







Idiomas
Páginas
Jurídico


Síndrome Anémico
M en C. Alejandro Morales de la Vega
Laboratorio Central, UMAE Hospital de Cardiología del CMN Siglo XXI, IMSS.

Síndrome Anémico
• Se caracteriza por la disminución por abajo de los límites normales de la masa eritrocitaria.

Anemia
• Puede definirse como una reducción en la capacidad normal de la sangre en transportar oxígeno.

Anemia
• La Anemia es un problema de salud pública en países en desarrollo.
• Las causas más comunes: las deficiencias nutricionales (hierro, ácido fólico y vitamina B12).

Anemia
• Cuando la Hb y el Hto están abajo del límite inferior del intervalo de referencia del 95 % correspondiente a la edad, sexo y localización geográfica.

Anemia
• Participación del Laboratorio Clínico:
• Comprobar la existencia de la anemia.
• Definir la causa.

Anemia
• En la mayoría de los casos la anemia se puede diagnosticar y con pocos recursos, establecer la causa:
• Cuantificación de Hemoglobina.
• Valor Hematocrito.
• Frotis de Sangre Periférica.

EVALUACION DE LA ANEMIAHEMATOLOGIA
• HEMATOCRITO• HEMOGLOBINA• RECUENTO ERITROCITARIO• INDICES ERITROCITARIOS• RECUENTO LEUCOCITARIO• RETICULOCITOS• RECUENTO PLAQUETARIO• VEL. SED. GLOBULAR• HEMOGRAMA


AnemiaDiagnóstico
• Historia Clínica
• Examen Físico
• Estudios de Laboratorio

AnemiaLaboratorio
• Reticulocito: Eritrocito inmaduro, anuclear que contiene organelos y un sistema ribosómico extenso para la síntesis de Hb.
• Estancia en médula ósea: 2 – 3 días
• Formas de reportarlos:
• Porcentaje (%)
• Valor Absoluto: Rt x 109/L

AnemiaLaboratorio
• Reticulocitos Corregidos (%).
• Hto del paciente x C Rt (%)
• Hto Ref (0.45)
• Índice de Producción de Reticulocitos.
• Hto del Paciente x C Rt (%) x 1
• Hto de Ref (0.45) Tiempo de maduración
con base al Hematocrito
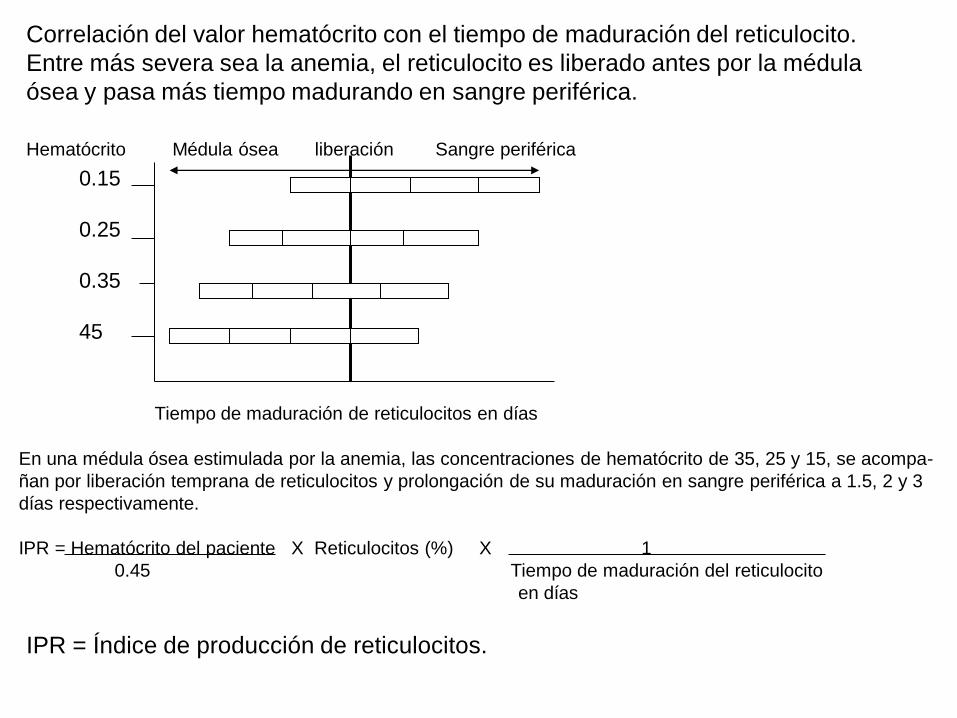
Correlación del valor hematócrito con el tiempo de maduración del reticulocito.
Entre más severa sea la anemia, el reticulocito es liberado antes por la médula
ósea y pasa más tiempo madurando en sangre periférica.
Hematócrito Médula ósea liberación Sangre periférica
Tiempo de maduración de reticulocitos en días
En una médula ósea estimulada por la anemia, las concentraciones de hematócrito de 35, 25 y 15, se acompa-
ñan por liberación temprana de reticulocitos y prolongación de su maduración en sangre periférica a 1.5, 2 y 3
días respectivamente.
IPR = Hematócrito del paciente X Reticulocitos (%) X 1
0.45 Tiempo de maduración del reticulocito
en días
0.15
0.25
0.35
45
IPR = Índice de producción de reticulocitos.

INDICES ERITROCITARIOS
VGM = Htc (L/L) (88 +/- 10 fL)
RE (x 1012/L)
HCM = Hb (g/L) (29.5 +/- 2.5 pg)
RE (x 1012/L)
CMHC = Hb (g/L) (32 +/- 2.5 g/dL)
Htc (L/L)
Hto = RE X VGM (L/L)
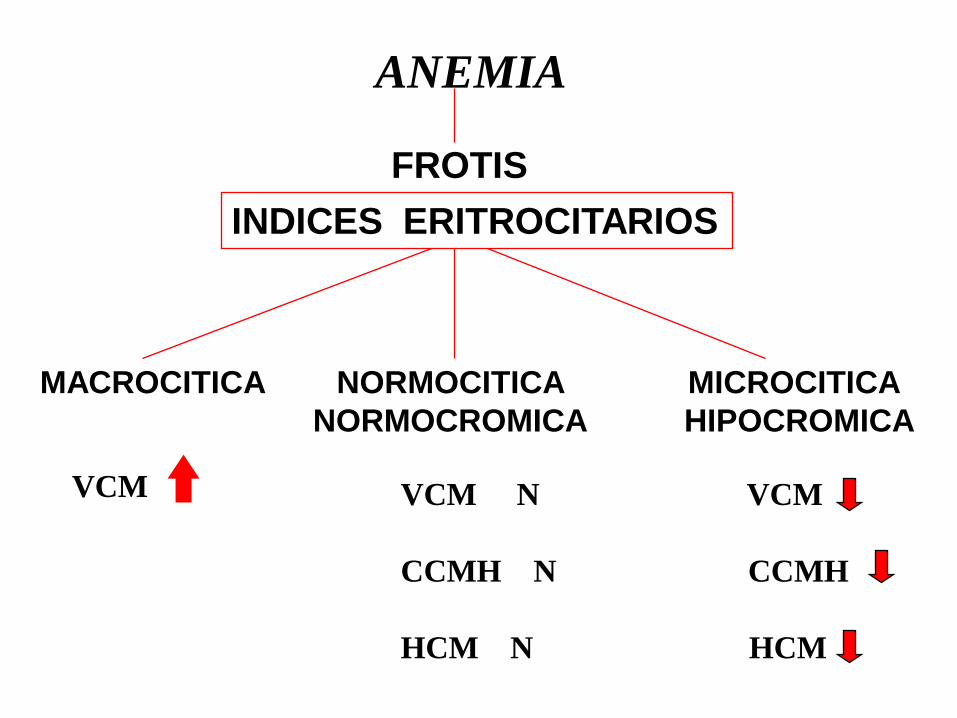
ANEMIA
FROTIS
INDICES ERITROCITARIOS
MACROCITICA NORMOCITICA MICROCITICA
NORMOCROMICA HIPOCROMICA
VCM VCM N VCM
CCMH N CCMH
HCM N HCM
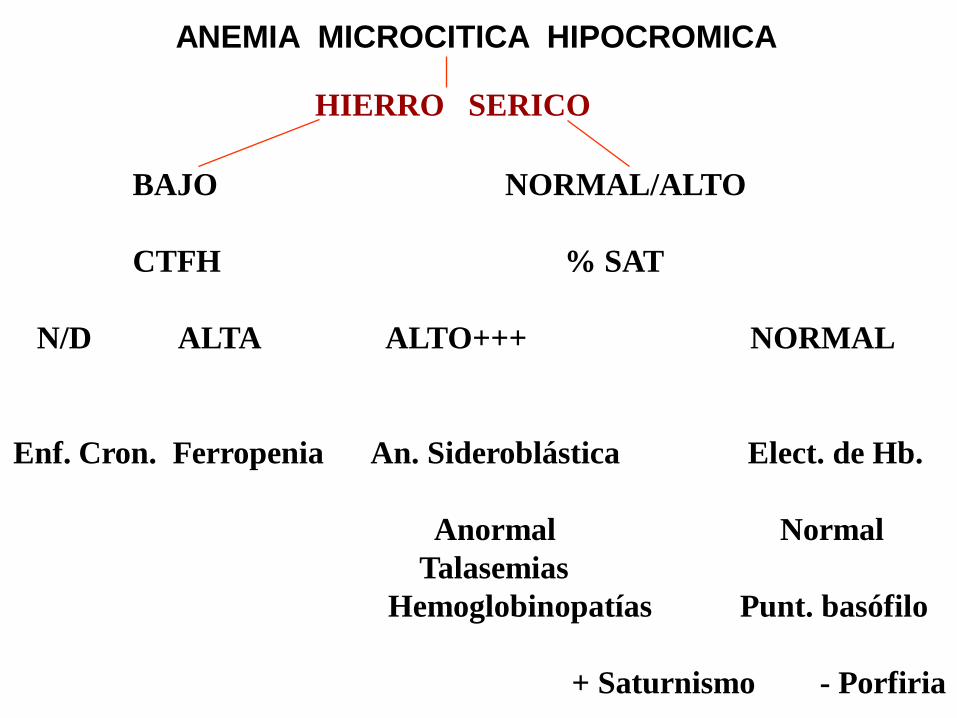
ANEMIA MICROCITICA HIPOCROMICA
HIERRO SERICO
BAJO NORMAL/ALTO
CTFH % SAT
N/D ALTA ALTO+++ NORMAL
Enf. Cron. Ferropenia An. Sideroblástica Elect. de Hb.
Anormal Normal
Talasemias
Hemoglobinopatías Punt. basófilo
+ Saturnismo - Porfiria
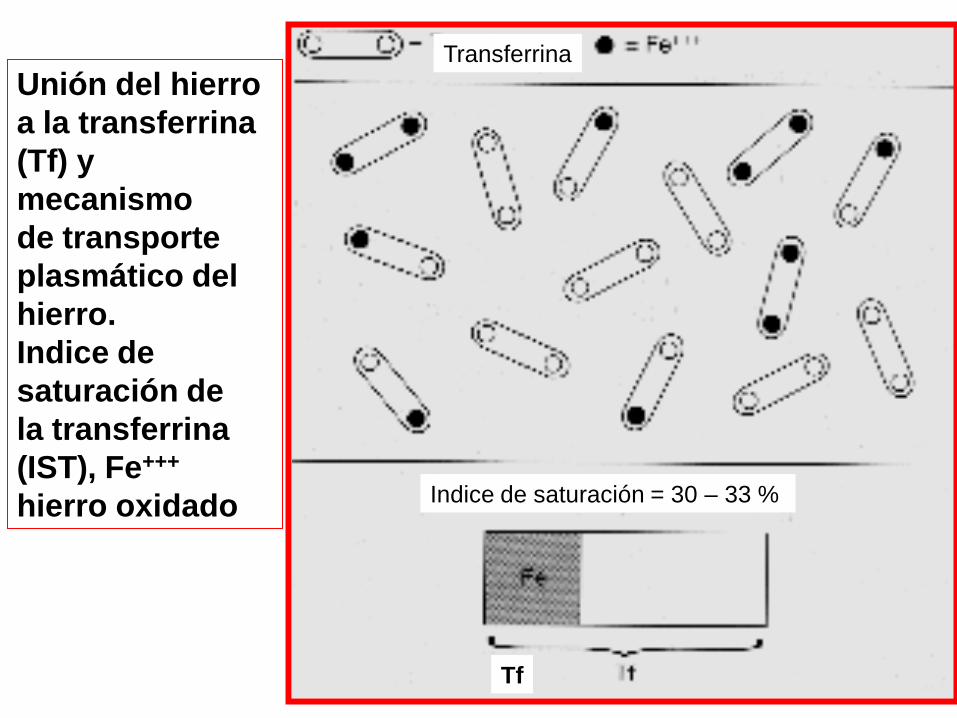
Tf
Indice de saturación = 30 – 33 %
Transferrina
Unión del hierro
a la transferrina
(Tf) y
mecanismo
de transporte
plasmático del
hierro.
Indice de
saturación de
la transferrina
(IST), Fe+++
hierro oxidado

HierroUTILIZACIÓN
• Todas las células del organismo poseen receptor de transferrina (R-Tf).
• En mayor número: precursores eritroides y hepatocitos (mecanismo de reutilización).

HierroUTILIZACIÓN
• La concentración plasmática de RTf es de 200 – 300 ng/mL.
• Se determina por anticuerpos monoclonales

Anemia FerropénicaFerropenia prelatente
• Desaparición del hierro de reserva (medular)
• Sideroblastos < 5 %
• Hierro sérico normal
• Ferritina disminuida

Anemia FerropénicaFerropenia latente
• IST disminuido < 12 % (N = 30 – 35 %)
• Hierro variable (N o disminuido)
• CTST incrementada
• Disminución discreta del VCM
• ADE incrementada

Anemia FerropénicaEritropoyesis ferropénica
• Hemoglobina disminuida
• Microcitosis e hipocromia
• Hierro sérico disminuido
• Ferritina disminuida
• IST disminuido

ANEMIA FERROPENICABIOMETRIA HEMATICA
• VCM = 55 - 74 fL
• CMHC = 22 - 31 g/dL, HCM = 14 - 26 pg
• Anisocitosis, hipocromía, poiquilocitosis (ADE )
• Hb , Rec. eritrocitario y Hto no son confiables.
• Reticulocitos = normales o ligeramente
• Plaquetas = variables, incrementadas pero inferiores a 700 x 109/L
• Neutrófilos hipersegmentados a veces

ANEMIA FERROPENICAESTUDIOS DE HIERRO
Las pruebas bioquímicas dependen de la etapa evolutiva del desarrollo de la anemia. Son útiles para el estados ferropénico pero pocas veces una ferropenia latente.
HIERRO SÉRICO = < 30 mg/dL
CAPACIDAD DE SATURACIÓN DE LA TRANSFERRINA= < 15%
FERRITINA = DISMINUIDA

Hierro sérico
• VR. 140 – 1200 mg/L (< 19 años)
• 450 – 1700 mg/L (> 20 años)
• IST: 30%
• Ferritina (RIA, ELISA, IRMA)
• Ferritina – Ac anti ferritina.
• VR. 7 – 340 mg/L mujeres
• 11 – 400 mg/L hombres
• Receptor soluble de la transferrina.

ANEMIA FERROPENICAMEDULA OSEA
Hiperplasia eritroide moderada
Relación M:E disminuida
Eritroblastos con Hb disminuida
Diseritropoyesis ocasional
Tinción de Perls: macrófagos con hierro
disminuido, sideroblastos disminuidos o
ausentes.
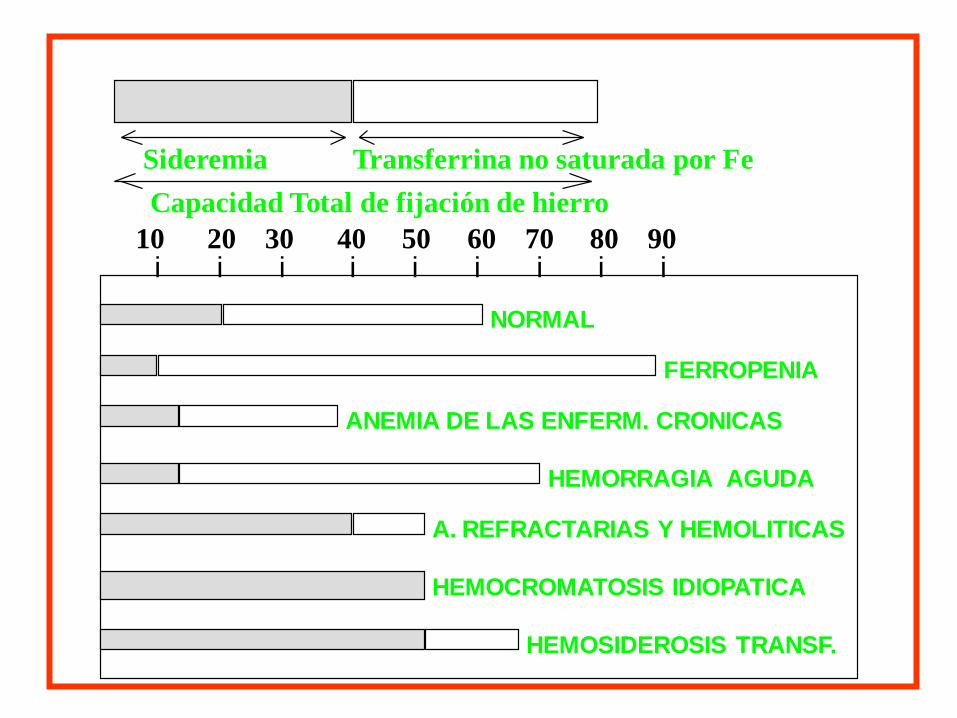
Sideremia Transferrina no saturada por Fe
Capacidad Total de fijación de hierro
i i i i i i i i i10 20 30 40 50 60 70 80 90
NORMAL
FERROPENIA
ANEMIA DE LAS ENFERM. CRONICAS
HEMORRAGIA AGUDA
A. REFRACTARIAS Y HEMOLITICAS
HEMOCROMATOSIS IDIOPATICA
HEMOSIDEROSIS TRANSF.

Talasemias
• El síndrome talasémico es una alteración congénita de la síntesis de hemoglobina caracterizada por una disminución parcial o total de la producción de una o varias cadenas globínicas.

Talasemias
• Se les denomina según el tipo de cadena de globina cuya síntesis se afecte:
• a: alfa talasemia
• b: beta talasemia
• d y b: delta y beta talasemia
• Conduce a la acumulación de cadenas que no se combinan

TalasemiasFisiopatología
• A) Deficiencia en la hemoglobinización, microcitosis, disminución de la CMHC (hipocromia).
• B) Precipitación de las cadenas sobrantes en el citoplasma de los eritroblastos, lisis de los eritroblastos (eritropoyesis ineficaz).
• C) Alteraciones morfológicas de los eritrocitos que comprometen seriamente su supervivencia en la circulación (hemólisis).

Talasemias
• Las mejor caracterizadas y clínicamente más importantes son:
• a
• b Talasemias
• db

a Talasemia
• Las formas más frecuentes se asocian a anemias microcíticas e hipocrómicas no ferropénicas sin expresividad electroforética (microcitosis familiares atípicas).
• Las formas graves: hemoglobinopatía H (Hb H) b4 y la hidropesía fetal (Hb Bart) g4.

HEMOGLOBINAS NORMALES(ADULTO)
• HEMOGLOBINA A1 (α2β2) 97%
• HEMOGLOBINA A2 (α2 d2) 2.5%
• HEMOGLOBINA FETAL (α2 g2) < 1%

TALASEMIAS INTERMEDIAS Y MAYORESCARACTERÍSTICAS DIAGNÓSTICAS
• Hipocromia y microcitosis acentuadas.
• Poiquilocitosis muy acentuada, codocitos, microesferocitos.
• Punteado basófilo
• Basofilia difusa
• Presencia de eritroblastos
• Cuerpos de Howell Jolly
• Reticulocitos “Aumentados”
• En la enfermedad por Hb H: Tinción de Hb H : positiva
•

ANEMIA MACROCITICA
RETICULOCITOS
ESFEROCITOS
ESQUIZOCITOS
Hemorragia Hemólisis
Intravascular Extravascular
Haptoglobina B. I.
Hb libre Urobilinógeno
Hemosiderina
Hb orina
Hiperregenerativas Hiporregenerativas
VB12, Folatos
Cambios Megalobl.
An. Meg. Leucocitos
Plaquetas
Méd. Osea N o
Aplasia Medicamentos
S. Mielodisp. Enf. hepática
Alcoholismo
Hipotiroidismo

Anemias Macrocíticas
• Las anemias macrocíticas definidas por un volumen corpuscular medio (VCM) superior a 103 fL, son en un 95% de los casos megaloblásticas.

Anemia Macrocítica
• El término “megaloblasto” fue introducido por Erlich para describir morfológicamente la disociación madurativa entre el núcleo y el citoplasma debida a su vez, a un retraso en la síntesis del DNA.

Anemia Megaloblástica
Deficiencia de:
Vitamina B 12
Ácido Fólico

Anemias Macrocíticas
• Tanto el ácido fólico como la V B12 son esenciales para la síntesis de DNA y deben ser aportados necesariamente por los alimentos.
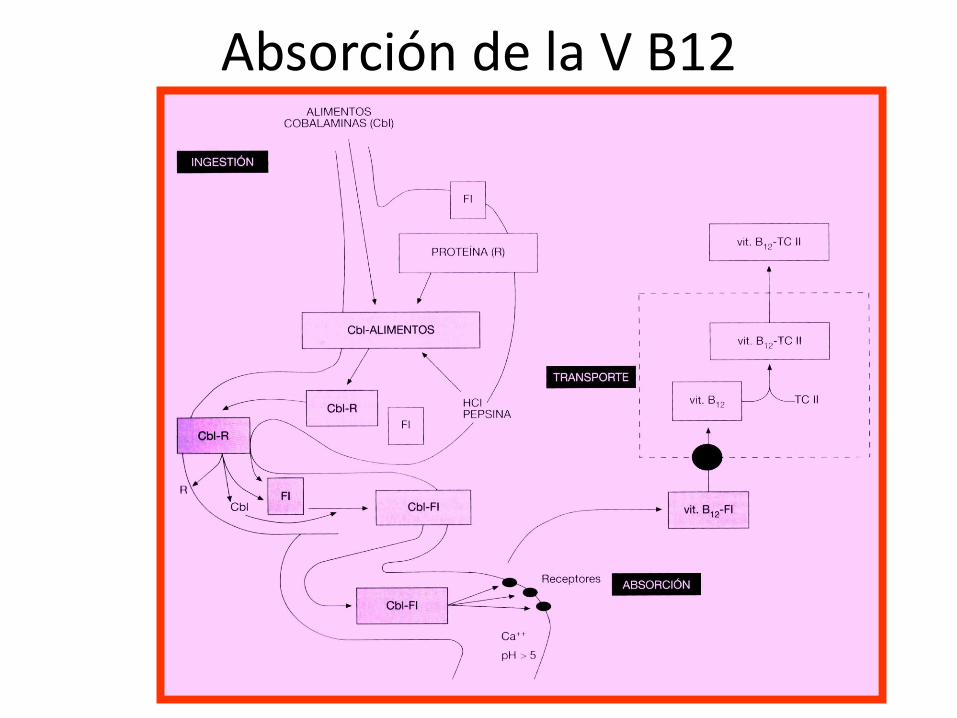
Absorción de la V B12
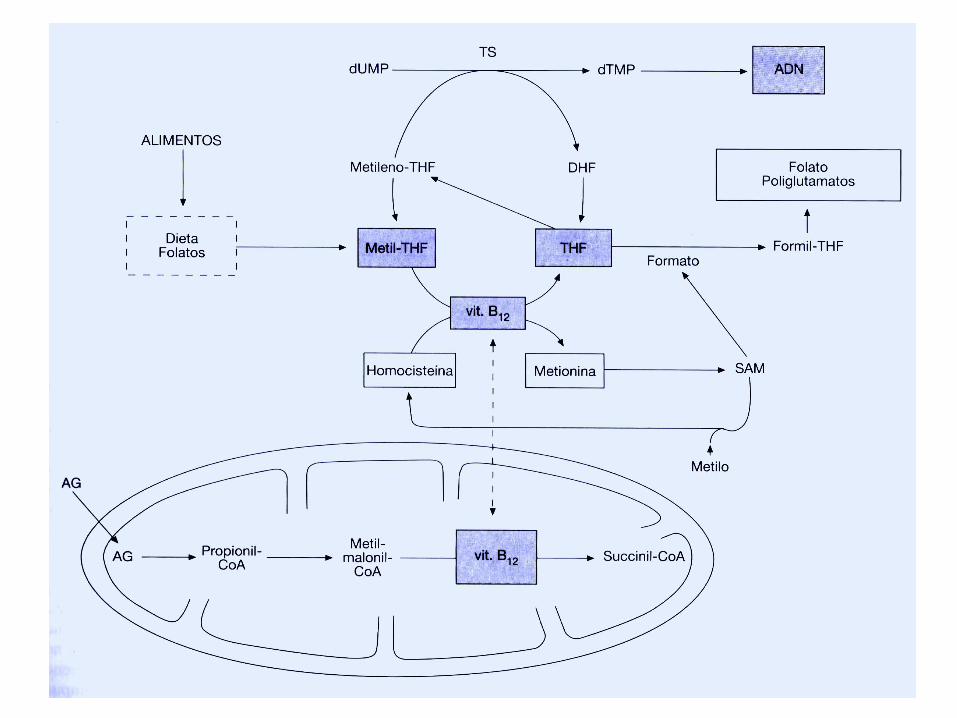

Anemias MegaloblásticasManifestaciones Clínicas
• Síndrome Anémico
• Trastornos en las Mucosas
• Manifestaciones Neurológicas
• Manifestaciones Psiquiátricas
• Ictericia ligera
• Esplenomegalia
• Púrpura trombocitopénica
• Infecciones

Anemia MegaloblásticaHemograma
• Anemia Macrocítica Normocrómica
• Eliptocitosis, anisopoiquilocitosis, macrocitos (ovalocitos), ertroblastos, diseritropoyesis (anillos de cabot).
• Reticulocitos normales o disminuidos (IPR < 2)
• Leucocitos normales o disminidos
• Macropolcitos
• Trombocitopenia leve

Anemia MegaloblásticaMielograma
• Hipercelularidad (hiperplasia eritroide)
• Asincronismo madurativo nucleocitoplasmático
• Series mielode megacariocítica con megalblastosis
• Sideroblastos y hierro macrofágico incrementado

Anemias MegaloblásticaVitamina B12 y Folatos en condiciones normales y patológicas
Cobalamina
sérica (pg/mL)
Folato sérico
(ng/mL)
Folato
eritrocitario
(ng/mL)
Normal 200 - 900 5 - 30 150 – 800
Déficit de VB12 < 100 N o > 30* < 150
Déficit de folato N o < 200** < 3 < 100
Déficit de
ambos factores < 100 < 3 < 150
* Aumentado en un 25% de los casos, ** en el 50% de los casos.

Anemia Macrocítica
• La macrocitosis no megaloblástica, a diferencia de la megaloblástica suele observarse como un fenómeno aislado, sin anemia y se caracteriza por una síntesis normal de ADN con macrocitosis normocítica.

Causas de Anemia Macrocítica No Megaloblástica
• Alcoholismo crónico• Enfermedad hepática crónica• Reticulocitosis importante• Aplasia medular• Embarazo• Deficiencia hereditaria de LCAT• Recién nacidos• Ictericia obstructiva• Síndrome Mielodispásico• Postesplenecomía

ANEMIA NORMOCITICA NORMOCROMICA
RETICULOCITOS
Historia, curso, frotis, Leucocitos, Plaquetas
pigmentos biliares
Hemorragia Hemólisis Médula Osea
-Lesión medular
-Infiltración
-Precursores
afectados
Normal
Hierro/CCTH
-Enf. Crónica
Normal
PFR
Anormal Normal
Médula Osea
- Aplasia Eritroide-Enf. Renal
Intravascular Extravascular
-Haptoglob. -B. I.
-Hb libre -Urobilinógeno
-Hemosid.
-Hb en orina

ANEMIAS HEMOLITICAS CONGENITAS
HISTORIA CLINICA
• HEMOLISIS AGUDA O CRONICA
• EDAD
• ORIGEN ETNICO
• SINTOMAS Y SIGNOS
• INGESTA DE MEDICAMENTOS O COMIDA (HABAS)
• ANTECEDENTES PATOLOGICOS Y FAMILIARES

ANEMIAS HEMOLITICAS CONGENITASEXPLORACION FISICA
• Ictericia sin coluria
• Palidez, taquicardia
• Esplenomegalia (formas crónicas)
• Desarrollo esquelético alterado (neonatos)
• Hemosiderosis (formas graves)

MEMBRANOPATIAS
*Anemias hemolíticas por anormalidades en
las proteínas y los lípidos constitutivos de la
membrana.
*Ambos pueden alterar su estabilidad,
deformabilidad y/o permeabilidad.
*Resulta en hemólisis extravascular.

MEMBRANOPATIAS
• ESFEROCITOSIS
• ELIPTOCITOSIS
• Común
• Estomatocítica
• Piropoiquilocitosis
• ESTOMATOCITOSIS
• Xerocitosis
• Hidrocitosis

ESFEROCITOSIS HEREDITARIA
• Es un trastorno heredado en el que hay hemólisis de intensidad variable, con esferocitos y una incrementada fragilidad eritrocitaria.

Membranopatías.
Esferocitosis hereditaria.
*Autosómico dominante, recesivo o mutación
espontánea.
*Deficiencia primaria o secundaria de espectrina.
*Interacción vertical defectuosa entre
proteínas y lípidos.
*Pérdida de la capa lipídica doble.
*Esferocitos.

Membranopatías
Eliptocitosis hereditaria
*Autosómico dominante, recesiva.
Deficiencia de espectrina, deficiencia
o defecto en la banda 4.1, proteínas
integrales anormales.
*Interacciones horizontales defectuosas,
membrana inestable y permeabilidad
alterada.
* Eliptocitos

Membranopatías
Piropoiquilocitosis hereditaria
* Autosómico recesivo
* Deficiencia de a espectrina y presencia de
una espectrina mutante.
* Interacciones verticales y horizontales alteradas.
Membrana inestable.
* Esquizocitos.

Membranopatías
Acantocitosis (abetalipoproteinemia)
* Autosómico recesivo
* Incremento de esfingomielina que puede ser
secundario a una composición anormal de
lípidos de membrana.
* Expansión de la capa lipídica externa
(forma anormal), aumento de la viscosidad
de la membrana y disminución
de la fluidez / deformabilidad.
* Acantocitos

Membranopatías
Hemoglobinuria paroxística nocturna
* Adquirida
* Deficiencia del FAD (factor acelerador
del decaimiento) y de la C8bp (proteína fijadora
del complemento).
* Incremento en la sensibilidad a la lisis
por complemento.
* Anemia normocítica, microcítica o macrocítica.
Puede ser hipocrómica.

Membranopatías
Xerocitosis y Estomatocitosis
Alteraciones en la permeabilidad de la
membrana (Na+) y (K+) consumo
de ATP vida media acortada.

Membranopatías
Laboratorio
* Hb variable
* Reticulocitosis
* Basofilia difusa
* CHCM incrementada (> 36 g/dL) - eferocitosis -
* Fragilidad osmótica incrementada - esferocitosis,
eliptocitosis -
* Coombs negativo
* Autohemólisis incrementada - esferocitosis,
eliptocitosis.

Membranopatías
Laboratorio
* Médula ósea con hiperplasia eritroide, Fe de
reserva incrementado
* Resistencia térmica (45 - 460 C) -
piropoiquilocitosis -
* Apolipoproteína B, colesterol y fosfolípidos
disminuidos en acantocitosis
* HAM y sacarosa positivas en HPN
* Esferocitos, eliptocitos, estomatocitos,
xerocitos, esquizocitos.

ESFEROCITOSISLABORATORIO
• Hb: variable
• Esferocitos, células pinzadas, policromasia, eritroblastos
• DHL y Bil. Indirecta incrementadas
• Urobilinógeno urinario incrementado
• Haptoglobina disminuida
• Fragilidad a las soluciones hipotónicas aumentada, a 37o/ 24 hrs más notoria

Esferocitosis
• Morfología e índices eritrocitarios
• Esferocitos: incremento de la CMHC
• Resistencia Osmótica Eritrocitaria: disminución de la ROE directa y/o incubada
• Prueba del glicerol acidificado (+++)
• Prueba de la autohemólisis (+++)
• Ektacitometría: disminución de la deformabilidad
• Permeabilidad Iónica: aumento de la permeabilidad pasiva al sodio

ELIPTOCITOSIS HEREDITARIALABORATORIO
• Eliptocitos > 40 %
• FOE: normal en casos leves, incrementada en casos severos
• Estabilidad térmica disminuida en piropoiquilocitosis hereditaria

Esferocitosis, Laboratorio.Diagnóstico Molecular
• 1.- Electroforesis de proteínas en SDS-PAGE y análisis densitométrico de las fracciones.
• Visualización de las alteraciones cualitativas o cuantitativas de las proteínas.
• 2.- Extracción de las espectrina a 00 C y electroforesis en gel de poliacrilamida y SDS (SDS-PAGE)
• Demostración de la capacidad de la espectrina para formar tetrámeros.
• 3.- Tripsinización de la espectrina y electroforesis bidimensional .• Identificación de péptidos de espectrina• 4.- Análisis de ADN con técnica de Southern, PCR o secuenciación• Identificación de la mutación

Anemias hemolíticas
Eritroenzimopatías
*Embden - Meyerhof: Piruvato cinasa, glucosa 6 - P - isomerasa,
hexocinasa,
fosfoglicerato cinasa, triosa - P - isomerasa
* Hexosa monofosfato:Glucosa - 6 - P - deshidrogenasa, glutatión sintetasa,
glutamil - cisteína - sintetasa
* Nucleótidos:Pirimidina - 5 - nucleotidasa

LAS ALTERACIONES EN ESTA ENZIMA ESTA LIGADA AL Cr. X
AL IGUAL QUE CUALQUIER SINTESIS PROTEICA, ESTA MOLECULA
ES SUSEPTIBLE DE PRESENTAR MULTIPLES VARIANTES DEBIDAS
A LA SUSTITUCION DE AMINOACIDOS, LO CUAL MODIFICARA SUS
PROPIEDADES FISICOQUIMICAS (funcionalidad y corrimiento elec-
troforético)
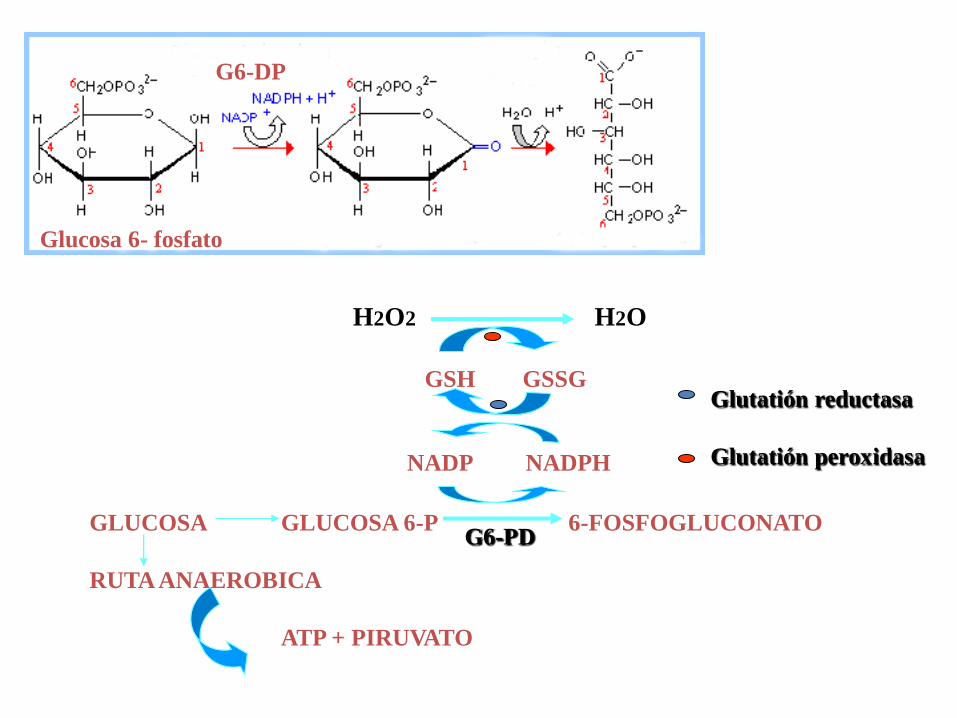
Glucosa 6- fosfato
G6-DP
GLUCOSA GLUCOSA 6-P 6-FOSFOGLUCONATO
RUTA ANAEROBICA
ATP + PIRUVATO
NADP NADPH
G6-PD
GSH GSSG
H2O2 H2O
Glutatión reductasa
Glutatión peroxidasa

SE HAN ESTABLECIDO LAS SIGUIENTES VARIANTES:
CLASE I CRISIS HEMOLITICAS SEVERAS
CLASE II CRISIS HEMOLITICAS MODERADAS
CLASE III CRISIS HEMOLITICAS LEVES
CLASE IV ACTIVIDAD NORMAL (sustitución de aa sin efecto)
CLASE V INCREMENTO DE ACTIVIDAD
CON EXCEPCION DE LA CLASE V EL EXCESO DE H2O2 CONDICIONA
LA FORMACION DE CUERPOS DE HEINZ QUE SON RECONOCIDOS
POR LOS MACROFAGOS.

Eritroenzimopatías
Glucosa - 6 - P - deshidrogenasa
Más común, ligada a cromosoma X
estrés oxidante
H2O2 ------ glutatión peroxidasa ------> H2O
GSSGH GSSG
glutatión reductasa
NADP NADPH
Glucosa
G - 6 - P G6PD 6 - fosfogluconato
Vía glucolítica
2 ATP
2 piruvatos

Eritroenzimopatías
Características
*Autosómicas recesivas
* Anemia normo - normo
* Reticulocitosis
* Hiperbilirrubinemia
* Coombs negativo
* No anormalidades morfológicas
* Policromasia
* Medición espectrofotométrica de la enzima
* Mancha fluorescente (+) G6PD y otras (NADPH)
* Punteado basófilo (P5’N)












Top Related