
Monografía de producto - Amazon Simple Storage Service · órganos de donantes excede, con mucho,...
68
Monografía de producto
Transcript of Monografía de producto - Amazon Simple Storage Service · órganos de donantes excede, con mucho,...

Monografía de producto

2

Resumen del producto 3
Los trasplantes en la actualidad 4Introducción: La revolución de los trasplantes de órganos; su impacto
en el hombre
Objetivos del tratamiento en trasplantes
Objetivos clínicos
Objetivos socioeconómicos
Aproximaciones actuales a la terapia farmacológica en transplantes
Limitaciones de las terapias actuales utilizadas en trasplantes
Limitaciones de los anticuerpos policlonales
Breve revisión de la inmunología en trasplante 12Activación de los linfocitos T
Interleuquina-2 y el receptor de IL-2
Anticuerpos monoclonales 17Anticuerpos monoclonales como concepto terapéutico en trasplante
Limitaciones de los anticuerpos monoclonales murinos como opciónterapéutica en trasplantes
Mejoras en el diseño de los anticuerpos monoclonales
Anticuerpos monoclonales quiméricos
Anticuerpos monoclonales humanizados
Zenapax®: un anticuerpo monoclonal diseñado a medida
Desarrollo de Zenapax® (daclizumab, anti-Tac humanizado) 22Descubrimiento y aislamiento
Humanización
Fabricación
Características químicas y físicas
Resumen
Estudios de toxicología y otros estudios preclínicos 24Genotoxicidad
Ensayo de mutagenicidad de Ames
Ensayo de genotoxicidad V79
Estudios de toxicidad aguda y subaguda
Estudios de toxicidad crónica y otros estudios toxicológicos
1
I n d i c e

Inmunogenicidad preclínica
Modelos animales en indicaciones diferentes al trasplante
Uveitis en el mono
Artritis inducida por colágeno
Resumen
Farmacología 28Farmacodinamia
Actividad biológica de Zenapax®
Mecanismo de acción/efectos inmunosupresores
Receptor alfa de IL-2 soluble (Tac)
Subpoblaciones de linfocitos en sangre periférica
Saturación de Tac
Farmacocinética
Estudios en animales
Estudios en humanos y razón de ser de la dosis
Resumen
Experiencia clínica 37Eficacia en trasplante renal
Estudio de la búsqueda de dosisEstudios Multicéntricos Comparativos Fase III
Experiencia clínica en el trasplante renal pediátrico
Cell Cept® y Zenapax® en com bi n ac i ó n : f a rm acoc i n é tica y ef i c acia en la prevención delrechazo del aloinjerto renal
Zenapax® en el tratamiento o la prevención de la EICH
Eficacia en el tratamiento o la prevención de la EICH
Tolerancia y seguridad de Zenapax®
Tolerancia y seguridad de Zenapax® en trasplante renal
Tolerancia y seguridad de Zenapax® en combinación con CellCept® en
trasplante renal
Tolerancia y seguridad de Zenapax® en trasplante de médula ósea
Resumen
Información Farmacéutica 54
Uso de Zenapax® : Ficha Técnica 55
Resumen 62
Bibliografía 63
2
Indice(continuación)

Zenapax® (daclizumab; anti-Tac humanizado) es un anticuerpomonoclonal IgG1 humanizado, obtenido mediante ingeniería genética,que se une a la subunidad TAC (p55, CD25) del receptor de interleuqui-na-2 (IL-2), expresado en los linfocitos T humanos activados. Al unirsea la subunidad TAC del receptor de IL-2, Zenapax® bloquea la unión deIL-2 al receptor de IL-2 de alta afinidad (IL-2R) y la activación subsi-guiente por IL-2.
Zenapax® está indicado para la prevención del rechazo agudo en pacientes receptoresde trasplantes renales alogénicos. La dosis recomendada de Zenapax® es de 1,0 mg/kg,administrada por vía intravenosa durante 15 minutos. La primera dosis de Zenapax® sedebe administrar hasta 24 horas antes del trasplante. La segunda dosis se debe administrar14 días después de la primera y cada dosis subsiguiente de Zenapax® se debe administrar aintervalos de 14 días, hasta un total de cinco dosis. Las dosis subsiguientes se deben admi-nistrar un día antes o un día después de la administración programada. Zenapax® se debeutilizar en combinación con un régimen inmunosupresor que contenga un inhibidor de lacalcineurina.
La eficacia clínica y la seguridad de Zenapax® en trasplante renal se ha establecido endos ensayos clínicos de fase III, a gran escala, controlados con placebo, que incluyeron 535pacientes adultos que recibían su primer trasplante renal de donantes fallecidos. Zenapax®reduce, hasta en un 40%, la incidencia de rechazo agudo durante los 6 meses posteriores altrasplante, mejora claramente la supervivencia y la función del injerto, mejora la supervi-vencia del paciente, reduce la necesidad de administrar una terapia inmunosupresorasecundaria y limita el uso de esteroides (es decir; permite administrar dosis menos acumu-ladas de esteroides).
Zenapax® se tolera muy bien y posee un perfil de efectos secundarios, efectos adver-sos y toxicidad favorable. No se ha demostrado que el uso de Zenapax® produzca unaumento de las infecciones o aumente el riesgo de que se desarrollen neoplasias.
3
Resumen del pro d u c t o
Zenapax® es bien tolerado y posee un perfil de efectos adversos y toxicidad favorable. No se ha demostrado que el uso de Zenapax®
produzca un aumento de las infecciones o aumente el riesgo de que sedesarrollen neoplasias.

Introducción: La revolución de los trasplantes deórganos; su impacto en el hombre
De s de que en 1954 se realizó el pri m er tra s p l a n te renal con éxito, el tra s p l a n te de órga-nos ha mej orado la calidad de vida y pro l on gado la vida de mu chos pac i en tes con pato l og í a sc a rd í ac a s ,h ep á ti c a s , renales y pulmon a res crónicas y debi l i t a n te s . Tal y como se puede ob s er-var en la Ta bla 1, casi dos tercios de los tra s p l a n tes nu evos son tra s p l a n tes ren a l e s .
Antes de la introducción de los agen-tes inmunosupresores, raras veces se reali-zaba un trasplante de órganos, porque elrechazo agudo del injerto en los primerosdías o semanas posteriores al trasplanteconstituía un problema importante. Sinembargo, los avances significativos en losconocimientos de la tipificación tisular y lainmunosupresión, así como la disponibili-dad de agentes inmunosupresores más efi-caces, han tenido un impacto considerableen el éxito de los trasplantes clínicos. Porejemplo, la introducción de la ciclosporinaen 1983 revolucionó el trasplante de órga-nos; las tasas de supervivencia del injertorenal aumentaron, aproximadamente, del50% al 80%-85% durante el primer año deltrasplante. A pesar de la terapia inmunosu-presora continuada, el fracaso del injerto, debido principalmente a rechazo crónico, siguesiendo un problema clínico grave: la pérdida del injerto es, aproximadamente, del 5% poraño en el segundo año y en los años posteriores al t rasplante y se ha estimado que la vidamedia de un injerto renal es, aproximadamente, de 7,2 años (Flechner 1994). Además, lamayoría de los agentes inmunosupresores actuales presentan dos elementos de toxicidad,que están relacionados con la dosis y el tipo no específico de la inmunosupresión actual:
• Toxicidad relacionada con la inmunosupresión, ej. infección y neoplasias.
• Otras toxicidades, incluidas nefrotoxicidad, hipertensión y leucopenia.
Objetivos del tratamiento en trasplantes
Objetivos clínicos
Los objetivos clínicos principales de la terapia inmunosupresora en los trasplantes sonlos siguientes:
4
Los tra s p l a n t e sen la actualidad
Organo trasplantado Nº de pacientes
Riñón 25.649
Riñón-páncreas 915
Corazón 3865
Pulmón 1207
Hígado 7189
TOTAL 38.825
Fuente: World Transplant Centre Directory, Terasaki 1996
Tabla.1N ú m ero de pa ci en tes que re ci bi eron tra s pl a n tes nu evos en 1996.

• Prevenir o tratar el rechazo agudo del injerto en el momento en que se produce ymejorar la supervivencia a largo plazo del injerto y del paciente, proporcionando unainmunosupresión adecuada.
• Disminuir la toxicidad del régimen inmunosupresor.
• Evitar las infecc i ones u otras com p l i c ac i ones debidas a una inmu n o su presión exce s iva .
Los estudios de seg u i m i en to a largo plazo y los análisis retro s pectivos de los re su l t ado sdel tra s p l a n te ren a l , han su geri do que el rech a zo agudo puede estar correl ac i on ado con elrech a zo crónico y el fracaso del injerto (Ba s adoona et al. 1 9 9 3 ; Cecka 1991; Gulanikar et al.1 9 9 2 ; Matas et al. 1 9 9 4 ; Fer g u s on et al. 1 9 9 2 ) . Por con s i g u i en te , un obj etivo cl í n i co pri n c i p a les limitar la frec u en c i a , i n ten s i d ad y du ración de los ep i s odios de rech a zo agudo, redu c i en-do, por lo tanto, el ri e sgo de que se produzca rech a zo crónico y pérdida del injerto.
Objetivos socioeconómicos
Los obj etivos soc i oecon ó m i cos de la terapia inmu n o su pre s ora en tra s p l a n te incluyen :
• Procurar que la terapia tenga la mejor relación coste-eficacia posible.
• Minimizar la morbilidad del paciente.
• Maximizar la supervivencia del injerto.
Aunque la terapia inmunosupresora es cara, el uso precoz de un agente inmunosu-presor eficaz y menos tóxico, que produzca menos episodios de rechazo agudo y menos
rech a zo crónico, p u ede of receruna serie de beneficios. No sólopuede mejorar la calidad de vida,sino también podría proporcionarun ahorro de los costes, en térmi-nos de los costes directos totalesac u mu l ados por la pérdida delinjerto, el diagnóstico y el trata-miento de los episodios de recha-zo, el tratamiento de las infeccio-nes, la hospitalización, el uso adi-c i onal de proced i m i en to s , t a l e scomo diálisis, y nuevos trasplan-te s . Adem á s , la redu cción delnúmero de episodios de rechazodel injerto debería permitir queun mayor número de pacientesrecibiesen trasplantes de órganosde los escasos donantes disponi-bles. En efecto, un factor limitantei m port a n te en el tra s p l a n te deórganos es que la demanda deórganos de donantes excede, conmucho, el número de pacientesque necesitan un trasplante (Figu-ra 1).
5
Los trasplantes en la actualidad
The Handbook of Transplant Immunology,Wood K (ed.)MedSci Publications,1995.

Aproximaciones actuales a la terapia farmacológica en trasplantes
El tratamiento inmunosupresor actual tiene dos aplicaciones principales en el tras-plante: prevención del rechazo agudo/crónico y tratamiento del rechazo agudo. La expe-riencia clínica establece que la clasificación terapéutica de un agente inmunosupresor estádeterminada por la eficacia de su aplicación, en vez de por su clase bioquímica.
• La terapia de prevención reduce la incidencia y la frecuencia del rechazo. Los agentesinmunosupresores profilácticos se administran en el período inmediatamente poste-rior al trasplante y se administran crónicamente para el mantenimiento (frecuente-mente en diferentes regímenes de administración). Los agentes con utilidad demos-trada para la prevención del rechazo agudo incluyen CellCept® (micofenolato mofe-til), Sandimmune® y Neoral® (ciclosporina), Prograf® (tacrolimus o FK506), Imurel®(azatioprina) y corticosteroides.
• La terapia de rescate es el tratamiento de un episodio de rechazo agudo mediante laadministración de fármacos con eficacia demostrada en rescate. Este tipo de terapiaconsiste en la adición de una de estas drogas a un régimen inmunosupresor de pre-vención del rechazo, hasta que se produzca la resolución del episodio de rechazoagudo. La terapia de rescate de primera línea consiste normalmente en la administra-ción de un corticosteroides por vía intravenosa, mientras que los agentes de segundalínea incluyen anticuerpos policlonales, tales como globulina antitimocítica (ATG) oglobulina antilinfocitaria (ALG) y el anticuerpo monoclonal de origen murinoOrthoclone OKT® 3 (muromonab-CD3). Algunos de los fármacos inmunosupresoresutilizados en la prevención del rechazo agudo, también son eficaces para el tratamien-to del rechazo agudo, ej. tacrolimus y CellCept®.
La terapia de inducción consiste en administrar agentes de inversión, ej. ATG yOKT®3 como parte del tratamiento de prevención del rechazo. La administración de estetipo de terapia es inmediata: en el período próximo a la intervención quirúrgica y durantelos días inmediatamente posteriores al trasplante. Se ha demostrado que la eficacia de ATG,ALG y OKT®3 en este entorno terapéutico sólo retrasa la aparición del episodio de recha-zo, pero no se reduce ni la frecuencia ni la incidencia del rechazo agudo. La utilización deestos agentes de reversión como terapia de inducción puede tener un efecto rebote sobre elrechazo; por consiguiente, este tipo de terapia no se puede considerar como una verdaderaterapia profiláctica y se sigue utilizando fuera de indicación en la mayoría de los países.
La terapia de inducción también se puede utilizar para retrasar la administración dela primera dosis de agentes inhibidores de la calcineurina, preservando de esa forma elaloinjerto de los efectos nefrotóxicos de ciclosporina o tacrolimus en el período del pos-trasplante inmediato.
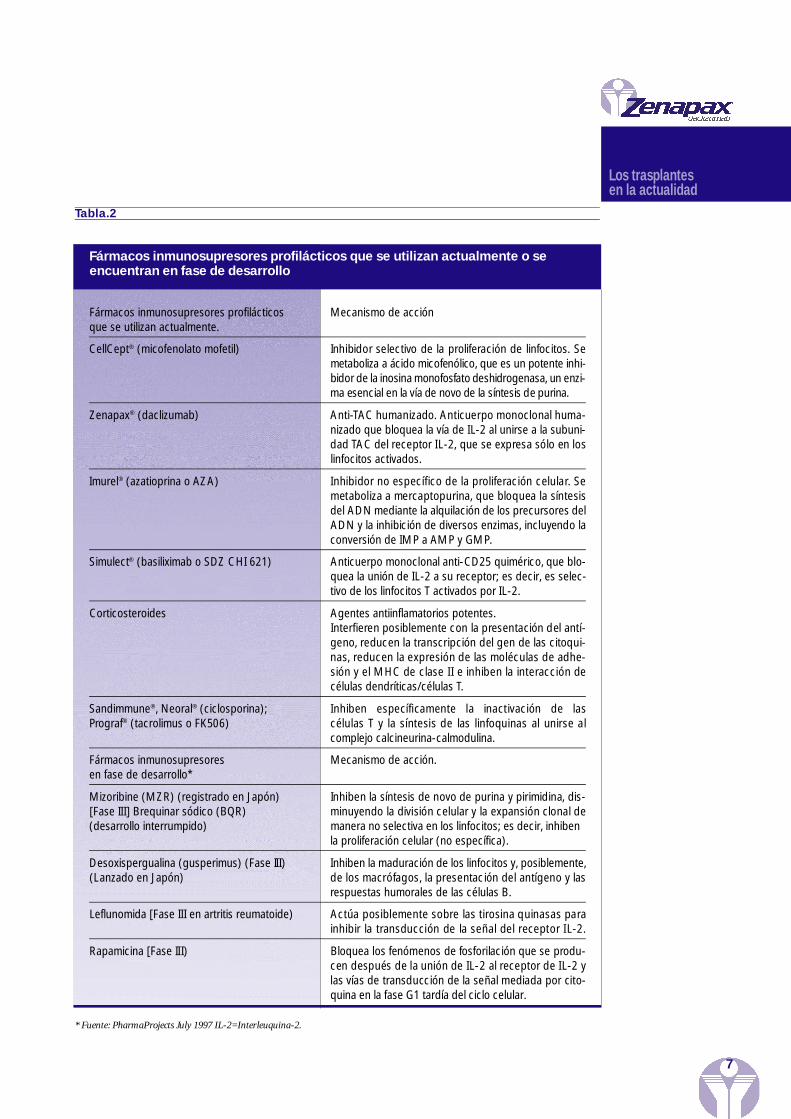
En las Ta blas 2 y 3 se enu m eran diversos fárm acos inmu n o su pre s ores que se uti l i z a nactu a l m en te o se en c u en tran en fase de de s a rro ll o, s egún la ef i c acia de su aplicación tera p é uti c a .
6
Los trasplantes en la actualidad
La terapia de inducción consiste en la administración de agentes derescate, ej. ATG y OKT®3 en un entorno profiláctico. La utilización
de este tipo de terapia se realiza en el período previo a la intervenciónquirúrgica y durante el postrasplante inmediato.
7
Los trasplantes en la actualidad
Tabla.2
* Fuente: PharmaProjects July 1997 IL-2=Interleuquina-2.
Fármacos inmunosupresores profilácticos que se utilizan actualmente o seencuentran en fase de desarrollo
Fármacos inmunosupresores profilácticos Mecanismo de acciónque se utilizan actualmente.
CellCept® (micofenolato mofetil) Inhibidor selectivo de la pro l i fe ración de linfocitos. Semetaboliza a ácido micofenólico, que es un potente inhi-bidor de la inosina monofo s fato deshidro g e n a sa, un enzi-ma esencial en la vía de novo de la síntesis de purina.
Zenapax® (daclizumab) A n t i -TAC humanizado. Anticuerpo monoclonal huma-nizado que bloquea la vía de IL-2 al unirse a la subuni-dad TAC del receptor IL-2, que se expre sa sólo en loslinfocitos activados.
Imurel® (azatioprina o AZA) Inhibidor no específico de la pro l i fe ración celular. Semetaboliza a mercaptopurina, que bloquea la síntesisdel ADN mediante la alquilación de los precursores delADN y la inhibición de diversos enzimas, incluyendo laconversión de IMP a AMP y GMP.
Simulect® (basiliximab o SDZ CHI 621) Anticuerpo monoclonal anti-CD25 quimérico, que blo-quea la unión de IL-2 a su receptor; es decir, es selec-tivo de los linfocitos T activados por IL-2.
Corticosteroides Agentes antiinflamatorios potentes.I n t e r f i e ren posiblemente con la presentación del antí-geno, reducen la transcripción del gen de las citoqui-nas, reducen la expresión de las moléculas de adhe-sión y el MHC de clase II e inhiben la interacción decélulas dendríticas/células T.
Sandimmune®, Neoral® (ciclosporina); Inhiben específicamente la inactivación de las Prograf® (tacrolimus o FK506) células T y la síntesis de las linfoquinas al unirse al
complejo calcineurina-calmodulina.
Fármacos inmunosupresores Mecanismo de acción.en fase de desarrollo*
Mizoribine (MZR) (registrado en Japón) Inhiben la síntesis de novo de purina y pirimidina, dis-[Fase III] Brequinar sódico (BQR) m i n u yendo la división celular y la expansión clonal de (desarrollo interrumpido) manera no selectiva en los linfocitos; es decir, inhiben
la proliferación celular (no específica).
Desoxispergualina (gusperimus) (Fase III) Inhiben la maduración de los linfocitos y, posiblemente, (Lanzado en Japón) de los macrófagos, la presentación del antígeno y las
respuestas humorales de las células B.
Leflunomida [Fase III en artritis reumatoide) Actúa posiblemente sobre las tirosina quinasas parainhibir la transducción de la señal del receptor IL-2.
Rapamicina [Fase III) Bloquea los fenómenos de fo s forilación que se pro d u-cen después de la unión de IL-2 al receptor de IL-2 ylas vías de transducción de la señal mediada por cito-quina en la fase G1 tardía del ciclo celular.

En la mayoría de las unidades de trasplantes, se utilizan combinaciones de agentesinmunosupresores indicados en la prevención del rechazo agudo, con o sin un agente deinducción. La terapia de combinación permite administrar dosis más bajas de cada agente;por consiguiente, se minimizan los efectos secundarios/toxicidades, a la vez que el efectoinmunosupresor puede ser aditivo. La terapia inicial consiste normalmente en la adminis-tración de ciclosporina en combinación con CellCept® o azatioprina y corticosteroides(terapia triple). La terapia cuádruple incluye una terapia de inducción con anticuerpos,tales como ALG, ATG o muromonab-CD3, al inicio del tratamiento.
En algunos centros de trasplantes se utiliza una terapia secuencial, que consiste enretrasar la introducción de los inhibidores de la calcineurina hasta que termine el ciclo deALG, ATG o muromonab-CD3 y se obtenga una función renal adecuada.
El nivel de monitorización de los pacientes sometidos a trasplante renal está en fun-ción de la toxicidad y la ventana, o índice terapéutico, de los fármacos utilizados. Por con-siguiente, aunque se ha demostrado que ciclosporina y tacrolimus son eficaces en trasplan-te renal, el riesgo de aparición de efectos secundarios (particularmente nefrotoxicidad) sedebe minimizar mediante una monitorización constante.
La introducción de Zenapax® supone la posibilidad de utilización de un anticuerpocon eficacia demostrada en la prevención del rechazo agudo en trasplante. Zenapax® estáindicado para la prevención del rechazo agudo y proporciona una inmunosupresión eficazdurante todo el período de mayor riesgo ( 120 días) después del trasplante, reduciendo laincidencia del rechazo. Los pacientes sometidos a trasplante renal que reciben Zenapax® nopresentan un aumento del riesgo de experimentar efectos adversos asociados con la terapiainmunosupresora, ni requieren una monitorización adicional a consecuencia de su admi-nistración; esto es un resultado de su eficacia bien establecida y al excelente perfil de segu-ridad del fármaco.
En el entorno actual, el siguiente desafío es controlar la incidencia de rechazo cróni-
8
Los trasplantes en la actualidad
Tabla.3
Terapias biológicas que se utilizan actualmente o se encuentran en fase de desarrollo en trasplante renal.
Terapia biológica Mecanismo de acción
Anticuerpos policlonales Contiene una combinación de anticuerpos contra los Globulina antilinfocítica (ALG) linfocitos B y T; inactiva todos los linfocitos.
Produce la depleción de los linfocitos mediante lisisdependiente del complemento o por opsonización yfagocitosis subsiguiente.
Atgam®, Thymoglobulin® Contiene una combinación de anticuerpos contra los(globulina antitimocítica o ATG) linfocitos B y T. Inactiva todos los linfocitos. Produce la
depleción de los linfocitos mediante lisis dependientedel complemento o por opsonización y fagocitosis sub-siguiente.
Anticuerpos monoclonales Anticuerpo monoclonal anti-CD 3 murino. Bloquea la OTK®3 (muromonab-CD3) función de las células T mediante una interacción espe-
cífica con la molécula CD3 del complejo del receptor delas células T. Produce la lisis de las células T.
Leukotac® (inolimomab o BT563) Anticuerpo monoclonal anti-CD-25 murino, que blo-(Fase III) quea la unión de I L-2 a su receptor

co. La nefrotoxicidad de ciclosporina y tacrolimus y la tendencia de todos los agentes inmu-nosupresores disponibles hasta el momento a aumentar el riesgo de infecciones y neopla-sias, representan también aspectos importantes.
Limitaciones de las terapias actualesutilizadas en trasplantes
Todos los fármacos inmunosupresores disponibles anteriormente tienen una serie delimitaciones:
• Carecen de selectividad inmunológica - No son específicos del aloantígeno; es decir, nosólo tienen como objetivo las células implicadas en la respuesta inmune dirigida por elaloantígeno del donante, sino también los leucocitos involucrados en otras respuestasinmunes relacionadas con los agentes infecciosos. Aunque el anticuerpo monoclonalmuromonab CD3 es específico del antígeno, puesto que hace que todas las células por-tadoras del antígeno CD3; es decir, todas las células T maduras,sean incapaces de pro-ducir respuestas inmunológicas normales,la administración inicial también da lugar auna rápida depleción de las células T en sangre periférica, incluídas las dirigidas a losantígenos ajenos al injerto, por ejemplo, virus.
• Su eficacia es limitada - El rechazo agudo y crónico sigue constituyendo un problemaclínico, especialmente el rechazo crónico: la vida media de los trasplantes después delprimer año se mantiene inalterada, a pesar de la administración de ciclosporina enterapia triple o cuádruple.
• Producen inmunodeficiencia - El uso concomitante de diversos agentes inmunosupre-sores puede producir inmunosupresión excesiva, que puede dar lugar a infeccionesoportunistas y a un aumento de las neoplasias inducidas por virus.
• Producen efectos adversos - Cada fármaco tiene un perfil de toxicidad único que puedelimitar su utilidad.
Por ejemplo:
— Ciclosporina y tacrolimus pueden ser nefrotóxicos y se sabe que producen hiper-tensión. Se sabe que la ciclosporina produce la regulación al alta de la expresión deTGF-ß, que puede tener influencias adversas en la función renal y la incidencia depérdida del injerto (Pankewycz et al.1996). Cualquier régimen inmunosupresor quelimite el uso de ciclosporina será beneficioso.
— Micofenolato mofetil provoca trastornos gastrointestinales y existen indicios de quesu administración produce una frecuencia más alta de determinados tipos de infec-ciones.
— Los corticosteroides producen múltiples efectos secundarios, incluyendo cicatriza-ción inadecuada de las heridas, osteoporosis y cataratas; por consiguiente, cualquierrégimen inmunosupresor que limite el uso de esteroides sería beneficioso.
— Azatioprina produce efectos secundarios, particularmente en los tejidos de divisiónrápida, tales como el tracto gastrointestinal y es mielosupresor.
— Ninguno de los agen tes inmu n o su pre s ores dispon i bles actu a l m en te indu cento l erancia a los aloa n t í genos del don a n te en el tra s p l a n te . Se cree que algunos agen-tes (por ej em p l o, c i cl o s porina) inhiben la indu cción de la to l erancia en mode -los animales y, por con s i g u i en te , requ i eren ad m i n i s tración a largo plazo (inde -
9
Los trasplantes en la actualidad

finida) después del tra s p l a n te , que puede dar lu gar a una morbi l i d ad sign i f i c a -tiva .
Limitaciones de los anticuerpos policlonales
Los anticuerpos policlonales, tales como ALG y ATG, son sueros con actividad inmu-nosupresora potente, que se producen en conejos y caballos. Producen una linfopenia pro-funda y una depleción de los linfocitos T prolongada, efectos que dan lugar a un aumentodel riesgo de que se desarrollen neoplasias e infecciones oportunistas. ALG o ATG se utili-zan, en ocasiones, en el período inicial postrasplante para evitar la nefrotoxicidad inducidapor ciclosporina, pero se utilizan con más frecuencia para el tratamiento de los episodiosde rechazo agudo resistente a esteroides. Entre los pacientes sometidos a trasplante renalque reciben inmunosupresión basada en ciclosporina,aproximadamente el 85% de los epi-sodios de rechazo resistente a esteroides se tratan con éxito con ALG (Simpson & Monaco1995). En algunos estudios en los que se utilizó ALG como terapia de inducción, se hancomunicado mejoras en la supervivencia del injerto y una reducción del número, la severi-dad y el tiempo hasta el primer rechazo (Shield et al. 1997). Sin embargo, en otros estudiosse ha observado que este tratamiento no afecta a la supervivencia global del injerto, ni a laincidencia del rechazo (Simpson y Monaco 1995).
Las desventajas de los anticuerpos policlonales son las siguientes:
• Se deben administrar en infusión intravenosa lenta en un vaso sanguíneo grande, conflujo sanguíneo adecuado, durante un período de 4-6 horas.
• La administración de ALG o ATG está asociada con un aumento de la incidencia deneoplasias e infecciones.
• Los efectos secundarios graves significativos son frecuentes, ej. trombocitopenia.
• Tienen acciones no específicas; es decir, producen la depleción de todos los linfocitos,no sólo de los que son activados por la respuesta inmune al aloinjerto.
• Aproximadamente sólo el 2% de los anticuerpos presentes están dirigidos contra loslinfocitos; por consiguiente,se deben administrar grandes cantidades de proteína hete-róloga.
• Se pueden desarrollar anticuerpos contra ALG o ATG y neutralizar sus efectos, limi-tando el número de veces que se pueden utilizar estos agentes.
• Los diferentes preparados tienen una potencia variable y los pacientes muestran unasensibilidad variable.
• Son laboriosos de preparar.
10
Los trasplantes en la actualidad
Los anticuerpos policlonales, tales como ALG y ATG, son sueroscon actividad inmunosupresora potente, que se producen en conejos
y caballos. Producen una linfopenia profunda y una depleción de linfoctios T prolongada, efectos que dan lugar a un aumento del riesgo
de que se desarrollen neoplasias e infecciones oportunistas.

La incidencia y la severidad de las reacciones adversas asociadas con ALG o ATG difie-re en cada paciente,según el lote específico del anticuerpo utilizado en la infusión y la espe-cie animal utilizada para preparar el producto. En general, los productos de origen equinose toleran peor que los procedentes del conejo (Granjard et al. 1989). La mayoría de lospacientes (hasta el 80%) experimentan escalofríos y fiebre durante la administración de laprimera dosis y, ocasionalmente, durante las dosis subsiguientes.Se cree que estos síntomasse deben a la lisis de los linfocitos,la liberación de citoquinas pirogénicas y la activación noespecífica de las células T, con producción de citoquinas. Las erupciones cutáneas tambiénson frecuentes y se deben, probablemente, a una reacción de hipersensibilidad de tipo tar-dío. La trombocitopenia se manifiesta hasta en el 50% de los pacientes y puede ser sufi-c i en tem en te grave para requ erir la su s pensión del tra t a m i en to y tra n s f u s i ó n .Ocasionalmente (5%-10% de los pacientes), se pueden manifestar signos de enfermedaddel suero hacia el final de la primera semana de tratamiento. Las reacciones anafilácticas ode hipersensibilidad aguda son menos frecuentes (<5% de los pacientes). Generalmente,los síntomas se observan con más frecuencia después de la primera inyección y disminuyenen el transcurso del tratamiento.
Uno de los problemas principales derivados de la administración de anticuerpos poli-clonales es la inmunosupresión excesiva, que está asociada con la falta de especificidad delos anticuerpos. Se puede manifestar en forma de infección, particularmente, de tipo viral,por ejemplo citomegalovirus (CMV), y da lugar a una morbilidad, e incluso mortalidad,significativa del paciente. Para reducir el riesgo de inmunosupresión excesiva,se recomien-da efectuar una monitorización diaria mediante citometría de flujo, del recuento de eritro-citos, plaquetas y leucocitos del paciente, realizando ajustes de dosis apropiados, si es nece-sario. Estas medidas pueden servir de ayuda, pero no erradican el problema y una monito-rización tan intensiva resulta extremadamente cara. Aunque muromonab-CD3 es un anti-cuerpo monoclonal, se observan problemas similares asociados con la inmunosupresiónexcesiva, debido a la amplia expresión del antígeno CD3.
11
Los trasplantes en la actualidad
Uno de los problemas principales derivados de la administración de anticuerpos policlonales es la inmunosupresión excesiva, que está
asociada con la falta de especificidad de los anticuerpos. Se puedemanifestar en forma de infección, particularmente, de tipo viral, por
ejemplo, citomegalovirus (CMV), y da lugar a una morbilidad,e incluso mortalidad,significativa del paciente.

La función esencial del sistema inmunológico es distinguir lo propio de lo ajeno y estadistinción la realizan principalmente los linfocitos T.
El rechazo del trasplante de órganos está asociado con una respuesta inmune com-pleja, específica del antígeno, al antígeno del donante, que está mediada por los linfocitosT y B (Figura 2). Los linfocitos T son esenciales para la respuesta inmune y, una vez activa-dos por el antígeno, inducen el reclutamiento, la diferenciación y la activación de otrasmuchas células. Los linfocitos T activados también desempeñan un papel esencial en laenfermedad injerto contra huésped (EICH) y las enfermedades autoinmunes.
Los linfocitos T son los responsables principales de la inmunidad mediada por lascélulas, que es el componente esencial de la respuesta inmune implicada en el rechazoagudo, mientras que los linfocitos B son responsables de la inmunidad humoral o media-da por anticuerpos, que puede desempeñar un papel en el rechazo agudo y crónico. Los lin-focitos T también regulan la actividad de los linfocitos B. Además de la respuesta inmuneespecífica del antígeno, una respuesta inflamatoria mediada por otras células, tales comolos macrófagos, las células polimorfonucleares y las células Natural Killer (NK), contribu-yen al rechazo del aloinjerto.
Activación de los linfocitos T
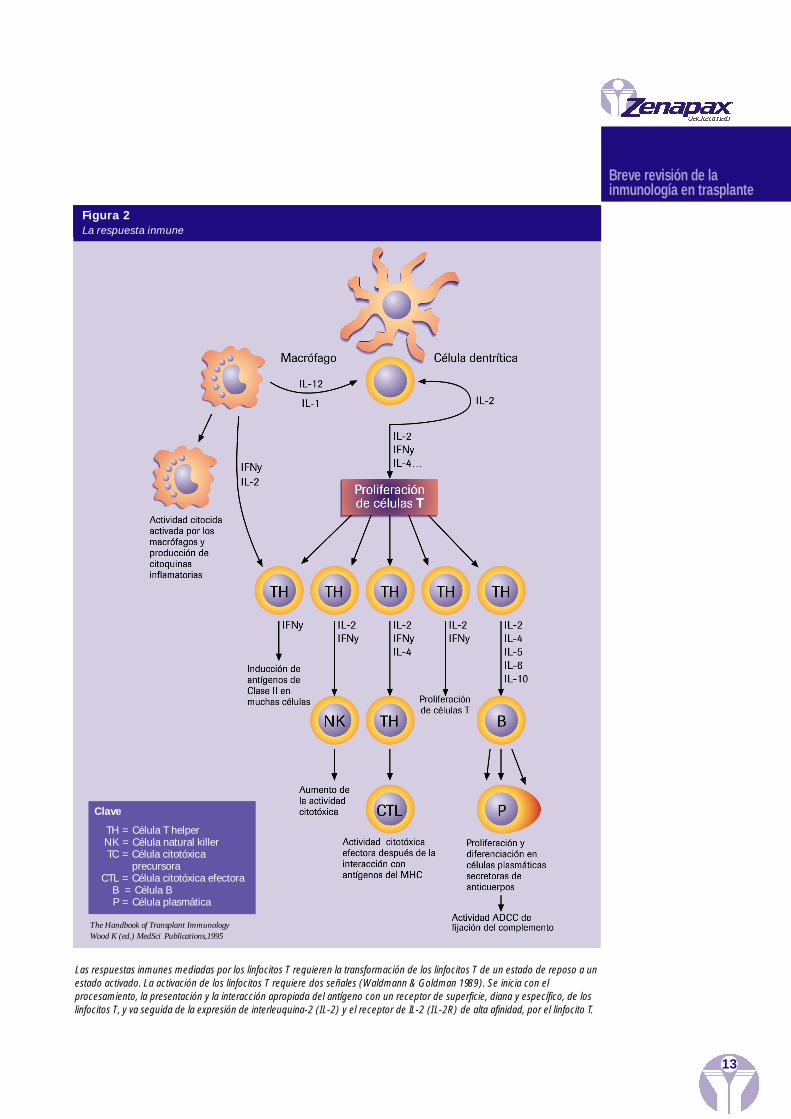
Las respuestas inmunes med i adas por los linfoc i tos T requ i eren la tra n s form ación de losl i n foc i tos T de un estado de reposo a un estado activado. Pa ra que se produzca la activac i ó nde los linfoc i tos T, s on nece s a rias dos señales (Waldmann & Goldman 1989). Se inicia con elproce s a m i en to, la pre s en t ación y la interacción aprop i ada del antígeno con un receptor desu perf i c i e , diana y espec í f i co, de los linfoc i tos T y va seguida de la ex presión de interl eu qu i-na-2 (IL-2) y el receptor de IL-2 (IL-2R) de alta afinidad , por parte del linfoc i to T.
La presentación del antígeno tiene lugar cuando los fragmentos de los péptidos de losantígenos del donante se asocian con las proteínas (de clase I o II) del complejo mayor dehistocompatibilidad (MHC) en la superficie de las células presentadoras del antígeno(APCs), tales como las células dendríticas, del donante o del receptor. El péptido conteni-do en la proteína del MHC es entonces reconocido por el receptor de células T específicodel antígeno (TCR), que está compuesto de dos cadenas (a y b), que forman un complejocon diversas cadenas del complejo CD3. La especificidad por el antígeno del TCR está defi-nida por regiones determinantes de la complementariedad (CDRs),localizadas en la regiónvariable o el dominio N-terminal de cada cadena del receptor. El complejo CD3 transduceuna señal de activación al linfocito T. En esta interacción entre los TCRs y las APCs, tam-bién están implicadas las moléculas de adhesión y las moléculas señalizadoras adicionales(ej. CD4 en las células helper, CD8 en las células citolíticas y supresoras), que sirven decoestimuladores o segundas señales (Figura 3) (Waldmann & Cobbold 1993).
12
B re ve revisión de la inmunología en tra s p l a n t e
13
Breve revisión de la inmunología en trasplante
Clave
TH = Célula T helperNK = Célula natural killerTC = Célula citotóxica
precursora CTL = Célula citotóxica efectora
B = Célula BP = Célula plasmática
Las respuestas inmunes mediadas por los linfocitos T requieren la transformación de los linfocitos T de un estado de reposo a unestado activado. La activación de los linfocitos T requiere dos señales (Waldmann & Goldman 1989). Se inicia con elprocesamiento, la presentación y la interacción apropiada del antígeno con un receptor de superficie, diana y específico, de loslinfocitos T, y va seguida de la expresión de interleuquina-2 (IL-2) y el receptor de IL-2 (IL-2R) de alta afinidad, por el linfocito T.
The Handbook of Transplant ImmunologyWood K (ed.) MedSci Publications,1995
Figura 2La respuesta inmune

La interacción del antígeno con el TCR específico inicia la activación de los linfocitosT, incluida la inducción de la expresión de IL-2 e IL-2R de alta afinidad. IL-2 produce launión del IL-2R de alta afinidad en los linfocitos T activados, estimulando la proliferaciónde los linfocitos T y su diferenciación en linfocitos T helper, supresores o citotóxicos,dependiendo, esta última, de la presencia de otras citoquinas en el entorno. Un requisitoadicional para que tenga lugar la activación y la diferenciación de los linfocitos T es la con-tribución de otros linfocitos T CD4+ sensibles al mismo antígeno.
Además de las señales del antígeno recibidas a través del TCR,los linfocitos T tambiénreciben señales a través de la unión de una red de mediadores solubles, las citoquinas, areceptores celulares de superficie específicos de citoquinas. Estas citoquinas actúan paracomunicarse entre las células en las respuestas inmunes e inflamatorias. Las citoquinasregulan la magnitud y el tipo de respuesta inmune, activando/induciendo la diferenciaciónde los linfocitos T CD4+ helper en células TH1 o TH2, que secretan diferentes patrones decitoquinas. Los linfocitos TH1 tienden a iniciar una respuesta mediada por la célula, mien-tras que los linfocitos TH2 dirigen el sistema inmunológico hacia una respuesta dominadapor el anticuerpo.
14
Breve revisión de la inmunología en trasplante
Existen tres tipos de interaccionesmoleculares entre las células T y lascélulas presentadoras del antígeno(APC). Primero, el receptor de las
células T específico (TCR)reconoce el antígeno extraño, enforma de péptido procesado [elpéptido del antígeno (Ag)], que
está unido en el surco de lasmoléculas del complejo mayor dehistocompatibilidad (MHC) de lasAPC. En segundo lugar, una seriede moléculas de adhesión que seencuentran en la Célula T se unen
a sus ligandos en las APC. Estosincluyen CD4 y CD8,
correceptores de las moléculas delMHC y CD28, que interaccionan
con el ligando B7 de APC paraproporcionar una
"coestimulación". En tercer lugar,la Célula T expresa receptores de
diversos factores que regulan elcrecimiento y la diferenciación
(citoquinas), tales como lainterleuquina 2, algunos de los
cuales son producidos por otrascélulas T activadas y forman la
base de "ayuda" y colaboración.Aunque la unión al receptor de las
células T proporciona la señalprimaria para activar las células T,
el resultado, que puede ser unarespuesta proliferativa o lainducción de un estado no
sensible, depende de las señalesadicionales procedentes de estas
moléculas de adhesión y losreceptores del factor de
crecimiento, para situar elreconocimiento del antígeno "en el
mismo contexto".
Reimpresión de: Trends Pharmacol Sci, Vol 14,Waldmann H. y Cobbold S. The use of monoclonal antibodies to achieve inmunological tolerance, p143-8,Copyright 1993, con permiso de Elsevier Science.
Figura 3Moléculas de interacción con los linfocitos T/Células presentadoras de antígeno.
Moléculas “de adhesión”
LinfocitoT
Células presentadorasdel antígeno
LFA-1, CD2CD4, CD8LFA-3, ICAM-1CD28, B7 etc.
Receptores delfactor de crecimiento
Factor decrecimiento
Péptido Ag
Otras células
Interleuquinas 1-12InterferonesFactores de necrosistumoral, etc.
Receptores de:

Una vez activados, los linfocitos T pueden iniciar, principalmente mediante la elabo-ración de citoquinas, la diferenciación terminal y la proliferación de diversas células hema-topoyéticas, tales como los linfocitos B, los linfocitos T citotóxicos, las células NK y losmacrófagos. Estas células pueden actuar de mediadoras en las funciones efectoras que danlugar al tipo de daño tisular asociado con el rechazo del injerto.
Interleuquina-2 y el receptor de IL-2
La IL-2 es una de las citoquinas más importantes producidas por los linfocitos T hel-per. Interacciona con receptores de superficie específicos en los linfocitos B y T activados,para facilitar la formación de anticuerpos y las respuestas inmunes mediadas por las célu-las. También puede estimular la activación de las células efectoras citolíticas no específicas,incluidas las células NK y las células killer activadas por linfoquina (LAK), que se creedesempeñan un papel en las defensas iniciales del huésped,antes de que se desarrolle inmu-nidad específica (Waldmann 1991).
El IL-2R de alta afinidad se encuentra en los linfocitos activados. El IL-R2 está com-puesto por tres subunidades de proteínas, tal y como se muestra en la Figura 4.
• IL-2Ra, un péptido de 55 kD, que también se conoce como CD25 o antígeno de acti-vación de las células T (Tac); es exclusivo del receptor de IL-2.
• IL-2Rb, un péptido de 75 kD.
• IL-2Rg, un péptido de 64 kD, que desempeña un papel al facilitar la unión de IL-2inducida por el péptido b y en la señalización del receptor.
La mayoría de los linfocitos T y B y las células NK en reposo expresan un complejodel receptor de afinidad intermedia, IL-2Rb/IL-2Rg, pero no IL-2Ra (Tac), que sólo seexpresa después de que las células son activadas por un antígeno extraño o IL-2. Cuandoestá aislado, el péptido Tac muestra una unión del IL-2 de baja afinidad, pero no tiene capa-cidad de transducción de la señal. Las células que expresan el complejo del receptor IL-2Rb/IL-2Rg, tales como los linfocitos granulares grandes, unen IL-2 con afinidad interme-dia. Cuando las células T son activadas por el antígeno, se produce la regulación al alta dela expresión de IL-2 y Tac.La subunidad Tac se asocia rápidamente con las subunidades IL-2Rb/IL-2Rg para formar un complejo IL-2R de alta afinidad (Waldmann et al. 1993).
La interacción de IL-2 con su IL-2R de alta afinidad de s en c adena la pro l i feración y la
15
Breve revisión de la inmunología en trasplante
Una vez activados, los linfocitos T pueden iniciar,principalmente mediante la elaboración de citoquinas, la diferencia-ción terminal y la proliferación de diversas células hematopoyéticas,
tales como los linfocitos B,los linfocitos T citotóxicos,las células NK ylos macrófagos.Estas células pueden actuar de mediadoras en las
funciones efectoras que dan lugar al tipo de daño tisular asociado conel rechazo del injerto.

d i feren c i ación de los linfoc i to sT, que culmina en la apari c i ó nde los linfoc i tos T efectore s ,que actúan de med i adores enlas funciones indu ctora s , su-pre s oras o citotóxicas (Wa l d-mann et al. 1 9 9 2 ) . Adem á s , l aseñal med i ada por el receptorde IL- 2 , produ ce la ex pre s i ó nad i c i onal de IL- 2 , a m p l i f i c a n-do, por con s i g u i en te , la re s-puesta inmu n e .
Aunque los linfocitos Tnormales en reposo no expre-san Tac, este receptor se ex-presa en las células malignasde algunos cánceres linfoides,tales como la leu cemia decélulas T adulta, el linfoma decélulas T cutáneo y la enfer-medad de Hodgkin. La expre-sión elevada o anormal de Taco la expresión de una formasoluble de Tac, también estánasociadas con muchas patolo-gías autoinmunes, tales comoartritis reumatoide, lupus eri-tematoso sistémico, así comocon el rechazo del trasplantede órganos y la EICH (Wald-mann et al.1993).En estas pa-
tologías, una proporción de células anormales expresan el antígeno Tac y la concentraciónsérica de Tac soluble liberado de las células activadas es elevada.
La expresión de Tac en los linfocitos T activados por el aloinjerto de pacientes queexperimentan rechazo de aloinjerto, pero no en los linfocitos T normales en reposo, pro-porciona el fundamento para las estrategias terapéuticas que eliminan las células que expre-san Tac, o impiden la interacción de IL-2 con su receptor de alta afinidad y, por consi-guiente, interfieren en esta vía de IL-2 y van dirigidas sólo contra los linfocitos T implica-dos en la respuesta de rechazo.
16
Breve revisión de la inmunología en trasplante
(Waldmann 1991)
Figura 4Representación esquemática del receptor de IL-2.

Anticuerpos monoclonales como conceptoterapéutico en trasplante
Desde principios de los años 80,se han desarrollado una serie de anticuerpos mono-clonales dirigidos contra varios procesos de la activación de los linfocitos T. La mayoríaestán dirigidos a los receptores celulares de superficie, tales como el complejo TCR/CD3,los receptores de citoquinas (ej. anti-IL-2R) o las moléculas de adhesión. Los anticuerposmonoclonales se han investigado experimentalmente, obteniéndose resultados variables, enterapia de inducción para la prevención del rechazo agudo, y en terapia de rescate para eltratamiento de los episodios de rechazo agudo resistente a esteroides.
Mu rom onab-CD3 fue el pri m er anti c u erpo mon ocl onal que se utilizó en tra s p l a n te ei n i c i a l m en te se pensaba que era la "bala mágica" de la inmu n o su pre s i ó n . Es un anti c u erpoa n ti-CD3 mon ocl onal mu ri n o, con ef i c acia dem o s trada para retrasar el rech a zo en el per í o-do po s tra s p l a n te inicial e invertir los ep i s odios de rech a zo agudo, i n cluso en pac i en tes que nore s pon den a esteroi des a dosis altas y A LGs po l i cl onales (Abra m owicz et al. 1 9 9 2 ;Abra m owicz & Goldman 1995; De Ma t tos & Norman 1993; Norman et al. 1 9 9 3 ) . Sin em b a r-go, el co s te y los efectos sec u n d a rios asoc i ados con mu rom onab-CD3 ll eva ron a mu chos cen-tros de tra s p l a n tes a re s tri n gir su uso a terapia para pac i en tes en los que los ep i s odios derech a zo no se habían pod i do revertir con corti co s teroi de s . Adem á s , los análisis simu l t á n eo sde los datos de pac i en tes som eti dos a tra s p l a n te ren a l ,i n clu i dos en en s ayos pro s pectivo s , ra n-dom i z ado s , de Bélgica y EE.UU, m o s tra ron qu e , en com p a ración con cicl o s pori n a , la tera p i ade indu cción con mu rom onab-CD3 no mej ora la su pervivencia del injerto a largo plazo enreceptores de injertos renales de cad á vere s , que pre s entaban un ri e sgo bajo de ex peri m en t a rrech a zo (ti em po de isqu emia fría < 24 horas y/o <2 rech a zos por incom p a ti bi l i d ad HLA- D R ) ;sin em b a r go, ex i s ten indicios que su gi eren que la terapia de indu cción con mu rom on a b - C D 3m ej ora los re su l t ados en pac i en tes de ri e sgo alto (Abra m owicz et al. 1 9 9 5 ) . Adem á s , p u edeex i s tir un antagonismo inmu n o l ó gi co en tre mu rom onab-CD3 y cicl o s pori n a , que puedelimitar la ef i c acia de una com bi n ación de estos agen tes (Abra m owicz & Goldman 1995).
Desafortunadamente, el uso de muromonab-CD3 está asociado con efectos secunda-rios producidos por la liberación de citoquina (Kreis 1993; Abramowicz & Goldman 1995).Los efectos secundarios más frecuentes experimentados después de la inyección inicial sonfiebre, escalofríos, cefalea, mialgia, náuseas, vómitos y diarrea. Raras veces se manifiestanefectos secundarios graves, tales como edema pulmonar, encefalopatía, meningitis aséptica,convulsiones y trombosis de los vasos del injerto. La inmunosupresión no selectiva produ-cida por muromonab-CD3, también da lugar a una incidencia alta de infecciones oportu-nistas y linfomas (Abramowicz & Goldman 1995).
Un agen te inmu n o su pre s or ideal estaría diri gi do sel ectiva m en te a los linfoc i tos T de s ti-n ados a participar en la re acción inmune al antígeno del don a n te . Pu e s to que Tac es sel ectivo
17
Anticuerpos monoclonalesMuromonab-CD3 fue el primer anticuerpo monoclonal
que se utilizó en trasplante e inicialmente se pensabaque era la "bala mágica" de la inmunosupresión.

p a ra las células T activad a s , un anti c u erpo mon ocl onal que inactivase sel ectiva m en te estas célu-las inhibiría las respuestas inmunes incipien tes o en curs o, sin su primir la inmu n i d ad natu ra lo la capac i d ad para producir una respuesta inmune específica a un antígeno nu evo, una ve zcom p l et ada la terapia anti - TAC . Adem á s , TAC no ti ene función señalizadora , de manera qu eun anti c u erpo anti - TAC no debería dar lu gar a los efectos sec u n d a rios graves asoc i ados con lal i beración de citoquina producida por la terapia con el anti c u erpo mon ocl onal TC R / C D 3 .
Limitaciones de los anticuerpos monoclonalesmurinos como opción terapéutica en trasplante
Se han desarrollado diversos anticuerpos monoclonales contra el IL-2R de alta afini-dad; son específicos de diferentes epítopes presentes en la cadena α (CD25, Tac). Aunqueestudios experimentales en animales demostraron que algunos de estos anti-IL-2R muri-nos eran eficaces, los resultados obtenidos en pacientes han sido variables y, en algunoscasos, decepcionantes (Amlot 1995).
• El anti-TAC murino (MAT) es un anticuerpo monoclonal IgG2a de ratón, que reco-noce la proteína Tac IL-2R e inhibe las respuestas biológicas mediadas por IL-2 de lascélulas linfoides activadas. En un ensayo clínico randomizado, 40 pacientes recibieronMAT en combinación con una terapia inmunosupresora triple. Este ensayo demostróuna reducción de los episodios de rechazo prematuro del aloinjerto renal en recepto-res de aloinjertos renales de donantes fallecidos y los primeros episodios de rechazoposteriores, pero no demostró una mejora de la supervivencia del injerto o del pacien-te (Kirkman et al. 1989; 1991). La eficacia de MAT estuvo limitada por su inmunoge-nicidad; se desarrollaron anticuerpos anti-ratón humanos (HAMAs) en 7 de los 10pacientes estudiados. Zenapax® (daclizumab) se derivó de este anticuerpo murino.
• 33B3.1 es un anticuerpo monoclonal IgG2a de rata que bloquea la unión de IL-2 al IL-2R. En un ensayo clínico randomizado inicial, se comunicó que 33B3.1 era tan eficazcomo ATG para prevenir el rechazo del aloinjerto renal (la tasa de rechazo agudo fue,aproximadamente, del 30%) y dio lugar a menos infecciones y efectos secundarios(Soulillou et al. 1990). Sin embargo, estudios subsiguientes han demostrado que ATGtenía una eficacia superior a 33B3.1 (Cantarovich et al. 1994; Hourmant et al. 1994).Cantarovich et al. (1994) estudiaron 40 pacientes sometidos a trasplante primariocadavérico o renal y pancreático combinado, que se randomizaron para recibir 33B3.1o ATG, así como azatioprina, corticosteroides a dosis bajas y ciclosporina. Este estudiomostró que,aunque la incidencia de rechazo no difirió significativamente en el primer,segundo y tercer mes del período postoperatorio, el número total de pacientes tratadoscon 33B3.1 que experimentaron rechazo durante el período de seguimiento fue signi-ficativamente más alto que el observado en el grupo que recibió ATG (p<0,02). Elmismo grupo estudió pacientes sometidos a un segundo trasplante renal. Ningúnpaciente experimentó episodios de rechazo celular durante los 10 días de tratamientocon ATG, mientras que el 50% de los episodios se manifestaron durante la terapia con33B3.1 (media del comienzo: 8 días) (Hourmant et al. 1994). Además, el mismo estu-dio mostró que el rechazo del injerto se producía antes en el grupo de pacientes trata-dos con el anticuerpo monoclonal: 39 días en el grupo que recibió ATG, en compara-ción con 29 días en el grupo tratado con el anticuerpo monoclonal.
• Leukotac® (inolimomab o BT563) es un anticuerpo monoclonal murino dirigido con-tra el IL-2R para el tratamiento de la EICH aguda resistente a esteroides y la preven-ción del rechazo en trasplante hepático, cardíaco y renal.
18
Anticuerposmonoclonales

Los anticuerpos monoclonales mencionados anteriormente parecen estar desprovis-tos de efectos secundarios significativos y no producen un aumento de las complicacionesinfecciosas.
En resumen, los anticuerpos monoclonales que se han utilizado en trasplante estánlimitados por su diseño; las limitaciones terapéuticas parecen deberse al hecho de que losanticuerpos son de origen murino o porque no están dirigidos contra un antígeno apro-piado. Las consecuencias de estas limitaciones del diseño son:
• Los anti c u erpos mon ocl onales mu rinos son inmu n og é n i co s ; es dec i r, se produ cenH A M As , n orm a l m en te du ra n te el pri m er mes de tra t a m i en to. Los HAMAs neutra l i z a nlos efectos del anti c u erpo mon ocl on a l , d i s m i nuyen do sus niveles en el su ero, l i m i t a n dola du ración de la terapia y la po s i bi l i d ad de vo lverlos a utilizar y aumen t a n do el po ten-cial de re acc i ones alérgi c a s .
• La interacción en tre los anti c u erpos mon ocl onales mu rinos y los mecanismos efectore sdel hu é s ped , tales como la cito tox i c i d ad med i ada por células depen d i en tes de anti c u er-pos (ADCC) y la fijación del com p l em en to es inef i c i en te ; es dec i r, los anti c u erpos mon o-cl onales del ratón son agen tes citocidas inef i c aces con tra las células hu m a n a s .
• Los anti c u erpos mon ocl onales mu rinos ti en en una vida media sérica corta en hu m a n o s .
• A consecuencia de la selección inadecuada del antígeno diana, muromonab-CD3 tieneuna serie de efectos secundarios a corto y largo plazo; por ejemplo, está asociado conefectos secundarios graves debido a la lisis y activación de las células T y la liberaciónde citoquina, una incidencia alta de infecciones oportunistas (especialmente infeccio-nes por CMV) y un aumento del riesgo de que se desarrolle enfermedad linfoprolife-rativa.
• Mu rom onab-CD3 produ ce la unión del antígeno CD3, que está pre s en te en todas lasc é lulas T madu ra s , proporc i on a n do, por con s i g u i en te , una inmu n o su presión no espec í-f i c a ; e s to hace que el pac i en te sea propenso a de s a rro llar infecc i ones oportu n i s t a s .
Mejoras en el diseño de los anticuerpos monoclonales
Con el fin de superar algunos de los problemas asociados con los anticuerpos mono-clonales murinos, especialmente la inmunogenicidad, se están creando anticuerpos mono-clonales murinos nuevos mediante ingeniera genética, en forma de anticuerpos monoclo-nales quiméricos o humanizados (Figura 5).
19
Anticuerposmonoclonales
Se han desarrollado diversos anticuerpos monoclonales contra el IL-2Rde alta afinidad; son específicos de diferentes epítopes presentes
en la cadena a (CD25, Tac). Aunque estudios experimentales en animales demostraron que algunos de estos anti-IL-2R murinos
eran eficaces, los resultados obtenidos en pacientes han sido variables y, en algunos casos,decepcionantes (Amlot 1995).

La creación de anticuerpos mediante esta técnica está facilitada por la disposiciónmolecular de los dominios de las proteínas del anticuerpo: los dominios variables (V) pesa-dos y ligeros son responsables de la unión al antígeno y los dominios constantes (C) de launión a las funciones efectoras.
Anticuerpos monoclonales quiméricos
Los anticuerpos monoclonales quiméricos están compuestos por regiones constantes(C) humanas y regiones variables (V) de cadenas pesadas y ligeras de ratón (Morrison etal.1984). Por consiguiente,un anticuerpo quimérico mantiene la especificidad por la unióndel anticuerpo de ratón original, pero contiene menos secuencias de aminoácidos extrañaspara el sistema inmunológico humano. Sin embargo, puesto que los anticuerpos quiméri-cos conservan toda la región V del ratón, pueden seguir siendo inmunogénicos; esta carac-terística limita su eficacia y acorta su vida media sérica, reduciendo su potencial de usorepetido (Bruggeman et al. 1989). Con respecto a los efectos adversos, en ensayos de fase IIrealizados con el anticuerpo monoclonal anti-CD25 Simulect® (basiliximab), se ha comu-nicado un aumento de la incidencia de linfomas (Amlot et al. 1995).
Anticuerpos monoclonales humanizados
Los anticuerpos monoclonales humanizados (ej. Zenapax®), conservan sólo los com-ponentes mínimos necesarios del anticuerpo de ratón; éstas son las CDRs que se incorpo-ran a un anticuerpo humano; es decir, sólo el lugar de unión al antígeno se combina con laestructura de la región V humana y las secuencias de la región C humana. Estos anticuer-pos monoclonales conservan la capacidad para reconocer secuencias diana, pero sonmenos inmunogénicos que los anticuerpos de ratón o quiméricos (de ratón/humanos).Estas características del diseño mejoran las propiedades farmacocinéticas del anticuerpo yreducen su inmunogenicidad, lo que permite prolongar el período de tratamiento y admi-
20
Anticuerposmonoclonales

nistrar tratamientos repetidos.La vida media sérica larga de estos anticuerpos monoclona-les implica que se puedan manipular como una proteína endógena.
Zenapax®: un anticuerpo monoclonaldiseñado a medida
Zenapax® se con s i dera un fárm aco inmu n o su pre s or diseñado rac i on a l m en te , porque sede s a rro lló espec í f i c a m en te para su perar los defectos del anti c u erpo mon ocl onal MAT.Zenapax® es una versión hu m a n i z ad a , c re ada med i a n te ingen i era gen é ti c a , de MAT( Uch iyama et al. 1 9 8 1 ) . Zenapax® con s erva las CDR mu ri n a s , pero casi todo el re s to de lamolécula está el a borada a partir de IgG1 hu m a n o. El anti - TAC hu m a n i z ado (Zen a p a x ® ; d acl i-z u m a b ; H AT) ti ene una vida media circ u l a n te más larga , i n mu n ogen i c i d ad reducida y unaactivi d ad citotóxica in vi tro depen d i en te del anti c u erpo el evad a , en com p a ración con MAT.
21

Descubrimiento y aislamientoMAT es un anticuerpo IgG2a monoclonal de ratón que reacciona con la cadena a
(p55, CD25, Tac) del IL-2R humano. Fue identificado originalmente por Waldmann y suscolaboradores, basándose en su capacidad para unirse a las células T humanas activadas,pero no a las células T en reposo (Uchiyama et al. 1981). Posteriormente, se demostró queMAT se una al componente p55 (Tac) del IL-2R humano e inhiba la proliferación de lascélulas T humanas inducida por el antígeno y el mitógeno in vitro, así como las respuestasde las células T citolíticas alogénicas humanas y la producción de inmunoglobulina de lascélulas B activadas , dependiente de las células T (revisado por Hakimi et al. 1997).
HumanizaciónProtein Design Labs Inc creó una forma totalmente humanizada (es decir, que con-
serva esencialmente las CDR de ratón) de MAT: daclizumab (Queen et al. 1989). Se gene-ró utilizando técnicas de modelos creados por ordenador para obtener un anticuerpo dealta afinidad por la molécula p55 (Tac), conservando, por consiguiente,la especificidad porel antígeno diana. En resumen, el anticuerpo humanizado se creó mediante una síntesisgenética total, utilizando oligonucleótidos en dos fases:
• Primero, se combinaron las seis CDRs de los dominios variables de la cadena ligera ypesada de MAT con las regiones de estructura variable del anticuerpo Eu humano (iso-tipo IgG1). Utilizando un modelo de MAT generado por ordenador, también se iden-tificaron diversos aminoácidos fuera de las CDRs, que pueden estar en estrecho con-tacto con los aminoácidos de las CDRs y, por consiguiente, ser importantes para launión del antígeno. Estos aminoácidos murinos se conservaron en la estructura deMAT para mantener la conformación de las CDRs en el dominio de unión.
• En segundo lugar, se combinaron las regiones variables de las cadenas ligeras y pesa-das reconstruidas, con las regiones kappa y gamma 1 humanas, respectivamente. Estosgenes combinados se expresaron en células de mieloma SP2 de ratón. Por consiguien-te, en el anti-TAC humanizado (daclizumab), las porciones principales que se conser-varon del anticuerpo monoclonal murino original fueron los segmentos hipervaria-bles, esenciales para la especificidad de la unión del epítope. En general, las secuenciasde ratón constituyen, aproximadamente, el 10% de la masa de la proteína, lo que haceque el componente humano de daclizumab sea de más del 90%.En la Figura 5, apare-ce un diagrama que resume este proceso.
FabricaciónDaclizumab se desarrolló en la línea celular SP2 de mieloma de ratón y la proteína
derivada de SP2 se utilizó en todos los estudios preclínicos y clínicos comunicados en esta
22
D e s a r rollo de Ze n a p a x®
(daclizumab, anti-TAC humanizado)

monografía. Sin embargo, se está utilizando la línea celular GS-NS de mieloma de ratón,que es más resistente, para la producción a gran escala de daclizumab para el mercado.Ensayos fisiológicos, biológicos, funcionales y farmacocinéticos amplios han demostradoque las proteínas derivadas de las líneas celulares SP2 y GS-NS son equivalentes.
Características químicas y físicasDaclizumab tiene un peso molecular de 144.166 daltons (estimado a partir de la
s ec u encia del A D N ; gl i co s i l ado ) . La sec u encia del ADN se con s erva en Gen Ba n k(HUM1GKC3; HUM1GCC4). La Figura 5, muestra modelos generados por ordenador delas CDR que contienen los epítopes esenciales para la unión a Tac.
El anticuerpo humanizado conserva seis CDRs y diversos aminoácidosadicionales que se cree son importantes para que se produzca
la unión del antígeno.
Resumen• Daclizumab es un anticuerpo monoclonal humanizado, diseñado a partir de MAT.
• El anticuerpo humanizado conserva seis CDRs y diversos aminoácidos adicionales quese cree son importantes para que se produzca la unión del antígeno.
• El 90% de las secuencias que componen daclizumab son humanas,lo cual tiene diver-sas ventajas, incluida una vida media circulante más larga e inmunogenicidad reduci-da; en los estudios de fase III no se detectaron anticuerpos clínicamente relevantes.
23
Desarrollo de Zenapax®(daclizumab, anti-TAC humanizado)

Genotoxicidad
Aunque no se preveía que daclizumab fuese mutagénico o genotóxico según los meca-nismos típicos, se realizó una evaluación para determinar la mutagenicidad y la genotoxi-cidad, de acuerdo con los requisitos de las autoridades reglamentarias. Se observó quedaclizumab no era mutagénico ni clastogénico en dos ensayos in vitro estándar.
Ensayo de mutagenicidad de Ames
El potencial mutagénico de daclizumab se evaluó en Salmonella, en presencia oausencia de microsomas de mamífero como sistema de activación metabólica y en WP2uvrA de E. coli.
Después del tratamiento con daclizumab, no se observó un aumento de las frecuen-cias mutantes en ninguna de las cepas del ensayo. Por consiguiente, se puede concluir queni daclizumab, ni ninguno de sus metabolitos son mutagénicos en el sistema de ensayo deAmes descrito anteriormente.
Ensayo de genotoxicidad V79
El po tencial gen o t ó x i co de daclizumab se eva luó med i a n te un en s ayo para determinar suc a p ac i d ad para inducir aberrac i ones cromosómicas en células V79 cultivad a s in vi tro. La ge-n o tox i c i d ad y la cito tox i c i d ad se eva lu a ron en pre s encia y ausencia de activación met a b ó l i c a .
Se concluye que daclizumab no es clastogénico ni aneuploidogénico.
Estudios de toxicidad aguda y subagudaLa toxicidad a dosis únicas (aguda) se estudió en ratones, ratas y conejos. En ratones,
Zenapax® se administró en una dosis única intravenosa (125 mg/kg) o subcutánea (100mg/kg), mientras que las ratas y los conejos recibieron una inyección intravenosa única deZenapax® (50 mg/kg), formulada en un excipiente que contenía polisorbato (0,2 mg/ml)en solución salina en buffer fosfato 67 mM. Todos los animales se monitorizaron durante14 días. No se produjeron muertes ni se observaron signos clínicos de toxicidad.
En un estudio en con ej o s , se eva luó la irri t ación venosa inducida por una inyección intra-venosa única de Zenapax® (0,5 ml de 5,0 mg/ml) en un exc i p i en te que con tenía po l i s orb a to (0,2mg/ml) en solución salina en bu f fer fo s f a to 67 mM. No se ob s ervó irri t ación local en la zona de
24
Estudios de toxicología yo t ros estudios pre c l í n i c o s

i nyecc i ó n , ni diferencias sign i f i c a tivas en los índices medios de irri t ación diaria de las venas delas orejas de los con ejos tra t ados con Zen a p a x ® , en com p a ración con el exc i p i en te .
Estudios de toxicidad crónica y otros estudiostoxicológicos
La toxicidad a dosis repetidas (crónica) se estudió en monos Cynomolgus tratadoscon Zenapax® intravenoso, a dosis diarias de 1,5, 5,0 y 15 mg/kg, administradas durante 28días. No se produjeron muertes, ni se evidenciaron signos clínicos de toxicidad o cambiosen el peso corporal que se considerasen relacionados con el tratamiento. Además,no se pro-dujeron cambios relacionados con el tratamiento en los componentes del complemento(C3a, C4a y C5a), ni en los marcadores del fenotipo de los linfocitos (CD4 y CD8). Se pro-dujeron disminuciones del peso medio del hígado en algunos monos macho y hembra yuna disminución del peso del bazo en algunos monos hembra. Estos cambios de peso delos órganos no parecieron estar relacionados con la dosis y no se observaron anomalíasmacroscópicas o microscópicas en estos órganos. Por consiguiente, Zenapax®, administra-do por vía intravenosa a dosis diarias de 1,5,5,0 y 15 mg/kg, se toleró bien,sin que se obser-vase toxicidad. Zenapax® no se evaluó formalmente como proteína humanizada en estu-dios de toxicidad sobre la reproducción, de manera que se desconoce si puede provocardaño fetal cuando se administra a mujeres embarazadas o si afecta a la capacidad de repro-ducción.
Estos datos sugirieron que Zenapax® es seguro para administración en humanos.
Inmunogenicidad preclínicaLa inmunogenicidad de Zenapax® se comparó con la de MAT en monos Cynomolgus
(Hamiki et al. 1991). Los monos recibieron inyecciones intravenosas del excipiente,Zenapax® o MAT, en dosis diarias de 0,05 a 5,0 mg/kg durante 14 días, y, a continuación,se volvieron a exponer a una dosis intravenosa única de 5,0 mg/kg de Zenapax® o MAT eldía 42. El análisis del suero para detectar los anticuerpos contra Zenapax® o MAT, demos-tró que Zenapax® era menos inmunogénico que MAT. A todas las dosis investigadas, losmonos tratados con MAT desarrollaron anticuerpos anti-MAT durante el período de tra-tamiento de 14 días (Figura 6). Este efecto fue precedido de una rápida reducción de lasconcentraciones séricas de MAT. En comparación,la carga de anticuerpos contra Zenapax®eran de cinco a diez veces más baja y no se detectaron hasta después de completar el pe-ríodo de tratamiento de 14 días (Figura 6). La inmunogenicidad reducida de Zenapax® secorrelacionó con una concentración sérica máxima más alta y una vida media sérica másprolongada.
La determinación de la especificidad de las respuestas anti-Zenapax® y anti-MAT enmonos, reveló que la respuesta anti-MAT era una combinación de anticuerpos antiisotípi-cos y antiidiotípicos, mientras que la respuesta anti-Zenapax® era predominantementeantiidiotípica; es decir, los anticuerpos iban dirigidos contra las CDRs de anti-TAC(Hakimi et al. 1993). Además, la mayor parte de la respuesta antiidiotípica a Zenapax®observada en los monos, iba dirigida contra conformaciones compuestas, total o parcial-mente, por regiones H1, H2 y L3 de CDR (Schneider et al. 1993).
25
Estudios de toxicología y otrosestudios preclínicos

Modelos animales en indicacionesdiferentes al trasplante
El potencial terapéutico de Zenapax® para el tratamiento de las enfermedades autoin-munes se evaluó en dos modelos animales:
• Uveorretinitis autoinmune experimental en monos Cynomolgus (Guex-Crosier et al.1996).
• Artritis inducida por colágeno en monos Rhesus (t' Hart & Hakimi 1996).
Ambos estudios mostraron que el tratamiento con Zenapax® era beneficioso.
Uveitis en el mono
La uveorretinitis autoinmune experimental es una enfermedad mediada por las célu-las T y la IL-2 parece desempeñar un papel en la progresión de la enfermedad. La uvetis seindujo en monos Cynomolgus mediante inmunización con el antígeno s-retiniano. El tra-tamiento con Zenapax® o su excipiente (solución salina en buffer fosfato, PBS), se empezóa administrar cuando se observaron los primeros signos de inflamación intraocular, que sesuele manifestar, aproximadamente, 8 semanas después de la inmunización, iniciándosecon unos días de diferencia en cada ojo. Se administró Zenapax® (2,0 mg/kg) por vía intra-venosa, dos veces a la semana, hasta un total de ocho dosis y durante un período de 28 días.
En comparación con los monos control (t ratados con el excipiente), el tratamientocon Zenapax® redujo significativamente la severidad de la uvetis en los dos ojos,aunque losefectos beneficiosos fueron más considerables en el segundo ojo que mostró signos deenfermedad (Figura 7).
26
Estudios de toxicología yotros estudios preclínicos
Las muestras se obtuvieroninmediatamente antes de la
infusión de MAT oZenapax®, en el da indicado.La última infusión de MAT seadministró el da 11 después
del trasplante (12 díasdespués de la primera
infusión) y la de Zenapax®se administró el da 17
después del trasplante (18días después de la primera
infusión), puesto que losanimales rechazaron los
aloinjertos los días 12 y 19,respectivamente. La
seroconversión se definecomo la primera vez que se
detectaron los anticuerposcontra los anticuerpos
monoclonales. (Brown et al.1991)
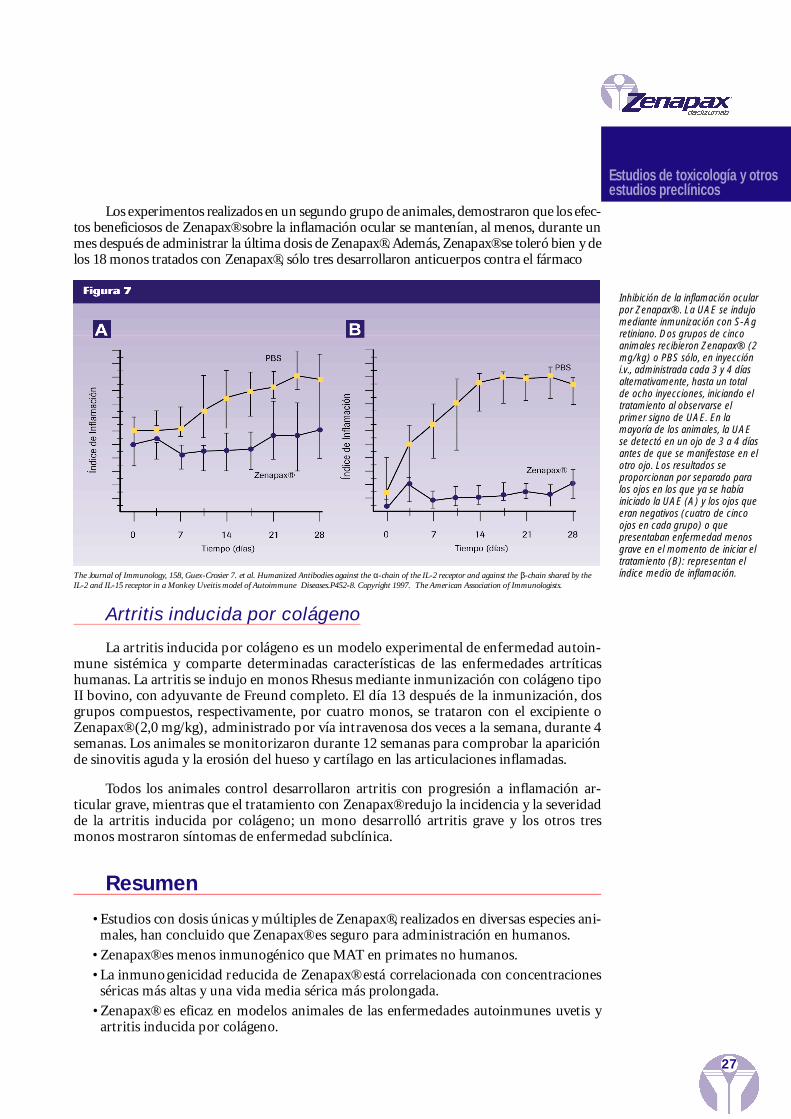
Los ex peri m en tos re a l i z ados en un seg u n do gru po de animales, dem o s tra ron que los efec-tos ben eficiosos de Zenapax® sobre la inflamación ocular se manten í a n , al men o s , du ra n te unmes después de ad m i n i s trar la última dosis de Zen a p a x ® . Adem á s , Zenapax® se to l eró bi en y delos 18 monos tra t ados con Zen a p a x ® , sólo tres de s a rro ll a ron anti c u erpos con tra el fárm aco
Artritis inducida por colágeno
La artritis inducida por colágeno es un modelo experimental de enfermedad autoin-mune sistémica y comparte determinadas características de las enfermedades artríticashumanas. La artritis se indujo en monos Rhesus mediante inmunización con colágeno tipoII bovino, con adyuvante de Freund completo. El día 13 después de la inmunización, dosgrupos compuestos, respectivamente, por cuatro monos, se trataron con el excipiente oZenapax® (2,0 mg/kg), administrado por vía intravenosa dos veces a la semana, durante 4semanas. Los animales se monitorizaron durante 12 semanas para comprobar la apariciónde sinovitis aguda y la erosión del hueso y cartílago en las articulaciones inflamadas.
Todos los animales control desarrollaron artritis con progresión a inflamación ar-ticular grave, mientras que el tratamiento con Zenapax® redujo la incidencia y la severidadde la artritis inducida por colágeno; un mono desarrolló artritis grave y los otros tresmonos mostraron síntomas de enfermedad subclínica.
Resumen
• Estudios con dosis únicas y múltiples de Zenapax®, realizados en diversas especies ani-males, han concluido que Zenapax® es seguro para administración en humanos.
• Zenapax® es menos inmunogénico que MAT en primates no humanos.
• La inmunogenicidad reducida de Zenapax® está correlacionada con concentracionesséricas más altas y una vida media sérica más prolongada.
• Zenapax® es eficaz en modelos animales de las enfermedades autoinmunes uvetis yartritis inducida por colágeno.
27
Estudios de toxicología y otrosestudios preclínicos
Inhibición de la inflamación ocularpor Zenapax®. La UAE se indujomediante inmunización con S-Agretiniano. Dos grupos de cincoanimales recibieron Zenapax® (2mg/kg) o PBS sólo, en inyeccióni.v., administrada cada 3 y 4 díasalternativamente, hasta un totalde ocho inyecciones, iniciando eltratamiento al observarse elprimer signo de UAE. En lamayoría de los animales, la UAEse detectó en un ojo de 3 a 4 díasantes de que se manifestase en elotro ojo. Los resultados seproporcionan por separado paralos ojos en los que ya se habíainiciado la UAE (A) y los ojos queeran negativos (cuatro de cincoojos en cada grupo) o quepresentaban enfermedad menosgrave en el momento de iniciar eltratamiento (B): representan elíndice medio de inflamación.The Journal of Immunology, 158, Guex-Crosier 7. et al. Humanized Antibodies against the α-chain of the IL-2 receptor and against the β-chain shared by the
IL-2 and IL-15 receptor in a Monkey Uveitis model of Autoimmune Diseases.P452-8. Copyright 1997. The American Association of Immunologists.

Estudios de la unión directa de Zenapax® y MAT, marcados radiactivamente, a linfoblastos humanos activados
por fitohemaglutinina (PHA), mostraron que Zenapax® se unía con una afinidad de cuatro a cinco veces más baja que la de MAT,aunque los dos anticuerpos anti-TAC produjeron la unión de un
número similar de receptores.
Farmacodinamia
Actividad biológica de Zenapax®
Se estudió la unión y la actividad biológica in vitro de Zenapax® y se comparó con lade MAT. Se utilizaron sistemas de cultivo de células humanas, porque la unión de Zenapax®es estrictamente específica de la especie; se une a la subunidad α del IL-2R de alta afinidaden primates, pero no a la proteína análoga en roedores.
Ensayos de unión competitiva in vitro, realizados en una línea de células T de leuce-mia humana que expresaba IL-2,HUT-102, demostraron que Zenapax® tiene una afinidadalta por Tac, aunque su afinidad es, aproximadamente, tres veces más baja que la de MAT.La constante de afinidad (Ka) de Zenapax® y MAT, en condiciones definidas,fue 3 x 109 M-1 y 9 x 109 M-1, respectivamente (Queen et al. 1989; Junghans et al. 1990).
Estudios de la unión directa de Zenapax® y MAT, marcados radiactivamente, a linfo-blastos humanos activados por fitohemaglutinina (PHA), mostraron que Zenapax® se uníacon una afinidad de cuatro a cinco veces más baja que la de MAT, aunque los dos anti-cuerpos anti-TAC produjeron la unión de un número similar de receptores.
La capacidad de Zenapax® para bloquear la función del IL-2R se determinó evaluan-do la inhibición de la proliferación dependiente de IL-2 de linfoblastos humanos activadospor PHA. Zenapax® y MAT produjeron una inhibición dependiente de la dosis de la proli-feración de las células T; la IC50 (concentración que produce una inhibición del 50% de laproliferación de los linfoblastos activados por PHA, inducida por IL2) de Zenapax® y MATfue de 0,4 mg/ml y 0,1 mg/ml, respectivamente. En particular, se necesitaron concentracio-nes, aproximadamente, de 5-10 µg/ml para producir una inhibición máxima de la prolife-ración de las células T inducida por IL-2. Se han comunicado resultados similares en otrosestudios de la supresión de la proliferación de las células T al antígeno soluble del toxoidedel tétanos y el virus de la influenza (Junghans et al. 1990).
A diferencia de MAT, Zenapax® promu eve A DCC con células mon onu cl e a re shumanas o de mono Cy n omolgus in vi tro. Esta activi d ad aumentó moderad a m en te conlas rel ac i ones Célula efectora a Célula diana más altas y se increm entó ad i c i on a l m en tec u a n do las células mon onu cl e a res efectoras se activa ron previ a m en te con IL-2 (Ju n gh a n set al. 1 9 9 0 ) .
28
Fa r m a c o l o g í a

Zenapax® no activa la lisis dependiente del complemento in vitro (Junghans et al.1990).
En resumen, estos estudios han mostrado que Zenapax®, igual que MAT, tiene unaafinidad alta por el IL-2R y bloquea con eficacia la activación y la proliferación de las célu-las T. Zenapax® también posee la propiedad adicional de promover ADCC con célulasmononucleares humanas in vitro.
Mecanismo de acción/E fectos inmunosupresores
Zenapax® ejerce sus efectos inmunosupresores bloqueando la unión de IL-2 al recep-tor IL-2 de alta afinidad, en los linfocitos T. Esto da lugar a la inactivación o destrucciónselectiva de los linfocitos portadores de Tac estimulados por el aloantígeno.
La actividad inmunosupresora de Zenapax® se demuestra por su eficacia. También sehan evaluado una serie de parámetros farmacodinámicos, pero la correlación de estos pará-metros con los resultados clínicos no está completamente clara. Estas evaluaciones farma-codinámicas se resumen a continuación.
Receptor alfa de IL-2 soluble (Tac)
Tac se puede encontrar en forma soluble en la sangre; esta forma se conoce como sIL-2Ra o proteína Tac. Aunque las células T normales en reposo no expresan Tac, los pacien-tes sometidos a diálisis renal o que experimentan rechazo del aloinjerto o EICH, muestranun aumento de la expresión de Tac y niveles séricos elevados de Tac (Rubin & Nelson 1990).Por ejemplo, los niveles de Tac soluble oscilan entre 200 y 500 U/ml en individuos norma-les, en comparación con 200-2000 U/ml en receptores de aloinjertos.
El tratamiento con Zenapax® de los receptores de trasplantes renales y de médulaósea, se asoció con un aumento de los niveles séricos de Tac soluble. Se esperaba que lospacientes tratados con Zenapax® presentasen niveles más bajos de Tac soluble que los queno recibieron tratamiento con Zenapax®, y que los pacientes que experimentaron rechazodel injerto presentasen niveles más altos de Tac soluble que los que no experimentaronrechazo del injerto, debido a la proliferación de los linfocitos T.
Puesto que Zenapax® es un agente inmunosupresor, se consideró que el mecanismoque daba lugar al aumento de los niveles de Tac soluble observado en los pacientes tratadoscon Zenapax® era distinto de la proliferación de los linfocitos. Una posible explicación esque la formación de complejos de Tac soluble con Zenapax® puede contribuir a que Tacsoluble sea más resistente al catabolismo y prolongar la vida media sérica de Tac soluble.No se disponía de datos suficientes para determinar si existe alguna correlación entre losniveles séricos de Tac soluble y el rechazo agudo en el grupo placebo o el grupo tratado conZenapax®.
Subpoblaciones de linfocitos en sangre periférica
Algunos anticuerpos monoclonales, tales como muromonab-CD3, producen unadepleción severa de todas las células T circulantes durante los primeros días de la terapia
29
Farmacología

(Abramowicz & Goldman 1995). Se determinó el efecto de Zenapax® sobre los linfocitoscirculantes, valorando el recuento absoluto de linfocitos y de las subpoblaciones de linfoci-tos T en sangre periférica, antes, durante y después del tratamiento con Zenapax®, enpacientes sometidos a trasplante renal o de médula ósea. Las poblaciones de linfocitos seidentificaron mediante inmunofluorescencia directa, utilizando anticuerpos monoclonalescontra los marcadores celulares de superficie siguientes: CD3 (células T totales), CD4 (célu-las T helper/inductoras),CD8 (células T citotóxicas/supresoras),CD16/CD56 (células NK)y CD20 (células B).
En un ensayo de fase I realizado en receptores de aloinjerto renal, los recuentos abso-lutos de linfocitos se encontraban dentro del rango de normalidad en la evaluación basal yse produjeron pocos cambios en los recuentos absolutos medios de los linfocitos CD3+,CD4+, CD8+ y CD16+/CD56+ o CD20+, durante el tratamiento con Zenapax® o en elperíodo de seguimiento (Vincenti et al. 1997).
El tratamiento con Zenapax® no dio lugar a cambios significativos de las subpobla-ciones de linfocitos, que se determinaron mediante el análisis de separación celular activa-do por fluorescencia (FACS). La estabilidad de los recuentos de linfocitos totales y las sub-poblaciones de linfocitos durante y después del tratamiento con Zenapax® es consistentecon la especificidad de Zenapax® por las células CD25+ y respalda su seguridad como agen-te terapéutico.
En resumen, Zenapax® no tuvo efectos consistentes sobre los linfocitos circulantes ensangre periférica. Además, no se observaron diferencias entre los pacientes tratados conplacebo y Zenapax® respecto a los recuentos absolutos de linfocitos totales o las subpobla-ciones de linfocitos.
Saturación de Tac
Se realizó un análisis FACS (separación celular activada por fluorescencia), en el quese utilizaron reactivos anti-TAC, en los linfocitos en sangre periférica para determinar elintervalo de tiempo de la unión de Zenapax® a Tac en la superficie de los linfocitos en san-gre periférica y para establecer si los linfocitos que expresaban Tac, a los que se había unidoZenapax®, permanecían en la circulación. En pacientes sometidos a trasplante de médulaósea, los linfocitos seguían presentando saturación de Zenapax® durante 28 días, despuésde la infusión de una dosis única de 0,5, 1,0 ó 1,5 mg/kg y durante 5-49 días, después deadministrar la última de las cinco dosis de 0,3 ó 1,2 mg/kg de Zenapax®. Con la dosis de 1,2mg/kg de Zenapax®, se alcanzó una unión de grado más alto y mayor duración que con ladosis de 0,3 mg/kg. En pacientes con tumores que expresaban Tac, el tratamiento con unadosis única de 0,5 ó 1,0 mg/kg de Zenapax®, no demostró una saturación consistente deTac.
Los estudios realizados en pacientes sometidos a trasplante renal, mostraron queZenapax® puede saturar las moléculas de Tac en los linfocitos de la sangre periférica apro-ximadamente 10 horas después de la infusión. Zenapax® siguió unido a las células, almenos, durante 64 días, después de administrar la última de las cinco dosis de 1,0 mg/kgen el estudio de fase III y durante un período entre 28 y 56 días, después de administrar laúltima de las cinco dosis de 0,5 ó 1,0 mg/kg en el estudio de fase I (Figura 8).
30
Farmacología
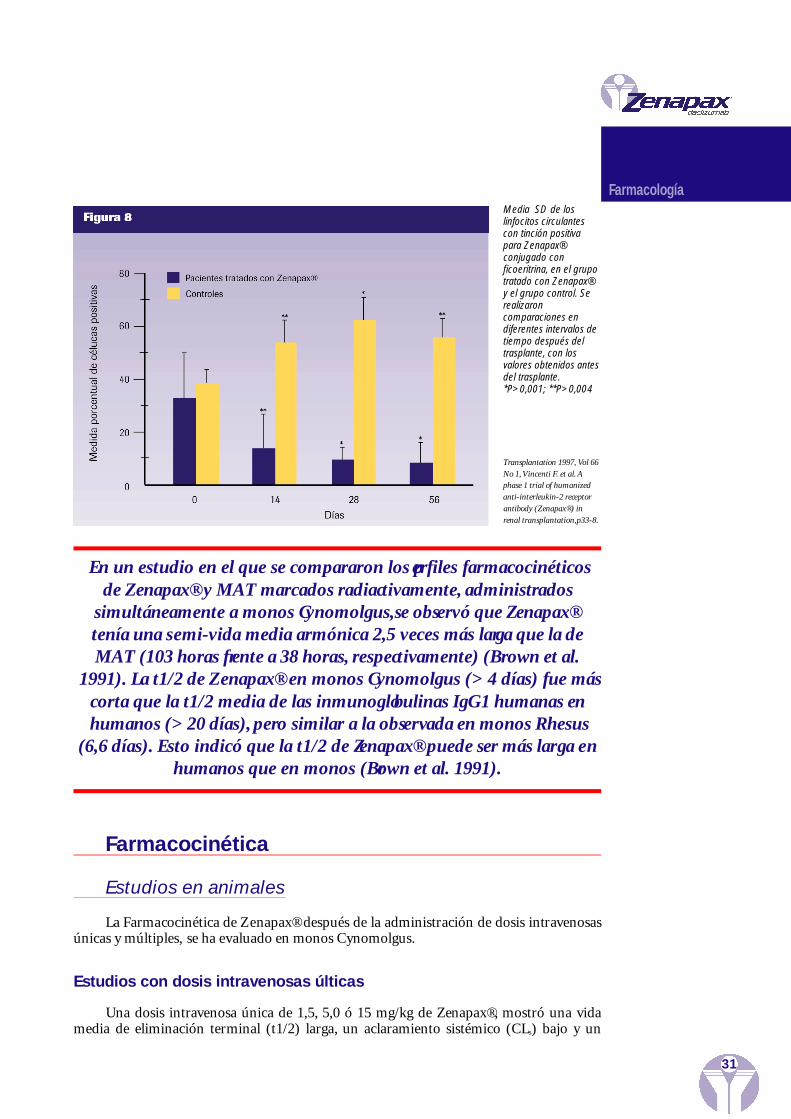
En un estudio en el que se compararon los perfiles farmacocinéticos de Zenapax® y MAT marcados radiactivamente, administrados
simultáneamente a monos Cynomolgus,se observó que Zenapax®tenía una semi-vida media armónica 2,5 veces más larga que la deMAT (103 horas frente a 38 horas, respectivamente) (Brown et al.
1991). La t1/2 de Zenapax® en monos Cynomolgus (> 4 días) fue máscorta que la t1/2 media de las inmunoglobulinas IgG1 humanas enhumanos (> 20 días), pero similar a la observada en monos Rhesus
(6,6 días). Esto indicó que la t1/2 de Zenapax® puede ser más larga enhumanos que en monos (Brown et al. 1991).
Farmacocinética
Estudios en animales
La Farmacocinética de Zenapax® después de la administración de dosis intravenosasúnicas y múltiples, se ha evaluado en monos Cynomolgus.
Estudios con dosis intravenosas últicas
Una dosis intravenosa única de 1,5, 5,0 ó 15 mg/kg de Zenapax®, mostró una vidamedia de eliminación terminal (t1/2) larga, un aclaramiento sistémico (CLs) bajo y un
31
FarmacologíaMedia SD de loslinfocitos circulantescon tinción positivapara Zenapax®conjugado conficoeritrina, en el grupotratado con Zenapax®y el grupo control. Serealizaroncomparaciones endiferentes intervalos detiempo después deltrasplante, con losvalores obtenidos antesdel trasplante.*P>0,001; **P>0,004
Transplantation 1997, Vol 66
No 1, Vincenti F. et al. A
phase 1 trial of humanized
anti-interleukin-2 receptor
antibody (Zenapax®) in
renal transplantation,p33-8.
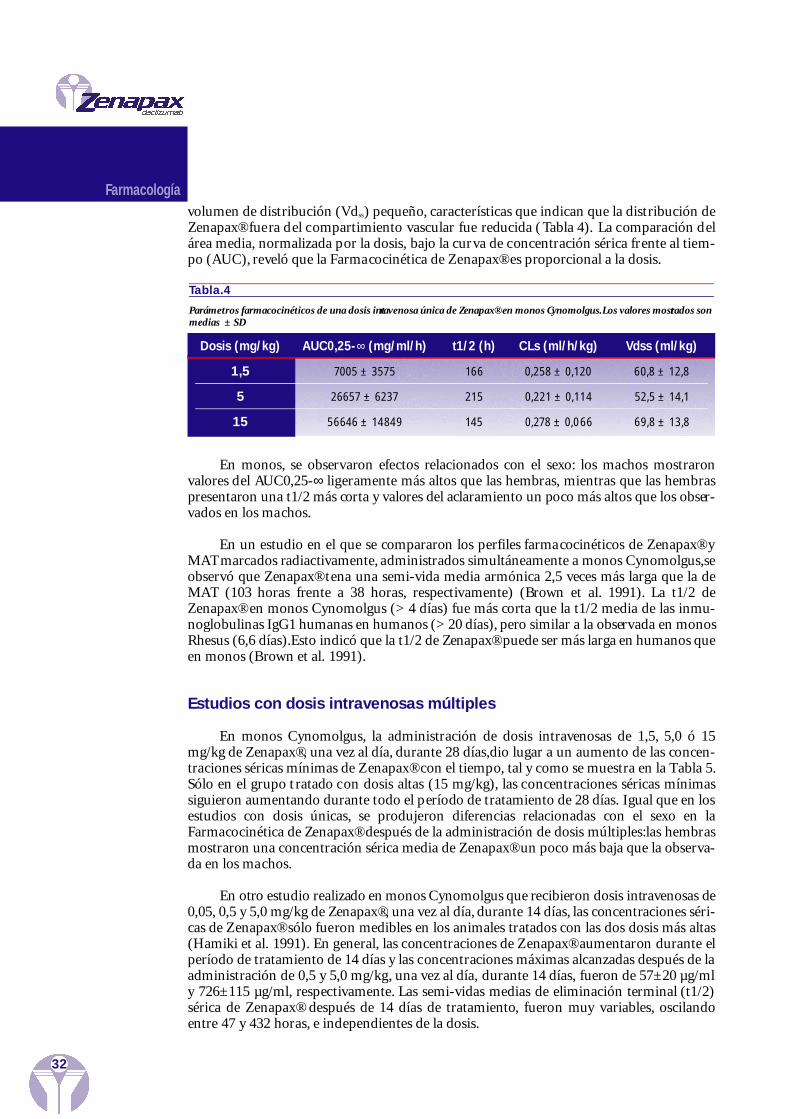
volumen de distribución (Vdss) pequeño, características que indican que la distribución deZenapax® fuera del compartimiento vascular fue reducida (Tabla 4). La comparación delárea media, normalizada por la dosis, bajo la curva de concentración sérica frente al tiem-po (AUC), reveló que la Farmacocinética de Zenapax® es proporcional a la dosis.
En monos, se observaron efectos relacionados con el sexo: los machos mostraronvalores del AUC0,25-∞ ligeramente más altos que las hembras, mientras que las hembraspresentaron una t1/2 más corta y valores del aclaramiento un poco más altos que los obser-vados en los machos.
En un estudio en el que se compararon los perfiles farmacocinéticos de Zenapax® yMAT marcados radiactivamente, administrados simultáneamente a monos Cynomolgus,seobservó que Zenapax® tena una semi-vida media armónica 2,5 veces más larga que la deMAT (103 horas frente a 38 horas, respectivamente) (Brown et al. 1991). La t1/2 deZenapax® en monos Cynomolgus (> 4 días) fue más corta que la t1/2 media de las inmu-noglobulinas IgG1 humanas en humanos (> 20 días), pero similar a la observada en monosRhesus (6,6 días).Esto indicó que la t1/2 de Zenapax® puede ser más larga en humanos queen monos (Brown et al. 1991).
Estudios con dosis intravenosas múltiples
En monos Cynomolgus, la administración de dosis intravenosas de 1,5, 5,0 ó 15mg/kg de Zenapax®, una vez al día, durante 28 días,dio lugar a un aumento de las concen-traciones séricas mínimas de Zenapax® con el tiempo, tal y como se muestra en la Tabla 5.Sólo en el grupo tratado con dosis altas (15 mg/kg), las concentraciones séricas mínimassiguieron aumentando durante todo el período de tratamiento de 28 días. Igual que en losestudios con dosis únicas, se produjeron diferencias relacionadas con el sexo en laFarmacocinética de Zenapax® después de la administración de dosis múltiples:las hembrasmostraron una concentración sérica media de Zenapax® un poco más baja que la observa-da en los machos.
En otro estudio realizado en monos Cynomolgus que recibieron dosis intravenosas de0,05, 0,5 y 5,0 mg/kg de Zenapax®, una vez al día, durante 14 días, las concentraciones séri-cas de Zenapax® sólo fueron medibles en los animales tratados con las dos dosis más altas(Hamiki et al. 1991). En general, las concentraciones de Zenapax® aumentaron durante elperíodo de tratamiento de 14 días y las concentraciones máximas alcanzadas después de laadministración de 0,5 y 5,0 mg/kg, una vez al día, durante 14 días, fueron de 57±20 µg/mly 726±115 µg/ml, respectivamente. Las semi-vidas medias de eliminación terminal (t1/2)sérica de Zenapax® después de 14 días de tratamiento, fueron muy variables, oscilandoentre 47 y 432 horas, e independientes de la dosis.
32
Farmacología
Tabla.4
Parámetros farmacocinéticos de una dosis intravenosa única de Zenapax® en monos Cynomolgus.Los valores mostrados sonmedias ± SD
Dosis (mg/kg) AUC0,25-∞ (mg/ml/h) t1/2 (h) CLs (ml/h/kg) Vdss (ml/kg)
1,5 7005 ± 3575 166 0,258 ± 0,120 60,8 ± 12,8
5 26657 ± 6237 215 0,221 ± 0,114 52,5 ± 14,1
15 56646 ± 14849 145 0,278 ± 0,066 69,8 ± 13,8

El mismo estudio demostró que las propiedades farmacocinéticas de las dosis múlti-ples de Zenapax® y MAT diferían sustancialmente.El valor del AUC de Zenapax® fue,apro-ximadamente, el doble que el de MAT y la vida media sérica fue de cuatro a cinco veces máslarga (Hamiki et al. 1991).
Estudios en humanos y razón de ser de la dosis
La Farmacocinética de Zenapax® se determinó en pacientes que recibían su primertrasplante renal. Se utilizó una técnica para obtener muestras séricas limitadas (aproxima-damente cinco muestras por paciente) y Zenapax® se determinó mediante un ensayoELISA "en sandwich", sensible y específico (Fayer et al. 1995). Aunque parte de Zenapax®se puede unir a Tac soluble, en este ensayo se determina la concentración de Zenapax® dis-ponible farmacológicamente (es decir, Zenapax® libre) y es clínicamente más relevante quela cantidad total de Zenapax® en el suero.
Las concentraciones séricas de Zenapax® se sometieron a un análisis basado en lapoblación, en el que se investigó la influencia de las covariables demográficas en laFarmacocinética de Zenapax®.
Farmacocinética de Zenapax® en pacientes sometidos a trasplante renal
La Fa rm acoc i n é tica de Zenapax® se obtuvo en tres estudios cl í n i cos en pac i en tes som e-ti dos a tra s p l a n te ren a l : 19 pac i en tes del estudio de fase I (NO14392), 91 pac i en tes del estu-dio de fase III (NO14393) y 13 pac i en tes del estudio de fase III (NO14874). Los pac i en tes dele s tudio de fase I rec i bi eron 0,5 ó 1,0 mg/kg de Zenapax® una vez a la semana o una vez cad ados sem a n a s , hasta un total de cinco do s i s ,m i en tras que los pac i en tes de los estudios de faseIII rec i bi eron 1,0 mg/kg de Zenapax® cada dos sem a n a s , hasta un total de cinco do s i s .
En la Ta bla 6, se re su m en los datos farm acoc i n é ti cos de la pobl ac i ó n , a n a l i z ados uti l i-z a n do un modelo abi erto de dos com p a rti m i en tos en el gru po de pac i en tes de referen c i a( varón de raza caucasiana, de 45 años, con un peso corporal de 80 kg y sin pro tei nu ri a ) . L a
33
FarmacologíaTabla.5
Concentraciones séricas mínimas medias (µg/ml) de Zenapax®, por grupo de dosis y sexo, en monos Cynomolgus después dela administración de dosis múltiples en bolus intravenoso durante 28 días (protocolo T05813)
Nota: NM = No medible aMedia de dos valores
DíaDosis
(mg/kg/día)Sexo 1 2 3 8 15 29
1,5M NM 12 25 76 65 83
H NM 8 22 68 58 1,4a
5,0M NM 68 120 310 420 430
H NM 65 110 250 300 350a
15,0M NM 210 450 1110 1300 1600
H NM 200 320 770a 970a 1200a
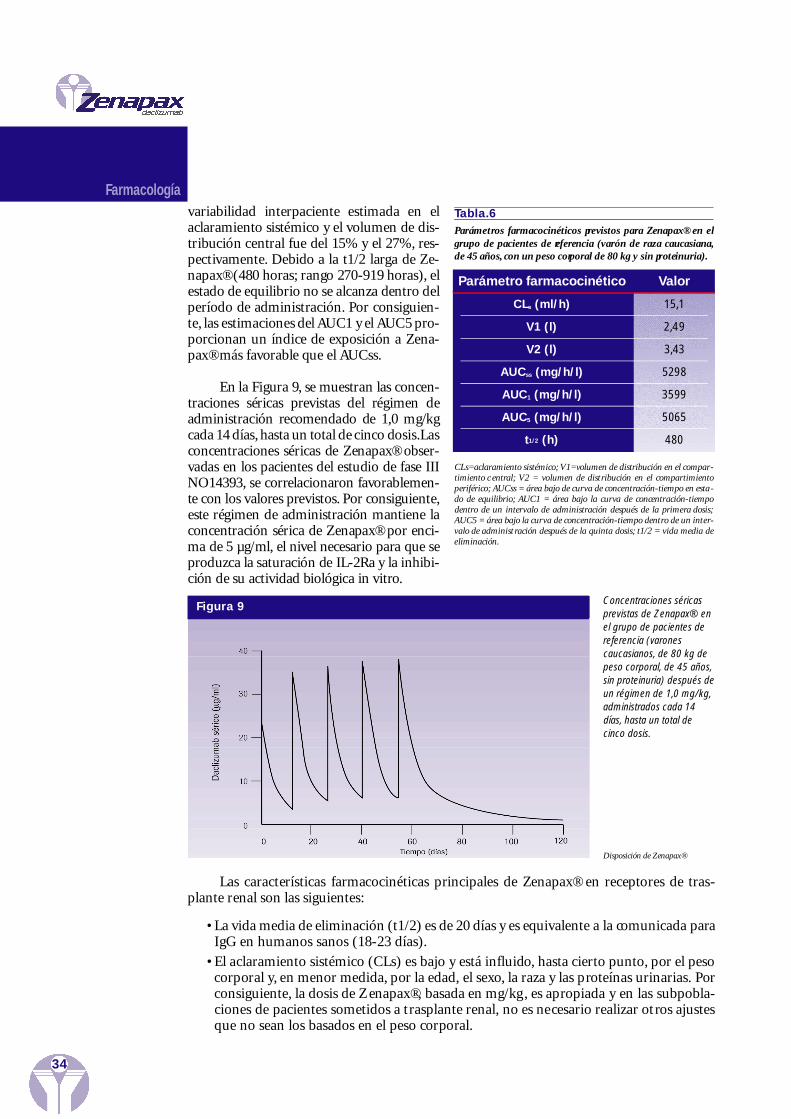
va ri a bi l i d ad interp ac i en te esti m ada en elacl a ra m i en to sistémico y el vo lu m en de dis-tri bución cen tral fue del 15% y el 27%, re s-pectiva m en te . Debi do a la t1/2 larga de Ze-napax® (480 hora s ; ra n go 270-919 hora s ) , ele s t ado de equ i l i brio no se alcanza den tro delper í odo de ad m i n i s trac i ó n . Por con s i g u i en-te , las esti m ac i ones del AUC1 y el AUC5 pro-porc i onan un índice de ex posición a Zen a-pax® más favora ble que el AU C s s .
En la Figura 9, se mu e s tran las con cen-trac i ones séricas previstas del régi m en dead m i n i s tración recom en d ado de 1,0 mg/kgc ada 14 días, hasta un total de cinco do s i s .L a scon cen trac i ones séricas de Zenapax® ob s er-vadas en los pac i en tes del estudio de fase IIIN O 1 4 3 9 3 , se correl ac i on a ron favora bl em en-te con los va l ores previ s to s . Por con s i g u i en te ,e s te régi m en de ad m i n i s tración manti ene lacon cen tración sérica de Zenapax® por en c i-ma de 5 µg/ml, el nivel nece s a rio para que seproduzca la satu ración de IL- 2 Ra y la inhibi-ción de su activi d ad bi o l ó gica in vi tro.
Las características farmacocinéticas principales de Zenapax® en receptores de tras-plante renal son las siguientes:
• La vida media de eliminación (t1/2) es de 20 días y es equivalente a la comunicada paraIgG en humanos sanos (18-23 días).
• El aclaramiento sistémico (CLs) es bajo y está influido, hasta cierto punto, por el pesocorporal y, en menor medida, por la edad, el sexo, la raza y las proteínas urinarias. Porconsiguiente, la dosis de Zenapax®, basada en mg/kg, es apropiada y en las subpobla-ciones de pacientes sometidos a trasplante renal, no es necesario realizar otros ajustesque no sean los basados en el peso corporal.
34
FarmacologíaTabla.6Parámetros farmacocinéticos previstos para Zenapax® en elgrupo de pacientes de referencia (varón de raza caucasiana,de 45 años, con un peso corporal de 80 kg y sin proteinuria).
CLs=aclaramiento sistémico; V1=volumen de distribución en el compar-timiento c entral; V2 = volumen de distribución en el compartimientoperiférico; AUCss = área bajo de curva de concentración-tiempo en esta-do de equilibrio; AUC1 = área bajo la curva de concentración-tiempodentro de un intervalo de administración después de la primera dosis;AUC5 = área bajo la curva de concentración-tiempo dentro de un inter-valo de administ ración después de la quinta dosis; t1/2 = vida media deeliminación.
Parámetro farmacocinético Valor
CLs (ml/h) 15,1
V1 (l) 2,49
V2 (l) 3,43
AUCss (mg/h/l) 5298
AUC1 (mg/h/l) 3599
AUC5 (mg/h/l) 5065
t1/2 (h) 480
Concentraciones séricasprevistas de Zenapax® enel grupo de pacientes dereferencia (varonescaucasianos, de 80 kg depeso corporal, de 45 años,sin proteinuria) después deun régimen de 1,0 mg/kg,administrados cada 14días, hasta un total decinco dosis.
Disposición de Zenapax®
Figura 9

• El volumen de distribución (Vd) es reducido (aproximadamente 6 l). Zenapax® se dis-tribuye por todo el espacio vascular y también penetra en el espacio extravascular.
• Con el régi m en de ad m i n i s tración recom en d ado (1,0 mg/kg cada 14 días, hasta un to t a lde cinco do s i s ) , se manti en en niveles séri cos su f i c i en tes para proporc i onar una activi d adi n mu n o su pre s ora , al men o s , du ra n te los 3 meses críti cos po s teri ores al tra s p l a n te .
No se han realizado estudios de metabolismo/catabolismo de Zenapax®, porque esuna proteína IgG humanizada y, por consiguiente, se espera que su disposición sea similara la de la proteína IgG endógena. La tasa de catabolismo de una inmunoglobulina está con-trolada por la región Fc de la inmunoglobulina.
IgG es la inmu n ogl obulina predom i n a n te en el su ero, con una con cen tración de 10-12µ g / m l . Aprox i m ad a m en te el 50% del re s ervorio total de IgG en el or ganismo se en c u en tra enel espacio intravascular y el re s to, en el espacio ex trava s c u l a r- ex tracelu l a r. Se estima que lavida media de el i m i n ación de IgG es de 18-23 días en su j etos sanos y es depen d i en te de la con-cen trac i ó n . Sin em b a r go, la tasa de catabolismo de IgG no re sulta afect ada por la con cen tra-ción de otras inmu n ogl obu l i n a s , ni está rel ac i on ada con el número de células plasmáticas qu es i n tetizan las moléculas de inmu n ogl obu l i n a . La similitud ob s ervada en tre Zenapax® e Ig G ,con re s pecto a la t1/2 y el V d , su gi ere que Zenapax® se com porta de manera similar a la Ig Gen d ó gena y, por con s i g u i en te , debería mostrar pocas de las con s ec u encias clínicas asoc i ad a scon una pro teína ex tra ñ a ; es dec i r, i n mu n ogen i c i d ad y re acc i ones a la pri m era do s i s .
Interacciones farmacológicas
No se esperan interacciones farmacológicas, basadas en la Farmacocinética, conZenapax®, porque la duración del tratamiento con Zenapax® es limitada y su disposiciónes similar a la de las moléculas de IgG endógena.
El po tencial de las interacc i ones farm aco l ó gicas se ha eva lu ado med i a n te una mon i to-ri z ación de los niveles plasmáti cos de cicl o s porina en los dos estudios de fase III re a l i z ados enreceptores de tra s p l a n te renal y en un estudio de fase I de interacción to l era n c i a / Fa r-m acoc i n é tica con micofen o l a to mofetil (NO15301) en pac i en tes som eti dos a tra s p l a n te ren a l .E s tos estudios indican que Zenapax® no pre s enta interacc i ones farm acoc i n é ticas con cicl o s-porina o ácido micofen ó l i co (MPA ) , el met a bo l i to activo de micofen o l a to mofeti l .
Razón de ser de la dosis
Los datos preclínicos y clínicos siguientes proporcionan la justificación para el régi-men de administración recomendado para Zenapax®, 1,0 mg/kg administrado cada 14días, hasta un total de cinco dosis:
• En el modelo de trasplante cardíaco en el mono, la eficacia de Zenapax® se demostróa una dosis de 1,0 mg/kg administrada cada dos días; este régimen mantuvo las con-centraciones séricas mínimas por encima de 10 mg/ml en monos y proporcionó unaexposición al fármaco de 4000 mg/h/ml.
• Los en s ayos in vi tro de la unión de IL-R2 y la pro l i feración de linfobl a s tos depen d i en-tes de IL- 2 , i n d i c a ron que las con cen trac i ones de 5 -10 µg/ml eran su f i c i en tes no sólop a ra satu rar los receptores de IL- 2 Ra , sino también para inhibir la activi d ad bi o l ó gi c ade los linfobl a s tos activado s . Un régi m en de 1,0 mg/kg, ad m i n i s trado cada 14 días, h a s t aun total de cinco do s i s , m a n ti ene las con cen trac i ones séricas por encima de 5 µg/ml, a lm en o s , du ra n te 120 días después del tra s p l a n te .E s te régi m en también proporc i ona unaex posición al fárm aco en receptores de tra s p l a n te ren a l , a prox i m ad a m en te , de 3600-
35
Farmacología

5100 mg/h/ml, que es similar a la asoc i ada con la ef i c acia en el estudio en tra s p l a n te car-d í aco en el mon o.
• La frecuencia de administración cada 14 días se basó en la estimación preliminar de lavida media sérica de Zenapax®, obtenida del estudio de fase I en pacientes sometidosa trasplante renal (Vincenti et al. 1997). Un régimen que contenga un total de cincodosis proporciona terapia inmunosupresora, al menos, durante 3 meses después deltrasplante, que es el período más crítico para que se produzca rechazo agudo.
Inmunogenicidad
Los ensayos clínicos de Zenapax® administrado a pacientes sometidos a trasplanterenal, tratados con terapia inmunosupresora doble o triple, han mostrado que no se detec-taron anticuerpos que afectasen a los niveles de eficacia o seguridad de Zenapax® en elsuero, ni cualquier otro parámetro clínicamente relevante. En la Tabla 7, se resume la inci-dencia de pacientes receptores de trasplante renal que desarrollaron títulos confirmables deanticuerpos idiotípicos anti-daclizumab, durante o después del tratamiento con Zenapax®.
Resumen• Estudios in vitro han mostrado que Zenapax® tiene una afinidad alta por Tac y blo-
quea eficazmente la activación y la proliferación de las células T.
• Zenapax® satura el Tac de los linfocitos circulantes durante un período de hasta 4semanas, después de una dosis única.
• Zenapax® tiene una vida media de eliminación terminal larga,aproximadamente de 20días, que es equivalente a la de IgG endógena.
• Zenapax® ti ene un acl a ra m i en to sistémico bajo y un vo lu m en de distri bución redu c i do.
• El régimen de administración recomendado (1,0 mg/kg cada 14 días, hasta un total decinco dosis) proporciona niveles séricos de Zenapax® adecuados, durante los 3 mesescríticos posteriores al trasplante.
• No se detectaron anticuerpos contra Zenapax® clínicamente relevantes.
36
Farmacología
Tabla.7
Incidencia global de anticuerpos idiotípicos anti-daclizumab en pacientes sometidos a trasplante renal,a los que se les admi-nistraron cinco perfusiones de Zenapax® o placebo
Pacientes tratados Pacientes tratadoscon Zenapax® con placebo
Nº Dosis de Número de Número de Número de Número deestudio/fase daclizumab pacientes pacientes con pacientes pacientes con
(mg/kg) evaluables anticuerpos evaluables anticuerpospositivos positivos
confirmados confirmados
NO14392/0,5, 1,0 18 0 - -Fase I
NO14393/1,0 83 10 (12,0%) 82 0Fase III
NO14874/1,0 125 22 (17,6%) 121 0Fase III

Eficacia en trasplante renal
Estudio de búsqueda de dosis
En un ensayo de fase I, randomizado, abierto, se evaluaron los efectos de Zenapax® en19 pacientes que reciban su primer trasplante renal (donante vivo relacionado, n=10;donante cadáver, n=2) (Vincenti et al. 1997).
Zenapax® se ad m i n i s tró uti l i z a n do uno de los cuatro reg í m en e s : 0,5 mg/kg ó 1,0 mg/kgtodas las semanas o cada dos sem a n a s , hasta un total de cinco do s i s . Todos los pac i en tes rec i-bi eron como terapia inmu n o su pre s ora cicl o s pori n a , a z a ti oprina y pred n i s on a .
Sólo un paciente que había recibido un injerto procedente de cadáver, experimentó un epi-sodio de rechazo agudo 7 días después de recibir la primera dosis de Zenapax® (0,5 mg/kguna vez cada dos semanas), durante los primeros 3 meses de seguimiento; este episodio derechazo se resolvió completamente con terapia antirrechazo y no se produjo recurrencia.En el seguimiento a un año, todos los pacientes estaban vivos y los injertos presentaban unafunción adecuada (Vincenti et al. 1997).
El modelo farmacocinético estableció el régimen de administración de Zenapax® de 1,0 mg/kg cada dos semanas,
hasta un total de cinco dosis.
Estudios Multicéntricos Comparativos Fase III
Se realizaron dos ensayos clínicos fase III, a gran escala, Multicéntricos, randomiza-dos, doble ciego, controlados con placebo, en pacientes de, al menos, 18 años de edad, paraevaluar la eficacia y la seguridad de Zenapax® en la prevención del rechazo agudo durantelos 6 meses posteriores al trasplante, en pacientes que habían recibido el primer trasplanterenal de donante cadáver. En ambos estudios, Zenapax® fue administrado a la dosis de1mg/kg una vez cada dos semanas, hasta completar un total de 5 dosis
En el estudio (NO14393) se comparó Zenapax® más terapia inmunosupresora triple(ciclosporina, prednisona, azatioprina), con terapia triple sola. Este estudio se realizó en 17centros (11 de EE.UU., 3 de Canadá, 3 de Suecia) e incluyó 260 pacientes; 126 pacientes serandomizaron para recibir Zenapax® y 134 placebo. En el segundo estudio (NO14874), secomparó Zenapax® más terapia inmunosupresora doble (ciclosporina, prednisona) conterapia doble sola. Este estudio se realizó en 19 centros (15 de Europa, 2 de Canadá, 2 deAustralia) e incluyó 275 pacientes; 141 pacientes se randomizaron para recibir Zenapax® y134 placebo. Las características demográficas y las indicaciones para el trasplante eran com-parables en los grupos Zenapax® y placebo, en los dos estudios.
37
Experiencia clínica

Resultados
• Rechazo agudo
El tratamiento con Zenapax® redujo significativamente, del 37% al 40%, la inci-dencia de rechazo agudo demostrado mediante biopsia, en los 6 meses posteriores al tras-plante.
En el estudio de terapia triple (NO14393), la tasa de rechazo agudo demostradomediante biopsia fue del 22% en los pacientes tratados con Zenapax®, en comparación conel 35% en los pacientes que recibieron placebo (p=0,03). Del mismo modo, en el estudiode terapia doble (NO14874), la tasa fue del 47% y el 28% en los pacientes tratados con pla-cebo y Zenapax®, respectivamente (p=0,001).
38
Experiencia clínica
Tabla.8a
Rechazo agudo en pacientes trasplantados renales que recibieron tratamiento con Zenapax® y placebo, a los 6 mesesde seguimiento
*Rechazo demostrado mediante biopsia o presuntivo
Estudio NO14393 Estudio N014874Triple terapia Doble terapia
Zenapax® Placebo Valor de Zenapax® Placebo Valor de(n=126) (n=134) p (n=141) (n=134) p
Rechazo demostradomediante biopsia a los 28 (22%) 47 (35%) 0,03 39 (28%) 63 (47%) 0,0016 meses
Primer rechazop re s u n t i vo o demostra d o 32 (25%) 52 (39%) 0,035 48 (34%) 67 (50%) 0,006mediante biopsia
Número de episodiosde rechazo por paciente* 23 (18%) 34 (25%) 0,001 30 (21%) 38 (28%) 0,004Uno 9 (7%) 18 (14%) 18 (13%) 29 (22%)Dos o más 0,33 0,57 0,51 0,83
Número medio
Tiempo medio hasta el primer rechazo (días) 54 20 0,008 36 15 0,0001

En los dos estudios, el número de pacientes que experimentaron episodios múltiplesde rechazo fue más elevado en el grupo tratado con placebo que en el grupo que recibióZenapax®; el número medio de episodios de rechazo por paciente también fue más bajo enel grupo tratado con Zenapax® (Tabla 8a). Además, el tiempo medio hasta el primer recha-zo fue más prolongado en los pacientes tratados con Zenapax® que en los pacientes querecibieron placebo.
Esta reducción significativa del rechazo agudo confirmado por biopsia a los 6 mesesde seguimiento, observada en los pacientes tratados con Zenapax®, también se demuestraen las estimaciones de Kaplan-Meier, tal y como se muestra en la Figura 11.
El tratamiento con Zenapax® redujo la incidencia de rechazo demostrado mediantebiopsia en el primer año después del trasplante (Tabla 8b). En el estudio de terapia triple(NO14393), la tasa de rechazo demostrado mediante biopsia fue del 28% frente al 38% enlos pacientes tratados con Zenapax® y placebo, respectivamente. El rechazo agudo confir-mado mediante biopsia también se redujo significativamente en el ensayo de terapia doble(NO14874), con una incidencia del 28% en el grupo tratado con Zenapax®, comparadocon el 49% en el grupo que recibió placebo (p=0,001).
39
Experiencia clínica
Tabla.8b
Datos a 12 meses de incidencia de rechazo agudo en pacientes tratados con Zenapax® frente a placebo en trasplante renal.
Estudio NO14393 Estudio N014874Triple terapia Doble terapia
Zenapax® Placebo Valor de Zenapax® Placebo Valor de(n=126) (n=134) p (n=141) (n=134) p
Rechazo agudoconfirmado por biopsia 35 (28%) 51 (38%) 0,09 39 (28%) 66 (49%) 0,00112 meses de seguimiento
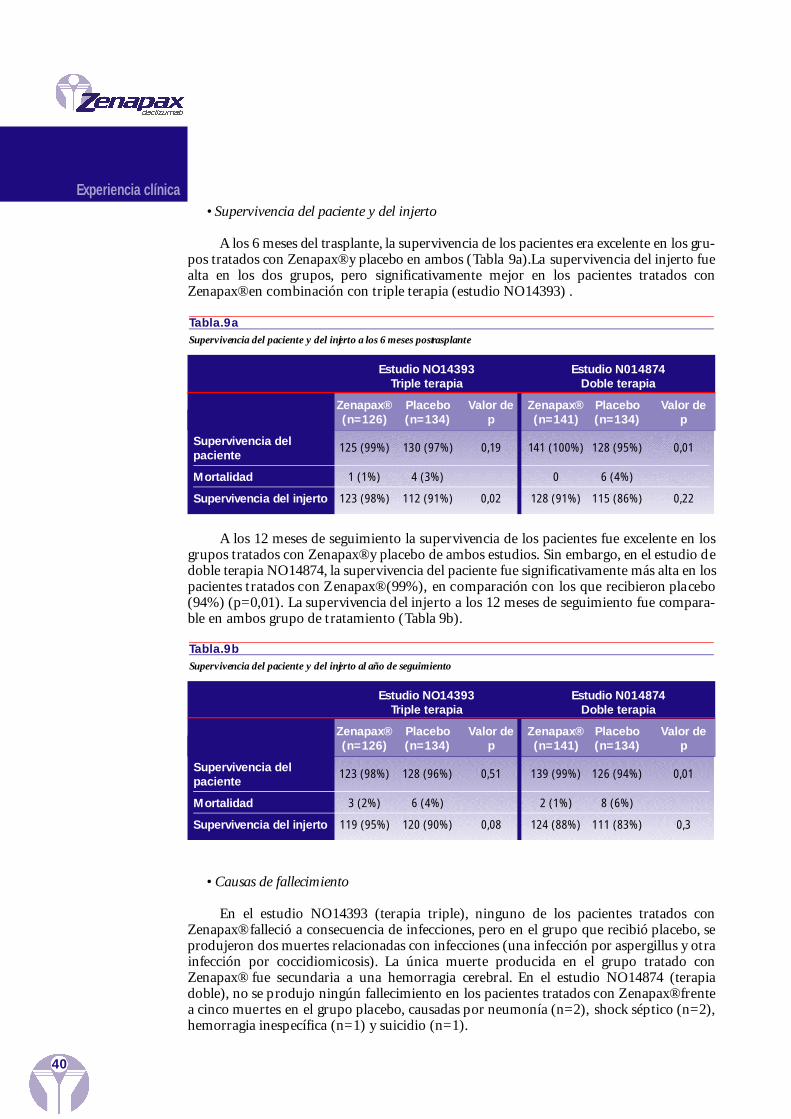
• Supervivencia del paciente y del injerto
A los 6 meses del trasplante, la supervivencia de los pacientes era excelente en los gru-pos tratados con Zenapax® y placebo en ambos (Tabla 9a).La supervivencia del injerto fuealta en los dos grupos, pero significativamente mejor en los pacientes tratados conZenapax® en combinación con triple terapia (estudio NO14393) .
A los 12 meses de seguimiento la supervivencia de los pacientes fue excelente en losgrupos tratados con Zenapax® y placebo de ambos estudios. Sin embargo, en el estudio dedoble terapia NO14874, la supervivencia del paciente fue significativamente más alta en lospacientes tratados con Zenapax® (99%), en comparación con los que recibieron placebo(94%) (p=0,01). La supervivencia del injerto a los 12 meses de seguimiento fue compara-ble en ambos grupo de tratamiento (Tabla 9b).
• Causas de fallecimiento
En el estudio NO14393 (terapia triple), ninguno de los pacientes tratados conZenapax® falleció a consecuencia de infecciones, pero en el grupo que recibió placebo, seprodujeron dos muertes relacionadas con infecciones (una infección por aspergillus y otrainfección por coccidiomicosis). La única muerte producida en el grupo tratado conZenapax® fue secundaria a una hemorragia cerebral. En el estudio NO14874 (terapiadoble), no se produjo ningún fallecimiento en los pacientes tratados con Zenapax® frentea cinco muertes en el grupo placebo, causadas por neumonía (n=2), shock séptico (n=2),hemorragia inespecífica (n=1) y suicidio (n=1).
40
Experiencia clínica
Tabla.9aSupervivencia del paciente y del injerto a los 6 meses postrasplante
Tabla.9bSupervivencia del paciente y del injerto al año de seguimiento
Estudio NO14393 Estudio N014874Triple terapia Doble terapia
Zenapax® Placebo Valor de Zenapax® Placebo Valor de(n=126) (n=134) p (n=141) (n=134) p
Supervivencia del 125 (99%) 130 (97%) 0,19 141 (100%) 128 (95%) 0,01paciente
M o r t a l i d a d 1 (1%) 4 (3%) 0 6 (4%)
S u p e r v i vencia del injerto 123 (98%) 112 (91%) 0,02 128 (91%) 115 (86%) 0,22
Estudio NO14393 Estudio N014874Triple terapia Doble terapia
Zenapax® Placebo Valor de Zenapax® Placebo Valor de(n=126) (n=134) p (n=141) (n=134) p
Supervivencia del 123 (98%) 128 (96%) 0,51 139 (99%) 126 (94%) 0,01paciente
M o r t a l i d a d 3 (2%) 6 (4%) 2 (1%) 8 (6%)
S u p e r v i vencia del injerto 119 (95%) 120 (90%) 0,08 124 (88%) 111 (83%) 0,3
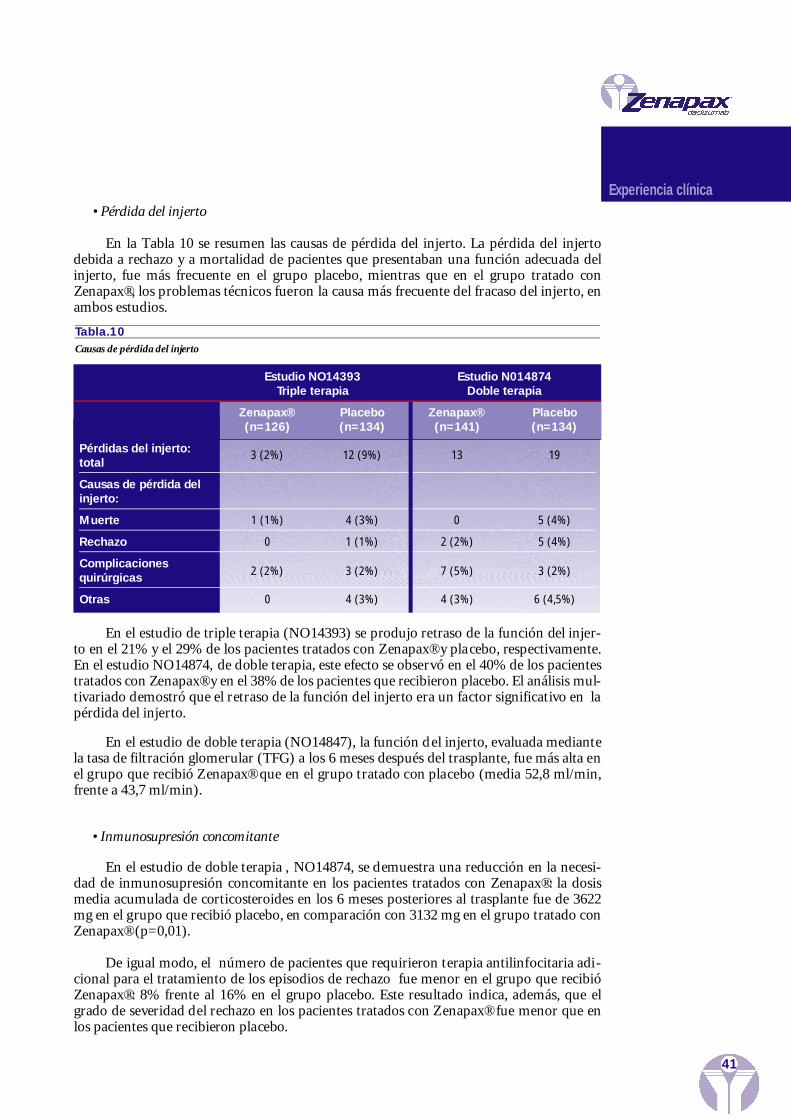
• Pérdida del injerto
En la Tabla 10 se resumen las causas de pérdida del injerto. La pérdida del injertodebida a rechazo y a mortalidad de pacientes que presentaban una función adecuada delinjerto, fue más frecuente en el grupo placebo, mientras que en el grupo tratado conZenapax®, los problemas técnicos fueron la causa más frecuente del fracaso del injerto, enambos estudios.
En el estudio de triple terapia (NO14393) se produjo retraso de la función del injer-to en el 21% y el 29% de los pacientes tratados con Zenapax® y placebo, respectivamente.En el estudio NO14874, de doble terapia, este efecto se observó en el 40% de los pacientestratados con Zenapax® y en el 38% de los pacientes que recibieron placebo. El análisis mul-tivariado demostró que el retraso de la función del injerto era un factor significativo en lapérdida del injerto.
En el estudio de doble terapia (NO14847), la función del injerto, evaluada mediantela tasa de filtración glomerular (TFG) a los 6 meses después del trasplante, fue más alta enel grupo que recibió Zenapax® que en el grupo tratado con placebo (media 52,8 ml/min,frente a 43,7 ml/min).
• Inmunosupresión concomitante
En el estudio de doble terapia , NO14874, se demuestra una reducción en la necesi-dad de inmunosupresión concomitante en los pacientes tratados con Zenapax®: la dosismedia acumulada de corticosteroides en los 6 meses posteriores al trasplante fue de 3622mg en el grupo que recibió placebo, en comparación con 3132 mg en el grupo tratado conZenapax® (p=0,01).
De igual modo, el número de pacientes que requirieron terapia antilinfocitaria adi-cional para el tratamiento de los episodios de rechazo fue menor en el grupo que recibióZenapax®: 8% frente al 16% en el grupo placebo. Este resultado indica, además, que elgrado de severidad del rechazo en los pacientes tratados con Zenapax® fue menor que enlos pacientes que recibieron placebo.
41
Experiencia clínica
Tabla.10Causas de pérdida del injerto
Estudio NO14393 Estudio N014874Triple terapia Doble terapia
Zenapax® Placebo Zenapax® Placebo(n=126) (n=134) (n=141) (n=134)
Pérdidas del injerto: 3 (2%) 12 (9%) 13 19total
Causas de pérdida delinjerto:
Muerte 1 (1%) 4 (3%) 0 5 (4%)
Rechazo 0 1 (1%) 2 (2%) 5 (4%)
Complicaciones quirúrgicas 2 (2%) 3 (2%) 7 (5%) 3 (2%)
Otras 0 4 (3%) 4 (3%) 6 (4,5%)

Conclusiones
Estos dos ensayos de fase III en trasplante renal, han demostrado que Zenapax® (1,0mg/kg, administrados por vía intravenosa una vez cada dos semanas, hasta un total decinco dosis) en combinación con terapia inmunosupresora doble o triple, reduce significa-tivamente la incidencia de rechazo agudo en trasplante renal y mejora la supervivencia delinjerto a los 6 meses postrasplante.
En resumen, el tratamiento con Zenapax ® :
• Disminuye las tasas de rechazo agudo hasta en un 40%.
• Reduce el número de episodios de rechazo por paciente.
• Mejora la supervivencia del injerto.
• Mejora la función del injerto (es decir, la TFG).
• Reduce la necesidad de terapia inmunosupresora concomitante:permitiendo administrar dosis acumuladas menores de esteroidesy disminuir la uti l i z ación de anti c u erpos anti l i n focti cos para el tra t a m i en to del rech a zo.
• Mejora la supervivencia del paciente.
Experiencia clínica en trasplante renal pediátrico
Aunque la eficacia y seguridad de Zenapax® en pacientes pediátricos no ha sido aúnestablecida a través de Ensayos de Fase III, existe ya cierta experiencia clínica sobre su uti-lización en este tipo de pacientes.
Se ha re a l i z ado un estudio mu l ti c é n tri co americano incluyen do 54 pac i en tes ped i á tri co stra s p l a n t ados ren a l e s , en el que Zenapax® se añadió a la terapia inmu n o su pre s ora habi tual dec ada cen tro : 74% rec i bi eron cicl o s pori n a , 26% tac ro l i mus y 87% Mi cofen o l a to mofetilo
Los pacientes recibieron 1mg/kg de Zenapax®, administrado en perfusión intraveno-sa, antes del trasplante y cada dos semanas hasta completar un total de 5 dosis.
La edad media de los pacientes fue de 10 años (16 pacientes <1-5 años, 13 pacientes6-12 años y 25 pacientes 13-17 años)
Resultados
La incidencia de rechazo, tras un seguimiento medio entre 6 y 12 meses, fue de un5% (3 pacientes), siendo rechazos leves que respondieron a terapia antirechazo convencio-nal, y no produciéndose ninguna pérdida de injerto debida a rechazo.
Las causas de pérdida del injerto (2 injertos) fueron trombosis vascular (n=1) y escle-rosis focal recurrente (n=1).
En cuanto al perfil de seguridad :
— Ningún paciente presentó síntomas de liberación de citoquina o anafilaxis.
— Las infecciones graves fueron infrecuentes: Un 11% de los pacientes desarrollaroninfecciones del tracto urinario, un 6% bacteremia y sólo un 7% viremia por CMV.
— Ningún paciente desarrolló síndrome linfoproliferativo postrasplante.
Los datos de este estudio, muestran que la adición de Zenapax® a los protocolos
42
Experiencia clínica

actuales de inmunosupresión en trasplante renal pediátrico, puede disminuir la tasa derechazo agudo sin aumentar efectos adversos ni infecciones.
CellCept® y Zenapax® en combinación:fármacocinética y eficacia en la prevención delrechazo del aloinjerto renal
CellCept® es un agente inmunosupresor con eficacia y seguridad bien establecidas enla prevención del rechazo y como tratamiento de mantenimiento en trasplante renal cuan-do se utiliza en combinación con ciclosporina y corticosteroides (European MMFCooperative Study Group 1995; Sollinger 1995; Tricontinental Mycophenolate MofetilRenal Transplantation Study Group 1996). CellCept® ha reducido al 17%-20% las tasas derechazo agudo demostrado mediante biopsia y, puesto que no es nefrotóxico, no producedaños en el riñón trasplantado.
Actualmente se está realizando un estudio de fase I/II randomizado, doble ciego, con-trolado con placebo, con dosis múltiples (NO15301), en seis centros de EE.UU., para eva-luar la eficacia y la seguridad de Zenapax® y CellCept® cuando se utilizan en combinación(Kirkman et al. 1997). Setenta y seis pacientes sometidos a su primer trasplante renal dedonante vivos relacionado o donante cadáver sin HLA idéntico, recibieron terapia inmu-nusupresora triple (CellCept®, ciclosporina y esteroides) y se randomizaron para recibirplacebo (n=26) o 1,0 mg/kg de Zenapax® (n=50),una vez cada dos semanas,hasta un totalde cinco dosis, iniciando el tratamiento dentro de las 24 horas previas al trasplante. Lospacientes se monitorizaron durante 6 meses para determinar la incidencia de rechazoagudo, la supervivencia del paciente y del injerto, la función del injerto y el desarrollo deinfecciones y neoplasias. Los resultados de eficacia obtenidos hasta la fecha, muestran quela incidencia de rechazo agudo confirmado por biopsia o diagnosticado por signos y sínto-mas clínicos era del 20% en el grupo tratado con placebo y del 12% en el grupo que reci-bió Zenapax®. No se produjeron interacciones farmacológicas entre CellCept® y Zenapax®y los perfiles de seguridad fueron consistentes con los controles históricos.
Zenapax® en el tratamiento o la prevenciónde la EICH
Eficacia en el tratamiento o la prevención de la EICH
En dos ensayos abiertos de fase I (N3681 y NO14790) se evaluaron los efectos de unadosis única de Zenapax® en el tratamiento de 31 pacientes sometidos a trasplante de médu-la ósea, que manifestaban EICH aguda resistente a esteroides.
En el estudio N3681 (estudio de dosis ascendentes) se administraron dosis únicas deZenapax® de 0,5 mg/kg (8 pacientes), 1,0 mg/kg (4 pacientes) ó 1,5 mg/kg (12 pacientes),en infusión intravenosa de 2 horas. Se realizó un seguimiento de los pacientes durante 56días. Nueve pacientes que manifestaron una respuesta transitoria se volvieron a tratar unavez. Diez de 24 pacientes (42%) experimentaron mejoría global de la EICH aguda el día 56.Cuatro pacientes experimentaron resolución completa de la EICH aguda y seis pacientes
43
Experiencia clínica

manifestaron una respuesta parcial. Se observaron respuestas en todos los niveles de ladosis y en los tres sistemas de órganos (piel, hígado e intestino).
En el estudio NO14790 se trataron siete pacientes que manifestaban EICH resistentea esteroides, con una dosis única de 1,0 mg/kg de Zenapax®, administrada en infusiónintravenosa de 2 horas. Se realizó un seguimiento de los pacientes durante 56 días. Tres delos siete pacientes experimentaron mejora global de la EICH aguda hacia el día 28; todoslos pacientes manifestaron una respuesta parcial. Dos de los pacientes que manifestaronuna respuesta parcial el día 28, demostraron resolución completa de la EICH hacia el día56. Se observaron respuestas en los tres sistemas de órganos (piel, hígado e intestino).
Se realizó un ensayo de fase II/III a gran escala (NO14348) para evaluar la eficacia yla seguridad de Zenapax® en la prevención de la EICH en pacientes sometidos a su primertrasplante de médula ósea, procedente de un donante no relacionado. El ensayo se diseñócomo estudio multicéntrico, doble ciego, controlado con placebo, en el que los pacientesrecibieron placebo ó 0,3 ó 1,2 mg/kg de Zenapax® todas las semanas,hasta un total de cincodosis, además de un régimen inmunosupresor de ciclosporina y metotrexato. Este estudiose realizó en 12 centros (6 de EE.UU., 2 de Canadá, 4 de Europa) e incluyó 210 pacientes.Los pacientes iniciaron el tratamiento con Zenapax® un día antes del trasplante de médu-la ósea y se realizó un seguimiento durante un año, después del trasplante.
En este estudio NO14348, la adición de una dosis de 0,3 ó 1,2 mg/kg de Zenapax® a unr é gi m en inmu n o su pre s or, no disminuyó la incidencia de EICH du ra n te los 100 pri m eros díaspo s teri ores al tra s p l a n te . El ti em po medio hasta el de s a rro llo de EICH aguda aumentó en losp ac i en tes que rec i bi eron 0,3 ó 1,2 mg/kg de Zenapax® (33 y 34 días, re s pectiva m en te ) , encom p a ración con los pac i en tes tra t ados con placebo (26 días); sin em b a r go, esta diferencia nofue estad í s ti c a m en te sign i f i c a tiva . No se ob s erva ron diferencias estad í s ti c a m en te sign i f i c a tiva sen tre los gru pos tra t ados con Zenapax® y placebo, con re s pecto a la severi d ad de la EICH ola incidencia de EICH crónica. En los pac i en tes tra t ados con Zen a p a x ® , se ob s ervó una dis-m i nución de la incidencia de rec u rrencia de la en ferm ed ad pri m a ria en los 100 pri m eros díaspo s teri ores al tra s p l a n te de médula ósea, en com p a ración con los pac i en tes tra t ados con pla-cebo, pero esta diferencia no fue estad í s ti c a m en te sign i f i c a tiva .
La falta de ef i c acia estad í s ti c a m en te sign i f i c a tiva en el estudio NO145348 no es rel eva n-te para la ef i c acia de Zenapax® en tra s p l a n te ren a l ; e s to se debe a las diferencias inmu n o l ó gi-cas en tre los pac i en tes que ex peri m en t a ron rech a zo del tra s p l a n te renal y EICH y al tra t a-m i en to que rec i bi eron antes del tra s p l a n te . Los pac i en tes som eti dos a tra s p l a n te de médu l aósea rec i ben un tra t a m i en to de acon d i c i on a m i en to inten s ivo con fárm acos cito s t á ti cos e irra-d i ación antes de la operac i ó n , que produ ce una "torm enta de citoquinas" en todo el or ga n i s-m o ; e s to puede hacer más difícil que un agen te inmu n o su pre s or alcance la ef i c acia de s e ad a .Evi den tem en te , Zenapax® a esta dosis no previno la EICH en el estudio NO14348. E s tu d i o sde inve s ti gador en curso del tra t a m i en to con Zenapax® en pac i en tes con EICH, mu e s tran unaef i c ac i a , en gen era l , s i m i l a r, a la de ATG , pero con un perfil de seg u ri d ad muy mej orado.
Tolerancia y seguridad de Zenapax®
Tolerancia y seguridad de Zenapax® en trasplante renal
La seguridad de Zenapax® se determinó en cuatro estudios clínicos, tres de los cualesfueron ensayos clínicos randomizados, controlados, en 629 pacientes que recibieron aloin-jertos renales. De la muestra total, 336 pacientes recibieron Zenapax® y 293 placebo.
44
Experiencia clínica

La seguridad de Zenapax® se determinó en cuatro estudios clínicos,tresde los cuales fueron ensayos clínicos randomizados,controlados, en 629
pacientes que recibieron aloinjertos renales. De la muestra total, 336pacientes recibieron Zenapax® y 293, placebo.
Dos de los estudios fueron ensayos de fase III doble ciego, en los que se comparó unadosis de 1,0 mg/kg de Zenapax® con placebo, cuando se administraron, respectivamente,como parte de un régimen que contenía ciclosporina y corticosteroides (régimen inmuno-supresor de terapia doble) o ciclosporina, corticosteroides y azatioprina (régimen inmuno-supresor de terapia triple) para prevenir el rechazo agudo. El tratamiento se inició durantelas 24 horas anteriores al trasplante y las dosis subsiguientes se administraron cada 14 días,hasta un total de cinco dosis.
El tercer estudio fue un ensayo de fase I, doble ciego, con dosis múltiples, realizado encinco centros de EE.UU. Los pacientes se trataron con un régimen inmunosupresor de tera-pia triple, que incluyó CellCept®, ciclosporina y prednisona, más 1,0 mg/kg de Zenapax® oplacebo, administrados una vez cada 14 días hasta un total de cinco dosis, iniciando el tra-tamiento 24 horas antes del trasplante.
El cuarto estudio fue un ensayo de fase I abierto, realizado en dos centros de EE.UU.Los pacientes se trataron con un régimen inmunosupresor de terapia triple, que incluíaciclosporina, prednisona y azatioprina,así como 0,5 ó 1,0 mg/kg de Zenapax®, administra-do una vez cada 7 días,o una vez cada 14 días hasta un total de cinco dosis,iniciando el tra-tamiento 24 horas antes del trasplante.
En comparación con placebo, Zenapax® no aumentó significativamente el perfil detoxicidad del régimen inmunosupresor concomitante. Los efectos adversos comunicadosestaban relacionados con el procedimiento del trasplante y los otros fármacos del régimeninmunosupresor. (Remitirse a la ficha técnica de ciclosporina, corticosteroides, azatioprina yCellCept® para la información sobre los efectos adversos asociados con estos tratamientos).
Se comunicaron efectos adversos en el 95% de los pacientes del grupo tratado conplacebo y en el 96% de los pacientes del grupo que recibió Zenapax®. La proporción depacientes que se retiraron prematuramente de los estudios combinados, debido a los efec-tos adversos,fue del 8,5% en el grupo tratado con placebo y del 8,6% en el grupo que reci-bió Zenapax®.
La administración de Zenapax® no se asoció con un aumento de la incidencia de efec-tos adversos, efectos adversos graves, infecciones o neoplasias.
Los efectos adversos que se comunicaron con más frecuencia fueron trastornos gas-trointestinales, cuya frecuencia fue similar en los grupos de pacientes tratados conZenapax® (67%) y placebo (68%).
Zenapax® no aumentó la incidencia de episodios infecciosos (72% en el grupo place-bo, en comparación con el 68% en el grupo Zenapax®). Los tipos de infecciones comuni-cadas fueron similares en los grupos tratados con Zenapax® y placebo. Se comunicó infec-ción por CMV en el 16% de los pacientes del grupo placebo y en el 13% de los pacientesdel grupo tratado con Zenapax®.
45
Experiencia clínica

La adición de Zenapax® no aumentó el número de linfomas después del trasplante,que se produjeron con una frecuencia de menos del 1% en los grupos tratados con place-bo y Zenapax®.
En comparación con placebo, Zenapax® no aumentó s i gn i f i c a tiva m en teel perfil de toxi cidad del régi m en inmu n o su pre sor co n co m i t a n te .Lo s
efe ctos adversos co municados estaban rel a cionados con el pro ced i m i en tod el tra s pl a n te y los otros fárm a cos del régi m en inmu n o su pre so r.
Evaluación global de los efectos adversos en ensayos clínicos
Hasta la fecha, no se han observado efectos secundarios específicos ni efectos adver-sos graves asociados con Zenapax®.
La infusión intravenosa de Zenapax® no se asoció con ninguno de los efectos advers o si n m ed i a to s , tales como escalof r í o s ,f i ebre y cef a l e a , que se ob s ervan frec u en tem en te con otro sa n ti c u erpos mon ocl on a l e s . No se ob s erva ron diferencias sign i f i c a tivas en tre los gru pos tra t a-dos con placebo y Zen a p a x ® , re s pecto a la incidencia y el ti po de efectos adversos comu n i c a-dos u ob s ervados du ra n te los 3 meses po s teri ores al tra s p l a n te (Ta bla 11). Más de la mitad delos pac i en tes ex peri m en t a ron efectos adversos ga s troi n te s ti n a l e s , los más frec u en tes de loscuales fueron estre ñ i m i en to, náuseas y do l or abdom i n a l . Se ob s erva ron pocas diferencias enc ada estu d i o, re s pecto a la incidencia o el ti po de efectos advers o s , en tre los pac i en tes tra t ado scon Zenapax® y placebo. Se comu n i c a ron frec u en tem en te efectos adversos que afectaban als i s tema nervioso aut ó n om o, tra s tornos del sistema uri n a ri o, tra s tornos met a b ó l i cos y nutri-c i on a l e s , tra s tornos del sistema nervioso cen tral y peri f é ri co, tra s tornos gen erales que afect a-ban a todo el or ganismo y tra s tornos re s p i ra tori o s . Au n que la incidencia de algunos efecto sadversos indivi duales fue más alta en el estudio NO14393 (en com bi n ación con triple tera-pia) que en el estudio NO14874 ( doble tera p i a ) , los sistemas corporales afect ado s , así com oel ti po de efectos adversos comu n i c ado s , f u eron los mismos.
Zenapax® es el primer agente inmunosupresor que no da lugar a unaumento de la incidencia de infecciones cuando se añade a un régimen
inmunosupresor. De hecho, la incidencia de episodios infecciosos,incluida infección por CMV, fue generalmente más baja
en los pacientes tratados con Zenapax® que en los pacientes que recibieron placebo.
Inmunogenicidad
La incidencia de anticuerpos anti-daclizumab en los dos ensayos clínicos de fase III enpacientes receptores de trasplante renal fue del 15%, que fue más alta de lo esperado en un
46
Experiencia clínica
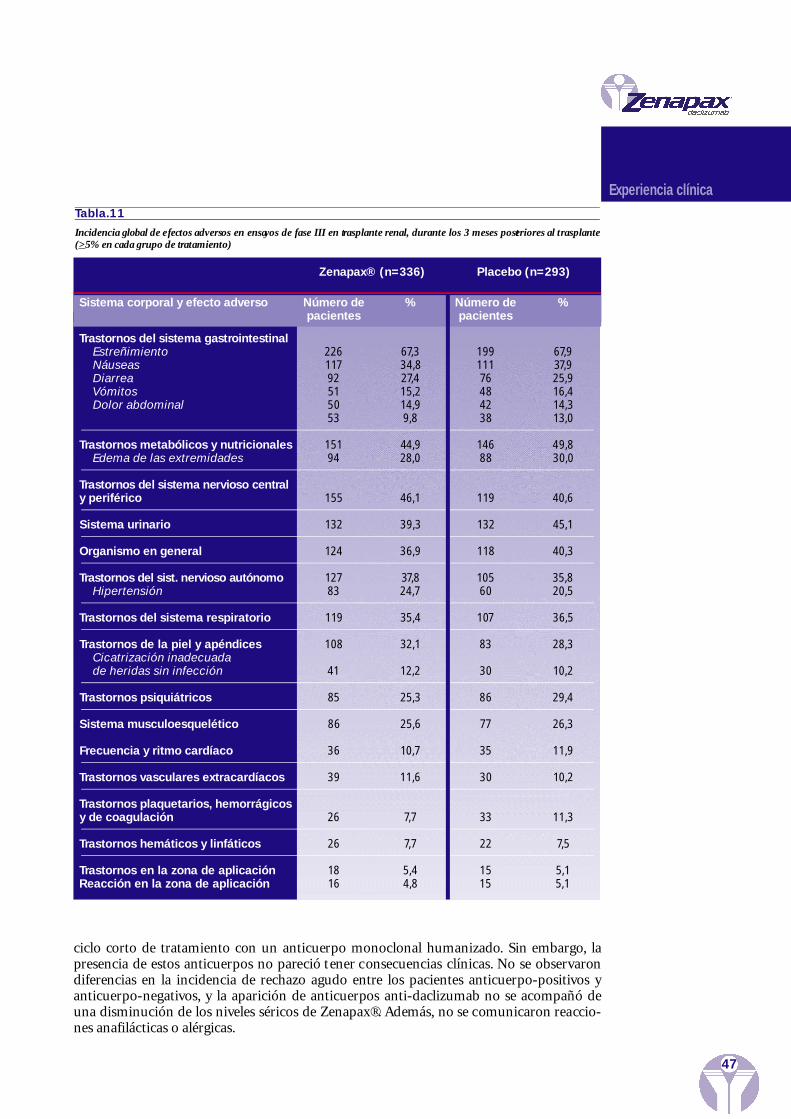
ciclo corto de tratamiento con un anticuerpo monoclonal humanizado. Sin embargo, lapresencia de estos anticuerpos no pareció tener consecuencias clínicas. No se observarondiferencias en la incidencia de rechazo agudo entre los pacientes anticuerpo-positivos yanticuerpo-negativos, y la aparición de anticuerpos anti-daclizumab no se acompañó deuna disminución de los niveles séricos de Zenapax®. Además, no se comunicaron reaccio-nes anafilácticas o alérgicas.
47
Experiencia clínicaTabla.11
Incidencia global de efectos adversos en ensayos de fase III en trasplante renal, durante los 3 meses posteriores al trasplante(>5% en cada grupo de tratamiento)
Zenapax® (n=336) Placebo (n=293)
Sistema corporal y efecto adverso Número de % Número de %pacientes pacientes
Trastornos del sistema gastro i n t e s t i n a lEstreñimiento 226 67,3 199 67,9Náuseas 117 34,8 111 37,9Diarrea 92 27,4 76 25,9Vómitos 51 15,2 48 16,4Dolor abdominal 50 14,9 42 14,3
53 9,8 38 13,0
Trastornos metabólicos y nutricionales 151 44,9 146 49,8Edema de las extremidades 94 28,0 88 30,0
Trastornos del sistema nervioso centra ly periférico 155 46,1 119 40,6
Sistema urinario 132 39,3 132 45,1
Organismo en general 124 36,9 118 40,3
Trastornos del sist. nervioso autónomo 127 37,8 105 35,8Hipertensión 83 24,7 60 20,5
Trastornos del sistema respiratorio 119 35,4 107 36,5
Trastornos de la piel y apéndices 108 32,1 83 28,3Cicatrización inadecuada de heridas sin infección 41 12,2 30 10,2
Trastornos psiquiátricos 85 25,3 86 29,4
Sistema musculoesquelético 86 25,6 77 26,3
Frecuencia y ritmo cardíaco 36 10,7 35 11,9
Trastornos vasculares extracardíacos 39 11,6 30 10,2
Trastornos plaquetarios, hemorrágicos y de coagulación 26 7,7 33 11,3
Trastornos hemáticos y linfáticos 26 7,7 22 7,5
Trastornos en la zona de aplicación 18 5,4 15 5,1Reacción en la zona de aplicación 16 4,8 15 5,1
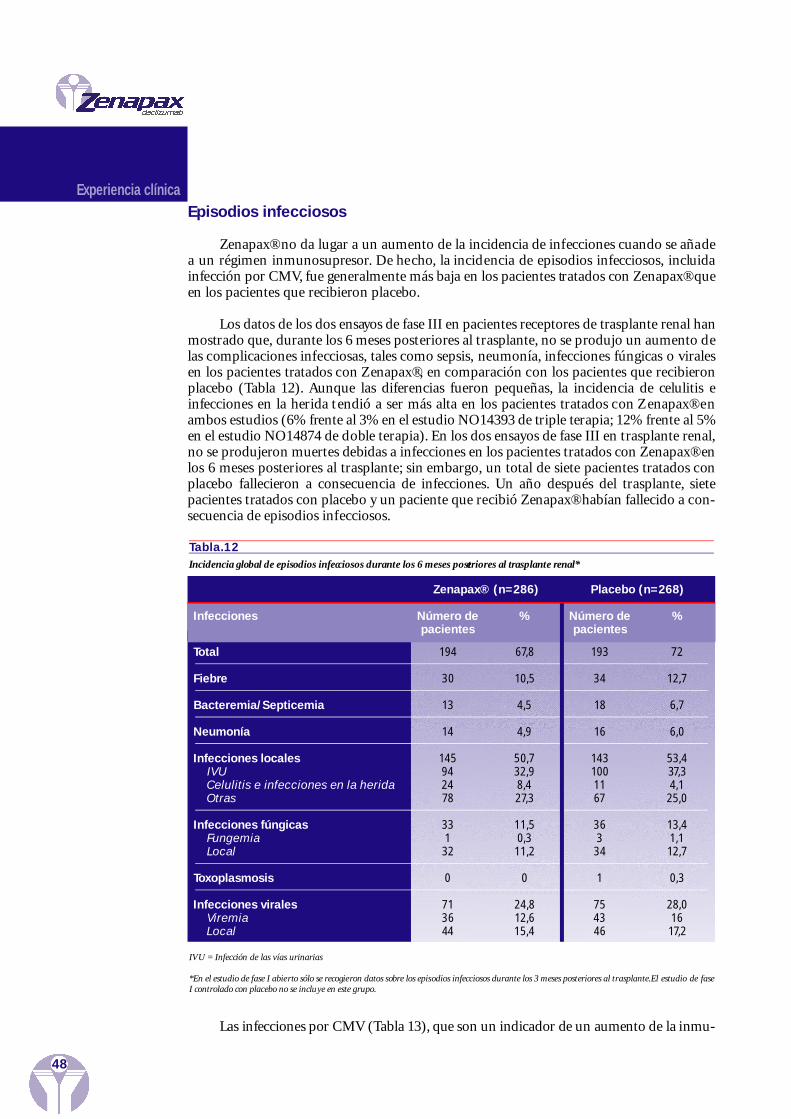
Episodios infecciosos
Zenapax® no da lugar a un aumento de la incidencia de infecciones cuando se añadea un régimen inmunosupresor. De hecho, la incidencia de episodios infecciosos, incluidainfección por CMV, fue generalmente más baja en los pacientes tratados con Zenapax® queen los pacientes que recibieron placebo.
Los datos de los dos ensayos de fase III en pacientes receptores de trasplante renal hanmostrado que, durante los 6 meses posteriores al trasplante, no se produjo un aumento delas complicaciones infecciosas, tales como sepsis, neumonía, infecciones fúngicas o viralesen los pacientes tratados con Zenapax®, en comparación con los pacientes que recibieronplacebo (Tabla 12). Aunque las diferencias fueron pequeñas, la incidencia de celulitis einfecciones en la herida tendió a ser más alta en los pacientes tratados con Zenapax® enambos estudios (6% frente al 3% en el estudio NO14393 de triple terapia; 12% frente al 5%en el estudio NO14874 de doble terapia). En los dos ensayos de fase III en trasplante renal,no se produjeron muertes debidas a infecciones en los pacientes tratados con Zenapax® enlos 6 meses posteriores al trasplante; sin embargo, un total de siete pacientes tratados conplacebo fallecieron a consecuencia de infecciones. Un año después del trasplante, sietepacientes tratados con placebo y un paciente que recibió Zenapax® habían fallecido a con-secuencia de episodios infecciosos.
Las infecc i ones por CMV (Ta bla 13), que son un indicador de un aumen to de la inmu-
48
Experiencia clínica
Tabla.12Incidencia global de episodios infecciosos durante los 6 meses posteriores al trasplante renal*
IVU = Infección de las vías urinarias
*En el estudio de fase I abierto sólo se recogieron datos sobre los episodios infecciosos durante los 3 meses posteriores al trasplante.El estudio de faseI controlado con placebo no se incluye en este grupo.
Zenapax® (n=286) Placebo (n=268)
Infecciones Número de % Número de %pacientes pacientes
Total 194 67,8 193 72
Fiebre 30 10,5 34 12,7
Bacteremia/Septicemia 13 4,5 18 6,7
Neumonía 14 4,9 16 6,0
Infecciones locales 145 50,7 143 53,4IVU 94 32,9 100 37,3Celulitis e infecciones en la herida 24 8,4 11 4,1Otras 78 27,3 67 25,0
Infecciones fúngicas 33 11,5 36 13,4Fungemia 1 0,3 3 1,1Local 32 11,2 34 12,7
Toxoplasmosis 0 0 1 0,3
Infecciones virales 71 24,8 75 28,0Viremia 36 12,6 43 16Local 44 15,4 46 17,2
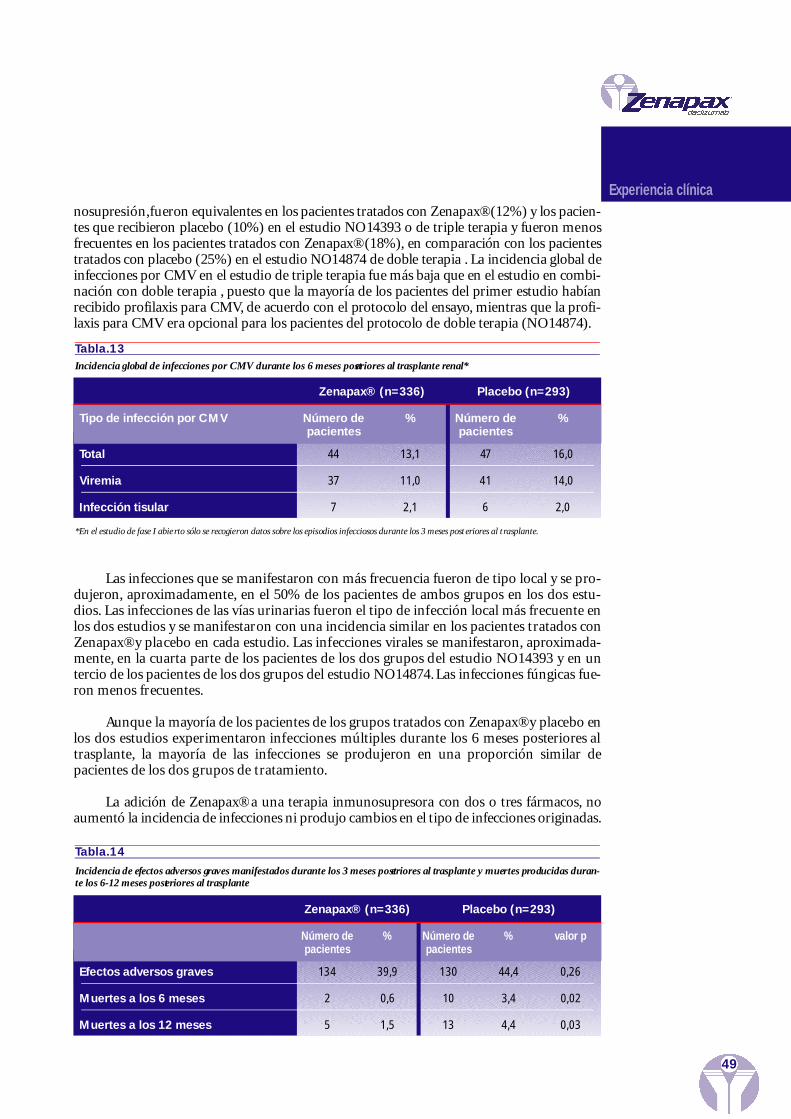
n o su pre s i ó n ,f u eron equ iva l en tes en los pac i en tes tra t ados con Zenapax® (12%) y los pac i en-tes que rec i bi eron placebo (10%) en el estudio NO14393 o de triple terapia y fueron men o sf rec u en tes en los pac i en tes tra t ados con Zenapax® (18%), en com p a ración con los pac i en te stra t ados con placebo (25%) en el estudio NO14874 de doble terapia . La incidencia gl obal dei n fecc i ones por CMV en el estudio de triple terapia fue más baja que en el estudio en com bi-n ación con doble terapia , p u e s to que la mayoría de los pac i en tes del pri m er estudio habíanrec i bi do profilaxis para CMV, de ac u erdo con el pro tocolo del en s ayo, m i en tras que la prof i-laxis para CMV era opc i onal para los pac i en tes del pro tocolo de doble terapia (NO14874).
Las infecciones que se manifestaron con más frecuencia fueron de tipo local y se pro-dujeron, aproximadamente, en el 50% de los pacientes de ambos grupos en los dos estu-dios. Las infecciones de las vías urinarias fueron el tipo de infección local más frecuente enlos dos estudios y se manifestaron con una incidencia similar en los pacientes tratados conZenapax® y placebo en cada estudio. Las infecciones virales se manifestaron, aproximada-mente, en la cuarta parte de los pacientes de los dos grupos del estudio NO14393 y en untercio de los pacientes de los dos grupos del estudio NO14874. Las infecciones fúngicas fue-ron menos frecuentes.
Aunque la mayoría de los pacientes de los grupos tratados con Zenapax® y placebo enlos dos estudios experimentaron infecciones múltiples durante los 6 meses posteriores altrasplante, la mayoría de las infecciones se produjeron en una proporción similar depacientes de los dos grupos de tratamiento.
La adición de Zenapax® a una terapia inmu n o su pre s ora con dos o tres fárm aco s , n oa u m entó la incidencia de infecc i ones ni produjo cambios en el ti po de infecc i ones ori gi n ad a s .
49
Experiencia clínica
Tabla.13Incidencia global de infecciones por CMV durante los 6 meses posteriores al trasplante renal*
*En el estudio de fase I abie rto sólo se recogieron datos sobre los episodios infecciosos durante los 3 meses post eriores al trasplante.
Zenapax® (n=336) Placebo (n=293)
Tipo de infección por CMV Número de % Número de %pacientes pacientes
Total 44 13,1 47 16,0
Viremia 37 11,0 41 14,0
Infección tisular 7 2,1 6 2,0
Tabla.14
Incidencia de efectos adversos graves manifestados durante los 3 meses posteriores al trasplante y muertes producidas duran-te los 6-12 meses posteriores al trasplante
Zenapax® (n=336) Placebo (n=293)
Número de % Número de % valor ppacientes pacientes
Efectos adversos graves 134 39,9 130 44,4 0,26
Muertes a los 6 meses 2 0,6 10 3,4 0,02
Muertes a los 12 meses 5 1,5 13 4,4 0,03

Efectos adversos graves
No se observaron diferencias significativas entre los grupos tratados con Zenapax® yplacebo, respecto a la incidencia de efectos adversos graves posible o probablemente rela-cionados con la medicación del estudio, 3 meses y 6-12 meses después del trasplante renal.En la Tabla 14 se resume la incidencia de efectos adversos graves y muertes.
Neoplasias
De los 535 pacientes incluidos en los ensayos de fase III de Zenapax® en estudios detrasplante renal, seis pacientes (1,1%) desarrollaron neoplasias. De éstos, un paciente delestudio NO14393 tratado con Zenapax® y terapia inmunosupresora triple y otro pacientedel estudio NO14784 tratado con placebo y terapia inmunosupresora doble, desarrollaronlinfoma después del trasplante. El paciente del estudio NO14393, que haba recibido cincodosis de Zenapax®, desarrolló linfoma de células B en la piel de la parte posterior de la rodi-lla 3 meses y medio después del trasplante, con recurrencia en los testículos 8 meses des-pués del trasplante. El paciente tratado con placebo en el estudio NO14784 desarrolló lin-foma en el aloinjerto renal, 49 días después del trasplante.
Dos pacientes tratados con Zenapax®, uno en el estudio NO14393 (el mismo pacien-te que desarrolló linfoma) y uno en el estudio NO14874, así como tres pacientes que reci-bieron placebo (uno en el estudio NO14393 y dos en el estudio NO14874), desarrollarontumores cutáneos de tipo no melanoma, durante los 6 meses posteriores al trasplante.
Ningún paciente del estudio de fase I NO14392 desarrolló trastornos linfoproliferati-vos durante el primer año después del trasplante.
Estos datos sugieren que los pacientes sometidos a trasplante renal tratados conZenapax®, no parecen presentar un aumento del riesgo de desarrollar neoplasias relacio-nadas con la inmunosupresión. Sin embargo, sería necesario tratar a miles de pacientespara demostrar esta observación.
Resultados de las pruebas de laboratorio
En los estudios de fase III NO14393 y NO1487 en tra s p l a n te ren a l , no se ob s erva rond i ferencias en tre los pac i en tes tra t ados con Zenapax® y los que rec i bi eron placebo : los re su l-t ados medios de las pru ebas re a l i z adas para determinar los parámetros hem a to l ó gi co s , bi o-qu í m i cos y uri n a rios fueron similares en los pac i en tes tra t ados con Zenapax® y placebo.
Tolerancia y seguridad de Zenapax® en combinación conCellCept® en trasplante renal
La seguridad y la tolerancia de Zenapax® se evaluó en comparación con placebo en elestudio NO15301, en el que se administró en combinación con CellCept®, ciclosporina yesteroides. El perfil de seguridad y tolerancia de Zenapax® observado en este estudio fuesimilar al comunicado en el ensayo de fase III de Zenapax® en trasplante renal, NO14393en combinación con triple terapia.
50
Experiencia clínica

Zenapax® (1,0 mg/kg), administrado cada dos semanas, hasta un total de cinco dosis,a pacientes que reciban su primer aloinjerto renal, fue seguro y bien tolerado en combina-ción con CellCept®, ciclosporina y esteroides.
La mayoría de los pacientes de los grupos tratados con placebo yZenapax® experimentaron un efecto adverso durante los 3 meses
posteriores al trasplante (98% y 100%, respectivamente). Sin embargo,sólo el 40% de los pacientes de los dos grupos experimentaron un efecto
adverso que se consideró remota, posible o probablementerelacionado con el tratamiento.
Trece pacientes (3 del grupo placebo; 10 del grupo Zenapax®) se retiraron prematu-ramente del estudio y recibieron menos de cinco dosis completas. De los 40 pacientes ran-domizados al tratamiento con Zenapax® y que recibieron cinco dosis completas deZenapax®,sólo cuatro (10%) experimentaron rechazo agudo y sólo dos (5%) desarrollaronuna infección por CMV. Tres de los 50 pacientes (6%) del grupo tratado con Zenapax®experimentaron pérdida del injerto; todos estos pacientes presentaban factores de conco-mitantes que precipitaron la pérdida del injerto.
La mayoría de los pacientes de los grupos tratados con placebo y Zenapax® experi-mentaron un efecto adverso durante los 3 meses posteriores al trasplante (98% y 100%,res-pectivamente). Sin embargo, sólo el 40% de los pacientes de los dos grupos experimenta-ron un efecto adverso que se consideró remota, posible o probablemente relacionado conel tratamiento. Sólo el 4% de los pacientes de los dos grupos de tratamiento experimenta-ron un efecto adverso que se consideró posible o probablemente relacionado con el trata-miento, y menos del 3% de los efectos adversos comunicados en los dos grupos de trata-miento se consideraron graves. Los efectos adversos que se comunicaron con más frecuen-cia en los grupos tratados con Zenapax® (78%) y placebo (80%) fueron de tipo gastroin-testinal. Los únicos efectos adversos que se consideraron posible o probablemente relacio-nados con el fármaco del ensayo fueron diarrea e hipoestesia (ambos en un paciente trata-do con Zenapax®) e hipertensión pulmonar en un paciente tratado con placebo.
Aunque la incidencia de efectos adversos graves fue más alta en el grupo tratado conZenapax® (36%) que en el grupo que recibió placebo (28%), ninguno de ellos se conside-ró posible o probablemente relacionado con el fármaco del ensayo. Un total de cuatropacientes se retiraron prematuramente del estudio debido a los efectos adversos (tres delgrupo tratado con Zenapax®; uno del grupo que recibió placebo).
La incidencia global de los seis tipos de infecciones oportunistas (infección por CMV,neumonía, infecciones fúngicas no cutáneas, infecciones herpéticas, sepsis y meningitis)que se manifestaron durante los 6 meses posteriores al trasplante, fue similar en los grupostratados con Zenapax® (20%) y placebo (16%).
La adición de Zenapax® a una terapia inmunosupresora de tres fármacos, que inclu-yó CellCept®, fue segura y bien tolerada por los receptores del primer aloinjerto renal.
51
Experiencia clínica

Tolerancia y seguridad de Zenapax®
en trasplante de médula osea
Zenapax® pareció seg u ro y bi en to l erado en los estudios N3681 y NO14790. Au n qu etodos los pac i en tes ex peri m en t a ron efectos advers o s , los inve s ti gadores con s i dera ron qu ela mayoría de ellos no estaban rel ac i on ados con el tra t a m i en to con Zen a p a x ® . En el estu-dio N3681, los únicos efectos adversos rel ac i on ados con Zenapax® comu n i c ados fuerontem bl or leve en un pac i en te y aumen to de la su doración en otro pac i en te ; a m bos pac i en-tes se tra t a ron con 0,5 mg/kg de Zen a p a x ® . En el estudio NO14790, el único efecto adver-so que se con s i deró rel ac i on ado con el tra t a m i en to con Zenapax® fue tem bl or leve en unp ac i en te .
Tal y como se preveía en pacientes sometidos a trasplante de médula ósea, todos lospacientes del estudio NO14348 experimentaron efectos adversos múltiples; sin embargo,no se observó una relación aparente entre la incidencia de efectos adversos y la dosis deZenapax®.
Inmunogenicidad
En estudios de pacientes sometidos a trasplante de médula ósea, tratados conZenapax®, no se observaron anticuerpos anti-daclizumab detectables.
Episodios infecciosos
En el en s ayo de fase I/II a gran escala, N O 1 4 3 4 8 , la adición de Zenapax® al régi -m en inmu n o su pre s or no dio lu gar a un aumen to de la incidencia de infecc i on e s . L am ayoría de los pac i en tes del estudio NO14348 ex peri m en t a ron ep i s odios infecc i o s o sdu ra n te los 100 días po s teri ores al tra s p l a n te de médula ósea. La incidencia gl obal de ep i-s odios infecciosos en los dos gru pos de pac i en tes tra t ados con Zenapax® fue similar a ladel gru po que rec i bió placebo (99% en el gru po que rec i bió 0,3 mg/kg de Zen a p a x ® ;97% en el gru po tra t ado con 1,2 mg/kg de Zen a p a x ® ; 95% en el gru po tra t ado con pla-cebo ) .
La incidencia de fallecimientos producidos a consecuencia de infecciones fue similaren los tres grupos de tratamiento: (13% de los pacientes tratados con 0,3 mg/kg deZenapax®; 12% de los pacientes que recibieron 1,2 mg/kg de Zenapax®; 11% de los pacien-tes tratados con placebo). Las causas más frecuentes de las muertes fueron neumonía einfecciones fúngicas sistémicas.
Resultados de las pruebas de laboratorio
En la mayoría de los pacientes de los estudios N3681 y NO14790, se observaron ano-malías notables en los parámetros hematológicos, bioquímicos y urinarios. Todas las ano-malías de laboratorio fueron consistentes con las enfermedades subyacentes y la medica-ción concomitante de los pacientes, así como las observadas en el estudio NO14348, en elque la incidencia fue similar en los pacientes tratados con Zenapax® y placebo.
52
Experiencia clínica

Resumen• Zenapax®, utilizado conjuntamente con terapia inmunosupresora doble o triple, redu-
ce significativamente la incidencia de rechazo agudo.
• Zenapax® es eficaz y se tolera bien cuando se administra a una dosis de 1,0 mg/kg cada2 semanas, hasta un total de cinco dosis, en pacientes sometidos a trasplante renal alo-génico.
• Zenapax® reduce hasta en el 40% la tasa de rechazo agudo de los aloinjertos renales ymejora la supervivencia del injerto y del paciente.
• La adición de Zenapax® a una terapia farmacológica inmunosupresora doble o tripleno aumenta la incidencia de episodios infecciosos.
• La administración de Zenapax® no da lugar a la producción de anticuerpos clínica-mente relevantes.
• Se ha demostrado que Zenapax®, utilizado en combinación con ciclosporina, corti-costeroides y CellCept®, tiene un perfil de tolerancia y seguridad favorable.
53
Experiencia clínica

PresentaciónZenapax®, 25 mg (en 5 ml), se presenta en viales de vidrio tipo flint. Zenapax® está
disponible en envases de uno y tres viales
Compatibilidad con los equipos de infusiónNo se ha observado ninguna incompatibilidad entre Zenapax® y las bolsas de cloru-
ro de polivinilo o los equipos de perfusión.
Almacenamiento y período de validez— Conservar entre 2-8ºC.— No congelar.— Proteger los viales de la luz— Período de validez: 18 meses
Estabilidad Se ha demostrado la estabilidad química y física de la solución preparada para usar,
durante 24 horas a 2-8ºC, o durante 4 horas a temperatura ambiente. Sin embargo, desdeun punto de vista microbiológico, el producto diluido debe usarse inmediatamente. No sedebe guardar el producto tras su dilución a menos que ésta se haya realizado en condicio-nes asépticas controladas y validadas. Si no se emplea inmediatamente, el usuario es res-ponsable del tiempo y de las condiciones de conservación previas a su administración.
Instrucciones de uso, manipulacióny eliminación (en su caso)
Zenapax® NO debe inyectarse directamente. Debe diluirse en 50 ml de una soluciónestéril de cloruro sódico al 0,9% antes de su administración intravenosa a los pacientes.Para mezclar la solución, no la agite para evitar que se forme espuma, sino que invierta labolsa suavemente. Procure mantener la esterilidad de la solución preparada, puesto que elmedicamento no contiene conservantes antimicrobianos ni sustancias bacteriostáticas.Zenapax® es una solución incolora que se suministra en viales de un solo uso, deberá dese-charse cualquier porción remanente del medicamento. Los productos de uso parenteraldeben inspeccionarse visualmente antes de su administración para descartar la presencia departículas o cambios de coloración. Una vez preparada la perfusión, se administrará inme-diatamente por vía intravenosa. Si se diluye en condiciones asépticas, puede conservarsedurante 24 horas en el frigorífico a una temperatura de 2-8ºC.
No se debe añadir ni tampoco perfundir otros medicamentos / sustancias de formasimultánea a través de la misma vía intravenosa.
54
I n formación fa r m a c é u t i ca

1. Denominación del medicamentoZenapax®, 5mg/ml concentrado para solución para perfusión.
2. Composición cualitativa y cuantitativaCada vial contiene 25 mg de daclizumab (DCI) en 5 ml de solución (5 mg/ml).
3. Forma farmacéuticaConcentrado para solución para perfusión.
4. Datos clínicos
4.1. Indicaciones terapéuticas
Zenapax® está indicado en la profilaxis del rechazo agudo en pacientes que no esténhiperinmunizados, que reciben por primera vez un trasplante renal alogénico. Se adminis-tra de forma simultánea con el tratamiento inmunosupresor que incluye ciclosporina y cor-ticosteroides.
4.2. Posología y forma de administración
Zenapax® debe ser prescrito únicamente por especialistas debidamente cualificadosen el uso de tratamientos inmunosupresores para trasplante de órganos.
La dosis recom en d ada de Zenapax® es de 1 mg/kg. El vo lu m en de Zenapax® que con-ti ene la dosis aprop i ada se añade a 50 ml de solución salina estéril al 0,9%, y se ad m i n i s tra porvía intravenosa du ra n te 15 minuto s . Pu ede ad m i n i s tra rse a través de una vena peri f é rica o cen-tra l .
Zenapax® se debe administrar, inicialmente, dentro de las 24 horas previas al tras-plante. La dosis siguiente y cada una de las sucesivas se aplicarán a intervalos de 14 días,hasta completar un total de 5 dosis.
Uso pediátricoPor el momento, no han finalizado los estudios adecuados y bien controlados en
pacientes pediátricos. Los datos farmacocinéticos de los que se dispone son muy limitados.
Pacientes ancianosLa ex peri encia con Zenapax® en pac i en tes ancianos (mayores de 65 años) es limitad a ,
55
Uso de Ze n a p a x®
(ficha técnica )

d ado el escaso número de los mismos que se som eten a tra s p l a n te ren a l , pero no hay evi den-cia de que los pac i en tes ancianos nece s i ten una dosis diferen te a la de los pac i en tes jóven e s .
Pacientes con alteración renal graveNo es necesario ajustar la Posología en los pacientes con alteración renal grave.
Pacientes con alteración hepática graveNo existen datos relativos a los pacientes con alteración hepática grave.
4.3. Contraindicaciones
Zenapax® está contraindicado en los pacientes con hipersensibilidad conocida aldaclizumab o a cualquiera de los componentes del producto (Véase apartado 6.1 "Relaciónde excipientes").
Zenapax® esta contraindicado durante el embarazo y la lactancia.
4.4. Advertencias y precauciones especiales de empleo
No hay experiencia del uso de Zenapax® en pacientes que estén hiperinmunizados.
Pueden producirse reacciones anafilácticas después de la administración de proteí-nas. En raras ocasiones se han comunicado reacciones graves de hipersensibilidad tras laadministración de Zenapax®. Por tanto, se dispondrá de la medicación necesaria para t ra-tar, de forma inmediata, las reacciones graves de hipersensibilidad.
Los pac i en tes som eti dos a tra s p l a n te que rec i ben tra t a m i en to inmu n o su pre s ormu e s tran mayor ri e sgo de en ferm ed ades linfopro l i fera tivas (ELPs) e infecc i ones opor-tu n i s t a s . Aun sien do Zenapax® un med i c a m en to inmu n o su pre s or, no se han de s c ri tohasta la fecha aumen to de ELPs o infecc i ones oportunistas en pac i en tes tra t ados con elm i s m o.
No hay experiencia de la exposición a segundos o subsiguientes tratamientos com-pletos con Zenapax® en pacientes sometidos a trasplante.
4.5. Interacciones con otrosmedicamentos y otras formas de interacción
Debido a que Zenapax® es una inmunoglobulina, no se espera que se produzcaninteracciones metabólicas fármaco-fármaco.
En en s ayos cl í n i cos con Zen a p a x ® , se ad m i n i s tra ron los siguien tes med i c a m en tos parael tra s p l a n te , sin que se produ j eran interacc i on e s : c i cl o s pori n a ,m i cofen o l a to mofeti l o, ga n c i-cl ovi r, ac i cl ovi r, t ac ro l i mu s , a z a ti opri n a , i n mu n ogl obulina anti ti m oc í ti c a , mu rom on a b - C D 3(OKT3) y corti coi de s .
4.6. Embarazo y lactancia
Zenapax® está contraindicado durante el embarazo y la lactancia. Debido a su poten-
56
Uso de Zenapax®(ficha técnica)

cial inmunosupresor tiene efectos potencialmente peligrosos sobre el desarrollo de la ges-tación y sobre el lactante expuesto al daclizumab presente en la leche materna.
4.7. Efectos sobre la capacidadpara conducir vehículos y utilizar máquinas
No existen pruebas de que Zenapax® altere la capacidad para conducir vehículos outilizar maquinaria.
4.8. Reacciones adversas
El perfil de seguridad de Zenapax® se ha investigado, en comparación con placebo,en enfermos que recibieron de forma simultánea tratamiento inmunosupresor con ciclos-porina y corticoides únicamente, con la adición de azatioprina o con la adición de micofe-nolato mofetilo.
Se noti f i c a ron acon tec i m i en tos adversos en aprox i m ad a m en te el 95% del gru po dep ac i en tes que rec i bió placebo y aprox i m ad a m en te el 96% del gru po tra t ado con dacl i z u m a b.Las re acc i ones adversas mas comunes (>20%) se comu n i c a ron con una frec u encia similar enlos pac i en tes tra t ados con Zenapax® y en los tra t ados con placebo. E n tre estas re acc i ones sei n cluyen tra s tornos dige s tivos (estre ñ i m i en to y nauseas), tra s tornos del met a bo l i s m o, edem a speri f é ri co s , tra s tornos del sistema nervioso cen tral y peri f é ri co, tra s tornos del apara to uri n a-ri o, re acc i ones sistémicas - tra s tornos gen erales (do l or po s t - tra u m á ti co ) , tra s tornos del siste-ma nervioso aut ó n omo (hiperten s i ó n ) , tra s tornos del apara to re s p i ra tori o, tra s tornos de lap i el y anejos cut á n eo s , a l terac i ones psíquicas y tra s tornos del apara to locom o tor.
Se describieron acontecimientos adversos graves en aproximadamente el 44,4 % delgrupo de pacientes que recibió placebo y el 39,9 % del grupo tratado con Zenapax®.
Las muertes ocurridas durante los primeros 6 meses post-trasplante se comunicaronen el 3,4% del grupo de pacientes que recibió placebo y en el 0,6% del grupo tratado conZenapax®. La mortalidad a los 12 meses fue del 4,4% en el grupo de pacientes que recibióplacebo y del 1,5% en el grupo tratado con Zenapax®.
Se comunicaron episodios de infecciones en un 72% de los pacientes tratados conplacebo y en un 68% de los pacientes tratados con Zenapax®. El tipo de infecciones comu-nicadas fue similar en los grupos tratados con Zenapax® y con placebo. La frecuencia deinfección por citomegalovirus se comunicó en el 16% de los pacientes del grupo placebo yen el 13% de los pacientes del grupo del Zenapax®.
Los trastornos linfoproliferativos posteriores al trasplante se dieron con una fre-cuencia inferior al 1% tanto en el grupo de pacientes que recibió placebo como en el grupotratado con Zenapax®.
En ra ras oc a s i ones se han comu n i c ado re acc i ones graves de hipers en s i bi l i d ad tras la ad-m i n i s tración de Zenapax® (ver apart ado 4.4 Advertencias y prec a u c i ones especiales de em p l eo )
Pacientes pediátricos: Los acontecimientos adversos comunicados con mayor fre-cuencia fueron hipertensión (53%), dolor en el post-operatorio (post-traumático) (45%),diarrea (43%), y vómitos (32%). Las tasas de hipertensión y deshidratación comunicadasfueron mayores en pacientes pediátricos que en pacientes adultos.
57
Uso de Zenapax®(ficha técnica)
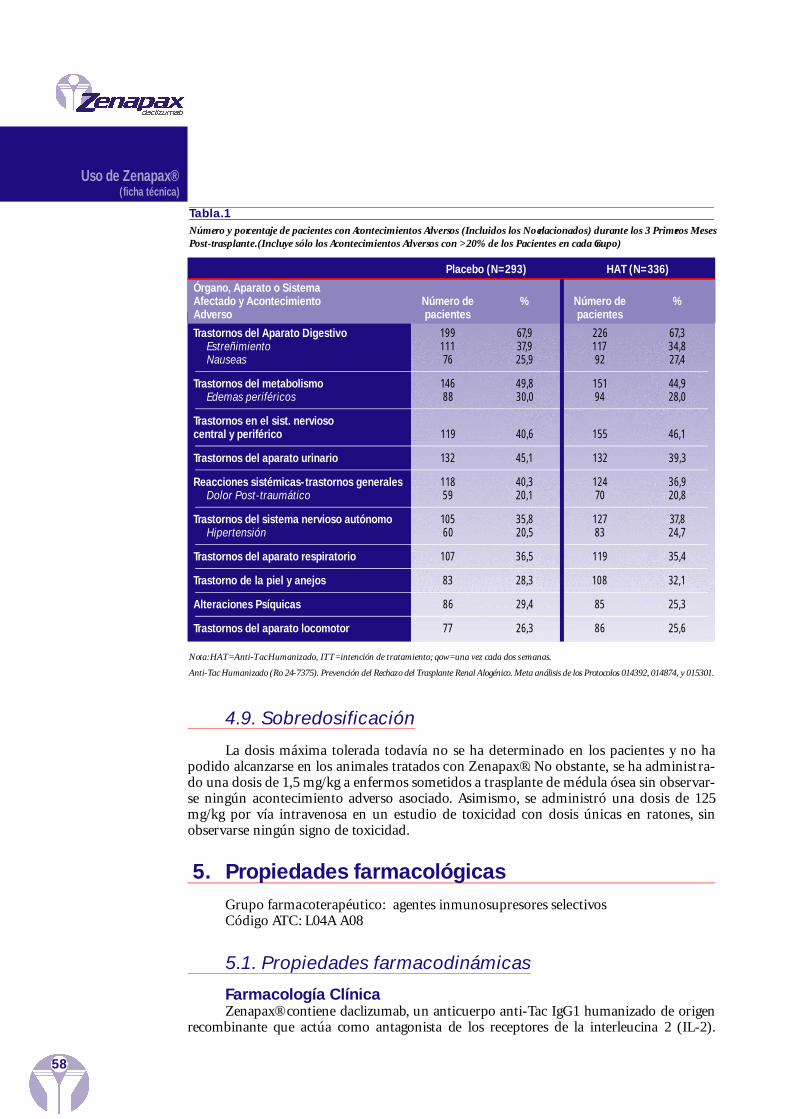
4.9. Sobredosificación
La dosis máxima tolerada todavía no se ha determinado en los pacientes y no hapodido alcanzarse en los animales tratados con Zenapax®. No obstante, se ha administra-do una dosis de 1,5 mg/kg a enfermos sometidos a trasplante de médula ósea sin observar-se ningún acontecimiento adverso asociado. Asimismo, se administró una dosis de 125mg/kg por vía intravenosa en un estudio de toxicidad con dosis únicas en ratones, sinobservarse ningún signo de toxicidad.
5. Propiedades farmacológicasGrupo farmacoterapéutico: agentes inmunosupresores selectivos Código ATC: L04A A08
5.1. Propiedades farmacodinámicas
Farmacología ClínicaZenapax® con ti ene dacl i z u m a b, un anti c u erpo anti - Tac IgG1 hu m a n i z ado de ori gen
recom bi n a n te que actúa como antagonista de los receptores de la interl eucina 2 (IL- 2 ) .
58
Uso de Zenapax®(ficha técnica)
Tabla.1Número y porcentaje de pacientes con Acontecimientos Adversos (Incluidos los No relacionados) durante los 3 Primeros MesesPost-trasplante.(Incluye sólo los Acontecimientos Adversos con >20% de los Pacientes en cada Grupo)
Nota: HAT=Anti-Tac Humanizado, ITT=intención de tratamiento; qow=una vez cada dos s emanas.
An ti - Tac Humanizado (Ro 24-7375). Preven ción del Re ch a zo del Tra s pl a n te Renal Al o g é n i co. Meta análisis de los Proto colos 014392, 0 1 4 8 7 4 , y 015301.
Placebo (N=293) HAT (N=336)
Órgano, Aparato o SistemaAfectado y Acontecimiento Número de % Número de %Adverso pacientes pacientes
Trastornos del Aparato Digestivo 199 67,9 226 67,3Estreñimiento 111 37,9 117 34,8Nauseas 76 25,9 92 27,4
Trastornos del metabolismo 146 49,8 151 44,9Edemas periféricos 88 30,0 94 28,0
Trastornos en el sist. nervioso central y periférico 119 40,6 155 46,1
Trastornos del aparato urinario 132 45,1 132 39,3
Reacciones sistémicas-trastornos generales 118 40,3 124 36,9Dolor Post-traumático 59 20,1 70 20,8
Trastornos del sistema nervioso autónomo 105 35,8 127 37,8Hipertensión 60 20,5 83 24,7
Trastornos del aparato respiratorio 107 36,5 119 35,4
Trastorno de la piel y anejos 83 28,3 108 32,1
Alteraciones Psíquicas 86 29,4 85 25,3
Trastornos del aparato locomotor 77 26,3 86 25,6

D aclizumab se une con gran espec i f i c i d ad a la su bu n i d ad alfa o Tac del com p l ejo receptorI L-2 de alta afinidad (ex pre s ado en células T activadas) e inhibe la unión y la activi d ad bi o l ó-gica de la IL- 2 . La ad m i n i s tración de Zenapax® inhibe la activación de los linfoc i tos med i ad apor la IL- 2 , una vía determ i n a n te en la respuesta inmu n i t a ria celu l a r, i m p l i c ada en el rech a zodel aloi n j erto. D aclizumab satu ra el receptor Tac du ra n te aprox i m ad a m en te 90 días en lam ayoría de los pac i en te s , con la pauta po s o l ó gica recom en d ad a . Se de s a rro ll a ron anti c u erpo sf ren te a Daclizumab en aprox i m ad a m en te el 9% de los pac i en tes tra t ados con Zenapax® ene s tudios cl í n i co s ; sin em b a r go, e s to no pareció afectar a la ef i c ac i a , la seg u ri d ad , la con cen tra-ción sérica de Dacl i z u m a b, ni a ningún otro parámetro cl í n i co rel eva n te ex a m i n ado.
Mediante el análisis de clasificación de células activadas por fluorescencia (CCAF)no se observaron grandes cambios en el recuento de los linfocitos circulantes ni en los feno-tipos celulares, a parte de los descensos transitorios esperados de células Tac+.
Tratamiento Combinado en Pacientes Receptoresde Aloinjerto RenalEn los ensayos en Fase III, se añadió Zenapax® al régimen de inmunosupresión
estándar con ciclosporina (5 mg/kg), esteroides (prednisona o metilprednisolona), con osin adición de azatioprina (4 mg/kg).
Zenapax® demostró una superioridad estadísticamente significativa frente al place-bo en la reducción de la tasa de rechazo agudo, confirmado por biopsia, a los seis mesessiguientes al trasplante renal alogénico. El tiempo de rechazo también fue significativa-mente superior. No se observó rechazo como efecto rebote.
En la base de datos referentes a seguridad de la Fase III, la tasa de supervivencia a los12 meses fue del 95% en el grupo tratado con placebo y del 99% en el grupo tratado conZenapax®.
No se ob s ervó el síndrome de liberación de citoquinas tras la ad m i n i s tración de Zen a p a x ® .
5.2. Propiedades farmacocinéticas
Durante los ensayos clínicos efectuados en enfermos sometidos a trasplante renalalogénico tratados con 1 mg/kg de Zenapax® cada 14 días hasta completar 5 dosis, el pro-medio de las concentraciones séricas (media desviación estándar) aumentaron entre la pri-mera (21 14 µg/ml) y la quinta dosis (32 22 µg/ml).La concentración sérica mínima mediala desviación estándar fue de 7,6 4,0 (g/ml antes de la quinta dosis). Se necesitan concen-traciones séricas de 0,5 a 0,9 µg/ml para saturar los receptores de IL-2, y concentracionesséricas de 5-10 µg/ml para inhibir la actividad biológica mediada por IL-2.El régimen reco-mendado de Daclizumab mantendrá las concentraciones séricas suficientes para saturar enla mayoría de los pacientes los alfa receptores IL-2R de los linfocitos T activados durantemás de 90 días posteriores al trasplante. Estos tres primeros meses constituyen el períodomás crítico post-trasplante.
La semivida estimada de eliminación terminal del Daclizumab osciló entre 270 y 919horas (promedio de 480 horas) en los enfermos que recibieron un trasplante renal alogé-nico y es equivalente a la descrita para la IgG humana la cual osciló entre las 432 y las 552horas (promedio de 480 horas). Esto es debido a la humanización de la proteína.
El análisis farmacocinético poblacional demostró que la eliminación sistémica delDaclizumab depende del peso corporal total, la edad, el sexo, la proteinuria y la raza.
59
Uso de Zenapax®(ficha técnica)

El efecto con oc i do del peso corporal sobre la el i m i n ación sistémica ju s tifica qu eZenapax® se do s i f i que en mg/kg y manti ene la ex posición al fárm aco den tro del 30% de laex posición de referencia en un gru po de pac i en tes con un amplio ra n go de caracter í s ti c a sdem ogr á f i c a s . No es nece s a rio realizar aju s tes de po s o l ogía basados en otras cova ri a n te si den ti f i c adas (sexo, pro tei nu ri a , raza y ed ad) en los pac i en tes con tra s p l a n te renal alog é n i co.
Pacientes pediátricos: Los resultados farmacocinéticos preliminares de un estudioen marcha de 30 pacientes pediátricos indican que las concentraciones séricas de Da-clizumab son comparables a las observadas en los pacientes adultos sometidos a trasplantey en tratamiento con el mismo régimen de dosificación. La subunidad Tac del receptor IL-2 se saturó inmediatamente después de la primera dosis de 1,0 mg/kg de Daclizumab y per-maneció saturada durante al menos los primeros tres meses del período post-trasplante. Lasaturación de la subunidad Tac del receptor IL-2 fue similar a la observada en pacientesadultos tratados con el mismo régimen de dosificación.
No existen interacciones farmacocinéticas entre Zenapax® y el ácido micofenólico,metabolito activo del micofenolato mofetilo (CellCept®).
5.3. Datos preclínicos sobre seguridad
D aclizumab fue bi en to l erado tras su ad m i n i s tración en bo lus intravenoso único o porvía su bc utánea a ra ton e s , ratas y con ej o s , en dosis de 50 a 125 mg/kg, y después de 28 días desu ad m i n i s tración a monos en dosis de 15 mg/kg. Uno de los 18 monos tuvo una re acc i ó na n a f i l á ctica al Dacl i z u m a b. Se mantuvi eron unas con cen trac i ones séricas de Daclizumab apre-c i a bl e s , exceptu a n do en 2 monos de los 18 que de s a rro ll a ron anti c u erpos anti - D acl i z u m a b.D aclizumab no pre s entó re activi d ad cru z ada in vi tro fren te a los cortes humanos de cri ocon-gel ación (de 28 órganos) a con cen trac i ones de hasta 56 mg/ml, que dem o s tra ron la ausen c i ade uniones no espec í f i c a s . D aclizumab no ha re su l t ado gen o t ó x i co en los en s ayos estándar.
6. Datos farmacéuticos6.1. Relación de excipientes
Polisorbato 80Cloruro sódicoFosfato sódico monobásicoFosfato sódico dibásicoÁcido clorhídricoHidróxido sódicoAgua para inyección
6.2. Incompatibilidades
No se ha observado ninguna incompatibilidad entre Zenapax® y las bolsas de cloru-ro de polivinilo o los equipos de perfusión.
6.3. Período de validez
18 meses.
60
Uso de Zenapax®(ficha técnica)

6.4. Precauciones especiales de conservación— Conservar entre 2-8ºC.— No congelar.— Proteger los viales de la luz.— Se ha demostrado la estabilidad química y física de la solución preparada para
usar, durante 24 horas a 2-8ºC, o durante 4 horas a temperatura ambiente. Sin embargo,desde un punto de vista microbiológico, el producto diluido debe usarse inmediatamente.No se debe guardar el producto tras su dilución a menos que ésta se haya realizado en con-diciones asépticas controladas y validadas. Si no se emplea inmediatamente, el usuario esresponsable del tiempo y de las condiciones de conservación previas a su administración.
6.5. Naturaleza y contenido del recipienteZenapax®, 25 mg (en 5 ml), se presenta en viales de vidrio tipo flint. Zenapax® está
disponible en envases de uno y tres viales
6.6. Instrucciones de uso,manipulación y eliminación (en su caso)
Zenapax® NO debe inyectarse directamente. Debe diluirse en 50 ml de una soluciónestéril de cloruro sódico al 0,9% antes de su administración intravenosa a los pacientes.Para mezclar la solución, no la agite para evitar que se forme espuma, sino que invierta labolsa suavemente. Procure mantener la esterilidad de la solución preparada, puesto que elmedicamento no contiene conservantes antimicrobianos ni sustancias bacteriostáticas.Zenapax® es una solución incolora que se suministra en viales de un solo uso, deberá dese-charse cualquier porción remanente del medicamento. Los productos de uso parenteraldeben inspeccionarse visualmente antes de su administración para descartar la presencia departículas o cambios de coloración. Una vez preparada la perfusión, se administrará inme-diatamente por vía intravenosa. Si se diluye en condiciones asépticas, puede conservarsedurante 24 horas en el frigorífico a una temperatura de 2-8ºC.
No se debe añadir ni tampoco perfundir otros med i c a m en tos / sustancias de form as i multánea a través de la misma vía intraven o s a .
7. Titular de la autorizaciónRoche Registration Limited40 Broadwater RoadWelwyn Garden City,Hertfordshire, AL7 3AY, Reino Unido
8. Números del registro comunitariode medicamentosEU/1/99/098/001 (envase de 1 vial) EU/1/99/098/002 (envase de 3 viales)
9. Fecha de la revisión del texto26.02.1999
10. PrecioZenapax® (5mg/ml) 1 vial de 5ml P.V.L.: 50.400 Ptas / 302,91 Euro. PVP MR: 77.326
Ptas / 464,74 Euro. PVP IVA: 80.419 Ptas / 483,33 Euro.Zenapax® (5mg/ml) 3 viales de 5 ml P.V.L.: 148.400 Ptas / 891,9 Euro. PVP MR:
227.683 Ptas / 1.368,4 Euro. PVP IVA: 236.790 Ptas / 1.423,14 Euro.
61
Uso de Zenapax®(ficha técnica)
Para más información contactar con Productos Roche, S.A.

Los datos globales obtenidos en ensayos clínicos controlados, han demostrado queZenapax® es eficaz y se tolera bien. En combinación con terapia inmunosupresora,Zenapax® reduce la incidencia de rechazo agudo en pacientes que reciben su primer tras-plante renal, sin aumentar la incidencia de efectos adversos.
Se pueden extraer las conclusiones siguientes con respecto al uso a corto plazo deZenapax®, conjuntamente con terapia inmunosupresora, en pacientes adultos receptoresdel primer aloinjerto renal:
• La dosis recomendada de Zenapax® es de 1,0 mg/kg, administrada en infusión intra-venosa en 50 ml de solución salina, en las 24 horas anteriores al trasplante, y, poste-riormente, cada dos semanas, hasta un total de cinco dosis.
• Zenapax® reduce hasta el 40% la incidencia de rechazo agudo del injerto durante los 6meses posteriores al trasplante.
• En estudios piloto, se ha demostrado que Zenapax®:
— mejora la supervivencia y la función del injerto
— mejora la supervivencia del paciente (un estudio)
— es menos inmunogénico que sus predecesores murinos
— tiene una vida media larga
— reduce la necesidad de terapia inmunosupresora secundaria
— permite administrar dosis acumuladas menores de esteroides.
• Zenapax® se tolera bien
• Zenapax® tiene un perfil de toxicidad favorable
• Zenapax® no parece aumentar la incidencia de efectos secundarios cuando se añade alas combinaciones existentes
• Zenapax® no da lugar a un aumento de la incidencia de las infecciones oportunistas
• Zenapax® no produce un aumento del riesgo de desarrollar neoplasias
62
R e s u m e n

Abramowicz D, Goldman M,de Pauw L etal.The long-term effects of prophylacticOKT3 monoclonal antibody in cadaverkidney transplantation – a single center,prospective, randomized study.Transplantation 1992; 54: 433-437.
Abramowicz D, Goldman M.The use ofOKT3 in clinical transplantation. In:Monoclonal Antibodies in Transplantation.RG Landes Co., Austin, Texas,1995: 99-135.
Abramowicz D, Norman D, Goldman M etal.OKT3 prophylaxis improves long-termrenal graft survival in high-risk patientsas compared to cyclosporine: combinedresults from the prospective, randomizedBelgian and US studies. Transplant Proc1995; 27: 852-853.
Amlot PL, Rawlings E, Fernando ON etal. Prolonged action of a chimeric IL-2(CD-25) monoclonal antibody used incadaveric renal transplantation.Transplantation 1995; 60: 748-756.
Amlot PL.The clinical and experimentaluse of monoclonal antibodies to theIL-2 receptor. In: Chatenoud L (ed.).Monoclonal Antibodies inTransplantation, RG Landes Co.,Austin, Texas,1995: 53-98.
Basadonna GP, Matas AJ,GillinghamKJ. Early versus late acute renal allo-graft rejection: impact on chronic rejec-tion. Transplantation 1993; 55: 993.
Brown PS, Parenteau GL, Dirbas FM etal. Anti-Tac,a humanized antibody tothe interleukin 2 receptor, prolongs pri-mate cardiac allograft survival. ProcNatl Acad Sci USA 1991; 88: 2663-2667.
Bruggemann M, Winter G, Waldmann H,Neuberger MS.The immunogenicity ofchimeric antibodies.J Exp Med 1989;170: 2153-2157.
Cantarovich D, Le MauffB, Hourmant Met al. Prevention of acute rejectionepisodes with an anti-interleukin-2 recep-tor monoclonal antibody. I. Results aftercombined pancreas and kidney transplan-tation. Transplantation 1994; 57: 198-203.
Cecka JM. Early rejection: determiningthe fate of renal transplants. TransplantProc 1991; 23: 1263-1264.
European MMF Cooperative StudyGroup. Placebo-controlled study ofmycophenolate mofetil combined withcyclosporine and corticosteroids for pre-vention of acute rejection. Lancet 1995; 345: 1321-1325.
Fayer BE,Soni PP, Binger MH et al.Determination of humanized-anti-Tac inhuman serum by a sandwich enzymelinked immunosorbent assay. J ImmunolMethods 1995; 186: 47-54.
Ferguson RM, Henry M,Elkhammas EA,Tesi RJ. Acute rejection episodes - bestpredictor of long-term primary cadavericrenal transplant survival. Am Soc TransplantSurgeons,18th Annual ScientificMeeting,Chicago, 1992: 83 (Abstract).
Flechner SM. Current status of renaltransplantation.The Urologic Clinics ofNorth America 1994; 21: 265-282.
Granjard P, Eygonnet JP, Defayolle M etal. Tolerance of equine (lympho) andrabbit (thymo) antilymphocyte globulin(ALG) in renal allografts: a prospectivemulticentre study. In: Touraine JL, ed.International Course on Transplantationand Clinical Immunology. Elsevier,Amsterdam,1989: 175.
Guex-Crosier Y, Raber J,Chan C-C, et al.Humanized antibodies against the a-chainof the IL-2 receptor and against the b-chainshared by the IL-2 and IL-15 receptors in a
monkey uveitis model of autoimmunediseases.J Immunol 1997; 158: 452-458.
Gulanikar AC, MacDonald AS,Sungurtekin U, Belitsky P. The incidenceand impact of early rejection episodeson graft outcome in recipients of firstcadaver kidney transplants.Transplantation 1992; 53: 323-328.
Hakimi J,Chizzonite R, Luke DR et al.Reduced immunogenicity and improvedpharmacokinetics of humanized anti-TACin cynomolgus monkeys.J Immunol 1991;147: 1352-1359.
Hakimi J, Ha VC, Lin P et al. HumanizedMikb1,a humanized antibody to the IL-2receptor b-chain that acts synergisticallywith humanized anti-Tac.J Immunol1993; 151: 1075-1085.
Hakimi J, Mould D, Waldmann TA et al.Development of Zenapax®: a humanizedanti-Tac antibody. Chapter 12; Harris WJ,Adair JR (eds) In Antibody TherapeuticsCRC Press 1997 pp277-300.
t‘Hart BA, Hakimi J. Evaluation of theeffect of humanized anti-TAC (HAT) andhumanized Mikb1 on collagen-inducedarthritis in the rhesus monkey.Unpublished ResearchReport No. 138779,1996.
Hourmant M, Le MauffB,Cantar ovich Det al. Prevention of acute rejectionepisodes with an anti-int erleukin-2 recep-tor monoclonal antibody. II. Results after asecond kidney transplantation.Transplantation 1994; 57: 204-207.
Junghans RP, Waldmann TA, Landolfi NFet al. Anti-Tac,a humaniz ed antibody tointerleukin 2 receptor with new featuresfor immunotherapy in malignant andimmune disorders.Cancer Res 1990; 50:1495-1502.
63
B i b l i o g ra f í a

Kirkman RL, Shapiro ME,Carpenter CB etal. Early experience with anti-Tac in clini-cal renal transplantation. TransplantationProceedings 1989; 21: 1766-1768.
Kirkman RL, Shapiro ME,Carpenter CBet al.A randomized prospective trial ofanti-TAC monoclonal antibody in humanrenal transplantation. Transplantation1991; 51: 107-113.
Kirkman R, Vincenti F, Pescovitz M et al.A Phase I/II randomized,double-blind,placebo controlled study of Zenapax® incombination with CellCept, Neoral andsteroids.[Abstract 1997]
Kreis H. Adverse effects associated with OKT3 immunosuppression in the preven-tion or treatment of allograft rejection.Clin Transplantation 1993; 7: 431-446.
Matas AJ,Gillingham KJ, Payne WD,Najarian JS.The impact of an acuterejection episode on long-term renalallograft survival (t1/2). Transplantation1994; 57: 857-859.
Morrison SL, Johnson MJ, Herzenberg LA,Oi VT. Chimeric human antibody molecules:mouse antigen-binding domains withhuman constant region domains. Proc NatlAcad Sci USA 1984; 81: 6851-6855.
Nashan B, et al. Reduction of AcuteRenal Allograft Rejection by Daclizumab.Transplantation,1999; 67: 110-115.
Nevis T, et al. Daclizumas (Zenapax) inPediatric Renal Allografts: Final Data.ASTP1999. Abstract 469
Norman DJ, Kahana L, Stuart FJ et al.A randomized clinical trial of inductiontherapy with OKT3 in kidney transplantation.Transplantation 1993; 55: 44-50.
Pankewycz, OG, Miao L, Isaacs R et al.Increased renal tubular expression of
transforming growth factor beta in humanallografts correlates with cyclosporinetoxicity. Kidney Int 1996; 50: 1634-1640.
Queen C,Schneider WP, Selick HE et al.A humanized antibody that binds to theinterleukin 2 receptor. Proc Natl AcadSci USA 1989; 86: 10029-10033.
Rubin LA, Nelson DL.The soluble inter-leukin-2 receptor: biology, function and clinical application. Ann Intern Med1990; 113: 619.
Schneider WP, Glaser SM, KondasJA, Hakimi J.The anti-idiotypic response bycynomolgus monkeys to humanized anti-TAC is primarily directed to complemen-tarity-determining regions H1,H2 and L3.J Immunol 1993;150: 3086-3090.
Shield,CF, Edwards,EB, Davies,DB et al.Antilymphocyte induction therapy incadaver renal transplantation.Transplantation 1997; 63: 1257-1263.
Simpson MA, Monaco AP. Clinical uses of polyclonal and monoclonal antilym-phoid sera. In: Chatenoud L (ed.)Monoclonal Antibodies inTransplantation, RG Landes Co, Austin,Texas,1995: 1-19.
Sollinger HW. Mycophenolate mofetil forthe prevention of acute rejection in pri-mary cadaveric renal allograft recipients.Transplantation 1995; 60: 225-232.
Soulillou JP, Cantarovich D, Le Mauff Bet al. Randomized controlled trial ofa monoclonal antibody against theinterleukin-2 receptor (33B3.1) ascompared with rabbit anti-thymocyteglobulin for prophylaxis againstrejection of renal allografts.N Engl J Med 1990; 322: 1175-1182.
Tricontinental Mycophenolate Mofetil
Renal Transplantation Study Group.A blinded, randomized clinical trial ofmycophenolate mofetil for the preventionof acute rejection in cadaveric renaltransplantation. Transplantation 1996;61: 1029-1037
Uchiyama T, Nelson DL,Fleisher TA,Waldmann TA.A monoclonal antibody(anti-Tac) reactive with activated andfunctionally mature human T cells. JImmunol 1981; 126: 1398-1403.
Vincenti F, Lantz M,Birnbaum J et al. Aphase I trial of humanized anti-interleukin-2 receptor antibody (HAT) in renal trans-plantation. Transplantation 1997; 63: 33-38.
Vincenti F, Kirkman R, Light S, et al.Interlenkin-2-receptor blockade withdaclizumab to prevent acute rejection inrenal transplantation. Daclizumab TripleTherapy Study Group. N Engl J Med 1998;338: 161-165
Waldmann TA.The interleukin-2 receptor.J Biol Chem 1991; 266: 2681-2684.
Waldmann H, Cobbold S.The use ofmonoclonal antibodies to achieveimmunological tolerance. TrendsPharmacol Sci 1993; 14: 143-148.
Waldmann TA, Goldman CK.The multi-chain interleukin-2 receptor: a target forimmunotherapy of patients receiving allo-grafts. Am J Kidney Dis 1989; XIV (No 5Suppl 2): 45-53.
Waldmann TA, Goldman C, Top L et al.The interleukin-2 receptor: a target forimmunotherapy. Adv Exp Med Biol 1992;323: 57-66.
Waldmann TA,White JD, Goldman CK etal.The interleukin-2 receptor: a target formonoclonal antibody treatment of humanT-cell lymphotrophic virus I-induced adultT-cell leukemia.Blood 1993; 82: 1701-1712.
64
Bibliografía(continuación)

65

Josefa Valcárcel, 42. 28027 MadridTeléf.: 91 324 81 00. Fax 91 324 83 50www.roche.es

















