Reacciones Unimoleculares y de Asociación
8
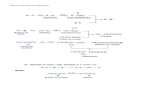
Reacciones unimoleculares y de asociación CH 3 CN CH 3 NC C 2 H 6 2 CH 3 C 2 H 5 Cl C 2 H 4 + HCl isomerización disociación asociación activación estabilización Los reactivos deben ser excitados y los productos estabilizados para que la reacción se produzca Reacciones unimoleculares y de asociación Las colisiones intermoleculares son impres- cindibles: a) para estabilizar una molécula que recién se acaba de formar b) para activar térmicamente una molécula que isomeriza o se disocia en forma unimo- lecular Para un tratamiento avanzado: http://physchem.ox. ac.uk/~vallance/pdfs/UnimolecularReactionNotes.pdf H + H H 2 ; estimación de k La asociación de átomos de H es muy poco efectiva y aumenta con la presión total: H + H + M E P r 0 = 4161 cm 1 0 = 1,25 10 14 s 1 c = 5 10 9 s 1 (1 atm) z 2 10 13 dm 3 s 1 k = z. c / 0 = 8 10 18 dm 3 s 1 = 4,8 10 6 M 1 s 1 colisión con un tercer cuerpo M H + H H 2 * H 2 = 0 Mecanismo de asociación: H + H H 2 La asociación de átomos no es estrictamente bimolecular E P r colisión con un tercer cuerpo M H + H H 2 * H 2 = 0 2 2 2 1 2 1 2 M H M * H H H * H * H H H k k k 0 c 1 2 1 2 1 2 1 1 2 2 1 2 1 2 2 1 ] M [ ] H [ ] M [ ] M [ ] H [ ] M [ ] M [ ] H [ z k k k k k k k k k k k k k k v
Transcript of Reacciones Unimoleculares y de Asociación

Reacciones unimoleculares y de asociación
CH3CN CH3NC C2H6 2 CH3
C2H5Cl C2H4 + HCl
isomerización disociaciónasociación
activa
ción
esta
bili
zaci
ón
Los reactivos deben ser excitados y los productos estabilizados para que la reacción se produzca
Reacciones unimoleculares y de asociación
Las colisiones intermoleculares son impres-cindibles:
a) para estabilizar una molécula que recién se acaba de formar
b) para activar térmicamente una molécula que isomeriza o se disocia en forma unimo-lecular
Para un tratamiento avanzado: http://physchem.ox.ac.uk/~vallance/pdfs/UnimolecularReactionNotes.pdf
H + H H2; estimación de k
La asociación de átomos de H es muy poco efectiva y aumenta con la presión total: H + H + M
EP
r
0 = 4161 cm 1
0 = 1,25 1014 s 1
c = 5 109 s 1 (1 atm)
z 2 10 13 dm3s 1
k = z. c / 0
= 8 10 18 dm3s 1
= 4,8 106 M 1s 1
colisión con untercer cuerpo M
H + HH2*
H2
= 0
Mecanismo de asociación: H + H H2
La asociación de átomos no es estrictamentebimolecular
EP
r
colisión con untercer cuerpo M
H + HH2*
H2
= 0222
12
12
MHM*H
HH*H
*HHH
k
k
k
0
c
1
21
2
1
21
1
221
21
221
]M[
]H[]M[
]M[]H[
]M[]M[
]H[
zkkk
k
kkk
kk
k
kkk
kv

Mecanismo de asociación: A + B AB
2
1
1
MABM*AB
BAAB*
*ABBA
k
k
k
]M[]M[
]A][B[]M[
]M[
21
21bim
21
21
kkkk
k
kkkk
v
kbim
[M]00
k1 k1k2/k 1
]M[1
21bim k
kkk 1bim kk
Las reacciones de asociación son de orden 3 a bajas presiones y de orden 2 a altas presiones
orden 3 orden 2
Reacciones unimoleculares
La activación puede producirse por colisiones, por irradiación con luz o partículas, por ultrasonido, etc.
activa
ción
CH3CN + Ar CH3NC* + Ar
CH3 + CF3 CH3CF3*
H2CO + h H2CO* H2 + CO
activación térmica (colisional)
activación química (colisional)
activación fotoquímica
Desarrollo de la teoría
Perrin (1919) Activación por radiación IR
Languir (1920) Crítica al argumento de Perrin
Lindemann (1922) Activación por colisiones
Hinshelwood (1926) Activación vibracional
Kassel / Rice-Ramsperger (1928) Concentración de energía en un oscilador crítico
Marcus (1952) Extensión de la teoría conside-rando la entropía de activación
Reacciones unimoleculares: AB Productos
2
1
1
ProductosAB*
MBAM*AB
M*ABMAB
k
k
k
21
21uni
21
21
]M[]M[
]AB[]M[
]M[
kkkk
k
kkkk
v
kuni
[M]00
k1
orden 2 orden 1
]M[1uni kk kkuni
Las reacciones unimoleculares son de orden 2 a bajas presiones y de orden 1 a altas presiones
k = k1k2/k 1
k
Teoría de Lindemann (activación térmica)
k /2
[M]½
[M]½ =k /k1

Reacciones unimoleculares: AB Productos
Graficando 1/kuni vs. 1/[M]se obtienen k1 y k = k1k2/k 1
2
1
1
ProductosAB*
MBAM*AB
M*ABMAB
k
k
k
]M[111
1uni kkk21
21uni ]M[
]M[kk
kkk
1/kuni
1/[M]00
1/k
1/k1
Interpretación de k1, k 1 y k2
2
1
1
ProductosAB*
MBAM*AB
M*ABMAB
k
k
k
)/exp( B01 TkEzk
zk 1
constante2k
AB se activa por colisiones y AB* se desactiva con una colisión o reacciona con k2 independiente de E
E0
k1 k 1
k2
E
Fallas de la teoría de Lindemann
El mecanismo de Lindemann no representa correctamente los resultados experimentales
1/kuni
1/[M]00
1/kLa representación no es lineal y el cambio de orden se produce a mayor presión; además [M]½ k /k1
2/k
1/[M]½
2/k
1/k
1/[M]½
1
2
)/exp( B01 TkEzk
Teoría de Lindemann-Hinshelwood
Además de las colisiones, la energía vibracional contribuye a superar la barrera de activación
)/exp()()!1(
1)( B
B
1
TkETk
Es
EPs
s
E0 E
P(E)P(E) es la función de distribución de la energía en s modos norma-les de vibración clásicos
)/exp()!1(
1)( B0
1
B
00 TkE
TkE
sEEP
s
E0 >> kBT > h
1

Teoría de Lindemann-Hinshelwood
La teoría justifica adecuadamente los altos valores de k1 encontrados experimentalmente
2
1
1
ProductosAB*
MBAM*AB
M*ABMAB
k
k
k
)/exp( B01 TkEzk
zk 1
)/exp()!1(
1B0
1
B
01 TkE
TkE
szk
s
La energía contenida en s osci-ladores contribuye a superar la barrera de activación
1
E0 >> kBT > h
Teoría de Rice-Ramsperger-Kassel
La teoría calcula la probabilidad de que la energía vibracional se concentre sobre la unión a romper
)(11 Ekk
zk 1
)(22 Ekk1
02 1.)(
s
EE
Ek
La energía vibracional migra entre los osciladores hasta que se concentra en un oscilador crítico cuya frecuencia es
)(ProductosAB*
MBAM*AB
)(M*ABMAB
2
1
1
Ek
k
Ek2
Teoría de Rice-Ramsperger-Kassel
Esto explica por qué la representación 1/kuni vs. 1/[M] no es lineal como predice la teoría de Lindemann
E
f(E)
E0
P(E)
)(ProductosAB*
MBAM*AB
)(M*ABMAB
2
1
1
Ek
k
Ek2
A bajas presiones no semantiene el equilibrio térmico y la reacción sevuelve más lenta
10
2 1.)(s
EE
Ek
[M]00
kuni
Límite de alta presión
10
2 1.)(s
EE
Ek
)(ProductosAB*
MBAM*AB
)(M*ABMAB
2
1
1
Ek
k
Ek
)/exp()()!1(
1)( B
B
1
TkETk
Es
EPs
s
)/exp(d)()( B021
21
0
TkEEEkEPk
kkk
E
El factor preexponencial en el límite de alta presión es la frecuencia del oscilador crítico
)!1(d)exp(0
1 sxxxs
TkEEx B0 /)(

Resumen
)(ProductosAB*
MBAM*AB
)(M*ABMAB
2
1
1
Ek
k
Ek
E0
k1(E) z
k2(E)E
1/kuni
1/[M]00
1/k
)/exp( B0 TkEk
A alta presión:
A baja presión no hay equilibrio para E > E0 y la constante uni-molecular es menor que la pre-dicha por Lindemann
Fallas de la teoría de Rice-Ramsperger-Kassel
36117,21400-2300C3H8 CH3 + C2H5
26115,61308-1498c-C4H8 2 C2H4
37616,0CH3OH CH3 + OH
23013,01100-1540(CHO)2 2CO + H2
27615,5690-1038c-C3H6 C3H6
37216,7840-913C2H6 2CH3
E(kJ/mol)
logAT(K)
Reacción
El factor preexponencial en el límite de alta presión es mayor que una frecuencia típica, 1013-1014 s 1
Teoría de Rice-Ramsperger-Kassel-Marcus
El factor entrópico, que puede ser mucho mayor que 1, justifica los altos valores del factor preexponencial
La teoría considera explícitamente las vibraciones y las rotaciones moleculares aplicando conceptos similares a los de la teoría del complejo activado.
En el límite de alta presión:
)/exp(
)/exp(
B
0Avvr
"vrB
RShTek
A
RTNqqqq
hTk
k
1,7 1013 (T/300 K)s 1
Entropías de activación
7417,21400-2300C3H8 CH3 + C2H5
29N2O5 NO2 + NO3
30cis-C6H8 1,3-c-C6H8 (1)
4515,61308-1498c-C4H8 2 C2H4
513,01100-1540(CHO)2 2CO + H2
4315,5690-1038c-C3H6 C3H6
6616,7840-913C2H6 2CH3
S(J.mol 1K 1)
logAT(K)
Reacción
La entropía de activación da cuenta de la dureza del complejo activado
(1) cis-hexatrieno 1,3 ciclohexadieno

Reacciones en solución
En lugar de colisiones existen “encuentros”
El solvente actúa como baño térmico
Se estabilizan las especies cargadas
La fuerza iónica y la constante dieléctrica influyen sobre las reacciones entre iones
La presión influye sobre la velocidad si el complejo se forma con cambio de volumen
El solvente puede participar como reactivo o catalizador
Colisiones y encuentros: efecto caja
La viscosidad limita el número de encuentros pero dentro de ellos se producen muchas colisiones
gas líquido
ttcolisiones
Colisiones y encuentros: efecto caja
En fase líquida no se produce CH3CD3 porque la recombinación de radicales en la caja es muy rápida
Experiencia de Lyon y Levy (1961)
CH3N2CH3 [CH3…N2…CH3] [C2H6…N2] C2H6 + N2
CD3N2CD3 [CD3…N2…CD3] [C2D6…N2] C2D6 + N2
2CH3 + N2
2CD3 + N2
CH3CD3 gas líquido
Control químico y control difusional
En una reacción controlada por difusión se consumen rápidamente las moléculas de reactivo cercanas
Sólo unas pocas colisiones son reactivas
La reacción se producedurante el primer encuentro
t t
Control químico Control difusional

Reacciones controladas por difusión
Estas ecuaciones valen para reactivos neutros; por ello la constante de H+ + OH es mayor
A
rc
DDrtn
dd
)(4dd B
BA2B1a. ley de Fick
rrA+rB B
BABBA
BBA2B d)(
4
ddd
c
crr rr
cDDr
rtn
constante = 0
))((4 BABAAvdif DDrrNkEn agua:
rA+rB=5Å
DA+DB=10 5cm2s 1
kdif = 4 109 M 1s 1 pero kH++OH = 1,5 1011 M 1s 1
B
Constantes de velocidad en gases y líquidos
Para una reacción cualquiera en fase gaseosa a baja presión:
k = zgas.Preac
k = zliq.Preac zgas.Preac << kdif
k kdif
k zgas
Constante de velocidad límite en fase gaseosa:
Para una reacción “lenta” en fase líquida (control químico):
Constante de velocidad límite en fase líquida (control difusional):
Se supone zliq zgas; Preac es la probabilidad de que los reactivos reaccionen antes de separarse
El solvente como baño térmico
Se mantiene siempre equilibrio entre reactivos y complejo; la reacción no ocurre adiabáticamente
gas líquido
activa
ción
desactivació
n
Constante dieléctrica y fuerza iónica
Menor interacción a mayor constante dieléctrica y mayor fuerza iónica
+

Efecto de la constante dieléctrica
RTG
hTk
k expB
AzA + BzB Productos
G = G quím. + G eléctr. = G quím. + zAzB/ r
log k = log k( = ) zAzB/ r
+
r
La constante dieléctrica tiene efecto positivo o negativo dependiendo de la carga de los reactivos
Efecto de la fuerza iónica
La fuerza iónica tiene efecto positivo o negativo dependiendo de la carga de los reactivos
BBAABBBAA 'cc
KhTkcc
Kcv
AzA + BzB Productos
BBAA ccc
K
BAB 'KhTk
k IzA i2
DHilog
IzzAIkk BADHi 2)0(loglog
Presión y volumen de activación
El efecto positivo de la presión sobre la velocidad de reacción es típico en las cicloadiciones
V < 0*
CC
C
CC
C
.. +
RTG
hTk
k expB
RTV
pkln
V = 20 a 40 cm3.mol 1