
M IOPATIAS INFLAMATORIAS - NeuroMuscular Sant … · I I CURSO DE FORMACION EN ENFERMEDADES...
12
II CURSO DE FORMACION EN ENFERMEDADES NEUROMUSCULARES MIOPATIAS INFLAMATORIAS Dr. Eduard Gallardo Unidad de Enfermedades Neuromusculares Institut de Recerca Hospital de la Santa Creu i Sant Pau, Barcelona Dr. Jordi Díaz Manera Unidad de Enfermedades Neuromusculares Servicio de Neurología Hospital de la Santa Creu i Sant Pau, Barcelona Las miopatías inflamatorias son un grupo heterogéneo de enfermedades de origen autoinmune, con una evolución desde aguda o subaguda hasta crónica y entamente progresiva. Se caracterizan todas ellas por la existencia de debilidad l muscular y por la presencia de signos inflamatorios en la biopsia muscular. xiste un número importante de entidades que pueden presentar infiltrados nflamatorios en la biopsia muscular: E i Polimiositis idiopática primaria Dermatomiositis idiopática primaria Polimiositis o dermatomiositis asociadas a cáncer Dermatomiositis juvenil Polimiositis o dermatomiositis asociadas a otras enfermedades del tejido conectivo Miositis por cuerpos de inclusión Formas raras de miositis idiopática: Miositis granulomatosa Miositis eosinofílica Miositis focal Miositis orbital. Miositis vasculítica Miositis ossificans Fascitis macrofágica Miopatías necrotizantes inmunomediadas Sarcoidosis, vasculitis Miositis infecciosas (triquinosis, piomiositis...) Algunas distrofias musculares (FSHD, Duchenne, disferlinopatías...)
Transcript of M IOPATIAS INFLAMATORIAS - NeuroMuscular Sant … · I I CURSO DE FORMACION EN ENFERMEDADES...

I I CURSO DE FORMACION EN ENFERMEDADES NEUROMUSCULARES
M
IOPATIAS INFLAMATORIAS
Dr. Eduard Gallardo Unidad de Enfermedades Neuromusculares Institut de Recerca Hospital de la Santa Creu i Sant Pau, Barcelona Dr. Jordi Díaz Manera Unidad de Enfermedades Neuromusculares Servicio de Neurología Hospital de la Santa Creu i Sant Pau, Barcelona Las miopatías inflamatorias son un grupo heterogéneo de enfermedades de origen autoinmune, con una evolución desde aguda o subaguda hasta crónica y entamente progresiva. Se caracterizan todas ellas por la existencia de debilidad lmuscular y por la presencia de signos inflamatorios en la biopsia muscular. xiste un número importante de entidades que pueden presentar infiltrados nflamatorios en la biopsia muscular: Ei
Polimiositis idiopática primaria Dermatomiositis idiopática primaria Polimiositis o dermatomiositis asociadas a cáncer Dermatomiositis juvenil Polimiositis o dermatomiositis asociadas a otras enfermedades del tejido conectivo
Miositis por cuerpos de inclusión Formas raras de miositis idiopática:
Miositis granulomatosa Miositis eosinofílica Miositis focal Miositis orbital. Miositis vasculítica Miositis ossificans Fascitis macrofágica
Miopatías necrotizantes inmunomediadas Sarcoidosis, vasculitis Miositis infecciosas (triquinosis, piomiositis...) Algunas distrofias musculares (FSHD, Duchenne, disferlinopatías...)

En esta revisión nos centraremos en la dermatomiositis, miositis por cuerpos de inclusión, polimiositis, las distrofias musculares por déficit de disferlina y las miopatías necrotizantes inmunomediadas. Dermatomiositis: La dermatomiositis se caracteriza por la presencia de un rash cutáneo acompañado de debilidad muscular. La afectación cutánea puede presentarse de diversas formas. El heliotropo es un eritema violáceo que afecta al párpado superior acompañándose o no de un eritema que afecta los pómulos, el cuello (especialmente la nuca) y la espalda. No es extraño encontrar un rash descamativo en codos, rodillas y orejas. En las manos, es característico el eritema de Gottron que afecta solamente a los nudillos y respeta a las falanges de los dedos. Puede existir afectación ungueal en forma de uñas frágiles y dolorosas al tacto o a la presión, y en ocasiones una alteración de los pulpejos que presentan una piel engrosada y resquebrajada. La clínica dermatológica suele preceder a la debilidad muscular, que es de evolución aguda o subaguda, afectando al compartimento proximal de las extremidades superiores e inferiores y que en casiones puede producir una disfunción bulbar con disfagia e insuficiencia espiratoria. or
Heliotropo Eritema de Gottron La DM se asocia con frecuencia a una neoplasia de base. El diagnóstico del tumor puede preceder o seguir a la aparición de los síntomas típicos de la DM. En mujeres el cáncer de ovario y en hombres el de estómago son los que con más frecuencia se detectan, si bien otros como el cáncer de pulmón, mama o el linfoma no Hodgkin deben descartarse. En todo paciente diagnosticado de DM es preciso la realización de pruebas complementarias encaminadas a descartar la xistencia de un tumor concomitante durante un período mínimo de 4 años edesde el diagnóstico. La respuesta inmunológica detectada en la DM es el clásico ejemplo de inmunidad humoral. Clásicamente se propone que la diana de la respuesta autoinmune es algún antígeno todavía desconocido del endotelio de los capilares situados en el endomisio de los músculos. La enfermedad se inicia posiblemente cuando anticuerpos dirigidos contra el endotelio activan la cascada de complemento que acaba en el depósito del complejo de ataque de membrana sobre las células endoteliales y su progresiva destrucción. La necrosis vascular se acompaña de inflamación perivascular, isquemia y destrucción de las fibras
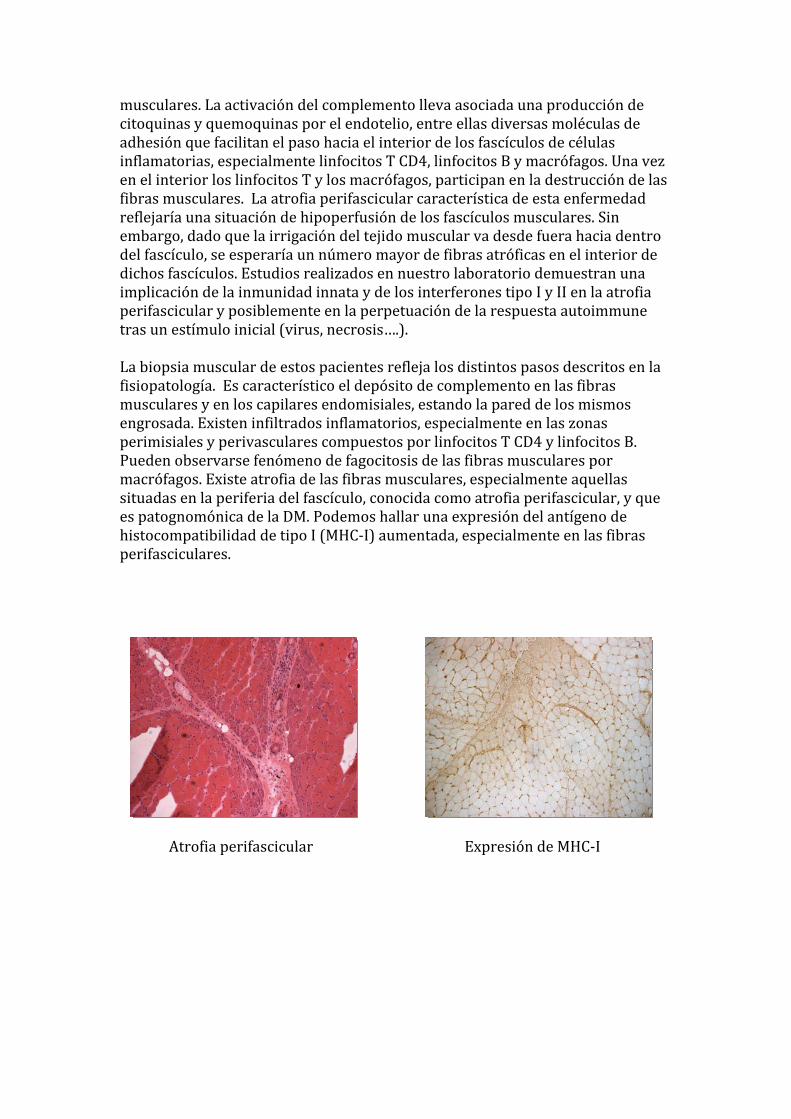
musculares. La activación del complemento lleva asociada una producción de citoquinas y quemoquinas por el endotelio, entre ellas diversas moléculas de adhesión que facilitan el paso hacia el interior de los fascículos de células inflamatorias, especialmente linfocitos T CD4, linfocitos B y macrófagos. Una vez en el interior los linfocitos T y los macrófagos, participan en la destrucción de las fibras musculares. La atrofia perifascicular característica de esta enfermedad reflejaría una situación de hipoperfusión de los fascículos musculares. Sin embargo, dado que la irrigación del tejido muscular va desde fuera hacia dentro del fascículo, se esperaría un número mayor de fibras atróficas en el interior de dichos fascículos. Estudios realizados en nuestro laboratorio demuestran una implicación de la inmunidad innata y de los interferones tipo I y II en la atrofia erifascicular y posiblemente en la perpetuación de la respuesta autoimmune ptras un estímulo inicial (virus, necrosis….). La biopsia muscular de estos pacientes refleja los distintos pasos descritos en la fisiopatología. Es característico el depósito de complemento en las fibras musculares y en los capilares endomisiales, estando la pared de los mismos engrosada. Existen infiltrados inflamatorios, especialmente en las zonas perimisiales y perivasculares compuestos por linfocitos T CD4 y linfocitos B. Pueden observarse fenómeno de fagocitosis de las fibras musculares por macrófagos. Existe atrofia de las fibras musculares, especialmente aquellas situadas en la periferia del fascículo, conocida como atrofia perifascicular, y que es patognomónica de la DM. Podemos hallar una expresión del antígeno de istocompatibilidad de tipo I (MHC‐I) aumentada, especialmente en las fibras erifasciculares. hp
Atrofia perifascicular Expresión de MHC‐I

Miositis por cuerpos de inclusión Se trata de una enfermedad con patología mixta autoimmune y degenerativa. Se inicia típicamente en fases medias o avanzadas de la vida, a partir de los 50 años de edad. El curso es siempre lentamente progresivo estableciéndose el diagnóstico diferencial con las distrofias musculares de inicio tardío. Los pacientes desarrollan de forma característica debilidad muscular que afecta fundamentalmente a los músculos flexores de los dedos de las manos, al uádriceps y a la musculatura distal anterior de las piernas. La mayoría de los
e cpacientes desarrollan una disfagia que es incapacitante precisando soportnutricional o sonda nasogástrica. Los datos que apoyan una base inmune son la presencia de infiltrados de linfocitos CD8, pero también el reciente descubrimiento de nódulos de células plasmáticas CD138 activas que producen y secretan inmunoglobulinas en el interior del músculo. Existen anticuerpos en sangre periférica específicos de la enfermedad, como el anti‐IBM‐43 presente en hasta un 50% de los pacientes. Sin embargo, la existencia de vacuolas ribeteadas que contienen material de degeneración mionuclear y el depósito de material amiloide en el citoplasma de as fibras musculares apoya un origen degenerativo de la enfermedad. La etección de depósitos de la proteína TDP‐43 en el citoplasma es altamente specífico de la IBM y su presencia asegura el diagnóstico.
lde
IBM: cambios distróficos + vacuolas
Ribeteadas + fibras anguladas + infiltrados inflamatorios La biopsia muscular muestra una patología mixta, en la que destaca una expresión difusa de MHC‐I e infiltrados inflamatorios a base de linfocitos T‐CD8 y macrófagos, con fenómenos frecuentes de fagocitosis. Además, existen evidencias de una respuesta humoral contra antígenos musculares y una incidencia de paraproteinemia del 23%. Estos hallazgos se acompañan de datos degenerativos como la presencia de una variabilidad en el tamaño de las fibras musculares, la existencia de vacuolas ribeteadas en las fibras musculares y la presencia de depósitos de amiloide y de diversas proteínas, destacando entre ellas la TDP‐43, en el sarcoplasma. Asimismo no es infrecuente encontrar fibras anguladas que sugieren una denervación reciente, y fibras raggedred o COX

negativas, consecuencia de alteraciones en la función mitocondrial descritas en estos pacientes. Así pues la MCI se considera una enfermedad muscular multifactorial en la que tendrían lugar dos procesos paralelos. 1) Por una parte un proceso degenerativo que incluiría: susceptibilidad genética (transtiretina‐ acúmulo de β‐Amiloide), agentes infecciosos, factores ambientales, envejecimiento celular (acúmulo de proteínas anormales, errores de transcripción, mutaciones en UBB, se acumula/ inhibición del proteasoma), stress oxidativo que conducirían a una degeneración, atrofia y muerte de la fibra muscular. 2) Por otra parte un proceso inmunomediado que incluiría tanto inmunidad humoral con presencia de utoanticuerpos, como una respuesta celular inflamatoria por linfocitos T CD8+ macrófagos. ay Polimiositis La PM se caracteriza por una debilidad muscular aguda o subaguda que afecta de forma fundamental al compartimento proximal de las extremidades superiores e inferiores. En general la musculatura facial y ocular no se afecta. En casos graves uede producir, al igual que en la DM, una debilidad bulbar que cursa con pdisfagia, disartria y disnea. La enfermedad puede presentarse sola, o con más frecuencia, asociada a otras enfermedades del tejido conectivo como la esclerodermia o el lupus. También se an descrito casos en el contexto de una infección por HIV o en una enfermedad hde Lyme. Una de las complicaciones característica de la PM es la enfermedad pulmonar intersticial que afecta hasta un 45% de los pacientes. Esta complicación puede ser aguda en la que el paciente desarrolla fiebre, tos no productiva, disnea y se demuestran infiltrados pulmonares en las bases, o puede ser crónica cursando con una disnea de lenta instauración asociada a una fibrosis pulmonar de redominio en bases. La existencia de anticuerpos anti‐Jo1en sangre de los ppacientes se asocia frecuentemente a esta complicación. El síndrome antisintetasa es otra de las complicaciones frecuentes de la PM, pudiéndose observar también en pacientes con una DM. Consiste en la asociación de miositis, una poliartritis que afecta a pequeñas articulaciones, fiebre, fenómeno de Raynaud, enfermedad intersticial pulmonar y manos de mecánico aracterizadas por un engrosamiento cutáneo con resquebrajamiento de los cpulpejos de los dedos. Desde el punto de vista fisiopatológico predomina una respuesta de la inmunidad celular. En este caso, linfocitos CD8 invaden y destruyen fibras musculares que expresan MHC‐I de forma difusa. No se conoce qué mecanismo desencadena la reacción, ahora bien, se postula que las fibras musculares presentan antígenos a los linfocitos a través de la molécula MHC‐I en conjunción con otras como la proteína BB1. A favor de esta hipótesis, se ha descrito que los infiltrados presentes en el músculo de estos pacientes pueden presentar una
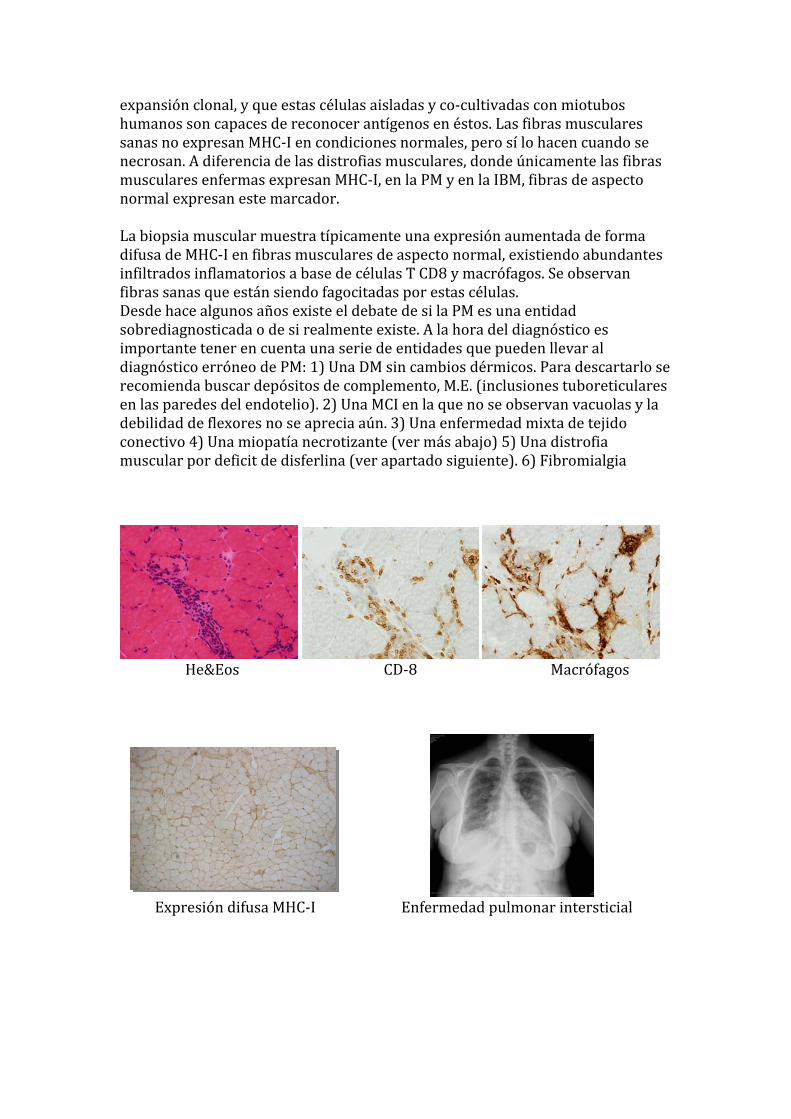
expansión clonal, y que estas células aisladas y co‐cultivadas con miotubos humanos son capaces de reconocer antígenos en éstos. Las fibras musculares sanas no expresan MHC‐I en condiciones normales, pero sí lo hacen cuando se necrosan. A diferencia de las distrofias musculares, donde únicamente las fibras usculares enfermas expresan MHC‐I, en la PM y en la IBM, fibras de aspecto m
normal expresan este marcador. La biopsia muscular muestra típicamente una expresión aumentada de forma difusa de MHC‐I en fibras musculares de aspecto normal, existiendo abundantes
servan infiltrados inflamatorios a base de células T CD8 y macrófagos. Se obfibras sanas que están siendo fagocitadas por estas células. Desde hace algunos años existe el debate de si la PM es una entidad sobrediagnosticada o de si realmente existe. A la hora del diagnóstico es importante tener en cuenta una serie de entidades que pueden llevar al diagnóstico erróneo de PM: 1) Una DM sin cambios dérmicos. Para descartarlo se recomienda buscar depósitos de complemento, M.E. (inclusiones tuboreticulares en las paredes del endotelio). 2) Una MCI en la que no se observan vacuolas y la debilidad de flexores no se aprecia aún. 3) Una enfermedad mixta de tejido onectivo 4) Una miopatía necrotizante (ver más abajo) 5) Una distrofia uscular por deficit de disferlina (ver apartado siguiente). 6) Fibromialgia
cm
He&Eos CD‐8 Macrófagos
Expresión difusa MHC‐I Enfermedad pulmonar intersticial

Distrofia muscular por déficit de disferlina Tal como se ha comentado anteriormente, la PM puede ser diagnosticada erróneamente en casos de distrofia muscular por déficit de disferlina. El diagnóstico precoz de disferlinopatia es importante en estos casos ya que el tratamiento con corticoides no sólo no es beneficioso sino que puede causar efectos deletéreos. El diagnóstico de disferlinopatia puede realizarse utilizando sangre periférica ya que los monocitos también expresan disferlina. Las disferlinopatías presentan una importante variabilidad fenotípica que va desde formas que cursan con debilidad de cinturas hasta formas distales pasando por casos de hiperCKemia asintomática. Una de las principales funciones de la disferlina es su implicación en la reparación del sarcolema después de una lesión. La incapacidad para sellar la membrana después de un daño llevaría a la degeneración del músculo esquelético.
Tinción de H&E en cortes criostáticos de biopsia muscular de dos pacientes con déficit de disferlina en la que se observan infiltrados endomisiales que podrían conducir a un diagnóstico erróneo de PM.
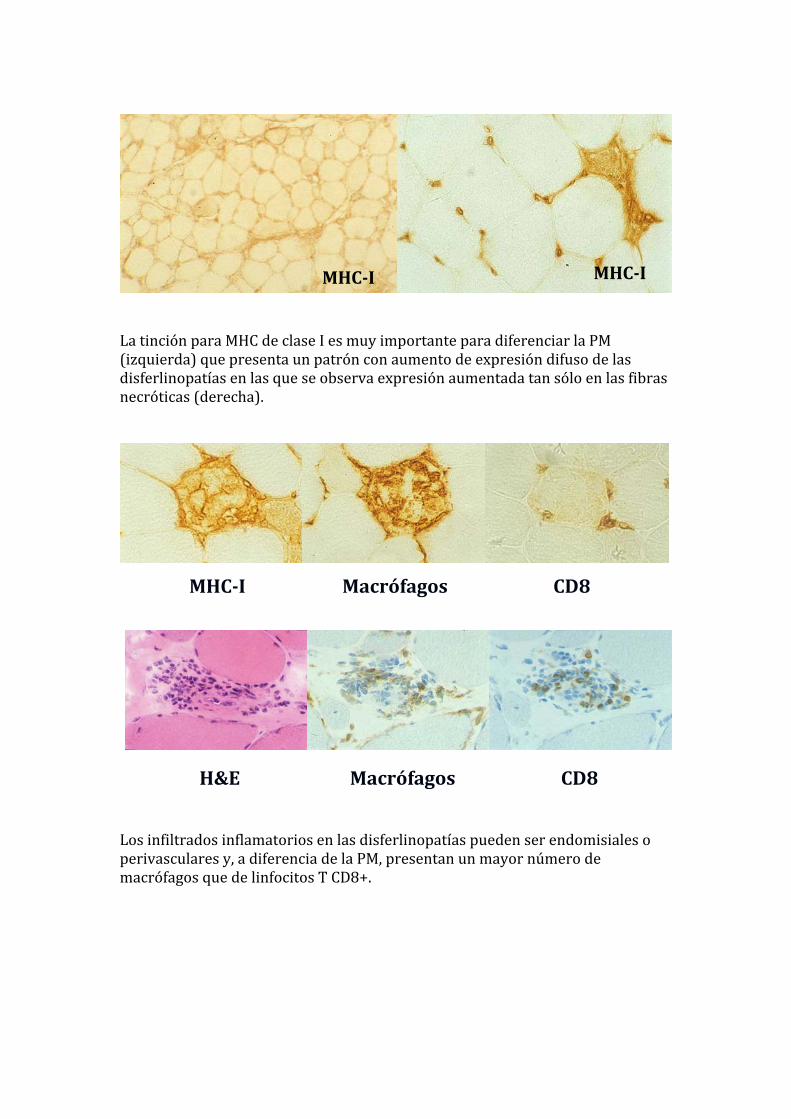
La tinción para MHC de clase I es muy importante para diferenciar la PM (izquierda) que presenta un patrón con aumento de expresión difuso de las isferlinopatías en las que se observa expresión aumentada tan sólo en las fibras ecróticas (derecha).
MHCI MHCI
dn
MHCI Macrófagos CD8
H&E Macrófagos CD8 Los infiltrados inflamatorios en las disferlinopatías pueden ser endomisiales o perivasculares y, a diferencia de la PM, presentan un mayor número de macrófagos que de linfocitos T CD8+.

Miopatías necrotizantes inmunomediadas Las miopatías necrotizantes inmunomediadas (MNI) se caracterizan desde el punto de vista clínico por una debilidad muscular de instauración subaguda asociada a cambios patológicos típicos consistentes en la presencia de abundantes fibras necróticas y un escaso infiltrado inflamatorio a base de macrófagos. Se trata de un grupo heterogéneo de enfermedades con características miopatológicas específicas. Hasta el momento se han descrito los siguientes subtipos: MNI asociada a anticuerpos antiSRP. Se presenta con debilidad de la musculatura proximal, simétrica de inicio en la quinta década de la vida, CKs siempre elevadas y puede asociar problemas cardíacos, pulmonares o de la piel. Presencia de anticuerpos anti‐SRP. La biopsia muscular muestra una miopatía necrotizante con abundantes macrófagos en perimisio y endomisio. La expresión de MHC de clase I y depósitos de complemento en los capilares se presentan de forma variable. No se observan linfocitos T CD8+ a diferencia de la PM, ni en este subgrupo ni en ninguno de los que se describen más abajo. No se observa atrofia perifascicular. MNI asociada a anticuerpos antiHMGCR. Se presenta con debilidad de la musculatura proximal, simétrica de inicio en la sexta década de la vida, CKs siempre elevadas. No se asocia a patología en otros órganos. En la serie más larga descrita los autores reportaron que un 66.7% de los pacientes con anticuerpos anti‐HMGCR habían tomado estatinas. Es importante destacar que un 33% de los pacientes no habían sido tratados con estatinas y sin embargo presentaban una miopatía inmuno‐mediada indistiguible de la de los pacientes que sí habían recibido tratamiento. La biopsia muscular muestra una miopatía necrotizante con abundantes macrófagos en perimisio y endomisio. La expresión de MHC de clase I y depósitos de complemento en los capilares se presentan en la mitad de los pacientes. MNI asociada a síndrome antisintetasa Se presenta con debilidad de la musculatura proximal, especialmente la cintura escapular (hombro) de inicio en la quinta década de la vida con niveles de CKs siempre elevados. Los pacientes pueden presentar fiebre, pérdida de peso y malestar general. La miopatía es a menudo subsidiaria a un síndrome reumatológico. La miopatía va característicamente asociada a enfermedad pulmonar intersticial, artritis y manifestaciones dérmicas. Se asocia a anticuerpos del grupo de los anti‐sintetasa (Jo‐1, PL‐7, PL‐12, OJ, KS, Zo, YRS). La biopsia muscular muestra una miopatía necrotizante con abundantes macrófagos en perimisio y endomisio. La expresión de MHC de clase I y depósitos de complemento en los capilares está muy incrementada. En algunos casos se observa atrofia perifascicular. MNI con capillares “pipestem” Se presenta con debilidad muscular proximal simétrica con implicación de otros órganos (piel, pulmón). Sólo se han descrito 6 casos hasta el momento. Todos los pacientes presentan enfermedades sistémicas graves tipo neoplasias, vasculitis y enfermedad intersticial pulmonar. Las CKs están generalmente elevadas. La biopsia muscular muestra una miopatía necrotizante con abundantes macrófagos en perimisio y endomisio. Algunos pacientes muestran una expresión de MHC de clase I muy incrementada y abundantes depósitos de

complemento en los capilares. La microscopia electrónica muestra acúmulo de un material amorfo en la pared y membrana basal de los vasos. MNI paraneoplásica Se presenta con debilidad de la musculatura proximal, especialmente la cintura escapular (hombro) de inicio en la séptima década de la vida. Aunque raramente, se puede observar debilidad facial. Las CKs están siempre elevadas Se puede
. La erpos.
asociar a cambios dermatológicos (distintos de DM) y afección pulmonarmiopatía es subsidiaria a una neoplasia. Raramente se asocia a autoanticuLa biopsia muscular muestra una miopatía necrotizante con abundantes macrófagos en perimisio y endomisio. Algunos pacientes muestran una xpresión de MHC de clase I muy incrementada y abundantes depósitos de omplemento en los capilares. ec
MNI: H&E Destrucción muscular difusa
Infiltrado de macrófagos
Tratamiento Dermatomiositis y polimiositis Una vez establecido el diagnóstico de la enfermedad y descartada la presencia de una enfermedad asociada, especialmente un tumor en el caso de la DM, se inicia el tratamiento con prednisona a dosis de 1 mg/kg y día. La respuesta suele ser claramente positiva en un período no superior al mes. En ese caso a partir del primer o segundo mes se inicia una reducción progresiva de las dosis. Recomendamos utilizar una pauta de seguimiento de corticoides a días alternos, reduciéndolos de forma lenta a un ritmo de 10 mg/mes hasta llegar a los 40 g/48 horas, y posteriormente a un ritmo de 5 mg/mes hasta llegar a la dosis m
mínima eficaz o a retirar la medicación de forma completa. Es frecuente que algunos pacientes afectos de una DM presenten recidivas durante la reducción de las dosis de corticoides o bien que los síntomas no mejoren de forma completa con los corticoides. En ambos casos puede lantearse tratamiento con IgIV ya que los estudios realizados hasta el momento pdemuestran una mejoría considerable de los síntomas a corto plazo. Si tras varios meses en tratamiento con corticoides la mejoría es escasa o inexistente, el diagnóstico debe replantearse. En estos casos se debe insistir

también en la búsqueda de un tumor coexistente. Puede ser preciso añadir fármacos de segunda línea. En el caso de la PM, la azatioprina y el metotrexate han demostrado una alta efectividad terapéutica. En el caso de la DM, los estudios realizados con azatioprina y ciclosporina han sido exitosos en el control de los síntomas. Otros fármacos de tercera línea, como el micofenolato, el tacrolimus, los anti‐TNFα como etanercept o infliximab y el anti‐CD20 rituximab. n aquellos pacientes con una PM que desarrollan una enfermedad pulmonar ntersticial se recomienda tratamiento con ciclofosfamida. Ei Miositis por cuerpos de inclusión: La respuesta obtenida con inmunosupresores en este caso es pobre o nula y no justifica su administración de forma crónica. Los estudios realizados con corticoides u otros inmunosupresores como la azatioprina han fracasado. Únicamente las IvIg consiguieron una cierta mejoría de la disfagia. El tratamiento de los pacientes se basa en la rehabilitación, colocación de órtesis si es preciso y n medidas paliativas de la disfagia, como la colocación de una sonda de astrostomía. eg Miopatía necrotizante: No existen estudios randomizados publicados por lo que las recomendaciones se basan en los casos publicados o en pequeñas series de pacientes. Si existen enfermedades asociadas (neoplasia, HIV o estatinas) es necesario tratarlas de forma concomitante o retirar la medicación. En nuestra experiencia la respuesta a los diferentes tratamientos es pobre y precisa de la asociación de varios fármacos. Nosotros iniciamos prednisona a dosis de 1 mg/kg/día siendo preciso no obstante asociar un segundo fármaco con frecuencia como azatioprina, etotrexate o micofenolato. Existe en el momento actual un ensayo clínico con ituximab que está en fase de reclutamiento. mr
ibliografía relevante: B
1. Dalakas, M. and Hohlfeld, R. “Polymiositis and dermatomyositis”. Lancet
2. 2003. 362: 971‐8
2. Greenberg, S. A. “Inclusion body myositis”. Current Opinion in
4‐78. Rheumatology. 2011. 23: 57
3. Engel, W.K. and Askanas, V. “Inclusion body myositis: clinical, diagnostic
y. 2006. 66 (Supplement 1) S20‐29. and pathological aspects”. Neurolog
4. Stenzel W., Goebel H.H., Aronica E. “Review: Immunemediated necrotizing
myopathies a heterogeneous group of diseases with specific

myopathological features”. Neuropathology and Applied Neurobiology.
2‐646. 2012. 38: 63
5. Dalakas, M. “Pathogenesis and therapies of immunemediated myopathies.”
ty reviews. 2012. 11: 203‐06. Autoimmuni
6. Pestronk, A. “Acquired immune and inflammatory myopathies: pathologic
t Opinion in Rheumatology. 2011. 23: 595‐604. classification.” Curren
7. Amato A.A., Griggs C. “Unicorns, dragons, polymyositis and other
ythological beasts”. Neurology. 2003. 61: 288‐290. m


















