







Idiomas
Páginas
Jurídico


Métodos bioinformáticos para el estudio de interacciones proteína-Métodos bioinformáticos para el estudio de interacciones proteína-ligandoligando
El problema del plegamientoEl problema del plegamiento
Universidad Nacional Autónoma de México
Rogelio Rodríguez-Sotres
Facultad de Química

Rodríguez-Sotres (2004) Educación Química 16(1): 56-62.Rodríguez-Sotres (2004) Educación Química 16(1): 56-62.
Las proteínas son heteropolímeros complejos de 20 monómerosLas proteínas son heteropolímeros complejos de 20 monómeros

Su estructura tridimensionalSu estructura tridimensional(3D) es determinante de la(3D) es determinante de la
funciónfunción
Algunas patologías sonAlgunas patologías sonresultado de proteínasresultado de proteínas
mal plegadasmal plegadas
La interacción entreLa interacción entreuna proteína ysusuna proteína ysusligandos suele estarligandos suele estarmediada por restos demediada por restos deA.A. no consecutivosA.A. no consecutivos

Conocer la secuencia de AA y suConocer la secuencia de AA y suestructura 3D de una proteína esestructura 3D de una proteína esesencial para entender la funciónesencial para entender la función
La secuencia puede deducirse de los datos degenómica y transcriptómica.
La determinación experimental es laboriosa yrequiere cantidades grandes de proteína.
En consecuencia, contamos con 100 veces mássecuencias que entradas en el PDB.

La estructura 3D está determinada porLa estructura 3D está determinada porla secuenciala secuencia
La informaciónque determina elplegamiento finalestá en lasecuencia deaminoácidos
Esto lo demostróChristian B.Anfinsen, premioNobel de química1972
Experimento de Anfinsen, 1961Experimento de Anfinsen, 1961

La ruta de plegamiento no es fácil de explorarLa ruta de plegamiento no es fácil de explorar
La determinación de estructura requiere:
tiempos largos de toma de datos
conformaciones estables
El plegamiento final se alcanza en 0.01 a 1 s.
No es posible resolver la estructura completa de losintermediarios de plegamiento.
Los datos espectroscópicos y calorimétricos de los intermediariosen la ruta de plegameinto dan información parcial de suestrucutra1
11Chung HS , Piana-Agostinetti S , Shaw DE , & Eaton WA (2015) Structural origin ofChung HS , Piana-Agostinetti S , Shaw DE , & Eaton WA (2015) Structural origin ofslow diffusion in protein folding. Science 349(6255):1504-1510. slow diffusion in protein folding. Science 349(6255):1504-1510.

No ha sido posible conocer las clavesNo ha sido posible conocer las clavesdel plegamientodel plegamiento
No sabemos cuantos plegamientos distintospueden existir
No sabemos el camino por el que se pliega unaproteína
No sabemos que parte de la conservación desecuencia es determinante: del plegamiento finalde la función y la dinámicade la ruta de plegamiento
No ha sido posible correlacionar secuencia yplegamiento

A través de la evolución, lasA través de la evolución, lasproteínas sufren cambios en suproteínas sufren cambios en susecuencia (mutaciones en genessecuencia (mutaciones en genescodificantes). codificantes). Sólo algunos cambios sonSólo algunos cambios sontolerables y persisten en latolerables y persisten en laevolución.evolución.Las secuencias de proteínasLas secuencias de proteínasemparentadas (homólogas)emparentadas (homólogas)muestran conservaciónmuestran conservación

La conservación en las secuencias de aminoácidos La conservación en las secuencias de aminoácidos
Obedece a lanecesidad demantener:
Estructura
Función
Ruta deplegamiento
Flexibilidadconformacional
Estas fuerzas actúan juntas.Estas fuerzas actúan juntas.Disecar que conservación se relacionaDisecar que conservación se relacionacon cada tipo de presión de seleccióncon cada tipo de presión de selecciónno es sencillo.no es sencillo.
1 0 2 0 3 0 4 0 5 0 6 0 7 0 8 0 9 0 1 0 0. . . . | . . . . | . . . . | . . . . | . . . . | . . . . | . . . . | . . . . | . . . . | . . . . | . . . . | . . . . | . . . . | . . . . | . . . . | . . . . | . . . . | . . . . | . . . . | . . . . |
1 k v 5 A - - - - - - - - - - - - - - - - - - - M S K P Q P I A A A N W K C N G S Q Q S L S E L I D L F N S - - - T S I N H D V Q C V V A S T F V H L A M T K E R L S H - - - - P K F V I A A Q N A I A K S - G A1 n e y A - - - - - - - - - - - - - - - - - - - - - A R T F F V G G N F K L N G S K Q S I K E I V E R L N T - - - A S I P E N V E V V I C P P A T Y L D Y S V S L V K K - - - - P Q V T V G A Q N A Y L K A S G A1 r 2 r A - - - - - - - - - - - - - - - - - - - A P S R K F F V G G N W K M N G R K K N L G E L I T T L N A - - - A K V P A D T E V V C A P P T A Y I D F A R Q K L D - - - - - P K I A V A A Q N C Y K V T N G A1 t p h 1 - - - - - - - - - - - - - - - - - - - A P - R K F F V G G N W K M N G D K K S L G E L I H T L N G - - - A K L S A D T E V V C G A P S I Y L D F A R Q K L D - - - - - A K I G V A A Q N C Y K V P K G A1 m o 0 A M S Y Y H H H H H H L E S T S L Y K A G L T R K F F V G G N W K M N G D Y A S V D G I V T F L N A - - - S A D N S S V D V V V A P P A P Y L A Y A K S K L K - - - - - A G V L V A A Q N C Y K V P K G A1 m 6 j A - - - - - - - - - - - - - - - - - - - M G A G K F V V G G N W K C N G T L A S I E T L T K G V A A S V D A E L A K K V E V I V G V P F I Y I P K V Q Q I L A G E A N G A N I L V S A E N A W T K S - G AC o n s e n s u s N K N G A N G A
1 1 0 1 2 0 1 3 0 1 4 0 1 5 0 1 6 0 1 7 0 1 8 0 1 9 0 2 0 0. . . . | . . . . | . . . . | . . . . | . . . . | . . . . | . . . . | . . . . | . . . . | . . . . | . . . . | . . . . | . . . . | . . . . | . . . . | . . . . | . . . . | . . . . | . . . . | . . . . |
1 k v 5 A F T G E V S L P I L K D F G V N W I V L G H S E R R A Y Y G E T N E I V A D K V A A A V A S G F M V I A C I G E T L Q E R E S G R T A V V V L T Q I A A I A K K L K K A D W A K V V I A Y E P V W A I G1 n e y A F T G E N S V D Q I K D V G A K Y V I L G H S E R R S Y F H E D D K F I A D K T K F A L G Q G V G V I L C I G E T L E E K K A G K T L D V V E R Q L N A V L E E V K - - D F T N V V V A Y E P V X A I G1 r 2 r A F T G E I S P G M I K D C G A T W V V L G H S E R R H V F G E S D E L I G Q K V A H A L S E G L G V I A C I G E K L D E R E A G I T E K V V F E Q T K V I A D N V K - - D W S K V V L A Y E P V W A I G1 t p h 1 F T G E I S P A M I K D I G A A W V I L G H S E R R H V F G E S D E L I G Q K V A H A L A E G L G V I A C I G E K L D E R E A G I T E K V V F E Q T K A I A D N V K - - D W S K V V L A Y E P V W A I G1 m o 0 A F T G E I S P A M I K D L G L E W V I L G H S E R R H V F G E S D A L I A E K T V H A L E A G I K V V F C I G E K L E E R E A G H T K D V N F R Q L Q A I V D K G V - - S W E N I V I A Y E P V W A I G1 m 6 j A Y T G E V H V G M L V D C Q V P Y V I L G H S E R R Q I F H E S N E Q V A E K V K V A I D A G L K V I A C I G E T E A Q R I A N Q T E E V V A A Q L K A I N N A I S K E A W K N I I L A Y E P V W A I GC o n s e n s u s T G E D L G H S E R R E K A G V C I G E T V Q A Y E P V A I G
2 1 0 2 2 0 2 3 0 2 4 0 2 5 0 2 6 0 2 7 0 2 8 0. . . . | . . . . | . . . . | . . . . | . . . . | . . . . | . . . . | . . . . | . . . . | . . . . | . . . . | . . . . | . . . . | . . . . | . . . . | . . . . | . . . . | .
1 k v 5 A T G K V A T P Q Q A Q E A H A L I S S W V S S K I G A D V A G E L R I L Y G G S V N G K N A R T L Y Q Q R D V N G F L V G G A S L K P - E F V D I I K A T Q - - - - - - - -1 n e y A T G L A A T P E D A Q D I H A S I R K F L A S K L G D K A A S E L R I L Y G G S A N G S N A V T F K D K A D V D G F L V G G A S L K P - E F V D I I N S R N - - - - - - - -1 r 2 r A T G K T A T P Q Q A Q E V H E K L R G W L K S N V S D A V A Q S T R I I Y G G S V T G A T C K E L A S Q P D V D G F L V G G A S L K P - E F V D I I N A K Q - - - - - - - -1 t p h 1 T G K T A T P Q Q A Q E V H E K L R G W L K T H V S D A V A Q S T R I I Y G G S V T G G N C K E L A S Q H D V D G F L V G G A S L K P - E F V D I I N A K H - - - - - - - -1 m o 0 A T G K T A S G E Q A Q E V H E W I R A F L K E K V S P A V A D A T R I I Y G G S V T A D N A A E L G K K P D I D G F L V G G A S L K P - D F V K I I N A R S T A L S C T C W1 m 6 j A T G K T A T P D Q A Q E V H Q Y I R K W M T E N I S K E V A E A T R I Q Y G G S V N P A N C N E L A K K A D I D G F L V G G A S L D A A K F K T I I N S V S E K L - - - - -C o n s e n s u s T G A A Q H A R I Y G G S D G F L V G G A S L F I I

Si la secuencia y la estructuraestán vinculadas...
Debería ser posible determina la estructuraconociendo la secuencia de AA.
Sólo que ...¡se trata de un problema muy muy muy
complejo! veamos...

¿Cuántas proteínas distintas hay?– Existen 20×20 posible dipéticos (400).
– Existen 2010 decapéptidos (10.2×1012)
● Tomaría 32 años escribirlas todas, a una tasa de 10000/s, sin repetir.
– Las proteínas promedio tienen ~300 aa's.
– Las combinaciones posibles de secuencias de 300 aa sonmuchos órdenes de magnitud mayores que:
– # protones del universo × Vida del universo en µs
– Es decir, péptidos podrían ser producidos por el hombre queexistieran POR PRIMERA VEZ en toda la historia natural deluniverso.
¿quíen dijo yo?¿quíen dijo yo?

¿Cuántos plegamientos adoptauna proteína?
● Restrinjamos a sólo 3-conformaciones cadaAA:
Una proteína de 100 AA tendría:
3100 = 3×10130 confórmeros
● De nuevo, más que
# protones del universo × Vidadel universo en µs
Los plegamientos observados(PDB) se agrupan en ~ 1000categorías.
● =>La naturaleza no explorado todas lasLa naturaleza no explorado todas lasformas posibles ni siquiera de una proteínaformas posibles ni siquiera de una proteína.

Primeras conclusiones
● La naturaleza no a agotado su exploraciónde la diversidad en las proteínas.
● Las proteínas no se pliegan explorandotodas las conformaciones posibles, siguen uncamino definido por conformaciones"preferenciales"
Desafortunadamente explorar la ruta deplegamiento es mucho más difícil queresolver la estructura.
No sabemos cuales son las conformaciones"preferenciales"

Preguntas:Preguntas:
¿Cómo predecir la estructura tridimensional deuna proteína?
¿Cómo saber si la predicción es confiable?

Las bases del modeladoLas bases del modelado 1) La información necesaria está en la secuencia
2) A cada estructura se asocia un valor de energía
3) Las estructuras estables son mínimos energéticos local (quasiestable) global (permanentemente estable).
El problema se reduce a explorar el espacioconformacional hasta encontrar el mínimo de energía
E=℘(descriptores estructurales)
min(E)=℘(descriptores estructurales nativos)

LimitantesLimitantes El desconocer la ruta de plegamiento, obliga a un
muestreo "ciego" del paisaje enegético-conformacional
El espacio confromacional es abrumadoramente grande
La función de energía no se conoce en forma exacta

QuímicaComputacional
➲ En la química computacional se buscacalcular las propiedades de átomos ymoléculas usando las teorías de lafísica de partículas.
OJO: estos dibujos son tan malosOJO: estos dibujos son tan maloscomo cualesquiera otros, pues elcomo cualesquiera otros, pues elátomo no es como lo pintan.átomo no es como lo pintan.
??

QuímicaComputacional
➲ Los cálculos requiren lidiar con laspropiedades cuantizadas de laenergía a escalas atómicas y conel principio de incertidumbre deHeisemberg
La ecuación que describe estos sistemas es laLa ecuación que describe estos sistemas es laecuación de Shöedingerecuación de Shöedinger
algunas peculiaridades de esta ecuación dieron legar a laalgunas peculiaridades de esta ecuación dieron legar a lallamada "paradoja del gato de Schrödinger":llamada "paradoja del gato de Schrödinger": "Un gato en una caja que puede estar vivo o muerto, estará"Un gato en una caja que puede estar vivo o muerto, estará
en ambos estados superpuestos hasta que alguien abra laen ambos estados superpuestos hasta que alguien abra lacaja y lo observe"caja y lo observe"

QuímicaComputacional
Sir, this gift arrivedSir, this gift arrivedfrom Wernerfrom WernerHeisenberg. Heisenberg. It’s for youIt’s for you
Thanks James, I’llThanks James, I’llopen it, when I find outopen it, when I find outif its contents are deadif its contents are dead
or alive... This fellow and his or alive... This fellow and his freaky uncertainties!freaky uncertainties!
mmm...mmm...I suspect of aI suspect of a
hidden CAT inside hidden CAT inside
Schödinger's cat mysteries.. Soon at a theaterSchödinger's cat mysteries.. Soon at a theaternearby!nearby!

Resolver la ecuación de Schrödinger...*
Well, that Well, that makes it formakes it forthe H atom,the H atom,and I still and I still ignore if ignore if
Heisenberg’sHeisenberg’shidden CAT ishidden CAT isalive or dead...alive or dead...Probably dead Probably dead
by now anyway, or half so.by now anyway, or half so.se logra, en forma exacta,se logra, en forma exacta,para un sistema de: para un sistema de:
¡2 partículas!¡2 partículas!

pervertum magister est natura
Good for you -you understand H! Good for you -you understand H! Now let's try something simple... mmm... Now let's try something simple... mmm...
RNAase: C RNAase: C575575HH907907NN171171OO192192SS1212 ? ?
...very simple: 100 ...very simple: 100 conformations...conformations...
What?What?
E. ShrödingerE. Shrödinger
the monkey isthe monkey isout of its mind!out of its mind!
I’d better go back to I’d better go back to the CAT’s the CAT’s problemproblem
En todos los otros casos se requiere simplificar y aproximar.En todos los otros casos se requiere simplificar y aproximar.Entre mayor el problema menos confiable la aproximación...Entre mayor el problema menos confiable la aproximación...

¿Qué podemos hacer?
Simplifica compadre, no seasSimplifica compadre, no seasheavy!heavy!
Schödinger es la netaSchödinger es la netadel planeta, simplificardel planeta, simplificar
es ignorarloes ignorarlo

Para sistemas de pocos electrones
➲ Se consideran los núcleos como puntos cargados fijos(aproximación de Born-Oppenheimer).
➲ Se usan métodos numéricos para aproximar funciones deonda para los orbitales atómicos y/o moleculares:
Métodos más usados: Hartree-Fock (HF) - self-consistent field (SCF) Density Functional’s theory (DFT) Møller-Plesset perturbation’s theory (MP)
WATER molecular orbitalsWATER molecular orbitals

Sistemas medianos (40-100 átomos tipo CHONPS)
➲ Métodos semiempíricos:Se simplifica el Núcleo y los electrones internos a
una esfera cargada.Se calcula la contribución de electrones de
valencia en los orbitales molecularesSe emplean parámetros ajustables (empíricos)
para que los cálculos reproduzcanexperimentos de referencia
métodos más empleados: ZINDO, MNDO, AM1 PM3, PM6 PM6-adh2, PM7

Sistemas enormes... adios a Schrödinger (never mind the cat).
E total = E covalente + E no covalenteE total = E covalente + E no covalente
E no covalente = E coulomb + E VdWE no covalente = E coulomb + E VdW
Mecánica MolecularMecánica Molecular
Se ignoran los efectos Se ignoran los efectos cuánticos y electrónicoscuánticos y electrónicos
++¡Es rápida de calcular!++¡Es rápida de calcular!
Se representa al sistema poliatómico como pelotas (atómos) conSe representa al sistema poliatómico como pelotas (atómos) conmasa y carga, unidas por resortes de elasticidad perfectamasa y carga, unidas por resortes de elasticidad perfecta(enlaces) y sometidas a potenciales clásicos (continuos).(enlaces) y sometidas a potenciales clásicos (continuos).
--¡Es muy poco realista, por tanto incierta!--¡Es muy poco realista, por tanto incierta!
Ups!Ups!

Mecánica Molecular
--Tiene un --Tiene un importanteimportanteerrorerrorsistemáticosistemático
Campos de fuerza = conjunto deCampos de fuerza = conjunto deparámetros y ecuacionesparámetros y ecuaciones
++Los parámetros++Los parámetrosse ajustanse ajustanempíricamenteempíricamentepara reproducirpara reproducirdatos dedatos dereferenciareferencia

ÁtomoÁtomo44
DinámicaMolecular
ÁtomoÁtomo11
ÁtomoÁtomo22
ÁtomoÁtomo44
Átomo 3Átomo 3
Átomo 5Átomo 5
++
--El intervalo de--El intervalo deintegración (integración (∆t∆t) debe) debeser del orden de 2x10ser del orden de 2x10--
1212 ps, ps, i.e.i.e. para simular para simular1x101x10-8-8 s hay que s hay quecalcular 5x10calcular 5x1066 estados estados
Programas:Programas:Algoritmos deAlgoritmos deintegración numéricaintegración numérica
++Se asignan velocidades a los átomos y se++Se asignan velocidades a los átomos y secalculan la fuerzas al tiempo t.calculan la fuerzas al tiempo t.+++ Se determinan las nuevas posiciones al+++ Se determinan las nuevas posiciones altiempo t+∆ttiempo t+∆t++++ el proceso se repite n veces++++ el proceso se repite n veces
vv ttf1f1tt
f2f2tt
tiempo ttiempo t
tiempotiempot+∆tt+∆t
vv t+∆tt+∆t
f1f1t+∆tt+∆t
f2f2t+
∆t
t+∆
t
vvt+∆tt+∆t
vvtt

Campos de fuerzas popularesCampos de fuerzas populares
AMBER: (variantes amber94, amber99SB,GAFF).
CHARMM (charmm27 charmm31).
GROMOS (gromos87, 96, 53a6)
OPLS (opls-aa)
MMFF
TRIPOS
ENCAD

Programas de MM/MDProgramas de MM/MD
AMBER
GROMACS
NAMD
TINKER
CHARMM

Métodos de modeladoMétodos de modelado
A pesar de ser un problema NP-completo (muydifícil), se han logrado aproximaciones al problema:Ab initio. Usamos los principios de la física y de la
química (o aproximaciones a los mismos) parapredecir la conformación estable de la proteína(estado de baja energía).
Comparativo. Usamos datos de estructuras 3Dconocidas (moldes) que “en teoría” tengan unplegamiento “comparable” a de la secuencia deaminoácidos que se desea modelar ( secuenciaobjetivo).

Métodos de modelado Métodos de modelado ab initioab initio:: El éxito es limitado y se requiere alguna forma de
confirmación del resultado (de preferenciaexperimental).
Aplicando principios de la mecánica cuánticaSoluciones completas, sólo en péptidos pequeñosMétodos “divide and conquer” y “kernels” se
require un modelo aproximado para empezar. Aproximaciones de mecánica molecular y
simulaciones de dinámica molecular y Monte Carlo.son más rápidos, pero tienen un error implícito
que hace incierto el resultado.para plegar completamente una proteína pequeña
se requieren miles de horas de CPU.hay diferentes campos de fuerzas - ¿cuál usar?

Modelado comparativo: Modelado comparativo: ¿Cómo saber que nuestra secuencia¿Cómo saber que nuestra secuenciaobjetivo tiene una estructura comparable a un cierto molde 3D?objetivo tiene una estructura comparable a un cierto molde 3D?
A) Datos espectroscópicos
i.e. se require información experimental, no siempre disponible
B) El molde y el objetivo son proteínas homólogas(modelado por homología), por tanto, su plegamiento separece en medida de su similitud en secuencia.
esto no siempre es cierto: proteínas homólogas pueden tener plegamientos distintos proteínas heterólogas pueden tener plegamientos semejantes
entre menos similitud exista la correlación estructural es másdébil... ¿en qué nivel de similitud un molde ya no es útil?

El problema del enhebrado (threading)El problema del enhebrado (threading)
Modelado por enhebrado es el alinear losátomos de una secuencia de aminoácidos a lasposiciones atómicas de un motivo deplegamiento tridimensional de una proteína. el motivo molde puede ser experimental o teórico.el problema es NP-completo (muy difícil).¿cómo saber a que posición del motivo
corresponde cada aminoácido?¿qué hacer si la longitud de la secuencia objetivo
es más larga o más corta que el molde?

Alineamiento de secuenciaAlineamiento de secuencia
Una manera de atacar el problema deenhebrado es aprovechar la similitud desecuencia...¿cómo lograr la mejor correspondencia?
de nuevo el problema es NP-completo (muy difícil) ¿hasta que punto la concordancia de propiedades de
dos aminoácidos es suficiente para hacerlosequivalentes en la estructura?
¿cómo garantizar que al realizar dicho alineamiento setendrá la mejor concordancia de contactos vecinos anivel tridimensional?
¡Una manera de responder sería evaluar elresultado!

Evaluación de la CalidadEvaluación de la Calidad
¿Está el modelo realmente en un mínimoenergético?
¿Existen otros mínimos energéticos?
¿qué tanto se desvía esta conformación es laque encontramos en la naturaleza (la cual esdesconocida)?
¿es posible aproximarnos a la estructura nativasin saber cual es?

Evaluación de la calidadEvaluación de la calidad1) 1) Consistencia estructuralConsistencia estructural
Consistencia con los principios básicos defísica y química:
Desviación respecto a valores canónicos en:distancia de enlaceángulos de enlaceángulos de torsión (dihedrales+impropios)
Una forma clásica de evaluación es lacomparación con las conformacionesobservadas en estructuras experimentales paracada tipo de residuo.
Interacciones no covalentes favorables

Consistencia estructural ...Consistencia estructural ...
Desviación de los valores canónicos
Interacciones no covalentes (vecinos en elespacio no unidos por enlaces)impedimentos estéricosrepulsión/atracción electrostática
fuerzas de dispersión (contactos de Var der Walls) puentes de hidrógeno puentes salinos
efectos entrópicos libertad conformacional efecto hidrofóbico

Consistencia estructural... Consistencia estructural... mediante MMmediante MM
La mecánica molecular provee cálculosenergéticos que consideran:Uniones covalentes y su geometríainteracciones no covalentes
Los valores absolutos no se parecen a lasenergías experimentales
Pero las energías elevadas indican "sitiosinestables", físicamente improbables.

EjemploEjemplo
Descargue Swiss PDB viewer
Portal http://spdbv.vital-it.ch/
Siga las instrucciones de instalación
abra el programa
Elija una estructura del depósito de Swiss model
Portal: http://swissmodel.expasy.org/repository/ repository query: “Palabra de búsqueda” elija un resultado descargue y descomprima el modelo (casi al final de la
página) Abralo en Swiss PDB viewer
File/open PDB file... elija el archivo PDB
Tools/compute/energy (Forcefield)... palomita a todo...OK
Analice el resultado...
Elija un modelo. Hay dos ejemplos paraElija un modelo. Hay dos ejemplos paradescargar en la página del curso y tambiéndescargar en la página del curso y tambiénpueden usar los modelos realizados en sesionespueden usar los modelos realizados en sesionesanterioresanteriores

Ejemplo...Ejemplo...
Analice el resultado: busque energías extremas
encuentre los sitios con peor energética (más alta)
pique sobre la línea y vea en la estructura el residuoseleccionado
Pase al panel de control (si no está abierto:wind/control panel)
elija el residuo en cuestión+ 2 antes y 2 después
Select/Neighbors of seleted residues
Presione la tecla enter (con foco en la ventana gráfica)
Describa los problemas más evidentes
tools/fix selected sidechains/simulated anealing
experiments:3 ... OK
calcule de nuevo la energía y compare los reportes

Ejemplo ...Ejemplo ... Abra nuevamente Swiss PDB viewer
Lea el primer modelo (File open)
Calcule la energía (Forcefield), no olvide poner todas las palomitas.
Analice el reporte y busque sitios en donde la energía es positiva y alta
Identifique colapsos de átomos (Select/Residues Making clashes)
Si no son demasiados puede intentar mejorarlos con Simulated Anealing
Vamos ahora a Swiss model Assesment http://swissmodel.expasy.org/workspace/index.php?func=tools_structureassessment1
Carge su pdb en el sitio (browse)
seleccione varias herramientas de arriba (ANOLEA, Gromos, Qmean6,Dfire, Prockeck...)
pique SUMBIT
Analice el resultado, busque los sitios en donde las energías ANOLEAsean elevadas (ROJO). Estos sitios son sitios posiblemente defectuosos.Busque desviaciones de los valores canónicos (Prockeck & Dfire).

Evaluación de la calidadEvaluación de la calidadRelevancia BiológicaRelevancia Biológica
¿Realmente representa el modelo generado lamanera en que la secuencia objetivo se pliegaen los sistemas biológicos?
¿Cómo contestar a esta pregunta sidesconozco la estructura final?Se puede contestar a esta pregunta si encuentro
alguna característica en las proteínas conestructura conocida, que permita establecer unacorrespondencia clara entre las coordenadasatómicas de la estructura y su secuencia.
Si tal característica existe, puede evaluarse en losmodelos

Una limitación mayor:Una limitación mayor: ¿Cómo saber si el modelo sirve sin recurrir ¿Cómo saber si el modelo sirve sin recurrira datos experimentales (X-ray, NMR)?a datos experimentales (X-ray, NMR)?
Concursos CASP: critical assesment of the structure ofproteins
La estructura es conocida pero sólo por los jueces
Los concursantes someten sus propuestas pueden ser máquinas (servidores) pueden ser grupos humanos
Los jueces evalúan la calidad de las propuestas
Programas de evaluación de calidad
Consistencia estructural
Relevancia biológica

A pesar de los CASP,A pesar de los CASP,seguimos sin conocer lasseguimos sin conocer las
claves del plegamiento de lasclaves del plegamiento de lasproteínasproteínas
????
-10 -10 Å RMSD from target !Å RMSD from target !You failed miserably!You failed miserably!
-I Made 3500000 energy-I Made 3500000 energyevaluations... It was hard workevaluations... It was hard workPlease don't fail me!Please don't fail me!
ProfessorProfessor Dr. CASPer AngerDr. CASPer Anger
Contestant (student)Contestant (student)MMr. Armando Malo Pliegor. Armando Malo Pliego

Gracias al CASP varios métodos handemostrado cierta eficacia
● I-Tasser (http://zhanglab.ccmb.med.umich.edu/I-TASSER/)
● HHpred (http://toolkit.tuebingen.mpg.de/hhpred)
● Prob-Cons (http://pcons.net/)
● Robetta (http://robetta.bakerlab.org/)
● Phyre2 (http://www.sbg.bio.ic.ac.uk/phyre2/)
● Rapper (http://mordred.bioc.cam.ac.uk/~rapper/)
● PMP (http://www.proteinmodelportal.org/)
● 3D-Jigsaw (http://bmm.crick.ac.uk/~3djigsaw/)
● QUARK (http://zhanglab.ccmb.med.umich.edu/QUARK/)
● LOMETS (http://zhanglab.ccmb.med.umich.edu/LOMETS/)
● MULTICOM (http://sysbio.rnet.missouri.edu/multicom_toolbox/)

Varios son totalmenteautomáticos
● Introduzcan la secuencia● Ármense de paciencia● Descarguen la predicción
I-TASSER bestI-TASSER best
PHYRE-2 bestPHYRE-2 bestSuperdiferentes...Superdiferentes...Y ahora ¿qué Y ahora ¿qué "&@%*$""&@%*$" hago? hago?

¿Qué hacer si no soy concursante delCASP?
¿Debo usar un servidor, o hacer mipropio intento de modelado?
¿Puede alguien decirme si mipredicción aprueba o reprueba aún sin
conocer le plegamiento correcto?

La relevancia biológica
● Si en la realidad, el moldeelegido y la proteína objetivo nose pliegan igual, la predicciónestaría "reprobada" por losprofesores del CASP.
Sin embargo,
● Identificar ese caso, es comosaber si un examen de opciónmúltiple está aprobado o no, sinsiquiera haber leído laspreguntas.

Basados en una estratagema estadística, nosotrosgeneramos un método para
"calificar"cuantitativamente la relevancia biológicasin conocer a priori el plegamiento nativo

¿Cómo puede ser esto posible?

La búsqueda de un algoritmo paracalificar un plegamiento
esqueleto coordenadas atómicas
3D
Secuencia algoritmo inverso (AI)
algoritmo directo (AD)
La mayoría de los intentos de modelado de la estructurade proteínas, se han enfocado en el AD, pero resulta mássencillo determinar que aminoácidos son compatibles con
cada posición en un plegamiento ... o sea el AI.

● Rosetta design (Rd)Rosetta design (Rd)‡‡ ( David Baker, University of Washington, Department of Biochemistry ).( David Baker, University of Washington, Department of Biochemistry ).Asigna residuos al esqueleto con gran concordancia con las estructurasAsigna residuos al esqueleto con gran concordancia con las estructurasexperimentalesexperimentales
((‡‡)) Kuhlman et al. Kuhlman et al. (2003). Design of a novel globular protein fold with atomic-level accuracy (2003). Design of a novel globular protein fold with atomic-level accuracy ScienceScience302, 1364-8.302, 1364-8.((‡‡) ) Misura et al.Misura et al. (2006). Physically realistic homology models built with ROSETTA can be more accurate (2006). Physically realistic homology models built with ROSETTA can be more accuratethan their templates than their templates Proc Natl Acad Sci U S A 103, 5361-5366. Proc Natl Acad Sci U S A 103, 5361-5366. ((‡‡) ) Rothlisberger et al.Rothlisberger et al. (2008) Kemp elimination catalysts by computational enzyme design. (2008) Kemp elimination catalysts by computational enzyme design. NatureNature28(836):online-10.1038/nature0687928(836):online-10.1038/nature06879((‡‡) ) Jiang et al.Jiang et al. (2008) De Novo Computational Design of Retro-Aldol Enzymes. (2008) De Novo Computational Design of Retro-Aldol Enzymes. ScienceScience319(5868):1387-1391319(5868):1387-1391
Ha tenido mucho éxito en elHa tenido mucho éxito en elrediseño de proteínas, al permitirrediseño de proteínas, al permitirhacer cambios dirigidos en lashacer cambios dirigidos en lasproteínas si afectar elproteínas si afectar elplegamientoplegamiento

Coordenadas 3DCoordenadas 3D
1 secuencia1 secuencia
algunas secuenciasalgunas secuencias
muchas secuenciasmuchas secuencias
1 solución posible 1 solución posible información precisainformación precisa
Algunas secuenciaAlgunas secuenciainformación valiosainformación valiosa
casi gran cantidad decasi gran cantidad desecuenciassecuenciasinformación escasainformación escasa
AI: tres posibles casos de solución
(*) se refiera a la degeneración del(*) se refiera a la degeneración delcódigo de informacióncódigo de información

Propiedades esperadas para unalgoritmo inverso
● Se han observado plegamientos muy semejantescon secuencias diferentes, i.e.
debe ser posible obtener más de unasolución por plegamiento.
● Pero hay una conservación significativa en lassecuencias de proteínas con el mismo plegamiento,i.e.
no cualquier solución soporta el plegamiento● Efectivamente, Rd no arroja varios cientos de
soluciones posibles y diferentes.

1 0 2 0 3 0 4 0 5 0 6 0 7 0 8 0 9 0 1 0 0. . . . | . . . . | . . . . | . . . . | . . . . | . . . . | . . . . | . . . . | . . . . | . . . . | . . . . | . . . . | . . . . | . . . . | . . . . | . . . . | . . . . | . . . . | . . . . | . . . . |
1 e 9 g 2 0 0 1 0 . p d b N F T V A K V G A P G A E N F A T A V R L N G F P V S A H H D I P L H Y D E N N N K F L T V I E I P K G Q Q A D L R I M L W Y Q L D P I M P A R Q N G Q L R F E K K V A D A E G A E T N F G A L P E T H1 e 9 g 4 0 0 2 2 . p d b N F T V A I V G A E N A E N F A T A V R L N G F P V S A H H D I P L H Y D E N N N L F L V V I E I P K G Q Q A D K R I I L W Q P L D P I R I A R Q N G Q L R F R K K V A D H E G A D S N E G A L P E T H1 e 9 g 3 0 0 0 5 . p d b N F T V D K V G A P G A E N F R T A V R L N G F P V S A H H D I D A H Y D E N N N I F K T V I E R P K G Q Q A D K R I L L W E P G D P I G I A R E N G Q L R F E K K V A D Q E G A N S N E G A L P E T H1 e 9 g 3 0 0 2 9 . p d b N F T V I K V G A P G A E N F R T M V A L N G F P V S A H H A I P A H A D E N N N I F K T V I E I P K G Q Q A D K R I L L W E P G D P I G I A R Q N G Q L R F E K K V A D Q E G S N S N R G A L P R T L1 e 9 g 2 0 0 0 5 . p d b N Y T V E K I G A P G A E N F R T H V R L N G F P V S A H H D I D A H Y D E N N N K F K T V I E I P K G Q Q A D K R I L L W E P G D P I G I A R Q N G Q L R F D K K V A D Q E G A N T N F G A L P E T L1 e 9 g 6 0 0 1 3 . p d b N F T V E K I G A P G A E N F A T H V R L N G F P V S A H H D I D A H Y D E N N N E F K V V I E I P K G Q Q A D L T I A L W Y P G D P I T P S R Q N G Q L R F R K K V A D A E G S T S N T G A L P E T H1 e 9 g 6 0 0 2 5 . p d b N F T V E I I G A P G A E N F A T H V R L N G F P V S A H H D I D A F A D K N N N I F K V V I E R P K G Q Q A D K S I A L W Y V G D P I T I R R Q N G Q L R F D K K V L D A V G D T S N S G S L P R T H1 e 9 g 2 0 0 5 8 . p d b N F T T A E I G A P G A E Q Y A T H V R L N G F P V S A H H D I P A H Y D E N N H K F L T V I T I P K G Q Q A D L Q I I L W Q V G D P I R P A R Q N G Q L R F E K K V A D H E G S D S N R G H L P E T H1 e 9 g 6 0 0 1 5 . p d b N F T V A K I G A E G A E N F L T H V R L N G F P V S A H H D I P A H Y D E N N N L F L T V I E I P K G Q Q A D K A I L L W Y P G D P I G I R R R N G Q L R F E K K V A D H E G S D S N R G A L P E T H1 e 9 g 6 0 0 7 9 . p d b N F T T E E I G A K G A E N Y A T H V R L N G F P V S A H H D I P A H Y D E N N K L F L T V I E I P K G Q Q A D L R I M L W Y P G D P I M P A R E N G Q L R F E K K V A D H E G M D V N W G A L P E T HC o n s e n s u s N T G A A E T V L N G F P V S A H H I D N N F V I P K G Q Q A D I L W D P I R N G Q L R F K K V D G N G L P T
1 1 0 1 2 0 1 3 0 1 4 0 1 5 0 1 6 0 1 7 0 1 8 0 1 9 0 2 0 0. . . . | . . . . | . . . . | . . . . | . . . . | . . . . | . . . . | . . . . | . . . . | . . . . | . . . . | . . . . | . . . . | . . . . | . . . . | . . . . | . . . . | . . . . | . . . . | . . . . |
1 e 9 g 2 0 0 1 0 . p d b L K S N R V H N N T N A P G A G V A I L V S V H S N R V G E T G K V T V V I A L G I T F L I E N G Y A S P V V H A V E V E A P L A N L L T S A S D I Q I F H P G Q L E N D L K F Y W T K E I A E G Q P P1 e 9 g 4 0 0 2 2 . p d b L K S N E V H D E T N S P G S G E A I R V S V H S N D V G Y T G A V I V T E A L G I T H L I R N G Y S D P V V H A V R V E D D L S S L L T S A N D I Q I F H P G Q L E N D L K F H W T E E I A E G Q P P1 e 9 g 3 0 0 0 5 . p d b N K S N E V V N Q T N S P G D G T A S H V E V H S N E V G E T G R V E T V K A T G V T H E E N N G Y D D D N V H A I R T D A P L A D L V T S A N D N Q I F F P G Q E N N D K K F H I T R E I A H G Q P P1 e 9 g 3 0 0 2 9 . p d b L K S N E V W N N T N A P G S G T A I H V S V H S N R V G E T G K V T E V K A T G I T L L E E N G Y A S P V V H A I E V E A P L A D L V T S A N D N Q I F F P G Q E E N D L K F F W T K E I A E G Q P P1 e 9 g 2 0 0 0 5 . p d b N K S N E V D D Q T N S P G N G T A S L V S V H S N R V G E T G A V I V V I A T G T T H L I R N G Y N A D I I H A I E V K A P L A N L V T S A N D V R I F F P G Q E D N D K K F W I T K E I A E G Q P P1 e 9 g 6 0 0 1 3 . p d b L K S N E V H D N T N S P G S G T A I L V S V L S N R V G E T G A V I V T R L T G V M L L I R N G Y S A P V L F A I E V K A P L A D L V T S A D D V K I F F P G A L D N A K K F W I T S E I A E G Q P P1 e 9 g 6 0 0 2 5 . p d b L K S N E V H D E T N S P G S G T A I N V S V H S N R V G Y T G A V I K T K A T G I T H L T R N G Y S N P V V H A I E V K A P L A D L V T S A N D N Q I F F P G Q E N N D K K F Y I T S E I A E G Q P P1 e 9 g 2 0 0 5 8 . p d b L K S N E V H D N T N A P G S G T A I H V S V H S N L V G Y T G A V I V V I A T G I T F E I R N G Y S H P V V H A I S T T A P L S S L V T S A N D V Q I F F P G Q E N N D K K H H I T H E I A E G Q P P1 e 9 g 6 0 0 1 5 . p d b M K S N E V H D Q N N S P G A G Q A A H V S V H S N L V G E T G A V I V V Y A T G V T H E I R N G Y S H D V V H A I S T T A P L A N L V T S S S D N R I F F P G Q E D N D K K H H I T H E I A E G H P P1 e 9 g 6 0 0 7 9 . p d b L K S N E V V D N T N A P G S G T A I N V A V V S N L V G E T G A V I V V Y A T G I I L L I R N G Y A D P V V I A I S T T A P L S S L V T S A N D I Q I F F P G L L E N I L K H H I T E E I A H G H P PC o n s e n s u s K S N V N P G G A V V S N V G T G V G N G Y A L L T S D I F P G N K T E I A G P P
¿Que hacer ahora con las soluciones?

la estadística al rescateSupongamos que tenemos las respuestas y las calificacionesSupongamos que tenemos las respuestas y las calificacionespara alumnos en el grupo, excepto UNO y quiero saber quepara alumnos en el grupo, excepto UNO y quiero saber quetan probable es que ese alumno aprobara.tan probable es que ese alumno aprobara.
Como vemos, el alumno sin calificación sólo tiene dos coincidenciasComo vemos, el alumno sin calificación sólo tiene dos coincidenciascon la respuesta más frecuente para los aprobados ... con la respuesta más frecuente para los aprobados ...
1 2 3 4 5 6 7 8 9 10
A D B A B A B C D AA C D A B C C C D AA D D C B A A C D AD D D A A C C D D AB D D A B A B B D DA D D A B A ? C D A
A C C B B C B A C B ??
B C A A C A C C A ?B C A A A C B A A AB C C A C D B C E CA D D A C A C A A DD C A B B A C A A BB A A A C A C A A B
Pregunta número
Respuesta ResultadoAprobadoAprobadoAprobadoAprobadoAprobado
↑Más frecuente
↓Más frecuenteReprobadoReprobadoReprobadoReprobadoReprobado

Con alta probabilidad estáreprobado
Pero quizá nos gustaría dar un valorcuantitativo para esa probabilidad...
¿es 90%, 80% o cuánto?

Cada estructura es como un largoexamen
● El número de preguntas es tan largo como lala secuencia de la proteína
● La posibles opciones de respuesta son 20(los aminoácidos)
● Las secuencias "aprobadas" son las querealmente pueden adoptar el plegamientopropuesto
● Todas las demás (que se pliegan de otromodo) están REPROBADAS.

En el caso de la predicción
● Los ejemplos de secuencias aprobadas(según ROSETTA, claro) son las solucionesde Rd.
● Las secuencias reprobadas se puedenobtener al "tin marín dedo pingüe" es decirson secuencias tomadas al azar.
● El examen desconocido es la secuenciaobjetivo y como es natural, está incluido enlas bases de datos (Uniprot, RefSeq, NR,etc).

Un método estadístico adecuado para estas
situaciones son los modelos ocultos de Markov El Programa HMMER* analiza el conjunto de secuencias yresume la probabilidad de encontrar cada aminoácidoaminoácido en cada una de las posiciones.
Luego permite analizar otras secuencias y determina sicoincide con "la respuesta más frecuente" estableciendo unaprobabilidad de aprobación y en su caso un "score"
Secuencias de ROSETTA design → alumnos aprobadosSecuencias tomadas al azar → Alumnos reprobadosExámenes sin calificar → Todas las secuencias conocidasde las proteínas naturales (ahí está también mi objetivo).
(*) (*) Eddy SR(2004) Nature Biotech. 22:1315.Eddy SR(2004) Nature Biotech. 22:1315.

RD·HMMer
1 0 2 0 3 0 4 0 5 0 6 0 7 0 8 0 9 0 1 0 0. . . . | . . . . | . . . . | . . . . | . . . . | . . . . | . . . . | . . . . | . . . . | . . . . | . . . . | . . . . | . . . . | . . . . | . . . . | . . . . | . . . . | . . . . | . . . . | . . . . |
1 e 9 g 2 0 0 1 0 . p d b N F T V A K V G A P G A E N F A T A V R L N G F P V S A H H D I P L H Y D E N N N K F L T V I E I P K G Q Q A D L R I M L W Y Q L D P I M P A R Q N G Q L R F E K K V A D A E G A E T N F G A L P E T H1 e 9 g 4 0 0 2 2 . p d b N F T V A I V G A E N A E N F A T A V R L N G F P V S A H H D I P L H Y D E N N N L F L V V I E I P K G Q Q A D K R I I L W Q P L D P I R I A R Q N G Q L R F R K K V A D H E G A D S N E G A L P E T H1 e 9 g 3 0 0 0 5 . p d b N F T V D K V G A P G A E N F R T A V R L N G F P V S A H H D I D A H Y D E N N N I F K T V I E R P K G Q Q A D K R I L L W E P G D P I G I A R E N G Q L R F E K K V A D Q E G A N S N E G A L P E T H1 e 9 g 3 0 0 2 9 . p d b N F T V I K V G A P G A E N F R T M V A L N G F P V S A H H A I P A H A D E N N N I F K T V I E I P K G Q Q A D K R I L L W E P G D P I G I A R Q N G Q L R F E K K V A D Q E G S N S N R G A L P R T L1 e 9 g 2 0 0 0 5 . p d b N Y T V E K I G A P G A E N F R T H V R L N G F P V S A H H D I D A H Y D E N N N K F K T V I E I P K G Q Q A D K R I L L W E P G D P I G I A R Q N G Q L R F D K K V A D Q E G A N T N F G A L P E T L1 e 9 g 6 0 0 1 3 . p d b N F T V E K I G A P G A E N F A T H V R L N G F P V S A H H D I D A H Y D E N N N E F K V V I E I P K G Q Q A D L T I A L W Y P G D P I T P S R Q N G Q L R F R K K V A D A E G S T S N T G A L P E T H1 e 9 g 6 0 0 2 5 . p d b N F T V E I I G A P G A E N F A T H V R L N G F P V S A H H D I D A F A D K N N N I F K V V I E R P K G Q Q A D K S I A L W Y V G D P I T I R R Q N G Q L R F D K K V L D A V G D T S N S G S L P R T H1 e 9 g 2 0 0 5 8 . p d b N F T T A E I G A P G A E Q Y A T H V R L N G F P V S A H H D I P A H Y D E N N H K F L T V I T I P K G Q Q A D L Q I I L W Q V G D P I R P A R Q N G Q L R F E K K V A D H E G S D S N R G H L P E T H1 e 9 g 6 0 0 1 5 . p d b N F T V A K I G A E G A E N F L T H V R L N G F P V S A H H D I P A H Y D E N N N L F L T V I E I P K G Q Q A D K A I L L W Y P G D P I G I R R R N G Q L R F E K K V A D H E G S D S N R G A L P E T H1 e 9 g 6 0 0 7 9 . p d b N F T T E E I G A K G A E N Y A T H V R L N G F P V S A H H D I P A H Y D E N N K L F L T V I E I P K G Q Q A D L R I M L W Y P G D P I M P A R E N G Q L R F E K K V A D H E G M D V N W G A L P E T HC o n s e n s u s N T G A A E T V L N G F P V S A H H I D N N F V I P K G Q Q A D I L W D P I R N G Q L R F K K V D G N G L P T
1 1 0 1 2 0 1 3 0 1 4 0 1 5 0 1 6 0 1 7 0 1 8 0 1 9 0 2 0 0. . . . | . . . . | . . . . | . . . . | . . . . | . . . . | . . . . | . . . . | . . . . | . . . . | . . . . | . . . . | . . . . | . . . . | . . . . | . . . . | . . . . | . . . . | . . . . | . . . . |
1 e 9 g 2 0 0 1 0 . p d b L K S N R V H N N T N A P G A G V A I L V S V H S N R V G E T G K V T V V I A L G I T F L I E N G Y A S P V V H A V E V E A P L A N L L T S A S D I Q I F H P G Q L E N D L K F Y W T K E I A E G Q P P1 e 9 g 4 0 0 2 2 . p d b L K S N E V H D E T N S P G S G E A I R V S V H S N D V G Y T G A V I V T E A L G I T H L I R N G Y S D P V V H A V R V E D D L S S L L T S A N D I Q I F H P G Q L E N D L K F H W T E E I A E G Q P P1 e 9 g 3 0 0 0 5 . p d b N K S N E V V N Q T N S P G D G T A S H V E V H S N E V G E T G R V E T V K A T G V T H E E N N G Y D D D N V H A I R T D A P L A D L V T S A N D N Q I F F P G Q E N N D K K F H I T R E I A H G Q P P1 e 9 g 3 0 0 2 9 . p d b L K S N E V W N N T N A P G S G T A I H V S V H S N R V G E T G K V T E V K A T G I T L L E E N G Y A S P V V H A I E V E A P L A D L V T S A N D N Q I F F P G Q E E N D L K F F W T K E I A E G Q P P1 e 9 g 2 0 0 0 5 . p d b N K S N E V D D Q T N S P G N G T A S L V S V H S N R V G E T G A V I V V I A T G T T H L I R N G Y N A D I I H A I E V K A P L A N L V T S A N D V R I F F P G Q E D N D K K F W I T K E I A E G Q P P1 e 9 g 6 0 0 1 3 . p d b L K S N E V H D N T N S P G S G T A I L V S V L S N R V G E T G A V I V T R L T G V M L L I R N G Y S A P V L F A I E V K A P L A D L V T S A D D V K I F F P G A L D N A K K F W I T S E I A E G Q P P1 e 9 g 6 0 0 2 5 . p d b L K S N E V H D E T N S P G S G T A I N V S V H S N R V G Y T G A V I K T K A T G I T H L T R N G Y S N P V V H A I E V K A P L A D L V T S A N D N Q I F F P G Q E N N D K K F Y I T S E I A E G Q P P1 e 9 g 2 0 0 5 8 . p d b L K S N E V H D N T N A P G S G T A I H V S V H S N L V G Y T G A V I V V I A T G I T F E I R N G Y S H P V V H A I S T T A P L S S L V T S A N D V Q I F F P G Q E N N D K K H H I T H E I A E G Q P P1 e 9 g 6 0 0 1 5 . p d b M K S N E V H D Q N N S P G A G Q A A H V S V H S N L V G E T G A V I V V Y A T G V T H E I R N G Y S H D V V H A I S T T A P L A N L V T S S S D N R I F F P G Q E D N D K K H H I T H E I A E G H P P1 e 9 g 6 0 0 7 9 . p d b L K S N E V V D N T N A P G S G T A I N V A V V S N L V G E T G A V I V V Y A T G I I L L I R N G Y A D P V V I A I S T T A P L S S L V T S A N D I Q I F F P G L L E N I L K H H I T E E I A H G H P PC o n s e n s u s K S N V N P G G A V V S N V G T G V G N G Y A L L T S D I F P G N K T E I A G P P
R.dR.d HMMERHMMERResumenResumenestadístico deestadístico deprobabilidadesprobabilidades
Comparación deComparación denuevas secuenciasnuevas secuenciascontra el resumencontra el resumen
Elección deElección desecuenciasecuencia"aprobadas""aprobadas"
¿Está mi objetivo¿Está mi objetivoentre lasentre las"aprobadas"?"aprobadas"?
Y permite buscar secuencias queY permite buscar secuencias queresponden a la predicción (aprobadas)responden a la predicción (aprobadas)

Una nota de advertenciaUna nota de advertencia
El índice Rd.HMM no da falsos positivos, perolos falsos negativos son frecuentes. i.e. su modelo puede declararse irrelevante, cuando en
realidad es parcialmente relevante (aproximado).
Los modelos relajados con ROSETTA RELAXo provenientes de ROSETTA AB INITIO tienenun sesgo.Su “SCORE” Rd.HMM se acerca a la longitud de su
secuencia.Asegúrese de calificar su modelo después de
haberlo relajado con algún otro método también

EjemplosEjemplos

Tomemos un ejemploTomemos un ejemplo

Apirasa humana (pdbid 1S1D, 1S18)Apirasa humana (pdbid 1S1D, 1S18) Rd·Hmm de 13 intermediarios X 77 reconstrucciones = 1001 secuencias.
hmmsearch - search a sequence database with a profile HMMHMMER 2.3.2 (Oct 2003)Sequence database: NCBI-nr Total sequences searched: 5878462Query HMM: cpx_1S1D_a-O [HMM was calibrated; E-values empirical estimates]Scores for complete sequences (score includes all domains): : 110 hitsSequence Description Score E-value N -------- ----------- ----- ------- ---gi|47168960|pdb|1S18|A Chain A, Structu 334.4 1.3e-94 1gi|112491029|pdb|2H2N|A Chain A, Crystal 334.4 1.3e-94 1gi|20270339|ref|NP_620148.1| calcium activate 334.4 1.3e-94 1gi|22218108|gb|AAM94564.1|AF328554_1 soluble calcium- 334.4 1.3e-94 1gi|114670750|ref|XP_523734.2| PREDICTED: calci 333.8 1.9e-94 1gi|112491033|pdb|2H2U|A Chain A, Crystal 333.8 1.9e-94 1...gi|74764847|sp|O96559|APY_CIMLE Apyrase precurso 135.8 7.5e-35 1gi|119609954|gb|EAW89548.1| calcium activate 130.4 3.3e-33 1gi|77696457|gb|ABB00907.1| 35.3 kDa salivar 120.9 2.4e-30 1...gi|10443907|gb|AAG17637.1|AF261768_1 salivary apyrase 109.6 5.9e-27 1gi|61817259|gb|AAX56357.1| 35.5 kDa salivar 106.2 6.1e-26 1gi|4928274|gb|AAD33513.1|AF131933_1 putative apyrase 103.4 4.5e-25 1...gi|67465798|ref|XP_649057.1| apyrase [Entamoe 33.5 1.3e-05 1gi|67472519|ref|XP_652062.1| apyrase [Entamoe 5.4 0.0025 1gi|165900885|gb|EDR27110.1| Apyrase precurso 0.7 0.0062 1gi|71280063|ref|YP_271339.1| response regulat -55.1 2.3e+02 1...gi|119358104|ref|YP_912748.1| lipase, class 3 -55.7 2.5e+02 1gi|163799550|ref|ZP_02193457.1| AMP-dependent sy -55.8 2.6e+02 1gi|31455467|dbj|BAC77359.1| putative NFkB ac -55.8 2.6e+02 1...
primer hit
primer hit

NCBI-nr: las 5 secuencias más cercanas >gi|47168960|pdb|1S18| Structure And Protein Design Of Human Apyrase ...>gi|112491029|pdb|2H2N|A Crystal Structure Of Human Soluble Calcium-Activated Nucleotidase >gi|20270339|ref|NP_620148.1| calcium activated nucleotidase 1 [Homo sapiens]...>gi|22218108|gb|AAM94564.1|AF328554_1 soluble calcium-activated nucleotidase 1 [Homo sapiens]>gi|114670750|ref|XP_523734.2| PREDICTED: calcium activated nucleotidase 1 isoform 4
[Pan troglodytes]gi|114670752|ref|XP_001158629.1| ...
Última secuencia recuperada con significancia estadísticagi|165900885|gb|EDR27110.1| Apyrase precurso 0.7 0.0062 1

Probando el algoritmo Rd·hmm con modelos sometidosProbando el algoritmo Rd·hmm con modelos sometidosal concurso CAPSal concurso CAPS
Se tomaron modelos de los concursos CASP (2006 &2007, si la estructura llegó al PDB)
Se eligieron modelos buenos y malos según loscriterios de calidad del CASP. Sus Rd·hmm recupera:
su secuencia con un log(E) << -10, para modelosmuy buenos.
su secuencia con un E significativo, pero no tanpequeño, para modelos intermedios
Nada, o a veces, el molde empleado para hacer elmodelo, en los modelos erróneos.
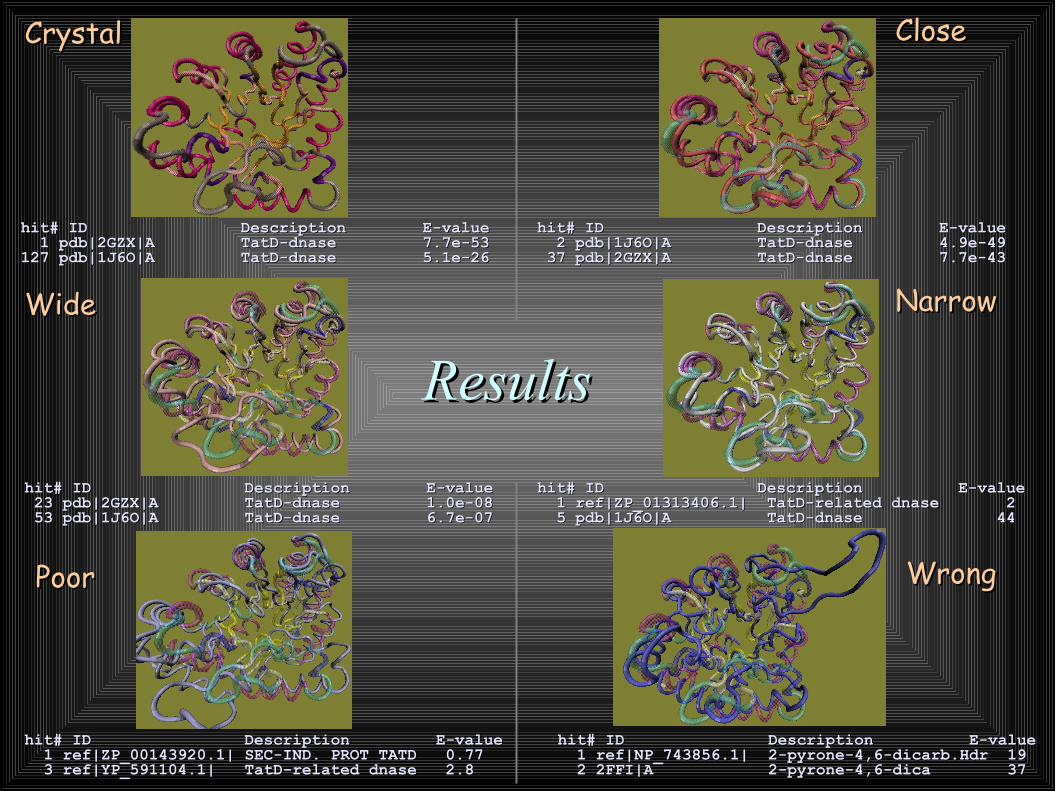
ResultsResults
hit# ID Description E-value hit# ID Description E-value 1 pdb|2GZX|A TatD-dnase 7.7e-53 1 pdb|2GZX|A TatD-dnase 7.7e-53 127 pdb|1J6O|A TatD-dnase 5.1e-26 127 pdb|1J6O|A TatD-dnase 5.1e-26
hit# ID Description E-value hit# ID Description E-value 2 pdb|1J6O|A TatD-dnase 4.9e-49 2 pdb|1J6O|A TatD-dnase 4.9e-49 37 pdb|2GZX|A TatD-dnase 7.7e-43 37 pdb|2GZX|A TatD-dnase 7.7e-43
hit# ID Description E-value hit# ID Description E-value 23 pdb|2GZX|A TatD-dnase 1.0e-08 23 pdb|2GZX|A TatD-dnase 1.0e-08 53 pdb|1J6O|A TatD-dnase 6.7e-07 53 pdb|1J6O|A TatD-dnase 6.7e-07
hit# ID Description E-value hit# ID Description E-value 1 ref|ZP_01313406.1| TatD-related dnase 2 1 ref|ZP_01313406.1| TatD-related dnase 2 5 pdb|1J6O|A TatD-dnase 44 5 pdb|1J6O|A TatD-dnase 44
hit# ID Description E-value hit# ID Description E-value 1 ref|ZP_00143920.1| SEC-IND. PROT TATD 0.77 1 ref|ZP_00143920.1| SEC-IND. PROT TATD 0.77 3 ref|YP_591104.1| TatD-related dnase 2.8 3 ref|YP_591104.1| TatD-related dnase 2.8
hit# ID Description E-value hit# ID Description E-value 1 ref|NP_743856.1| 2-pyrone-4,6-dicarb.Hdr 19 1 ref|NP_743856.1| 2-pyrone-4,6-dicarb.Hdr 19 2 2FFI|A 2-pyrone-4,6-dica 37 2 2FFI|A 2-pyrone-4,6-dica 37
PoorPoor WrongWrong
CrystalCrystal CloseClose
WideWide NarrowNarrow

1J601J602GZX2GZX
T0315_pT0315_p
El modelo cercano es intermedio entreEl modelo cercano es intermedio entrelos cristales 1J60 y 2GZXlos cristales 1J60 y 2GZX
El modelo cercano es intermedio entreEl modelo cercano es intermedio entrelos cristales 1J60 y 2GZXlos cristales 1J60 y 2GZX

El modelo incorrecto y su “pariente” estructuralEl modelo incorrecto y su “pariente” estructuraldistante 2FFIdistante 2FFI
Secuencias no relacionadas, peroSecuencias no relacionadas, peroestructuras 3-D ...estructuras 3-D ...


Valor de Rd.HMMValor de Rd.HMM
El algoritmo Rg·Hmm puede identificaresqueletos 3·D-de proteínas con plegamientos“tipo cristal”.
La predicción de la estructura a partir de lasecuencia sigue siendo empírica y azarosa,pero si se logra un modelo correcto es posibleidentificarlo de entre varios candidatos.
En el peor escenario, si ningún modelo escercano, se evita perder tiempo tratando devalidar modelos incorrectos.

ComparaciónComparación
Top Related