
18. Miopatías
45
MIOPATÍAS 2014 Julio César Alfaro Mantilla
-
Upload
jhoselyn-de-la-torre -
Category
Documents
-
view
63 -
download
1
Transcript of 18. Miopatías

MIOPATÍAS2014
Julio César Alfaro Mantilla

MIOPATÍASCONTENIDO
Fisiología muscular Manifestaciones clínicas Exámenes auxiliares Clasificación Distrofias musculares Miopatías inflamatorias Miopatías endócrinas Miopatías tóxicas Miotonías Parálisis periódicas

EL MÚSCULO
Está compuesto de fibras musculares La fibra muscular es la unidad fisiológica Las fibras musculares están compuestas de
miofibrillas La miofibrilla es la unidad contráctil Las miofibrillas están compuestas de
miofilamentos de actina y miosina

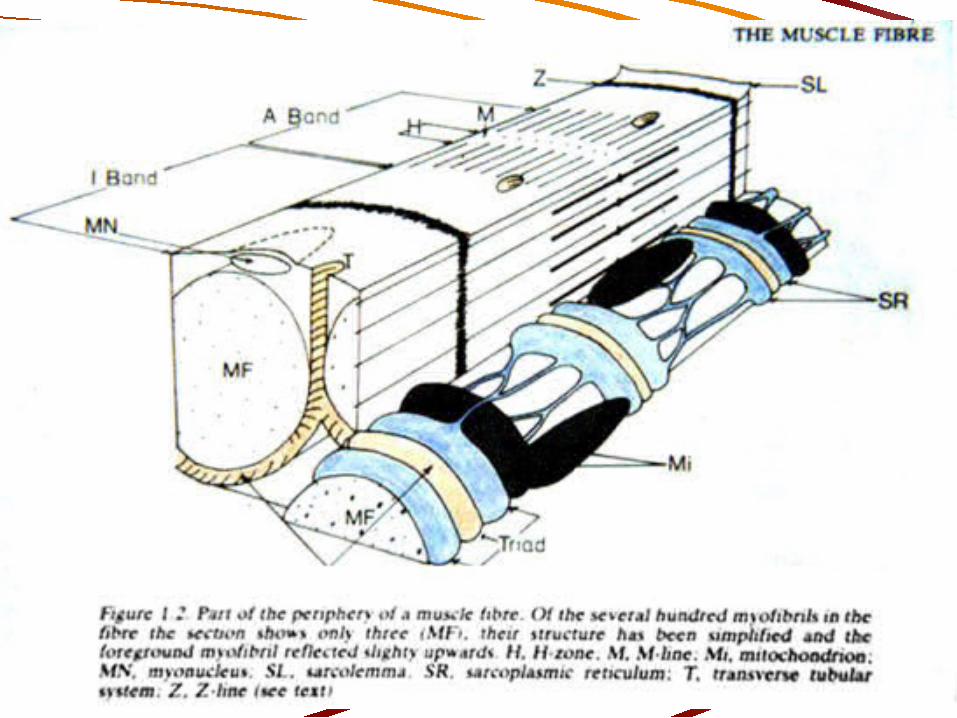
LA FIBRA MUSCULAR
Su membrana es el sarcolema Contiene numerosos núcleos que se ubican en
su periferia Contiene muchas mitocondrias Está formada de miofibrillas cuya disposición le
da un aspecto estriado Contiene dos sistemas de túbulos: Tubos T y
tubos del retículo sarcoplásmico

Potencial de acción muscular

MITOCONDRIAS

MIOPATÍAS. MECANISMOS PATOGENÉTICOS
Deficiencia de proteínas del sarcolema. Distrofinopatías. Sarcoglicanopatías
Anomalías estructurales en las fibras musculares: miopatías congénitas
Miopatías inflamatorias. Infecciosas, autoinmunes
Miopatías endócrinas, tóxicas (alcohol, cortisona, estatinas
Canalopatías. Parálisis periódicas, Miotonías, Hiperpirexia maligna
Miopatías mitocondriales. Deficiencia de carnitina o palmitilcarnitina

MIOPATÍASSÍNTOMAS
Debilidad y atrofia muscularMiotoníaParálisis periódicasIntolerancia al ejercicio–Dolor–Contractura–Mioglobinuria

MIOPATÍASCARACTERÍSTICAS CLÍNICAS
Inicio insidiosoDebilidad a predominio proximal (signo de Gowers)Anadeo, dificultar para subir escalerasAtrofia selectiva de ciertos músculosDebilidad desproporcionada al volumen muscularSeudohipertrofiaReflejos conservadosSensibilidad normalAusencia de fasciculacionesMiotonía, calambres y dolor muscular episódico (miopatías raras)

MIOPATÍASEXÁMENES AUXILIARES
CreatinfosfokinasaElectromiografíaBiopsia muscular


MIOPATÍASHALLAZGOS ELECTROMIOGRÁFICOS
Velocidad de conducción nerviosa normalUnidades motoras polifásicas de amplitud y duración disminuida, de aspecto “miopático”Patrón de interferencia completoAusencia de fibrilaciones y ondas positivasAusencia de fasciculaciones

Distrofia muscular

Miopatía nemalínica
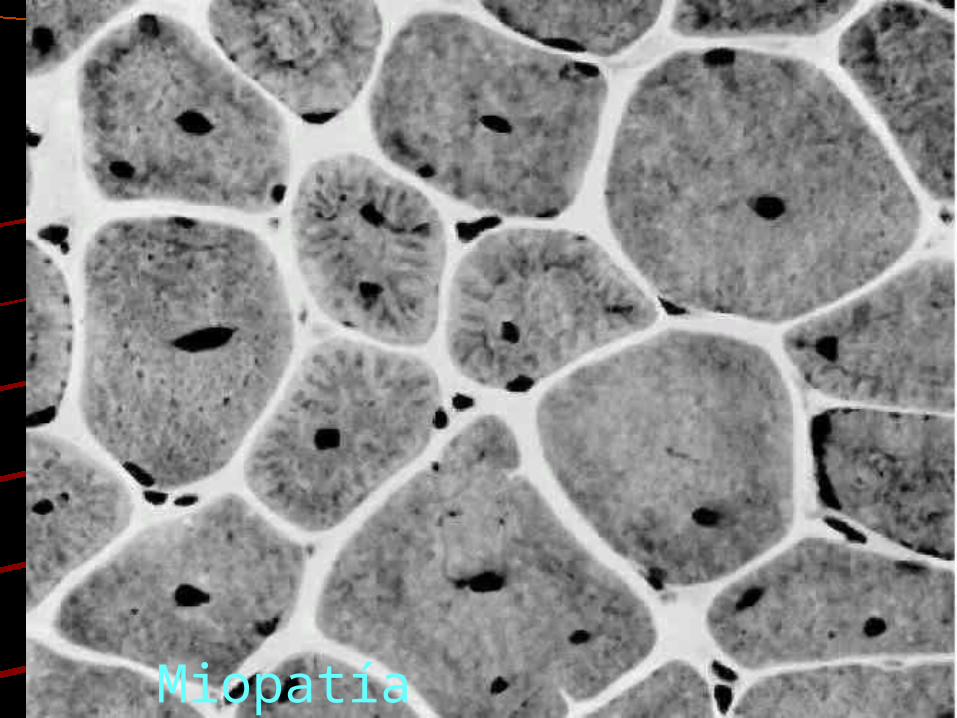
Miopatía Centronuclear

MIOPATÍAS CLASIFICACIÓN
Distrofias muscularesMiopatías congénitasMiopatías metabólicasMiopatías endócrinasMiopatías inflamatoriasMiopatías tóxicasMiotoníasParálisis periódicas

DISTROFIAS MUSCULARESCLASIFICACIÓN
Seudohipertrófica de DuchenneTipo BeckerFacio escápulo humeralEscápulo humeral (de las cinturas)Miopatía ocularMiopatía oculofaríngeaDistal

Distrofia muscular

DISTROFIA MUSCULAR TIPO DUCHENNE
Ausencia de distrofinaTransmisión recesiva ligada al sexoComienzo entre los 2 y 6 años de edadDistribución selectiva pelvipectoralPseudohipertrofia de músculosCurso rápidamente progresivo:– Incapacidad alrededor de los 10 años– Muerte alrededor de los 20 años

DISTROFIA MUSCULAR TIPO BECKER
Igual que el Duchenne, excepto inicio entre los 5 y 45 añosCurso más benignoIncapacidad a los 25 a 30 añosMuerte en la quinta década

Distrofia escapulohumeral

DISTROFIA FASCIOESCAPULOHUMERAL
-Transmisión autosómica dominante-Edad de inicio variable (2da. década)-Distribución selectiva en cara y cintura
escapular-Formas frustras frecuentes-Curso lento
DetencionesNo invalidanteEspectancia de vida: promedio normal
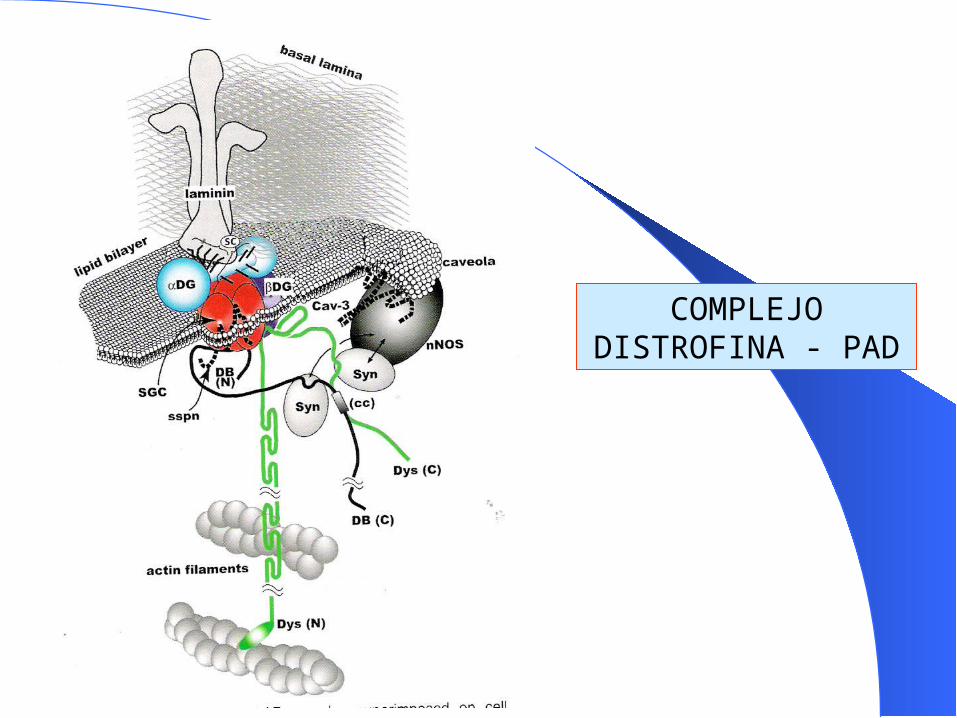
COMPLEJO DISTROFINA - PAD

DISTROFIA ESCÁPULO HUMERAL(Limb Girdle)
Transmisión variable.Frecuentemente autosómica recesivaInicio: adolescencia o edad adultaDistribución: cintura escapular y pelvianaCurso relativamente benigno
Progresión lenta Expectancia de vida disminuidaDiagnóstico diferencialEnfermedades neurogénicas
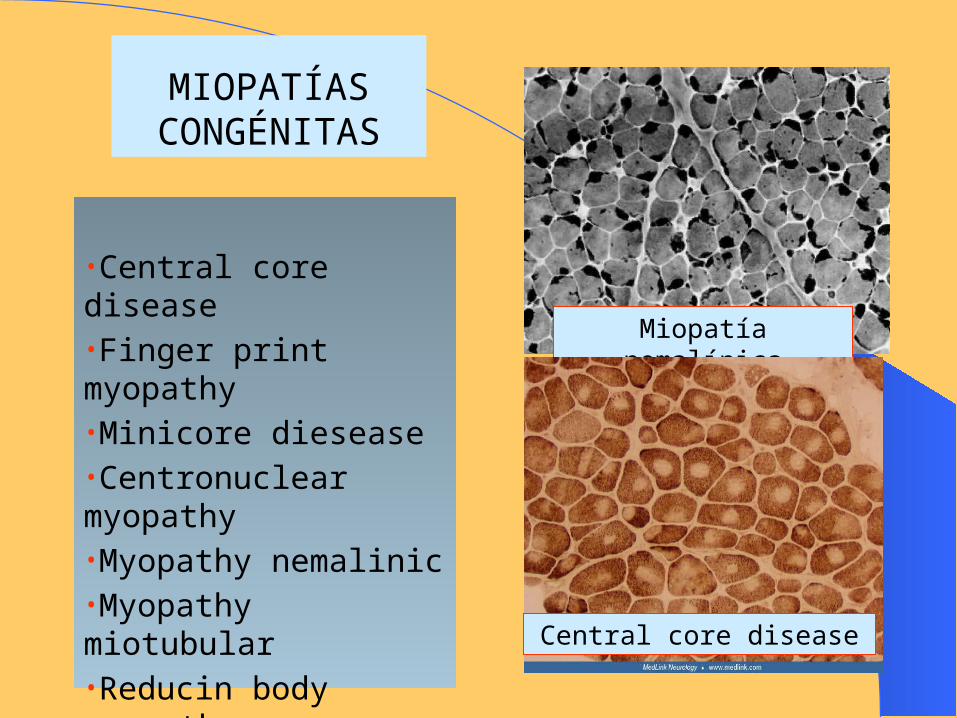
MIOPATÍAS CONGÉNITAS
•Central core disease•Finger print myopathy•Minicore diesease•Centronuclear myopathy•Myopathy nemalinic•Myopathy miotubular•Reducin body myopathy
Miopatía nemalínica
Central core disease

MIOPATÍAS INFLAMATORIASCLASIFICACIÓN
IDIOPÁTICASPolimiositisMiositis por cuerpos de inclusiónSarcoidosis
INFECCIOSASVirales (Coxsackie, influenza, Bornholm)Bacterianas ( piógena, TBC)Parasitarias (toxoplasmosis, cisticercosis, triquinosis)

MIOPATÍAS INFLAMATORIAS
DermatomiositisPolimiositisMiositis con cuerpos de inclusión

POLIMIOSITISDebilidad muscular proximal simétricaEnzimas musculares aumentadasEMG característicoHistología característica

DERMATOMIOSITIS
MiositisRash típicoHistología característicaAsociación con neoplasias

DERMATOMIOSITISRASH TÍPICO
Eritema facialRash en heliotropoPápulas de Gottron

POLIMIOSITIS. Infiltración linfocitaria
DERMATOMIOSITIS Atrofia perifascicular

POLIMIOSITISHALLAZGOS EMG
• Signos de miopatía• Unidades motoras polifásicas de amplitud y duración disminuida
• Patrón de interferencia completo
• Fibrilaciones y ondas positivas• Descargas seudomiotónicas

INMUNOTERAPIA
PrednisonaAzathioprinaMethotrexateCyclophosphamideCyclosporinePlasmaferesisInmunoglobulinas

MIOSITIS POR CUERPOS DE INCLUSIÓN
Debilidad proximal y distal en m. inferioresCompromiso selectivo del cuadrícepsDebilidad en antebrazosHistología característica (vacuolas)Falta de respuesta al tratamiento

MIOPATÍAS ENDÓCRINAS
TirotoxicosisMixedemaInsuficiencia suprarenalCushingAcromegaliaHipercalcemia

MIOPATÍAS TÓXICASETIOLOGÍA
CloroquinaAlcoholCortisonaVincristine Zidovudine (AZT)CiclosporinaAgentes colesteropénicos

AGENTES COLESTEROPÉNICOS QUE PUEDEN PRODUCIR MIOPATÍA
DERIVADOS DEL ACIDO FÍBRICO– Fenfibrato– Clofibrate (Atromid)– Gemfibrosil (Lopid)– Benzafibrate
ACIDO NICOTÍNICOINHIBIDORES DE LA HMG.Coa.REDUCTASA– Lovastatina (Mevacor)– Sinvastatina (Zocor)– Pravastatina (Prevachol

MIOPATÍA POR CORTISONA
Debilidad muscular proximalMejora al suspender o disminuir la dosis de cortisonaEMG normal o miopático, sin fibrilacionesEnzimas musculares normales

MIOTONÍA
Anomalía de la membrana muscularMembrana muscular anormalmente excitableA un estímulo responde en forma repetitivaDescarga de potenciales de alta frecuencia con “waxing and waning”


MIOTONÍASCLASIFICACIÓN
Distrofia miotónicaMiotonía congénitaParamiotonía congénita

DISTROFIA MIOTÓNICA(Enfermedad de Steinert)
-Trasmisión: Autosómica dominante-Inicio: Adolescencia o edad adulta Características clínicas Atrofia selectiva de ciertos músculos (Elevador de los párpados, faciales, maseteros,
esternocleidomastoideo, distales en m. superiores, miocardio)
-Compromiso extramuscular Calvicie frontal, cataratas, cardiopatía, atrofia
testicular, alteraciones endócrinas, deficiencia mental
-Miotonía-Curso lento: Expectancia de vida disminuida

MIOTONÍA CONGÉNITA(Enfermedad de Thomsen)
TRANSMISIÓNAutosómica dominanteAutosómica recesivaINICIO EN LA INFANCIAMANIFESTACIONES CLÍNICASMiotoníaFrecuentemente hipertrofia muscularTRATAMIENTOQuinina, fenitoina, ACTH, CortisonaDiamoxDIAGNOSTICOElectromiografía: Descargas miotónicas

PARÁLISIS PERIÓDICASFISIOPATOLOGÍA
Anomalía de la membrana muscularDepolarización espontánea de la membrana muscularPérdida del potencial de membranaMembrana muscular inexitable. En período refractario absoluto.










![New II. Miopatías asociadas a estatinas [actualizado a 15-ago-2015]evalmedicamento.weebly.com/uploads/1/0/8/6/10866180... · 2019. 5. 21. · Sección 3: Efectos adversos. II. Miopatías](https://static.fdocuments.co/doc/165x107/5ff09b3eb78e17674c649011/new-ii-miopatas-asociadas-a-estatinas-actualizado-a-15-ago-2015-2019-5-21.jpg)







