







Idiomas
Páginas
Jurídico


Neuropatía PeriféricaDr.Sittenfeld
Son variables en su forma de presentación y etilogía, se pueden dividir desde el punto de vista histopatológico en:
Desmielinizante Degeneración axonal Vascular Mixtas (la mayoría)
DESMIELINIZANTESPueden presentarse en parches, teniendo etiología inflamatoria, o ser uniformes cuando la causa metabólica o hereditarias.
Se pierde la mielina pero el axón está preservado produciendo problemas de velocidad de conducción y no de amplitud.
Puede haber remielinización dependiendo de la severidad. Algunas neuropatías desmielinizantes pueden tener componente
neuroaxonal. La desmielinización limita la conducción nerviosa y produce debilidad pero
no atrofia, como si lo hacen las neuroaxonales.
DEGENERACIÓN AXONALPrincipalmente de causa metabólica, pero también puede ser genética, tóxica, nutricional y física, ocasionada por el frío o traumas.
Los nervios largos son los más afectados. Clínicamente se tiene
Debilidad distal Atrofia Pérdida sensitiva
Si se preserva la neurona hay posibilidad de regeneración.
VASCULARESExiste afección de vasa nervorun que lleva a infartos de nervios y degeneración axonal distal al sitio del infalto. Las principales causas están asociadas a vasculitis y DM.La mononeuritis del III par craneal, es de las más frecuentes en la neuropatía vascular diabética. Se tiene aparición súbita de diplopia, desviación de la mirada hacia fuera, abajo y arriba de un lado + párpado caído. La estructura anatómica que se debe revisar es la a. cerebral comunicante posterior. Un tercer par compresivo o completo afecta oculomotores, elevadores del párpado y pupila, se tiene midriasis. En cambio cuando hay afectación de vasa nervorum las pupilas son isocóricas normoreactivas. Recordar que los músculos inervados por el III par craneal son recto superior, recto inferior, recto interno y oblicuo inferior, más elevador del párpado, más la pupila. Si esta pupila no está dilatada y es diabético, se tiene un III Par incompleto secundario a una mononeuritis vascular diabética.

Características clínicas Sensitivas o motoras Usualmente mixtas: dependiendo de la causa puede predominar una o la
otra. Dependen del proceso patológico y de la rapidez de instauración. De tipo axonal : atrofia y pérdida de ROT. Desmielinizantes : sin atrofia y con disminución de ROT. También pueden haber manifestaciones autonómicas como pérdida de
sudoración, viscerales como estreñimiento, problemas de vaciamiento vesical, problemas de erección.
Patrones de presentaciónNeuropatía simétrica distal
Forma más frecuentes entre neuropatías axonales. Afección distal: piernas > brazos. Puede ser sensitiva (en raros casos) o sensitiva motora Neuropatía de guante – calcetín. Usualmente primero pies y manos.
Neuropatías multifocales Asimétrico. Neuropatías desmielinizantes y vasculíticas.
Mononeuropatías Afección de un solo nervio. Usualmente asociadas a compresión en sitios de
atrapamiento. Por ejemplo túnel carpal donde hay afectación del n. mediano; parálisis del surco ulnar, parálisis del sábado por la noche en donde se afecta el n. radial cuando se queda dormido en la silla y despierta con la mano caída. Otro caso es en las personas que acostumbran cruzar las piernas y pierden peso y de repente pie caído.
Mononeuritis múltiple Afectación de varios nervios. Sugiere etiología vasculítica.

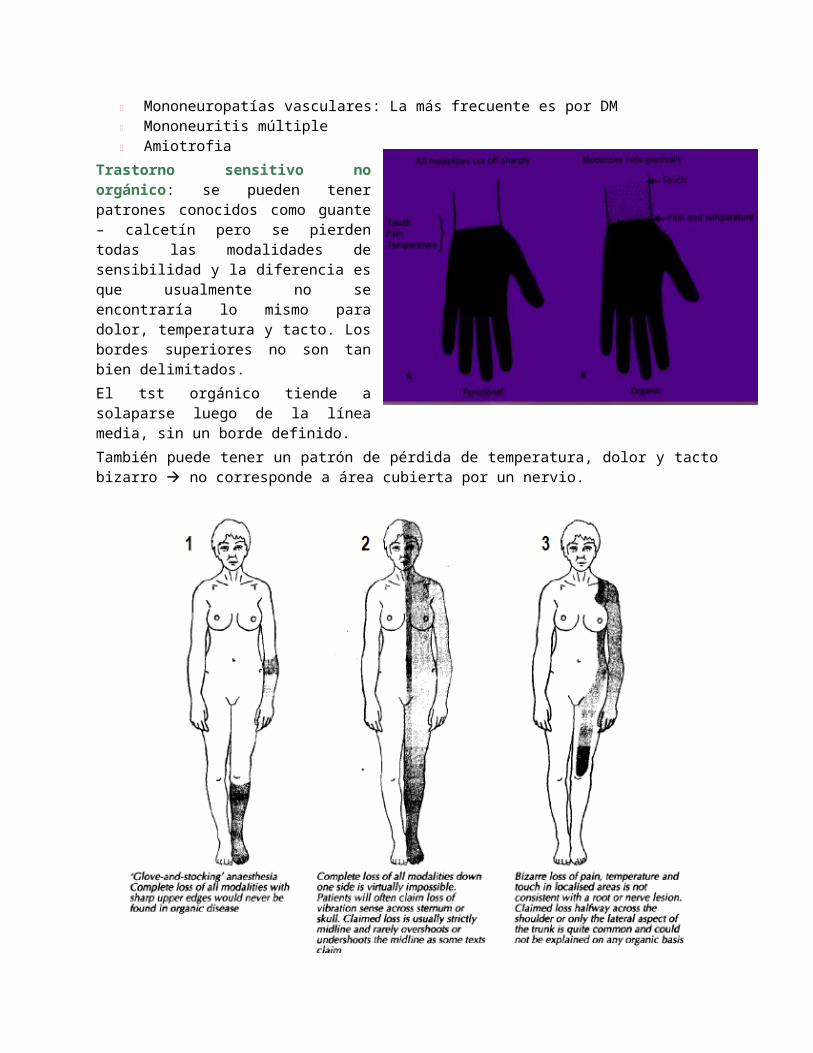
1) Nervio mediano, 2) Nervios ulnar y peroneal, 3) Neuropatía multifocal, 4) Guante y calcetín.
Factores asociados con neuropatíasEnfermedades sistémicas
DM: más frecuente. Aproximadamente 50% desarrollan neuropatía después de 10a de evolución.
Cáncer Enfermedades del colágeno: LES, AR, enfermedad mixta del colágeno. Enfermedades inflamatorias crónicas: sarcoidosis. HIV: por el virus o medicamentos antirretrovirales. Uremia Hipotiroidismo Polineuropatía del paciente crítico
Deficiencias Tiamina (B1): principalmente en alcohólicos. Pelagra (niacina): raro, el doctor solo vió un caso cuando era residente. Piridoxina (B6) Anemia perniciosa (B12): es la más frecuente. Son más tempranas las
manifestaciones neurológicas que la aparición de la anemia megalobástica que puede ser compensada con exceso de ácido fólico. Los niveles que se usan en neurología son mayores que los utilizados para el diagnóstico de anemia. La neuropatía usualmente es multifocal o polineuropatía distal simétrica sensitivo – motora; puede tener degeneración combinada subaguda de médula que afecta cordones posteriores y laterales, afectando propiocepción. Al final combina afectación corticoespinal lateral, ataxia propioceptiva, piramidalismo y neuropatía periférica.
Deficiencia de folatos Deficiencia de Vitamina E
Tóxicos Etanol: Es la causa más
frecuente de neuropatías toxica. Arsénico Oxido de etileno Plomo: Pacientes que
trabajaban con baterías de automóvil o pinturas.
Mercurio Organofosforados Talio
Drogas Amioradona: antiarrítmico Cisplatino Dapsona: antimalárico, lepra,
enfermedad de Hansen y enfermedades reumatológicas.
Oro: AR Isoniacida: TB Metotrexate: cáncer Talidomina: naúseas y mareos
asociados al embarazo, provocaba focomelia.
Vincristina: cáncer.
Relación entre la presentación y la etiología

Es importante establecer el patrón de distribución, una vez hecho esto se debe clasificar neurofisiológicamente (desmielinizante o axonal), después dependiendo de la rapidez de presentación, si es agudo, subagudo o crónico, nos orienta hacia el diagnóstico.
Patrón Neurofisiología
Aguda Subaguda Crónica
Distal simétrica
Axonal (hay atrofia y ausencia de reflejos)
ToxinasPorfiria
Enfermedades sistémicasDeficienciasToxinasCáncerAmiloidosis
Enfermedad sistémicaDeficienciasToxinasCáncerAmiloidosisHMNS2
Distal simétrica
Desmielinizante
SGBdifteria
CIDP CIDPHMNS1
MultifocalMononeuritis múltiple
Axonal Vasculitis DMSarcoidosis
Lepra
MultifocalMononeuritismúltiple
Desmielinizante
SGB CIDP CIDP
SGB : síndrome de Guillain Barré.CIDP : poliradiculoneuropatía idiopática crónica difusa (es como un Guillain Barré crónico)HMNS : hereditary motor sensitive neuropathy, tipo 1 (tipo desmielinizante con disminución de velocidades motoras) y tipo 2 (conocido antes como Charcot-Marie-Tooth)
En estudios neurofisiológicos se puede diferenciar si es axonal, desmielinizante o mixta. Se valoran velocidades motoras, cada nervio tiene una velocidad específica que es normalmente superior a 40m/s, en las axonales van de 30 a 35, en las desmielinizantes llegan a 10 o menos. En la forma desmielinizante la velocidad de conducción esta disminuida, en axonal levemente disminuida o normal. La amplitud de los potenciales varia, en la forma desmielinizante estaría poco afectada, en la axonal disminuida. Para decir que definitivamente es axonal hay que demostrar que hay denervación.
NEUROPATÍA PERIFÉRICASignos clásicos motores: atrofia, debilidad, calambres musculares, fatiga y fasciculaciones.Diagnostico diferencial
Miopatía: usualmente con debilidad proximal. Los reflejos no están abolidos. Miastenia (placa neuromuscular): debilidad fluctuante con distribución
ocular, bulbar; respeta extremidades. Enfermedad de la motoneurona inferior: fasciculaciones y gran atrofia.
Síntomas sensoriales Positivos

Parestesias: sensación de hormigueo Disestesias: dolor de tipo calambre eléctrico, como agujas. Hiperestesias Alodinia: estimulo doloroso producido por un estímulo que usualmente
no es doloroso Negativos
Hipoestesia o anestesia Síntomas varían grandemente dependiendo de la causa.
Diferencias entre niños y adultosEn los niños hay ausencia de síntomas subjetivos, se tiene que poner atención a retraso motor y torpeza. Es importante hacer diagnóstico diferencial con distrofias musculares por que la mayoría son proximales y hereditarias.En el adulto se tiene signos típicos: parestesias periféricas, pérdida de la sensibilidad y torpeza.
Datos de interés por recolectar Historia familiar: algunas neuropatías son familiares. Exposición a drogas y tóxicos. Hábitos alimenticios pensando en déficit nutricional. Ahora los alcohólicos no
suelen tener deficiencias nutricionales por que el los bares sirven bocas. Los que si la pueden tener son los vegetarianos estrictos.
Procedimientos quirúrgicos: muchas mononeuritis agudas se dan después de un procedimiento.
Enfermedades sistémicas
Datos por establecer ¿Es sensitiva-motora-mixta? ¿Qué patrón de distribución tiene?
Mononeuritis : ¿Qué nervios están involucrados? Por ejemplo en el túnel carpal hay compresión del n. mediano en el retináculo de la muñeca. Pero si el túnel es bilateral se debe de pensar en enf sistémicas como hipotiroidismo, acromegalia, AR y DM. ¿Sitios de atrapamiento? Túnel carpal, subulnar, canal de Guyon.
Mononeuritis múltiple Distal y simétrica (guante – calcetín)
¿Compromiso autonómico? importante revisar hiptensión ortostática y FC que no modifican con ejercicio, problemas GI
¿Amiotrofia distal o proximal? Daño desmielinizante o neuroaxonal por estudios neurofisiológicos: EMG-
VCN.
Estudios de laboratorio y gabinete Hemograma : valorar presencia de anemia. VES : para valorar inflamación. Se puede encontrar elevada VES con relación
a neuropatías: en enfermedades neoplásicas, autoinmunes o infecciosa (TB).

Glicemia : tomar glicemias en ayudas y postprandial. Se considera que es diabético con 126mg/dL en ayunas. Valorar también Hb glicosilada, para valorar glicemia a través del tiempo.
N 2U, Creatinina: IR, nefropatía urémica. Proteínas totales y fraccionadas : elevada en mieloma y tumores que
produzcan gammapatías monoclonales. TSH-T4 : hipo/hipertiroidismo. Nivel de vitamina B12 y acido fólico Estudio por colagenopatías : FR, ANA, Cels LE. Velocidad de conducción nerviosa (VCN): decide si es motora, sensitiva o
ambas: neurografía motora y sensitiva. Electromiografía Biopsia de nervio : en casos muy aislados. Se utiliza nervio Sural, por ser
sensitivo y distal. Estudios de conducción nerviosa Se pone una corriente eléctrica de 10mAmp sobre un nervio conocido, y otro electrodo sobre un músculo conocido se obtiene una respuesta motora (M), esta se mide en diferentes sitios, se calcula la diferencia y se saca la velocidad de la conducción para ese nervio.El estudio puede realizarse sensitivo, ortodrómico o antidrómico. En el antidrómico el estimulo es proximal y la respuesta distal. El ortodrómico es en el sentido natural de la conducción sensitiva, el estimulo es distal al nervio y la respuesta es proximal.
Fenómeno de bloqueo de conducción: si se estimula distalmente y le da 8mV (normal), todo está bien, pero se estimula a nivel proximal y resulta que hay una diferencia significativa en la amplitud entre distal y proximal (cae más de un 50%), es decir la conducción no se da en forma adecuada. Eso es un bloqueo. Pero si hay caída de pico en diferentes sitios puede ser que tenga algún grado de bloqueo de conducción difusa pero el diagnóstico diferencial es con una lesión de la

motoneurona, y la única manera de de demostrar la lesión de la motoneurona, o del axón en sí, es demostrando que hay un patrón de denervación, por lo tanto hay que hacer una prueba de agujas, y lo que demuestra es un patrón de denervación activa.
Defectos de Conducción Nerviosaa) Normalb) Degeneración axonal: velocidad mantenida + caída de amplitudes
debilidad.c) Lesión desmielinizante: velocidad disminuida, puede haber bloqueo de
conducción.d) Vascular o traumática aguda: no hay conducción ni respuesta. Diagnóstico
diferencial sería bloqueo de tipo inmunológico como Guillain Barre, poliradiculoneuritis que produce bloqueos de conducción.
Electromiografía Si se tiene músculo inervado por un nervio normal en reposo la EMG tendrá
un patrón silencioso. Si se le pide al paciente que contraiga se observará la activación de unidades
motoras. Los potenciales tienen menos de 5 deflexiones por unidad motora, < 4 – 6mV
de tamaño.

Patrón de desmielinización: disminución del patrón normal, no hay actividad espontánea ni potenciales de denervación, disminución de las unidades motoras.
Degeneración axonal: fascículos un patrón de denervación, patrón de inserción aumentada, fibrilaciones y ondas positivas agudas. Las fibrilaciones tienen un pico para arriba y otro para abajo. Las ondas positivas agudas sólo tiene uno para abajo, todo depende de la aguja.
Fasciculaciones: contracciones espontáneas de una unidad motora, en EMG se ve reclutación de la unidad motora completa. No tienen validez en el EMG excepto que se acompañen de fibrilaciones y ondas positivas agudas. Una tercera parte de la población reporta fasciculaciones espontáneamente. Del personal médico, 60 – 70% las presenta. Son malignas si se asocian con debilidad o atrofia.
La denervación produce de forma aguda actividad espontánea en reposo fibrilaciones positivas agudas y actividad disminuida. Meses después de la lesión inicial se tiene un patrón de renervación crónica donde hay potenciales gigantes y polifásicos.
Miopatías: si es agudo se tienen fibrilaciones, pero a la hora que se contrae, las fibras lo hacen simultáneamente, y encontramos potenciales pequeñitos, que rápidamente llenan la pantalla.
Formas de neuropatías

Polineuropatía sensitivo periférico: leve, arreflexia, hipoestesia distal. En guante – calcetín.
Polineuropatía sensitivo motora Neuropatía autonómica Mononeuropatías compresivas Mononeuropatías vasculares: La más frecuente es por DM Mononeuritis múltiple Amiotrofia
Trastorno sensitivo no orgánico: se pueden tener patrones conocidos como guante – calcetín pero se pierden todas las modalidades de sensibilidad y la diferencia es que usualmente no se encontraría lo mismo para dolor, temperatura y tacto. Los bordes superiores no son tan bien delimitados.El tst orgánico tiende a solaparse luego de la línea media, sin un borde definido.También puede tener un patrón de pérdida de temperatura, dolor y tacto bizarro no corresponde a área cubierta por un nervio.

Polineuropatías donde la biopsia del nervio es útil Inflamatorias: SGB, poliarteritis nodosa (mono neuritis múltiple), sarcoidosis
y lepra Hereditarias: HMSN I-II-III Metabólica: amiloidosis Toxica (grande axonal): N-hexano, acrilamida, metilbutilketona
Causas de Neuropatía Periférica: hereditarias, infecciosas: Lepra (presentan zona de anestesia local), metabólicas: DM; tóxicas y desórdenes oscuros
Polineuropatías (PN) dolorosas PN predominantemente sensitivas
Diabética sensorial Alcohólica Porfiria aguda Amiloidea Enfermedad de Fabry SGB Asociada a AR Isquémica Subaguda paraneoplásica Mielomatosa sensitivo motora Arsénico Cloranfenicol Metronidazole Misonidazole Organofosforados Talio (Se usa en AR)
Diabética distal sensorial Alcohólica Amiloidosis Enfermedad de Tangier Exceso de piridoxina HMNS I-II-III-IV Neuropatía subaguda sensorial
(ganglionitis dorsal) AR Lepra Cloranfenicol Etionamida Glutatemida Metronidazole Oxido nitroso (gas de la risa, que
utilizan los odontólogos como sedante)
Cisplatino Arsénico Talio
PN predominantemente motoras PN desmielinizantes SGB: AIDP, CIDP Porfirio aguda intermitente Amiotrofia diabética HMNS I-II Nitrofurantoina Dapsona (Se usa en Lepra) Mercurio Neuropatía subaguda motora Neuropatía motora multifocal
Síndrome de Guillain Barre Polineuropatia crónica
inflamatoria: CIDP Diftérica Leucodistrofia metacromática:
Trastorno genético del metabolismo
Enfermedad de Krabe: Trastornos genético del metabolismo
Enfermedad de Refsum: Trastornos genético del metabolismo. Se asocia a exceso de ácido fitánico.
Polineuropatías con nervios gruesos palpablesCuando hay desmielinización y remielinización se forman “nódulos” en el nervio.
Infiltración inflamatoria: SGB, neuropatía crónica inflamatoria, lepra. Hipertrofia de la cobertura de los nervios: HMNS I-II, enfermedad de
Refsum: trastorno del metabolismo del ácido teitanico Depósitos intraneurales: neuropatía amiloidea
SÍNDROME DE GUILLAIN BARRÉ Características
2/100000/año Enfermedad monofásica, poliradiculoneuropatía predominantemente motora
y desmielinizante

Se desarrolla en días a 4 semanas, usualmente con un pico máximo entre el 7 – 10d.
Se tiene debilidad leve a severa con parálisis respiratoria (10-20%), se debe monitorizar la capacidad vital, porque pueden hacer falla respiratoria.
Signos sensoriales leves ROT desaparecen, hay arreflexia o hiporreflexia. Afectación del SNA frecuente: se manifiesta con arritmias, hipo o
hipertensión. Se tiene un paciente previamente sano que tuvo un cuadro viral que en cuestión de 2 – 3S empieza con cuadro de debilidad progresiva de abajo hacia arriba patrón del Guillain Barré clásico, si no se presenta de esta manera solo se llama poliradiculoneuritis aguda.
70% está asociada a infección: CMV, Mononucleosis, Campylobacter jejuni que produce una forma más severa y de tipo axonal.
LCR: disociación albumino citológica. Se tiene aumento importante de proteínas en LCR pero no hay aumento franco de la celularidad (leucocitos). +50 células en sospecha de GB hacer TAC.
Manejo1. Sostén: medición de Capacidad Vital (1 litro ventilación). < 20cc/kg se
tiene en unidad de vigilancia y < 15 cc/kg hay que intubar con ventilación mecánica.
Cuando llegan los pacientes con formas severas y dificultad para movilización, se brindan medidas para cambian el curso natural de la enfermedad. Se pueden hacer hasta 1S después de iniciados los síntomas:
2. Plasmaféresis 3. Inmunoglobulinas 0.9mg por kg.
Pronóstico: mortalidad 5% (arritmias, TEP, Infecciones), déficit motor 20%, recurrencia 3%.Variantes:
Síndrome de Miller Fisher: arreflexia, oftalmoplejia y ataxia. CIDP: progresiva y recurrente
NEUROPATÍAS HEREDITARIASa. Neuropatías sensitivo/motorasHMSN I (Charcot Marie Tooth-Levy
Rousy) HMSN II (Charcot Marie Tooth)
Autosómico dominante o recesivo (GEN1)
Signos aparecen a cualquier edad Cifoescoliosis + Pes cavus o pie de
Friedreich’s (aumento del arco del pie)
Usualmente obvia a los 10 – 20a
Autosómica dominante. Cifoescoliosis + pes cavus o pie de
Friedreich’s. Usualmente obvia 20 – 30a. Atrofia de músculos de la
pantorrilla más déficit motor que sensorial.

Produce atrofia de músculos de pantorrillas (pierna en garza o en copa de champaña) más déficit motor sensorial.
Hiporreflexia VCN muy enlentecida (10-20m/s) Engrosamiento de los nervios en
50% Es de predomino desmielinizante.
Hiporeflexia Daño neuroaxonal. VCN: caídas de amplitud y
denervación.
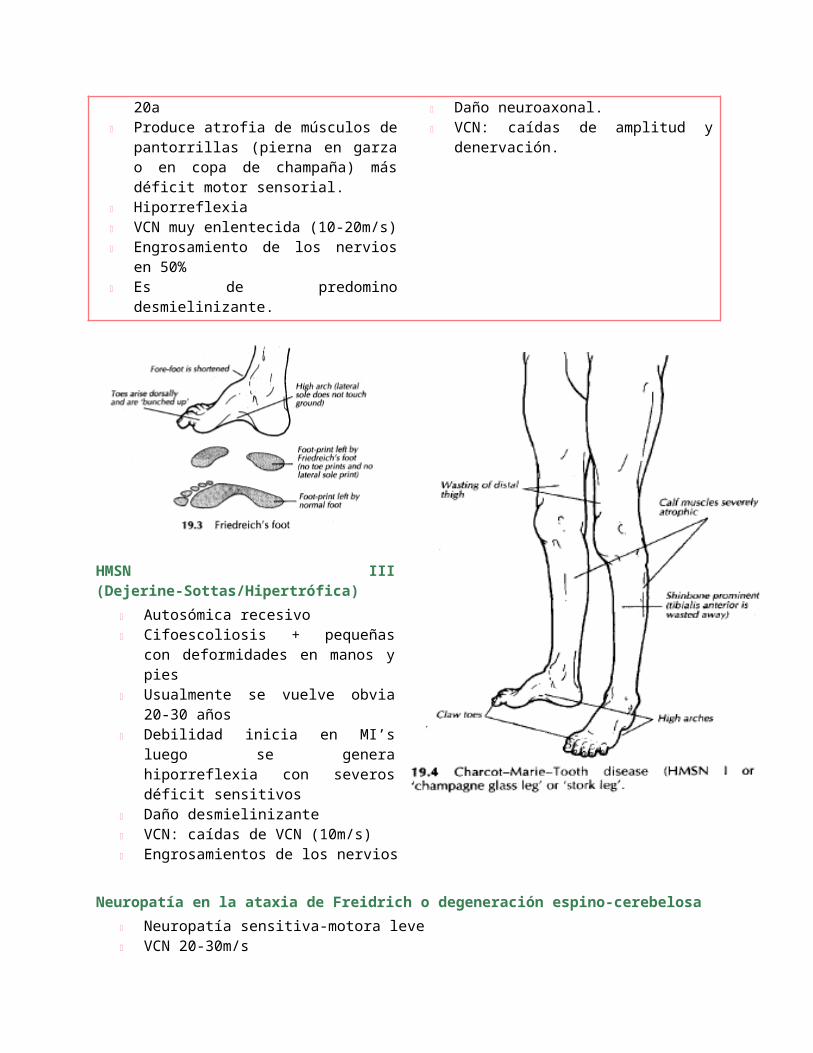
HMSN III (Dejerine-Sottas/Hipertrófica)
Autosómica recesivo Cifoescoliosis + pequeñas con
deformidades en manos y pies Usualmente se vuelve obvia 20-
30 años Debilidad inicia en MI’s luego se
genera hiporreflexia con severos déficit sensitivos
Daño desmielinizante VCN: caídas de VCN (10m/s) Engrosamientos de los nervios
Neuropatía en la ataxia de Freidrich o degeneración espino-cerebelosa Neuropatía sensitiva-motora leve VCN 20-30m/s Deformidades en los pies Ataxia Disartria
b. Neuropatías hereditarias puramente sensitivasHSN I (Jenny Brown)
Rara autosómica dominante 10 – 20 años

Afecta sobre todo a fibras no mielinizadas, por lo que ataca a las de dolor y temperatura
HSN II (congénita): rara autosómica recesiva e inicia en la infancia.HSN III (Riley Day disautonomía familiar): rara, autosómica recesiva (judíos)
Congénita con ausencia de ganglios autonómicos y dorsales Neuropatía autonómica severa
HSN IV (congénita con pérdida de la sudoración): rara, autosómica recesiva. Desde la infancia no controla la temperatura
Neuropatías disparadas por desordenes metabólicasAmiloidea
Forma familiar Depósitos de amiloide en las coberturas de los nervios y microvascular 4 tipos de neuropatías hereditarias Daños de fibras pequeñas: hay perdida de dolor y temperaturas y
neuropatía autonómica No familiar o sistémica
Mieloma Neuropatía sensorio motora que evoluciona a autonómica
Desordenes de lipoproteínasLipoidosis cerebrales
Leucodistrofia metacromática Enfermedad de Krabbe
Neuropatía por deficiencia de glicoproteínas Enfermedad de Tangier Bassen Kornzweig (A-beta lipoproteinemia) produce acantositosis
Refsum (deficiencia de acido fitanico): rara, retinitis pigmentosa, sordera y ataxia.Enfermedad de Fabry: ligada al X, inicio en la niñez (lesiones telangiectasias), dolorosaPorfiria aguda intermitente
Defecto uroporfirinogeno 1 sintetasa Autosómica dominante Incidencia: 1.5/100000 Drogas provocativas: sulfas y barnitúricos. Dolores abdominales (autonomica) Neuropatías ascendente (como el SGB) Detección de porfobilinogeno + acido aminolevolínico Disfunción de la ADH: síndrome de hiponatremia Si ponemos a la luz la orina de un paciente con porfiria se vuelve de color
morado.
Neuropatías infecciosasLepra (enfermedad de Hansen): lesiones hipoestésicas + hipopigmentadas.
Herpes Zoster: puede dar neuropatía postherpética. Se da en raíces produciendo un brote + dolor + hipoestesia.Diftérica: toxina que inhibe síntesis de mielina y produce poliradiculoneuropatía 4 semanas después, pueden hacer miocarditis.Sarcoidosis: Granulomatosis crónica con problemas pulmonares, 5% dan neuropatía periférica. Puede presentar parálisis de Bell bilateral y recurrente.
Neuropatías tóxicas: arsénico, plomo, talio, trio-ortocresyl-fosfato, solventes orgánicos, acrilamida.
Neuropatías inducidas por drogas: talidomida, oro, metronidazole, nitrofurantoina, cloranfenicol, drogas antineoplasicas (alcaloides de la vinca), drogas anti HIV
Neuropatías más frecuentesAlcohólica: toxica vs deficiencia de B1. Son dolorosas en miembros inferioresDiabética
Más frecuente en países desarrollados Relacionada con la duración de la enfermedad: 50% DM con más de 20 años
de evolución tiene neuropatía diabética. Más prevalente en hombre DM adulta puede ser la forma de presentación Todas las fibras son afectadas: motoras, sensitivas y autonómicas Cambios patológicos:
Degeneración axonal, desmielinización secundaria, lesiones vasculares proximales y aparición de parálisis compresivas en los sitios de pobre vascularidad.
Usualmente se presenta de forma crónica en donde la mayoría de las fibras afectadas son las pequeñas tanto mielinizadas como las amielínicas, por lo que el dolor y la temperatura se afectan inicialmente. La afección motora usualmente es tardía y rara.
Las formas más agudas generalmente se dan en sitios de atrapamiento o por compresión externa en sitios de pobre vascularidad
Formas de neuropatía diabética PN sensorial periférica Polineuropatia sensitivo motora:
Distal sensorial, progresiva Arreflexia generalizada Dolorosa Pérdida de dolor profundo: artropatía de Charcot
Neuropatía autonómica: Cardiovascular : hipotensión postural o taquicardia persistente. Gastrointestinal : diarreas o constipación, o lo hacen de forma
alternante. Urogenital : impotencia, vejiga neurogénica.

Otro : constricción pupilar. Mononeuropatías compresivas Mononeuropatías vasculares Mononeuritis múltiple Amiotrofia diabética: se manifiesta con pacientes diabéticos que empiezan
a perder peso y masa muscular proximal.Neuropatía diabética periférica
Polineuropatía: crónica. La mayoría de fibras afectadas son pequeñas tanto mielinizadas como amielínicas, por lo que el dolor y la temperatura se afectan inicialmente.
Mononeuropatías: agudas. Sitios de atrapamiento o compresión externa. Vascular
Patrón clínico de las Polineuropatías (Resumen)Tipo Clínico Manifestaciones CausasAgudo Ascendente o descendente
con parálisis pudiendo involucrar PC o músculos respiratorios
- Guillain Barre- Difteria- Porfiria- Tonínas: nitrofurantoina,
dapsona, talio, arsénicoSub Aguda Distal simétrica
Sensitivo motora Predominio MI
- Metabólica: DM, uremia- Nutricional: complejo B- Inflamatoria: LES, AR- Paraneoplásica: Ca, mieloma,
linfomas- Toxinas: drogas, arsénico,
metales pesados, toxinas industriales, antineoplásicas
Crónica lenta progresiva
Distal simétrica Mixta con pérdida
sensitiva
- HMSN I, II, III, IV- HSN I, II, III, IV- Polineuropatía inflamatoria
crónica- Enf de Tangier- Amiloidosis- Lepra
Crónica recurrente
Distal simétrica Motora Pérdida sensitiva
- CIDP- Variante de GB
Neuropatías por Compresión Síndrome del túnel carpal Nervio ulnar (codo-sulcus ulnaris) Femoral cutáneo lateral: En esta entidad se da una sensación de parestesia
en la cara lateral del muslo, puede dar también disestesias o alodinia en esa región. El femoral cutáneo lateral sale por el ligamento inguinal. No tiene

componente motor, es puramente sensitiva. La fuerza del cuádriceps y del ilio-psoas se encuentran conservados, el reflejo rotuliano se encuentra normal. Es puramente sensitiva. Se ve usualmente en gente que usa ropa interior tallada y aumentan de peso rápidamente.
Peroneal (cabeza del peroné) Radial: mano caída Mononeuritis múltiple
Tipo de lesión
Daño estructural Tasa de recuperación
Neuropraxia Daño en la mielina pero el axón está intacto
Recuperan a las 2-12 semanas
Axonotmesis Pérdida de la contigüidad del axón pero con el epineuro intacto
Se regenera a 1mm/d
Neurotmesis Hay separación del tronco No hay regeneración
Lesiones vasculares nerviosas Parálisis oculomotoras recurrentes IIIPC incompleto VI PC Dermatomas torácicos: dolor súbito e hipo estesia intercostal y parálisis de
m. interostal. Recuperables
Dolor neuropático

Prevalencia de dolor neuropático: los más prevalente son 1) Sds lumbosacros, 2) Neuropatía diabética, 3) Neuralgia posherpética y neuropatías relacionadas con neoplasias.Manejo de dolor neuropático: antidepresivos tricíclicos, antiepilépticos, neuromoduladores: Amiptriptilina, carbamazepina-oxacarbazepina, fenitoína, gabapentina, pregabalina, topiramato y capsaicina tópica.
Resumen
Top Related