
Casa abierta II btmw UNIVERSIDAD AUTONOMA ...148.206.53.84/tesiuami/UAM LOTE 5/UAM20503.pdfem...
39
L. -. c Casa abierta II btmw UNIVERSIDAD AUTONOMA METROPOLITANA ( 1 0 .TIPO DE SERVICIO SOCIAL: ( x I INTERNO EXTERNO A Lh FEDERACION AILASMIENTO Y PURIFICACION DE PROTEINAS NOMBRE DEL PROYECTO: -*- I ---_.-I. .--. ASESOR RESPONSABLE:------ - ---..I DRA. SILVfA SOLIS'MENDIOLA INVESTIGADORA 7-24-46-74 CARGO:-----.-- ---.._.- ~I___ TELEFONO: ---------.-.---- LUGAR DE REAL I ZAC I ~ N : - - L - - - - . - DOMICILIO: AV. M1C - X LA p - COL 09340 MEXICO, D.F. 5-ENERO-90 . 15-JULIO-98 FECHA DE INICIO: _I CBCHA DE TERMiNACiÓN* -- I I! I I -dl-J 24 1 I nm . . UNIDAD IZTAPALAPA Av. Michoacán y la Purísima, Col. Vicentina, 09340 México, D.F., Tel.: 724-4600, Telefax: (5) 612-0885
Transcript of Casa abierta II btmw UNIVERSIDAD AUTONOMA ...148.206.53.84/tesiuami/UAM LOTE 5/UAM20503.pdfem...

L.
-.
c
Casa abierta II btmw
UNIVERSIDAD AUTONOMA METROPOLITANA
( 1 0 .TIPO DE SERVICIO SOCIAL: ( x I INTERNO EXTERNO A Lh FEDERACION
A ILASMIENTO Y P U R I F I C A C I O N DE PROTEINAS NOMBRE DEL PROYECTO: -*- I ---_.-I. .--. ASESOR RESPONSABLE:------ - ---..I DRA. S I L V f A SOL IS 'MENDIOLA
INVESTIGADORA 7-24-46-74 CARGO:-----.-- ---.._.- ~ I _ _ _ TELEFONO: ---------.-.---- LUGAR DE REALIZACI~N:--L----.
- DOMICILIO: AV . M1C-X L A p- COL
09340 MEXICO, D . F .
5-ENERO-90 . 15-JULIO-98 FECHA DE INICIO: _I CBCHA DE TERMiNACiÓN* -- I I ! I
I -d l - J 24 1
I n m
. .
UNIDAD IZTAPALAPA Av. Michoacán y la Purísima, Col. Vicentina, 09340 México, D.F., Tel.: 724-4600, Telefax: (5) 612-0885

.
cmia*iidiinpo
UNIVERSIDAD AUTONOMA M€l'ROPOLITANÁ.
A QUIEN CORRESPONDA:
Por medio de la presente, me permito hacer constar que la alumna FRANCISCO MÁRQUEZ MiSAELA, con matrícula 93323165 inscrita en la Licencia- en QUIMICA, ha concluido satisfactoriamente su SERVICIO SOCIAL, requerjdo dentro del Plan de Estudios.
A solicitud de la interesada y para los fines que estime convenientes, se extiende la presente, en la Ciudad de México, Distrito Federal a los nueve dias del mes de junio de mil novecientos noventa y nueve.
A T E N T A M E N T E
DR RICHARD S. RUiZ MARTíNEZ. SECRETARIO ACADÉMICO.
* JUN. 9 1999 * S E R V I C I O S O C I A L
C B l
RSRM*llc.
UNIDAD IZTAPALAPA '
Av. Michoacán y la Purísima, Col. Vicentina. 09340 México, D.F., Tel.: 724-4600, Telefax: (5) 612-0885 -

Casa abierta al tiempo
UNIVERSIDAD AUTONOMA METROPOLITANA D I U SlLVlA SOLIS MENDIOLA
DEPARTAMENTO DE QUlMlCA AREA DE BIOFICICOQUIMICA
Mayo 21, 1999
DRA. MARIA JOSE ARROYO PANIAGUA Directora de la División de C. B. I. Unidad lztapalapa P r e s e n t e .
. . ,
. ..
Por medio .de este conducto’ me dirijo a usted para informarle que, la. alumna MISAELA FRANCISCO MARQUE2 con matrícula 93323165, de. la Licenciatura em Química, realizó su Servicio Social Bn apoyo a la investigación, en el proyecto intitulado “~ISLAMI~N‘íO Y PURlFlCAClON DE’ PROTEINAS” en el áree de Biofisicoquímica, del Departamen@ de buímica de la Divisian de Ciencias Básicas e Ingeniería de esta Unidad, el cual fue realizado durante el periodo del 5 de enero- al 15 de julio de 1998.
Agradeciendo su atención. Is envío un cordial saludo.
A T EN TAM E N T,’E. . TASA ABIERTA AL TIEMPO
&A%. DRA. SILVIA SOLIS MEND LA Profesora del Departamento de Química Titular “C”
Av. Michoacan y la Purisima IztApalapa O9340 México. D.F. A.P. 55-534 Tel. 724-46-74 Fax: 724-46-66 e-mail:dssm~xanum.uam.mx

--I- , _ _ _ _ ^ -_-_ _,..-- -__.
INFORME FINAL DE SERVICIO SOCIAL
1. DATOS GENERALES.
NOMBRE Misaela Francisco Márquez.
MATRICULA 93323 165. ..
' 'Química. , . .
LICENCIATURA . .
. . . . . . . I
. . .. 2. ' LUGAR Y PERIODO DE REALIZACION.
Universidad Autónoma .Metropolitana-Unidad Iztapalapa, División de
CBI, Departamento de Química,' Area de Biofisicoquímica (R-209). El
período que comprende del 5 de'Enerd de 1998 . . al t5.de.julio de 1998. .'
I
. . ., .
3.. NOMBRE DEL PROYECTO.
Aislamiento y purificación de proteínas.
4. ASESOR RESPONSABLE.
D
Dra. Silvia Solís Mendiola

5. INTRODUCCION
DEFINICION Y CLASlFlCAClON DE LA CROMATOGRAFIA
Lacromatografía [l] es un método físico de separación, en el cual los
componentes que se van a separar se distribuyen entre dos fases, una de
. estas fases contribuyen en una capa estacionaria de gran área superficial,
la'otra es un fluido que eluye a través de la fase estacionaria.
La fase estacionaria puede ser líquida o sólida y la fase móvil puede ser
líquida o gas de este modo se tiene cuatro tipos de cromatografías: líquido-
solido, gas-sólido, Iíquido-líquido y gas-líquido.
En estas técnicas, los solutos que se van a separar migran a .lo largo
. de la columna (o del equivalente físico de una columna 'en las
cromatografíasde papel y de capa fina), la separación sé basa en las
diferentes velocidades de migración de los diferentes solutos. El resultado
de ésto es, que uno tiende a mover el soluto y otro lo retarda. En el
proceso original de Tswett, la tendencia que tiene los* solutos de
adsorberse sobre la fase sólida retarda su movimiento, mientras que su solubilidad en la fase móvil líquida los mueve a lo largo de la columna. Una
pequeña diferencia en la firmeza de adsorción de dos solutos y su -

interaccidn cuando las moléculas del soluto se distribuyen repetidas veces
entre las dos fases una y otra vez en toda la longitud de la columna.
CROMATOGRAFIA DE INTERCAMBIO IONIC0
La purificación de las prateínas es de gran importancia ya que para realizar
cualquier estudio sobre ellas, es necesario considerar su pureza, con 10
cual nos aseguramos que. los resultados obtenidos corresponden a la
proteína estudiada.
La cromatografía [l
modernos y eficaces para
y 21 iónica esta relacionada con los métodos
la separación y determinación de iones que se , .
basa en el uso de resinas de intercambio iónico. La cromatografía iónica
empezó a desarrollarse a mediados de los años setenta, cuando se
demostró que mediante el intercambio aniónico y catiónico, se podía
separar mezclas de cationes y aniones.
Esta técnica presenta una gran cantidad de ventajas con respecto a la
cromatografía tradicional, por ejemplo, versatilidad, rapidez,
reproducibilidad, estabilidad y sensibilidad.
Algunos de los componentes del equipo de HPLC son:
1. Sistema de entrega del solvente.
2. Válvula de inyección para la muestra.
3. Detector

I_- I _- __----- P
Y
-. .
I
I
..'.
.*
f I.
.... c
-. c
_-
c
4. Registrados o computadora para exhibir y almacenar los resultados.
La cromatografía de intercambio iónico se basa en el equilibrio de los
de soluto entre el solvente y los sitios fijos cargados de la fase
estacionaria. En los intercambiadores aniónicos, los grupos con carga
positiva están unidos covalentemente a la fase estacionaria. Los aniones
del soluto son atraídos hacia estos sitios. Los intercambiadores catiónicos
contienen sitios de carga negativa, unidos covalentemente, los cuales
retienen los cationes del soluto [l y 21.
La resolución de la cromatografía de líquidos mejora al disminuir el
tamaño de.partícula de la fase estacionaria. El precio de utilizar partículas
muy finas es la resistencia al flujo. Por lo tanto es necesario emplear alta
presión para forzar el paso líquido a través de la columna. Normalmente se
requiere presiones aproximadamente de 7 a 40 MPa (70 a 400 atm.) para
obtener gastos de 0.5 a 5 ml/min.
La celulosa, dextran e intercambiadores relacionados son adecuados
para el interca,mbio iónico de macromoleculas, como las proteínas. El
segundo (dextrán), en los enlaces cruzados de glicerina, se vende con
nombre de Sephadex. Otros intercambiadores iónicos macroporosos se
basan en el pQl¡SaCandQ agarosa y en la poliacrilamida. Los geles de
intercambio iónico se +tiliza en el caso de moléculas grandes (proteínas y
ácidos nlrcleicgs), las cuales pueden penetrar en los poros de la resina. A
menwdo, las moléculas grandes tienen cargas tan altas que si pudieran
penetrar en 'las resinas, quedarían retenidas tan fuertemente que no

podrían eluirse. En la cromatografía de intercambio iónico se debe tomar
. en cuenta tanto el punto isoelectrico (pi) como la carga neta de las
proteínas a separar.
INTERCAMBIO CATIONIC0
Cuando la proteína a separar tiene carga positiva neta, se trata de
intercambio catiónico, por lo tanto la fase estacionaria (gel, resina, etc.)
debe tener carganegativa neta. En este tipo de intercambio iónico el pH de
la fase móvil (regulador) será menor que el valor .del punto isoelectrico de’
la proteína. El valor del pH debe estar alejado del punt8 isoeléctrico, ya
que en este punto las fuerza$ de repulsión so61 mínimas lo que hace que
las proteínas tiendan a agregarse con su consecuente precipitación final.: 9
o
INTERCAMBIO ANIONIC0
Cuando la proteína a separar tiene carga negativa neta se trata de
intercambio aniónico, por lo tanto la fase estacionaria debe tener carga
positiva neta. El pH de la fase móvil (regulador) será mayor que el valor del
punto isoeléctrico de la proteína a separar.
. . . .
a

CROMATOGRAFIA DE FILTRACION EN GEL (CFG)
La cromatografía de exclusión molecular también se denomina
comúnmente cromatografía de filtración en gel o cromatografía de
permeaciói en gel. Esta técnica [2], en la cual las moléculas se separan
por su tamaño, se utiliza ampliamente .en bioquímica para separar
moléculas grandes como proteínas y carbohidratos. La fase estacionaria
contiene pequeños poros en los que pueden penetrar las molécvlas de
tamaño reducido, pero no así las grandes. Por lo tanto, el volumen
disponible es mayor para moléculas pequeñas que para las más grandes.
En consecuencia, las moléculas de mayor tamaño se eluirán de la columna
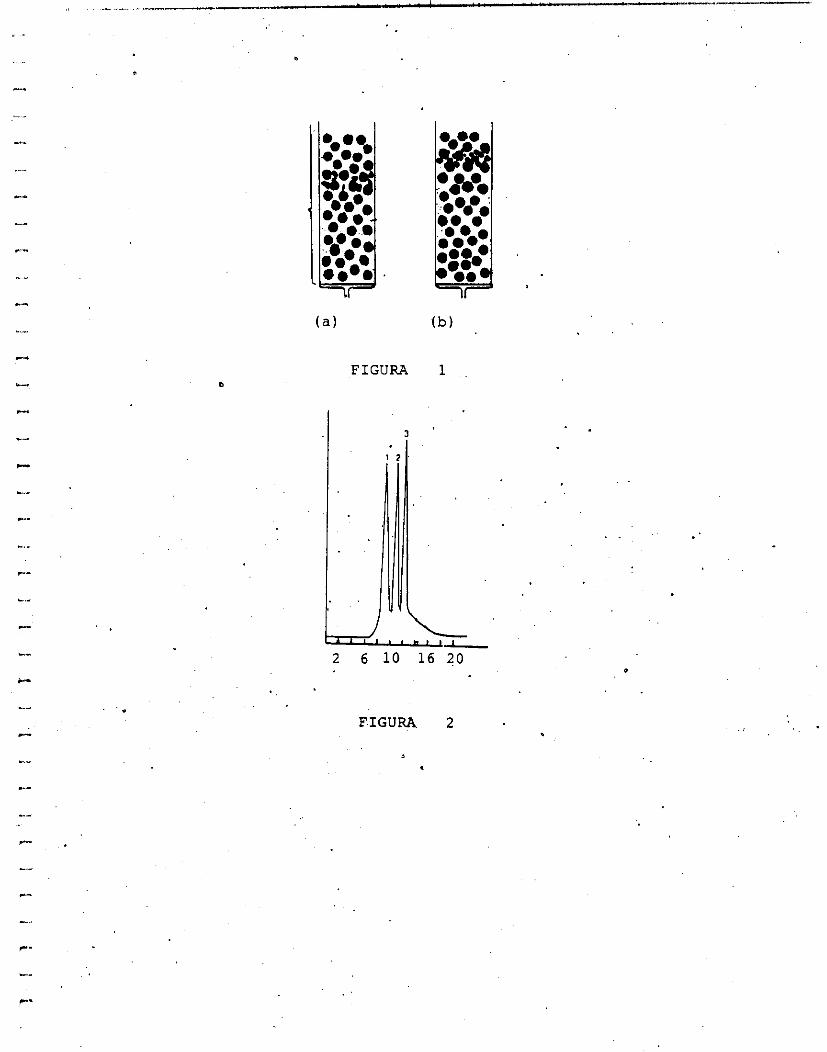
antes que las pequeñas, como se muestra en la figura 1. Desde otro punto .
de vista, puede considerarse que las moléculas grandes pasan todo el
,tiempo en la fase móvil, mientras que las de menor tamaño pasan sólo una
fracción del tiempo en esta fase. Así las moléculas pequeñas se
transportan más lentamente.
Lo; geles más utilizados en la exclusión molecular son de la clase de
Sephadex. El tamaño de poro de los geles es controlado por el grado de.
formación de enlaces cruzados en el proceso de fabricación. Cada gel se
encuentra disponible en varios tamañus de partícula. A menor tamaño de
'partícula, mayor resolución y meno; gasto de columna. La filtración en gel
se utiliza principalmente para .separar mezclas de moléculas con distinta
masa mo¡ecular. Se puede separar una mezcla de tres moléculas, por
ejemplo, una mezcla de papaína, quimopapaína y proteinasa; cuyas
masas moleculares son 23,350, 24,000, 23,900 Da respectivamente [3].
Tomando en cuenta las masas moleculares es posible identificar en un
e .

cromatograma. que pico corresponde a cada proteína, ya que primero se
eluye la de mayor masa molecular que en este caso sería quimopapaína,
después la proteinasa y por Último la papaína, como se puede observar en
al figura 2.
La cromatografía de filtración en gel también se utiliza para
determinar masas moleculares. Se realizan una gráfica de logaritmo de la
masa molecular de proteínas ya conocidas contra el coeficiente de
distribución (Kd) de cada proteína. El coeficiente de distribución se
determina por la relación:
Donde:
o
. . Ve - Vv
. Vm - Vit . . Kd =
. . . . . .. . - -. .
.. Ve = Volumen de elucion de la partícula.
Vv = Volumen vacío de la co1umn.a.
Vm = Volumen máximo de elución.
Comparando al Kd de la proteína desconocida con la de una serie de
patrones, es posible estimar la masa molecular de la proteína desconocida
(figura 3).
r-
FIGTJRA
3
2 6 10 16 20
íb 1
1
F.IGURA 2

. .. . . . . .
.,.*
. .
,--
r-
-...
r-
,^_
. ." p<."
-.- I-
,-. -.
. . F-
.
,.-
L.,.
.- &..
5.0
L a
L.6
L.L
' . L.i
. . I 1
1) dirnero de albumina ( 1 36000) 2) albúmina (68000)
3) ovalbumina (43000) 4) & p i n a (23350)
. 5) miogiobina (16950)
' 6) iisozima ( 1 4300)
- .
0.2 Oh
.. "
Figura 3. Grcifica del logaritmo de la masa molecular contra .el
coeficiente de distribución (Kd) para proteínas, obtenida por cromatografía . " . de filtración en gel.
~

o
ELECTROFORESIS
A fin de purificar. una proteína es esencial disponer de un método
para detectar y cuantificar dicha proteína en presencia de muchas
proteínas más. Las proteínas pueden caracterizarse por electroforesis.
Además de la cromatografía existe otro conjunto importante de métodos
para la separación de proteínas basado en el desplazamiento de las
proteínas cargadas en un campo eléctrico, proceso denominado
electroferesis.
. . La electroforesis.es muy útil como método analítico [4]:Su ventaja es que al tiempo que se separan tambiéri se puede visualizar, permitiendo al
investigador hacer una estimación rápida sobre el número de' proteínas en
una mezcla o el grado de pureza de una preparación proteica determinada;'
La ,electroforesis también permite la determinación. de propiedades
cruciaies de una proteína, tales como su punto isoeiéctrico y la masa
molecular aproximada
..
. . .
. . b , . . . . . . a
. . . . . En la 'electroforesis, la fuerza . . que.remueve la macromolécula es el
potencial eléctrico E. En el'caso de proteínas se lleva a cabo normalmente.
en geles formados por el polímero entrecruzado poliacrilamida: Un método
electoforético comúnmente utilizado para le estimación de la .pureza y la
masa molecular se utiliza el detergente dodecil sulfato sódico (SDS). . - 4
# '
O '
. .

O II '
II : o
Na O-S-O-(CHa)llCH3
dodecil sulfato sódico
- , , El SDS se une a ¡a mayoría de las proteínas. en una cantidad
c aproximadamente proporcional 'a .la. masa molecular de la proteína
c alrededor de una molécula por cada dos residuos de aminoácidos (2 y 41. I La electroforesis en' presencia de SDS separa, las proteínas casi
' .. exclusivamente en función'de la masa (masa molecular relativa) de forma
'' que los péptidos pequeños se desplazan . , más rápidamente. Después de la
electroforesis las proteínas se visualizan ahadiendo- un colorante tal como
el azul Comassie que se fija a las proteínas pero no al gel, la intensidad de
. . ~ * . ' ' la coloración retenida es proporcional a. la cantidad de proteína.. Este tipo
,. - . de gel prbporciona un método para seguir el progreso en el aislamiento d'e . la .proteína ya qge el número de bandas proteicas ha de disminuka~.'
medida, que transcurre la.pÚrificaciÓn.' .
r-
. c
id
. c
I
c
L_
- . . .. - . * .
. . .I 1
I
. . c a . -
c- En nuestro caso, después .de haber separado las proteínas
(quimopapaína y proteinasa) por métodos cromatográficos, podemos
emplear electroforesis para verificar .la pureza de dichas proteínas. Como
nuestra proteína (caricaína) tiene carga positiva, emigrará hacia el polo
negativo. Otro método para verificar la pureza de kcaricaína, es reaiizarle
una recromatografía en el HPLC.
. . c
. . c
I
C .
- .- 7
I
c

-. ..
.-
Con la electroforesis también podemos determinar la masa molecular
de la caricaína, comparado las posiciones a las que se desplazan en el gel
proteínas de masa molecular conocida con la posición de la caricaína
(caricaína desconocida) figura 4.
. . . < . '
DESNATURALIZACION TERMICA . . . .
, . a
La desnaturalización [5 y 61 es un proceso por el cual la proteína
pierde sus estructuras cuateroarias, terciarias y secundaria 'y < . se transforma
en una distribuclón de conformaciones aleatorias de .aminoácidos, sin
embargo la estructura 'primaria no se altera, pero pierde su actividad
enzimática así ,como sus propiedades. físicas y ~ químicas. y como' .
consecuencia la disminución en la solúbilidad de la molécula, porque los
grupos hidrofobicos ubicados en 'el. .interior:quedan expuestos a'l solvente o .
'O . ' ~
, acuoso
. .
. .
. - . : 8
TITULAC ION
La titulación [4] 8s un método. que se basa en al escala del pH,
'constituye una forma conven¡ente.de designar la - concentración -_ real de H'
(y por consiguiente de OH) en cualquier solución acuosa entre 1 .OM de H'
y 1 .OM de OH-, el término pM se d e f i n m n t e la expresión:
'
pH = -log [H']
c

Las soluciones que tienen pH superiores a 7 son alcalinas o básicas;
la concentración de OH- es mayor que la de H', inversamente, las
soluciones con un pH inferior a 7 son ácidas.,
Las determinaciones precisas del pH en el laboratorio químico se
hacen con un electrodo de vidrio que tienen una sensibilidad selectiva para
la concentración de H' pero que. es insensible al Na', K' y otros cationes.
En un pHmetro se amplifica la señal de un electrodo de este tipo y se
compara cÓn la señal generada por una solución d e un pH conocido con
exactitud.
La medición de pH 'es una de las 'operaciones más importantes y
utilizadas en bioquímica con más frecuencia. El pH afecta a la estru-ctura y . * . . a la actividad de macromoléculas biológicas.
. .
'
Los aminaácidos pueden actuar como ácidos y como bases,
conociendo estas propiedades se efectúa la titulación: Esto implica la
adición o eliminacibo gradual de protones.
Se utiliza la ecuación de Henderson-Hasselbalck:
. . .
[aceptor de protones]
[dador de protones] pH = pK, + log
Esta ecuación nos permite calcular el pK, de cualquier ácido a partir
de la relación molar entre dador y aceptor de protones a cualquier valor de -
PH-

. <
0
i.8
ú .6
4.2
f 4 C
albúmina (68000)
yglobulina H (50000)
a-1 -giicqxoteina (44000)
ovalbumina (43000)
tripsinogeno (23991)
y-giobulina L (23500) , '
citwromo c (12,384)
iisoeima (14300) . .
I .
2 3 & 5 o kMl)
Figura. 4. Gráfica-del logaritmo de la ,masa' molecular contra el 0 .
9
desplazamiento de las proteínas, (rnm), obtenida por electroforesis. .

r-
6: OBJETIVOS .- c
Para determinar el método experimental, para llevar a cabo el
-- I..
proceso de la purificación de las proteínas, es conveniente realizar ciertos
estudios de la proteína, para poder elegir las condiciones de trabajo'. .- r-
De la muestra es necesario conocer su punto isoeléctrico, para elegir
la columna y el pH adecuado. De acuerdo a los datos determinados,
podremos seleccionar las soluciones reguladoras y escoger la columna de
I-
- b-
I , intercambio iónico adecuado.
I
Para obtener la muestra pura es necesario seguir los siguientes I
pasos: "I
a. Purificar por el método de cromatografía de líquidos, intercambio
b. Dializar: Método y proceso de separación de partículas de sal a través
de uria membrana porosa que permite el paso de sustancias de baja
masa molecular.
0 -
catiónico.
c. Concentrar la muestra.
d. Verificar la pureza de las 'proteínas obtenidas por el método de
recromatograf ía y electroforesis.
e: Diálisis: eliminar el exceso de la sal de la muestra colectada.
f. Liofilizar: Deshidratación por sublimación de una proteína congelada
dentro de un matraz bola en que se práctica el vacío.
b
.

/.""
g. Electroforesis. -. c h. Seguir la desnaturalización térmica de la cari'caína por dicroísmo
- - circular
7. METODOLOGIA UTILIZADA - -
c
I
c-
..- c
I-
C
a. Aistar la caricaína y la quimopapaína con el método de cromatografía
b. Verificar la pureza de las proteínas obtenidas por el método de
. c. E!imirrar el exceso de la sal de proteína colectada,
d. Utilizar el método de liofilización para obtener la proteína eh polvo.
e. Realizar estudios de desnaturalkación térmica, por dicroísmo circular
f: Estudios de protonación por el método de titulación potenciométrica.
de intercambio iónico
recromatografía y electroforesis.
8. ACTIVIDADES REALIZADAS
Se utilizó quimopapaína lote. 124F80751 Sigma (EC 3.4.22,6) y un
cromatógrafo de líquidos de alta presión (HPLC) Varian 9012/9050. Las
muestras de quimopapaína y caricaína se 'aislaron por cromatografía de
intercambio iónico, se utilizó una columna Bio-Gel SP-5-PW de 7.5 cm de
alto y 7.5 mm de diámetro de la siguiente forma: .
.I.
Se preparon dos soluciones reguladoras (A y B) de fosfatos 0.05M,
pH 7.0, al 0.02% de NaN, a la solución B se le adicionó extra NaCl 1M. -_ - P-

...
C I
. c
. .-
..*
. ..~
z...
Para preparar la myestra se utilizó quimopapaína parcialmente purificada,
del látex de papaya, la concentración utilizada fue de 7.5 mg/ml, esta
Concentración se determinó haciendo pruebas a concentraciones
diferentes.
Con el fin de impedir la autólisis de la proteína durante la purificación,
se procedió a bloquear su sitio activo de la siguiente manera: la
quimopapaína se disolvió en la solución A regulador de fosfatbs, se le
adicionó mercaptoetanol O.lM, después de una hora se le agregó-
iodoacetamida 0.1 M, dejando reaccionar durante 30 minutos, por Último
se fiitró la muestra. Es importante hacer notar que la muestra se mantuvó
en hielo par evitar la hidrók’sis de !a proteína.
Para obtener una buena resolución en los cromatogramas se partió . e de las condiciones de elución reportadas [7] con anterioridad, una vez
probadas y establecidas las nuevas condiciones se procedió a purificar la
muestra a un flujo 0.5 ml/min.
I ’
-. - 0 . . c
I Después de un cierto tiempo se obtuvó una buena cantidad de cada
una de las muestras separadas (quimopapaína y caricaína), se procedió a
investigar su grado de pureza, para esto se realizaron recromatografías.
Por otro lado, se utilizó la elecaroforesis con el mismo fin. La
-_ recromatografía se realizó para cada upa de las proteínas en el
- r-
- _ c-
-- cromatógrafo de líquidos de alta presión (HPLC) y se usaron exactamente
las mismas condiciones y soluciones de fosfatos 0.05 M. - _ -.
c -

La électroforesis se llevó a cabo en un sistema Phast-System
(Pharmacia-LKB), en una placa de gel homogéneo 12.5% de
poliacrilamida, con dimensitines 0:45 x 43 x 50 mm. La concentración de
las muestras analizadas fue de 1 mglml y se colocaron 8 pI de las
,muestras en cada una de las ranuras del gel y se aplicó un voltaje de 12
V/cm, durante aproximadamente 1 hora, a 15 'C. El tratamiento posterior
para el gel consistió en" aplicarle una solución fijadora que es una m&la
de 0.1% de phast gel blue R, 30% de metana1.y '10% .de ácido acético,
posteriormente se le hace pasar 'una solución de desteñido,,que es una
'mezcla de 30% de metano¡. y 10%. de ácido 'acético, por último una
solución preservadora, la cual es'una mezcla.de glicerol'al: 10%; al 10% de
ácido acético y al 10% de agua. . .
D
Para obtener una rápida desalinización de,las proteínas,aisladas, se
utilizó una columna PD-10, 'contiene Sephadex . G-2 M, con las . dimensiones de 5, cm de altura, 1.5 cm de diámetro interno y un voíumen
, de 9.1 ml. El método consistió. en*eluir. primero. et exceso de l.íqwido ,
contenido en la columna; para esto se agregaron 2.5'ml. de regulador de,
fosfatos, después se añadieron 2.5 mi de la muestra y por último 2.5 ml del
mismo regulador y se colectó la cancaína sin sal (NaCI).
Una vez purificadas y dializadas las proteínas, se liofilizaron. El , . . .- ' procedimiento consta esencialmente ~ de dos fases, congelación y
r- desecación. _ _ r-.
1. La muestra se congeló con nitrógeno'iíquido, en el mismo recipiente
donde tuvó lugar la desecación. e -
- _ *-.

2. El material congelado se sometió a alto vacío por medio de una
bomba de vacío de alta potencia, mientras el condensador se
mantuvó durante la operación a temperaturas muy bajas. En estas
condiciones el líquido pasó directamente de la fase sólida a la fase de
vapor y este Último se condensó, inmediatamente en el condensador,
obteniéndose de nuestras muestras un sólido poroso.
Los experimentos de desnaturalización térmica 'se realizaron en el
equ'ipo de dicroísmo circular. JASCO. modelo J-50.0A; y la celda, utilizada . . . ,
fue de 1 mm. de recorrido Óptico, la muestra utilizada fue caricaína en
regulador de glicina 0.05 M it pH 2.5:
'
. . . . - .
Par.a determinar la condiciones adecuadas antes.de realizar nuestros
experimentos, primero se calibró el equipo con regulador de glicina 0.05 M ' , . '
a pH 2.5 a temperatura ambiente, para obtener la línea base. La..
concentración de la caricaína se obtuvó espectrofotométricamente a 280 . '
0. . .
. .
nm,
2.5,
utilizando un coeficiente de extinción A'% 1.85.
Se realizaron estudios de desnaturalización de la caricaína a un pH
se colocaron en la celda Óptica 0.87 ml de la muestra y 1.23 ml de
regulador de glicina 0.05 M, para obtener una concentiación aproximada
de 0.1 1 mglml. Se. hizó un barrido de 190 a 260'. nm a temperatura
ambiente, se eligió el vdlor adecuado de la elipticidad para continuar los
siguientes. experimentos.
o

I. - ~
Se hicieron estudigs de transicióR con la caricaína a diferentes
,.-
I _ _
velocidades'de calentamiento (0.1, 0.3 0.5 1.0 y 1.5 "C/min), a un pH de
2.5. Se colocaron en la celda Óptica 0.87 ml de caricaína y 1.23 ml de
regulador de glicina 0.05M, para obtener una concentración aproximada de * ..
, 0.1 1 mgíml, después se procedió a calentar de manera continua hasta
llegar a la temperatura deseada. Se controló la velocidad de calentamiento
con el baño HAAKE NK-22. La temperatura de la celda se midió con un
L..
* . " termómetro digital 08402-29 Cole-Parmer. ,.̂
Antes 'de realizar la protonación se calibró el pHmetro con
' reguladores estándares a . pH's (7, 4, 10 y j ) para obtener los mejores
valores en la medición del pH. La titulacion de la caricaína Se siguió
'potenciométricamente, se preparon 2 ml, de muestra .en w+iadw da. - g&cm&&+, a una.conce,ntraciÓn inicial de la caric8ína.de.4 mg/ml y un.
pH de '6.0, 'para segu,ir la' protonación, . . el sisxema .se adaptó. .. a" un
termómetro para mantener la' temperatura a 25 OC, con agitación constante
para que la mezcla se mantuviera homogénea. Se siguieron los ca.mbios
"""
-..*
. -y+U- 0ri.d :A, P... c
.- . . . . c-
,. . o , ' --
. . L
r-
-- de pH, se añadieron volúmenes muy pequeños de HCI 0.1 M, de acuerdo
a la tabla I, en.htervalos aproximadamente de 5 minutos hasta llegar a un
pH de 2.
c
I
F-
I . . .I_
C.
_.
c.

TABLA I
Volumen de HCI agregado
PH Proteinasa
6.229 5.654 5.055 4.745 4.437 4.176 3.982 3.829 3.704 ' 3.49 3.319 3.182 3.069 2.973 2.892 2.821 2.759 2.65 2.563 2.494 2.437 ,
2.379 2.334 2.29 2.249 2.218 2.183 . 2.159 2.129 2.104 2.077 2.055 2.035 2.014 , ' ''
Volumen1 ,
HCI O.. 1 M Proteinasa
2.00E-03 2.00E-03 2.01 E-03 2.01E-O3 2.01 E-03
" 2.01E-03 2.02E-03 2.02E-03 2.02E-03
.2.03E-03 2,03503 2.04E-03
2.05E-03 2.05E-03 2.06E-03 2.06E-03 2.07E-03 2.08E-03 2.09E-03
. 2.10E-03 Z.11E-03
, 2.12E-03 2.1 3E-03 2.14E-03 2.15E-03 2.1 6E-03 2.17E-03
. 2.18E-03 2.1 9E-03 2.20E-03 2.21E-03
o 2.22E-03 2.23E-03
. .
' 2.04503 '
. . 0

I-
.I
.~ - -- .
c
- - .
ic
.--
. ..
9. OBJETIVOS ALCANZADOS
Para la cromatografía de intercambio iónico se encontró el sistema
adecuado para la purificación, bajo estas condiciones se aisló y purificó la
caricaína.
Fueron .satisfactorios los resultados .de los métodos de
recromatografía y electroforeCis para detectar y cuantificar la caricaína en . presencia de otras proteínas. . I a
~ '.
. . . . .
Se. obtuvieron las proteinas quimopapaína y caricaína puras,' como
solidos po,roSos. Posteriormente, se guardaron en refrigeración, para , . ' 0
' estudios posteriores. . .
. . .
En la desnaturalización térmica de la proteína por . , dicrqísmo circular
se observó ,la Péi'dida be la estructura secundaria a altas temperaturas. ,
: Además; se,comprobó .la irreversibilidad de la caricaína. . . ~ . . ~
Con el método .de titu1,aciÓn se obtuvó el húmero de grupos . *
c protonadais.
. . . . , . .. . . .
. '. , . . . .
6 * . . Y-
. . . . . . . o . . . . . . .
.s

r -
..- P..
_ _ e.-
10. RESULTADOS Y CONCLUSIONES
La quimopapaína parcialmente purificada esta compuesta por
quimopapaína y caricaína de las cuales el punto isoeléctrico es 10.3 y
11.0, respectivamente [3]. Las proteínas a separar tienen una carga
positiva neta, por lo tanto se empleó una columna 'para intercambio
catiónico 610 GEL SP-5-PW, tamaño de 75 x 7..5 ,mm. El pH de la fase
móvil debe ser menor que 1.0.3 y 11, por . . esto, se tomaron más 'de 3
pi, p o r y e . puede retenerse demasiado la . muestra en' la columna, o
2, . desnaturalizarse antes de. 1.a; purificación, tornando. en cuenta todbs. estos ,
d&alles se tra.bajÓ a un valor de pH 7,O;
. . . . '. unidades.de diferencia, no es conveniente tomar .valores : muy.a.lejados . . . . del. . ' '
, .
. . . . -.
. .
. .
Se utilizó una fase móvil de un sistema binario, un regulador de
. . fosfatos 0.05 M a un de pH 7.0, que se consideró como solución A y la misma solución pero con NaCl 1 M como eluyente B.
. . . .
Lac condiciones experimentales más adecuadas para la separación
fueron: regulador de fosfatos 0.05M pH 7:0, flujo 0.05 ml/min, longitud de
onda 280 n.m, concentración de la muestra 7.5 mg/ml y el programa final '
de acuerdo al gradiente salino, se muestra en la tabla .1. . .
. _ - . . . . P . .
. . a

. . TABLA I
. .. ~
. .
, . . . .. 0 . , -
' .. Con este programa se realizó el proceso de purificacióq de la : , ,
quimopapaína, parcialmente purificada, '. lote. 124F8Of51 Sigma (EC 3.4.22.6) del látex de papaya, de esta forma se obtuvó el cromatograma de
la figura 5, donde se observa la.separación'de las diferentes proteínas.
En la figura 5 se observa la separación de la quimopapaína de la
. caricaína, estas tienen formas diferentes la de mayor tamaño es la
' , quimopapaína (I) y la otra de dos picos es caricaina (11).
En las siguientes cromatogramas (figuras, 6 y 7) tenemos las
. recromatografías de la quimopapaína y caricaína, respectivamente, los cuales se realizaron exactamente a las mismas condiciones de la purificación, variando únicamente el programa del gradiente salino de acuerdo la tabla II. Con esta técnica nos aseguramos que realmente
obtuvimos la pureza que esperabamos.
.I.
*

. . O
N m
' 4 I
m "3 O io N
P r- 4
I
io n n
O N
N
m 03
n io m
n * N n
N *
. . . . Ftgura 5. Cromatograma de la quimopapeína parcialmente purificada
con regulador de fosfatos 0.05 M a pH 7.0, flujo 0.5 ml/min del eluyente. La
zona I es la quimopapaína y la zona I1 es la caricaína.

O W N
4
,. o UIi
c N
. .
. .
.
TIEMPO (min)
Figura 6. Oromatograma de la quimopapaína a un flujo 0.5 ml/min
. 'COR el mismo eluyeflte, el pico pequeiiolndiCa que hay impureza y el de
mayor tamaño es la proteína que se esperaba.

. .
E E:
O W N
4
4 u z
a
H
m z
' r n 4
. . 4
TIEMPO (min )
Figura 7Cromatograma de la caricaína a un flujo de 0.5 mlímin con el
. mismo eluyente. El pico indica que la muestra esta pura.

Tiempo (min)
O
3
30
35
40 50
La figura 8 muestra un espectro de dicroísmo circular de la caricaína
nativa en regulador de glicha 0.05 M a un pH de 2.5, a temperatura
ambiente, .en. un intervalo de longitud' de. onda 1'90 a:260 nm. Esta región
espectral (190 a 260) es. el reflejo de la estructura secundaria de las
.. proteínas.'.Se observan 2 mínimos en- 2.10 y '220 nm, los cuales son de
&teréS para seguir físicamente" los cambios. de 'desnaturalización .de la.
caricaína. . .
. . . . -., . . . .
%A %B
1 O0 O
1 O0 O
60 40
60 40
1 O0 O
1 O0 O
' En la figura 9 presentamos. los resultados de desnaturalización
téfmica de la caricaína a diferentes velocidades de calentamiento O. 1, 0.3. 0.5 1 .O y 1.5 OC/min. Estos estudios se realizaron por la técnica de DC, en
regulador de glicina 0.05 M a un pH de 2.5, midiéndo de manera continua
el cambio de elipticidad a 220 nm, variando la temperatura de ambiente a
80 OC. A partir de los valores de elipticidad, éstos fueron transformados a
fracción desnaturalizada de. la caricaína. Esta gráfica muestra la transición
del estado nativo al' estado desnaturalizado contra la temperatura, en
donde se observa que las curvas son fuertemente afectadas por la
.

..
r..
_I.
-..
velocidad de calentamiento, proceso que se encuentra bajo control cinético
debido a la presencia de una reacción irreversible [9]. I
Los cambios de elipticidad observados a diferentes temperaturas se -.
usaron para obtener la fracción de la proteína desnaturalizada fD, de
acuerdo a la siguiente ecuación. - -
8 - 8N 80 - 8N
fD =
c
I
I .donde 8 es una propiedad física que cambia durante la transición: 0~ y 80
' son . los' valores de 'dich,a ,propiedad en los estados .'nativo. y c : . . desnaturalizado, respectivamente.. . - ~ . ~
c *
-._
C^
A partir de los valares de pH se'obthii, :el, valor 'de lbs.. grupos ' . protonados de. acuerdo a siguier$e ecuación:.
. . * ' - . . .
. . ILi . - ..- - .-
AH' qH+ =
' fcaricaína] . .
. . .
I qH+ = número de grupos protonados
r- A H = H ; - H , + ' - -
0 .
c..
-- 0
v
._ AH' es la diferencia de la concentración de H' en solución.
L _

- I i3
O
o aJ W
E .CI
CI E
o E
o
W
bD
a n U
O
-2000
-4000
-
-6000
-8000
- 1.oooo
O
l l
O
O \ I
O
O \ O
\
e \
o i O
\ .. O
\
pH2.5' 7 -
O I i
O I o
I O I
I O
190 200, 210 220 '230 240 250 260' .
LONGITUD DE ONDA (nm)
Figura 8. Espectro de tlicroísmo circular de la cancaína nativa en
regulador de glicina a pH 2.5, a temperatura ambiente.

1 .o
0.8
0.6
-0.4
0.2
0.0
-0.2 30 40 * 50 60 70 80
TÉMPERATURA O c
.
< .
I . . . . - , ..
Figura 9. Fracción desnaturalizada 'en funcih 'de la temperatura, con
una dependencia de la elipticidad de 220 nm. Caricaina en regulador de
glicina 0.05 M y uri pH de 2.5, a diferentes velocidades de calentamiento.

r.
-- I
c.
[caricaína] = mgiml, esta concentración cambia en cada punto de adición
de HCI O. 1 M. Los resultados de la protonación se resumen en la tabla IV
La secuencia de aminoácidos de la caricaína tiene tres grupos
aspartatos y once grupos glutamatos, de estos grupos no todos se
protonan.en el intervalo de pH 6 a 2. En la tabla V se menciona algunas
propiedades de estos grupos carboxilícos y el la tabla VI se muestra la
secuencia de 'aminoácidos. Como podemos observar, la molécula de caricaina tiene,un.nÚmero grande de pares iónicos, un total de 14 grupos
cirboxíticos y I I únicamente ~ éstan' i&oiucridos en la titulación,
' probablemente algunos pares iónicos tengan baja accesibilidad al ' ' ''
solvente.
. .
. . r.
-..
-.
.

PH 'roteinasa
6.229 5.654 5.055 4.745 4.437 4.1'76 3.982 3.829 3.704 3.49 3:319 3:182 3.069 2.9'73 2892 2.821 2.759 2.65 2.563 2.494 2.437 2.379 2.334 ' 2.29 2.249 2.218 2.183 2.159 2.129 2.104 2.077 2.055 2.035 2.014
Va (litros) HCI O. 1 N
Proteinasa
O 2.50E-06 5.00E-06 7.50E-06 1.00E-05 1.25E-05 1.50E-05 1.75E-05 2.00E-05 2.50E-05 3.00E-05 3.50E-05 4.00E-05 4.506-85 5.00E-05 5.50E-05 6.00E-05
. 7.00E-05 8.00E-05
' '9.00E-05 1 .OOE-04 1.1 OE-O4 .í .20E-04 1.30E-04 1.40E-04 1.50E-04 1.60E-04 1.70E-04 1.80E-04 1.90E-04 2.00E.04 2.10E-04 2.20E-04 2.30E-04
P
lolumen I iCI 0.1M 'roteinas
2.00E-03 2.00E-03 2.01 E-O3 2.01E-03 2.01E-03 2.01 E-O3 2.02E-03 2.02E-03. 2.026-03 2.03E-03 2.03E-03 2.04E.03 2.04E-03 2.05E-03 2.05E-03 2.06E-03 2.06E-03 2.07E-03 2.Q8E.03 2.09E-03 2.1 OE-03 2.11E-03 2.12E-03 2.13E-03 2.14E-03 2.15E-03 2.16E-03 2.1 7E-03 2.18E-03 2.19E-03 2.20E-03 2.21 E-03 2.22E-03 2.23EL03
'
TABLA IV RESULTADOS DE LA CARlCAlNA
n&vaNac Nac= 0.1 1
O.OOE+OO 2.75E-07. 5.5OE-O7 8.25E-O7 1.10E-06 1.38E-06 1.6%-06 1,93E-06 2.20E-06 2.75E-06 3.30E-06 3.85E-06 4.40E-06, 4.95E-06 5.50E-06 .6.05E-06 6.60E-06 7.70E-06 8.80E-06 9.9OE-06 1.1 OE-05 1.2.1 E-O5 1.32E-05 1.43E-05 1 ..%E305 1.65E-05- 1.76E-05 1.87E-05 1.98E-05 2.09E-05, 2320E-O5 2.31 €-OS 2.42E-95 2.53E-O5
ip=na-(nf-ni)
2.72E-07 5:34E-07 7.90E-07
' 1.03E-06 1.24E-06 1.44506 1.63E-06 1.80E-06 2.10E-06 2.33E-06 2.51E-06 2.66E06 2.78E-06 2.87E-06 2.95E-06 3.01 E-06
. 3.07E-06 3.1 1 E-06 3.20E-06 3.32E-06. 3.28E-06 3.38E-06 3.38E-06 3.34E-06
'-3.49E-06 ;. 3.43E-06'
3:65E-06' . 3.60E-06 3.66E-06 3.58E-06
' 3.63E-06 3.72E-06 3.71 E-06
np = concentraci6n de H'
ni = concentracidn inicial deli'
nf = concentracibn final de H'
na = concentraci6n de HCI
n proteina
3.38E-07
vi (litros) 2.00E-03
ni (proteinasa)
ni=ViHi
1,18E-09
npln proteina
0.80395995 1,57844785 2.33747949 3.04052026 3.67451343 4.26376757 4.81 38531 9. 5.33086549 6.20089718 6.88556657 7.4344577
.7.87235553; 8.2 1 O08263 8.49823286 8.721791 97 8.91 43735 9.07404629 9.20657234 9.4676495 9.833401.71 9.71881558 9.98847841 9.991 81 338 9.87968807 10.3146497 10.1 43321 4 10.8101787 10.6608379 10,8428332 10.5785095 10,7396459 11.0061974 10.9721426'
nf-ni 'roteinasa
3.26E-09 1.65E-08 3.49E-08 7.23E-08 1.33E-07 2.09E-07 2.98E-07 3.98E-O7 6.54E-07 9.73E-07 1.34E-06 1.74E-Q6 2:17E-06' 2.63E-06 3.10E-06. 3.59E-O6 4.63E-06 5.69E-06 6.íOE-06. 7.68E-06 8.82E-06 9.82E-06 1.09E-05 1.21 E-O5 1.30E-05 1.42E-05 1.5OE-05 1.62E-05 1.72E-05 1.84E-05 1.95E-05 2.05E-05 2.1 SE-O5
npin protelnas = número de grupos protonados
. . .

Aminoácidos
Asp.ato o
Glutamato
.”
- Pr PK Nombres
abreviados , . .
Asp D 2.77 2.09 y 3.86 carboxilos a y p Glu E 3.22 2.1 9 y 4.25 carboxilos 01 y z

TABLA VI Secuencia de aminoácidos de la caricaína [7]
1 5 10 15 20
-Leu-Pro-G/ccAsn-VaI-AspTrp-ArgLys-Lys-Gly-Ala-Val-Thr-Pro-Val-~gHis-Gln-Gly
25 30 35 40
-Ser-Cys-Gly-Ser-Cys-Trp-Ala-Phe-Ser-Ala-Val-Ala-Thr-Val-G/u-Gly-llsA~-Lys-lle
45 50 55 60
-Arg-Thr-Gly-Lys-Leu-Val-G/ccLeu-Ser-G/u-Gln-G/ccLeu-Val-AspCys-G/u-Arg-Arg-Ser
65 70 75 80 -His-Gly-Cys-Lys-Gly-Gly-Tyr-Pro-Pro-Tyr-Ala-Leu- a/u-Tyr-Val-Ala-Lys-Asn-Gly-lle
. . 85 90 95 ' ' ' 100
-His-Leu-Arg-Ser-Lys-Tyr-Pro-Tyr-'L~-Ala-Lys-~n-Ql~Thr-Cys-ArgAla-Lys-Gl~-Val
. . . . 105 110 115 120
, . -Gly.GLY-Pro-lle-Val-Lys-Thr-Ser-Gl)r-Val-Gly-Ar~Val-Gln~Pro-A~-A~-G/ccGly-Asn '
125 130 '135 140 .-Leu-Leu-~sn-Ala-lle-Ala-Lys-Gln-Pro-Val-Ser-Val-Val-Val-G/~Ser-Lys-Gly-ArgPro
145 150 155 1 160
-Phe-Gln-Leu-Tyr-Lys-Gly-Gly-lle-Phe-G/u%ly-Pro-Cys-Gly-Thr-Lys-Val -AspHis-Ala
165 170 175 i80
-Val-Thr-Ala-VaCGl~Tyr~~-Ly~er-Gly~ly-Ly~Gly-Tyr-llsLeu-llsLys-Asn-Ser
1 a5 190 195 200
-Trp-Gly-Thr-Ale-Trly-G/ccLys-Gly-Tyr- IleArg-¡lsLys-Arg-Ala-Pro-Gly-Asn-Ser
205 21'0 21 5
-Pro-Gly-Val-Cys-Gly-Leu-TyrrLys-S~-Ser-Tyr-Tyr-Pro-Thr-Lys-Asn o

BIBLIOGRAFIA
1.
2.
3.
.4.
5.
6.
7.
8.
Day R. A. y -.iderwoc 4. L.(1989)química , .nalítica Cuantitativa,
5 ed., PRETICE-HALL-HISPANOAMERICANA S. A., México.
Daniel C. Harris, Análisis químico Cuantitativo, Grupo Editorial
Iberoamérica (1 972)
S Solís Mendiola, Tesis de Maestría en Química, México, junio
de 1089
Lehninger, A.L., Nelson D. L. y Cox M. M., Principios de
Bioquímica( 1993), Ediciones Omega, S. A., Barcelona
Shirley, B. A., (1995) Methods in Molecular Biology, 40., Totowa,
New Jersey.
Ghélis, C., Yon J.,(1982) Protein Folding Academic Press,lnc,
U.S.A.
Solís Mendiola, (junio 1989) Tesis demaestría, México, D. F.
Solís-Mendiola, S., Gutiérrez-González, L. H., Aroyo-Reyna, A:,
Padilla-Zúniga, J., Rojo-Domínguez, A y Hernández-Arana, A.
I .
. (1998) Biochimica et Biophysica Acta 35765, 1-10.
9. Arroyo-Reyna, A., Hernández-Arana, A. (1 995) Biochim.
Biophys. Acta 1248, 123-1 28.










![Tromboembolia [autoguardado] bn](https://static.fdocuments.co/doc/165x107/55acb5421a28ab045a8b471e/tromboembolia-autoguardado-bn.jpg)







