
Hemofilia Laaaab
55
HEMOFILIA. .Centro de Bachillerato, Tecnológico industrial y de servicios 03
-
Upload
karenciita-xd -
Category
Education
-
view
182 -
download
0
Transcript of Hemofilia Laaaab

HEMOFILIA.
.Centro de Bachillerato, Tecnológico industrial y de servicios 03

introducción
La hemofilia es una enfermedad de origen genético, recesiva y ligada al cromosoma X, en el cual se encuentran los genes que codifican los factores hemostáticos VIII y IX. Algunas alteraciones estructurales o moleculares de dichos genes condicionan una deficiencia cuantitativa o funcional del factor VIII (FVIII) en la HA, llamada también «hemofilia clásica», y del factor IX (FIX) en la HB o «enfermedad de Christmas». La enfermedad es heredada en el 70% de los casos; en el otro 30% es consecuencia de una mutación de novo cuyo propositus la heredará a su descendencia con el mismo patrón recesivo ligado a X.

Debido a que la hemofilia está ligada a este cromosoma con un patrón recesivo, se manifiesta clínicamente solo en los varones .
las mujeres son las portadoras
. Las alteraciones cromosómicas son, generalmente,
o mutaciones puntuales en 46% de los casos,
o rearreglos (inversiones) en 42%,
o deleciones en 8%,
o mutaciones no identificadas en 4%.


epidemiologia
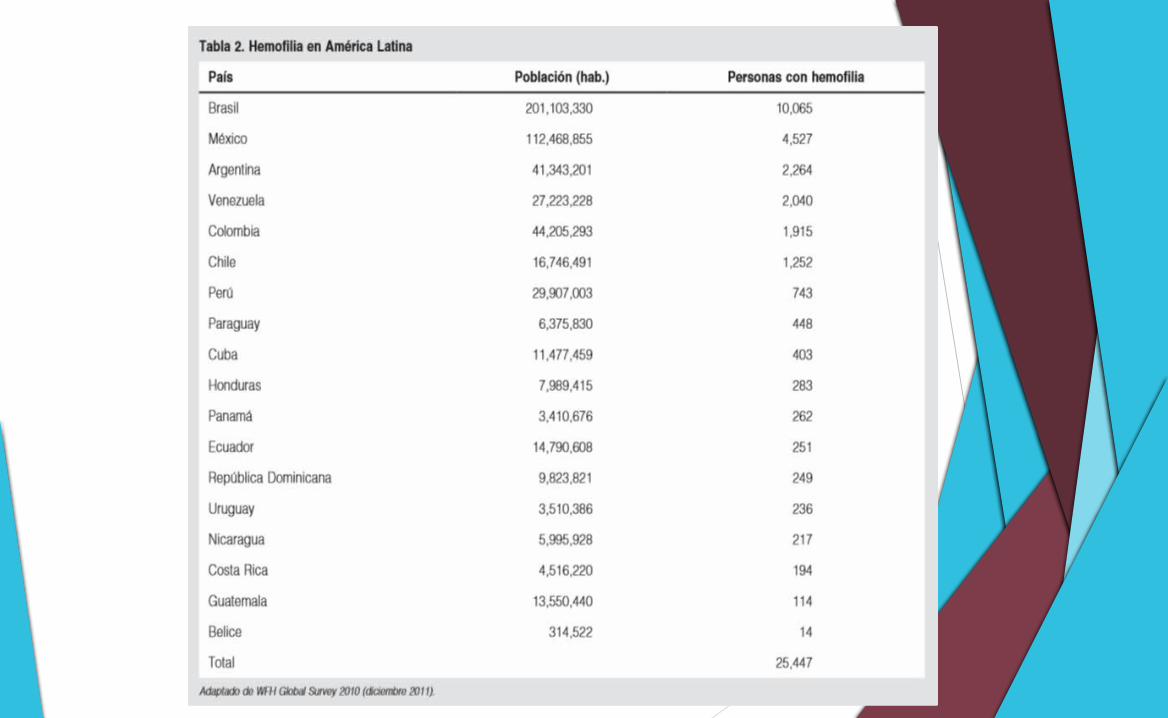
La prevalencia mundial aproximada es de:
o 1 caso/10,000 varones para la HA y de
o 1/50,000 para la HB.
De acuerdo con estas cifras, se calcula que en el mundo hay cerca de 400,000 personas con hemofilia
Para las personas con hemofilia, la enfermedad representa una limitante en todos los aspectos de su vida biológica, psicológica y social.
A pesar de tener una prevalencia baja, tiene un impacto alto en la sociedad y los sistemas de salud del país.

Historia En los tiempos de la reina Victoria, se creía que se trataba de una
enfermedad «real», prevalente solo en las monarquías, ya que el fenotipo se expresaba notoriamente entre las familias reales .Hoy sabemos que la hemofilia tiene una distribución casi mundial con casos documentados en todas las razas y etnias.
Los registros más antiguos se remontan al Talmud y a los papiros egipcios, en los cuales se describen casos que, a la luz del conocimiento actual, podrían ubicarse como sujetos con hemofilia. Desde entonces, poco se avanzó hasta mediados del siglo XX, cuando se dilucidó la diferencia entre la HA y la HB; también por esa época se consolidó al plasma como una terapia efectiva, un avance terapéutico notable con repercusiones en el pronóstico del paciente.
En la década de 1940 se avanzó en el entendimiento de la genética, fisiopatología y epidemiología, pero sobre todo en el tratamiento.

HEMOFILIA
La hemofilia es una enfermedad genética recesiva que impide la buena coagulación de la sangre. Está relacionada con el cromosoma X y existen tres tipos:
la hemofilia A, cuando hay un déficit del factor VIII de coagulación.
la hemofilia B, cuando hay un déficit del factor IX de coagulación.
la hemofilia C, que es el déficit del factor XI.

HERENCIA
Las hemofilia A y la hemofilia B son de herencia gonosómica (sexual, ligada al cromosoma X); el gen alterado en la hemofilia A se localiza en el locus XQ28 , y el de la hemofilia B, en XQ27.1-Q27.2.

Con un padre con hemofilia y madre sana no portadora: el 100% de sus hijas serán portadoras sanas (heredan el alelo mutado del padre), y el 100% de los hijos serán sanos no portadores (no tienen de quién recibir el X mutado).
Con un padre con hemofilia y madre sana portadora (heterocigota): el 50% de las hijas serán portadoras sanas y el 50% de las hijas serán hemofílicas. En cuanto a los hijos varones, el 50% serán pacientes con hemofilia (pues reciben un único X materno, que en este caso es el mutado) y el 50% serán sanos no portadores (han recibido el X sin defecto).
Con un padre sano* y madre portadora sana: el 50% de las hijas serán sanas no portadoras, y el 50% serán sanas portadoras. En cuanto a los hijos varones, al igual que en el caso anterior, el 50% serán pacientes con hemofilia y el 50% serán sanos no portadores.
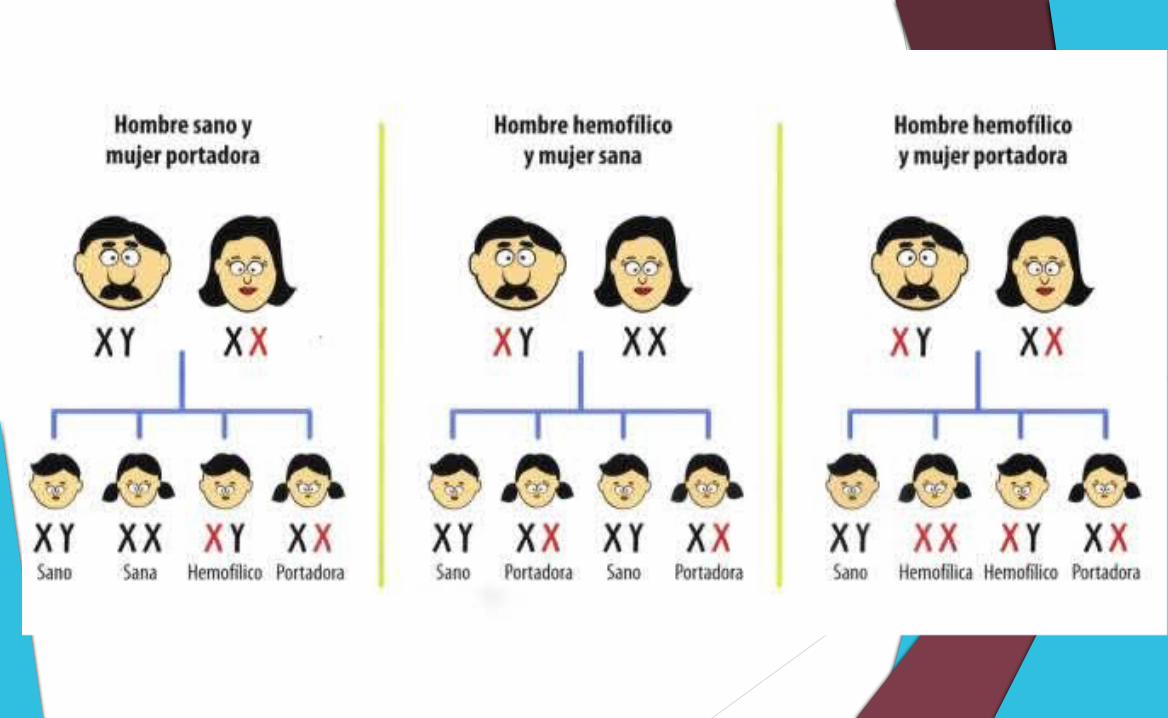

La hemofilia es una afección que padecen casi exclusivamente los varones, y casi todos los pacientes con hemofilia son hijos de madres sanas, portadoras del gen, es decir que en sus antepasados existió algún paciente, pero en casi un tercio de los pacientes ha ocurrido sin que haya un historial familiar. En estos casos, la hemofilia es producida por una mutación en el gen de la madre o del niño.

La transmisión de la Hemofilia se dice que es recesiva y no dominante ya que puede que no aparezca en una generación siguiente (salto de generación) por la simple razón de que se den portadoras sanas o varones sanos, y sí aparezca en otra generación posterior.

CAUSAS DEL MAL FUNCIONAMIENTO DEL FACTOR DE LA COAGULACIÓN. La causa de que un factor no funcione es que
el organismo lo sintetice defectuoso y como se trata de una enfermedad hereditaria esto significa que el defecto se encuentra en una región del ADN (gen) que da lugar a una proteína que es el factor. En cualquier caso el factor defectuoso es así porque antes se han producido cambios en ese gen.

Por lo tanto…
La intensidad clínica de la enfermedad dependerá del:
tipo de mutación.
Funciones moleculares perturbadas.
En todos lo miembros de una familia se produce una misma mutación, dando lugar a expresiones clínicas muy similares.

HEMOFILIA A

Causas de la Hemofilia A

La Hemofilia es una enfermedad que no se adquiere o se contrae como la gripe o cualquier otra infección o enfermedad por un accidente traumático; se trata de una enfermedad que se hereda, se transmite de padres a hijos y sucesivas generaciones.
La hemofilia es una enfermedad de origen genético recesiva y ligada al cromosoma X, en el cual se encuentran los genes que codifican los factores hemostáticos VIII y IX.
Algunas alteraciones estructurales o moleculares de dichos genes codifican una deficiencia cuantitativa o funcional del factor VIII en HA

Es, por tanto, una enfermedad hereditaria cuyo defecto se encuentra en el cromosoma X, es decir, el cromosoma que se relaciona con el sexo por lo que es una enfermedad hereditaria pero además ligada al sexo lo que significa que en el caso concreto de la Hemofilia la transmiten las mujeres (portadoras) y la padecen los hombres debido a la dotación de dos cromosomas X (XX) de la mujer y una dotación XY en el hombre
La causa de que un factor no funcione es que el organismo lo sintetice defectuoso y como se trata de una enfermedad hereditaria esto significa que el defecto se encuentra en una región del ADN (gen) que da lugar a una proteína que es el factor.
El defecto más habitual es un gran cambio, llamado inversión del intrón 22.
Debido a: Mutaciones puntuales
Deleciones
inserciones

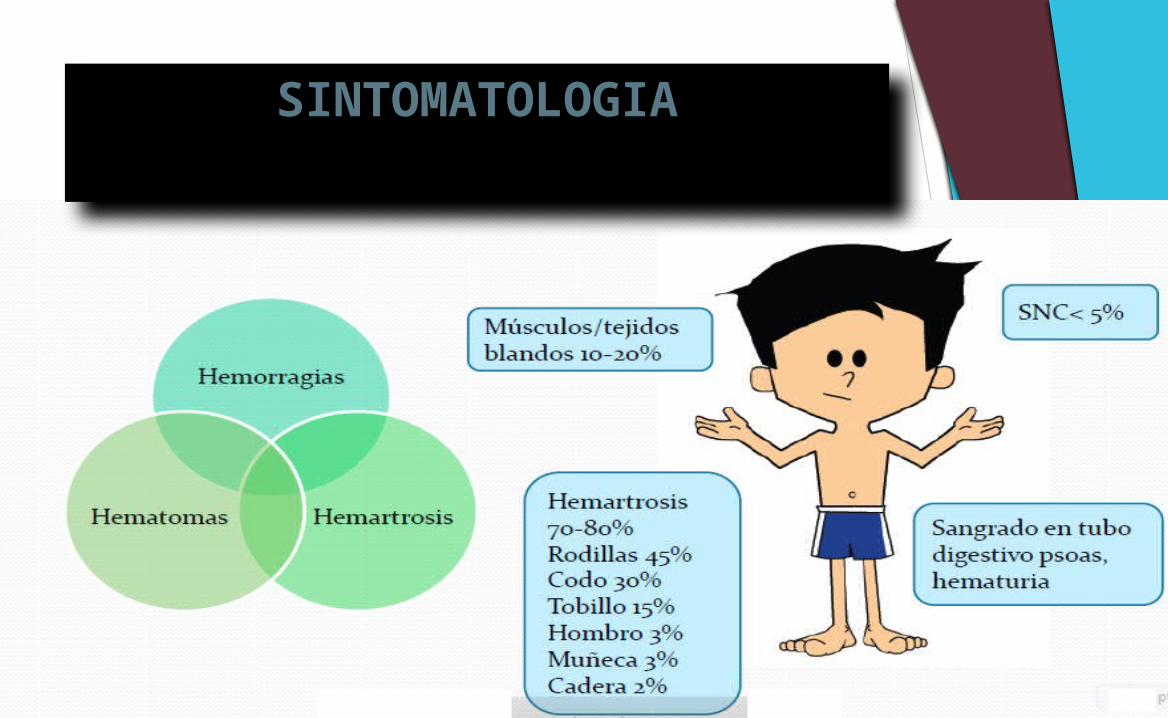
SINTOMATOLOGIA
Hemartrosis, hematomas musculares profundos y hemorragias cerebrales constituyen 95% de las hemorragias del hemofílico, aunque pueden afectar a cualquier parte del cuerpo, potencialmente.
Las hemorragias más frecuentes son, por mucho, las hemartrosis (en las articulaciones de carga: rodillas, tobillos y codos), y le siguen los hematomas musculares superficiales y profundos.

La hemorragia de la hemofilia suele ser tardía, es decir, no sigue inmediatamente a la lesión, sino que inicia unos minutos después del traumatismo.
En los casos graves, la hemorragia suele ocurrir en forma espontánea y reiterativa (sobre todo articular)
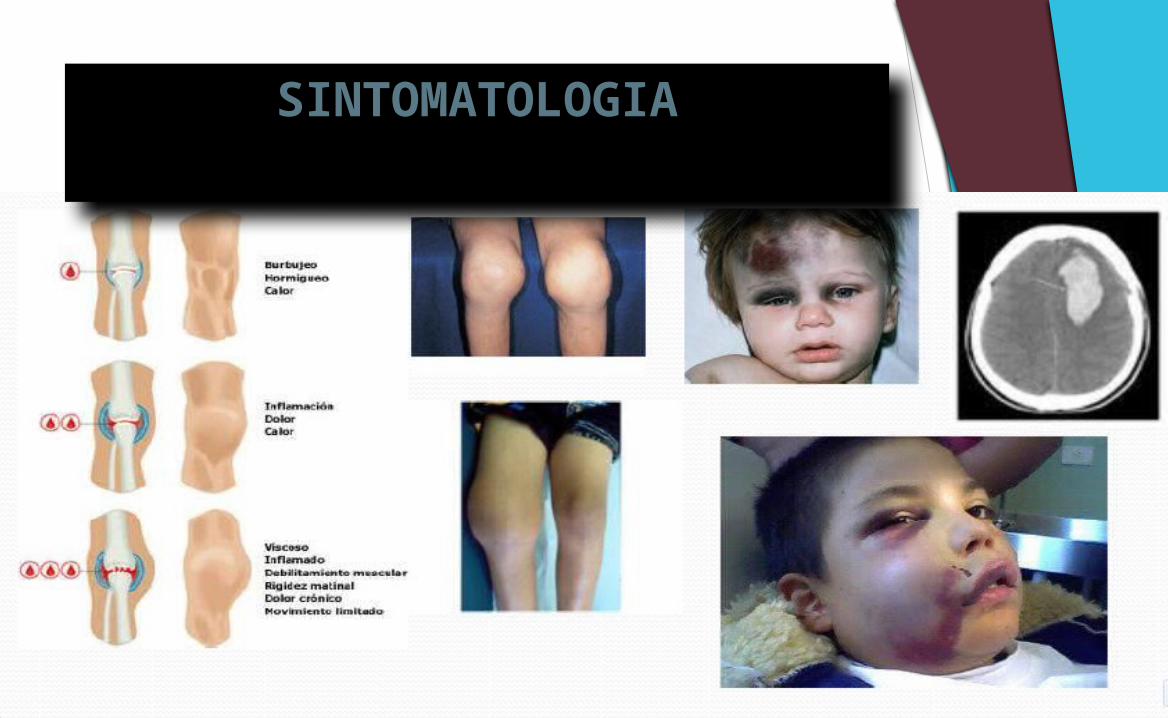
SINTOMATOLOGIA

Las hemofilias se caracterizan clínicamente por una tendencia hemorrágica proporcional al grado de deficiencia del factor hemostático, aunque suele haber excepciones.
En la hemofilia grave, las hemartrosis inician generalmente en los primeros 2 años de vida como efecto del aumento de la movilidad y estrés articular.
SINTOMATOLOGIA
SINTOMATOLOGIA
SINTOMATOLOGIA

Datos de laboratorio Hemofilia A
Es necesario realizar estudios de coagulación:
Actividad baja del factor VIII sérico
Tiempo de protrombina: normal
Tiempo de trombina: normal
Tiempo de sangría: normal
Nivel normal de fibrinógeno
Tiempo parcial de tromboplastina (TPT): prolongado
Número de plaquetas: normales
Análisis de factor VII: disminuido menor a 0.5 u/LValores normales factor VII:
0.6-2.0 U/L

TRATAMIENTO Y PRONOSTICO

TRATAMIENTO
El tratamiento estándar implica la reposición del factor de coagulación faltante. La cantidad de concentrados del factor VIII que se necesita depende de la gravedad y sitio del sangrado, al igual que de la talla del paciente.
La hemofilia leve se puede tratar con:
Desmopresina (DDAVP)
Administrar concentrados del factor VIII

Las personas con formas graves de la enfermedad pueden requerir
un tratamiento preventivo regular.
Dependiendo de la gravedad de la enfermedad, se puede
administrar concentrado de factor VIII o desmopresina (DDAVP).

Es necesaria la vacunación contra la hepatitis B, dado que hay un aumento en el riesgo de
exposición al virus de la hepatitis, debido a las frecuentes infusiones de
sangre.
Los pacientes que desarrollan un inhibidor para el factor VIII pueden
requerir tratamiento con otros factores de la coagulación, tales como el factor VIIa, que pueden
ayudar a la coagulación incluso sin ningún factor VIII.

PRONOSTICO
El desenlace clínico generalmente es bueno con tratamiento. La mayoría de las personas con hemofilia son capaces de llevar vidas relativamente normales.
Deben establecer una atención regular con
un hematólogo
La capacidad de tener acceso fácil y rápido a
historias clínicas
niveles del factor IX, transfusiones de
factores

COMPLICACIONESSe pueden presentar deformidades articulares crónicas a raíz del sangrado dentro de las articulaciones
Las transfusiones repetitivas pueden incrementar ligeramente el riesgo de contraer: VIH y hepatitis

HEMOFILIA B

CAUSAS
Es un trastorno hemorrágico hereditario causado por una falta del factor IX de coagulación de la sangre.
Las mujeres tienen dos copias del cromosoma X, de modo que si el gen del factor IX en uno de los cromosomas es defectuoso, el gen en el otro cromosoma puede hacer el trabajo de producir suficiente factor IX.
Los hombres, tienen únicamente un cromosoma X.
Si una mujer tiene un gen defectuoso del factor IX, se considera una portadora .

Los niños nacidos de tales mujeres tienen un 50% de probabilidad de padecer hemofilia B y las niñas tienen un 50% de probabilidad de ser portadoras.
Todas las hijas de hombres hemofílicos son portadoras del gen defectuoso.
Los factores de riesgo para la hemofilia B abarcan:
*Antecedentes familiares de sangrado
*Ser hombre

Sintomatología Hemofilia tipo B

Hemofilia las manifestaciones clínicas varias con la cantidad de factor presente
Se considera que los pacientes con :
menos de 0.01 U/mL (0.1%) tienen la enfermedad grave.
De 0.01 a 0.05 U/mL (1 a 5 %) presentan un cuadro clínico moderado.
De 0.05 a a.25 U/mL (5 a 25%) cursan con enfermedad leve.
Y del factor IX de =.5 a 1.5 U/mL (50ª 150%)

Hemofilia grave:El dato característico más importante es el sangrado espontáneo. Suele presentarse en edad temprana; la mayoría de los pacientes graves son diagnosticados antes del primer año de vida, bien al nacimiento si ya había antecedentes familiares o bien en el periodo perinatal por sangrados graves. Hay que detallar de forma precisa el tipo de sangrado: si son muco-cutáneo (epistaxis, gingivorragias, hemorragias digestivas, equimosis, preferentemente osteo-muscular (hemartrosis, hematomas musculares
Hemofilia moderada: Se caracteriza por no haber sangrado espontáneo. Casi siempre hay un antecedente traumático en mayor o menor grado. Suele ser frecuente cuando se realiza la historia encontrar con un sangrado anterior que no fue debidamente investigado pero que llamó la atención, . Su frecuencia de sangrado puede ser una vez al mes, sin embargo varia de unas personas a otras una persona que posee una hemofilia moderada puede sangran espontáneamente
Hemofilia leve: Se considera leve cuando una persona padece una hemofilia en la que los sangrados son con poca frecuencia, solo estas son importantes cuando sufren una extracción dental o cuando tienen una operación o sufren un gran accidente.

Los síntomas principales son:
Sangrado dentro de los músculos y las articulaciones, particularmente en las rodillas, los tobillos y los codos.
Hematomas extensos.
Sangrado espontáneo sin que haya un motivo claro.
Sangrado durante mucho tiempo tras cortarse, extraerse una muela o someterse a una cirugía.
Hemorragia interna grave en órganos vitales, por lo general después de un traumatismo serio.
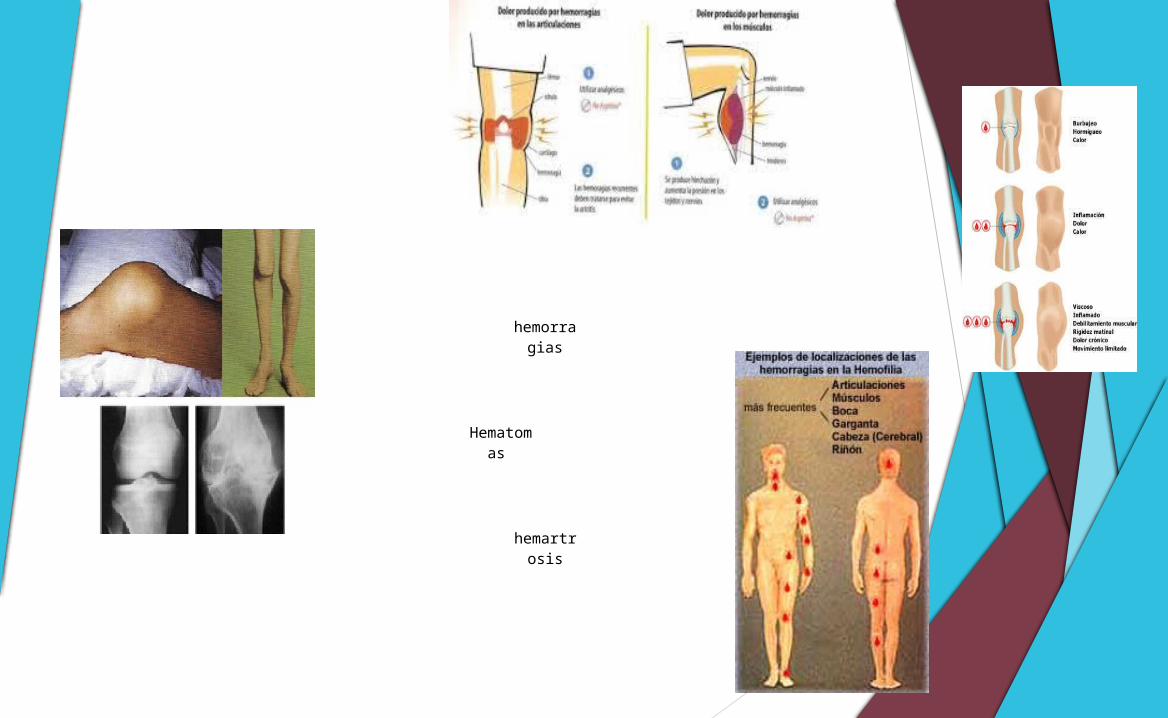
hemorragias
Hematomas
hemartrosis

Hemorragias graves con compromiso de la vida.
Equimosis
hemorragias musculares.
Hematuria.
Sangrados intestinales y cerebrales
no común hemartrosis
Hemofilia tipo C

DATOS CLINICOS
Los resultados de los exámenes de sangre pueden abarcar:
Tiempo parcial de tromboplastina (TPT) prolongado
Tiempo de protrombina normal
Tiempo de sangría normal
Nivel de fibrinógeno normal
Factor IX bajo
Conteo de plaquetas normal

Análisis de los factores de la coagulación

TRATAMIENTOEl tratamiento incluye la reposición del factor defectuoso de la coagulación. Usted recibirá concentrados del factor IX. La cantidad que reciba depende de: La gravedad del sangrado El sitio del sangrado Su peso y talla
Para prevenir una crisis hemorrágica, a las personas con hemofilia y a sus familias se les puede enseñar la forma de administrar concentrados del factor IX en sus
hogares, ante los primeros signos de sangrado. Las personas con formas graves de la enfermedad pueden requerir infusiones preventivas continuas.

Si usted padece hemofilia grave, es posible que también necesite tomar concentrado del factor IX antes de una cirugía o de determinados tipos
de trabajos de odontología. A usted le deben aplicar la vacuna contra la hepatitis B, dado que las personas con hemofilia son más propensas a padecer hepatitis, debido a
que pueden recibir hemoderivados. Evitar el consumo de aspirina.

PRONOSTICO
Con tratamiento, la mayoría de las personas con hemofilia son capaces de llevar vidas
relativamente normales. Sin embargo, puede producirse la muerte por pérdida extrema de
sangre.
Si tiene hemofilia, debe someterse chequeos regulares con un hematólogo.

POSIBLES COMPLICACIONES
Las complicaciones pueden abarcar:
• Problemas articulares crónicos, lo cual puede requerir una artroplastia
• Sangrado en el cerebro (hemorragia intracerebral)• Trombosis debido al tratamiento• Las transfusiones repetitivas pueden incrementar
ligeramente el riesgo de contraer el VIH y hepatitis; sin embargo, el mejoramiento continuo en los procedimientos de análisis de la sangre hace que los hemoderivados sean más seguros como nunca antes.

HEMOFILIA C

• La deficiencia de factor XI es un trastorno hereditario de la coagulación.
• Se transmite de padres a hijos en la concepción. • Cada célula del cuerpo contiene estructuras llamadas
cromosomas. Un cromosoma es una larga cadena formada por miles de unidades llamadas genes.
• Los genes determinan las características físicas, como el color de ojos. El trastorno es causado por un gen anormal llamado
una mutación. Diferentes tipos de mutaciones pueden ser responsables por el defecto genético que causa la deficiencia
de factor XI.
CAUSAS

• Esto explica, al menos en parte, la gran variabilidad de los síntomas en personas con deficiencias que son similares en
apariencia a la deficiencia de factor XI.• Cada individuo tiene un duplicado de cada uno de los genes
después de recibir una copia del par de cada padre.• Él podría recibir uno o dos genes defectuosos de una sola
pareja.• Los individuos que recibieron dos genes defectuosos tienen
una forma más grave de la enfermedad que los que reciben sólo un único gen.

Niveles de factor XI de sangre son significativamente inferiores a lo normal en las personas con dos genes
defectuosos, y ligeramente inferior en las personas con sólo una. diferencia de la hemofilia clásica (deficiencia del
factor VIII), que afecta principalmente a los niños, la deficiencia de factor XI no tiene distinción de género, y afecta tanto a niños y niñas. El gen de la deficiencia de
factor XI se encuentra en el cromosoma 4 y se encuentra tanto en hombres como en mujeres.

Datos clínicos HC

Pacientes homocigoto se afectan con síntomas de hemorragia excesiva.
Pacientes heterocigotos son asintomáticos
La deficiencia del factor XI tiene una alta frecuencia en la raza judía Ashkenazi: cerca de 0.2% son homocigotos y 11% son heterocigotos
Con frecuencia la enfermedad no se sospecha hasta que se practican pruebas de laboratorio pre quirúrgicas

No hay correlación entre la intensidad clínica y el valor de la actividad del factor XI mediante in vitro
Las pruebas de detección de laboratorio revelan un TTPa prolongado; otras pruebas son normales.
Cuando la única prueba anormal es TTPa, se considera que la anormalidad proviene de los factores del sistema intrínseco XII, XI, precalicreìnia , cininògeno de alto peso molecular
La historia clínica familiar es útil para llegar a una decisión sobre sobre cual es el análisis de factor que debe practicarse

El análisis especifico para el factor XI es la prueba definitiva.
Los pacientes homocigotos tienen menos de 0.01 a 0.1 U/mL (1 a 10% ) de actividad del factor XI
Las pruebas de laboratorio para la actividad del factor XI requieren precauciones en la colección y manejo de la muestra.

Los pacientes pueden necesitar tratamiento, normalmente con concentrados de factor XI o plasma fresco congelado.
Los antifibrinolíticos (ácido aminocaproico, ácido tranexámico) también se utilizan para prevenir la hiperfibrinolisis, que puede conllevar el déficit de factor FXI. El pronóstico es favorable, las hemorragias son por lo general moderadas.
TRATAMIENTO HEMOFILIA C


















