







Idiomas
Páginas
Jurídico


EDICIÓN 2015 1
INSTITUTO POLITÉCNICO NACIONAL
ESCUELA NACIONAL DE CIENCIAS
BIOLÓGICAS
DEPARTAMENTO DE QUÍMICA ORGÁNICA
ACADEMIA DE Q.B.P
MANUAL DE PRÁCTICAS DE
QUÍMICA ORGÁNICA
EDICIÓN 2015

EDICIÓN 2015 2
MANUAL DE PRÁCTICAS
DE QUÍMICA ORGÁNICA
QUÍMICO BACTERIÓLOGO PARASITÓLOGO
EDICIÓN 2015

EDICIÓN 2015 3
CALENDARIO DE PRÁCTICAS DE QUÍMICA ORGÁNICA QUÍMICO BACTERIÓLOGO PARASITÓLOGO
Introducción al laboratorio de química orgánica
1. Destilación simple y destilación fraccionada
2. Destilación por arrastre de vapor
3. Separación de una mezcla ternaria por destilación
E X A M E N
4. Recristalización
5. Extracción líquido – líquido
6. Cromatografía
E X A M E N
7. Síntesis a microescala de ácido fumárico
8. Síntesis de ciclohexeno
9. Síntesis de cloruro de ter-butilo y Cinética química: efecto del disolvente en
la velocidad de una reacción de solvólisis.
E X A M E N
10. Síntesis de orto y para-nitrobromobenceno
11. Transposición bencílica.
E X A M E N

EDICIÓN 2015 4
TEMARIO SEGÚN PLAN DE ESTUDIOS DE LA ASIGNATURA DE QUÍMICA ORGÁNICA PARA LA CARRERA DE QUÍMICO BACTERIÓLOGO PARASITÓLOGO. Fundamentación: Requiere, conocimientos de química general y se correlaciona con fisicoquímica y química bioorgánica, brindando a su vez sustento para asignaturas como bioquímica general, fisiología humana, microbiología general, genética microbiana y biología molecular, entre otras. Objetivos Generales: Enseñar los conceptos básicos de Química Orgánica, aplicar dichos conceptos en el estudio de diferentes tipos de reacción. Aprender y adquirir habilidad para la solución de problemas en química orgánica. Relacionar los conocimientos adquiridos con otras áreas afines. Desarrollar hábitos de estudio. I.- INTRODUCCIÓN (18 HRS. Teoría) Objetivo Particular: Resaltar la importancia de la Química Orgánica en todos los aspectos de la vida cotidiana, sus relación con otras áreas de la carrera y a la práctica profesional, estudiar el carácter especial del carbono, aprender la estructura de diferentes grupos funcionales, aplicar las reglas IUPAC y trivial en la nomenclatura. 1. Desarrollo histórico e importancia
de la Química Orgánica 1.1. La Función social del profesional
de la química 1.2. Estructura Atómica características
del átomo de carbono en la formación de compuestos orgánicos.
1.3. Grupos Funcionales. Alcanos, Alquenos, Alquinos, Álenos, Árenos, Alcoholes, Fenoles, Éteres, Anhídridos, Óxidos, Furanos, Píranos, Amidas, Anilinas, Amidas, Imidas, Iminas, Nitrilos, Enaminas, Isonitrilos, Aminas heterocíclicas, Halogenuros de alquilo, Mercaptanos , Sulfuros, Disulfuros, Sulfóxidos, Sulfonas, Ácidos Sulfónicos, Peróxidos, Ozónidos, Polisulfuros, Oximas , Hidrazinas, Azidas, Azo compuestos, Diazo compuestos, Isocianatos, Cianatos, Tiocianatos, Ureas, Imidatos, Semicarbazidas, O y N carbamatos, Nitrocompuestos, C y N nitroso compuestos.
1.4. Nomenclatura Sistemática y Trivial
II.- PROPIEDADES DEL ENLACE QUÍMICO (22 HRS. Teoría)
(Objetivo Particular reafirmar y aplicar los conceptos del curso de Química General, relacionados al tema de enlace.) 2. Enlace Químico 2.1. Teoría de enlace por electrones de
valencia (TEEV) 2.2. Teoría de enlace por orbital
molecular 2.2.1. Hibridación de Orbitales 2.2.2. Diagramas de energía y formación
de Orbitales Moleculares. 2.2.3. Teoría de enlace por Orbitales
Frontera 2.3. Propiedades de los Orbitales
Moleculares. 2.3.1. Carga Formal. 2.3.2. Enlace Iónico, Enlace covalente,
Longitud de enlace, Ángulo de enlace, Momento Dipolar, Polarizabilidad, Entalpía de disociación, Potencial de ionización, Electroafinidad, Electronegatividad, Resonancia e Hiperconjugación
2.3.4 Fuerzas Moleculares, Fuerzas electrostáticas, ión-dipolo, dipolo-dipolo, Dipolo inducido-Dipolo.
2.3.5 Puente de hidrógeno 2.3.6 Complejos de transferencia de
carga 2.3.7 Propiedades Físicas dependientes
de las interacciones moleculares. Solubilidad, punto de fusión.
2.3.8 Métodos de aislamiento y purificación de compuestos orgánicos.
III.- PROPIEDADES MOLECULARES (26 HRS. Teoría) Objetivo Particular: Aprender y comprender la caracterización de las propiedades moleculares del los compuestos orgánicos, estudiar los

EDICIÓN 2015 5
fundamentos del análisis espectroscópico, propiedades conformacionales, configuracionales y sus efectos relacionados. 3. Propiedades Moleculares. 3.1 Constitución Molecular, Fórmula
mínima, Análisis Elemental, Determinación de la masa molecular
3.2 Conectividad Molecular, Isomerismo Estructural, Fórmula Molecular, Fórmulas Desarrolladas,
3.3 Configuración Molecular, Elementos de Simetría, Elementos de Quiralidad, Centro Asimétrico, Estereoisómeros, Diastereómeros y Enantiómeros, Descriptores Estereoquímicos, Fórmulas de Proyección
3.4 Conformación Molecular, Determinación de la Conformación Molecular, Análisis Conformacional del Etano, Análisis Conformacional del Butano, Análisis conformacional del Ciclohexano, Análisis Conformacional de Ciclohexanos monosustituidos, Confórmeros axiales y ecuatoriales.
IV.- REACCIONES QUIMICAS (24 HRS. Teoría) Objetivo Particular: Analizar y entender los diferentes tipos de ruptura de enlace, introducción a los mecanismos de reacción y sus métodos de estudio mediante el análisis de los principales tipos de reacción de los compuestos orgánicos.) 4. Propiedades de las Reacciones
Químicas 4.1 Termodinámica de las reacciones,
Entalpía de reacción, La función de Gibbs y la constante de equilibrio, reacciones reversible e irreversibles.
4.2 Comportamiento ácido-base, ácidos y bases conjugados, fuerza relativa de acidez (pKa), efectos inductivos y resonantes sobre la acidez de los enlaces C-H y O-H
4.3 Nucleófilos y Electrófilos, Nucleófilos y Electrófilos Duros y Blandos, Principios de reactividad entre Nucleófilos y Electrófilos.
4.4 Cinética de reacción, Velocidad de reacción, Orden de reacción, Molecularidad de reacción, Energía de Activación, Teoría del complejo activado.
4.5 Reactividad y Selectividad, Control Cinético y Control Termodinámico de la reacción
4.6 Mecanismo de Reacción, Tipos de ruptura de enlace, intermediarios de reacción
4.7 Evidencias Cinéticas, efecto isotópico
4.8 Evidencias no cinéticas, Reacciones cruzadas, Atrapamiento de intermediarios, Estereoquímica de la reacción
4.9 Tipos de Mecanismos de reacción,
4.10 Mecanismos de Eliminación, Eliminación unimolecular (E1) y bimolecular ( E2 )
4.11 Mecanismos de Sustitución, Sustitución Nucleofílica Unimolecular (SN1), Sustitución Nucleofílica Bimolecular (SN2) y Sustitución Electrofílica Aromática (SNAr).
4.12 Mecanismos de Transposición 4.13 Mecanismos de Oxido-reducción V.- REACCIONES DE ELIMINACION (5 HRS. Teoría) (Objetivo Particular: Analizar los diferentes mecanismos de eliminación con sus variables, estudiar las implicaciones estereoquímicas y factores que orientan la eliminación) 5.1 Vía iónica, Deshidratación de
alcoholes, Deshidrohalogenación. 5.2 Concertadas, Deshalogenación,
Eliminación de Hofmann, Pirólisis de esteres. Eliminación de Cope.
5.3 Aplicaciones sintéticas. VI.- REACCIONES DE SUSTITUCION (19 HRS. Teoría) Objetivo Particular; Estudio de los diferentes tipos de sustitución y las implicaciones estereoquímicas y factores que orientan la reacción. 6. Sustitución vía radicales libres,
halogenación de alcanos 6.1. Sustitución nucleofílica,
unimolecular (SN1), sustitución nucleofílica bimolecular (SN2),
Sustitución nucleofílica intramolecular, Sustitución nucleofílica con transposición, Formación de halogenuros de

EDICIÓN 2015 6
alquilo, alcoholes, esteres, éteres, aminas, ácidos carboxílicos, alcanos.
6.2. Sustitución Electrofílica Aromática ( SEAr), en mono y policiclos
6.3. Nitración, Halogenación, Sulfonación, Alquilacion y Acilación.
6.4. Efectos de orientación y reactividad. 6.5. Sustitución Nucleofílica
Aromática (SNAr), concertada (clásica), vía sales de diazonio, vía bencino, aplicaciones sintéticas.
VII.- REACCIONES DE OXIDO-REDUCCION.
(6 HRS. Teoría) Objetivo Particular: Aplicar el concepto de
óxido-reducción en el cálculo del número de oxidación y su aplicación en el balanceo de reacciones orgánicas.
7. Reacciones De óxido-reducción
7.1 Número de oxidación 7.2 Balanceo de ecuaciones 7.3 Reacciones de oxidación de
diferentes tipos de compuestos orgánicos.
VIII.- REACCIONES DE TRANSPOSICION (4 HRS. Teoría) Objetivo Particular: Analizar algunas reacciones de transposición que implican migraciones sigmatrópicas 1,2 así como sus implicaciones estereoquímicas. 8. Reacciones de Transposición. 8.1 Transposición de Wagner-
Meerwein 8.2 Transposición Pinacólica 8.3 Transposición de Hofmann. 8.4 Transposiciones de Curtius,
Schmidth, Losen. 8.5 Transposición de Beckmann 8.6 Aplicaciones sintéticas.

EDICIÓN 2015 7
REGLAMENTO INTERNO PARA LOS LABORATORIOS DEL DEPARTAMENTO DE QUÍMICA ORGÁNICA
I. GENERALIDADES. 1. Las disposiciones de este reglamento regirán todas las actividades de los
laboratorios del Departamento de Química Orgánica y serán obligatorias para los alumnos que cursen cualquier asignatura del departamento.
2. Los alumnos que deseen cursar el laboratorio, deberán reunir los requisitos que marca la E.N.C.B. así como los estipulados en el presente reglamento:
a) No será permitida la estancia a los alumnos que no porten bata y lentes de seguridad (los lentes serán proporcionados al inicio de cada práctica y su uso y cuidado serán responsabilidad de los alumnos; los lentes serán devueltos al termino de cada práctica )
b) Los alumnos que no mantengan el comportamiento adecuado en el laboratorio no podrán permanecer en él.
c) Para abandonar temporalmente el laboratorio durante el desarrollo de la práctica, se deberá solicitar el permiso correspondiente al profesor.
d) Al concluir la práctica, los alumnos deberán dejar completamente limpio su lugar
de trabajo y las áreas comunes como las campanas de extracción. 3. No se aceptarán alumnos condicionales.
4. Los alumnos a los que se les hayan autorizado baja en el curso, deberán presentar la constancia correspondiente; de no hacerlo, el curso se considerará reprobado.
II. ORGANIZACIÓN. 1. La hora de entrada será la indicada en el horario de cada grupo, dándose una
tolerancia máxima de 15 minutos, después de los cuales se pasará lista y no se permitirá la entrada al laboratorio. No habrá retardos.
2. El trabajo de laboratorio se realizará en el sitio indicado por el profesor. 2. Se formarán equipos de trabajo en el laboratorio, los cuales serán de dos o tres
alumnos según las características del grupo, siendo permanentes durante todo el curso.
4. El total de equipos formados por grupo, serán divididos en dos o tres secciones. 5. La sesión de laboratorio iniciará con un seminario, en donde se tomarán los puntos
teóricos necesarios, así como, las indicaciones de seguridad y de trabajo para la buena realización de la práctica; posteriormente se desarrollará la parte experimental de la misma.

EDICIÓN 2015 8
6. Cada equipo contará con la cantidad necesaria de reactivos para la realización de la práctica. No habrá reposición de los mismos, en caso de pérdida o accidente.
7. Cada equipo hará un vale al almacén, por el material que se requiera en la práctica,
debiendo hacer una revisión exhaustiva del mismo en el momento de recibirlo y reportando cualquier anomalía al almacenista antes de entregar el vale.
8. En caso de ruptura o pérdida del material, se dará un plazo máximo de 15 días
para reponerlo; de no hacerlo oportunamente, no se permitirá la realización de prácticas, las cuales serán calificadas con CERO. Si al final del semestre hay adeudo de material, la calificación del curso será reprobatoria.
9. Todo asunto relacionado con el material, se deberá tratar directamente con el
almacenista. 10. Cada equipo deberá traer el siguiente material: cerillos, detergente, escobillones,
franela, jerga, vaselina sólida, papel absorbente, aceite, perilla de seguridad o jeringa y espátula.
11. Al final de la práctica se entregará el material limpio; de no ser así, no será recibido
por el almacenista. 12. El material roto o restos de material quedará en poder del almacenista y será
destruido en presencia del alumno en el momento en que éste lo reponga en el almacén.
III. EVALUACIÓN.
1. Para acreditar el curso teórico, el alumno deberá aprobar el curso práctico, para lo cual requerirá:
a) Un mínimo de 80% de asistencias. b) Calificación final mínima de seis. c) No adeudar material
2. La evaluación del curso práctico se hará en la forma siguiente: a) Se realizarán cuatro exámenes parciales. No habrá examen final ni reposición.
b) La calificación promedio de los seminarios, contará como un 2º examen parcial y el promedio de calificaciones del trabajo de laboratorio, como un 3º examen parcial y se realizará un promedio de los reportes como un 4° examen.
La calificación final del curso de laboratorio, será el promedio de estas 4 evaluaciones:
CF. Calificación final PE. Promedio de los exámenes parciales PS. Promedio de calificaciones de seminarios PTL. Promedio de calificaciones del trabajo de laboratorio PR. Promedio de Reportes
PE+PR+PTL+PS
4= CF
EDICIÓN 2015 9
INSTITUTO POLITÉCNICO NACIONAL ESCUELA NACIONAL DE CIENCIAS BIOLÓGICAS
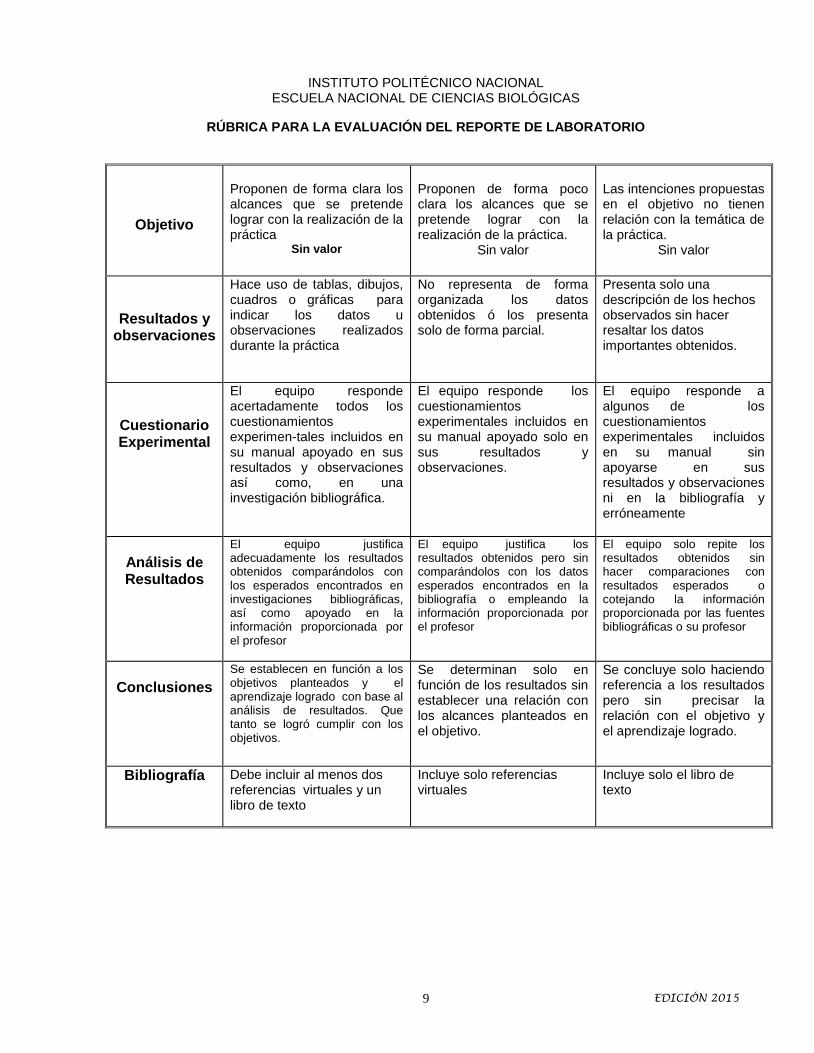
RÚBRICA PARA LA EVALUACIÓN DEL REPORTE DE LABORATORIO
Objetivo
Proponen de forma clara los alcances que se pretende lograr con la realización de la práctica
Sin valor
Proponen de forma poco clara los alcances que se pretende lograr con la realización de la práctica.
Sin valor
Las intenciones propuestas en el objetivo no tienen relación con la temática de la práctica.
Sin valor
Resultados y observaciones
Hace uso de tablas, dibujos, cuadros o gráficas para indicar los datos u observaciones realizados durante la práctica
No representa de forma organizada los datos obtenidos ó los presenta solo de forma parcial.
Presenta solo una descripción de los hechos observados sin hacer resaltar los datos importantes obtenidos.
Cuestionario Experimental
El equipo responde acertadamente todos los cuestionamientos experimen-tales incluidos en su manual apoyado en sus resultados y observaciones así como, en una investigación bibliográfica.
El equipo responde los cuestionamientos experimentales incluidos en su manual apoyado solo en sus resultados y observaciones.
El equipo responde a algunos de los cuestionamientos experimentales incluidos en su manual sin apoyarse en sus resultados y observaciones ni en la bibliografía y erróneamente
Análisis de Resultados
El equipo justifica adecuadamente los resultados obtenidos comparándolos con los esperados encontrados en investigaciones bibliográficas, así como apoyado en la información proporcionada por el profesor
El equipo justifica los resultados obtenidos pero sin comparándolos con los datos esperados encontrados en la bibliografía o empleando la información proporcionada por el profesor
El equipo solo repite los resultados obtenidos sin hacer comparaciones con resultados esperados o cotejando la información proporcionada por las fuentes bibliográficas o su profesor
Conclusiones
Se establecen en función a los objetivos planteados y el aprendizaje logrado con base al análisis de resultados. Que tanto se logró cumplir con los objetivos.
Se determinan solo en función de los resultados sin establecer una relación con los alcances planteados en el objetivo.
Se concluye solo haciendo referencia a los resultados pero sin precisar la relación con el objetivo y el aprendizaje logrado.
Bibliografía Debe incluir al menos dos referencias virtuales y un libro de texto
Incluye solo referencias virtuales
Incluye solo el libro de texto

EDICIÓN 2015 10
RUBRICA DE EVALUACIÓN DE LAS EXPOSICIONES ORALES
Aspecto a evaluar
Insuficiente
Deficiente Satisfactorio
Bueno
Excelente
Calif. Obs.
EXPRESIÓN ORAL
Intensidad de voz Ninguno de los integrantes expone con un volumen
adecuado
Uno de los integrantes expone con un volumen
adecuado
Dos de los integrantes expone con un volumen
adecuado
Tres de los integrantes expone con un volumen
adecuado
Todos los integrantes expone con un volumen
adecuado
Uso de muletillas Las emplean todos los integrantes
Las emplean tres de los integrantes
Las emplean dos de los integrantes
Las emplea uno de los integrantes
Ninguno de los integrantes las utilizan
No leer Todos los integrantes leen el contenido de la información en
las diapositivas
Tres de los integrantes leen el contenido de la
información en las diapositivas
Dos de los integrantes leen el contenido de la
información en las diapositivas
Un integrantes leen el contenido de la
información en las diapositivas
Ninguno de los integrantes leen el contenido de la
información en las diapositivas
Uso de lenguaje técnico apropiado
Ninguno de los integrantes hace uso de él
Uno de los integrantes hace uso de él
Dos de los integrantes hace uso de él
Tres de los integrantes hace uso de él
Todos los integrantes hace uso de él
Respeto al tiempo asignado de exposición
Rebasa el tiempo asignado por más de 15 minutos
Rebasa el tiempo asignado por más de 10
minutos
Rebasa el tiempo asignado por más de 7
minutos
Rebasa el tiempo asignado por más de 5
minutos
Cumple con exactitud el tiempo asignado
EN LA PRESENTACIÓN POWER POINT
Título de la exposición No la tiene Es inapropiado Es inadecuado sin embargo da idea
apropiada de lo que se pretende exponer
Es adecuado sin embargo es susceptible de mejora para hacerlo
más preciso
Es correcto
Nombre de los alumnos, en orden alfabético por
apellido
No lo tiene Contiene solo los nombres propios de los
estudiantes
Contiene los nombres y un solo apellido y sin
orden alfabético
Contiene los nombres y un solo apellido y en
orden alfabético
Contiene los nombres completos y en orden
alfabético
Numero de equipo No lo tiene Lo tiene
Materia No lo tiene Lo tiene
Grupo No lo tiene Lo tiene
Semestre No lo tiene Lo tiene
Citar fuentes en casos de cuadro y graficas
No contiene citas Cita incorrectamente en algunas ocasiones
Cita incorrectamente en algunos casos
Cita correctamente pero no en todos los casos
Cita correctamente en todos los casos
Las letras no se pierdan con el fondo de la
presentación
No cumple Cumple
Ortografía y escritura correcta de las sustancias
empleadas
Más de 5 errores 5 de errores 4 errores 2 errores Sin errores
Mecanismo de reacción No se incluye o se hace de forma incorrecta
Se incluye en más de una diapositiva y está
incompleto
No se incluyen todos los pasos importantes
Se incluye en más de una diapositiva y
contiene todos los pasos importantes
Se incluye en una sola diapositiva y contiene todos los pasos importantes
Justificación de cada uno de los pasos de la técnica
aplicada
No se incluye o se menciona de forma incorrecta
Se incluye la técnica pero no se justifica el por qué
de los pasos
Se incluye la técnica aplicada pero existen muchos errores en la
justificación de los mismos
Se incluye una breve explicación de los pasos aplicados en la síntesis
Se explica las razones de cada uno de los pasos
incluidos en la técnica de síntesis
Bibliografía Sólo se incluyen direcciones de páginas Web
La tiene de forma incompleta y no se ajusta
al estilo Vancouver
La tiene pero no se ajusta al estilo
Vancouver
La tiene de forma incompleta pero se
ajusta al estilo Vancouver
La contiene de forma completa y se ajusta al
estilo Vancouver

EDICIÓN 2015 11
RÚBRICA PARA EVALUAR UN DIAGRAMA DE FLUJO
CRITERIO
NIVEL DE DESEMPEÑO
BUENO
REGULAR
INSUFICIENTE
CONSTRUCCIÓN
Hay inicio, entrada de
información, preguntas,
actividades,
condiciones, flechas
y final
Falta uno de los
elementos
antes mencionados
Faltan 2 o más de los
elementos antes
mencionados
PROFUNDIZACIÓN
La información es
suficiente,
adecuada y pertinente a
cualquier situación
La información es
insuficiente, es menos
de la
mitad de la requerida
La información no es
suficiente y es
inadecuada
JERARQUIZACIÓN
La información está
bien
jerarquizada de tal
manera que
disipa toda duda del
proceso
La información falla en
dos o
tres jerarquizaciones,
causa
confusión
La información está mal
jerarquizada causa
dudas
ANÁLISIS
La toma de decisiones,
las
actividades, las
condiciones y las
flechas se relacionan
perfectamente
La toma de decisiones,
las
actividades, las
condiciones y
las flechas llegan a fallar
en
tres cinco ocasiones
La toma de decisiones,
las
actividades, las
condiciones
y las flechas no están
bien
relacionadas
SOLUCIÓN DEL
PROBLEMA
El planteamiento es
adecuado y
ofrece muchas
expectativas de
trabajo académico
El planteamiento tiene
pocos
errores, ofreciendo
pocas
expectativas de trabajo
académico
El planteamiento es
inadecuado
No ofrece expectativas
CREATIVIDAD Tiene colorido y la
información
está distribuida
adecuadamente
Falta color y espacios Es simple y todo
encimado

EDICIÓN 2015 12
SIMBOLOGÍA

EDICIÓN 2015 13
INTRODUCCIÓN AL LABORATORIO DE QUÍMICA ORGÁNICA
TRABAJO EN EL LABORATORIO DE QUÍMICA ORGÁNICA
OBJETIVO.
Conocer las normas y la metodología requeridas para el desempeño de las
actividades que se realizan en el laboratorio.
Desarrollar un criterio que le permita usar y comprender las operaciones y
procesos comunes de la Química Orgánica, así mismo, conocer las limitaciones
y riesgos que conlleva dicho trabajo.
Conocer el material de laboratorio, el equipo de vidrio, el manejo de los reactivos
y el montaje de aparatos a utilizar durante la realización de las prácticas.
Aprender a buscar información y a registrar las observaciones de manera
metódica, precisa, completa y reproducible.
INTRODUCCIÓN.
La Química Orgánica es una materia experimental, por lo que se requiere de disciplina y
metodología para la obtención de resultados confiables, así como de la aplicación de
las normas de seguridad apropiadas para evitar accidentes. La realización de este
trabajo implica el diseño experimental, la interpretación de resultados y el registro de
éstos.
NORMAS DE TRABAJO.
Procedimientos de operación en el Laboratorio de Química Orgánica.
El laboratorio de Química Orgánica es una área de alto riesgo, por lo cual
cualquier estudiante que sea sorprendido comportándose de manera inapropiada
y no observe las normas indicadas será dado de baja de la materia.
Actitud y Preparación.
El trabajo de laboratorio demanda del estudiante una actitud crítica, inquisitiva y una
cooperación ilimitada. Para lograr lo anterior es necesaria una participación activa en la
observación de las normas de trabajo que se han establecido para evitar accidentes y
así lograr un alto rendimiento en el trabajo de experimental.
Antes de realizar cualquier experimento, se deberán revisar los antecedentes teóricos
de la reacción a efectuar, el mecanismo de reacción, los fundamentos fisicoquímicos así
como los problemas de seguridad involucrados en el manejo de los reactivos.
La lectura previa y la comprensión de las indicaciones del experimento, permitirán que
el curso y el desarrollo de la práctica sean claros en todos sus detalles. Al ingresar al
laboratorio se deberá estar preparado físicamente; no hacer el trabajo de laboratorio

EDICIÓN 2015 14
con el estómago vacío o sin dormir. Se deberá llegar puntualmente ya que sólo se
permiten 15 minutos de tolerancia y se deberá estar preparado mentalmente para
estudiar el experimento y planear las actividades.
Seguridad y Normas de Trabajo para el Laboratorio de Química Orgánica.
Los reactivos usados en el laboratorio se convierten en un peligro cuando no se
manejan con cuidado, pero son inocuos cuando se manipulan precavidamente.
Se deberá usar bata para el trabajo de laboratorio la cual deberá estar siempre
protegiendo todo el cuerpo y deberá mantenerse limpia.
Se deberá usar ropa cómoda, incluyendo zapatos que sean confortables y que
permitan desplazarse rápidamente en caso de emergencia. El cabello deberá estar
recogido de manera que no obstruya la visión o que cuelgue sobre los matraces
de reacción. No se permite usar calzado o ropa que dejen al descubierto el pie y
las piernas.
El uso de lentes de seguridad es obligatorio siempre que se permanezca en el
laboratorio, independientemente de manejar los reactivos o no. Los lentes protegen de
proyecciones e impactos en caso de accidente y es necesario mantenerlos limpios y
desempañados. En caso de usar lentes de contacto se deberá usar lentes de seguridad
sellados con protecciones laterales.
No está permitido introducir alimentos ni comer, beber o fumar dentro del
laboratorio
Los compuestos orgánicos pueden absorberse por la piel, por lo que se deben evitar
derramamientos sobre ésta y se evitará el contacto de los reactivos directamente con
las manos. No se deben succionar los líquidos con la boca, se deberá emplear una
perilla de seguridad de acuerdo al procedimiento indicado en la figura 1.

EDICIÓN 2015 15
FIGURA 1.
Para protegerse de la absorción de productos químicos por la piel se deberán usar
guantes desechables de látex ó de polipropileno, manteniéndolos siempre limpios.
En caso de requerir oler algún reactivo, se debe atraer un poco de sus vapores pasando
rápidamente la mano por la boca del frasco de acuerdo a la Figura 2. Para evitar la
inhalación de vapores se deberá calentar o evaporar la mezcla de reacción dentro de la
campana de extracción. No se debe oler el reactivo directamente del frasco.
FIGURA 2.

EDICIÓN 2015 16
El Ambiente de trabajo.
Mantener la mesa de trabajo ordenada y limpia, sin productos o con agua derramados
sobre ésta. En caso de derrames se deberá limpiar rápidamente el lugar utilizando
papel absorbente, si el material es volátil se deberá colocar en la campana de
extracción.
Si se derrama un ácido concentrado sobre la mesa se deberá utilizar una solución de
bicarbonato de sodio para neutralizarlo, si es una base la que se ha derramado se
deberá utilizar ácido acético diluido.
Se deberán mantener limpias y ordenadas las áreas comunes, las áreas de
pesado de reactivos y las balanzas.
No contaminar los reactivos con espátulas o pipetas que tengan restos de otros
reactivos.
Material de vidrio.
No usar material de vidrio roto o en mal estado, revisar el material antes de utilizarlo.
Utilizar material de vidrio limpio y seco. No utilizar el termómetro como agitador.
Identificar cada uno de los materiales de vidrio por su nombre (Figura 3).
Muchos compuestos son inflamables y pueden producir fuego a altas temperaturas por
lo cual el trabajo con mecheros u otra flama abierta se realizara dentro del periodo de
laboratorio y bajo la supervisión del profesor. NO SE DEBE DEJAR EL MECHERO
ENCENDIDO SIN USO ALGUNO, EL MECHERO SE PRENDE CUANDO SE INICIA
EL CALENTAMIENTO Y SE APAGA CUANDO ÉSTE TERMINA, NO SE DEBE
DEJAR EL MECHERO ENCENDIDO SIN VIGILANCIA.
NO SE PERMITE NINGÚN EXPERIMENTO NO AUTORIZADO POR EL MAESTRO.

EDICIÓN 2015 17
FIGURA 3.
En caso de encender una flama buscar el lugar más retirado del material combustible.
Al usar el mechero Bunsen revisar que no haya fugas y que ninguna de las
pertenencias pueda ser alcanzada por el fuego (corbata, cabello o ropa).
Nunca evaporar materiales inflamables calentando con la flama del mechero.
Está estrictamente prohibido calentar un sistema cerrado, ya que éste puede ser
causa de una proyección que puede convertirse en explosión.

EDICIÓN 2015 18
En caso de producirse fuego, tener identificadas las ubicaciones de los extinguidores,
los botes de arena, y el material de auxilio, así como la salida más próxima.
Al calentar con baño de aceite, revisar que el recipiente donde se encuentra el aceite
esté totalmente seco ya que la presencia de agua provoca proyecciones de aceite
caliente.
El fuego de un tubo de ensaye o matraz puede sofocarse con un vidrio de reloj, con el
extintor o con arena.
En caso de fuego en la ropa en una persona, cubrirlo con una manta y evitar correr.
LOS DESECHOS SE COLOCARÁN EN LOS LUGARES DESTINADOS A ESTE FIN.
COLOCAR EL PAPEL Y LA BASURA EN LOS RECIPIENTES APROPIADOS, NO
TIRAR NINGÚN REACTIVO O DESECHO QUÍMICO EN LAS TARJAS.
En casos de tener alguna condición física que pueda afectar tu rendimiento o tu salud,
como alergias, embarazo, epilepsia, etc. informar al profesor; dicha información será
totalmente confidencial. En caso de accidente informar inmediatamente al profesor.
Desarrollo de la Práctica.
El trabajo de laboratorio no empieza en el momento que se entra al laboratorio, por el contrario, previamente se ha de realizar una investigación bibliográfica que cubra los siguientes aspectos:
Datos físicos de cada uno de los reactivos que se usen, punto de fusión, punto
de ebullición, solubilidad, etc.
Datos toxicológicos, precauciones relacionadas con el manejo de cada uno de
los reactivos.
Datos complementarios. Fundamentos fisicoquímicos, reacciones y mecanismos
de reacción involucrados en el desarrollo de la práctica, ecuación química
balanceada, e identificación del reactivo limitante. Productos y subproductos
esperados y precauciones que hay que considerar para el desarrollo exitoso de
la práctica.
Seminario.
El propósito del seminario es aclarar cualquier aspecto de la práctica que no esté
comprendido, por lo que se requiere de la participación de todos los estudiantes.

EDICIÓN 2015 19
Informe de resultados.
El profesor indicará las características que deberá contener cada informe.
1. Los alumnos deberán consultar y completar las fórmulas, las propiedades físicas, las propiedades químicas y la toxicidad de las sustancias a emplear en las prácticas.
1. Acetanilida.
2. Acetato de etilo.
3. Acetato cúprico.
4. Acetona.
5. Agua.
6. Ácido acético glacial.
7. Ácido benzoico.
8. Ácido clorhídrico.
9. Ácido fosfórico.
10. Ácido fumárico.
11. Ácido maleico.
12. Ácido nítrico.
13. Ácido oxálico.
14. Ácido pícrico.
15. Ácido salicílico.
16. Ácido sulfúrico.
17. Alcohol isoamílico.
18. Alcohol terbutílico.
19. Anilina.
20. Azul de metileno.
21. Benceno.
22. Bencilo.
23. Benzaldehido.
24. Benzoína.
25. Bicarbonato de sodio.
26. Bromo.
27. Carbón activado.
28. Carbonato de sodio.
29. Ciclohexanol.
30. Ciclohexeno.
31. Cloroformo.
32. Cloruro de calcio.
33. Cloruro de metileno.
34. Cloruro de sodio.
35. Cloruro de terbutilo.
36. Dibenzalacetona.
37. Etanol.
38. Eter etílico.
39. Fenol.
40. Fenolftaleína.
41. Glicerol.
42. Hexano.
43. Hidróxido de potasio.
44. Hidróxido de sodio.
45. Metanol.
46. Nitrato de amonio.
47. o-Nitrofenol.
48. p-Nitroacetanilida.
49. p-Nitrofenol.
50. Sacarosa.
51. Sulfato de sodio anhidro.
52. Tetracloruro de carbono.
53. Tolueno.
54. Yodo.

EDICIÓN 2015 20
2. Con la lista anterior, completa la tabla siguiente:
Disolventes Agentes Desecantes Reactivos Orgánicos Reactivos
Inorgánicos

EDICIÓN 2015 21
3. En conjunto los alumnos con ayuda de los profesores, definirán cada uno de los
siguientes términos:
Reactivos. ____________________________________________________________________________________________________________________________________________
Productos. ____________________________________________________________________________________________________________________________________________
Qué indica el subíndice en una reacción química. ____________________________________________________________________________________________________________________________________________
Qué indica el coeficiente en una reacción química. ____________________________________________________________________________________________________________________________________________
Peso molecular. ____________________________________________________________________________________________________________________________________________
Peso atómico. ____________________________________________________________________________________________________________________________________________
Reactivo en exceso. ____________________________________________________________________________________________________________________________________________
Reactivo limitante. ____________________________________________________________________________________________________________________________________________
4. Cálculos estequiométricos: El profesor explicará cómo se calcula la eficiencia, el
rendimiento teórico y práctico de reacción para diferentes tipos de reacciones,
así como la eficiencia.

EDICIÓN 2015 22
Etiquetado de seguridad de productos químicos. La etiqueta es, en general, la primera información que recibe el usuario y es la que permite identificar el producto en el momento de su utilización. Todo recipiente que contenga un producto químico deberá llevar una etiqueta visible en su envase que, contenga:
Nombre de la sustancia o del preparado.
Fecha de preparación u obtención.
Nombre de la persona o equipo que lo preparó, grupo y sección.
Símbolos e indicaciones de peligro para destacar los riesgos principales. Para manejar con seguridad las sustancias químicas se han ideado diversos códigos y pictogramas dependiendo de la casa fabricante. A continuación se muestra uno de los más usados. (Azul) (Rojo)
(Blanco) (Amarillo)
FIGURA 4.
Algunos de los pictogramas de peligro más utilizados se muestran a continuación en la siguiente Tabla

EDICIÓN 2015 23
TABLA 1. Pictogramas de peligrosidad.
E Explosivo
Clasificación: Sustancias y preparaciones que reaccionan exotérmicamente también sin oxígeno y que detonan según condiciones de ensayo fijadas, pueden explotar al calentar bajo inclusión parcial. Ejemplo: dicromato de amonio. Precaución: Evitar el choque, Percusión, Fricción, formación de chispas, fuego y acción del calor.
F Fácilmente inflamable
Clasificación: Líquidos con un punto de inflamación inferior a 21ºC, pero que NO son altamente inflamables. Sustancias sólidas y preparaciones que por acción breve de una fuente de inflamación pueden inflamarse fácilmente y luego pueden continuar quemándose o permanecer incandescentes. Precaución: Mantener lejos de llamas abiertas, chispas y fuentes de calor. A. Sustancias autoinflamables. Ejemplo: alquilos de aluminio, fósforo. Precaución. Evitar contacto con el aire B. Gases fácilmente inflamables. Ejemplo: butano, propano. Precaución. Evitar la formación de mezclas inflamables gas-aire y aislar de fuentes de ignición. C. Sustancias sensibles a la humedad. Productos químicos que desarrollan emanaciones de gas inflamable al contacto con el agua. Ejemplo: litio, borohidruro de sodio. Precauciones: evitar contacto con agua o con humedad.
F+ Extremadamente
inflamable
Clasificación: Líquidos con un punto de inflamación inferior a 0ºC y un punto de ebullición de máximo de 35ºC. Gases y mezclas de gases, que a presión normal y a temperatura usual son inflamables en el aire. Precaución: Mantener lejos de llamas abiertas, chispas y fuentes de calor.
C Corrosivo
Clasificación: Destrucción del tejido cutáneo en todo su espesor en el caso de piel sana, intacta. Ejemplo: bromo, ácido sulfúrico. Precaución: Mediante medidas protectoras especiales evitar el contacto con los ojos, piel e indumentaria. NO inhalar los vapores. En caso de accidente o malestar consultar inmediatamente al médico.
T Tóxico
Clasificación: La inhalación y la ingestión o absorción cutánea en pequeña cantidad, pueden conducir a daños para la salud de magnitud considerable, eventualmente con consecuencias mortales. Ejemplo: trióxido de arsénico, cloruro de mercurio (II). Precaución: evitar cualquier contacto con el cuerpo humano. En caso de malestar consultar inmediatamente al médico. En caso de manipulación de estas sustancias deben establecerse procedimientos específicos.
EDICIÓN 2015 24
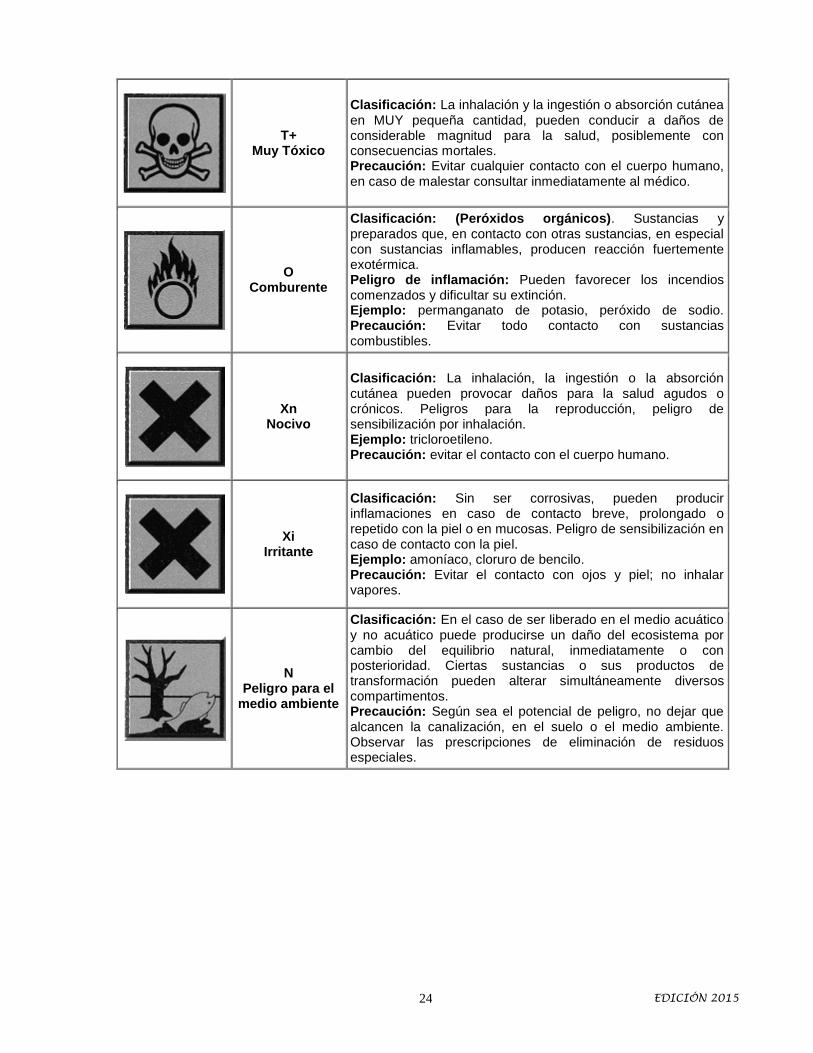
T+ Muy Tóxico
Clasificación: La inhalación y la ingestión o absorción cutánea en MUY pequeña cantidad, pueden conducir a daños de considerable magnitud para la salud, posiblemente con consecuencias mortales. Precaución: Evitar cualquier contacto con el cuerpo humano, en caso de malestar consultar inmediatamente al médico.
O Comburente
Clasificación: (Peróxidos orgánicos). Sustancias y preparados que, en contacto con otras sustancias, en especial con sustancias inflamables, producen reacción fuertemente exotérmica. Peligro de inflamación: Pueden favorecer los incendios comenzados y dificultar su extinción. Ejemplo: permanganato de potasio, peróxido de sodio. Precaución: Evitar todo contacto con sustancias combustibles.
Xn Nocivo
Clasificación: La inhalación, la ingestión o la absorción cutánea pueden provocar daños para la salud agudos o crónicos. Peligros para la reproducción, peligro de sensibilización por inhalación. Ejemplo: tricloroetileno. Precaución: evitar el contacto con el cuerpo humano.
Xi Irritante
Clasificación: Sin ser corrosivas, pueden producir inflamaciones en caso de contacto breve, prolongado o repetido con la piel o en mucosas. Peligro de sensibilización en caso de contacto con la piel. Ejemplo: amoníaco, cloruro de bencilo. Precaución: Evitar el contacto con ojos y piel; no inhalar vapores.
N Peligro para el
medio ambiente
Clasificación: En el caso de ser liberado en el medio acuático y no acuático puede producirse un daño del ecosistema por cambio del equilibrio natural, inmediatamente o con posterioridad. Ciertas sustancias o sus productos de transformación pueden alterar simultáneamente diversos compartimentos. Precaución: Según sea el potencial de peligro, no dejar que alcancen la canalización, en el suelo o el medio ambiente. Observar las prescripciones de eliminación de residuos especiales.

EDICIÓN 2015 25
PRÁCTICA 1
DESTILACIÓN SIMPLE Y DESTILACIÓN FRACCIONADA
OBJETIVOS.
1. Conocer y comprender la enorme importancia que poseen los métodos de
separación en química como herramientas indispensables para la separación,
purificación e identificación de compuestos orgánicos.
2. Conocer y aplicar las destilaciones simple y fraccionada en la separación y
purificación de líquidos.
3. Comparar la eficiencia de ambos tipos de destilación.
4. Conocer la influencia de la presión sobre el punto de ebullición de un líquido en
una destilación a presión reducida.
INTRODUCCIÓN.
Las destilaciones, simple y fraccionada son métodos de separación y purificación, que
pueden aplicarse con relativa facilidad en el aislamiento de líquidos provenientes de
una gran diversidad de mezclas líquido-líquido. El tipo de destilación a elegir dependerá
de las características individuales de cada mezcla a separar, dependiendo de si son
miscibles entre sí o no, de la separación de sus diferentes puntos de ebullición o de su
propensión a formar mezclas azeotrópicas.
FUNDAMENTO.
PUNTO DE EBULLICIÓN.
Las fuerzas entre las moléculas de un líquido en condiciones normales de presión y
temperatura, no son los suficientemente grandes para mantener a las moléculas
conformando un cristal definido, pero si para evitar que la sustancia se comporte como
un gas. El volumen de un líquido, es afectado por un cambio en la presión y se
modifica con los cambios de temperatura.

EDICIÓN 2015 26
Cuando un líquido se pone en un recipiente cerrado, algunas de sus moléculas escapan
a la fase gaseosa y otras regresan al seno del líquido, eventualmente se alcanza un
equilibrio donde las moléculas entran y salen a igual velocidad. Las moléculas en fase
de vapor ejercen una presión, la cual se denomina presión de vapor del líquido (presión
de equilibrio del líquido). La presión de vapor depende de la temperatura y su valor es
constante para una temperatura dada.
Cuando la temperatura de un líquido se eleva a tal grado que la presión de vapor iguala
la presión atmosférica, el líquido empieza a hervir. Así, el punto de ebullición de una
sustancia se define como la temperatura a la cual su presión de vapor se iguala con la
presión atmosférica.
El punto de ebullición es más sensible a los cambios de presión que el punto de fusión.
Para algunos líquidos disminuye alrededor de 0.5 ºC por cada decremento de 10 mmHg
en la presión, de tal modo que esta constante solo sirve como un criterio en la
identificación de una sustancia, cuando se especifica la presión a la cual ha sido
determinado.
Los compuestos en una serie homóloga, es decir, que poseen los mismos grupos
funcionales en su estructura pero mayor número de átomos de carbono con la misma
hibridación, muestran una relación definida entre el punto de ebullición y el peso
molecular. Las moléculas más pesadas necesitan mayor energía cinética para poder
salir de la fase líquida y pasar a la fase gaseosa, además, el incremento en la cadena
hidrocarbonada implica mayor número de interacciones.
A) DESTILACIÓN SIMPLE.
Cuando una mezcla de líquidos sufre únicamente una evaporación y una condensación,
hablamos de una destilación simple. La aplicación de esta técnica es muy limitada, ya
que una purificación con un 95 % de rendimiento requiere que la diferencia entre los
puntos de ebullición de ambos compuestos sea por lo menos de un 80 %.
El aparato utilizado en el laboratorio para una destilación simple se muestra en la figura
No. 4.

EDICIÓN 2015 27
FIGURA No. 4 DESTILACIÓN SIMPLE
B) DESTILACIÓN FRACCIONADA.
Si las presiones de vapor de dos o más componentes están próximas, es decir, si sus
puntos de ebullición son similares, la destilación simple resulta inadecuada para la
separación ya que su eficiencia es muy baja y debe utilizarse una columna de
fraccionamiento. Suponiendo que una mezcla de dos o más líquidos está en equilibrio
con el vapor que contiene ambos componentes, el vapor será más rico que el líquido en
su componente más volátil a una temperatura dada. Suponiendo que el vapor se
condensa por separado y que se alcanza un nuevo equilibrio vapor- líquido, ahora el
nuevo vapor será aún más rico en el componente más volátil. De esta forma se puede
conseguir una serie de evaporaciones y condensaciones sucesivas, es decir, equilibrios
que llevarán a obtener una muestra casi pura del componente más volátil, contaminada

EDICIÓN 2015 28
con una cantidad insignificante de la sustancia menos volátil. Dicha secuencia de
evaporaciones-condensaciones se puede llevar a cabo empleando columnas de
fraccionamiento de diferentes tipos.
La medida de la eficiencia de una columna de fraccionamiento se expresa en términos
de platos teóricos. Una columna en la que la mezcla producirá un destilado inicial con
una composición igual a la del vapor en equilibrio con la solución original, tendrá un
plato teórico. Un plato teórico corresponderá siempre a una longitud de columna en
centímetros.
La eficiencia en la separación de dos líquidos por destilación, depende de los siguientes
factores:
a) La diferencia en los calores de vaporización. Por ejemplo, la acetona tiene un
calor de vaporización Hº vap.= 7.35 kcal/mol y un punto de ebullición de 55 ºC,
por lo que fácilmente se separa del agua con punto de ebullición de 100 ºC y Hº
vap.= 9.72 kcal/mol, mediante una destilación simple, mientras que otro par de
líquidos con puntos de ebullición comparativamente diferentes, benceno p. eb. 80
ºC, Hº vap.= 7.35 Kcal/mol y tolueno p. eb. 110 ºC, Hº vap.= 7.07 k cal/ mol, se
separan con dificultad debido a su proximidad en calores de vaporización.
b) El número de platos teóricos de la columna de fraccionamiento, El número de
platos teóricos requeridos para la separación será mayor en la medida en que la
diferencia entre los puntos de ebullición sea menor.
c) Tiempo de destilación. Si la destilación se lleva a cabo muy rápidamente, el
sistema no podrá alcanzar el equilibrio por lo tanto la separación del componente
más volátil será deficiente, es necesario que se dé el tiempo suficiente para que la
fase líquida se pueda esparcir en la columna y se empaque completamente, de tal
forma que el intercambio con la fase gaseosa se facilite en un mayor grado y sea
lo más eficiente posible.
El aparato empleado para este tipo de destilación se presenta en la figura No. 5

EDICIÓN 2015 29
FIGURA No.5 DESTILACIÓN FRACCIONADA
C) DESTILACIÓN A PRESIÓN REDUCIDA.
Un gran número de compuestos no pueden purificarse por destilación a presión normal,
entre otras razones porque se descomponen por debajo de sus puntos normales de
ebullición. Otros presentan puntos de ebullición tan altos, que su destilación no resulta
conveniente e incluso se torna difícil, con frecuencia tales sustancias se pueden destilar
en una forma más fácil si se lleva a cabo una destilación a presión reducida.
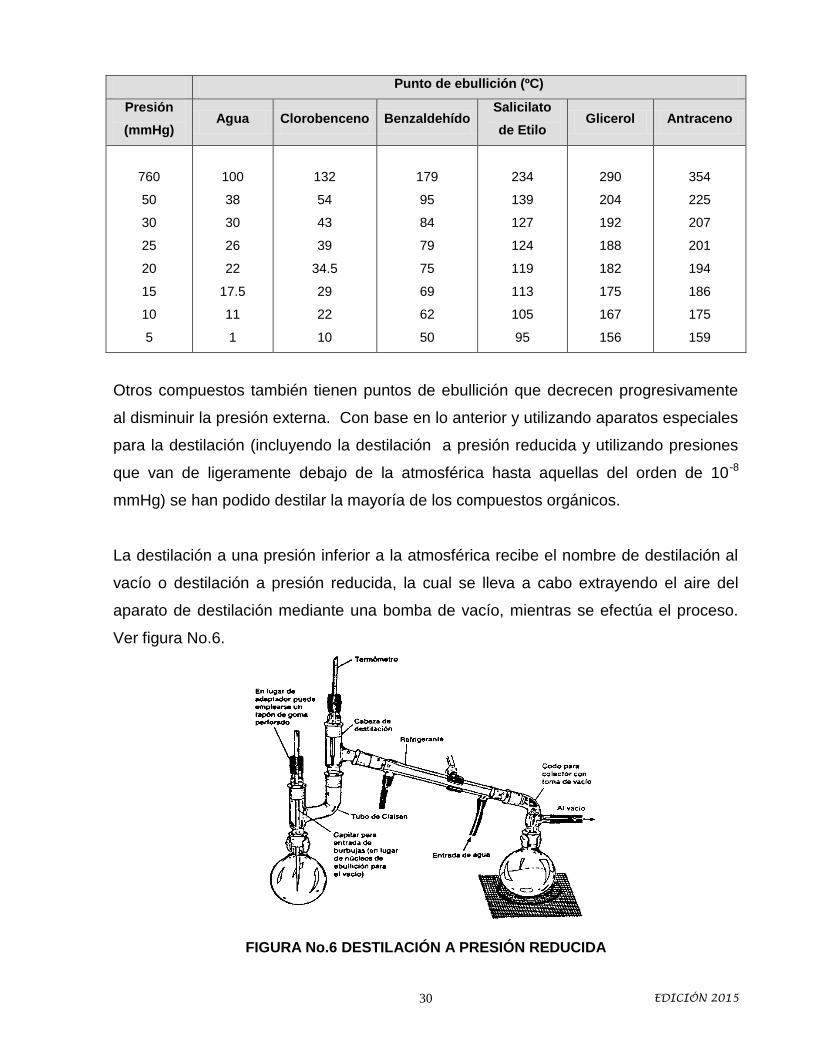
Un líquido comienza a hervir cuando la temperatura a la que su presión de vapor se
iguala a la presión exterior. En la tabla que se da a continuación puede observarse el
efecto que tiene la reducción de la presión exterior sobre los puntos de ebullición de
algunas sustancias.
EDICIÓN 2015 30
Punto de ebullición (ºC)
Presión
(mmHg) Agua Clorobenceno Benzaldehído
Salicilato
de Etilo Glicerol Antraceno
760
50
30
25
20
15
10
5
100
38
30
26
22
17.5
11
1
132
54
43
39
34.5
29
22
10
179
95
84
79
75
69
62
50
234
139
127
124
119
113
105
95
290
204
192
188
182
175
167
156
354
225
207
201
194
186
175
159
Otros compuestos también tienen puntos de ebullición que decrecen progresivamente
al disminuir la presión externa. Con base en lo anterior y utilizando aparatos especiales
para la destilación (incluyendo la destilación a presión reducida y utilizando presiones
que van de ligeramente debajo de la atmosférica hasta aquellas del orden de 10-8
mmHg) se han podido destilar la mayoría de los compuestos orgánicos.
La destilación a una presión inferior a la atmosférica recibe el nombre de destilación al
vacío o destilación a presión reducida, la cual se lleva a cabo extrayendo el aire del
aparato de destilación mediante una bomba de vacío, mientras se efectúa el proceso.
Ver figura No.6.
FIGURA No.6 DESTILACIÓN A PRESIÓN REDUCIDA
EDICIÓN 2015 31
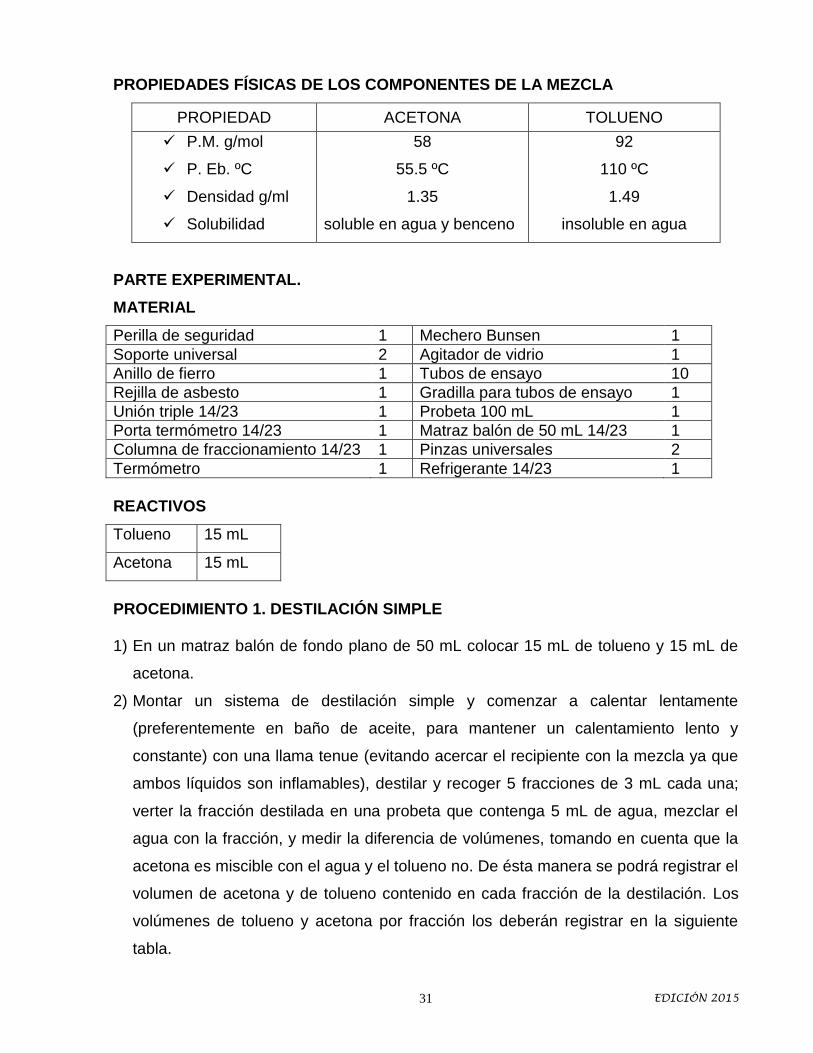
PROPIEDADES FÍSICAS DE LOS COMPONENTES DE LA MEZCLA
PROPIEDAD ACETONA TOLUENO
P.M. g/mol
P. Eb. ºC
Densidad g/ml
Solubilidad
58
55.5 ºC
1.35
soluble en agua y benceno
92
110 ºC
1.49
insoluble en agua
PARTE EXPERIMENTAL.
MATERIAL
Perilla de seguridad 1 Mechero Bunsen 1
Soporte universal 2 Agitador de vidrio 1
Anillo de fierro 1 Tubos de ensayo 10
Rejilla de asbesto 1 Gradilla para tubos de ensayo 1
Unión triple 14/23 1 Probeta 100 mL 1
Porta termómetro 14/23 1 Matraz balón de 50 mL 14/23 1
Columna de fraccionamiento 14/23 1 Pinzas universales 2
Termómetro 1 Refrigerante 14/23 1
REACTIVOS
Tolueno 15 mL
Acetona 15 mL
PROCEDIMIENTO 1. DESTILACIÓN SIMPLE 1) En un matraz balón de fondo plano de 50 mL colocar 15 mL de tolueno y 15 mL de
acetona.
2) Montar un sistema de destilación simple y comenzar a calentar lentamente
(preferentemente en baño de aceite, para mantener un calentamiento lento y
constante) con una llama tenue (evitando acercar el recipiente con la mezcla ya que
ambos líquidos son inflamables), destilar y recoger 5 fracciones de 3 mL cada una;
verter la fracción destilada en una probeta que contenga 5 mL de agua, mezclar el
agua con la fracción, y medir la diferencia de volúmenes, tomando en cuenta que la
acetona es miscible con el agua y el tolueno no. De ésta manera se podrá registrar el
volumen de acetona y de tolueno contenido en cada fracción de la destilación. Los
volúmenes de tolueno y acetona por fracción los deberán registrar en la siguiente
tabla.

EDICIÓN 2015 32
Fracción Volumen mL
Acetona Tolueno
1
2
3
4
5
El residuo del matraz de destilación, también se vierte en una probeta con 5 mL de agua, para medir el volumen de acetona y tolueno residuales.
PROCEDIMIENTO 2. DESTILACIÓN FRACCIONADA
1) Repetir el procedimiento anterior, colocando una columna de fraccionamiento
cuyo empaque será asignado por el profesor (vidrio, Porcelana, tezontle, fibra de
vidrio, aire). Si es necesario cubre la columna con una franela seca para evitar la
condensación de vapores. Anota y compara los resultados con los obtenidos
entre la destilación simple y la fraccionada. Realiza una tabla comparativa entre
los volúmenes de las sustancias separadas entre las diferentes fracciones, en
cada una de las destilaciones realizadas, indicando que tipo de material de
empacamiento empleaste.
CUESTIONARIO.
1. ¿Cuál es la razón de adicionar un volumen de agua a cada una de las fracciones
colectadas?
2. ¿Cuál de las sustancias a separar predomina en las primeras fracciones?
3. ¿En cuál de los dos procedimientos se logra mejor separación? explica tu
respuesta.

EDICIÓN 2015 33
4. Da tus conclusiones acerca de los diferentes tipos de destilación utilizadas, así
como de la utilidad de los distintos empaques en las columnas.
5. ¿Qué características debe tener una columna de fraccionamiento para que posea
la máxima eficiencia posible?
6. ¿Cuál es la limitante más importante que tienen las destilaciones como métodos
de separación?
7. ¿Por qué se debe calentar lentamente la mezcla de líquidos al iniciarse la
destilación?
8. Menciona otra aplicación que tiene la destilación a presión reducida como método
de separación a nivel industrial.

EDICIÓN 2015 34
PRÁCTICA 2
DESTILACIÓN POR ARRASTRE DE VAPOR
OBJETIVOS.
1. Aplicar la destilación por arrastre de vapor, en la separación de aceites esenciales.
2. Concluir sobre los resultados obtenidos en los diferentes tipos de destilación.
FUNDAMENTO. A) DESTILACIÓN POR ARRASTRE DE VAPOR.
Las destilaciones simple y fraccionada, se aplican a mezclas de componentes
miscibles. Cuando los componentes son inmiscibles entre sí, es posible separarlos
mediante otro tipo de destilación llamada destilación por arrastre de vapor, la cual se
basa en que una mezcla de líquidos inmiscibles, tendrá un punto de ebullición inferior al
punto de ebullición del componente más volátil cuando éste se encuentra puro. De ésta
manera, los compuestos de alto punto de ebullición, pueden ser aislados ó purificados,
combinándolos con algún líquido inmiscible de bajo punto de ebullición, generalmente el
agua, y sometiendo la mezcla a destilación. El proceso recibe el nombre de destilación
por arrastre de vapor. Se usa el agua como líquido de arrastre, porque posee varias
ventajas: Es de fácil acceso y por lo tanto barato; de bajo peso molecular; de fácil
manejo, etc. Cuando dos líquidos son inmiscibles, cada uno ejercerá su propia presión
de vapor, independientemente del otro, por lo que la presión total del sistema, será la
suma de las presiones parciales de acuerdo a la Ley de Dalton.
PT = PA + PB - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - (1)
En donde
PT = Presión total de la mezcla (A + B)
PA = Presión de vapor del componente A
PB = Presión de vapor del componente B

EDICIÓN 2015 35
Las proporciones molares de los dos componentes en el destilado, son iguales a sus
presiones de vapor en la mezcla en ebullición.
B
A
Pº
Pº
B moles
Amoles - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - (2)
En donde Pº = presión de vapor
OH delPº
compuesto del Pº
OH de moles
compuesto del moles
22
- - - - - - - - - - - - - (3)
Si molecular Peso
Pesomoles - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - (4)
Sustituyendo (4) en (3), tenemos que:
OH del molecular Peso
OH del Peso
compuesto del molecular Peso
compuesto del Peso
2
2
= OH del Pº
compuesto del Pº
2
De aquí que:
OH del Peso
compuesto del Peso
2
=OH Pº
compuesto del Pº
2
OH del molecular Peso
compuesto del molecular Peso
2
Una de las principales ventajas de este método, es que los compuestos de alto punto
de ebullición inestables a esa temperatura, pueden destilarse a temperaturas
suficientemente bajas como para evitar su descomposición. Debido a que es un
proceso muy eficiente y barato; se usa frecuentemente para:
a) Aislar ó purificar aceites esenciales de sus fuentes biológicas.
b) Purificar productos de reacción, sobre todo si la mezcla contiene ácidos ó álcalis
que a elevadas temperaturas descompongan los productos. (Anilina,
Nitrobenceno, bromo benceno, etc.)
c) Remover disolventes de alto punto de ebullición, tales como el cloro benceno,
siempre que el producto de interés no se altere por el agua.
d) La determinación de pesos moleculares, conociendo la proporción en peso, en la
que destilan los constituyentes de una mezcla y sabiendo además que la presión
de vapor de cada uno de ellos depende de la temperatura a la que es sometido.
EDICIÓN 2015 36
REACTIVOS
Clavo (especia), anís estrella o Canela
50 g
Hexano 30 mL
Sulfato de sodio anhidro 0.5 g
MATERIAL
1 Embudo de separación de 250 mL 1 Mechero Bunsen
2 Soporte universal 1 Agitador de vidrio
1 Anillo de fierro 1 Refrigerante 14/23
1 Rejilla de asbesto 2 Matráz Erlenmeyer de 500 mL
1 Unión triple 14/23 1 Tapón de hule horadado
1 Porta termómetro 1 Matraz balón de 50 mL 14/23
1 Columna de fraccionamiento 14/23 2 Pinzas universales
1 Termómetro
PARTE EXPERIMENTAL
A) DESTILACIÓN POR ARRASTRE DE VAPOR.
Colocar en un matráz Erlenmeyer de 500 mL 50 g del material elegido. Adicionar
300 mL de agua y montar un equipo de destilación simple para llevar a cabo la
destilación por arrastre de vapor. Calentar suavemente hasta que la mezcla empiece a
hervir y hacer pasar una corriente de vapor de agua a través del refrigerante hasta
colectar 200 mL de destilado o hasta que no se observen gotas de aceite en el
refrigerante. Terminada la colección, verter el destilado en un embudo de separación,
separar la fase orgánica en un vaso de precipitados y secarla sobre sulfato de sodio
anhidro. Éste aceite se guarda para el siguiente experimento (Destilación al vacío).
NOTA: La mitad de los equipos de cada sección seguirán la técnica como se ha
indicado y la otra mitad hará lo siguiente:
Después de haber separado la fase orgánica de la fase acuosa, lavar ésta última con 3
porciones de 10 mL de hexano y juntar los tres extractos orgánicos con el aceite

EDICIÓN 2015 37
obtenido anteriormente; secar la mezcla sobre sulfato de sodio anhidro y evaporar el
disolvente.
Todos deberán guardar en un vial, el aceite esencial obtenido, ya que se utilizará
en la práctica de cromatografía.
CUESTIONARIO
1 Indicar cómo se sabe cuándo termina la destilación por arrastre de vapor.
2 Explicar cuál es la diferencia fundamental entre el destilado que proviene de una
destilación por arrastre de vapor y el de cualquier otro tipo de destilación.
3 Explicar cómo se comportaría una mezcla de bromobenceno y p-dibromobenceno
en una destilación por arrastre de vapor.
4 Explicar la siguiente observación:
Una mezcla de para- y orto-nitrofenoles puede separarse por destilación por
arrastre de vapor. El orto-nitrofenol es más volátil que para-nitrofenol.
5 Indicar cuál es la estructura del componente principal del aceite esencial que aisló.
6 Calcular el peso del benceno, el cual destila con cada gramo de agua y el % de
composición del vapor producido durante la destilación. El punto de ebullición de la
mezcla es de 69.4 ºC, la presión de vapor del agua a 69.4 ºC es 227.7 mm Hg.

EDICIÓN 2015 38
7 Indicar qué características debe tener un líquido para que pueda separarse ó
purificarse por una destilación por arrastre de vapor.
8 Indicar cuáles son las principales ventajas y desventajas de la destilación al vacío
comparadas con la destilación simple.
9 Mencionar las precauciones que deben observarse cuando se usa una bomba de
agua en la destilación al vacío.
10 Indicar la presión que se debe alcanzar para que cada uno de los siguientes
compuestos: bromobenceno, anilina y benzoato de etilo destilen a una temperatura
de 100 ºC.
11 Seleccionar el tipo de destilación que se debe utilizar para separar los
componentes y constituyentes de los siguientes sistemas.
Sistemas Componentes Constituyentes Puntos de ebullición Tipo de destilación
A C1 + C2 30 - 150 oC
B C1 + C2 80 - 98 oC
C C1 + C2 40 - 100 oC
D C1 + C2 140 – 192 oC Termolábil ó inmiscible con H2O
12 Indicar cuál será el punto de ebullición del tolueno a 50 mm de Hg.

EDICIÓN 2015 39
PRÁCTICA 3
SEPARACIÓN DE UNA MEZCLA TERNARIA POR
DESTILACIÓN
OBJETIVOS
1. Utilizar los diferentes tipos de destilación para separar los componentes de una
mezcla.
2. Aplicar las propiedades físicas y químicas de algunos compuestos, para su
purificación.
INTRODUCCIÓN
La separación de mezclas es una tarea frecuente en Química Orgánica. Los métodos
utilizados en la práctica se basan principalmente en la naturaleza química de los
componentes de la mezcla a separar, considerando las diferencias en peso molecular,
polaridad, constantes físicas, acidez y basicidad.
La mezcla a separar está formada por anilina, alcohol iso-amílico y glicerol en
volúmenes iguales. El primer paso para conseguir la separación es una destilación por
arrastre de vapor, aprovechando la baja volatilidad del glicerol y su alta solubilidad en
agua, así como la comparativamente baja solubilidad en agua de la anilina y el alcohol
iso-amílico. En esta forma se separa el glicerol de la mezcla; la purificación total de éste
se consigue por medio de una destilación a presión reducida, debido a que una
destilación a presión normal trae consigo su descomposición (p. eb. 290 oC con
descomposición).
Se tiene ahora una mezcla de alcohol iso-amílico y anilina; para su separación se
aprovecha el carácter básico de la anilina, transformándola en su sal. La baja volatilidad
de la sal permite la separación por destilación del alcohol iso-amílico. La purificación de
la anilina implica el tratamiento de la sal formada, con un álcali, a fin de regenerarla.
Tanto la anilina como el alcohol iso-amílico se purifican totalmente por medio de una
destilación fraccionada o a presión reducida.

EDICIÓN 2015 40
PROPIEDADES FÍSICAS DE LOS COMPONENTES DE LA MEZCLA
OH OH
OH
NH2
OH
GLICEROL ANILINA ACOHOL ISOAMÍLICO
P.M. 92.1 93.2 88.2
p.f. 186 oC -6.2
oC
p.eb. 290 oC (760 mmHg) 184.3
oC 131
oC
Densidad 1.26 g/ml 1.022 g/ml 0.813 g/ml
Solubilidad agua y etanol Benceno, alcohol, éter y
cloroformo
agua, alcohol, éter, benceno
y cloroformo
Insoluble en cloroformo y tetracloruro de
carbono
Agua
MATERIAL Y REACTIVOS POR EQUIPO
REACTIVOS MATERIAL
Alcohol iso-amílico 5 ml Soporte 3 vaso de precipitados 3
Glicerol 5 ml Anillo 1 columna de
fraccionamiento
1
Anilina 5 ml Rejilla 1 embudo de separación 1
Ácido sulfúrico 2.5 ml pinzas nuez 3 matraz balón 1
Sulfato de sodio
anhidro
Lo que se
requiera
baño María 1 unión triple 1
Hidróxido de sodio al
40 %
mechero o parrilla 1 probeta 1
Agua Refrigerante 1 termómetro 1
otros, por cualquier eventualidad
matraz Erlenmeyer 2
portatermómetro
1
MATERIAL POR SECCIÓN
Bomba de vacío
Papel indicador de pH

EDICIÓN 2015 41
PARTE EXPERIMENTAL
En un matraz Erlenmeyer de 250 mL colocar 5 mL de alcohol iso-amílico, 5 mL de
anilina* y 5 mL de glicerol; adicionar 33 mL de agua y adaptar un aparato de destilación
por arrastre de vapor (el montaje se hará como el de una destilación simple). Destilar
hasta que el matraz de destilación contenga aproximadamente 12 mL, el destilado
contiene anilina, alcohol iso-amílico y agua; el residuo contiene glicerol impuro.
Transferir éste a un matraz para su posterior destilación.
Agregar al destilado anterior (el que contiene anilina y alcohol iso-amílico), lentamente y
con agitación, H2SO4 hasta pH entre 3 y 5 (aproximadamente 1.5 mL del ácido
concentrado); adicionar 15 mL de agua y efectuar una destilación por arrastre de
vapor (montaje de destilación simple).
Reacción:
NH2
NH3
H2SO
4++ + HSO4
-
+..
Bisulfato de anilonio
El destilado contiene alcohol iso-amílico y agua; transferirlo a un embudo de
separación, separar el alcohol presente en la fase orgánica y que por diferencia de
densidades debe ser la fase de arriba, y secar con Na2SO4 anhidro. (agregando la
cantidad requerida, cuando ya no se formen grumos en el líquido se encontrará éste
razonablemente seco).
* Produce anemia, irritabilidad y pérdida de peso en intoxicaciones graves.
Al residuo, que contiene la sal bisulfato de anilonio, agregarle una solución de NaOH al
40 % hasta alcalinidad (pH 9); agregar 10 ml de H2O y efectuar una destilación por
arrastre de vapor (montaje de destilación simple). Separar la anilina destilada,

EDICIÓN 2015 42
transfiriéndola a un embudo de separación (si se forma una emulsión y no se puede
observar bien a la anilina, adicionar una solución de cloruro de sodio saturada) y
posteriormente secarla con Na2SO4 anhidro, como se hizo con el alcohol iso-amílico.
Reacción
NH3HSO
4
NH2
Na2SO
4+
+ -
2NaOH + + 2 H2O
Como se menciona en la Introducción, tanto el alcohol iso-amílico como la anilina se
purifican totalmente por medio de una destilación fraccionada o a presión reducida.
TRATAMIENTO DE RESIDUOS
Los residuos se depositarán en los recipientes colocados para ello, en una determinada
área del laboratorio, haciendo la distinción entre residuos clorados y no clorados.
EDICIÓN 2015 43
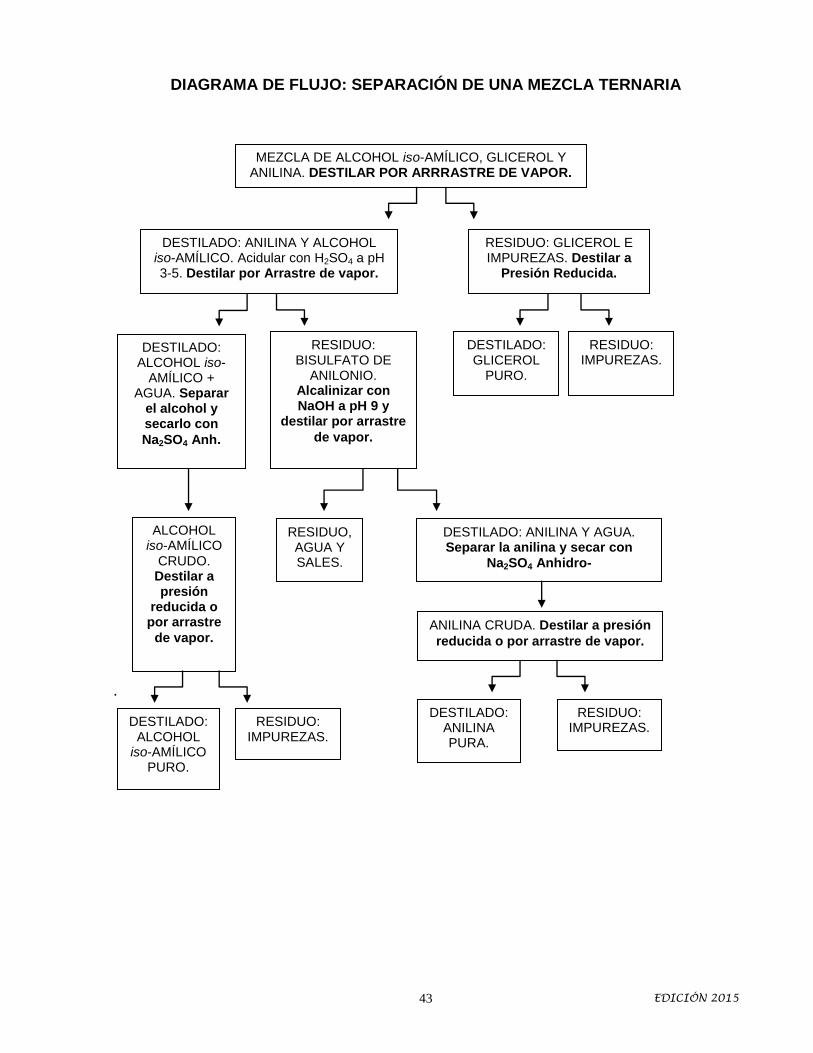
DIAGRAMA DE FLUJO: SEPARACIÓN DE UNA MEZCLA TERNARIA
.
MEZCLA DE ALCOHOL iso-AMÍLICO, GLICEROL Y ANILINA. DESTILAR POR ARRRASTRE DE VAPOR.
DESTILADO: ALCOHOL iso-
AMÍLICO + AGUA. Separar
el alcohol y secarlo con
Na2SO4 Anh.
RESIDUO: GLICEROL E IMPUREZAS. Destilar a
Presión Reducida.
DESTILADO: ANILINA Y ALCOHOL iso-AMÍLICO. Acidular con H2SO4 a pH 3-5. Destilar por Arrastre de vapor.
RESIDUO: BISULFATO DE
ANILONIO. Alcalinizar con NaOH a pH 9 y
destilar por arrastre
de vapor.
DESTILADO: GLICEROL
PURO.
RESIDUO: IMPUREZAS.
ALCOHOL iso-AMÍLICO
CRUDO. Destilar a presión
reducida o por arrastre
de vapor.
RESIDUO, AGUA Y SALES.
DESTILADO: ANILINA Y AGUA. Separar la anilina y secar con
Na2SO4 Anhidro-
ANILINA CRUDA. Destilar a presión
reducida o por arrastre de vapor.
RESIDUO: IMPUREZAS.
DESTILADO: ALCOHOL
iso-AMÍLICO PURO.
DESTILADO: ANILINA PURA.
RESIDUO: IMPUREZAS.

EDICIÓN 2015 44
CUESTIONARIO
1. Indicar a que se atribuye la baja volatilidad del glicerol
2. Explicar por qué las sales de los componentes orgánicos tienen puntos de ebullición
elevados
3. Indicar qué características debe tener una sustancia para purificarse con arrastre
con vapor de agua
4. Indicar cómo se sabe cuándo ha terminado una destilación por arrastre de vapor
5. Explicar en qué casos debe utilizarse la destilación a presión reducida
6. Proponer un procedimiento para separar cada una de las siguientes mezclas:
e) Ácido acético-acetona-octano
f) Anilina-cloruro de sodio-acetato de sodio
7. Sugerir un procedimiento diferente al empleado en el laboratorio para separar la
misma mezcla

EDICIÓN 2015 45
8. Explicar qué tipo de fuerzas intermoleculares actúan entre cada uno de los
componentes de la mezcla ternaria
9. Explicar qué tipo de fuerzas intermoleculares actúan en cada uno de los
componentes ya separados. (suponga una separación ideal).
BIBLIOGRAFÍA
William H. Brown. Química Orgánica
2a Ed. CECSA pp. 271-274
L.C. Wade J.R. Química Orgánica
2a
Ed. Prentice Hall pp. 24-36
Mary Ann Fox J.K.Whitesell. Química Orgánica
2a Ed. Pearson Education pp. 97-99
Morrison y Boyd Química Orgánica
5a Ed. Adison – Wesley – Iberoamericana pp. 938-946
John McMurry. Química Orgánica.
5a Ed. Thomson. Pp. 983-987
Hart – Craine Química Orgánica
9a Ed. McGraw Hill pp. 334-335

EDICIÓN 2015 46
PRÁCTICA 4
RECRISTALIZACIÓN.
OBJETIVOS.
Conocer y aplicar la técnica de recristalización para la purificación de
compuestos orgánicos.
Aplicar los conceptos relacionados con la estructura y la polaridad de los
compuestos.
Realizar la selección del disolvente en una recristalización.
ANTECEDENTES.
1.- Interacciones intermoleculares.
2.- Polaridad de las moléculas.
FUNDAMENTO.
Los compuestos orgánicos sólidos que se obtienen en una reacción o se aíslan de
alguna fuente natural suelen estar acompañados de impurezas que hay que eliminar
para poder disponer del producto deseado en el mayor grado de pureza posible. El
método más adecuado para la eliminación de las impureza que contamina un sólido es
por cristalizaciones sucesivas bien en un disolvente puro, o bien en una mezcla de
disolventes. Al procedimiento se le da el nombre genérico de recristalización.
La técnica de recristalización consiste en la formación de partículas sólidas en el seno
de una fase homogénea, basándose en las diferencias de solubilidad del sólido y sus
impurezas en diferentes disolventes. Considerando como impureza a toda sustancia
extraña cuya concentración no exceda del 5 %.
En la recristalización, el sólido impuro se disuelve en un volumen mínimo de disolvente
a ebullición; la mezcla caliente se filtra para eliminar todas las impurezas insolubles y la
solución se deja enfriar; al descender la temperatura, decrece la solubilidad del soluto y
cristaliza de la solución. En el caso ideal, la concentración de cualquier impureza no

EDICIÓN 2015 47
sobrepasa su punto de saturación en la solución fría y por ello permanecerá disuelta en
las aguas madres. Finalmente los cristales se separan por filtración y se dejan secar. En
la práctica, parte de las impurezas pueden cristalizarse con la sustancia deseada, por lo
que debe recristalizarse para obtener una purificación satisfactoria.
Cuando están presentes impurezas coloridas, éstas se eliminan agregando a la
solución una mínima cantidad de carbón activado que adsorbe las impurezas.
Los pasos para efectuar una recristalización, de acuerdo a lo anterior, son:
1. Elección del disolvente 2. Disolución de la sustancia en caliente 3. Si la solución tiene color, adicionar carbón activado y llevar a ebullición 4. Filtración de la solución en caliente 5. Enfriamiento para recristalizar 6. Separación de los cristales 7. Secado de los cristales
1. Elección del Disolvente.
Una estimación de la solubilidad de un sólido en un disolvente puede realizarse
considerando la estructura del sólido, la estructura del disolvente y la acción de las
fuerzas intermoleculares involucradas.
Así, los hidrocarburos son insolubles en agua, sin embargo, los alcoholes, los ácidos
carboxílicos y las amidas que tienen menos de 5 átomos de carbono pueden formar
puentes de hidrógeno con el agua y son solubles en ésta. En la Tabla No.1 se indican
algunos de los disolventes más empleados en la recristalización.
TABLA 1.La polaridad de los disolventes está indicada en orden descendente.
DISOLVENTE FÓRMULA PUNTO DE
EBULLICIÓN (ºC)
PUNTO DE
CONGELACIÓN (ºC)
INFLAMABI-
LIDAD
Agua H2O 100 0 0
Metanol* CH3OH 65 -198 ++++
Etanol* C2H5OH 78 -117 ++++
Acetona* CH3COCH3 56 - 95 ++++
Acetato de Etilo* CH3COOCH2CH3 77.2 - 84 ++++
Cloruro de Metileno CH2Cl2 40 - 97 0
Eter etílico* (C2H5)2O 35 -116 ++++
Cloroformo** CHCl3 61 - 64 0
Benceno*** C6H6 80 6 ++++
Tetracloruro de carbono** CCl4 76 - 23 0
Ligroína* Mezcla de Hidrocarburos C7 y C8 90-115 ++++
Hexano* Mezcla de Hidrocarburos C6H14 68 - 195 ++++
Eter de Petróleo* Mezcla de Hidrocarburos C5 y C6 35-60 ++++
Pentano* Mezcla de Hidrocarburos C5H12 36 -130 ++++

EDICIÓN 2015 48
NOTAS:
Todos los disolventes son tóxicos a excepción del agua, por lo cual deberán
usarse con precaución y con buena ventilación. Los disolventes clorocarbonados
producen daños hepáticos por contacto con la piel o por inhalación. Se ha
reportado que el CCl4 y El CH2Cl2 producen cáncer en animales de laboratorio y
han sido prohibidas por la FDA para emplearlos en cosméticos y drogas.
Se debe evitar el contacto de los disolventes con la piel así como su inhalación ya
que producen irritación de las mucosas.
La elección del disolvente para la purificación de un sólido se basa en las
características siguientes:
1. El material que se desea purificar deberá ser considerablemente más soluble en el
disolvente caliente que en frío.
2. Las impurezas deben ser o muy solubles o insolubles en el disolvente, o bien
deben poder eliminarse fácilmente con carbón activado.
3. El disolvente debe tener un punto de ebullición lo más bajo posible, para facilitar la
evaporación del mismo y el secado de los cristales.
4. El disolvente no debe reaccionar con el soluto.
5. Es también conveniente considerar el costo, toxicidad e inflamabilidad en la
elección de disolventes.
En ocasiones el disolvente más eficaz para la recristalización de un compuesto es una
mezcla de dos líquidos. Tales mezclas se usan cuando un sólido es soluble en un
disolvente e insoluble en el otro; en estas condiciones puede lograrse una cristalización
eficiente. El material a recristalizar se disuelve en el disolvente caliente en el cual es
más soluble, luego se agrega el otro disolvente lentamente, hasta que el soluto tienda a
separarse (la solución se volverá turbia). La mezcla se calienta de nuevo para disolver
todo el material (si es necesario, se añaden pequeñas cantidades del primer disolvente
para ayudar al proceso). Con la ayuda de un enfriamiento lento, se separará el producto
cristalino.
Entre los pares de disolventes más comunes se encuentran:
Metanol- Agua Éter etílico-Metanol Etanol - Agua Éter etílico-Acetona Acetona-Agua Éter etílico- Éter de petróleo

EDICIÓN 2015 49
2. Disolución de la sustancia en caliente.
La recristalización se basa en el principio de que la mayoría de los sólidos son más
solubles en un disolvente en caliente que en frío. De igual manera la solubilidad de un
sólido en un disolvente, está en función de su estructura química y de la temperatura.
Cuando un compuesto sólido se recristaliza en un disolvente apropiado, se forma una
solución saturada a temperatura elevada, de la cual al enfriarse se separa en forma
cristalina.
Una solución saturada se obtiene de la forma siguiente:
El soluto finamente pulverizado, se disuelve en una mínima cantidad de disolvente en
ebullición, calentando en un baño de vapor; a esta solución hirviente se le agrega más
disolvente en pequeñas porciones con agitación. Cuando el sólido se disuelve
totalmente, no debe agregarse más disolvente.
En la tabla No. 2 se aprecia el cambio de solubilidad en función de la temperatura.
TABLA No. 2
SOLUTO DISOLVENTE TEMPERATURA
(ºC) SOLUBILIDAD
(g/100 ml)
Ácido succínico Agua 20
100 7
121
Colesterol Etanol 17 78
11 130
En la Figura No. 1 se relaciona la solubilidad de una sustancia en función de la temperatura.
FIGURA 1.
Temperatura
A) Buen Disolvente
B) Mal Disolvente
C) Mal Disolvente
Solubilidad

EDICIÓN 2015 50
Se aprecia en la figura 1 que en una recta con baja pendiente (B) no es apropiada la
relación solubilidad temperatura, por lo cual el disolvente no es adecuado para efectuar
una recristalización. En la línea (C) se observa que se trata de un disolvente en el cual
la sustancia es demasiado soluble a cualquier temperatura y no es apropiado para
efectuar esta técnica, mientras que un disolvente que exhibe un comportamiento como
el indicado en (A) es ideal para efectuar una recristalización, ya que el sólido es muy
soluble a elevadas temperaturas y poco soluble a temperatura ambiente.
3.- Filtración de la solución en caliente.
En esta etapa, se pretenden eliminar las impurezas insolubles; esta filtración deberá
hacerse rápidamente empleando un embudo de tallo corto, pasando a través del papel
filtro, una pequeña cantidad de disolvente caliente para evitar que cristalice el
compuesto en el embudo. Para lograr una mayor rapidez en el filtrado, éste deberá
llevarse cabo doblando el papel filtro en la forma que se indica en la figura 2.
FIGURA 2.
4.- Precipitación o cristalización.
En esta etapa se busca obtener un sistema cristalino ordenado y de mayor pureza. Por
lo que las condiciones durante la formación de los nuevos cristales son fundamentales
ya que generarán un cristal (sólido puro con un ordenamiento geométrico importante), o
bien un amorfo (sólido desordenado). Es importante resaltar que con base en éstas

EDICIÓN 2015 51
características se pueden variar sus propiedades físicas, como la solubilidad,
resistencia, volumen, etc.
El proceso se inicia con la nucleación, que es básicamente la precipitación del primer
micro cristal sobre el cual posteriormente se apilaran las demás moléculas afines
permitiendo el crecimiento del cristal
Al enfriar la solución caliente, se pretende que se obtenga una máxima cristalización
con un mínimo de impurezas. Es preferible realizar este enfriamiento en un matraz
Erlenmeyer para evitar una gran evaporación.
Es conveniente que los cristales obtenidos sean de tamaño medio, ya que cristales muy
grandes o muy pequeños, pueden incluir o absorber cantidades apreciables de
impurezas.
El tamaño de los cristales se controla por la velocidad de cristalización; así una
cristalización rápida, favorece la formación de cristales pequeños y una cristalización
muy lenta origina cristales grandes. Se puede inducir la cristalización de las siguientes
formas:
a) Formando pequeños fragmentos de vidrio que actúan como núcleos de
cristalización, raspado las paredes del matraz donde se encuentre la solución a
cristalizar.
b) Añadiendo un pequeño cristal del producto, para sembrar la solución y provocar
la cristalización.
c) La solución se introduce en una mezcla frigorífica. En la tabla No. 3 se indican
algunas de las más empleadas.
TABLA No. 3
MEZCLA TEMPERATURA OBTENIDA (ºC)
Hielo - agua (v/v) 0
CaCl2 (250 g x 100 H2O) -8
NH4Cl (25 g x 100 g hielo) -15
NaCl (33 g x 100 g hielo) -20
CO2 Sólido/CCl4 -50
CO2/Acetona -70
5. Separación de los cristales.
En esta etapa se pretende separar los cristales formados eliminando al máximo el
disolvente; esta separación se puede llevar a cabo por filtración al vacío empleando un
embudo Büchner unido a un matraz Kitazato (filtración al vacío), o bien empleando un

EDICIÓN 2015 52
embudo de vidrio de tallo corto (filtración por gravedad). Los cristales así separados,
deben lavarse con una pequeña cantidad de disolvente frío. Ver figura 3.
6. Secado de los cristales.
Los cristales separados en la etapa anterior, se colocan sobre un papel filtro y se
presionan fuertemente para eliminar el disolvente, posteriormente se colocan en un
vidrio de reloj y se secan por alguna de las siguientes formas:
a) Secado al aire c) En una estufa
b) Empleando una desecadora a vacío d) Empleando una corriente de aire caliente
FILTRACIÓN POR GRAVEDAD FILTRACIÓN AL VACÍO
FIGURA 3.
PARTE EXPERIMENTAL. Esta práctica se divide en dos partes, en la primera se realizarán las pruebas de
solubilidad para elegir el disolvente adecuado para recristalizar cuatro diferentes
compuestos y en la segunda se sintetizará acetanilida y se purificará por
recristalización.
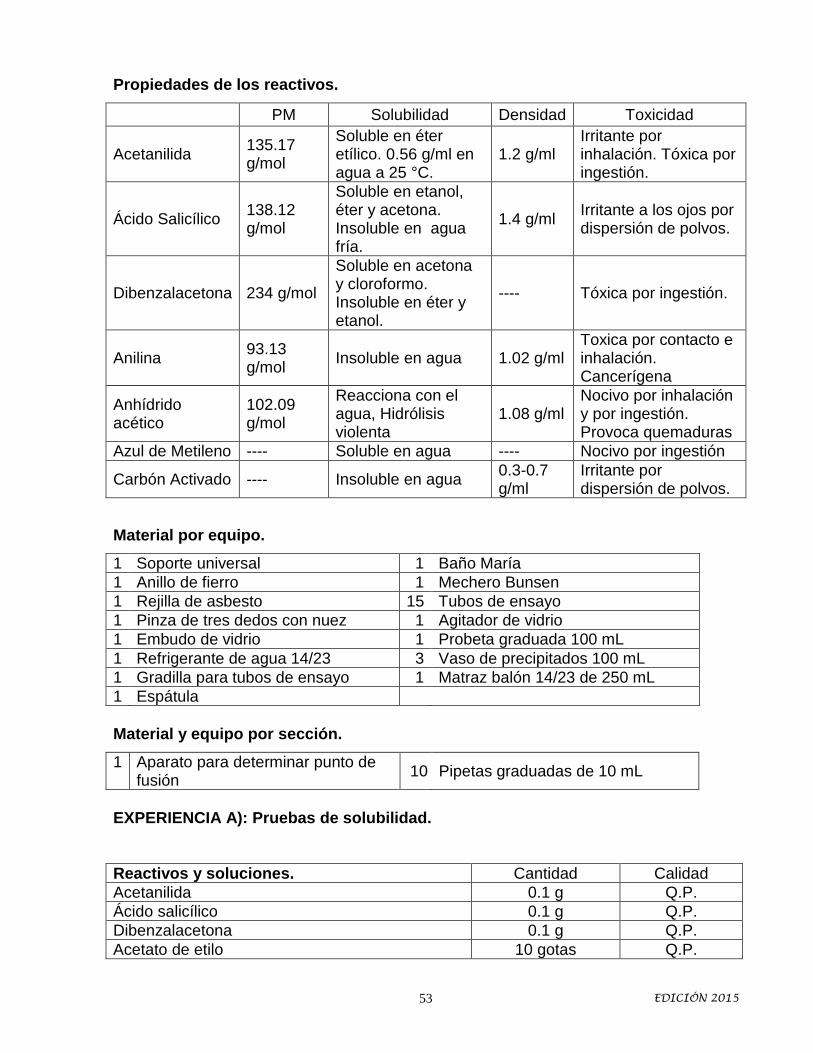
EDICIÓN 2015 53
Propiedades de los reactivos.
PM Solubilidad Densidad Toxicidad
Acetanilida 135.17 g/mol
Soluble en éter etílico. 0.56 g/ml en agua a 25 °C.
1.2 g/ml Irritante por inhalación. Tóxica por ingestión.
Ácido Salicílico 138.12 g/mol
Soluble en etanol, éter y acetona. Insoluble en agua fría.
1.4 g/ml Irritante a los ojos por dispersión de polvos.
Dibenzalacetona 234 g/mol
Soluble en acetona y cloroformo. Insoluble en éter y etanol.
---- Tóxica por ingestión.
Anilina 93.13 g/mol
Insoluble en agua 1.02 g/ml Toxica por contacto e inhalación. Cancerígena
Anhídrido acético
102.09 g/mol
Reacciona con el agua, Hidrólisis violenta
1.08 g/ml Nocivo por inhalación y por ingestión. Provoca quemaduras
Azul de Metileno ---- Soluble en agua ---- Nocivo por ingestión
Carbón Activado ---- Insoluble en agua 0.3-0.7 g/ml
Irritante por dispersión de polvos.
Material por equipo.
1 Soporte universal 1 Baño María
1 Anillo de fierro 1 Mechero Bunsen
1 Rejilla de asbesto 15 Tubos de ensayo
1 Pinza de tres dedos con nuez 1 Agitador de vidrio
1 Embudo de vidrio 1 Probeta graduada 100 mL
1 Refrigerante de agua 14/23 3 Vaso de precipitados 100 mL
1 Gradilla para tubos de ensayo 1 Matraz balón 14/23 de 250 mL
1 Espátula
Material y equipo por sección.
1
Aparato para determinar punto de fusión
10 Pipetas graduadas de 10 mL
EXPERIENCIA A): Pruebas de solubilidad.
Reactivos y soluciones. Cantidad Calidad
Acetanilida 0.1 g Q.P.
Ácido salicílico 0.1 g Q.P.
Dibenzalacetona 0.1 g Q.P.
Acetato de etilo 10 gotas Q.P.
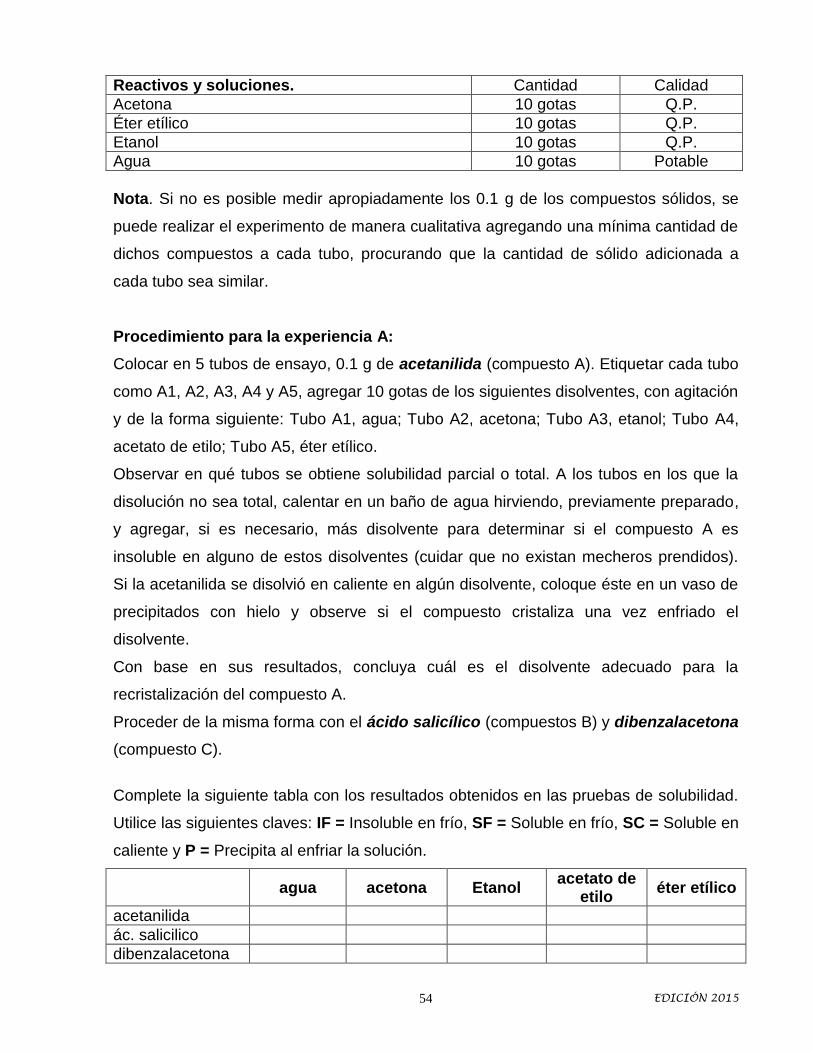
EDICIÓN 2015 54
Reactivos y soluciones. Cantidad Calidad
Acetona 10 gotas Q.P.
Éter etílico 10 gotas Q.P.
Etanol 10 gotas Q.P.
Agua 10 gotas Potable
Nota. Si no es posible medir apropiadamente los 0.1 g de los compuestos sólidos, se
puede realizar el experimento de manera cualitativa agregando una mínima cantidad de
dichos compuestos a cada tubo, procurando que la cantidad de sólido adicionada a
cada tubo sea similar.
Procedimiento para la experiencia A:
Colocar en 5 tubos de ensayo, 0.1 g de acetanilida (compuesto A). Etiquetar cada tubo
como A1, A2, A3, A4 y A5, agregar 10 gotas de los siguientes disolventes, con agitación
y de la forma siguiente: Tubo A1, agua; Tubo A2, acetona; Tubo A3, etanol; Tubo A4,
acetato de etilo; Tubo A5, éter etílico.
Observar en qué tubos se obtiene solubilidad parcial o total. A los tubos en los que la
disolución no sea total, calentar en un baño de agua hirviendo, previamente preparado,
y agregar, si es necesario, más disolvente para determinar si el compuesto A es
insoluble en alguno de estos disolventes (cuidar que no existan mecheros prendidos).
Si la acetanilida se disolvió en caliente en algún disolvente, coloque éste en un vaso de
precipitados con hielo y observe si el compuesto cristaliza una vez enfriado el
disolvente.
Con base en sus resultados, concluya cuál es el disolvente adecuado para la
recristalización del compuesto A.
Proceder de la misma forma con el ácido salicílico (compuestos B) y dibenzalacetona
(compuesto C).
Complete la siguiente tabla con los resultados obtenidos en las pruebas de solubilidad.
Utilice las siguientes claves: IF = Insoluble en frío, SF = Soluble en frío, SC = Soluble en
caliente y P = Precipita al enfriar la solución.
agua acetona Etanol acetato de
etilo éter etílico
acetanilida
ác. salicilico
dibenzalacetona

EDICIÓN 2015 55
EXPERIENCIA B): Purificación de un compuesto orgánico sólido obtenido por
síntesis.
Reactivos y soluciones. Cantidad Calidad
Anilina 2.5 mL Q.P
Anhídrido acético 4 mL Q.P
Agua 15 mL Destilada
SÍNTESIS DE ACETANILIDA.
La síntesis de acetanilida es la reacción que servirá de ejemplo para la obtención y
posterior purificación de un producto de síntesis, la cual consiste en la acetilación de
anilina con anhídrido acético. Esta es una reacción de sustitución nucleofílica, en la que
el grupo básico (-NH2), efectúa un ataque nucleofílico sobre el átomo de carbono
carbonílico del anhídrido, que es un centro electrofílico. De manera general, la reacción
transcurre muy rápidamente con cloruros de ácido, más lentamente con anhídridos de
ácido y muy lentamente con los ácidos carboxílicos, que en este último caso, para que
se produzca la reacción se requiere una temperatura elevada. Por ejemplo, la
fabricación industrial de la acetanilida se realiza calentando una mezcla de anilina y
ácido acético durante unas seis u ocho horas; en tanto que una solución caliente de
anhídrido acético en medio acuoso, reacciona con las aminas a una velocidad tal, que
es adecuada para su realización en el laboratorio.
Reacción.
O
O O
NH2
N
O
H
OH
O
+ +
Procedimiento para la experiencia B:
En un matraz balón de fondo plano colocar 2.5 mL de anilina, adicionar lentamente y
con precaución 4 ml de anhídrido acético (al agregar el anhídrido acético se observa
desprendimiento de calor). El anhídrido acético es un irritante fuerte, por lo que esta
operación debe llevarse a cabo en la campana. Adaptar al matraz un refrigerante de
agua en posición de reflujo y calentar la solución a ebullición durante 10 min. Enfriar un

EDICIÓN 2015 56
poco el matraz y verter el contenido en un vaso que contenga 15 mL de agua con hielo.
Agitar bien la mezcla, filtrar la acetanilida y lavar los cristales con pequeñas porciones
de agua fría. Tomar una pequeña muestra para determinar punto de fusión de la
acetanilida sin recristalizar (cruda).
Purificación de la acetanilida por recristalización.
En este punto se hará adicionalmente una demostración del uso del carbón activado
para eliminar impurezas coloridas. En un vaso de precipitados colocar toda la
acetanilida sintetizada en el experimento anterior. Agregar una gota de azul de metileno
como contaminante colorido y disolver en la mínima cantidad de agua calentando a
ebullición. Es importante recordar que se debe preparar una solución saturada, por lo
que todo el sólido se debe disolver en la mínima cantidad de agua a ebullición. Cuando
se haya disuelto totalmente la acetanilida, retirar brevemente el mechero y adicionar
una pequeña cantidad de carbón activado; reanudar el calentamiento hasta ebullición y
filtrar en caliente (esta técnica deberá ser ilustrada por los profesores del
laboratorio). Posteriormente dejar enfriar el filtrado hasta 40ºC aproximadamente
(temperatura a la cual se puede tomar el vaso de precipitados con las manos), y
sumergir en baño de hielo hasta la precipitación total de la acetanilida. Pesar el papel
filtro en donde se recuperará la acetanilida para facilitar su cuantificación. Separar por
filtración y secar. Determinar el rendimiento del producto obtenido y el punto de fusión
de la acetanilida recristalizada. Es importante recordar que este producto deberá ser
entregado en un frasco debidamente rotulado al profesor, ya que será empleado como
materia prima en prácticas subsecuentes.
Punto de fusión de:
Acetanilida cruda _________ Acetanilida recristalizada: _________
Cálculo de rendimiento de producto obtenido:
Rendimiento Teórico: _________
Rendimiento Experimental: _________

EDICIÓN 2015 57
Tratamiento de residuos.
Las aguas madres resultantes de la recristalización pueden ser vertidas en la tarja con
agua corriente, puesto que el exceso de anhídrido se hidroliza a ácido acético y éste se
encuentra diluido en agua.
Cuestionario.
1. Hacer un esquema general de la técnica de recristalización.
2. Indicar en qué casos y con qué finalidad se lleva a cabo una recristalización.
________________________________________________________________________________________________________________________________________________________________________________________________________________ 3. Explicar para qué sirve el carbón activado.
________________________________________________________________________________________________________________________________________________________________________________________________________________ 4. Indicar por qué es importante reducir al mínimo la evaporación durante la filtración de
una solución caliente.
________________________________________________________________________________________________________________________________________________________________________________________________________________ 5. En la purificación de un sólido por recristalización en un disolvente, explicar si es
aconsejable enfriar la solución rápida o lentamente.
________________________________________________________________________________________________________________________________________________________________________________________________________________ 6. Si los puntos de fusión determinados a los compuestos purificados, no coinciden con
los reportados. Indica qué interpretación se daría a este hecho y proponga qué
procedimiento seguiría con base en su interpretación.
________________________________________________________________________________________________________________________________________________________________________________________________________________

EDICIÓN 2015 58
7. Explicar por qué aumenta la solubilidad de un compuesto en un disolvente al
aumentar la temperatura.
________________________________________________________________________________________________________________________________________________________________________________________________________________ 8. Indicar qué condiciona que una sustancia (soluto) se solubilice en otra (disolvente). ________________________________________________________________________________________________________________________________________________________________________________________________________________ 9. Indicar en una recristalización qué ventajas tendrá el agua sobre el éter y el benceno. ________________________________________________________________________________________________________________________________________________________________________________________________________________ 10. Indicar qué ventajas tendrá el tetracloruro de carbono sobre el éter y el benceno en una recristalización. ________________________________________________________________________________________________________________________________________________________________________________________________________________ 11. Explicar por qué en condiciones de saturación ya no es posible disolver más
cantidad de soluto.
________________________________________________________________________________________________________________________________________________________________________________________________________________ 12. Explicar para qué se calienta a reflujo durante 10 minutos la mezcla de anilina,
anhídrido acético y agua.
__________________________________________________________________________________________________________________________________________________________________________________________________________________ 13. En la síntesis de acetanilida, explicar con qué fin se enfría la mezcla de reacción. __________________________________________________________________________________________________________________________________________________________________________________________________________________ Bibliografía.
Adams, Johnson. J. R. y Wilcox, C. H. F., Jr. Laboratory Experiments in Organic
Chemistry. McMillan. New York, 1970.

EDICIÓN 2015 59
PRÁCTICA 5
EXTRACCIÓN LÍQUIDO-LÍQUIDO
Objetivos:
General:
Conocer la técnica como método de separación y purificación de
sustancias integrantes de una mezcla
Particulares:
Aplicar la técnica en la purificación de compuestos orgánicos, tanto
neutros como ionizables.
Elegir los disolventes adecuados para un proceso de extracción.
Realizar diferentes tipos de extracción: múltiple y selectiva.
Introducción.
Extracción con disolventes orgánicos.
La extracción es una técnica de transferencia de un soluto de un disolvente a otro. El
soluto se extrae por un proceso de distribución.
Cuando una disolución (soluto A en un disolvente 1) se agita con un segundo disolvente
(Disolvente 2) con el cual es inmiscible, el soluto se distribuye entre las dos fases hasta
lograr una situación de equilibrio.
Donde [A]o es la concentración del analito en la fase orgánica y [A]aq es la
concentración del analito en la fase acuosa.
Al separarse las dos capas de los disolventes inmiscibles se determina la concentración
del soluto en cada capa, la relación de las concentraciones en cada fase es una
constante. Esta constante, es llamada coeficiente de distribución (o partición), K, la cual
es definida por:

EDICIÓN 2015 60
Donde C1 y C2 son las concentraciones en equilibrio, en gramos/litro, del soluto en el
disolvente 1 y en el disolvente 2 a una temperatura determinada.
Esta relación es independiente de la concentración total y de los volúmenes de los
disolventes. El coeficiente de distribución tiene un valor constante para cada soluto y es
dependiente de la naturaleza del disolvente utilizado en cada caso.
Con base en el coeficiente de distribución, no todo el soluto se transfiere al disolvente 2
en una sola extracción a menos que el valor de K sea muy alto. Generalmente se
requieren varias extracciones para eliminar el soluto del disolvente 1. Los disolventes
orgánicos utilizados en extracción deben tener baja solubilidad en agua, alta capacidad
de solvatación hacia la sustancia que se va a extraer y bajo punto de ebullición para
facilitar su eliminación posterior. La extracción tiene amplia aplicación en la Química
Orgánica. Se utiliza para extraer productos y eliminar impurezas de las mezclas de
reacción, se emplea también para extraer productos de tejidos animales o de plantas.
Para extraer un soluto de una disolución es más efectivo realizar varias extracciones
empleando volúmenes pequeños (extracción múltiple), que realizar una sola extracción
(extracción simple) mediante el empleo de un volumen grande de disolvente.
Matemáticamente esto se comprueba con la siguiente expresión:
n
nVKV
VWW
12
10
Donde:
Wn = g de soluto remanentes en la fase acuosa después de n extracciones.
Wo = g de soluto en fase acuosa.
K = coeficiente de partición
V1 = volumen total de la solución de la solución a extraer
V2 = volumen del disolvente de c/u extracción
n = No. De extracciones
Extracción selectiva con disolventes activos
La extracción selectiva se emplea para separar mezclas de compuestos orgánicos, en
función de la acidez, de la basicidad o de la neutralidad de éstos. Para realizar estas

EDICIÓN 2015 61
separaciones, es necesario utilizar disolventes activos, éstos pueden ser ácidos o
básicos. Los disolventes activos ácidos que más se utilizan son el HCl y el H2SO4 en
disolución acuosa del 5 al 10 %.
Los disolventes activos básicos pueden ser fuertes como NaOH, KOH o moderados
como NaHCO3 y Na2CO3 en disolución acuosa al 5 ó 10 %.
La extracción selectiva se basa en una reacción ácido-base entre el producto a separar
y el disolvente activo adecuado. Los compuestos iónicos son más solubles en agua que
los compuestos covalentes y éstos son más solubles en disolventes orgánicos que
aquéllos.
Los compuestos básicos como aminas, se extraen con disolventes activos ácidos (HCl
al 5 ó 10%); la reacción que ocurre es la siguiente:
R―NH2 + HCl R―NH3+ - Cl
Unión covalente Unión iónica
Menos polar Más polar
Soluble en disolventes orgánicos Soluble en agua
Los ácidos carboxílicos reaccionan con disolventes activos básicos como NaOH,
Na2CO3 o NaHCO3 al 5 ó 10 %.
Las reacciones ácido-base que se efectúan son las siguientes:
R―COOH + NaOH R―COO-+Na + H2O
Unión covalente Unión iónica
Menos polar Más polar
Soluble en disolventes orgánicos Soluble en agua
Los fenoles son compuestos menos ácidos que los ácidos carboxílicos. Por esta
característica, reaccionan únicamente con bases fuertes como NaOH a las mismas
concentraciones ya mencionadas, lo anterior constituye la base para separar ácidos de
fenoles.
Ar―OH + NaOH Ar―O-+Na + H2O
Fenol Fenóxido de sodio sal soluble en agua
La extracción selectiva también se utiliza para eliminar impurezas ácidas o básicas a un
producto aislado de una mezcla de reacción. Los compuestos que no tienen

EDICIÓN 2015 62
características ácidas o básicas los consideramos neutros. Los compuestos extraídos
pueden recuperarse por neutralización del disolvente activo para extraer.
Si un compuesto está disuelto en fase acuosa básica éste puede recuperarse
acidificando la disolución. Por el contrario, si el compuesto está disuelto en fase acuosa
ácida, puede recuperarse al agregar una base y modificar el pH hasta lo básico. Los
compuestos neutros disueltos en un disolvente orgánico, se recuperan al eliminar el
disolvente por destilación.
El embudo de separación
El embudo de separación es la pieza que se utiliza en la extracción. El tapón y la llave,
que deben estar bien ajustados, se lubrican con una “grasa” adecuada antes de usarlos.
El embudo debe agitarse moderadamente y purgarlo (aliviar la presión) con frecuencia,
para evitar la presión en su interior. La forma correcta de agitar el embudo se muestra
en la figura 1.
Figura 1. Método sugerido para la realización de la extracción líquido-líquido.
Después de agitar el embudo se deja reposar para la formación de una interface que da
la pauta para la separación de las fases. (Figura 2). En la identificación de fases es
importante conocer las densidades de los disolventes orgánicos para ubicar su posición
en el embudo con respecto a la fase acuosa. El número de extracciones en cada caso
depende del coeficiente de distribución del disolvente.

EDICIÓN 2015 63
Figura 2.
Separación de fases.
Emulsiones:
Con frecuencia se forman emulsiones durante el proceso de extracción. Éstas pueden
romperse mediante:
Un movimiento de giro suave al líquido del embudo.
Agitación vigorosa de la capa emulsionada.
Agitar la fase acuosa con una disolución saturada de cloruro de sodio.
Centrifugación.
Adición de una solución saturada de cloruro de sodio
Agentes desecantes químicos.
Un desecante debe reunir ciertas condiciones:
No reaccionar con la sustancia que se va a secar.
Ser eficaz, o sea, tener alto poder desecante; esto es, eliminar el agua
completamente o casi completamente cuando se alcanza el equilibrio.
Tener gran capacidad de desecación, es decir, eliminar una gran cantidad de
agua por unidad de peso de desecante.
Secar rápidamente (alcanzar rápido el equilibrio).
Separarse fácilmente de la sustancia una vez seca.

EDICIÓN 2015 64
Los desecantes químicos pueden dividirse en dos grupos:
a) Aquellos que reaccionan químicamente con el agua en un proceso no reversible,
lo cual da lugar a un nuevo compuesto libre de agua.
b) Los que se combinan reversiblemente con el agua para formar un hidrato.
Parte Experimental
Propiedades Físicas de los Reactivos
S a c a r o s a Á c i d o B e n z ó i c o
Peso Molecular 342.3 g/mol 122.1 g/mol
Punto de fusión 160-186 ºC 128.0 ºC
Punto de ebullición ------------------------- 249.0 ºC
pKa ------------------------- 4.20
Solubilidad s. en agua y etanol l. s. en éter
s. en etanol, éter y benceno l. s. en agua, tetracloruro de carbono
Reactivos Material
Sacarosa R.A. 1.0 g 1 Vaso de precipitados de 50 mL
Ácido benzoico R.A. 1.0 g 1 Vaso de precipitados de 100 mL
Éter etílico R.A. 30 mL 1 Embudo de separación con tapón esmerilado.
HCl al 10 % 10 mL 1 Probeta de 50 mL
NaOH al 10 % 10 mL 1 Pipeta graduada de 10 mL
Etanol 10 mL 1 1 Matraz balón de 250 mL 14/23
Hexano 10 mL 1 Refrigerante recto
Benceno 10 mL Papel indicador de pH 0-14
Metanol 10 mL 3 Mangueras de látex
Cloruro de metileno 10 mL 1 Mechero de Bunsen
Disoln. de NaCl sat. 20 mL 1 Tapón adaptador
Sulfato de sodio anhidro
Cuanto baste para secar
1 Baño María
1 Soporte universal y pinzas de tres dedos con nuez
1 Anillo y rejilla de asbesto

EDICIÓN 2015 65
Desarrollo Experimental
EXPERIENCIA A: Extracción
1. En un vaso de precipitados colocar 0.5 g de sacarosa y 0.5 g de ácido benzoico.
y solubilizarlos con 35 mL de agua.
2. Verter la disolución en un embudo de separación y agregar 10 mL de éter etílico.
3. Mezclar cuidadosamente, de acuerdo al procedimiento descrito en la figura,
dejando aliviar la presión interna del embudo de separación.
4. Dejar reposar el embudo con la disolución y observar cómo se separan las dos
capas. Drenar la fase acuosa en un matraz Erlenmeyer y la fase etérea en un
vaso de precipitados. Cubra el vaso con un trozo de papel aluminio.
5. Secar la fase orgánica con sulfato de sodio anhidro(cuanto baste para secar).
6. Decantar y evaporar el éter en un baño de agua caliente sin llegar a sequedad.
(PRECAUCIÓN: Los vapores de éter son inflamables, es recomendable
recuperar el éter mediante una destilación simple). Pesar el residuo sólido y
comparar con la mezcla original.
7. Calcule el rendimiento.
EXPERIENCIA B: Variación del pH
1. En un vaso de precipitados disolver una mezcla de 0.5 g de sacarosa y 0.5 g de
ácido benzoico y solubilizarlos con 25 mL de solución de NaOH al 10 % a pH =
12.
2. Verter la disolución en un embudo de separación y agregar 10 mL de éter etílico.
3. Mezclar cuidadosamente, dejando aliviar la presión interna del embudo.
4. Dejar reposar el embudo con la disolución y observar cómo se separan las dos
capas.
5. Drenar la fase acuosa en un matraz erlenmeyer y la fase etérea en un vaso de
precipitados, posteriormente evaporar la fase etérea.
6. Observar y concluir.
7. Escribir la reacción entre el ácido benzóico y el NaOH.

EDICIÓN 2015 66
EXPERIENCIA C: Extracción de Licopenos.
1. En un matraz balón 14/23 de 250 mL colocar 20 g de jitomate previamente
macerado en un mortero y agregar 10 mL de etanol al 95 %.
2. Adaptar un refrigerante 14/23 en posición de reflujo y calentar en un baño de
agua a ebullición durante 5 minutos.
3. Filtrar la mezcla en caliente y presionar el sólido suavemente, recogiendo el
filtrado amarillo en un matraz Erlenmeyer.
4. Transferir el residuo sólido de la filtración al matraz balón 14/23 de 250 mL y
agregarle 10 mL de cloruro de metileno.
5. Calentar la mezcla a reflujo en baño maría durante 3-4 min. (CH2Cl2 p.eb. 41 °C,
es un líquido altamente tóxico; EVITAR INHALAR LOS VAPORES).
6. Filtar el extracto y adicionar al residuo sólido en el papel 2 ó 3 porciones de 10
mL de CH2Cl2.
7. Reunir los extractos de CH2Cl2 con el primer extracto etanólico.
8. Pasar la solución a un embudo de separación, agitar y agregar 20 mL de una
disolución saturada de NaCl.
9. Separar la capa inferior colorida en un vaso de precipitados y secar la con
Na2SO4 anhidro (cuanto baste para que la fase orgánica sea trasnparente).
10. El licopeno obtenido es inestable a la luz y al aire, guardar el extracto para la
práctica de cromatografía en un frasco ámbar con tapa.
EXPERIENCIA D: Extracción de Carotenos.
Los carotenos son tetra-terpenos. Están presentes en la mayoría de las plantas
verdes. Los carotenos sirven como precursores de la vitamina A, ya que pueden ser
convertidores a ésta por enzimas del hígado.
1. Hacer un extracto de hojas de espinacas, macerándolas con 30 mL de una
mezcla benceno-hexano-metanol en partes iguales.
2. Verter la mezcla en un embudo de separación y lavar 2 veces la capa orgánica,
utilizando porciones de 30 mL de agua.
3. Separar la capa orgánica y secarla con sulfato de sodio anhidro.

EDICIÓN 2015 67
4. Decantar y destilar hasta obtener un residuo de 5 mL.
5. Guardar el extracto para la siguiente práctica en un frasco ámbar tapado.
Cuestionario:
1. Escriba las ecuaciones involucradas en el proceso de extracción selectiva.
2. En un proceso de extracción, investigue que es más eficiente, hacer cuatro
extracciones con 50 mL de éter ó dos extracciones con 100 mL del mismo.
______________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
3. Investigue como se determina el coeficiente de partición K de manera práctica.
______________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
4. Cuales serían los factores que pudieran intervenir en la eficiencia de la extracción
que usted realizó en el laboratorio.
______________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
5. Investigue la estructura de los carotenos y de los licopenos extraidos en la práctica,
así como su coeficiente de extinción molar.

EDICIÓN 2015 68
PRÁCTICA 6
CROMATOGRAFÍA
OBJETIVOS
1. Conocer la técnica de cromatografía y los factores experimentales que la afectan.
2. Aplicar y comparar los métodos cromatográficos de capa fina y columna, para
separar, identificar y purificar compuestos orgánicos.
3. Observar el efecto de diferentes fases móviles y estacionarias en la separación.
4. Determinar los valores de Rf, como un parámetro en la identificación de los
compuestos separados.
ANTECEDENTES
Según la definición dada por Keulemans, la cromatografía es un método en el que los
componentes a separar se distribuyen entre dos fases, una de las cuales constituye un
lecho estacionario de amplio desarrollo superficial y la otra es un fluido que pasa a
través o a lo largo del lecho estacionario.
TEORÍA
La cromatografía consiste en una serie de métodos que sirven para la separación de
una mezcla de solutos. Se basa en la diferente velocidad con que se mueve cada uno
de los solutos a través de un medio poroso, arrastrados por un disolvente en
movimiento.
El fundamento de la cromatografía consiste en el reparto o distribución diferencial de
dos o más compuestos (llamados solutos) entre dos fases, una de las cuales
permanece fija, por lo que se le denomina fase fija o estacionaria y otra que fluye a
través de ella, por lo que se le denomina fase móvil o eluyente. Como la fase
estacionaria debe permanecer fija, su estado físico se limita a sólidos y líquidos, en
tanto que la fase móvil requiere ser un líquido o un gas para poder fluir. Tomando en
consideración el estado físico de las fases, la cromatografía se clasifica en dos grandes
categorías y dos subcategorías cada una:

EDICIÓN 2015 69
1) CROMATOGRAFÍA DE GASES.
En ésta, la fase móvil es un gas y la estacionaria puede ser un líquido o un sólido, por lo
que existen dos variantes:
a) Cromatografía sólido-gas (CSG)
b) Cromatografía líquido-gas (CLG)
2) CROMATOGRAFÍA DE LÍQUIDOS.
En ésta, la fase móvil es un líquido y la estacionaria puede ser un sólido o un
líquido, por lo que existen dos variantes:
a) Cromatografía sólido-líquido (CSL)
b) Cromatografía líquido-líquido (CLL)
Cuando la fase estacionaria es sólida, la cromatografía puede ser en columna y en
placa (también llamada cromatografía de capa fina).
Las separaciones por cromatografía de reparto, son particularmente interesantes para
los compuestos solubles en agua. Asimismo, de acuerdo con el mecanismo
predominante en el reparto diferencial de solutos, las técnicas cromatográficas se
clasifican de la siguiente forma:
a) Adsorción Fenómeno de adhesión superficial cuyo mecanismo consiste
en interacciones bipolares.
b) Partición Fenómeno de reparto entre las dos fases debido a su
solubilidad. La solubilidad es una medida de la polaridad y
de otro tipo de interacciones moleculares.
c) Intercambio iónico Fenómeno superficial debido a interacciones entre iones de
diferente carga.
d) Ultra filtración Como su nombre lo indica, se debe a un fenómeno de
tamizado molecular.
Debido a su distribución diferencial entre las fases móvil y estacionaria, dos solutos
aplicados en un extremo del sistema cromatográfico recorrerán el mismo camino a
diferentes velocidades, arrastrados por la fase móvil, con su consecuente elución en

EDICIÓN 2015 70
diferentes tiempos o volúmenes, lo cual permite diferenciar a cada analito por sus
propiedades cromatográficas
Los sistemas cromatográficos pueden ser:
a) Cromatografía en columna.
El sistema cromatográfico en columna más sencillo consta de un reservorio para el
eluyente (fase móvil); una columna que debe empacarse homogéneamente, la fase
estacionaria en cuyo extremo superior se aplica la muestra por separar (solutos); un
sistema de control de flujo (llave de paso o pinzas) y los recipientes para recibir los
volúmenes eluídos, como se observa en la siguiente figura.
1. Adición de eluyente 2. Mezcla de A y B 3. Posición de A después de una elución parcial 4. Posición de B después de una elución parcial
5. Eluato
b) Cromatografía en placa fina.
Este sistema consta de un recipiente o cámara del tamaño adecuado para contener una
placa de vidrio ó aluminio adsorbida con sílica gel. En la cámara se coloca una muestra
del o los disolventes que servirán de fase móvil.
En la placa cromatográfica, que puede ser de tamaño variable, se coloca una muestra
de la mezcla de solutos, aproximadamente a 0.5 cm del extremo inferior de la placa.
Una vez que el eluyente (fase móvil) ha ascendido, por capilaridad hasta 0.5 cm antes

EDICIÓN 2015 71
del extremo superior de la placa, se saca de la cámara, se le evapora el disolvente y se
revela con luz ultravioleta (UV) si se han usado placas de sílica gel adsorbidas con
fluoresceína, en éste caso, el revelador será una lámpara de luz ultravioleta que opera
a una longitud de onda de 254 nm. También se puede revelar la placa cromatográfica
en una cámara de yodo ó por aspersión de un reactivo revelador como KMnO4 ó H2SO4
y calor:
Esquema gráfico que muestra la preparación de capilares para la aplicación de las muestras en la placa cromatográfica.

EDICIÓN 2015 72
Representación esquemática sobre la aplicación, elución y determinación de los Rf de los componentes de una muestra.

EDICIÓN 2015 73
Parámetros cromatográficos.
a) En el caso de la cromatografía en columna, éstos son:
i. Tiempo muerto (to). Tiempo que tarda un soluto idealmente no retenido, en
salir del sistema cromatográfico.
ii. Tiempo de retención (tr). Tiempo que tarda el 50 % de un soluto en eluir y
corresponde a la posición de máxima concentración en un cromatograma.
iii. Velocidad de flujo del disolvente (F).
iv. Volumen de retención (Vr): Volumen en el que eluye el 50 % de un soluto.
Se calcula con la ecuación:
Vr = tr F
Para procesos sistematizados (como son la Cromatografía de gases y la Cromatografía
de Líquidos a Alta Presión) encontramos que además de los incisos i a iv, dos
parámetros de gran importancia son:
v. Factor de selectividad (α): Medida relativa del grado de separación de dos
solutos. Se calcula mediante la ecuación:
oar
obr
tt
tt
)(
)(
vi. Resolución (Rs): Medida absoluta de separación. Se calcula mediante la
ecuación: 2)(
)(2 )()(
ba
brar
sWW
ttR
Donde Wa y Wb corresponden a los tiempos del ancho de un pico en su base.

EDICIÓN 2015 74
b) En el caso de la cromatografía en capa fina, los parámetros más usuales son:
i. Frente del disolvente (fd).
ii. Centro del soluto (fs).
iii. Relación de frentes (Rf): Cociente de la división fs/fd. Se utiliza con fines de
identificación de compuestos cuando se tienen patrones de referencia.
iv. Diferencia de Rfs. Medida cuantitativa del grado de separación.
REACTIVOS
Sílica-gel para columna 4 g
Hexano: Acetato de Etilo 1:1 30 mL
Acetato de Etilo: metanol 1:1 30 mL
Acetato de Etilo: metanol 7:3 10 mL
Acetato de Etilo: metanol 1:1 10 mL
Benceno 10mL
Acetona 100mL
MATERIAL
1 Soporte universal
1 Columna para cromatografía
1 Pinzas para bureta
2 Pipeta graduada de 5 mL
1 Pinzas universales
1 Vaso de precipitados de 100 mL
1 Matraz Erlenmeyer de 200 mL
10 Tubos de ensayo
1 Cámara cromatografíca
1 Capilares sin heparina
1 Cromatoplaca de sílica gel F254 de 5x2.5 cm
1 Agitador de vidrio
1 Probeta graduada de 100 mL
1 Gradilla para tubos de ensaye

EDICIÓN 2015 75
PARTE EXPERIMENTAL
EXPERIENCIA 1. CROMATOGRAFÍA EN PLACA FINA.
a. separación de carotenos
En una cromatoplaca de 5 2.5 cm, trazar con un lápiz de punta suave una línea a 0.5
cm de distancia del borde inferior; aplicar sobre ésta en dos puntos equidistantes, por
medio de una micropipeta previamente elaborada con un capilar (esquema pag. 66), la
muestra de carotenos. Dejar secar y desarrollar el cromatograma en una mezcla
hexano: acetona 6:1 (v/v), teniendo cuidado de que éste no rebase los puntos de
aplicación y de que eluya hasta 0.5 cm antes del borde superior de la placa. Retirar de
la cámara cromatográfica, marcar el centro de cada mancha observada y calcular los
valores de Rf. para cada componente.
b. Identificación de aceites esenciales por cromatografía en
placa fina.
Realizar el mismo procedimiento, pero ahora empleando como muestra los diferentes
aceites esenciales obtenidos en la práctica de arrastre de vapor, utilizando benceno
como fase móvil. Es importante mencionar que a diferencia de los carotenos, en este
caso se debe emplear un agente revelador para poder observar las diferentes manchas
en el cromatograma, para tal efecto, las placas se deberán sumergir en una solución de
permanganato de potasio al 10%, inmediatamente, las placas se enjuagaran con agua
hasta que se observen las manchas, correspondientes a los compuestos presentes en
la muestra. Calcular los Rf y compararlos con los de la siguiente tabla.

EDICIÓN 2015 76
EXPERIENCIA 2. SEPARACIÓN DE CAROTENOS POR CROMATOGRAFÍA EN
COLUMNA.
HACER EL SIGUIENTE PROCEDIMIENTO CON LA AYUDA DEL PROFESOR DE LA
SECCIÓN:
Colocar la columna para cromatografía con una pinza universal o con una pinza para
bureta e introducirle por medio de un agitador, un trozo de algodón hasta la base de la
misma, de tal manera que la sílica no se salga al momento de empacar la columna. Hay
que tomar cuenta que no debe ser mucho algodón, ya que si es así, la velocidad de
elución será muy lenta.
Empacar la columna con sílica gel adicionando por la parte superior una suspensión de
4 g de sílica gel en una mezcla de hexano: acetona 1:1 manteniendo un goteo uniforme
en la llave de salida del disolvente durante todo el procedimiento.
Dejar gotear el disolvente y esperar hasta que éste quede ligeramente arriba de la fase
estacionaria, una vez empacada la columna con la sílica, agregar un poco de cloruro de
sodio de tal forma que quede una capa de aproximadamente 1 cm; posteriormente
agregar 10 gotas del concentrado de espinacas con ayuda de una pipeta Pasteur,
cuidando de no derramarlo por las paredes de la columna ni que se mezcle con la fase
móvil; nunca permitir que el nivel del disolvente baje más allá de la superficie de la
sílice, ya que ésta se agrietaría y no permitiría un desarrollo homogéneo).
Mantener el goteo hasta que los pigmentos penetren en la sílice y llenar lentamente la
columna con la mezcla de hexano-acetona 1:1. Volver a abrir la llave para permitir el
desarrollo del cromatograma, manteniendo constante el volumen del eluyente.
Recoger la primera fracción colorida en tubos de ensayo numerados y cubiertos con
papel aluminio. Cuando termine de eluir esta primera fracción, cambiar el eluyente a
acetato de etilo: metanol 1:1 y recoger la siguiente fracción en tubos de ensayo
numerados y cubiertos con papel aluminio.

EDICIÓN 2015 77
EXPERIENCIA 2. SEPARACIÓN DE LICOPENOS.
Hacer la cromatografía en columna del extracto de jitomate para la separación de
licopenos, en la misma forma en que se efectuó para el extracto de espinaca.
RESULTADOS Y CONCLUSIONES.
El alumno deberá analizar y concluir cada una de las experiencias realizadas.
CUESTIONARIO
1. Indicar qué precauciones se deben tener al empacar una columna de cromatografía.
________________________________________________________________________________________________________________________________________________________________________________________________________________
2. Indicar qué fracciones se separaron por cromatografía en columna, de los extractos
de espinaca, jitomate y cómo se identificaron.
________________________________________________________________________________________________________________________________________________________________________________________________________________
3. Escriba ¿qué conclusiones se obtienen de la cromatografía en capa fina y de la
cromatografía en columna?
________________________________________________________________________________________________________________________________________________________________________________________________________________
4. Definir los siguientes términos: Adsorción, partición, eluyente, eluato, disolvente,
fase móvil, fase estacionaria, Rf.
________________________________________________________________________________________________________________________________________________________________________________________________________________
5. Especificar en qué parte de la práctica tienen aplicación los términos anteriores.
________________________________________________________________________________________________________________________________________________________________________________________________________________

EDICIÓN 2015 78
6. Hacer una comparación entre la cristalización, extracción, destilaciones y
cromatografía, en cuanto a la eficiencia como métodos de separación y purificación.
________________________________________________________________________________________________________________________________________________________________________________________________________________
7. Establecer una distinción clara entre cromatografía en columna y capa fina e indicar
en qué casos es preferible alguno de los dos métodos.

EDICIÓN 2015 79
PRÁCTICA 7
SÍNTESIS A MICROESCALA DE ÁCIDO FUMÁRICO
OBJETIVOS.
1.- Convertir un isómero cis- en un isómero trans-.
2.- Diferenciar los isómeros geométricos del ácido 2-butenodioico.
INTRODUCCIÓN.
El término estereoisómeros se utiliza para designar los compuestos que poseen igual
fórmula molecular e igual conectividad, pero que se distinguen entre sí por la relación
espacial de los átomos o grupos dentro de la molécula.
Esto origina dos tipos de estereoisómeros:
a) Los que guardan entre sí una relación objeto-imagen especular.
b) Aquellos en los que esta relación no existe.
Los primeros se denominan enantiómeros y los segundos diastereómeros. Un tipo de
diastereómeros es el de los isómeros geométricos, que deben su existencia a la
imposibilidad de rotación a través de un doble enlace o de un anillo. Al bisectar un doble
enlace con un plano que pase por el núcleo de los dos átomos de éste, dos grupos
diferentes entre si pueden ubicarse de dos maneras con relación al doble enlace.
1. Ambos del mismo lado del plano.
2. Uno a cada lado del plano.
El primer arreglo da lugar a la isomería cis- (del latín: a este lado), en tanto que el
segundo origina la isomería trans- (del latín: al otro lado).
De acuerdo con esto, existen 2 ácidos 2-butenodioicos: el cis-2-butenodioico y el trans-
2-butenodioico
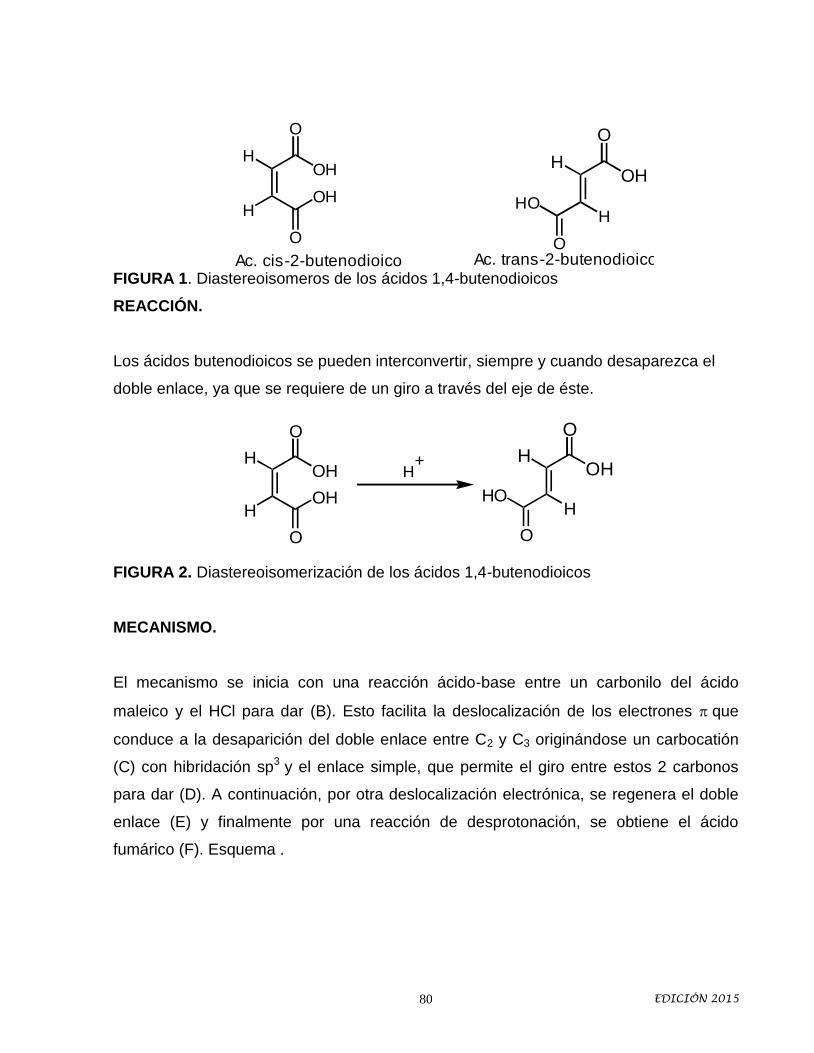
EDICIÓN 2015 80
FIGURA 1. Diastereoisomeros de los ácidos 1,4-butenodioicos
REACCIÓN.
Los ácidos butenodioicos se pueden interconvertir, siempre y cuando desaparezca el
doble enlace, ya que se requiere de un giro a través del eje de éste.
FIGURA 2. Diastereoisomerización de los ácidos 1,4-butenodioicos
MECANISMO.
El mecanismo se inicia con una reacción ácido-base entre un carbonilo del ácido
maleico y el HCl para dar (B). Esto facilita la deslocalización de los electrones que
conduce a la desaparición del doble enlace entre C2 y C3 originándose un carbocatión
(C) con hibridación sp3 y el enlace simple, que permite el giro entre estos 2 carbonos
para dar (D). A continuación, por otra deslocalización electrónica, se regenera el doble
enlace (E) y finalmente por una reacción de desprotonación, se obtiene el ácido
fumárico (F). Esquema .
OH
OH
O
O
H
H H
OH
O
H
O
HO
Ac. cis-2-butenodioico Ac. trans-2-butenodioico
OH
OH
O
O
H
H H
OH
O
H
O
HO
H
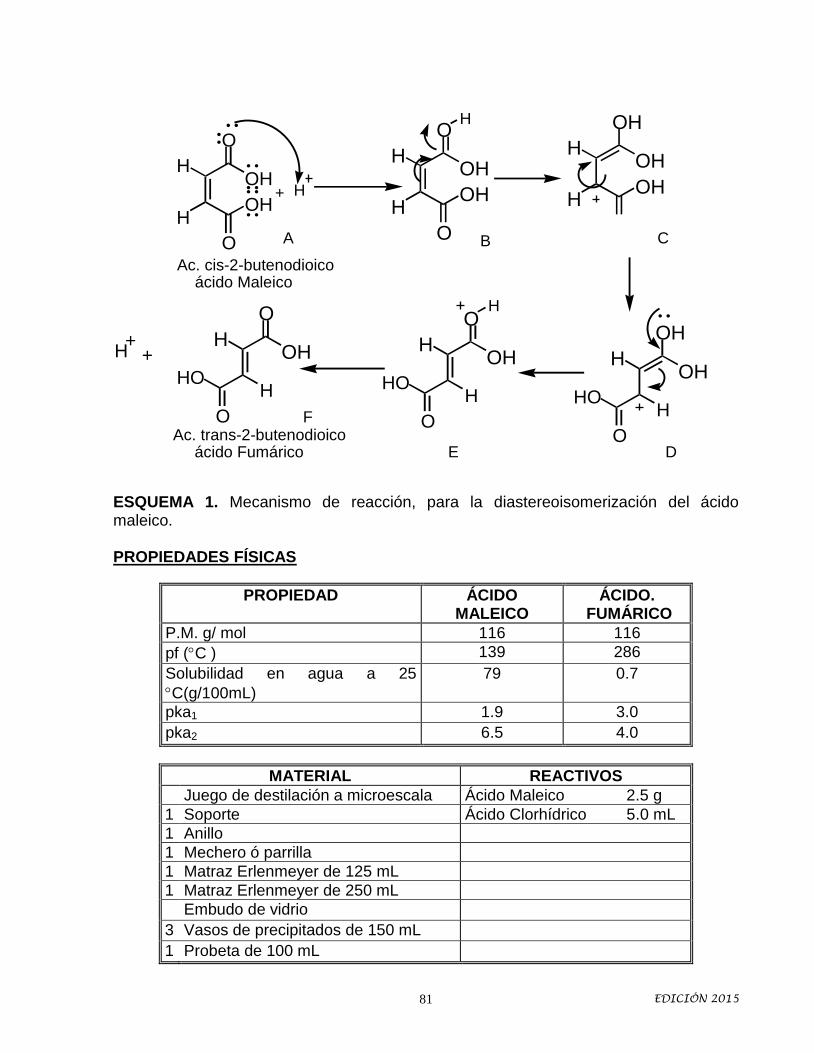
EDICIÓN 2015 81
ESQUEMA 1. Mecanismo de reacción, para la diastereoisomerización del ácido maleico. PROPIEDADES FÍSICAS
PROPIEDAD ÁCIDO MALEICO
ÁCIDO. FUMÁRICO
P.M. g/ mol 116 116
pf (C ) 139 286
Solubilidad en agua a 25
C(g/100mL)
79 0.7
pka1 1.9 3.0
pka2 6.5 4.0
MATERIAL REACTIVOS
Juego de destilación a microescala Ácido Maleico 2.5 g
1 Soporte Ácido Clorhídrico 5.0 mL
1 Anillo
1 Mechero ó parrilla
1 Matraz Erlenmeyer de 125 mL
1 Matraz Erlenmeyer de 250 mL
Embudo de vidrio
3 Vasos de precipitados de 150 mL
1 Probeta de 100 mL
OH
OH
O
O
H
H
H
B
OH
OH
OH
H
H
C
H
OH
OH
H
O
HO
D
H
OH
O
H
O
HO
H
E
H
OH
O
H
O
HO
H +
+
Ac. trans-2-butenodioico
ácido Fumárico
F
OH
OH
O
O
H
H
H +
Ac. cis-2-butenodioico
ácido Maleico
A

EDICIÓN 2015 82
PARTE EXPERIMENTAL
Preparar por sección una solución al 40 % de ácido maleico en agua, se puede calentar
para favorecer la solución. El volumen será determinado de acuerdo al número de
equipos por sección. El volumen que ocupará cada equipo será de 1ml.
1.- En el matraz de reacción verter 1 mL de la solución al 40 % de ácido maléico,
medido con pipeta graduada.
2.- Adicionar lentamente 2 mL de ácido clorhídrico concentrado por las paredes del
matraz de reacción (líquido irritante, evitar inhalación y el contacto con la piel).
3.- Adaptar el refrigerante en posición de reflujo
4.- Calentar la mezcla durante 30 minutos a reflujo moderado.
5.- Enfriar a temperatura ambiente y separar por filtración los cristales de ácido
fumárico.
6.- Recristalizar en agua y secar.
7.- Identificar el producto por su punto de fusión.
CUESTIONARIO EXPERIMENTAL.
1. Escribe tus observaciones del proceso de interconversión de isómeros.
__________________________________________________________________________________________________________________________________________________________________________________________________________________
2. Anota las características del producto crudo y el producto recristalizado incluyendo
el punto de fusión de éste último.
__________________________________________________________________________________________________________________________________________________________________________________________________________________
Responda las siguientes preguntas:
En la síntesis de ácido fumárico, indicar qué papel desempeña el ácido clorhídrico.

EDICIÓN 2015 83
3. Explicar cómo se pueden justificar:
a) La diferencia en los puntos de fusión de los isómeros estudiados.
b) La diferencia de solubilidad en agua.
c) La diferencia en la primera y segunda constantes de acidez.
d) Explicar a qué se debe que en el reflujo se observe formación y separación de
cristales.
4. Indicar porqué, de los isómeros cis- y trans- de esta práctica es la forma trans la que
predomina.
5. Indicar qué pruebas químicas se pueden llevar a cabo para evidenciar la presencia
del grupo funcional carboxilo y la insaturación en el ácido fumárico.

EDICIÓN 2015 84
B
A+ AB
Eliminación 1,2 o Beta
PRÁCTICA 8
SÍNTESIS DE CICLOHEXENO
OBJETIVOS:
1. Ilustrar las reacciones de eliminación, efectuando una
deshidratación de alcoholes.
2. Sintetizar un alqueno cíclico.
3. Aplicar una reacción de adición para identificar el producto
INTRODUCCIÓN. En Química Orgánica, el término eliminación se refiere a la pérdida de dos átomos ó
grupos de una molécula para formar enlaces múltiples mediante la pérdida de dos
grupos unidos a átomos adyacentes. El proceso se denomina eliminación 1,2 ó beta-
eliminación a causa de la disposición relativa de los dos grupos que se pierden, si la
molécula que se pierde es agua, la reacción se denomina deshidratación.
La deshidratación de alcoholes es una reacción de eliminación catalizada por ácidos, la
evidencia muestra que los alcoholes reaccionan en el orden terciario > secundario >
primario, ésta reactividad se relaciona directamente con la estabilidad del carbocatión
intermediario formado en la reacción. En las reacciones de deshidratación llevadas a
cabo en el laboratorio se usa generalmente ácido fosfórico ó ácido sulfúrico como
catalizador, sin embargo, en los procesos industriales de deshidratación se usa óxido
de aluminio ó sílica gel como ácidos de Lewis para ésta reacción.
En las reacciones de eliminación, al igual que las reacciones de sustitución nucleofílica,
interviene una especie con pares libres de electrones, por lo cual las reacciones de
eliminación se encuentran en competencia con las reacciones de sustitución
nucleofílica.

EDICIÓN 2015 85
La reacción que se realizará en esta práctica es una –eliminación, conducente a la
formación de un alqueno, ésta reacción está en competencia con la sustitución
nucleofílica, por lo que se deberán controlar las condiciones de reacción para favorecer
la primera.
Se consideran dos posibilidades mecanísticas para la reacción de Eliminación :
1) El Mecanismo E2
2) El Mecanismo E1
Mecanismo E2: Tanto el sustrato como el nucleófilo participan en el paso lento de la
reacción. La base ó el nucleófilo toman al protón ácido al mismo tiempo que el grupo X
sale de la molécula.
X
H
CH2
CH2
Base-
+ BH
La velocidad depende de la concentración de ambos reactivos, por lo cual es una
reacción de segundo orden.
V = k [Nu:] [sustrato] Mecanismo E1: En éste, sólo el sustrato participa en el paso que controla la velocidad.
Se propone la formación de un carbocatión intermediario.
X
HH
H2C CH
2CH
2
+
La velocidad de la reacción, depende de la concentración del sustrato, por lo cual la
reacción es de primer orden.
V = k [sustrato]
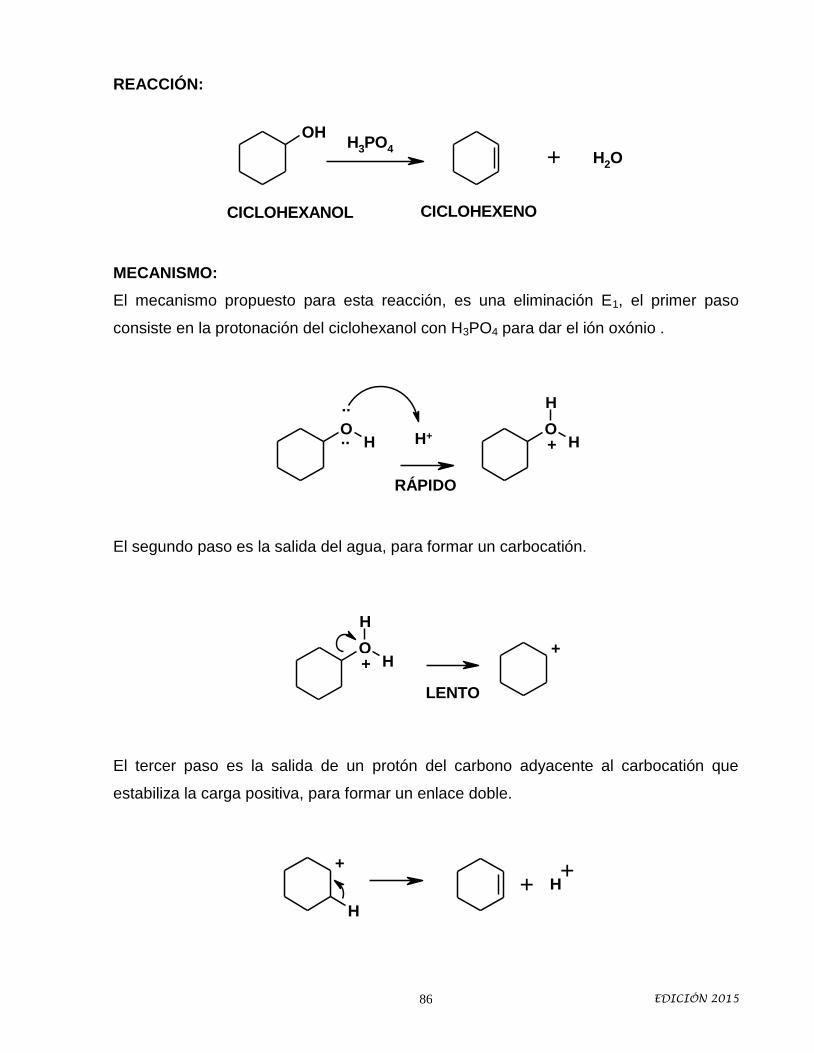
EDICIÓN 2015 86
REACCIÓN:
OHH3PO4
CICLOHEXANOL CICLOHEXENO
+ H2O
MECANISMO:
El mecanismo propuesto para esta reacción, es una eliminación E1, el primer paso
consiste en la protonación del ciclohexanol con H3PO4 para dar el ión oxónio .
O
H
HO
H
RÁPIDO
+H+
..
..
El segundo paso es la salida del agua, para formar un carbocatión.
O
H
H++
LENTO
El tercer paso es la salida de un protón del carbono adyacente al carbocatión que
estabiliza la carga positiva, para formar un enlace doble.
H
+
+ H+

EDICIÓN 2015 87
REACTIVOS MATERIAL
Ácido Fosfórico 2 mL 2 Soportes universales
Bromo CCl4 1 mL 1 Anillo de fierro
Ciclohexanol 5 mL 2 Pinza universal
Carbonato de sodio al 10% 4 mL 1 Vaso de precipitados de 50 mL
NaCl 1 Embudo de separación de 100 mL
Sulfato de sodio Anhidro 1 Baño María
1 Matraz balón de 50 mL 14/23
1 Matraz Erlenmeyer de 125 mL (matraz colector)
1 Refrigerante de aire 14/23
1 Refrigerante de agua 14/23
3 Tubos de ensayo de 10 mL
2 Pipeta graduada de 10 mL
1 T de destilación 14/23
1 Portatermómetro 14/23
1 Termómetro
PROPIEDADES FÍSICAS.
COMPUESTO P.M. (g/mol)
p. f. p. eb. ºC
DENSIDAD g/mL
SOLUBILIDAD TOXICIDAD
Ciclohexanol 100.16 23-
25ºC 161 0.962
s. en Etanol p.s. agua
Líquido irritante de ojos y nariz
s. soluble p.s. poco soluble
PARTE EXPERIMENTAL.
Preparación de Ciclohexeno:
1. Adaptar un aparato de destilación fraccionada con el matraz colector (matraz
Erlenmeyer de 125 mL) sumergido en hielo.
2. Colocar en el matraz de balón 5 mL de ciclohexanol y 2 mL de ácido fosfórico.
Precaución: El ácido fosfórico es un líquido corrosivo, irritante del tracto
respiratorio y de la piel.
3. Calentar lentamente el sistema hasta alcanzar 110-120 ºC y destilar el líquido hasta
que quede 1 mL de residuo en el matráz de reacción.
4. Adicionar al destilado NaCl hasta que desaparezca la turbidez y pasarlo a un
embudo de separación.

EDICIÓN 2015 88
5. Agregar carbonato de sodio al 10 % al ciclohexeno obtenido hasta alcanzar un pH
alcalino.
6. Eliminar la capa acuosa inferior y pasar el ciclohexeno a un vaso de precipitados de
50 mL.
7. Adicionar sulfato de sodio anhidro al ciclohexeno hasta que desaparezca la turbidez.
8. Destilar el ciclohexeno a 80 °C con un aparato de destilación fraccionada.
9. Identificar el producto colocando 0.5 mL de ciclohexeno en un tubo de ensayo y
adicionándole a éste unas gotas de Bromo en CCl4. Observar si la solución roja de
Br2/CCl4 cambia de coloración. Esto indica una prueba positiva
CUESTIONARIO EXPERIMENTAL. 1. Explicar qué líquido se obtiene en el primer destilado.
______________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
2. Explicar cuál es la razón de saturar el primer destilado con NaCl.
______________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
3. Explicar cuál es la razón de lavar con una solución de carbonato de sodio
______________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
4. Explicar para qué se agrega Na2CO3 al destilado.
______________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________

EDICIÓN 2015 89
Observaciones y Resultados:
Análisis de Resultados y Conclusiones:
Química Orgánica. Fox M. A. & Whitesell S. K., 2a edición Addison Wesley
Longman, México 2000.
Química Orgánica Experimental. Xorge A. Domínguez y Xorge A. Domínguez
S.,editorial Limusa 1a Edición, 1982,
Experiments in organic chemistry, J. S. Nimitz. Prentice-Hall Inc. 1991.

EDICIÓN 2015 90
PRÁCTICA 9
SÍNTESIS DE CLORURO DE ter-BUTILO
OBJETIVO:
1. Efectuar la síntesis de cloruro de ter-butilo, mediante una reacción de sustitución
nucleofílica unimolecular (SN1)
INTRODUCCIÓN:
Uno de los principales métodos de obtención de halogenuros de alquilo es la sustitución
nucleofílica alifática de alcoholes. Esta sustitución es posible realizarla con nucleófilos
(Nu), tales como PCl3, SOCl2, HCl, HBr, etc.
REACCIÓN:
ROH + Nu: R Nu + HOH
Dependiendo de las condiciones experimentales, las reacciones de sustitución
nucleofílica alifática, pueden ser de dos tipos
a) Sustitución Nucleofílica Unimolecular (SN1).
b) Sustitución Nucleofílica Bimolecular (SN2).
En las reacciones de SN1, la velocidad de la reacción depende de la concentración del
alcohol, siendo independiente del nucleófilo, teniendo influencia, la polaridad del
disolvente, la estructura del alcohol, la naturaleza del grupo saliente y el tipo de
nucleófilo.
REACCIÓN:
OHCl+ +HCl H2O

EDICIÓN 2015 91
MECANISMO:
La primera etapa, consiste en una reacción ácido-base entre él hidroxilo del alcohol y el
protón del ácido para dar la formación del intermediario (A). En la etapa siguiente, que
constituye el paso lento de la reacción, se lleva a cabo la formación del carbocatión (B),
mediante la pérdida de una molécula de agua. Finalmente el nucleófilo (cloruro)
reacciona con el carbocatión para formar el cloruro de ter-butilo (C).
Los disolventes polares como el agua, etanol, etc., favorecen la reacción, solvatando al
halógeno, facilitando la formación del carbocatión. De igual forma el empleo de ácidos
de Lewis y el efecto del ión común incrementa la velocidad de estas reacciones.
OH O
H
H
O
H
H
ClCl
+ H+ +
1) (A)
+2) + (B)
+ +-
3) (C)
MATERIAL REACTIVOS
1 Matraz Erlenmeyer de 250 mL Alcohol terbutílico
1 Probeta de 125 mL Ácido clorhídrico
1 Embudo de separación Cloruro de calcio
2 Vasos de precipitados de 100 mL Solución de bicarbonato de sodio al 5%.
1 Tapón de hule

EDICIÓN 2015 92
PARTE EXPERIMENTAL:
1. En un matraz Erlenmeyer de 250 mL colocar 10 mL de alcohol ter-butílico
(líquido inflamable e irritante) y 30 mL de ácido clorhídrico concentrado (Líquido
corrosivo).
2. Tapar el matraz y agitarlo vigorosamente, evitando cualquier exceso de presión
dentro del matraz.
3. Después de 20 minutos de agitación agregar 3 g de cloruro de calcio y disolver.
4. Colocar la mezcla de reacción anterior en un embudo de separación y eliminar la
fase acuosa, verter la fase orgánica en un vaso de precipitados y adicionar
lentamente 10 mL de una solución de bicarbonato de sodio al 5 %.
5. Cuando ya no se observe desprendimiento de bióxido de carbono, separa las
dos fases eliminando la capa acuosa utilizando el embudo de separación.
6. Finalmente secar el cloruro de ter-butilo, adicionando aproximadamente 1 g de
sulfato de sodio anhidro.
CUESTIONARIO:
1. ¿Cuál es el propósito de adicionar cloruro de calcio 20 minutos después de
iniciada la reacción?
2. Explica por qué se adiciona solución de bicarbonato de sodio al 5% cuando se
lava el cloruro de ter-butilo y porqué no se puede usar hidróxido de sodio.
3. Al adicionar bicarbonato de sodio se produce una efervescencia, indica de que
gas está formada y explica mediante una reacción, ¿Cómo se produce?

EDICIÓN 2015 93
CINÉTICA QUÍMICA:
EFECTO DEL DISOLVENTE EN LA VELOCIDAD
DE UNA REACCIÓN DE SOLVÓLISIS.
OBJETIVOS:
1. Conocer los métodos de estudio de los mecanismos de reacción.
2. Analizar los diferentes factores de importancia en el método cinético.
3. Relacionar el orden de reacción con los diferentes tipos de reacciones orgánicas.
4. Estudiar una reacción de solvólisis.
5. Comprobar el efecto de la polaridad del disolvente sobre la velocidad de una
reacción de primer orden.
INTRODUCCIÓN
La mayoría de las reacciones orgánicas son complejas, realizándose en varias etapas
que difieren por su velocidad. Para poner de manifiesto estos cambios existen
diferentes métodos de estudio que dan información acerca de la estructura de los
intermediarios y de los complejos activados, del orden y de la molecularidad de la
reacción y de la naturaleza de la etapa que controla la velocidad de la reacción, entre
otros. Los métodos de estudio de los mecanismos de las reacciones químicas se
clasifican de manera general en dos grupos:
MÉTODOS CINÉTICOS
MÉTODOS NO CINÉTICOS.

EDICIÓN 2015 94
MÉTODOS CINÉTICOS:
Como se ha indicado, las reacciones orgánicas se llevan a cabo en varios pasos; uno
de éstos es más lento que otros y este último es el que determina la velocidad total de
la reacción. Un estudio cinético proporciona datos sobre el número y la naturaleza de
las moléculas que intervienen en la etapa que controla la velocidad total.
La velocidad de una reacción se determina por el cambio de la concentración de
los reactivos a diferentes intervalos de tiempo; esta relación se conoce como orden de
reacción. Así en una reacción de primer orden, la velocidad de reacción es proporcional
a la concentración de sólo uno de los reactivos.
Esto es:
Algunos factores que modifican la velocidad de una reacción son la temperatura, la
fuerza iónica y la naturaleza del disolvente.
EFECTO DEL DISOLVENTE
El cambio en la polaridad del disolvente, puede afectar la velocidad de reacción y
también puede cambiar el orden de la misma. En general podemos considerar que:
a) Un aumento en la constante dieléctrica del disolvente, aumenta la velocidad de las
reacciones en las que la densidad de carga es más grande en el estado de
transición que en los reactivos.
b) Un aumento en la constante dieléctrica del disolvente, disminuye la velocidad de las
reacciones en las que la densidad de carga es menor en el estado de transición que
en los reactivos.
dt
AdAkvA
Av
k - constante de velocidad
- velocidad de la reacción de primer orden
- concentración molar del reactivo A
t - tiempo
A
donde
EDICIÓN 2015 95
En la siguiente tabla se indican ejemplos de algunos disolventes y sus polaridades.
NOMBRE DEL DISOLVENTE
CONSTANTE DIELÉCTRICA (ε)
MOMENTO DIPOLO (μ)
Disolventes próticos
Ácido acético 6.0 1.68
Etanol 26.0 1.66
Metanol 32.0 2.87
Agua 81.0 1.84
Ácido Fórmico 58.0 1.82
Disolventes apróticos
Ciclohexano 2.0 0.0
Benceno 4.0 0.0
Cloroformo 5.0 1.01
Los halogenuros de alquilo de tipo terciario R3CX presentan una cinética de primer
orden y el mecanismo que se lleva a cabo se conoce como Sustitución Nucleofílica
Unimolecular (SN1). Estas reacciones son posibles sólo si el nucleófilo es una base
sumamente débil, ya que una base fuerte daría la reacción de Eliminación. El agua y los
alcoholes son bases sumamente débiles, así que pueden ser empleados como
nucleófilos en las reacciones SN1.
Las reacciones que se llevan a cabo por un mecanismo SN1 son favorecidas en
disolventes polares próticos y con constante dieléctrica alta, ya que hay una
estabilización del carbocatión intermediario. Cuando el nucleófilo que se emplea es a la
vez el disolvente, se le llama REACCIÓN DE SOLVÓLISIS.
En esta práctica se pone de manifiesto el efecto del disolvente sobre la velocidad
relativa, en una reacción de solvólisis, en la cual se varía la naturaleza y proporción del
disolvente. La reacción se puede ejemplificar con el cloruro de ter-butilo y agua.
En esta reacción se produce alcohol ter-butílico y se libera una molécula de HCl;
así, el curso de la reacción se puede seguir por la acidez que se produce en la mezcla
de reacción, que se manifiesta al agregar unas gotas del indicador azul de bromotimol
que cambiará su color de azul verdoso (pH=7) a amarillo (pH 5.8).
(CH3)3CCl + H2O (CH3)3COH + HCl

EDICIÓN 2015 96
REACCIONES DE SOLVÓLISIS
Cloruro de ter-butilo
Metanol Etanol Propanol
Solubilidad insoluble en agua
soluble en agua
soluble en agua
Soluble en agua
P.eb. 73.3°C 65°C 78.5°C 97.4°C
MECANISMO (SN1):
El cloruro de ter-butilo, molécula polar (A), forma interacciones dipolo-dipolo con
moléculas del disolvente. En una etapa lenta se obtiene el carbocatión intermediario
(B), en el cual el momento dipolar es mayor que el del cloruro de ter-butilo original; un
disolvente polar estabilizará el estado de transición que lleva a la formación de este
carbocatión, ocasionando una disminución en la energía de activación.
Etapa 1:
Este carbocatión intermediario en una etapa rápida, interacciona con una
molécula de agua o de metanol, produciendo (C) o (C1) respectivamente.
Etapa 2:
Finalmente el oxonio resultante se desprotona generando los productos finales (D) o
(D1).
Cloruro de ter-butilo
Agua/Metanol Alcohol ter-butílico
ter-Butilmetiléter
(CH3)3CCl + H2O/CH3OH (CH3)3COH + (CH3)3COCH3 + HCl
agua
metanol
(CH3)3C+ O H
H
(CH3)3C O H
H
(rápida)
B
C
(CH3)3CH+ O CH3
H
(CH3)3C O CH4
H
(rápida)
B
C1
(CH3)3C Cl (CH3)3C+ + Cl (lenta)
A B

EDICIÓN 2015 97
Etapa final
En las dos rutas descritas se libera HCl. Este ácido es el producto que sirve de
referencia para poder apreciar la variación de la velocidad de reacción en función del
cambio y proporción del disolvente.
PARTE EXPERIMENTAL:
REACTIVOS MATERIAL
Cloruro de ter-butilo 6 Buretas (por sección)
Propanol 3 Pipetas graduadas de 10 mL
Etanol 95% 18 Tubos con tapón
Metanol 1 Termómetro
Fenolftaleína 1 Vaso de precipitados de 250 mL
Disolución de NaOH al 10% Baño maría
Gradilla
Cronómetro o reloj
Preparar tres sistemas de dos buretas cada uno, de la siguiente forma.
SISTEMA BURETA 1 BURETA 2
I Metanol Agua
II Etanol Agua
III Propanol Agua
(CH3)3C O H
H
+ HOH (CH3)3C O H + HOH
H
(rápida)
DC
(CH3)3C O CH3
H
+CH3OH (CH3)3C O CH3 +CH3OH
H
(rápida)
D1C1
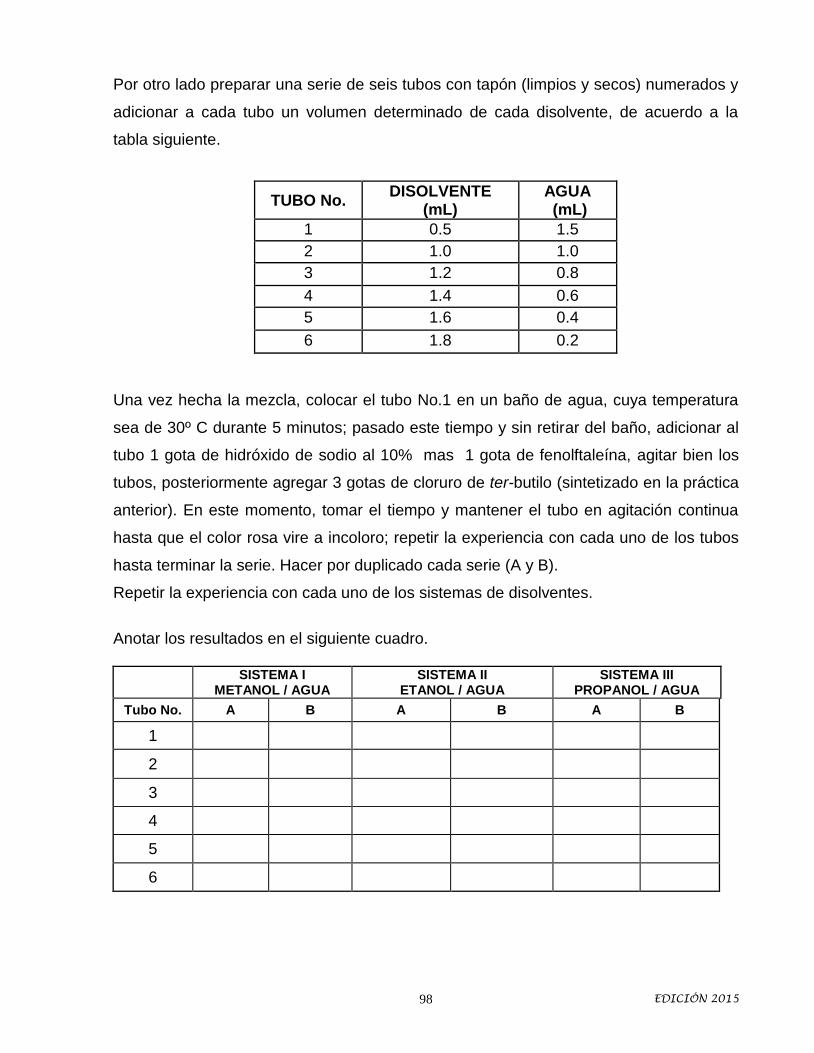
EDICIÓN 2015 98
Por otro lado preparar una serie de seis tubos con tapón (limpios y secos) numerados y
adicionar a cada tubo un volumen determinado de cada disolvente, de acuerdo a la
tabla siguiente.
TUBO No. DISOLVENTE
(mL) AGUA (mL)
1 0.5 1.5
2 1.0 1.0
3 1.2 0.8
4 1.4 0.6
5 1.6 0.4
6 1.8 0.2
Una vez hecha la mezcla, colocar el tubo No.1 en un baño de agua, cuya temperatura
sea de 30º C durante 5 minutos; pasado este tiempo y sin retirar del baño, adicionar al
tubo 1 gota de hidróxido de sodio al 10% mas 1 gota de fenolftaleína, agitar bien los
tubos, posteriormente agregar 3 gotas de cloruro de ter-butilo (sintetizado en la práctica
anterior). En este momento, tomar el tiempo y mantener el tubo en agitación continua
hasta que el color rosa vire a incoloro; repetir la experiencia con cada uno de los tubos
hasta terminar la serie. Hacer por duplicado cada serie (A y B).
Repetir la experiencia con cada uno de los sistemas de disolventes.
Anotar los resultados en el siguiente cuadro.
SISTEMA I
METANOL / AGUA SISTEMA II
ETANOL / AGUA SISTEMA III
PROPANOL / AGUA
Tubo No. A B A B A B
1
2
3
4
5
6

EDICIÓN 2015 99
TRATAMIENTO DE RESIDUOS
Los residuos del experimento son depositados en el frasco de “RESIDUOS
ORGÁNICOS CLORADOS”.
CUESTIONARIO:
1. Indicar por qué en las reacciones de solvólisis realizadas, es importante:
a) Controlar la temperatura a + 30 ºC
b) Trabajar con el material seco.
c) Medir exactamente los reactivos.
2. Escribir todas las reacciones de solvólisis que se realizaron con el cloruro de
terbutilo.
3. Definir los siguientes términos:
a) Orden de reacción
b) Molecularidad
c) Solvólisis
d) Par iónico
e) Constante dieléctrica ()
4. Ejemplificar con uno de los sistemas empleados, la reacción competitiva de la SN1.
5. Con los valores de la constante dieléctrica indicados ejemplificar una reacción de
solvólisis, diferente a la realizada en el laboratorio.
6. Dibujar un diagrama de energía con uno de los sistemas empleados, indicando los
siguientes términos: energía de activación, complejo activado, paso lento.
7. Con los datos obtenidos en la reacción de solvólisis, graficar de la siguiente forma:
abscisas -% de agua, ordenadas – tiempo.

EDICIÓN 2015 100
BIBLIOGRAFÍA
1. Laidler, K. J., Cinética de Reacciones, Vol. 1., Editorial Alhambra. Madrid, 1971, P.
1-10, 19 y 29.
2. Morrison, R. T. y Boyd, R. N., Química Orgánica, 3. edición Fondo Educativo
Interamericano, S. A. México, 1976, P. 473-475, 480-490.
3. Brewster, R. Q. y Vander Werf, C. A., Curso Práctico de Química Orgánica, 2ª
edición, Editorial Alhambra, España, 1970.
4. Moore, J. A. y Dalrymple, D. L., Experimental Methods in Organic Chemistry, 2.
edición, W. B. Sauders Company, U.S.A., 1976, P. 271-275.
5. Weininger, S. J., Química Orgánica, Editorial Interamericana, México, 1975,
Pags:137-140.

EDICIÓN 2015 101
PRACTICA 10
SÍNTESIS DE orto- y para-NITROBROMOBENCENO
OBJETIVO
1. Demostrar la reacción de sustitución electrofílica aromática (SEA) mediante la
nitración del bromobenceno.
2. Demostrar el efecto orientador del Bromo a las posiciones orto- y para- en las
reacciones de SEA
ANTECEDENTES
Reacciones de sustitución electrofílica aromática
INTRODUCCIÓN
Un doble enlace carbono-carbono (C=C) es una especie rica en densidad electrónica,
ésta densidad electrónica es la característica dominante en las reacciones que
experimenta dicho grupo funcional, sin embargo, los dobles enlaces C=C en los
sistemas aromáticos presentan diferente reactividades si se someten a reacciones bajo
las mismas condiciones. La razón de ésta diferente reactividad es la tendencia de los
dobles aromáticos a conservar la aromaticidad del sistema.
La falta de reactividad de los dobles enlaces aromáticos no implica que no existan
reacciones de sustitución aromática, como ya se mencionó, sino que el doble enlace
aromático es menos reactivo que un doble enlace aislado, en muchos casos es
necesaria la presencia de catalizadores ácidos para que el doble enlace aromático
reaccione con los electrófilos
El mecanismo por el cual reaccionan los dobles enlaces aromáticos frente a electrófilos,
es la Sustitución Electrofílica Aromática. El primer paso de la reacción es la adición del
electrófilo (E+) a un doble enlace (C=C) aromático, formándose un carbocatión
intermedio (A) Fig. 1, el cual se encuentra estabilizado por varias formas de resonancia
a través del anillo bencénico (B y C), pero se encuentra desestabilizado debido a que el
sistema, inicialmente aromático, ha perdido la deslocalización completa que va
asociada con la aromaticidad. La pérdida de un protón en la última etapa permitirá que

EDICIÓN 2015 102
el sistema aromático se regenere y, cuando ello ocurra, se volverá a ganar la energía
de resonancia.
La reacción ocurre del mismo modo en todos los casos en que se tenga una reacción
de tipo SEA, independientemente del electrófilo o del compuesto aromático que tome
parte en la reacción Los detalles que son distintos en cada ocasión son la identidad del
electrófilo (E+), la facilidad con que éste se forma, la velocidad con la que ataca al
compuesto aromático y la estabilidad del compuesto intermedio (A).
+ E+
E E E
A B C
E
A
H E
+ H+
Fig.1. Mecanismo general de una reacción de Sustitución Electrófilica Aromática.
En la nitración de compuestos aromáticos, el electrófilo (E+), es el ión nitrónio (+NO2), el
cual es generado a partir de la reacción entre el ácido nítrico (HNO3) y el ácido sulfúrico
(H2SO4) como se muestra en la siguiente reacción:
O N
OH
O O
S
O
HO OH+ N
O
O
+ HSO4- + H2O
Los grupos electrodonadores (-NH2, -OH, Cl, Br, -SH, etc) hacen mas rico en densidad
electrónica al sistema aromático, por lo que lo activan hacia la reacción de sustitución

EDICIÓN 2015 103
electrofílica aromática, y orientan la entrada del electrófilo preferentemente a las
posiciones orto- y para-.
PARTE EXPERIMENTAL
1. Material por equipo
3 vasos de precipitados de 125 mL 1 Mechero de Bunsen
1 Equipo de destilación simple 1 Anillo de fierro
1 Matraz Erlenmeyer 1 Rejilla de asbesto
1 Probeta de 50 mL 2 Pinzas universales con nuez
2 Pipetas graduadas de 10 mL 1 Baño María
1 Embudo de separación de 200 mL Tubería de hule
5 Tubos de ensayo 1 Capilares
2. Material y equipo por sección
4 cámaras cromatográficas Lampara U.V. de = 254 nm
Placas de sílica gel F254
3.-Reactivos y disolventes
Bromobenceno Etanol (96 %)
Ácido nítrico concentrado Cloruro de metileno
Ácido sulfúrico concentrado Hexano
Solución saturada de bicarbonato de sodio
Acetato de etilo
Solución saturada cloruro de sodio Sulfato de sodio anhidro
PROCEDIMIENTO
Precauciones: utilice guantes y efectúe todos los trasvases en campanas. No
invierta el orden de mezclado de los ácidos.
En un vaso de precipitados mezclar 7 mL de ácido nítrico concentrado seguido de 7 mL
de ácido sulfúrico concentrado. La mezcla de ácidos se lleva a un baño de hielo
agitando constantemente por un periodo de 10 min. A la mezcla de ácidos se le
adiciona poco a poco 5 mL de bromobenceno con agitación constante sin retirar la

EDICIÓN 2015 104
mezcla del baño de hielo. Una vez terminada la adición del bromobenceno, la mezcla se
retira del baño de hielo y se agita durante 15 minutos hasta la aparición de nódulos
blancos (o una masa amarilla), si la temperatura de la mezcla se eleva, llevarla
nuevamente al baño de hielo.
El sólido formado se disuelve con 20 mL de cloruro de metileno, la mezcla bifásica se
pasa a un embudo de separación (la fase orgánica se torna de un color ligeramente
amarillo) y se desecha la fase acuosa ácida (inferio). La fase orgánica se lava con 10
mL de agua, posteriormente se realizan 3 lavados con solución saturada de bicarbonato
de sodio y finalmente un lavado con 10 mL de solución saturada de cloruro de
sodio.(conforme se realizan los lavados, la fase acuosa se aclara, no perder de vista
que en estos lavados, la fase orgánica siempre queda abajo).
La fase orgánica se pasa a un vaso de precipitados limpio y perfectamente seco y se
seca sobre sulfato de sodio anhidro.
La fase orgánica se destila hasta sequedad sobre un baño de agua hirviendo para
eliminar el cloruro de metileno.
El sólido que queda después de la evaporación del cloruro de metileno se suspende en
40 mL de etanol al 96 % y se disuelve en un baño de agua hirviendo (verificar que no
existan mechero prendidos), una vez disuelto todo el sólido, se lleva a un baño de hielo
hasta que la solución etanólica tome la temperatura del hielo. El sólido formado es él
para-nitrobromobenceno, el cual se filtra y se lava con 5 mL de etanol frio.
El líquido filtrado se concentra en baño María hasta un cuarto del volumen inicial y se
coloca en un baño de hielo hasta que solidifique el orto-nitrobromobenceno de color
amarillo.
Realizar cromatografía en placa fina de los dos sólidos obtenidos y correrla en un
sistema de hexano:acetato de etilo (7:3); revelar mediante luz U.V y calcular los Rf.
Compuesto Rf= Hex:AcOEt (7:3)
para-nitrobromobenceno 0.8
orto-nitrobromobenceno 0.6
NOTA: DEPOSITAR LOS RESIDUOS DE CLORURO DE METILENO EN LOS
FRASCOS DISPUESTOS PARA ELLO. PREGUNTAR A SU PROFESOR LA
UBICACIÓN DE ÉSTOS.

EDICIÓN 2015 105
REPORTE
1.- Determina el punto de fusión de tus compuestos.
2.- Calcula el rendimiento de la reacción.
2.- Completa la siguiente tabla
Compuesto Cantidad
obtenida
(g)
P.M.
g/mol
Punto de fusión
Experimental/reportado.
Rf
Calculado
Br
NO2
Br
NO2
CUESTIONARIO:
1. Realiza el mecanismo de reacción de la nitración del bromobenceno para la síntesis
de orto- y para-nitrobromobenceno.
Br
H2SO4 + HNO3 +
Br
NO2
Br
NO2
+

EDICIÓN 2015 106
2. ¿Por qué la adición del bromobenceno a la mezcla de ácidos se debe hacer en baño
de hielo y no a temperatura ambiente o a reflujo?
3. ¿Por qué crees que el orto-nitrobromobenceno es el producto mayoritario?
4. ¿Por qué motivo los grupos con pares de electrones libres tales como el –OH, -NH2,
-X (Cl, Br), etc, son grupos que además de activar a la SEAr, dirigen la entrada del
E+, a las posiciones orto y para?
5. ¿Cómo crees que sean energéticamente los isómeros orto- y para- del
bromobenceno?
6. ¿Por qué motivo el para-nitrobromobenceno es el producto que cristaliza primero en
etanol y después el orto-nitrobromobenceno ?
7. Sí en dado momento nos interesara formar el 2,4,6-trinitrobromobenceno, ¿qué
modificación tendrías que hacer al protocolo seguido en ésta práctica?
8. Diseña una alternativa para sintetizar los compuestos de ésta práctica partiendo de
benceno.

EDICIÓN 2015 107
BIBLIOGRAFÍA 1. Becker, H.; et al, por Hazard, B. J., Organicum, Practical Handbook of Organic
Chemistry, Pergamon Press Ltd., Nueva York, EU, 1973
2. Durst, H. D.; Gokel, G. W. Química Orgánica Experimental, Reverte, España,
1985

EDICIÓN 2015 108
PRÁCTICA 11
TRANSPOSICIÓN BENCILICA
(Obtención de ácido bencílico y alcohol bencílico)
OBJETIVOS:
1. Ilustrar las reacciones de transposición o rearreglo.
2. Obtener un producto derivado de la transposición bencílica.
ANTECEDENTES:
1.- Estabilidad de carbaniones.
2.- Capacidad migratoria de carbaniones.
INTRODUCCIÓN:
La transposición bencílica es la reacción de re arreglo del bencilo con KOH para
producir ácido bencílico, dicha reacción la llevó a cabo por primera vez Justus Liebig en
1838 y es el tipo de reacción general que producen la 1,2-dicetonas en medio básico.
La reacción de transposición bencílica está relacionada con otros re arreglos, como la
reacción de Cannízaro y la transposición Pinacólica.
O
O OH
O
OH
BENCILO ÁCIDO BENCILICO
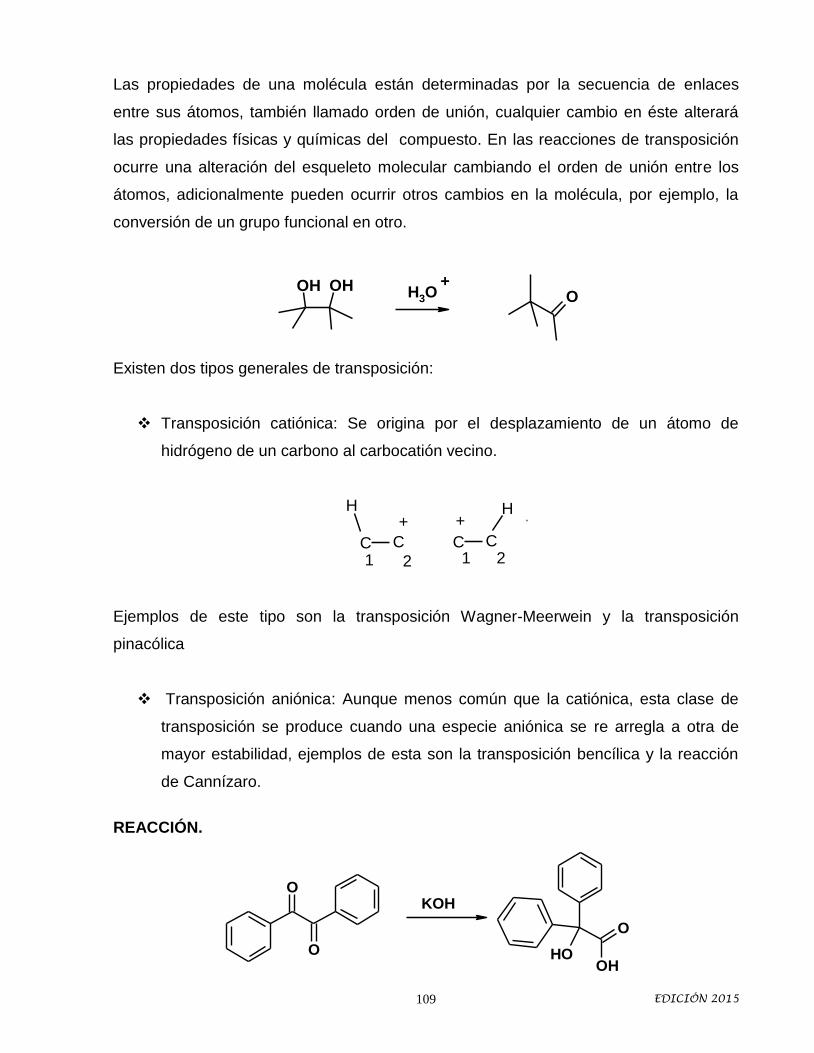
EDICIÓN 2015 109
Las propiedades de una molécula están determinadas por la secuencia de enlaces
entre sus átomos, también llamado orden de unión, cualquier cambio en éste alterará
las propiedades físicas y químicas del compuesto. En las reacciones de transposición
ocurre una alteración del esqueleto molecular cambiando el orden de unión entre los
átomos, adicionalmente pueden ocurrir otros cambios en la molécula, por ejemplo, la
conversión de un grupo funcional en otro.
OH OHOH3O
+
Existen dos tipos generales de transposición:
Transposición catiónica: Se origina por el desplazamiento de un átomo de
hidrógeno de un carbono al carbocatión vecino.
C C C C
H H+ +
1 2 1 2
Ejemplos de este tipo son la transposición Wagner-Meerwein y la transposición
pinacólica
Transposición aniónica: Aunque menos común que la catiónica, esta clase de
transposición se produce cuando una especie aniónica se re arregla a otra de
mayor estabilidad, ejemplos de esta son la transposición bencílica y la reacción
de Cannízaro.
REACCIÓN.
O
O OHOH
O
KOH
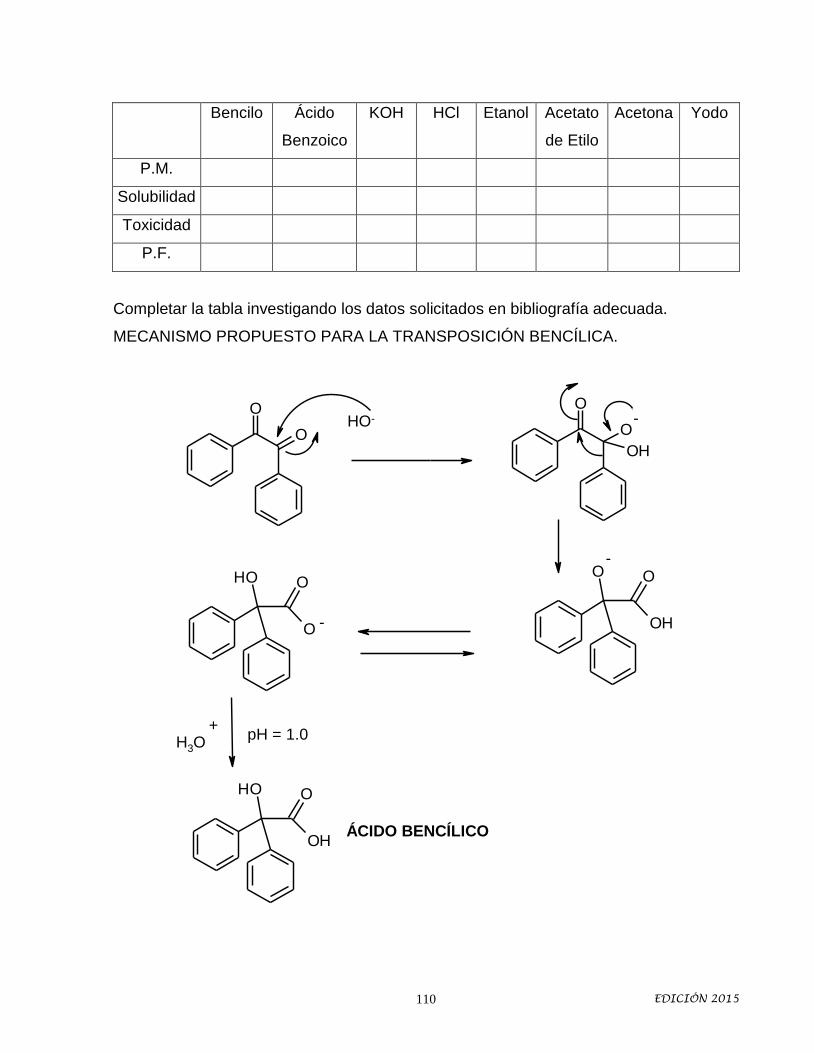
EDICIÓN 2015 110
Bencilo Ácido
Benzoico
KOH HCl Etanol Acetato
de Etilo
Acetona Yodo
P.M.
Solubilidad
Toxicidad
P.F.
Completar la tabla investigando los datos solicitados en bibliografía adecuada.
MECANISMO PROPUESTO PARA LA TRANSPOSICIÓN BENCÍLICA.
O
O
O
O
OH
OO
OH
OOH
O
OOH
OH
-
-
-
H3O+
pH = 1.0
HO-
ÁCIDO BENCÍLICO

EDICIÓN 2015 111
MATERIAL POR EQUIPO:
1 Matraz balón 14/23 50 mL 3 Papel filtro
1 Refrigerante 14/23 2 Soporte Universal
1 Baño María 1 Mechero de Bunsen
1 Unión Triple 14/23 1 Anillo
2 Vaso de precipitados 250 mL 2 Pinzas de 3 dedos con nuez
1 Embudo de filtración 1 Termómetro
1 Agitador de vidrio 1 Rejilla de asbesto
1 Capilar 1 Cromatoplaca de sílica gel F254 de 5 x 2 cm
1 Probeta 10 mL 1 Porta termómetro 14/23
Lámpara de ultravioleta 254 nm
MATERIAL POR SECCIÓN:
1 Embudo Büchner 1 Matraz Kitazato
1 Bomba de agua p/vacío 5 Cámara cromatográfica
1 Frasco boca ancha c/ tapa 100 mL 4 Pipetas 10 mL
1 Papel indicador de pH 0-14 1 Aparato de punto de fusión
REACTIVOS:
Cantidad Calidad
Bencilo 2.5g Grado reactivo
KOH 2.5g Grado reactivo
HCl 7 mL Grado reactivo
Etanol 15 mL Grado reactivo
Acetato de Etilo 8 mL Grado reactivo
Acetona 2 mL Grado reactivo
Yodo El necesario Grado reactivo
Sol, NaOH 40 % El necesario Grado reactivo

EDICIÓN 2015 112
DIAGRAMA DE FLUJO:
Disolver 2.5 g de Bencilo en 7.5 mL de
etanol
Disolver 2.5 g de KOH en 5 mL de agua
Mezclar las soluciones de Bencilo y KOH, reflujar por
30 min en baño maría
Destilar el etanol El etanol destilado, depositarlo en
un recipiente para su reciclaje
Residuo, adicionarle 35 mL de agua, calentar a ebullición por 5 min,
agitando vigorosamente
Filtrar en caliente
Lavar el precipitado con 10 mL de agua caliente y
posteriormente enfriar.
Acidular el filtrado con HCl conc. hasta pH = 1.0
Enfriar en baño de hielo, hasta precipitar los cristales
Filtrar a vacío en frío y lavar con agua helada
Sólido, recristalizar de agua, filtrar al vacío, secar, tomar P.F., calcular el rendimiento y hacer cromatografía en placa
fina eluyendo con etanol : acetona 8 : 2 y revelar con yodo.
Filtrado, neutralizar y depositar en desechos acuosos

EDICIÓN 2015 113
PARTE EXPERIMENTAL:
En un matraz balón de fondo plano de 50 mL disolver 2.5 g de bencilo en 7.5 mL de
etanol, agregar a ésta última una la solución previamente preparada de 2.5 g de KOH
en 5 mL de agua. Colocar el refrigerante al matraz balón en posición de reflujo y
calentar la mezcla en baño María durante 30 minutos. Pasado éste tiempo, colocar el
sistema para destilar el etanol por destilación simple. Después de la destilación de
etanol se le adicionan 35 mL de agua al residuo remanente en el matraz y se calienta el
mismo a ebullición por 5 minutos, agitando vigorosamente. La mezcla resultante se filtra
al vacío (usando un matraz Kitazato y un embudo buchner) y se lava el precipitado que
queda en el papel con 10 mL de agua caliente. El filtrado se transfiere a un vaso de
precipitados y se coloca en un baño de hielo, acidulando la solución con HCl
concentrado hasta pH=1.0 hasta completar la cristalización del producto. Los cristales
obtenidos se filtran al vacío, se lavan con agua helada* y se recristalizan de agua
caliente.
Análisis del producto:
1.- Determinar el punto de fusión de los cristales obtenidos antes y después de la
recristalización.
2.- Hacer cromatografía en placa fina del producto eluyendo con etanol : acetona 8 : 2,
revelar con yodo y con lámpara ultravioleta de longitud de onda de 254 nm, calcular el
Rf.
3.- Calcular el rendimiento de la reacción.
* Neutralizar el filtrado con NaOH al 40 % y depositar en desechos acuosos.

EDICIÓN 2015 114
CUESTIONARIO:
1. ¿Qué papel desempeña el hidróxido de potasio en esta reacción?
2. Señalar por qué es necesario destilar el exceso de etanol.
3. ¿Qué sustancias se disuelven en el agua caliente?
4. ¿Qué producto quedó en el papel filtro?
5. Dibujar la placa de cromatografía con el r.f. de cada mancha.
6. Anotar los cálculos de rendimiento, discutir si es adecuado y por que
7. Anotar el P. F. y compararlo con el encontrado en la bibliografía
BIBLIOGRAFÍA.
Química Orgánica. Fox M. A. & Whitesell S. K., 2a edición Addison Wesley
Longman, México 2000, págs 702-707.
Química Orgánica Experimental. Xorge A. Domínguez y Xorge A. Domínguez
S.,editorial Limusa 1a Edición, 1982, págs. 36-37, 88-97, 106-111, 113-126 y
352-353.
Experiments in organic chemistry, J. S. Nimitz. Prentice-Hall Inc. 1991, págs.
10-11 , 22-34, 61-71.

EDICIÓN 2015 115
FICHA DE EVALUACIÓN FINAL DE LABORATORIO DE QUÍMICA ORGÁNICA
DEPARTAMENTO DE QUÍMICA ORGÁNICA
ESCUELA NACIONAL DE CIENCIAS BIOLÓGICAS INSTITUTO POLITÉCNICO NACIONAL
NOMBRE DEL ALUMNO_____________________________________ GRUPO___________________________________________________ TURNO___________________________________________________ SEMESTRE ENERO-JULIO ( ) AGOSTO-DICIEMBRE ( ) AÑO______________________
CALIFICACIÓN ORDINARIA FINAL DE LABORATORIO*
_________________________________ LETRA NÚMERO
FIRMA DE ENTERADO DEL ALUMNO____________________________ NOMBRE Y FIRMA DEL PROFESOR______________________________
CALIFICACIÓN EXTRAORDINARIA DE LABORATORIO
_________________________________ Fecha de examen ___________ LETRA NÚMERO NOMBRE Y FIRMA DEL PROFESOR QUE APLICO EL EXAMEN EXTRAORDINARIO _____________________________________________ * Si la calificación es reprobatoria, anotar si es por inasistencias ó por examen.

EDICIÓN 2015 116
CALENDARIO DE PRÁCTICAS DE QUÍMICA ORGÁNICA QUÍMICO BACTERIÓLOGO PARASITÓLOGO
SEPTIEMBRE 2015-ENERO 2016
PRÁCTICA FECHA
Introducción al laboratorio de química orgánica (normas de
seguridad, criterios de evaluación, formación de secciones y
equipos, asignación de seminarios, explicación sobre cálculos
de rendimientos teóricos y experimentales )
8-11 Septiembre
Y
14-18
Septiembre
1. Destilación simple y destilación fraccionada 21-25 Septiembre
2. Destilación por arrastre de vapor 28 Sep-2 Oct
3. Separación de una mezcla ternaria por destilación 5-9 Octubre
E X A M E N (prácticas 1-3) 12-16 Octubre
4. Recristalización 12-16 Octubre
5. Extracción líquido – líquido 19-23 Octubre
6. Cromatografía 26-30 Octubre
E X A M E N (prácticas 4-6) 9-13 Noviembre
7. Síntesis a microescala de ácido fumárico 9-13 Noviembre
8. Síntesis de ciclohexeno 23-27 Noviembre
9. Síntesis de cloruro de terbutilo y Cinética química: efecto del
disolvente ente en la velocidad de una reacción de solvólisis.
30 Nov-4 Dic
E X A M E N (prácticas 7-9) 7-11 Diciembre
10. Síntesis de orto y para nitrobromobenceno 7-11 Diciembre
11. Transposición bencílica. 13-18 Diciembre
E X A M E N (prácticas 10-11) 13-18 Diciembre
Entrega calificaciones a Profesor de TEORIA 7-12 Enero
PRESIDENTE DE ACADEMIA: Angel Díaz Porras
Top Related