Resumen MGA
33
Contenido Resumen.....................................................3 Introducción................................................4 Capítulo I: Antecedentes....................................5 I.1. Farmacopea de los Estados Unidos Mexicanos..........5 I.1.1........................Métodos Generales de Análisis 6 I.2. Fármaco DCD........................................ 23 I.3. Características del área de trabajo y ubicación geográfica.............................................24 I.4. Problema de Resolver...............................26 I.5. Alcances y Limitaciones............................27 Justificación............................................ 28 Objetivos................................................ 29 Capitulo II: Materiales y Métodos..........................30 Descripción............................................30 Determinación de agua por Karl- Fischer...............31 Medición del pH........................................33 Cromatografía..........................................35 Disolución.............................................37 Uniformidad de dosis...................................40 Método de Análisis para el Fármaco DCD.................44 Capitulo III: Resultados y Discusión.......................48 Conclusiones...............................................60 Bibliografía...............................................61 Página 1
-
Upload
cristinamartinezg -
Category
Documents
-
view
260 -
download
1
description
MGA FEUM
Transcript of Resumen MGA

Contenido
Resumen..................................................................................................................................3
Introducción............................................................................................................................4
Capítulo I: Antecedentes.........................................................................................................5
I.1. Farmacopea de los Estados Unidos Mexicanos....................................................5
I.1.1. Métodos Generales de Análisis........................................................................6
I.2. Fármaco DCD.....................................................................................................23
I.3. Características del área de trabajo y ubicación geográfica.................................24
I.4. Problema de Resolver.........................................................................................26
I.5. Alcances y Limitaciones.....................................................................................27
Justificación.......................................................................................................................28
Objetivos...........................................................................................................................29
Capitulo II: Materiales y Métodos........................................................................................30
Descripción...................................................................................................................30
Determinación de agua por Karl- Fischer....................................................................31
Medición del pH............................................................................................................33
Cromatografía...............................................................................................................35
Disolución.....................................................................................................................37
Uniformidad de dosis....................................................................................................40
Método de Análisis para el Fármaco DCD...................................................................44
Capitulo III: Resultados y Discusión....................................................................................48
Conclusiones.........................................................................................................................60
Bibliografía...........................................................................................................................61
Anexos..................................................................................................................................62
Abreviaturas..........................................................................................................................68
Glosario.................................................................................................................................69
Página 1

Resumen
Los Laboratorios Senosiain es una compañía farmacéutica mexicana dedicada al
desarrollo, fabricación y comercialización de productos farmacéuticos de calidad, con la
producción de antibióticos y otros productos para el uso humano, la línea veterinaria así
como farmoquímicos, para el abastecimiento continúo de materias primas de alta calidad,
por lo que es necesario el desarrollo de pruebas y análisis para la determinación de
identidad, pureza y calidad así como la actualización constante de estos métodos.
En este trabajo se llevaron a cabo la actualización de los métodos correspondientes a la
actualización de la Farmacopea de los Estados Unidos Mexicanos 11a edición.
Como parte de este proyecto se realizó un análisis comparativo de las actualizaciones
con la versión anteriormente publicada de los métodos generales de análisis que son
utilizados en la validación y en los procesos de estabilidad para los fármacos valorados en
el área de Investigación y Desarrollo Farmacéutico.
Al concluir el análisis comparativo se elaboraron los protocolos para los métodos generales
y reporte de resultados para Pruebas de Estabilidad del Medicamento DCD, donde se
registraron los valores obtenidos de los lotes que se sometieron a evaluación, donde de
determino que el principio activo es estable durante los primero 6 meses.
Página 2

Introducción
La Farmacopea de los Estados Unidos Mexicanos (FEUM) es el documento expedido por
la Secretaria de Salud que establece los métodos de análisis y las especificaciones
requeridas para asegurar la identidad, pureza y calidad de los medicamentos, productos
biológicos y biotecnológicos, así como de sus materia primas en el caso de fármacos y
aditivos.
Los textos presentes en los Métodos Generales de Análisis (MGA) de la FEUM son
comúnmente referenciados dado por la consulta de sus monografías, lo cual exige de una
constante actualización con base a la Armonización Farmacopeica , que deriva
esencialmente de las observaciones realizadas a los contenidos ya existentes, o bien por la
inclusión de nueva información.
Los MGA son utilizados en todos los protocolos que se utilizan en la Validación y Pruebas
de Estabilidad en la evaluación de medicamentos y materias primas, esto implica el uso de
criterios como exactitud, precisión, sensibilidad, límites de detección, costos, número de
muestras a analizar, cantidad de muestra disponible, entre otros.
En necesaria la adición de nuevos métodos modificado otros; de acuerdo a los avances
científicos y tecnológicos, que permiten asegurar la calidad de las materias primas e
insumos para la salud en beneficio de la seguridad y eficiencia terapéutica. Con la correcta
actualización de los protocolos de los MGA se analizara si los medicamentos y materias
primas, cumplen con los parámetros de evaluación y se determina si son útiles para las
evaluaciones iniciales y a largo plazo. De tal manera que todos los resultados obtenidos
deber ser archivados y aprobados por los responsables del área involucrada en la redacción
de los protocolos de los métodos con las especificaciones requeridas para cada uno de los
medicamentos a analizar.
Página 3

Capítulo I: Antecedentes
I.1. Farmacopea de los Estados Unidos Mexicanos
En Septiembre 1984, una vez implementada la Ley General de Salud se creó la Comisión
Permanente de la Farmacopea de los Estados Unidos Mexicanos (CPFEUM) que da inicio a
la historia contemporánea de la Farmacopea de los Estados Unidos Mexicanos (FEUM).
Documento que se ha ido actualizando progresivamente, abarcando controles de calidad
para los fármacos y medicamentos así como para medicamentos alopáticos, homeopáticos,
herbolarios, dispositivos médicos y controles de calidad para los establecimientos como son
las farmacias en sus suplementos, el cual que se presentó bajo la definición final:
“Documento expedido por la Secretaría que consigna los métodos generales de análisis y
los requisitos sobre identidad, pureza y calidad de los fármacos, aditivos, medicamentos,
productos biológicos y demás insumos para la salud.” (FEUM, 2014)
La presentación de la información en la FEUM se divide en diversos apartados como:
Orden de los capítulos, Presentados en un orden lógico y no alfabético.
Orden de las monografías, La monografías se siguen en orden alfabético, en donde a las
preparaciones farmacéuticas, disoluciones, preparaciones o extractos naturales, se ha dado
prioridad al nombre del principio activo o fármaco seguido del tipo de preparación, en
cursivas, separado con una coma del resto del título.
Orden de las soluciones, apartado dividido en las soluciones de tipo
indicadoras (SI), amortiguadoras (SA), volumétricas (SV), y las soluciones reactivo (SR).
Soluciones empleadas en las monografías.
Orden de los métodos generales de análisis (MGA), ordenados de manera alfabética.
Los MGA en las monografías suelen indicarse únicamente por la clave o cuando sea
requerida la clave va seguida del título o el nombre de la prueba en particular, por ejemplo,
MGA XXXXX/ MGA XXXX, XXXX.
Página 4

Índices, Contenido de los índices analíticos detallados.
La FEUM como un documento oficial autorizado por la Secretaria de Salud establece los
requisitos mínimos de calidad que deben satisfacer los productos nacionales e
internacionales y, por lo tanto, no se permite comercializar los que no cumplan al menos los
requisitos que señala. (FEUM, 2014)
I.1.1. Métodos Generales de Análisis
Los Métodos Generales de Análisis establecen la metodología analítica para identificar y
valorar sustancias, así como pruebas límite y análisis oficiales, sobre los cuales se basan
las monografías contenidas en la FEUM, en las cuales normalmente, las pruebas deben
realizarse a una temperatura entre 15°C y 25°C, a menos que en la monografía se indiquen
otros valores, ya sea en cuestiones de temperatura u otros tratamientos específicos que
deban aplicarse a la muestra. (FEUM, 2014)
Como punto importante en la valoración de los Principios Activos y sus derivados en
Fármacos y Medicamentos son las sustancias de referencias (SRef) también conocidos
como estándares químicos de referencia de representan un factor clave en el desarrollo y
fabricación de productos farmacéuticos de alta calidad.
Este tipo de sustancias son muy importantes para la industria farmacéutica ya que
constituyen una herramienta práctica, directa y confiable en los dictámenes para garantizar
los resultados analíticos de laboratorios de control de calidad.
Las SRef requieren un almacenamiento y manejo estricto, de tal manera que deben ser
almacenadas en sus envases originales perfectamente cerrados y a temperaturas y
humedades de acuerdo a lo indicado en la etiqueta o certificado. (USP, 2015)
Por lo tanto la selección de un Método de Análisis se basa en criterios como exactitud,
precisión, sensibilidad, límites de detección, costos, numero de muestras a analizar,
cantidad de muestra disponible, entre otros, ocasionalmente la independencia de estos
parámetros no logra generar un equilibrio adecuado, de tal manera que es permitido hacer
uso de métodos no indicados por la Farmacopea, métodos que debieron haber pasado por
Página 5

una validación previa y se encuentren autentificados por la autoridad sanitaria, con
fundamentos técnicos y científicos. (FEUM, 2011)
Las monografías de los MGA contienen una serie de elementos comunes que facilitan la
identificación apropiada de los principios activos así como de los fármacos, los cuales se
desglosan en las siguientes características:
Título, el cual es asignado de acuerdo a los criterios descritos en el numeral del Orden de
las monografías correspondiente a la Denominación Común Internacional establecida por
la Organización Mundial de la Salud (OMS).
Fórmula desarrollada, fórmula condensada, peso o masa molecular, nombre químico,
donde las sustancias simples no combinadas (aditivos y fármacos) se les asigna la formula
desarrollada y el o los nombres correspondientes a la nomenclatura IUPAQ (International
Union Pure and Applied Chemistry) acompañada de la formula condensada y masa
molecular.
Contenido, determina los límites de la sustancia, preparación o producto biológico
referido en la monografía.
Sustancias de Referencia, las SRef será indicada por la monografía de referencia ya sea
haciendo uso de las que son establecidas por la FEUM, sin embargo, si estas no están
disponibles podrá hacerse uso de SRef que este avaladas por organismos nacionales.
Descripción, apartado destinado a la identificación de la sustancia, preparación o
producto referido de acuerdo a sus características físicas y organolépticas.
Ensayos de identidad, son pruebas que estrictamente no proporcionaran la confirmación
de la estructura química o composición de la sustancia, sin embargo, podrán corroborar
que una sustancia se ajusta a la descripción. Generalmente es utilizado el espectro IR.
Análisis y valoración, contiene las pruebas que se realizarán a la muestra a analizar para
determinar la cantidad de la sustancia de interés de acuerdo a los límites establecidos.
Página 6

Los MGA utilizados comúnmente para la Valoración y pruebas de estabilidad de las
muestras incluye los siguientes métodos:
Determinación de agua por Karl- Fischer (MGA 0041)
MGA para la determinación de agua de una muestra por el método Karl – Fischer.
Aplicable aquellos productos que sus monografía lo indique.
Fundamento. Se basa principalmente en la relación cuantitativa producida entre el agua y
un reactivo constituido por dióxido de azufre y yodo en piridina anhidra y metanol anhidro
siguiendo las siguientes reacciones:
3 C5 H 5 N+ I 2+H 2O+SO2→ 2C5 H5 N ∙ HI+C5 H5 N ∙ SO3
C5 H5 N ∙ SO3+CH 3 OH →C5 H5 N ∙ HSO4 CH3
Una vez que el agua reacciona con el yodo libre en la solución, se produce el cambio de
color y el punto final de la titulación puede determinarse electrométricamente utilizando un
microamprerimetro, dado por la diferencia de potencial en la reacción teniendo con especial
cuidado que los reactivos y los recipientes donde se lleve a cabo la reacción no tengas
contacto con la humedad atmosférica.
Debido a las condiciones bajo las que se lleva la reacción, la estequiometria no es exacta y
la repetabilidad depende de factores externos como la concentración de los ingredientes del
reactivo, la naturaleza del disolvente y la técnica utilizada en la determinación especifica.
Equipo. Se requiere de un aparato capaz de proporcionar la adecuada exclusión de
humedad atmosférica y la determinación del punto final de la reacción, donde el puto final
se determina eléctricamente con el empleo de un circuito eléctrico simple capaz de
registrar un voltaje aproximado de 200 mV de potencial aplicado entre los electrodos de
platino sumergidos en la solución titular.
Los aparatos comerciales consisten en un sistema cerrado con una o dos buretas
automáticas y un vaso de titulación cubierto herméticamente, equipado con los electrodos
Página 7

y un agitador magnético adicionado con un desecante y un vaso de titulación. (FEUM,
2010) (FEUM, 2014)
Medición del pH (MGA 0701)
El pH como indicador de la acides de una sustancia, de acuerdo a esto la escala de pH está
dada por una serie de valores que representan la concentración de los iones hidrógeno en
una solución expresados como la concentración de iones hidrogeno en moles/Lt. De esta
manera se define al pH como la actividad del ion hidrógeno
pH=−log¿¿
Fundamento. Esta prueba se basa en la determinación de la actividad de iones hidrógeno
mediante un instrumento denominado potenciómetro, este aparato detecta el potencial en
milivolts y en unidades de pH a través del par de electrodos, principalmente de vidrio
debido a la respuesta inmediata a los cambios rápidos de las concentraciones de iones
hidrogeno aun en soluciones poco reguladas.
Los valores de pH dependen de la temperatura, al ser esta una característica principal las
mediciones deben efectuarse a determinadas temperaturas constantes.
Equipo. El Potenciómetro (pH-metro), aparato empleado para la medición de pH, opera
sobre el principio de equilibrio cero al medir el potencial de la solución a través de los
electrodos en milivolts para transformar a unidades de pH.
Debido a variaciones en la naturaleza y la apropiada operación del potenciómetro, es
necesario examinar los electrodos observando si presentan el puente salino (de no ser así
verificar las indicaciones del fabricante) y hacer uso de dos Soluciones Amortiguadoras
(SA) para la calibración del equipo.
Las SA empleadas pueden ser preparadas de acuerdo al valor de pH requerido, como se
muestra a continuación:
Solución A. Tetraoxalato de potasio 0.05 M. Disolver 12.61 g de tetraoxalato de potasio
dihidratado [KH3(C2O4)2 ∙ 2H2O] en agua hasta obtener 1 000 mL.
Página 8

Solución B. Biftalato de potasio 0.05 M. Disolver 10.12 g de biftalato de potasio
(KHC8H4O4) previamente secado a 110°C durante 1 h, en agua hasta obtener 1 000 mL.
Solución C. Fosfato equimolal 0.05 M. Disolver 3.53 g de fosfato dibásico de sodio
(Na2HP04) y 3.39 g de fosfato monobásico de potasio (KH2P04), cada uno previamente
secado a 120°C durante 2 h en agua, hasta obtener 1 000 mL.
Solución D. Tetraborato de sodio 0.01 M. Disolver 3.80 g de tetraborato de sodio
decahidratado (Na2B4O7 ∙ 10H20) en agua hasta obtener 1 000 mL. Proteger la solución de
la absorción de bióxido de carbono.
Solución E. Hidróxido de calcio saturado. A 25°C. Agitar un exceso de hidróxido de
calcio [Ca(OH)2] en agua, decantar a 25°C antes de su uso. Proteger la solución de la
absorción de bióxido de carbono.
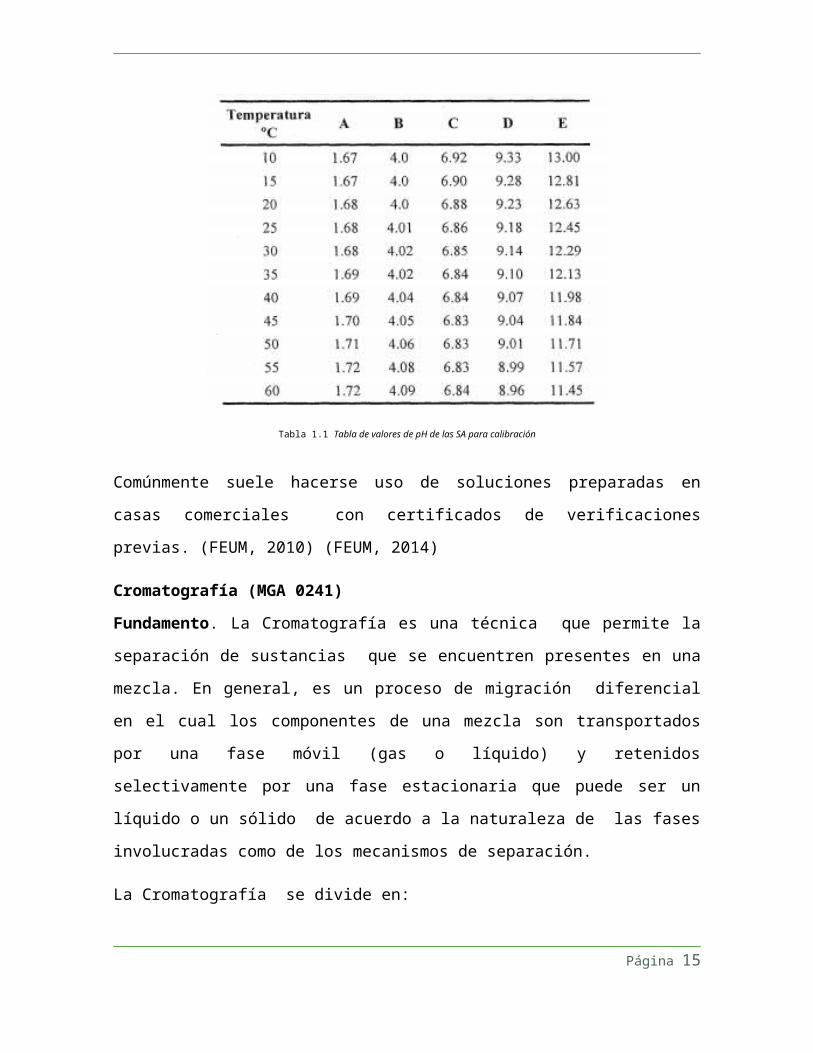
En la Tabla 1.1 Tabla de valores de pH de las SA para calibración se indican los valores de
pH que presentan las soluciones amortiguadoras en función de la temperatura. En caso
necesario las soluciones deberán ajustarse al pH indicado, con otra solución de referencia.
Tabla 1.1 Tabla de valores de pH de las SA para calibración
Página 9

Comúnmente suele hacerse uso de soluciones preparadas en casas comerciales con
certificados de verificaciones previas. (FEUM, 2010) (FEUM, 2014)
Cromatografía (MGA 0241)
Fundamento. La Cromatografía es una técnica que permite la separación de sustancias
que se encuentren presentes en una mezcla. En general, es un proceso de migración
diferencial en el cual los componentes de una mezcla son transportados por una fase móvil
(gas o líquido) y retenidos selectivamente por una fase estacionaria que puede ser un
líquido o un sólido de acuerdo a la naturaleza de las fases involucradas como de los
mecanismos de separación.
La Cromatografía se divide en:
I. Cromatografía de gases
En la cromatografía de gases se puede diferenciar como se muestra en la Tabla 1.2
Cromatografía de gases
Tabla 1.2 Cromatografía de gases
Es decir, en este tipo de cromatografía la fase móvil es un gas y la estacionaria es un sólido
(cromatografía gas – solido) o un líquido (cromatografía gas- líquido). En la primera, el
proceso de separación se lleva a cabo por absorción entre el gas que transporta al soluto
mientras que en la segunda, la partición se lleva a cabo entre una fase estacionaria liquida
que cubre a un sólido inerte y el gas transporta al soluto.
Cuando se introduce una sustancia en la corriente del gas, esta se volatiliza por la elevada
temperatura y de esta manera es transportada por el gas transportador a lo largo de la
Página 10

comuna donde se distribuye entre la fase sólida y liquida. Este proceso de partición o
reparto entre ambas fases está definido por el factor de capacidad K´, determinado por la
cantidad o el tiempo de residencia de la sustancia entre las fases respectivas.
Equipo. El Cromatógrafo de gases es un aparato simple en el cual el gas transportador,
generalmente está disponible en cilindros unidos a un manómetro para regular su presión,
lo que permite el control adecuado de la velocidad de flujo del gas requerida. Los gases
comúnmente utilizados como transportadores son el helio, el nitrógeno y algunos otros
gases inertes de acuerdo a las características del detector.
En este tipo de equipos las muestras se inyectan con una jeringa a través de un sello de
hule o silicón que se encuentran en la puerta de inyección y deben encontrarse equipados
con una columna de vidrio o metal, localizada en un horno capaz de mantener la
temperatura seleccionada ya que la salir de la columna los componentes saldrán de forma
individual para dirigirse al detector que podría ser de tipo de conductividad térmica,
ionización de flama alcalina, captura de electrones o espectrómetro de masas.
Una vez en el detector, la temperatura debe controlarse para evitar la condensación de la
muestra, con el fin de que este emita las correctas señales a través de un amplificador o
electrómetro conectado automáticamente a un aparato capaz de graficar la señal conocida
como cromatograma. (FEUM, 2010) (FEUM, 2014)
II. Cromatografía de líquidos de Alta Resolución
En la Cromatografía de Líquidos de Alta Resolución (CLAR), también conocida como
Cromatografía de Líquidos de Alta Presión (CLAP), los factores que generan éxito en la
técnica se basan en la preparación de la muestra, el tipo de columna, la velocidad de flujo
de la fase móvil, el tipo de detección, el algoritmo de detección entre otras características.
La separación de los componentes de una mezcla en la CLAR es resultado de la
interacción entre la fase estacionaria y la fase móvil. Dichos componentes se separan en la
columna y al salir de ésta son conducidos por la fase móvil en el orden en que surgieron,
hacia un detector donde se registra una respuesta proporcional a sus concentraciones y
tiempos de retención en la columna.
Página 11

El grafico resultante se denomina cromatograma, donde se muestra cada compuesto que
sale de la columna en forma de picos gausianos, es decir, de forma simétrica con un tiempo
de retención característico por lo que este tiempo puede emplearse para identificar el
compuesto. (FEUM, 2010) (FEUM, 2014).
Los mecanismos o procesos de separación surgen como resultado de la retención de las
moléculas presentes en la muestra por acción de la fase estacionaria se sugieren los
diferentes tipos de cromatografía de líquidos:
Partición (liquido- liquido), que consta de una fase estacionaria liquida de
composición diferente a la de la fase móvil e inmiscibles. Donde las moléculas de la
muestra se distribuyen entre ambas fases como sucede en la extracción líquido –
liquido.
Adsorción (liquido – solido), en este tipo de cromatografía se incluye partículas de
gran aérea superficial donde las moléculas son atraídas y a su vez retenidas.
Intercambio iónico, en la cual la fase estacionaria contiene grupos iónicos fijos como
–SO3, junto con iones de carga opuesta (contraion) que se encuentran presentes en la
móvil en forma de sales que al interaccionar con las moléculas de muestras iónicas
estas son retenidas en la columna por el intercambio iónico.
Exclusión molecular, el empaque es uno de los principales componentes para este tipo
de cromatografía, es decir el empaque es un material poroso. El poro al estar bien
definido permite que las moléculas de mayor tamaño al poro se extraen de las
partículas y salen rápidamente de la comuna mientras que las moléculas más pequeñas
entran en los poros aumentando su recorrido y tiempo de elución, logrando separar
compuestos por su peso molecular.
A partir de los tipos de cromatografía de generan modificaciones generando otros tipos de
cromatografías como:
Cromatografía de fases enlazadas, la fase estacionaria está unida químicamente a las
partículas del soporte. En donde el empaque es estable permitiendo que la fase
estacionaria se mantenga con el uso.
Página 12

La variante de esta cromatografía puede llevarse a cabo en fase normal o reversa
donde la fase normal utiliza empaques polares haciendo semejanza a la
cromatografía de adsorción mientras que la de fase inversa involucra una fase
estacionaria relativamente poco polar, como cadenas de hidrocarburos de 8 a 18
carbonos unidos a los grupos silano del soporte, con el uso de fases móviles muy
polares para la separación de componentes poco polares. (FEUM, 2010) (FEUM, 2014)
En la calidad de la CLAR existen factores que determinaran si el resultado obtenido es
satisfactorio tal es el caso de,
La Selectividad, que es una función de la retención que la molécula tiene a lo largo del
proceso de separación reflejado a través del factor de capacidad ( k´)
k ´ tt0
−1 o k ´t−t0
t 0
Mientras que la Selectividad de una columna también denominada como retención
relativa o separación entre picos (α), es la relación entre los factores de capacidad (k') de
dos picos adyacentes:
∝=k2
´
k1´
Por otra parte la Eficiencia, hace referencia al ensanchamiento del pico durante su
separación reflejado a través del número de platos teóricos (N) de la columna, proceso
que se determina a partir del gráfico:
N=16 ( tW )
2
donde, t= tiempo de retención d la sustancia
W= ancho de la base del pico obtenido
A partir de estos factores se determina la Resolución que se expresan en términos de
selectividad y eficiencia como se muestra a continuación:
Página 13

R=N4
(∝−1 )( k1+k )
Donde k es el promedio de k´1 y k´2, lo que permite controlar la resolución (R) variando
el factor selectividad (α), la eficiencia de la columna (N) o el factor de capacidad (k´)
mientras que el factor de separación se varía de acuerdo a la composición de la fase móvil
y/o estacionaria. Por otra parte la variación de la eficiencia se da de acuerdo a la longitud de
la columna, flujo del disolvente y tamaño de partícula. (FEUM, 2010) (FEUM, 2014).
Equipo. El cromatógrafo de líquidos de alta resolución comprendido por los siguientes
compartimentos:
a) Sistema de bombeo: Compartimento encargado impulsar la fase móvil a través de
la columna cumpliendo ciertas especificaciones como reproducibilidad y precisión
a través de un flujo laminar y velocidad constante. Mediante algunos tipos de
bombas donde se destacan las Bombas de flujo constante y las Bombas de Presión
Constante.
Página 14
Figura 1. Esquema de un Cromatógrafo de Líquidos
En el primer tipo de bombas se mantiene una velocidad de flujo de la fase móvil
constate y se encuentran las bombas reciprocantes que funcionan a base de pistones
en número par con el fin de impulsar el disolvente a en las cámaras cuando se
encuentra en volúmenes pequeños, sin embargo, las pulsaciones de la fase móvil
producen perturbaciones a la línea base que pueden ser corregidas con el uso de
dispositivos especiales.
Las bombas de desplazamiento positivo, son otro claro ejemplo de las bombas de
flujo constante, que presentan dos tipos de formas ya sea parecidas a una jeringa
debido a que actúa mediante una espiral que empuja al disolvente mientras que la
de amplificador hidráulico aumenta la presión del disolvente mediante un sistema
hidráulico, lo que reduce las pulsaciones del disolvente.
A diferencia de las Bombas de flujo constante en las Bombas de presión constante
se deben de controlar ciertos parámetros como la viscosidad del disolvente, la
temperatura de la columna y la presión constantes para el control de las pulsaciones.
La forma más sencilla de este tipo de bombas utiliza la presión de un gas inerte
para presurizar el disolvente con la desventaja ocasional cuando el gas se disuelve
en el disolvente formando burbujas en el sistema. Otro tipo de sistema en este tipo
de bombas emplea un amplificador neumático reduciendo el efecto del gas en el
pistón y a su vez reduciendo el contacto del gas comprimido con el disolvente.
Estos normalmente son sistemas isocráticos, es decir, que se mantiene constante la
proporción de los disolventes en la fase móvil con la desventaja de que esto
sistemas no son aplicables a separaciones en mezclas de solutos con valores muy
variables de k´, donde es necesario el uso de elución con gradiente con el uso de al
menos bombas binarias programables para modificar en forma lineal o exponencial
las proporciones de los disolventes. El uso de gradientes tiene como principal
inconveniente su compatibilidad con ciertos tipos de detectores como los
refractómetros. (FEUM, 2010) (FEUM, 2014).
b) Sistema de inyección: Es considerado como un factor importante para la resolución
en la separación ya que se genera a partir de la adecuada introducción de la muestra
Página 15

al sistema.
La manera más sencilla consiste en introducir la muestra mediante una jeringa, a
cual tiene que atravesar un septo y soportar la presión del sistema, en donde la
precisión del volumen de inyección depende de la jeringa empleada y del analista al
realizar el llenado de la misma y la inyección de la muestra.
Existe otro sistema que consiste en inyectores con asas intercambiables de
volumen fijo, las cuales suelen llenarse con un exceso de muestra, estos
dispositivos desvían el flujo del disolvente mientras se introduce la muestra
reanudándolo posteriormente a través del inyector y arrastrando un volumen
constante de muestra. Este fundamento define que este tipo de sistema de
inyectores son más precisos pero deben ser cambiados de acuerdo al volumen de
inyección en las diferentes corridas analíticas.
Un tercer sistema de inyección consiste en un inyector automático capaz de reducir
los errores en la medición de volumen y mantener la reproducibilidad entre
inyecciones entre cada muestra, como ventaja cuenta con la capacidad de realizar
diferentes tipos de inyección con alto grado de precisión y exactitud. (FEUM,
2010) (FEUM, 2014).
c) Detector: Hace referencia al dispositivo que permite medir, a la salida de la
columna, una propiedad física del eluyente. De tal manera que existen dos tipos de
detectores:
Tipo 1: Aquellos que miden alguna propiedad de la fase móvil.
Detector de Índice de Refracción.
Detector de Conductividad eléctrica.
Tipo 2: Miden alguna propiedad del anualito con alta especificidad.
Detector de luz UV/VIS (longitud fija o arreglo de diodos).
Detector de Radioactividad (con contador alfa, beta o gamma).
Detector de Fluorescencia (fijos o con monocromador de excitación y de emisión).
Detector Electroquímico (Amperométricos y Coulométricos).
Página 16

Espectrómetro de masas (sencillos o en "tandem"). (FEUM, 2010) (FEUM, 2014).
d) Columna: Parte fundamental de la Cromatografía, es en este apartado donde se
lleva a cabo la separación. De acuerdo al tipo de separación se consideraran
características como el material del empaque (Anexo A: Tipos de Soportes para
CLAR) y dimensiones de la columna.
Este accesorio de la Cromatografía de Líquidos suele ser de acero inoxidable con
presentaciones de vidrio con longitudes de 10 a 100 cm, lo que determina el
número de platos teóricos.
El relleno presente en las columnas es a base de partículas de cerámica inorgánica
(sílica o alúmina) o un polímero inorgánico (poliestireno – divinilbenceno o
metacrilatos) mientras que los tamaños de partícula disponibles estarán sujetos al
tipo de técnica cromatografía, es decir,
> 10 µm, para técnicas preparativas.
10 µm, cromatografía semi-preparativa.
5 µm, es el tamaño más común en técnicas analíticas.
3 µm para separaciones muy rápidas.
En el caso de los materiales empleados en la Cromatografía de Fase Reversa, se
deben considerar aspectos como la densidad de las cadenas alifáticas unidas a la
sílica base, las dimensiones de las columnas analíticas que van desde los 30 hasta
los 300 mm de longitud y de los 0.5 a los 4.6 mm de diámetro.
e) Registrador de señales: Una vez que el compuesto sale de la columna ya separado y
ha pasado al detector, la señal que este provoca es registrada por un graficador, es
decir, un integrador o un sistema computarizado para el procesamiento de los datos.
(FEUM, 2010) (FEUM, 2014).
Página 17

Disolución (MGA 0291)
Fundamento. La Disolución se denomina como una prueba de disolución aparente con el
fin de medir la liberación de un principio activo, a partir de la forma de dosificación que lo
contiene y la disolución de éste, en el medio de prueba.
El Método Disolución es empleado para determinar el cumplimiento de los requisitos de
disolución en tabletas o cápsulas establecidos en la monografía individual, excepto para
tabletas masticables de acuerdo a lo que establezca la monografía para el caso de cada
aparato. (FEUM, 2010) (FEUM, 2014).
Equipo.
Consta de un baño de agua o en su caso chaquetas de calentamiento y de seis unidades de
prueba, donde cada una está constituida por: Un vaso cilíndrico de fondo semiesférico, con
tapa, un eje transmisor, un regulador de velocidad de rotación y el aparato de disolución.
Vaso. Debe ser de vidrio o de otro material inerte y transparente. De forma cilíndrica y de
fondo semiesférico, de 160 mm a 210 mm de alto y de 98 mm a 106 mm de diámetro
interno, con capacidad para 1 000 mL. La tapa debe estar ajustada para retardar la
evaporación y permitir la inserción de un termómetro, así como la toma de la muestra. El
vaso debe estar firmemente ajustado, sumergido en el baño de agua, el cual debe mantener
la temperatura del medio de disolución a 37°C ± 0.5°C. El aparato debe permitir la
visualización del desarrollo de la prueba.
Eje transmisor. Eje de acero inoxidable tipo 316 y girar suavemente, sin bamboleo, de 6.3
mm a 6.5 mm o de 9.4 mm a 10.1 mm de diámetro. Debe estar colocado en el centro del
vaso, de tal manera que no quede a más de 2.0 mm de cualquier punto del eje vertical del
vaso.
Regulador de velocidad de rotación. Accesorio capaz mantener la velocidad constante de
acuerdo con lo indicado en la monografía del producto.
Página 18

Descripción de los aparatos
Aparato I: Canastillas
Consta de dos partes: la parte superior y la parte inferior.
Parte superior. Está unida al eje transmisor y es de acero inoxidable tipo 316, con un
orificio de salida de 2.0 mm ± 0.5 mm de diámetro; se ajusta a la parte inferior por medio
de 3 grapas o de un empaque para permitir que se coloque la muestra en el interior de la
canastilla y la sostenga firmemente, permitiendo que gire en forma concéntrica al eje del
vaso durante la rotación.
Parte inferior. De acero inoxidable tipo 316, soldado, formando un "cilindro de 37.0 mm ±
3 mm de alto por 22.2 mm ± 1.0 mm de diámetro externo del tamiz, con un borde angosto
de hoja de metal alrededor de la tapa, de 5.1 mm ± 0.5 mm de ancho, de malla número 40.
La distancia entre el fondo del vaso y el fondo de la canastilla, debe mantenerse constante a
25 mm ± 2.0 mm durante la prueba. Existen además canastillas con un recubrimiento de
oro de 2.5 µm de espesor.
Aparato II: Paletas
Hélice agitadora de 4 mm ± 1 mm de espesor y de 19 mm ± 0.5 mm de alto, en forma de
sección de un círculo de radio de 41.5 mm ± 1.0 mm y cuerdas paralelas subtendidas de 42
mm ± 1.0 mm y de 74.5 mm ± 0.5 mm, quedando la sección más pequeña hacia abajo. La
distancia de la base de la paleta al centro del círculo imaginario es de 35.8 mm ± 1.0 mm.
La línea central de la cuchilla pasa a través del eje transmisor de tal manera que la sección
de 42 mm de la misma quede perpendicular al eje transmisor al final del mango formando
una unidad que puede estar recubie11a con un polímero de fluorocarbono o de cualquier
otro material inerte.
Durante la prueba se debe mantener una distancia de 25 mm ± 2.0 mm entre la orilla
inferior de la propela y el fondo del vaso. Se puede utilizar un dispositivo de material no
reactivo, para mantener la muestra en el fondo del vaso y evitar que flote. Validar su
empleo. (FEUM, 2010) (FEUM, 2014)
Página 19
Anexo B. Aparatos de Disolución

Uniformidad de dosis (MGA 0299)
En el MGA de Uniformidad se definen como formas farmacéuticas que contienen una
única dosis o parte de una dosis de un fármaco en cada unidad.
La uniformidad de dosis se puede demostrar por los métodos de Variación de masa o el de
Uniformidad de contenido.
El método de Variación de masa se basa en la medición de la masa individual de las
unidades de dosis en prueba y el cálculo de la variación entre ellas, relacionada al contenido
del principio activo, y suponiendo una distribución homogénea. (FEUM, 2010) (FEUM,
2014).
Principalmente se aplica para las siguientes formas farmacéuticas: Cápsulas duras y tabletas
que contengan 25 mg o más de un principio activo y si éste constituye el 25 % o más de la
masa total de la unidad de dosis o del contenido de la cápsula en el caso de cápsulas duras,
Soluciones orales en envases de dosis única y en capsulas blandas. Solidos
(estériles o no) en envases de dosis única sin sustancias agregadas, ya sean activas
o inactivas.
Solidos (estériles o no) en envases de dosis única con o sin sustancias agregadas, ya
sean activas o inactivas que hayan sido preparadas a partir de soluciones
verdaderas o liofilizadas en el envase final y cuyas etiquetas indiquen el método de
preparación.
El método de Uniformidad de contenido se basa en la determinación cuantitativa del
contenido individual del principio activo en un cierto número de unidades de formas
farmacéuticas de dosis única, para determinar si la variación de los contenidos individuales
está dentro de los límites establecidos. Se puede aplicar a todas las formas farmacéuticas y
es necesaria en los casos que se describen a continuación:
Tabletas recubiertas, con excepción de las tabletas recubiertas con una película y
que contengan 25 mg o más de un principio activo que constituya el 25 % o más de
la masa total de la tableta.
Página 20

Suspensiones, emulsiones o geles en envases de dosis única o en cápsulas blandas,
destinadas exclusivamente para administración sistémica y no para los fármacos
destinados para administración externa, cutánea.
Sólidos y sólidos estériles en envases de dosis única con sustancias agregadas, ya
sean activas o inactivas cuando no se cumplen los requerimientos establecidos para
variación de masa.
Soluciones para inhalación envasada en unidades de dosificación previamente
medida (exceptuando las soluciones para inhalación envasadas en frasco ámpula de
vidrio o de plástico, destinadas para uso en nebulizadores). (FEUM, 2010) (FEUM,
2014).
Para una fácil determinación del método que se utilizara se puede consultar la Tabla
1.3 Requisitos para pruebas de Uniformidad de contenido (UC) y Variación de masa
(VM).
Página 21
Tabla 1.3 Requisitos para pruebas de Uniformidad de contenido (UC) y Variación de masa (VM).