrece la
temperatura. Esta transición de fase se produce en cada uno de los parches
y se manifiesta en la isotermas en forma de dos saltos abruptos, el
primero en el rango de cubrimiento 0,0 - 0,5 (condensación en el parche
-100 -80 -60 -40 -20 0 20 40 60 80
0,0
0,2
0,4
0,6
0,8
1,0
Figura 6.19: Isotermas obtenidas por simulación de Monte Carlo.
w/kT = -1,50 w/kT = -0,75 w/kT = 0,0 w/kT = 2,0 w/kT = 5,0
Red Triangular Grandes Parches ∆ε/kT = 48
θ
µ/kT

5
fuerte), y el segundo en el rango 0,5 - 1,0 (condensación en el parche
débil).
Cuando w/kT > 0, y en el régimen de altas temperaturas (cuadrados
vacíos) no se manifiesta ningún comportamiento particular en las
isotermas. Cuando la temperatura disminuye, o aumenta la relación w/kT,
como en la isoterma para w/kT = 5 (triángulos vacíos), se observan cinco
mesetas, cada una de las cuales corresponde a la formación de una
estructura diferente en el adsorbato. La primera meseta se produce en el
parche fuerte una vez que se alcanza la fase ordenada que se muestra en la
figura 6.20.a. Dicha fase aparece para un cubrimiento de 2/5 respecto del
número de sitios de ese parche, lo cual corresponde a 1/5 del cubrimiento
total. La segunda meseta aparece una vez que se arma, en el parche fuerte,
la fase ordenada que se muestra en la figura6.20.b. La meseta a θ = 0,5
corresponde al llenado total de los sitios fuertes. Aplicando iguales
argumentos a los sitios débiles se puede explicar el comportamiento de la
isoterma en el rango de cubrimiento de 0,5 a 1,0.
Fase ordenada a θθ = 2 / 5 Fase ordenada a θθ = 2 / 3
(a) (b)
Figura 6.20: Estructuras que se forman sobre la superficie cuando se adsorben dímeros coninteracciones repulsivas sobre una red triangular heterogénea: (a), para θ = 2/5 y (b), para θ= 2/3.
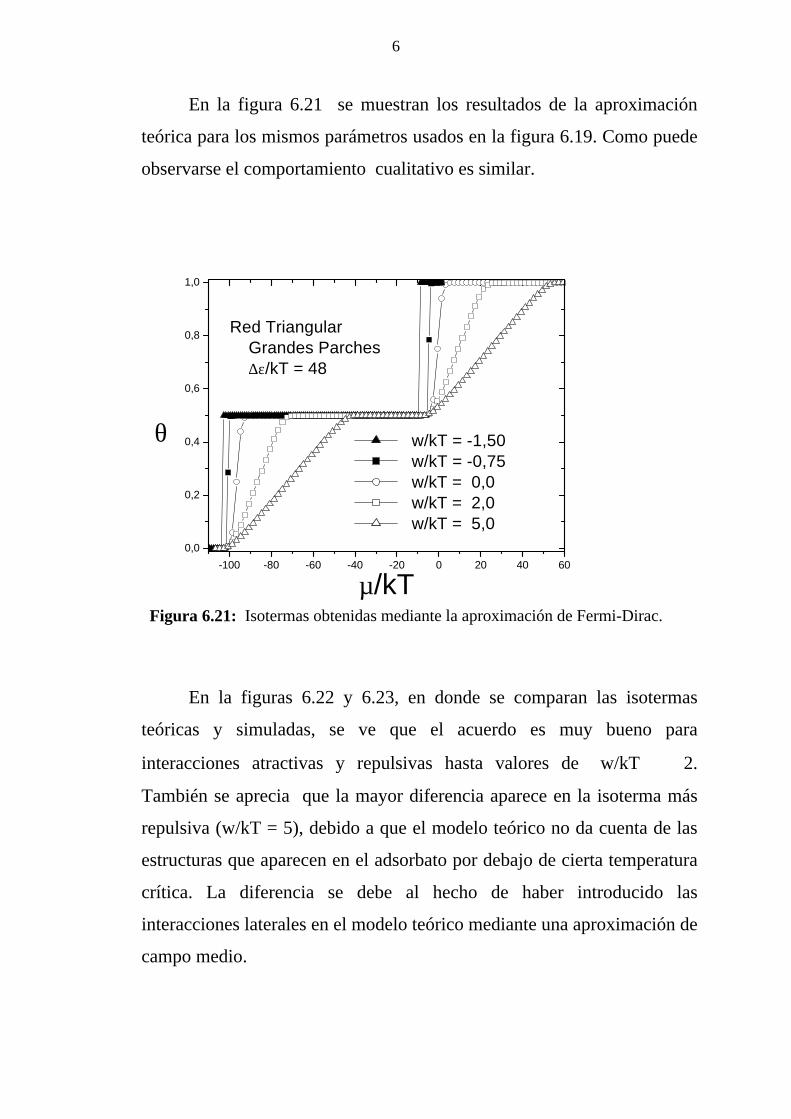
6
En la figura 6.21 se muestran los resultados de la aproximación
teórica para los mismos parámetros usados en la figura 6.19. Como puede
observarse el comportamiento cualitativo es similar.
En la figuras 6.22 y 6.23, en donde se comparan las isotermas
teóricas y simuladas, se ve que el acuerdo es muy bueno para
interacciones atractivas y repulsivas hasta valores de w/kT ≅ 2.
También se aprecia que la mayor diferencia aparece en la isoterma más
repulsiva (w/kT = 5), debido a que el modelo teórico no da cuenta de las
estructuras que aparecen en el adsorbato por debajo de cierta temperatura
crítica. La diferencia se debe al hecho de haber introducido las
interacciones laterales en el modelo teórico mediante una aproximación de
campo medio.
-100 -80 -60 -40 -20 0 20 40 60
0,0
0,2
0,4
0,6
0,8
1,0
Figura 6.21: Isotermas obtenidas mediante la aproximación de Fermi-Dirac.
Red Triangular Grandes Parches ∆ε/kT = 48
w/kT = -1,50 w/kT = -0,75 w/kT = 0,0 w/kT = 2,0 w/kT = 5,0
θ
µ/kT
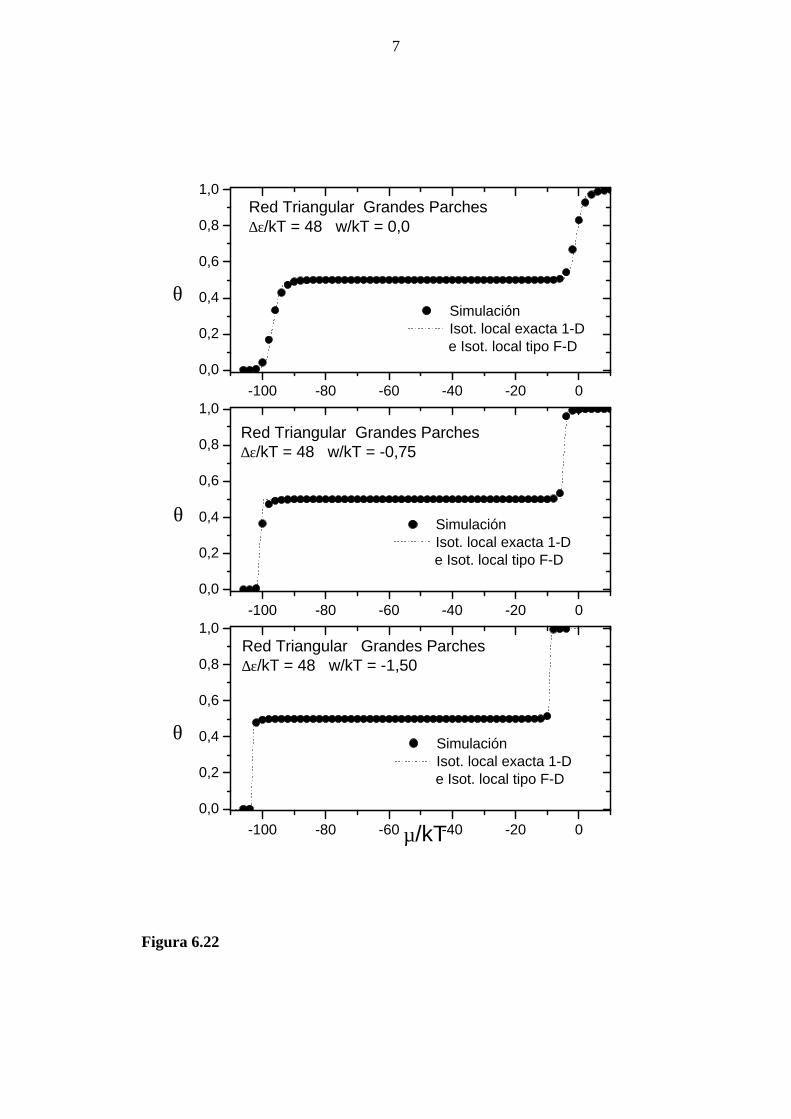
7
Figura 6.22
-100 -80 -60 -40 -20 0
0,0
0,2
0,4
0,6
0,8
1,0
-100 -80 -60 -40 -20 0
0,0
0,2
0,4
0,6
0,8
1,0
-100 -80 -60 -40 -20 0
0,0
0,2
0,4
0,6
0,8
1,0
Red Triangular Grandes Parches ∆ε/kT = 48 w/kT = 0,0
θ Simulación Isot. local exacta 1-D
e Isot. local tipo F-D
Red Triangular Grandes Parches ∆ε/kT = 48 w/kT = -0,75
θ Simulación Isot. local exacta 1-D
e Isot. local tipo F-D
Red Triangular Grandes Parches ∆ε/kT = 48 w/kT = -1,50
θ
µ/kT
Simulación Isot. local exacta 1-D
e Isot. local tipo F-D
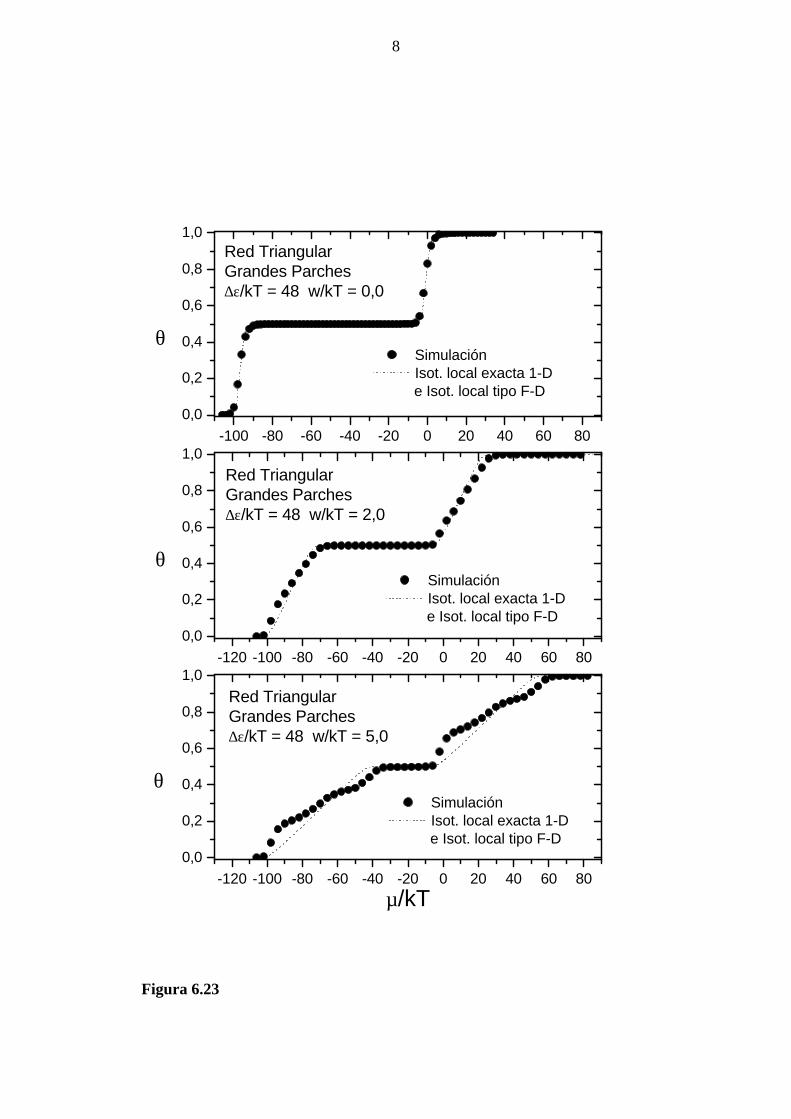
8
Figura 6.23
-100 -80 -60 -40 -20 0 20 40 60 80
0,0
0,2
0,4
0,6
0,8
1,0
-120 -100 -80 -60 -40 -20 0 20 40 60 80
0,0
0,2
0,4
0,6
0,8
1,0
-120 -100 -80 -60 -40 -20 0 20 40 60 80
0,0
0,2
0,4
0,6
0,8
1,0
Red Triangular Grandes Parches ∆ε/kT = 48 w/kT = 0,0
θ Simulación Isot. local exacta 1-D
e Isot. local tipo F-D
Red Triangular Grandes Parches ∆ε/kT = 48 w/kT = 2,0
θ Simulación Isot. local exacta 1-D
e Isot. local tipo F-D
Red Triangular Grandes Parches ∆ε/kT = 48 w/kT = 5,0
θ
µ/kT
Simulación Isot. local exacta 1-D
e Isot. local tipo F-D

9
En la figura 6.24 se muestran las isotermas obtenidas para un
sistema constituido por dímeros interactuantes adsorbidos sobre una red
triangular de sitios al azar. Las curvas corresponden a diferentes valores
de la energía de interacción lateral, siendo la diferencia de energía entre
los parches (∆ε/kT) constante e igual a 48.
Se observan acá dos diferencias principales respecto al caso de
grandes parches.
1) Las isotermas presentan un pequeño escalón en torno a θ = 0,5.
En el caso de grandes parches, los dímeros pueden adsorberse sobre
dos tipos de pares de sitios (débil-débil) y (fuerte-fuerte), y por lo
tanto las isotermas presentan sólo dos escalones asociados a la
heterogeneidad, en cambio, en una superficie de sitios al azar
existen tres pares de sitios posibles (débil-débil), (fuerte-fuerte) y
-100 -80 -60 -40 -20 0 20 40 60 80
0,0
0,2
0,4
0,6
0,8
1,0
Figura 6.24: Isotermas obtenidas por simulación de Monte Carlo.
Red Triangular Sitios al azar ∆ε/kT = 48
w/kT = -1,50 w/kT = -0,75 w/kT = 0,0 w/kT = 2,0 w/kT = 5,0
θ
µ/kT
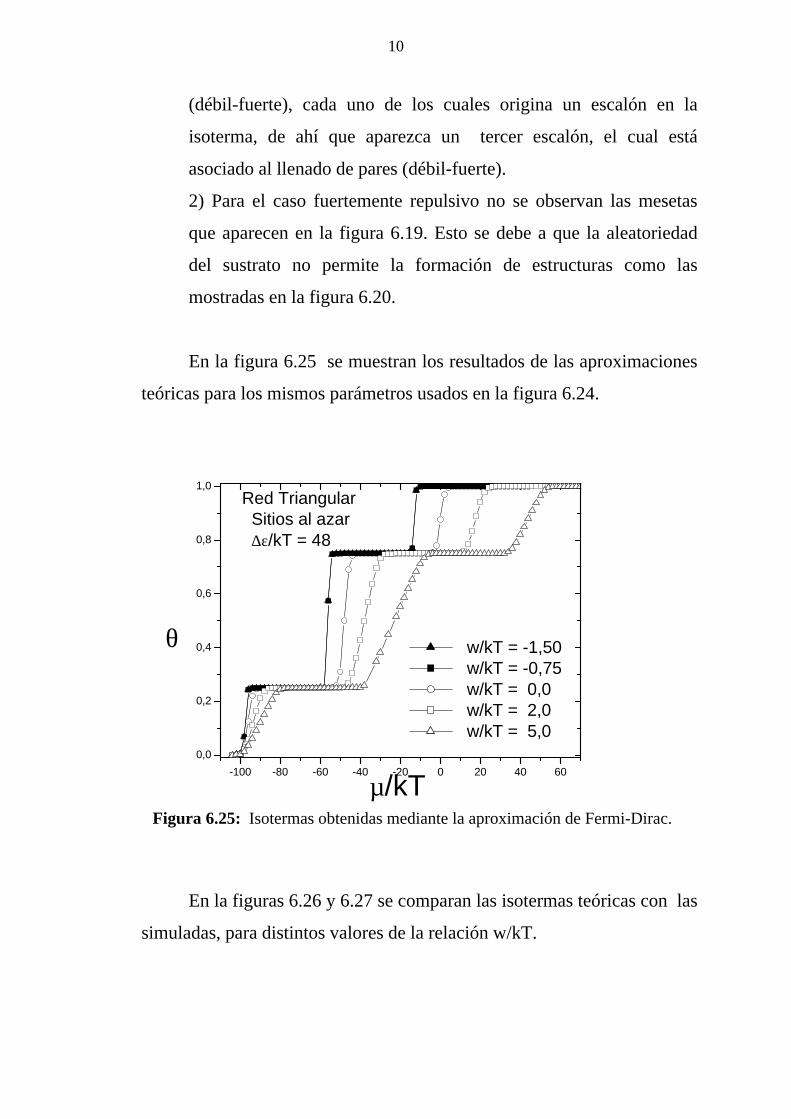
10
(débil-fuerte), cada uno de los cuales origina un escalón en la
isoterma, de ahí que aparezca un tercer escalón, el cual está
asociado al llenado de pares (débil-fuerte).
2) Para el caso fuertemente repulsivo no se observan las mesetas
que aparecen en la figura 6.19. Esto se debe a que la aleatoriedad
del sustrato no permite la formación de estructuras como las
mostradas en la figura 6.20.
En la figura 6.25 se muestran los resultados de las aproximaciones
teóricas para los mismos parámetros usados en la figura 6.24.
En la figuras 6.26 y 6.27 se comparan las isotermas teóricas con las
simuladas, para distintos valores de la relación w/kT.
-100 -80 -60 -40 -20 0 20 40 60
0,0
0,2
0,4
0,6
0,8
1,0
Figura 6.25: Isotermas obtenidas mediante la aproximación de Fermi-Dirac.
Red Triangular Sitios al azar ∆ε/kT = 48
w/kT = -1,50 w/kT = -0,75 w/kT = 0,0 w/kT = 2,0 w/kT = 5,0
θ
µ/kT
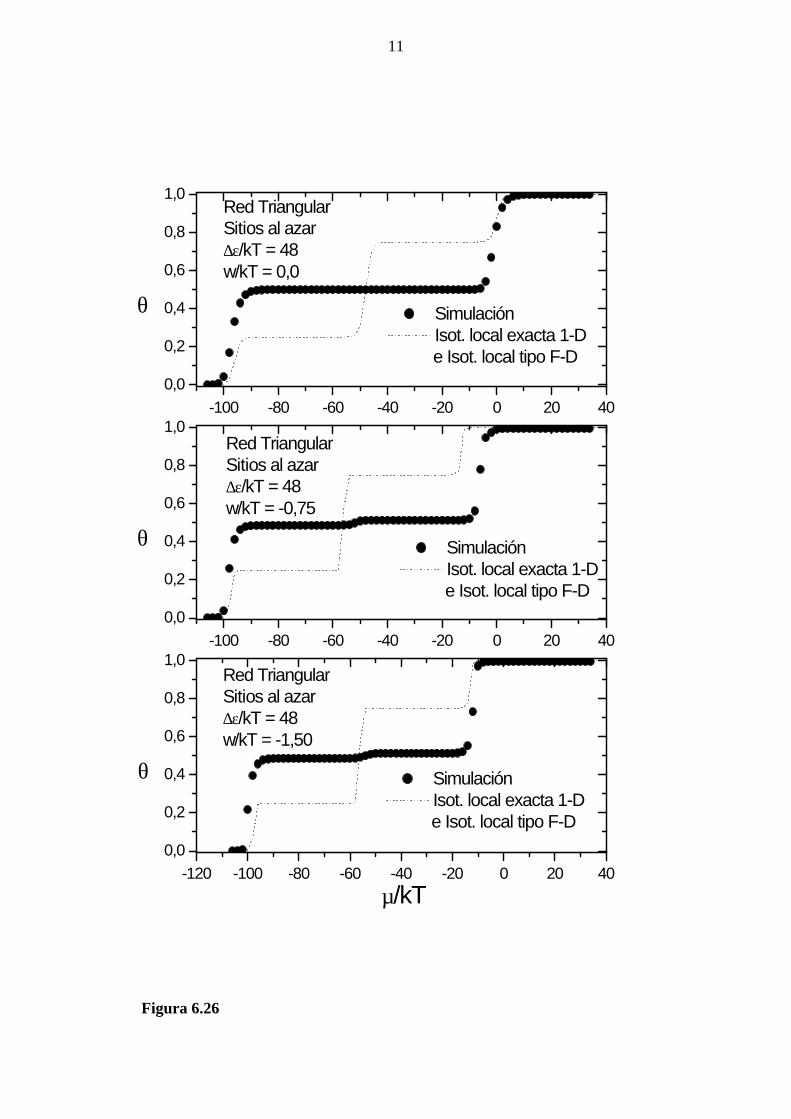
11
Figura 6.26
-100 -80 -60 -40 -20 0 20 40
0,0
0,2
0,4
0,6
0,8
1,0
-120 -100 -80 -60 -40 -20 0 20 40
0,0
0,2
0,4
0,6
0,8
1,0-100 -80 -60 -40 -20 0 20 40
0,0
0,2
0,4
0,6
0,8
1,0
Simulación Isot. local exacta 1-D
e Isot. local tipo F-D
Red Triangular Sitios al azar ∆ε/kT = 48 w/kT = 0,0
θ
Simulación Isot. local exacta 1-D
e Isot. local tipo F-D
Red Triangular Sitios al azar ∆ε/kT = 48 w/kT = -1,50
θ
µ/kT
Simulación Isot. local exacta 1-D
e Isot. local tipo F-D
Red Triangular Sitios al azar ∆ε/kT = 48 w/kT = -0,75
θ

12
Figura 6.27
-100 -80 -60 -40 -20 0 20 40 60
0,0
0,2
0,4
0,6
0,8
1,0-100 -80 -60 -40 -20 0 20 40 60
0,0
0,2
0,4
0,6
0,8
1,0-120 -100 -80 -60 -40 -20 0 20 40 60
0,0
0,2
0,4
0,6
0,8
1,0
Simulación Isot. local exacta 1-D
e Isot. local tipo F-D
Red Triangular Sitios al azar ∆ε/kT = 48 w/kT = 5,0
θ
µ/kT
Red Triangular Sitios al azar ∆ε/kT = 48 w/kT = 2,0
Simulación Isot. local exacta 1-D
e Isot. local tipo F-D
θ
Red Triangular Sitios al azar ∆ε/kT = 48 w/kT = 0,0
Simulación Isot. local exacta 1-D
e Isot. local tipo F-D
θ

13
En este caso las discrepancias entre el modelo teórico y los
resultados de Monte Carlo son mayores que en el caso de grandes parches
debido a que la suposición de independencia entre los tres tipos de pares
de sitios posibles, que se asocian a tres niveles de energía en la ecuación
(6.16), lleva a las isotermas teóricas a mostrar tres regímenes de adsorción
bien marcados, los cuales no aparecen en Monte Carlo. Estrictamente, los
tres niveles de energía no son independientes, sino que el número de
ocupación de uno afecta al número de ocupación de los otros, ya que los
distintos pares de sitios comparten unidades.
Si bien el modelo teórico se usó para tratar con dos casos
heterogéneos límite, podrían abordarse también sustratos con
correlaciones intermedias.
6.4.2 Adsorción sobre redes hexagonales
En primer término se considera la adsorción de dímeros sobre
redes hexagonales de grandes parches, como la representada en la figura
siguiente.
Figura 6.28: Red hexagonal de grandes parches.

14
En la figura 6.29 se pueden observar las isotermas obtenidas para un
sistema constituido por dímeros interactuantes adsorbidos sobre una superficie
hexagonal de grandes parches, para valores diferentes de la energía de
interacción lateral (w/kT), siendo la diferencia de energía entre los parches
(∆ε/kT) constante e igual a 48.
En el caso de w/kT = 0 (círculos vacíos) la isoterma presenta una
meseta para θ = 0,5, la cual separa dos regímenes de adsorción diferente:
1) Llenado del parche fuerte. 2) Llenado del parche débil.
Cuando w/kT < 0 (interacciones atractivas), y a medida que
disminuye la temperatura, en el adsorbato tiende a producirse una
transición de primer orden en cada uno de los parches, la cual se
-100 -80 -60 -40 -20 0 20 40 60
0,0
0,2
0,4
0,6
0,8
1,0
Figura 6.29: Isotermas obtenidas por simulación de Monte Carlo.
w/kT = -3,0 w/kT = -1,0 w/kT = 0,0 w/kT = 4,0 w/kT = 8,0
Red Hexagonal Grandes parches Simulación de Montecarlo ∆ε/kT = 48
θ
µ/kT

15
manifiesta en la isotermas en forma de dos saltos abruptos, el primero en
el rango de cubrimiento 0,0 - 0,5 (condensación en el parche fuerte), y el
segundo en el rango 0,5 - 1,0 (condensación en el parche débil).
Cuando w/kT > 0, y en el régimen de altas temperaturas (cuadrados
vacíos) no se manifiesta ningún comportamiento particular en las
isotermas. Cuando la temperatura disminuye, o bien aumenta la relación
w/kT, como en el caso de la isoterma para w/kT = 8 (triángulos vacíos),
se observan cinco mesetas, cada una de las cuales corresponde a la
formación de una estructura diferente en el adsorbato. La primera meseta
se produce en el parche fuerte una vez que se alcanza la fase ordenada que
se muestra en la figura 6.30. Dicha fase aparece para un cubrimiento de
5/9 respecto del número de sitios de ese parche, lo cual corresponde a
5/18 del cubrimiento total. La segunda meseta aparece una vez que se
arma, en el parche fuerte, la fase ordenada que se muestra en la figura
6.31. Esa fase se forma para un cubrimiento de 2/3 respecto del número
de sitios de ese parche, lo cual corresponde a 1/3 del cubrimiento total.
La meseta a θ = 0,5 corresponde al llenado total de los sitios fuertes.
Aplicando iguales argumentos a los sitios débiles se puede explicar el
comportamiento de la isoterma en el rango de cubrimiento de 0,5 a 1,0.
(6.30) (6.31)
Figuras 6.30 y 6.31: Estructuras que se forman sobre la superficie cuando se adsorbendímeros con interacciones repulsivas sobre una red hexagonal heterogénea: (6.30),para θ = 5/18 y θ = 7/9, y (6.31), para θ = 1/3 y θ = 5/6.
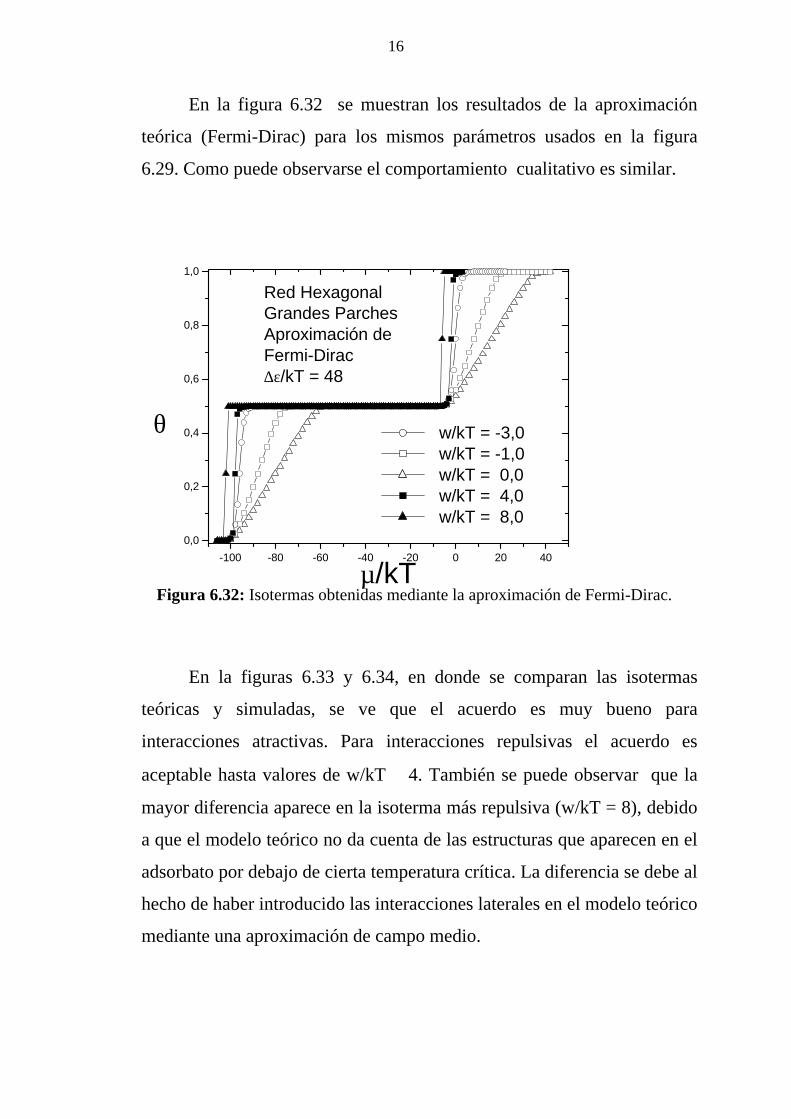
16
En la figura 6.32 se muestran los resultados de la aproximación
teórica (Fermi-Dirac) para los mismos parámetros usados en la figura
6.29. Como puede observarse el comportamiento cualitativo es similar.
En la figuras 6.33 y 6.34, en donde se comparan las isotermas
teóricas y simuladas, se ve que el acuerdo es muy bueno para
interacciones atractivas. Para interacciones repulsivas el acuerdo es
aceptable hasta valores de w/kT ≅ 4. También se puede observar que la
mayor diferencia aparece en la isoterma más repulsiva (w/kT = 8), debido
a que el modelo teórico no da cuenta de las estructuras que aparecen en el
adsorbato por debajo de cierta temperatura crítica. La diferencia se debe al
hecho de haber introducido las interacciones laterales en el modelo teórico
mediante una aproximación de campo medio.
-100 -80 -60 -40 -20 0 20 40
0,0
0,2
0,4
0,6
0,8
1,0
Figura 6.32: Isotermas obtenidas mediante la aproximación de Fermi-Dirac.
w/kT = -3,0 w/kT = -1,0 w/kT = 0,0 w/kT = 4,0 w/kT = 8,0
Red Hexagonal Grandes Parches Aproximación de Fermi-Dirac ∆ε/kT = 48
θ
µ/kT

17
Figura 6.33
-100 -80 -60 -40 -20 0
0,0
0,2
0,4
0,6
0,8
1,0
-100 -80 -60 -40 -20 0
0,0
0,2
0,4
0,6
0,8
1,0
-120 -100 -80 -60 -40 -20 0
0,0
0,2
0,4
0,6
0,8
1,0
Red Hexagonal Grandes Parches ∆ε/kT = 48 w/kT = -3,0
θ
µ/kT
Simulación Isot. local exacta 1-D
e Isot. local tipo F-D
Simulación Isot. local exacta 1-D
e Isot. local tipo F-D
Simulación Isot. local exacta 1-D
e Isot. local tipo F-D
Red Hexagonal Grandes Parches ∆ε/kT = 48 w/kT = 0,0
θ
Red Hexagonal Grandes Parches ∆ε/kT = 48 w/kT = -1,0
θ

18
Figura 6.34
-100 -80 -60 -40 -20 0 20 40
0,0
0,2
0,4
0,6
0,8
1,0
-100 -80 -60 -40 -20 0 20 40
0,0
0,2
0,4
0,6
0,8
1,0
-100 -80 -60 -40 -20 0 20 40
0,0
0,2
0,4
0,6
0,8
1,0
Simulación Isot. local exacta 1-D
e Isot. local tipo F-D
Simulación Isot. local exacta 1-D
e Isot. local tipo F-D
Simulación Isot. local exacta 1-D
e Isot. local tipo F-D
Red Hexagonal Grandes Parches ∆ε/kT = 48 w/kT = 4,0
θ
Red Hexagonal Grandes Parches ∆ε/kT = 48 w/kT = 8,0
θ
µ/kT
Red Hexagonal Grandes Parches ∆ε/kT = 48 w/kT = 0,0
θ

A continuación se considera la adsorción de dímeros sobre redes
hexagonales de parches distribuidos al azar, como la que se muestra en
la figura siguiente.
-100 -80 -60 -40 -20 0 20 40
0,0
0,2
0,4
0,6
0,8
1,0
Figura 6.36: Isotermas de adsorción obtenidas mediante simulación de Monte Carlo.
w/kT = -3,0 w/kT = -1,0 w/kT = 0,0 w/kT = 4,0 w/kT = 8,0
Red Hexagonal Sitios al azar Simulación de Montecarlo ∆ε/kT = 48
θ
µ/kT
Figura 6.35: Red hexagonal de parches al azar.

2
En la figura 6.36 se pueden observar las isotermas obtenidas por
simulación para un sistema constituido por dímeros interactuantes adsorbidos
sobre una superficie hexagonal de sitios al azar, para diferentes valores de la
energía de interacción lateral (w/kT), siendo la diferencia de energía entre los
parches (∆ε/kT) constante e igual a 48.
En la figura 6.37 se muestran los resultados de las aproximaciones
teóricas para los mismos parámetros usados en la figura 6.36.
En la figuras 6.38 y 6.39 se comparan las isotermas teóricas con las
simuladas, para distintos valores de la relación w/kT.
-100 -80 -60 -40 -20 0 20 40
0,0
0,2
0,4
0,6
0,8
1,0
Figura 6.37: Isotermas de adsorción obtenidas mediante la aproximación de Fermi-Dirac.
w/kT = -3,0 w/kT = -1,0 w/kT = 0,0 w/kT = 4,0 w/kT = 8,0
Red Hexagonal Sitios al azar Aproximación de Fermi-Dirac ∆ε/kT = 48
θ
µ/kT
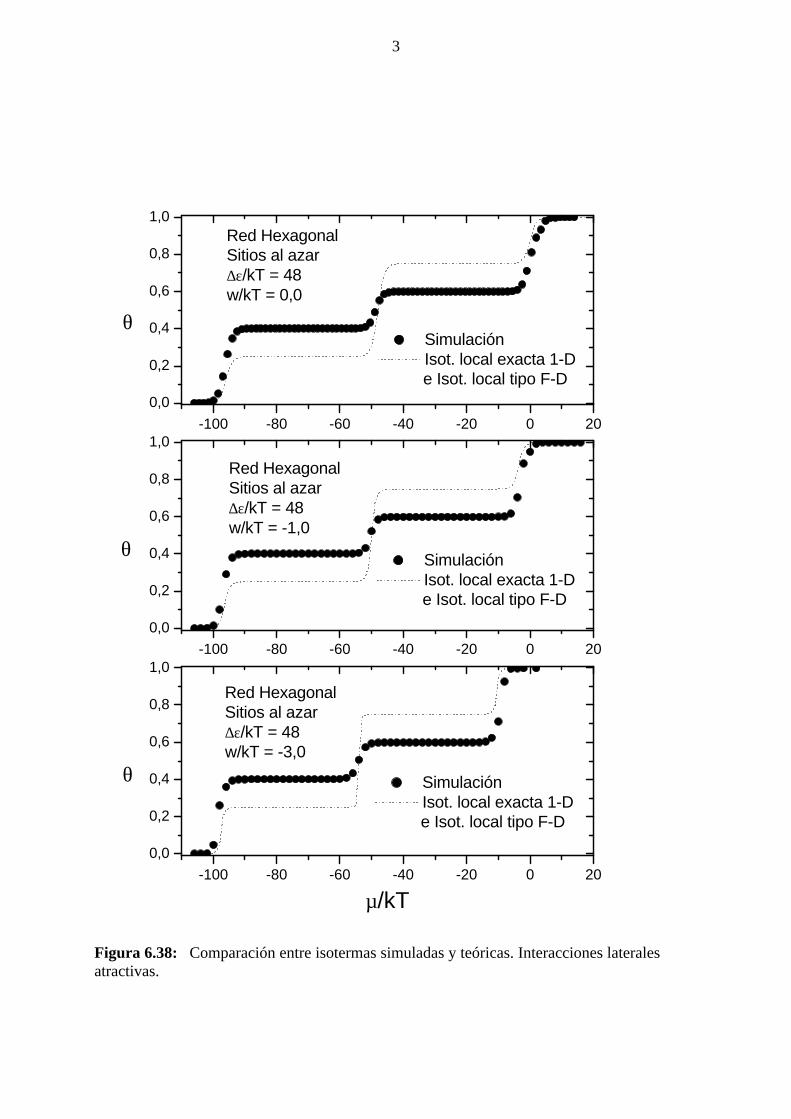
3
Figura 6.38: Comparación entre isotermas simuladas y teóricas. Interacciones lateralesatractivas.
-100 -80 -60 -40 -20 0 20
0,0
0,2
0,4
0,6
0,8
1,0-100 -80 -60 -40 -20 0 20
0,0
0,2
0,4
0,6
0,8
1,0-100 -80 -60 -40 -20 0 20
0,0
0,2
0,4
0,6
0,8
1,0
Red Hexagonal Sitios al azar ∆ε/kT = 48 w/kT = -3,0
Simulación Isot. local exacta 1-D
e Isot. local tipo F-D
θ
µ/kT
Red Hexagonal Sitios al azar ∆ε/kT = 48 w/kT = -1,0
Simulación Isot. local exacta 1-D
e Isot. local tipo F-D
θ
Simulación Isot. local exacta 1-D
e Isot. local tipo F-D
Red Hexagonal Sitios al azar ∆ε/kT = 48 w/kT = 0,0
θ
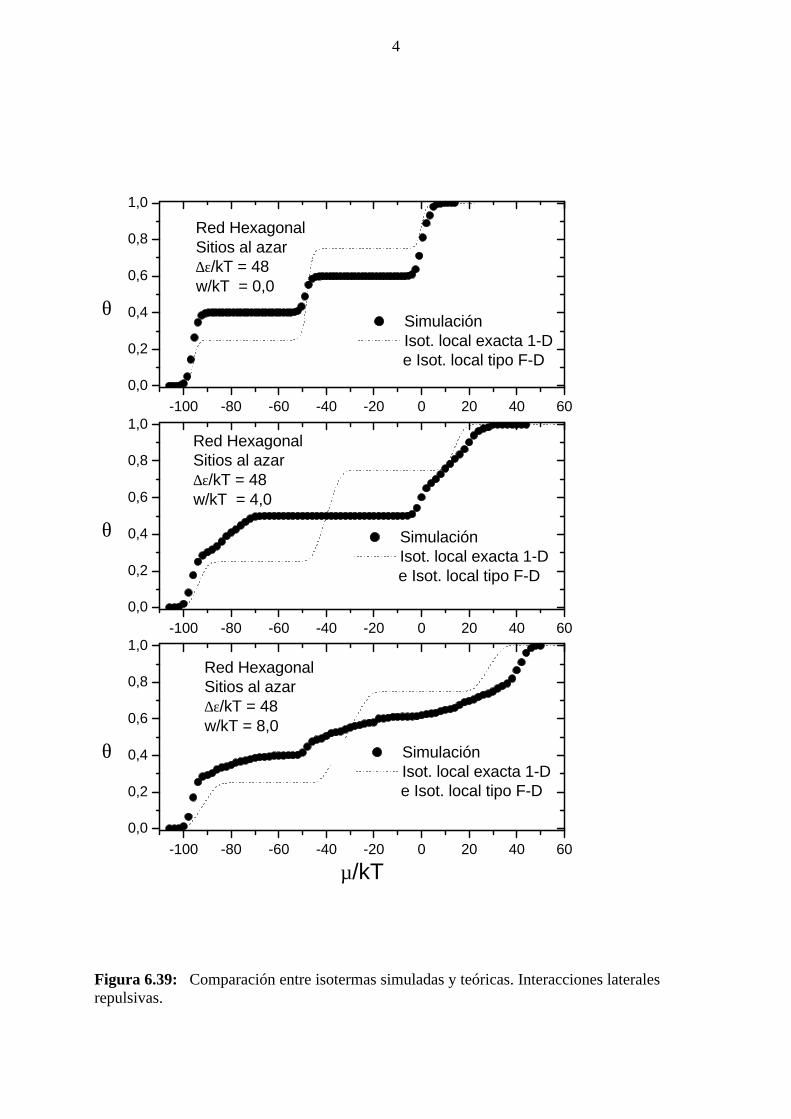
4
Figura 6.39: Comparación entre isotermas simuladas y teóricas. Interacciones lateralesrepulsivas.
-100 -80 -60 -40 -20 0 20 40 60
0,0
0,2
0,4
0,6
0,8
1,0
-100 -80 -60 -40 -20 0 20 40 60
0,0
0,2
0,4
0,6
0,8
1,0
-100 -80 -60 -40 -20 0 20 40 60
0,0
0,2
0,4
0,6
0,8
1,0
Red Hexagonal Sitios al azar ∆ε/kT = 48 w/kT = 4,0
Simulación Isot. local exacta 1-D
e Isot. local tipo F-D
θ
Red Hexagonal Sitios al azar ∆ε/kT = 48 w/kT = 8,0
Simulación Isot. local exacta 1-D
e Isot. local tipo F-D
θ
µ/kT
Simulación Isot. local exacta 1-D
e Isot. local tipo F-D
Red Hexagonal Sitios al azar ∆ε/kT = 48 w/kT = 0,0
θ

5
Como puede apreciarse en las figuras 6.38 y 6.39, las discrepancias entre
el modelo teórico y los resultados de Monte Carlo son mayores que las que se
observan en el caso de grandes parches, siendo las razones de las diferencias las
mismas que se indicaron para el caso de redes triangulares.
6.5 RESUMEN
En este capítulo se describieron en primer término los principales modelos
teóricos para estudiar la adsorción de dímeros sobre superficies heterogéneas,
sin interacciones laterales (Marczewski, Nitta y Nitta modificado en Ref. [66]),
y con interacciones laterales (Modelo tipo Fermi-Dirac para la isoterma local de
adsorción, y Modelo para la isoterma local basado en la isoterma homogénea
exacta en una dimensión [66]).
Luego se simuló el proceso de adsorción a través del método de Monte
Carlo en el Conjunto Gran Canónico, suponiendo al sustrato representado por
una red bidimensional, hexagonal o triangular, de L x L sitios de adsorción, con
condiciones periódicas de borde. La heterogeneidad se introdujo considerando
dos tipos de sitio de adsorción: débiles, con energías de adsorción ε1, y fuertes,
con energías de adsorción ε2, ambos en igual concentración (f1 = f2 = 0,5). La
superficie heterogénea se modeló como un conjunto de parches, que son
dominios de igual tamaño MP = l x l, distribuidos al azar, o en forma de tablero
de ajedrez. Después se compararon los valores que surgen de la aplicación de
las teorías mencionadas con los resultados obtenidos mediante simulación de
Monte Carlo.
Por último, se propuso un método para caracterizar la topografía
energética de sustratos que pueden aproximarse por medio de superficies
bivariadas, a través de medidas de adsorción de dímeros no interactuantes [74].
************************

CAPITULO 7
CONCLUSIONES Y PERSPECTIVAS
7.1 CONCLUSIONES
En el desarrollo de la presente tesis se estudió la adsorción de dímeros
interactuantes sobre superficies homogéneas y heterogéneas con geometrías
triangular y hexagonal. La metodología que se empleó en el análisis fue la
simulación de Monte Carlo en el conjunto Gran Canónico. Desde este punto de
vista, en el capítulo 3 se describieron las técnicas computacionales, se
presentaron las diferentes cantidades termodinámicas a medir por simulación y
por último, se efectuó el diseño de las redes triangular y hexagonal que se
usaron posteriormente para representar el sustrato en los procesos de adsorción.
Estas redes, junto a los programas computacionales respectivos, constituyen un
aporte importante de esta tesis ya que, como se indica más adelante, son
aplicables a otros procesos relacionados con la ciencia de superficies.
Los resultados obtenidos pueden agruparse en dos categorías, de acuerdo
al sentido en que fue utilizada la técnica numérica. En la primera de ellas, la
simulación se empleó para corroborar las teorías existentes referidas a múltiple
ocupación de sitios. En la segunda, los resultados computacionales permitieron
acceder a condiciones en la cuales la teoría no ofrece una descripción acabada
del sistema en estudio. En base a esta clasificación, se presentan a continuación
las principales conclusiones de la presente tesis.

2
1. Comparación de los resultados teóricos con aquellos provenientes de lassimulaciones de Monte Carlo
1.1. Por primera vez se corroboraron los resultados teóricos obtenidos
aplicando los modelos vistos en el capítulo 2 para dímeros interactuantes
adsorbidos en redes triangulares y hexagonales.
1.2. En el capítulo 6 se describieron en primer término los modelos teóricos
que estudian la adsorción de dímeros sobre superficies heterogéneas, sin
interacciones laterales (Marczewski, Nitta y Nitta modificado por Ramirez
Pastor), y con interacciones laterales (Modelo tipo Fermi-Dirac para la isoterma
local de adsorción, y Modelo para la isoterma local basado en la isoterma
homogénea exacta en una dimensión, ambos de Ramirez Pastor). Después se
compararon los valores que surgen de la aplicación de las teorías mencionadas
con los resultados obtenidos mediante simulación de Monte Carlo. De la
comparación se obtuvieron conclusiones sobre la validez y aplicabilidad de los
modelos analíticos.
2. Resultados obtenidos mediante simulación de Monte Carlo
2.1. En el capítulo 4 se estudió la adsorción de dímeros sobre superficies
homogéneas para interacciones laterales atractivas y repulsivas. Como aporte
importante de esta tesis, cabe destacar que en el caso particular de interacciones
laterales repulsivas y valores altos de la relación w/kT, se encontraron dos
nuevas fases ordenadas en el adsorbato, para θ1 = 5/9 y θ2 = 2/3. Esto indica
que el sistema experimenta una transición de fase, de estructuras desordenadas a

3
estructuras ordenadas, que denominamos FOBC (fase ordenada de bajo
cubrimiento) y FOAC (fase ordenada de alto cubrimiento), las cuales
reportamos por primera vez recientemente en la literatura [73]
También se estimaron las temperatura críticas correspondientes a los
cubrimientos críticos.
2.2. En el capítulo 5 se estudió la adsorción de dímeros sobre superficies
triangulares homogéneas. También acá, como para redes hexagonales, en el
caso particular de interacciones laterales repulsivas, y para valores altos de la
relación w/kT, se encontraron dos nuevas fases ordenadas en el adsorbato, para
θ1 = 4/5 y θ2 = 2/3, que muestran una transición de fase en el sistema, de
estructuras desordenadas a estructuras ordenadas, que llamamos FOBC (fase
ordenada de bajo cubrimiento) y FOAC (fase ordenada de alto cubrimiento), las
cuales reportamos por primera vez recientemente en la literatura [73].
Asimismo, se estimaron las temperaturas críticas correspondientes a los
cubrimientos críticos.
2.3. En la sección 5.2.3 se compararon los resultados obtenidos por simulación
para ciertas cantidades termodinámicas (isotermas de adsorción, calor
diferencial de adsorción, y entropía configuracional), para redes hexagonales y
triangulares, y valores iguales de interacción lateral repulsiva. Las diferencias
observadas se explicaron en función de las conectividades diferentes de las
redes.
2.4. Se simuló el proceso de adsorción a través del método de Monte Carlo en
el Conjunto Gran Canónico, suponiendo al sustrato representado por una red
bidimensional, hexagonal o triangular, de L x L sitios de adsorción, con
condiciones periódicas de borde. La heterogeneidad se introdujo considerando
dos tipos de sitio de adsorción: débiles, con energías de adsorción ε1, y fuertes,
con energías de adsorción ε2, ambos en igual concentración (f1 = f2 = 0,5). La
superficie heterogénea se modeló como un conjunto de parches, que son

4
dominios de igual tamaño MP = l x l, distribuidos al azar, o en forma de tablero
de ajedrez. Por último, y como aporte de esta tesis, se propuso un método para
caracterizar la topografía energética de sustratos que pueden aproximarse por
medio de superficies bivariadas, a través de medidas de adsorción de dímeros
no interactuantes [74].
7.2 PERSPECTIVAS
Los estudios comenzados en la presente tesis dejan un camino abierto para
su continuación, así como para la realización de estudios en otros procesos de la
ciencia de superficies. Entre las acciones que se pueden llevar a cabo para lograr
dichos fines se puede mencionar:
• Determinación de los parámetros de orden relacionados con las
estructuras ordenadas que aparecen en el adsorbato para interacciones
laterales repulsivas, llamadas FOBC (fase ordenada de bajo cubrimiento)
y FOAC (fase ordenada de alto cubrimiento), para redes triangulares y
hexagonales.
• Confirmación, mediante un escaleo de tamaño finito, de las temperaturas
críticas estimadas, correspondientes a la aparición de las fases ordenadas
mencionadas, para redes triangulares y hexagonales.
• Obtención de los diagramas de fases para la adsorción de dímeros sobre
redes hexagonales y triangulares, para interacciones laterales atractivas y
repulsivas.
• Ampliación de los estudios de simulación de Monte Carlo de adsorción de
dímeros homonucleares sobre redes hexagonales y triangulares, a dímeros

5
heteronucleares formados por segmentos diferentes entre sí, y a k-meros
homonucleares formados por 3 o más segmentos.
• Estudio del efecto de la heterogeneidad superficial sobre la adsorción de
dímeros en redes triangulares y hexagonales, para los casos de
topografías intermedias entre grandes parches y azar.
• Desarrollo de métodos para caracterizar la topografía energética de
sustratos heterogéneos que pueden aproximarse por medio de superficies
bivariadas, a través de medidas de adsorción de dímeros interactuantes.
• Aplicación de las redes triangular y hexagonal diseñadas, y de los
respectivos programas computacionales desarrollados, a la simulación de
procesos de difusión de dímeros homonucleares, sobre redes hexagonales
y triangulares, homogéneas y heterogéneas.
• Diseño de algoritmos y metodologías de simulación de Monte Carlo que
permitan estudiar la termodinámica de adsorción de dímeros
interactuantes sobre redes de geometría intermedia entre la de redes
hexagonal y triangular, considerando al sustrato representado por una red
bidimensional triangular heterogénea, de L x L sitios de adsorción, con
condiciones periódicas de borde, y dos tipos de sitio de adsorción,
representados por símbolos negros y blancos (figura 7.1), de energías de
adsorción ε1 y ε2, respectivamente, siendo 0 ≤ (ε1/ε2) ≤ 1,
correspondiendo los casos extremos a red hexagonal homogénea (figura
7.2), y a red triangular homogénea (figura 7.3), respectivamente.
6
Figura 7.1: Red triangular heterogénea (0 ≤ (ε1/ε2) ≤ 1)
Figura 7.2: Red hexagonal homogénea (ε1 = 0)

7
Figura 7.3: Red triangular homogénea (ε1 = ε2)
* * * * * * * * *

8
BIBLIOGRAFÍA
1. Zhdanov, V.P. Elementary Physicochemical Processes on Solid Surfaces,
Plenum Press: New York, 1991.
2. Luth, H. Surfaces and Interfases of Solids. Springer-Verlag: Berlin, 1993.
3. Ma, Y. H.; Clark, W. M. Adsorption News 1991, 2, 8.
4. Talu, O. Adsorption News 1990, 1, 3.
5. Sircar, S.; Myers, A.L. Surf. Sci. 1988, 205, 353.
6. Barton, S.S.; Dacey, J.R.; Quinn, D.F. Fundamentals of adsorption; Myers,
A.L.; Belfort, G., Eds.; 1983, p.p 65.
7. Otto, K. Proc. 4th International Conference on Alternative Energy Sources,
Florida, 1981, 6, 241.
8. Bonzel M. P. Surf. Sci. 1977. 68, 236.
9. Isaacson, M.; Kopf, D.; Utlaut. M.; Parker, N. W.; Crewe, A. V. Proc. Natl.
Acad. Sci. 1977, 74, 1802.
10. Binnig, G.; Rohrer, H. Helv.Phys. Rev. Lett. 1982, 55, 726.
11. Binnig, G.; Rohrer, H. Surf. Sci. 1983, 126, 236.
12. Binnig, G.; Rohrer, H.; Gerber, Ch.; Weibel E. Appl. Phys. Lett. 1982, 40,
178.
13. Binnig, G.; Rohrer, H.; Gerber, Ch.; Weibel E. Phys. Rev. Lett. 1983, 50,
120.
14. Binnig, G.; Rohrer, H. Sci. Amer. 1985, p.p 40.
15. Kellogg, G. L. Surf. Sci. Rep. 1994, 21, 1.
16. Turner, P.J.; Cartwright, P.; Southon, M.J.; van Oostrom, A.; Manley, B.W.
J. Phys. E (Sci. Instr.) 1969, 2, 731.
17. van Oostrom, A. Ned. Tijdschr. Vacumtech. 1971, 9, 13.
18. Mueller, E. W.; Tsong, T. T. Field Ion Microscopy, Principles and
Applications ; Elsevier: N. Y., 1969.

9
19. van Oostrom, A. Phys. Lett.. 1966, 22,137.
20. van Oostrom, A. J. Chem. Phys. 1967, 47, 761.
21. van Oostrom, A. Acta Electron. 1973, 16, 59.
22. van Oostrom, A. CRC Crit. Rev. Solid State Sci.. 1974, 4, 353.
23. Mueller, E. W.; Z. Phys. 1951, 131, 136.
24. Metropolis, N.; Ulam, S. J. Am. Stat. Ass. 1949, 44, 335.
25. Metropolis, N.; Rosenbluth, A.; Rosenbluth M.N.; Teller, A. W.; Teller, E.
J. Chem. Phys. 1953, 21, 1087.
26. Nicholson, D.; Parsonage, N. G. Computer. Simulation and the Statisticaal
Mechanics of Adsortion; Academic Press: London, 1982.
27. Binder, K. Applications of the Monte Carlo method in statistical physics.
Topics in current Physics Vol. 36; Springer; Berlin, 1984.
28. Binder, K. Monte Carlo method in statistical physics (2nd edn.). Topics in
current Physics Vol. 7; Springer; Berlin, 1986.
29. Allen, M.P.; Tildesley, D.J. Computer Simulation of Liquids; Clarendom
Press; Oxford, 1987.
30. Migone, A. D.; Kim, H.; Chan, M.H. W.; Talbot, J.; Tildesley, D. J.; Steele,
W. A. Phys. Rev. Lett. 1983, 51, 192.
31. Talbot, J.; Tildesley, D. J.; Steele, W. A. Molec. Phys. 1985, 51, 1331.
32. Talbot, J.; Tildesley, D. J.; Steele, W. A. Faraday Discuss. Chem. Soc. 1985,
80, 91.
33. Solkolowski S.; Steele, W. A. Molec. Phys. 1985, 54, 1453.
34. Wood, W. W. Proc. Enrico Fermi Summer School, Varenna, 1985, p.p. 3.
35. Ciccotti, G; Frenkel, D.; McDonald, I. R. Simulation of Liquid and Solids,
Molecular Dynamics and Monte Carlo Methods in Statistical Mechanics;
Elsevier: Amsterdam, 1988.
36. Clark, A. The Theory of Adsorption and Catalysis. Academic Press: New
York, 1970.

10
37. Steele, W. A. The interaction of gases with solid surfaces; Pergamon Press:
N.Y., 1974.
38. Hill, T. L. An Introduction to Statistical Thermodinamics.; Addison-Wesley
Publishing Company: Reading, Mass. 1960.
39. Domb, C.; Adv. Physics, 1960, 9, 149.
40. Ising, E. Z. Physik 1925, 31, 253; Onsager, L. Phys. Rev. 1944, 65, 117;
Baxter, R. J. Exactly solved models in statistical mechanics; Academic Press:
London, 1982; Landau, D. P.; Binder, K. Phys. Rev. B 1985, 31, 5946.
41. Ramirez-Pastor, A. J.; Nazzarro, M. S.; Riccardo J. L.; Pereyra, V. Surf. Sci.
1997, 391, 267.
42. Rudzinski, W.; Everett, D. H. Adsorption of Gases on Heterogeneous
Surfaces; Academic Press: New York, 1992;
43. Jaroniec, M.J.; Madey, R. Physical Adsorption on Heterogeneus Surfaces;
Elsevier: Amsterdam, 1988.
44. Rudzinski, W.; Steele, W. A.; Zgrablich, G. Equilibria and Dynamics of Gas
Adsorption on Heterogeneous Solid Surfaces; Elsevier: Amsterdan, 1996.
45. Zgrablich, G.; Ripa P. J. Chem. Phys. 1975, 79, 2118; Riccardo J.L.; Chade,
M. A.; Pereyra V.D.; Zgrablich, G. Langmuir 1992, 8, 1518.
46. O'Brien, J. A.; Myers, A. L. J. Chem. Soc., Faraday Trans. 1985, 81, 355.
47. Ramirez Pastor, A.J.; Nazzarro, M. S.; Riccardo, J. L.; Zgrablich, G. Surf.
Sci., 1995, 341, 249.
48. Zgrablich, G.; Mayagoitia, V.; Rojas, F.; Bulnes, F.; González, A. P.;
Nazzarro, M. S.; Pereyra, V.; Ramirez Pastor, A.J.; Riccardo, J. L.; Sapag, K.
Langmuir 1996, 12, 129.
49. Riccardo, J. L.; Tesis Doctoral, Universidad Nacional de San Luis, 1991.
50. Brunauer. S.; Emmett, P. H.; Teller, E. J. Am.Chem. Soc. 1938, 60, 309.
51. Hill, T.L., J..Chem. Phys. 1946, 14, 263.
52. de Boer, J.H. The Dynamical Character of Adsorption; Oxford University
Press: London, 1953.

11
53. Flory, P. J. Principles of Polymer Chemistry; Cornell Univ. Press: Ithaca, N.
Y., 1953.
54. Huggins, M. L. J. Chem. Phys. 1942, 46, 151.
55. Huggins, M. L. Ann. N.Y. Acad. Sci..1942, 41, 151.
56. Huggins, M. L. J. Am. Chem. Soc. 1942, 64, 1712.
57. Marczewski, A. W.; Derylo-Marczewska, A.; Jaroniec, M. J. Coll. Interface
Sci. 1986, 109, 310.
58. Nitta, T.; Kuro-Oka, M.; Katayama, K. J. Chem. Eng. Japan 1984, 17, 45.
59. Cooper, L.N. Phys. Rev. 1959, 104, 1189.
60. Tsong, T.T.; Casanova, R. Phys. Rev. 1980, 21, 4564.
61. Tsong, T.T.; Casanova, R. Phys. Rev. 1980, 22, 4632.
62. Noebl, C.; Benndorf, C.; Madey, T. Surf. Sci. 1985, 157, 29.
63. Bassett, D. W. J. Phys. C: Solid State Phys. 1976, 9, 2491.
64. McQuistan, R. B.; Hock, J. L. J. Math. Phys. 1986, 27, 599.
65. Phares, A. J.; Wunderlich, F. J.; Curley, J. D.; Grumbine, D. W. J. Phys. A:
Math. Gen. 1993, 26, 6847.
66. Ramirez Pastor, A. J, Tesis Doctoral, Univ. Nac.de San Luis, 1998.
67. Feibelman, P. J. Phys. Rev. Lett. 1987, 58, 2766.
68. Flory, P.J. J. Chem. Phyys. 1942, 10, 51.
69. Nitta, T.; Kuro-Oka, M.; Katayama, K. J. Chem. Eng. Japan 1984, 17, 45.
70. Bakaev,V.A; Steele, W.A. Langmuir 1992, 8,148.
71. D. Nicholson, N.G. Parsonage, Computer Simulation and the Statistical
Mechanics of Adsorption, Academic Press, London, 1982.
72. Romá, F. J, Tesis de Licenciatura, Univ. Nac. de San Luis, 2000.
73. Gonzalez, J.; Ramirez-Pastor, A.; Pereyra, V. Langmuir, 2001, 17, 6974.
74. Gonzalez, J.; Ramirez-Pastor, A.; Physica A, 2002, 3, 339.
75. Bulnes, F.; Ramirez-Pastor, A.;Zgrablich, G. J. Chem. Phys. 2001, 115,
1513.
* * * * * * * * *