
REVISTA CHILENA DE EPILEPSIArevistachilenadeepilepsia.cl/revistas/a_8_n1_diciembre2008.pdf ·...
70
REVISTA CHILENA DE EPILEPSIA Publicación Oficial de la Sociedad de Epileptología de Chile Capítulo Chileno de la ILAE http: //www.epilepsiadechile.com / E-mail: [email protected] Año 8, Nº 1, Diciembre de 2007 ISSN 0717-5337 Contenido Editorial 3 Trabajos originales Clínica y comorbilidades en niños con epilepsia fallecidos por muerte súbita 4 Cirugía de la epilepsia en niño. Hemisferectomía. Experiencia en el Instituto de Neurocirugía Asenjo 14 Sindrome de West: Presentación clínica y pronóstico a largo plazo 19 Trabajos de Revisión Epilepsia, sueño y trastornos del sueño 32 Factores desencadenantes de crisis epilépticas 38 Epilepsia Rolándica: Un amplio espectro 45 ¿Qué es la Comorbilidad? 49 Crónica Programa Coloquios en Epilepsia 52 Acreditados 2007. Ingresados 2006-2007. 54 Memoria 2007 55 Programa VIII Jornadas Invernales de Epilepsia 58 Trabajos presentados en las VIII Jornadas Invernales de Epilepsia 60 Congresos y Cursos 2008 69 Sugerencias para las contribuciones a los autores 70 70 S O C I E D A D D E E P I L E P T O L O G I A D E CH I L E
Transcript of REVISTA CHILENA DE EPILEPSIArevistachilenadeepilepsia.cl/revistas/a_8_n1_diciembre2008.pdf ·...

�
REVISTA CHILENA DE EPILEPSIAPublicación Oficial de la Sociedad de Epileptología de Chile
Capítulo Chileno de la ILAEhttp: //www.epilepsiadechile.com / E-mail: [email protected]
Año 8, Nº 1, Diciembre de 2007ISSN 0717-5337
Contenido
Editorial 3
Trabajos originalesClínica y comorbilidades en niños con epilepsia fallecidos por muerte súbita 4Cirugía de la epilepsia en niño. Hemisferectomía. Experiencia en el Instituto de Neurocirugía Asenjo 14Sindrome de West: Presentación clínica y pronóstico a largo plazo 19
Trabajos de RevisiónEpilepsia, sueño y trastornos del sueño 32Factores desencadenantes de crisis epilépticas 38Epilepsia Rolándica: Un amplio espectro 45¿Qué es la Comorbilidad? 49 CrónicaPrograma Coloquios en Epilepsia 52Acreditados 2007. Ingresados 2006-2007. 54Memoria 2007 55Programa VIII Jornadas Invernales de Epilepsia 58Trabajos presentados en las VIII Jornadas Invernales de Epilepsia 60Congresos y Cursos 2008 69Sugerencias para las contribuciones a los autores 70 70
SOCI
EDAD
DE EPILEPTOLOG
IA
DE CHILE

2
Revista Chilena de Epilepsia Año 8, Nº 1, Diciembre de 2007
SOCIEDAD DE EPILEPTOLOGIA DE CHILE
Capítulo Chileno de la Liga Internacional con-tra la EpilepsiaFundada el 13 de Marzo de 1999
DIRECTORIO 2005
PresidenteDr. Juan Salinas
VicepresidenteDr. Marcelo Devilat
Secretaria GeneralDra. Daniela Avila
TesoreraDra. Daniela Triviño
Past PresidentDr. Cayetano Napolitano
Editora de PublicacionesDra. Perla David
DirectorDra. Erna Rauch
Encargado de EventosDr. Marcelo Devilat
Delegados Anliche:Dr. Jorge FörsterDr. Manuel Campos
Comité EditorialEditora: Perla DavidComité Editorial: Marcelo Devilat, Fernando Ivanovic-Zuvic, Andrea Pérez, Erna Rauch, Juan Salinas y Daniela Triviño
Dirección:Av. Providencia 2315, Of. 215, Fonos: 231 0172, 235 1470, Fax 234 0671, Providencia, Santiago, Chile. E-mail: [email protected] ó[email protected]
Diseño GráficoJuan Silva / 635 2053 / [email protected]
DIRECTORIO ILAE 2005-2009
PresidentPeter Wolf
1st VicepresidentProf. Frederick Andermann, MD.
2nd VicepresidentProf. Frederick Andermann, M.
Treasurer Prof. Martin J. Brodie, M.D.
SecretarySolomon Moshe M.D.
Past PresidentProf. Giuliano Avanzini
EditorPhillip A. Schwartzkroin P.h.
InformationProf. Simon D. Shorvon
Ibe PresidentPhilip Lee
Ibe SecretaryDr. Esper A. Cavalheiro
Ibe, TreasurerJohan Falk-Pedersen

3
Editorial
Otro de los temas que se tocan tiene que ver con la frecuente Comorbilidad que acompaña a las personas que presentan epilepsia, este es un área de altísimo in-terés dado que frecuentemente nos vemos frente a la situación de un paciente que presenta una enfermedad comórbida, por una parte tenemos la posibilidad que la epilepsia aumente el riesgo de presentar algún tipo de patología, como lo vemos en casos como la de-presión o los trastornos hipercinéticos; pero también nos vemos a la situación del incremento en el riesgo para epilepsia asociada a algunas condiciones. Más allá de lo anterior, en muchas circunstancias la pre-sencia de Comorbilidad va a significar un problema no sólo en el control y pronóstico de quién presenta esta condición, sino que también un deterioro en la calidad de vida de los sujetos y los costos asocia-dos. Esto nos lleva a destacar la preocupación por ir acercándonos al conocimiento de la realidad de nuestros pacientes en cuanto al tipo y gravedad de la Comorbilidad que presentan.
Un tema importante, sin lugar a duda, es aquél que se refiere a la muerte súbita de las personas con epilepsia, afortunadamente la incidencia de muerte súbita e inesperada sin una causa evidente es baja; sin embargo, por la gravedad de la situación, es preciso abordar el punto conociendo nuestra realidad y en lo posible ser capaces de ir identificando los potenciales factores de riesgo, particularmente aquellos suscep-tibles de ser modificados y que permitan evitar un desenlace fatal en estas condiciones.
También en esta revista nos encontraremos con la revisión de dos importantes síndromes epilépticos de la niñez, el Síndrome de West y la epilepsia rolándica. Estos cuadros, sea por su frecuencia o su gravedad deben encontrarse permanentemente en nuestro ámbito de preocupaciones para insistir en su estudio y abordaje.
Felicito a la Dra. Perla David, por su permanente preocupación para mantener la excelencia de los artículos publicados, lo que se ve reflejado en las pre-sentaciones que encontrarán en la presente edición.
La Sociedad de Epileptología de Chile tiene como misión contribuir al desarrollo de la preparación de los profesionales que trabajan en el área de la Epilepsia, para ello desde su génesis, primero como grupo de estudio y posteriormente como Sociedad Científica ha contribuido a dicho propósito a través de diversas actividades como las reuniones mensuales de presentaciones de temas de interés, nuestras Jor-nadas Invernales que convocan a una gran cantidad de profesionales interesados en el tema de Epilep-sia, la capacitación entregada por los Coloquios en Epilepsia que ya cuenta con su segunda edición y la participación en la elaboración de Políticas y Normas que regirán la atención de nuestros pacientes.
Además de lo anterior contamos con la Revista Chi-lena de Epilepsia, publicación científica que destaca por la alta calidad de sus artículos, que dan cuenta de la preparación técnica de los profesionales chilenos y constituye una importante fuente de difusión de interesantes tópicos.
En la presente edición nos encontramos con diversos artículos de gran interés, los que por su excelencia, contribuyen de manera importante a la especializa-ción en el tema y representan una buena muestra de la capacidad de generar un movimiento hacia la mejor calidad en la atención de la población de personas con epilepsia, sus familias y la sociedad en general.
Entre los diferentes temas se plantea el de sueño y epilepsia, sin lugar a dudas, la relación entre ambos constituye un tema de alto interés para el clínico, desde esta perspectiva hay distintas situaciones que resultan de interés analizar, así podemos enfrentarnos a las frecuentes quejas sobre el sueño que presentan las personas con epilepsia; pero también tenemos la diferenciación de los fenómenos paroxísticos que se presentan durante el sueño y además podemos preocuparnos de la influencia del sueño en el estudio de la epilepsia o de los tipos de crisis que se presen-tarán en relación al ciclo sueño/vigilia. Por tanto, representa un área de interés esencial para el clínico y su inclusión nos alienta a considerarla como un tema a tener presente.

4
Revista Chilena de Epilepsia Año 8, Nº 1, Diciembre de 2007
Clínica y comorbilidades en niños con epi-lepsia fallecidos de muerte súbitaDr. Marcelo Devilat, Dr. Gianni Rivera, E.U. Verónica Gómez, Sr. Juan Pablo SepúlvedaCentro de Epilepsia Infantil. Servicio de Neurología y Psiquiatría. Hospital de Niños Luis Calvo Mackenna. Santiago, Chile. E-mail: [email protected]
Trabajos Originales
RESUMEN
IntroducciónLa muerte súbita inesperada en epilepsia (MSIEP) es una eventualidad que ha sido bien descrita en adultos. En niños la información es escasa y contradictoria. Objetivo. Determinar el riesgo de la MSIEP en ni-ños con epilepsia, describir algunas características clínicas y determinar las comorbilidades asociadas a la epilepsia. Pacientes y métodos. Desde 1996 a 2006 fallecieron 22 niños con epilepsia controlados en el Centro y entraron a un protocolo prospectivo. En 9 de ellos la causa de muerte fue MSIEP y son los que constituyen el grupo investigado. La infor-mación fue extraída del banco de datos, de la ficha clínica y de entrevistas con los padres. Resultados. La incidencia fue de 1.66 x 1.000. En sólo 2 niños se pudo determinar el tipo definitivo de MSIEP y en uno, se estableció una MSIEP posible. El resto de los pacientes falleció de MSIEP probable. Todos los niños, salvo 1, fallecieron en domicilio, la mayoría murió en el sueño y los que fallecieron en vigilia, lo hicieron después de una crisis epiléptica. Siete enfermos tuvieron epilepsias sintomáticas, activas o resistentes, con EEG específico, con múltiples esque-mas terapéuticos y daño neurológico. En 7 pacientes la comorbilidad neurológica más frecuente fue el retardo mental y la parálisis cerebral. Siete enfermos presentaron comorbilidad no neurológica siendo la más frecuente la bronconeumonía. Conclusión. El riesgo de MSIEP en niños con epilepsia pareciera ser moderado especialmente si presentan epilepsias sintomáticas de difícil manejo y asociadas a comor-bilidades neurológicas y no neurológicas.
Palabras clavesAspectos clínicos. Comorbilidades. Epilepsia. Muer-te súbita inesperada. Niños. Riesgo.
Palabras de cabeceraMuerte súbita inesperada en niños con epilepsia.
ABSTRACT
IntroductionSudden unexpected death in epilepsy (SUDEP) has been well described in adults. There is a few and con-tradictory information about children. Objetive. To determine the risk of SUDEP in children with epilepsy, to describe some clinical characteristics and to determine the comorbidities associated to epilepsy. Subjects and methods. Between 1996 and 2006, twenty two children with epilepsy treated at the Center died and entered to a prospective protocol. There were 9 patients who died of SUDEP, and they constituted the investigated group. The information was gathered from a data base, the clinical records and interviews with the parents. Results. The inci-dence was 1.66 x 1.000. In only 2 children it was possible to determine the definitive type of SUDEP and in one, it was stablished a possible SUDEP.The rest of the patients died of probable SUDEP. All of the patients, except one, died at home, most of them died during sleep, and those who died awake did so after a seizure. There were seven children who had symptomatic, active, or refractory epilepsy, with an specific EEG, treated with several drugs and with a neurologic damage. In seven patients the most common neurologic comorbidity was mental retar-dation and cerebral palsy. Seven children had non neurologic comorbidities, and the most frequent of if them was bronchopneumonia. Conclusions. It seems that the there is a moderate risk of SUDEP in children, specially if they have symptomatic, resistant epilepsy, and associated with neurological and non-neurological comorbidities.
Key wordsSudden unexpected death in epilepsy. Children. Risk. Clinical characteristics. Comorbidities.
INTRODUCCION
La mortalidad en niños con epilepsia ha recibido menos atención [1] que la de los adultos[2], a pesar

5
Clínica y comorbilidades en niños con epilepsia fallecidos de muerte súbita Marcelo Devilat et al
que los trabajos disponibles han comunicado para aquellos, tasas de mortalidad estándar (TME) de hasta 13.2 (IC 95%; 8,5-20,7)[3]. Esta cifra puede triplicarse en el caso de las epilepsias sintomáticas [4], especialmente las asociadas a neurodéficits [5].
Sin embargo, dado que los estudios son muy hete-rogéneos, la información obtenida puede llegar a tener una gran variabilidad [1,2,6,7]. El análisis de las causas de muerte es un ejercicio de utilidad pues permite detectar factores destinados a prevenirla [8,9]. Una de ellas es la muerte súbita inesperada, que constituye la mayor causa de muerte relacionada con la epilepsia [6,10-12]. De acuerdo a la literatura disponible [3,4,6,11,13-17], la incidencia de la muerte súbita inesperada en los niños varía de 0 a 0.2 x 1.000, lo que corresponde a un riesgo insignificante [18]. En los adultos la incidencia fluctúa entre 0 a 10 x 1000 [6,19,20] lo que constituye un riesgo moderado [18]. Las cifras dependen de la metodología de los estudios [1,6,21] encontrándose incidencias superiores en cohortes seleccionadas que en aquellas de comunidad [21]. Existe alguna información acerca de los factores de riesgo de muerte súbita en los niños con epilepsia. Sin embargo, aun los estudios son escasos y a nues-tro conocimiento deben haber alrededor 60 casos publicados [1,2,4,6].
Los pacientes con epilepsia presentan altos índices de comorbilidad, de acuerdo a trabajos en los que participan adultos [22], adultos y niños [23,24] y exclusivamente niños [25]. Ella ocasiona enormes gastos en salud y puede contribuir a la refractariedad [22,23].
Las personas con epilepsia que fallecen presentan también altas tasas de comorbilidad, superiores a las de la población general como ha sido comunicado para adultos[2,5,21,26], . En los niños con epilepsia que fallecen, las comorbilidades han sido menos estudiadas[1,3,4,17]. Para adultos con epilepsia, el retardo mental (RM), como comorbilidad, ha sido mencionado asociado con la muerte súbita inesperada [21], pero en niños no existe información sistemati-zada al respecto. El objetivo de esta investigación consistió en de-terminar el riesgo de muerte súbita inesperada en niños con epilepsia, describir algunas características
clínicas y determinar las comorbilidades.
PACIENTES Y METODO
Desde octubre de 1996 y hasta marzo de 2006, los 22 niños con epilepsia fallecidos en ese lapso y que estaban en control y tratamiento en el Centro, se ingresaron prospectivamente a un protocolo previo. Una parte de ellos fueron comunicados en un trabajo anterior [1]. De los 22 pacientes fallecidos hubo 9 que fallecieron de muerte súbita inesperada y son los que constituyen el motivo de este estudio. El Centro se informó de los fallecimientos a través de los padres por entrevista personal o telefónica y los datos se extrajeron de la base computacional y de la ficha médica de los pacientes. El diagnóstico de epilepsia, del tipo de crisis y de sín-drome se realizó de acuerdo a las recomendaciones de la Liga Internacional contra la Epilepsia [27,28]. Los conceptos de epilepsia remota sintomática y epilepsia sintomática fueron expuestos en un trabajo anterior [1]. El coeficiente intelectual fue determi-nado clínicamente y por evaluación psicométrica en algunos pacientes. El término de parálisis cerebral (PC) se utilizó para designar un control anormal del movimiento o de la postura, de inicio precoz y en ausencia de enfermedad progresiva subyacente [1]. Cuatro de los 9 pacientes tenían tomografía axial computada de cerebro. En el resto no se realizó el examen por considerarlo innecesario en dos niños y en 3 no se pudo realizar por diversas razones. En todos los pacientes los EEG fueron interictales y su descripción corresponde al último examen antes de morir. Se empleó un electroencefalógrafo de 12 cana-les con derivaciones en el sistema internacional 10-20 y las definiciones empleadas en los informes fueron detalladas en una comunicación anterior [1]. Se designó como politerapia al esquema terapéutico con 2 o más fármacos antiepilépticos (FAE). Los niveles de FAE se realizaron en todos los pacientes menos uno y fueron consignados como terapéuticos si estaban dentro del rango recomendado por el labo-ratorio. Estos exámenes corresponden a los últimos realizados antes del fallecimiento. Los conceptos de epilepsia activa, inactiva, resistente y catastrófica utilizados en esta investigación fueron desarrollados en una comunicación anterior [1]. Todos los decesos de los pacientes fueron comuni-

6
Revista Chilena de Epilepsia Año 8, Nº 1, Diciembre de 2007
cados al Centro por los padres, cuando ocurrió en domicilio. Una niña (caso 3) falleció en el Servicio de Urgencia durante una crisis tonicoclónica generaliza-da y estaba cursando una escarlatina. Esta paciente corresponde al caso 9 de nuestra comunicación anterior [1] y en esa oportunidad no fue incluida como muerte súbita inesperada, sin embargo, una revisión de los datos permiten ubicarla dentro de una muerte súbita posible, como se ha hecho en la presente comunicación. Se definió como comorbilidad a la co-ocurrencia de más de una enfermedad en la misma persona [22,24] y el criterio de diagnóstico de muerte súbita [19] fue el siguiente: 1. La persona cumple con los criterios de epilepsia. 2. La muerte ocurre repentina e ines-peradamente. 3. La muerte acaece en un razonable estado de salud, en benignas circunstancias y dentro del desarrollo de actividades normales. 4. No existe una causa médica obvia o razonable. El diagnóstico de muerte súbita definitiva incluyó los criterios señalados más una autopsia, que descartó una causa anatómica y toxicológica. Se designó como muerte súbita probable a aquellos que cumplieron con los 4 criterios clínicos descritos y muerte súbita posible a aquella en la cual las causas de muerte invocadas no son del todo seguras. El cálculo de la incidencia se realizó en base al nú-mero de pacientes en control en el Centro que fluctuó entre 402 para el año 1996 y 644 para el año 2006. RESULTADOS
Durante el lapso del estudio fallecieron 22 pacien-tes que estaban en control y tratamiento en nuestro Centro. De ellos, 9 (40.90%) lo hicieron debido a una muerte súbita inesperada. La incidencia reveló ser de 1.66 por 1.000 niños. El grupo se compone de 4 hombres y 5 mujeres. La edad de inicio de la epilepsia fue de 3.87 años en promedio (1 mes a 13 años). Cuatro niños iniciaron su enfermedad antes del primer año de vida. La edad promedio de la muer-te fue de 6 años 5 meses (5 meses a 19 años) En la Tabla 1 se muestran las circunstancias y tipo de muerte súbita en los 9 niños con epilepsia. En sólo 2 niños se pudo determinar el tipo definitivo y en uno, una muerte súbita posible, el resto de los pacientes falleció de muerte súbita probable. En el caso 1 la autopsia reveló un paro cardiorrespiratorio como la causa de muerte y en el caso 7 se informó que hubo
una hipoxia generalizada. Todos los pacientes, salvo 1, fallecieron en domicilio, la mayoría muere mien-tras duerme, en uno de los cuales se pudo detectar que estaba en prono. Tres de los 4 que fallecen en vigilia, lo hacen después de una crisis epiléptica en presencia de testigo. La Tabla 2 destaca que la mayoría de los niños pre-sentó crisis generalizadas en forma de crisis tonico-clónicas generalizadas aisladas (CTCG) o asociadas a crisis mioclónicas. Estas últimas se presentaron en 2 niños. Un tercio de los pacientes tenía crisis parciales. Seis de los niños tenían síndromes gene-ralizados, entre los que se destaca a 2 síndromes de West y 1 con epilepsia mioclónica juvenil. El resto presentaba síndromes parciales. Etiológicamente, 7 enfermos presentaban epilepsias sintomáticas. El resto, uno era idiopático y otro criptogénico. Desde el punto de vista evolutivo 7 niños tenían epilepsias activas, resistentes o catastróficas. Dos, las tenían inactivas. En la Tabla 3 se exhiben los resultados del EEG y los niveles de FAE, así como también el manejo medi-camentoso de los pacientes. Todos los niños salvo 2 (los con epilepsias inactivas) tenían EEG específico para epilepsia. Durante su evolución el grupo de niños había recibido en promedio 2.66 esquemas de tratamiento (1 a 8), en los que se utilizaron FAE de primera y segunda línea, así como de última gene-ración. Un paciente falleció durante el lapso en que recibía dieta cetógena. La tabla 4 informa que 5 pacientes presentaron una etiología probablemente responsable de su cuadro epiléptico como lo fueron infecciones del SNC, daños hipóxicos cerebrales y malformaciones encefálicas. Todos los niños, salvo 2, los correspondientes a aquellos que tenían epilepsias inactivas, presentaron comorbilidades neurológicas dobles, en las que en la mayoría se asoció RM y PC. Un niño tenía RM con autismo y otro, RM con accidente vascular cerebral. Las comorbilidades no neurológicas que se presen-tan en la tabla 4, ocurrieron en todos los pacientes, salvo en 2, destacándose las bronconeumonías, en un caso asociado a gastrostomía. Dos niños tenían desnutrición y enfermedad del tejido conectivo, y una paciente falleció estando con un embarazo de 21-22 semanas. La familia, especialmente la madre de 7 de los niños fallecidos recibió el apoyo por parte del personal del

7
Centro al conocerse la muerte, con el objeto de con-tribuir a la elaboración del duelo. Apoyo fue personal en 6 pacientes y en uno fue telefónico pues la familia era fuera de Santiago. En 2 familias no se pudo rea-lizar el apoyo. En una de ellas, que vivía alejada de la capital, no se pudo realizar contacto telefónico. En la otra familia tampoco se hizo apoyo por cuanto el padre, que se presentó personalmente al hospital a comunicar el fallecimiento de su de hijo, se retiró contrariado ante la negativa de recibir de nuestra parte el certificado de defunción. Por haber sido un fallecimiento en domicilio, sin controles regulares en el Centro, se le comunicó que debía efectuarse una autopsia en el Servicio de Medicina Legal. De este caso no tuvimos información posterior.
DISCUSION La muerte súbita inesperada es la más frecuente causa de muerte relacionada a epilepsia entre las personas que sufren la enfermedad [1,21], y su incidencia, tanto para adultos como para niños es muy variable con cifras desde 0 a 10 x 1.000 [6,21], dependien-do de la metodología empleada en las diferentes investigaciones, de la definición de muerte súbita inesperada y del origen de las muestras [1,21]. A pesar de lo anterior, la incidencia es varias veces superior a la de la población general [4,29]. Como en la mayoría de los trabajos publicados, en éste, un escaso número de pacientes tiene autopsia, lo que impide certificar una muerte súbita inesperada como definitiva [8,11,13,30]. En la presente casuística, de 22 casos de niños con epilepsia fallecidos, en 9 (40%), la muerte fue causada por MSIEP y se obtuvo una incidencia de 1.66 por 1.000 niños con epilepsia por año, revelándose como una cifra de magnitud si se la compara con otras investigaciones. En efecto, otros estudios [3,4,13,15] revelan frecuencias muy bajas, que han permitido sugerir a algunos [4] que la incidencia estimada para los niños con epilepsia sería de 0.13 x por 1.000, y globalmente de 1 a 2 x 10.000 [15] lo cual permitiría concluir que el problema sería irrelevante [18] y cuya explicación “debe ser investi-gada” [18]. Sin embargo, es posible acceder a cifras de incidencia superiores como las presentadas aquí, cuando se analizan investigaciones realizados en centros especializados, tal como lo revela el 1.24 por 1000 del un reciente trabajo holandés en pacientes adultos [31] y la cifra de 1.66 por 1000 de la presente investigación. Las dos incidencias señaladas sugie-ren que el problema se ubica al menos dentro de un riesgo moderado, como lo indica un meta análisis
que arroja 0 a 2.5 por 1000 [6]. Poco menos de la mitad de los niños iniciaron su enfermedad antes del año de vida y la edad promedio de inicio de la epilepsia es menor de 4 años, lo que podría sugerir que un inicio precoz de la epilepsia podría ser un factor de riesgo de muerte súbita ines-perada. Debido a la escasa información al respecto [3,26] es difícil proponer una conclusión, aunque se ha sugerido que podría existir una relación entre una precoz edad de inicio de la enfermedad y el riesgo de muerte, lo que indicaría la existencia de una base neuropatológica que se exprese tempranamente en los niños [1,5,6]. El tipo de muerte súbita inesperada de esta muestra revela una alta proporción de tipos probables y en sólo 2 pacientes se determinó que la forma definitiva, lo que revela como es habitual, la infrecuencia de la autopsia en las personas con epilepsia que fallecen [8,19]. Debido a lo anterior se ha propuesto, el uso de la autopsia verbal [30] en la que los familiares, responden a un cuestionario previamente preparado acerca de la enfermedad de la persona, los síntomas y las circunstancias de la muerte, entre otras pregun-tas. A pesar de lo anterior, la información disponible de autopsias en muerte súbita inesperada en niños, revela que de los casos comunicados en la literatura a disposición [1,4,17,14] más algunos anecdóticos [32], mayoritariamente corresponden a la forma definitiva. Todos los pacientes de esta serie, salvo 1, fallecieron en domicilio, la mayoría lo hizo en el sueño mientras dormía, uno de ellos en prono. La muerte en la cama mientras el niño duerme parece ser la circunstancia más frecuente como fue observado en esta serie y en otras [17,33] y en casos anecdóticos [11,32], corres-pondientes a niños. La mayor información disponible al respecto en adultos, permite destacar aún más este hecho [2,9,21,34,35]. Se ha podido establecer signos de crisis epiléptica reciente, previa a la muerte en la mayoría de los casos de las personas que fallecen durante el sueño [2,9], fase durante la cual se incre-menta el tono vagal, causando bradicardia relativa, lo que podría contribuir a la inestabilidad de los mecanismos autonómicos que ocurren en las crisis, predisponiendo a la muerte súbita inesperada [34]. Lo anterior es importante puesto que se ha sugerido la relevancia de la supervisión del sueño y su postura en supino en las personas con epilepsia [9,33], con el objeto de prevenir las muertes súbitas.
Clínica y comorbilidades en niños con epilepsia fallecidos de muerte súbita Marcelo Devilat et al

8
Revista Chilena de Epilepsia Año 8, Nº 1, Diciembre de 2007
Una proporción inferior de personas fallecen en vigi-lia generalmente luego de una crisis epiléptica como sucedió en 3 de nuestros pacientes y ante testigos presenciales. La proporción de muertes súbitas in-esperadas ante testigos se sitúa en el 10% [21] de los casos y generalmente asociadas a una crisis tónico-clónica generalizada. Sin embargo, es posible como ocurrió en nuestro caso 5, que acontezca la muerte sin una crisis epiléptica, tal como ha sido relatado en la literatura [17]. En nuestro caso, se agrega un factor, el temblor, cuya relación con la muerte súbita inesperada, no queda del todo claro, salvo postular un fenómeno causal cardiogénico [36]. La mayoría de los niños presentaron crisis generali-zadas y dentro de estas, crisis tonicoclónicas, lo que también ha sido comunicado por otros autores en casuísticas de niños [17] y en adultos [21], lo cual podría estar en relación a la falla de los mecanismos autonómicos que ocurren durante las CTCG [9,34]. Sin embargo, las personas que fallecen de muerte súbita inesperada también pueden ser portadoras de crisis epilépticas parciales como ocurrió en 3 de nuestros pacientes, hecho que sido citado en la lite-ratura dedicada a los niños [17]. En este sentido, los pacientes con crisis parciales secundarias a esclerosis hipocampal podrían fallecer de MSIEP por acción de una desregulación autonómica relacionada con la al-teración a ese nivel [37]. No parece estar establecido que la muerte súbita inesperada esté asociada a un tipo especial de epilepsia si se compara epilepsias generalizadas con parciales [21], lo que claramente puede ser explicado por lo heterogéneo de las mues-tras. Sin embargo, es destacable que sólo 1 paciente de esta muestra portaba una epilepsia idiopática. El resto tenían formas remotas sintomáticas o criptogé-nicas, que en su mayoría eran a epilepsias activas, refractarias o catastróficas [38] lo que revelaría un especial riesgo en esos tipos de epilepsias de acuerdo revisiones en investigaciones con adultos y niños [5,6,21,39]. Todos los niños salvo 2 presentaron interictalmente electroencefalogramas específicos para epilepsia, lo que por una parte confirma el diagnóstico y por otra sugiere que el cuadro epiléptico estaba activo y podría un factor de riesgo. En niños, salvo en nuestra casuística anterior [1], no se ha hecho referencia a este importante hallazgo. También en las muy es-casas las investigaciones con enfermos adultos, se revela que un tercio tiene actividad epiléptica [40], aunque para otros [39] la mayoría de los EEG son
epilépticos, en tanto algunos [36]) han comunicado que tanto los EEG ictales como los interictales son mayoritariamente normales. El cumplimiento con la medicación parece ser acep-table en nuestros pacientes, ya que todos se mane-jaban con niveles terapéuticos, lo que podría ser un factor protectivo que no se hizo efectivo en nuestros niños. En todo caso no existe suficiente experiencia como para evaluar esta situación, salvo mencionar que los niveles de FAE postmortem pueden resultar subterapéuticos [33,39]). Concordante con las carac-terísticas de los niños de esta investigación, la mayo-ría de ellos había tenido varios esquemas terapéuticos y estaban en politerapia. Salvo en una comunicación anterior nuestra [1], la literatura dedicada al tema en este punto en los niños no menciona este hecho. En adultos, los múltiples esquemas [2,35] y la politerapia [6,9], sugieren ser factores de riesgo, aunque para otros esta relación no es tan evidente [41]. El apoyo a la familia después de ocurrir el deceso es muy relevante porque contribuye a elaborar el duelo [1]. La mayoría de las familias recibió dicho apoyo personal por parte del profesional a cargo del niño y las entrevistas fueron de utilidad para disminuir la ansiedad y los sentimientos de desvalimiento y culpa que origina la muerte. Todos los pacientes salvo 2, portadores de epilepsia idiopática y criptogénica respectivamente, presenta-ron comorbilidad neurológica severa dentro de una epilepsia sintomática. Las comorbilidades en niños con epilepsia son frecuentes y variadas [25], pero no alcanzan el alto número de retardo mental (RM) y parálisis cerebral (PC) que presentan los niños de esta serie, debido probablemente a lo orgánico que era la mayoría de los pacientes. Por otra parte, la asociación de RM y epilepsia presenta altas tasas de mortalidad estandar [2], así como también cuando el RM se asocia a PC en series en la que intervienen adultos y niños con epilepsia para mortalidad en general y para muerte súbita inesperada [5,16,21]. Sin embargo si se revisan los trabajos de mortalidad en niños con epilepsia en los cuales se hace referen-cia a comorbilidad neurológica, sólo se encontraran 3 publicaciones [1,3,17] las que por otra parte, se refieren a mortalidad general por epilepsia y no es-pecíficamente a muerte súbita inesperada. Aunque sin exponer información, un autor [14], destaca que “La mayoría de las muertes (en niños con epilepsia)

9
son relacionadas con comorbilidades neurológicas”. Es relevante destacar que en las 3 publicaciones cita-das se menciona un alto número de comorbilidades neurológicas que van del 62 al 91%, lo que también se observa en la presente casuística.
Especialmente en el trabajo australiano [6], se señala que la comorbilidad en mortalidad por epilepsia en los niños que fallecen o no de muerte súbita inespe-rada es diferente a la de los adultos. Específicamente, en ese trabajo, la PC y la RM, también alcanzan como en el nuestro un alto número, en tanto que los adultos las comorbilidades representan una mayor gama de posibilidades [5,42]. Un hecho relevante en esta serie es que los pacientes con comorbilidad neurológica, la presentaron en los 7 casos en forma doble, con RM acompañado ya sea con PC, AVC o autismo, lo que sugiere una mayor severidad y puede constituir factores de riesgo no destacados en la literatura. La comorbilidad no neurológica en niños pacientes con muerte súbita inesperada ha sido muy infre-cuentemente comunicada [1,17], a pesar que en esta serie la presentaron 7 de 9 niños, constituyendo una magnitud muy superior a las 6 de 27 de otro autor [17]. En adultos, especialmente para mortalidad general en epilepsia, se han mencionado numerosas comorbilidades no neurológicas [2,5,42].
La presencia de bronconeumonías a repetición son una comorbilidad frecuente tanto en los niños que fallecen de muerte súbita inesperada como ocurrió en esta serie, como en adultos. Este mismo hecho se presenta cuando se analiza la mortalidad general de niños con epilepsia [1,17], representando también una importante causa de muerte [1,2]. Uno de los pacientes de esta serie falleció estando con gastros-tomía lo que también ha sido comunicado por otros autores [17] revelando la gravedad del compromiso neurológico y la falla en impedir una aspiración mediante el procedimiento gástrico. Una paciente (caso 1) falleció siendo portadora de un embarazo de 22 semanas, hecho que había sido reportado por nosotros, como caso único [1]. Sin embargo, un segundo caso ha sido comunicado [32] en una mujer joven con un embarazo de 8 meses y una epilepsia que se había activado y se había vuelto del sueño. Ambas pacientes eran jóvenes, fallecieron durante el sueño y la muerte súbita inesperada fue de-finitiva. A pesar de algunas similitudes, entre ambos casos, no tenemos explicación para esta asociación
entre embarazo y muerte súbita inesperada. Otra paciente (caso 5) falleció poco después de un temblor [1], pero aunque no sabemos la magnitud del impacto psicológico del temblor sobre la niña, existe información de cierta asociación entre stress emocional previo y muerte súbita inesperada en adul-tos [34]. Podría ser probable que un stress excesivo y brusco pudiera originar un desbalance autonómico [15][36] de tal magnitud que ocasione la muerte en personas predispuestas.
En conclusión, aunque esta serie informa de sólo 9 niños con muerte súbita inesperada se destaca que ella no es tan rara como ha sido mencionado [4,14,15,17] en consideración que se obtuvo un 1,66 x 1000 de incidencia. Se destaca que las epilepsias sintomáticas y refractarias, con frecuentes CTCG, de inicio precoz, con EEG específico, con varios cambios terapéuticos y frecuentes comorbilidades en niños con daños neurológicos son las características más relevantes de estos pacientes.
BIBLIOGRAFIA
1. Devilat-Barros M, Rivera-Gómez G, Gómez-Muñoz V, Sepúlveda-Olmos JP. Mortalidad en niños con epilepsia. Estudio clínico prospectivo. Rev Neurol 2004; 38: 607-4.
2. Gaitatzis A, Sander JW. The mortality of epilepsy revisited. Epileptic Disord 2004; 6: 3-13.
3. Harvey AS, Nolan T, Carlin JB. Community-ba-sed study of mortality in children with epilepsy. Epilepsia 1993; 34: 597-3.
4. Berg AT, Shinnar S, Testa FM, Levy SR, Smith SN, Beckerman B. Mortality in childhood–onset epilepsy. Ach Pediatr Adolesc Med 2004; 158: 1147-2.
5. Forsgren L, Hauser WA, Olafsson E, Sander JWAS, Sillanpää M, Tomson T. Mortal i ty of epilepsy in developed countries. Epilepsia 2005; 46 (Suppl 11): S18-7.
6. Tellez-Zenteno JF, Hernández Ronquillo L, Wiebe S. Sudden unexpected death in epilepsy: evidence-based analysis of incidence and risk factors. Epilepsy Research 2005; 65: 101-5.
7. Logroscino G, Hesdorffer, D. Methodologic is-sues in studies of mortality following epilepsy: measures, types of studies, sources of cases, cohort effects and competing risks. Epilepsia 2005; 46 (Suppl 11): S3-7.
8. Hanna NJ, Black M, Sander JWAS, Smithson
Clínica y comorbilidades en niños con epilepsia fallecidos de muerte súbita Marcelo Devilat et al

10
Revista Chilena de Epilepsia Año 8, Nº 1, Diciembre de 2007
WH, Appleton R, Brown S, et al. National Sen-tinel clinical audit of epilepsy-related death: epi-lepsy-death in the shadow. The stationary office. United Kingdom: Epilepsy Bereaved; 2002.
9. Langan F, Nashef L, Sander JW. Case-control study of SUDEP. Neurology 2005; 64: 1131-3.
10. Rufo-Campos M. Mortalidad en las epilepsias. Rev Neurol 2000; 30 (Supl 1): S 110-4.
11. Morentin B, Alcaraz R. Muerte súbita inesperada en epilepsia en niños y jóvenes. Rev Neurol 2002; 34: 462-5.
12. Gairazar C. Muerte súbita en epilepsia: la expe-riencia del clínico. Rev Neurol 2002; 34: 460-2.
13. Callenbach PMC, Westendorp RGJ, Geerts AT, Arts WFM, Peeters EAJ, van Donselaar CA, et al. Mortality risk in children with epilepsy: the dutch study of epilepsy in childhood. Pediatrics 2001; 107: 1259-3.
14. Camfield CS, Camfield P, Veugelers PL. Death in children with epilepsy: a population-based estudy. Lancet 2002; 359: 1891-5.
15. Camfield P, Camfield C. Sudden unexpected death in people with epilepsy: a pediatric pers-pective. Semin Pediatr Neurol 2005; 12: 10-4.
16. Appleton RE. Sudden,unexpected death in epi-lepsy in children. Seizure 1997; 6: 175-7.
17. Donner EJ, Smith CR, Snead OC. Sudden unex-plained death in children with epilepsy. Neuro-logy 2001; 57: 430-4
18. Hanna J. Epilepsy and sudden death: a personal view: Epilepsia 1997; 38 (Suppl 11): S3-5.
19. Nashef L, Shorvon SD. Mortality in epilepsy. Epilepsia 1997; 38 (Suppl 11): S 6-8.
20. Carpio A, Bharucha NE, Jallon P, Beghi E, Campostrini R, et al. Mortality of epilepsy in developing countries. Epilepsia 2005; 46 (Suppl 11): S28-2.
21. Tomson T, Walczak T, Sillanpää M, Sander JWAS. Epilepsia 2005; 46 (Suppl 11): S54-1.
22. Gaitatzis A, Caroll K, Majeed A, Sander JW. The epidemiology of the comorbidity of epilepsy in the general population. Epilepsia 2004; 45: 1613-2.
23. Van der Broek M, Beghi E. Morbidity in patients with epilepsy: type and complications: a european cohort study. Epilepsia 2004; 45: 71-6.
24. Tellez-Zenteno JF, Matijevic S, Wiebe S. Somatic comorbidity of epilepsy in the general population in Canada. Epilepsia 2005; 46: 1955-2.
25. Pellock JM. Understanding co-morbidities affec-ting children with epilepsy. Neurology 2004; 62
(Suppl 2): S17-3.26. Nilsson L, Farahmand BY, Person PG, Thiblin
I, Tomson T. Risk factors for sudden unexpected death in epilepsy: a case-control study. Lancet 1999; 353: 888-3
27. Commission on Classification and Terminology of the International League Against Epilepsy. Proposal for revised clinical and electroence-phalographic classification of epileptic seizures. Epilepsia 1981; 22: 489-01.
28. Commission on Classification and Terminology of the International League Against Epilepsy. Proposal for revised clinical and electroence-phalographic classification of epileptic seizures. Epilepsia 1989; 30: 389-9.
29 International League against Epilepsy. Epidemio-logy. Epilepsy: safety, excess mortality, and sudden death. Epilepsia 2003; 44 (Suppl 6): 19-0.
30. Aspray TJ. The use of verbal autopsy in attribu-ting cause of death from epilepsy. Epilepsia 2005; 46 (Suppl 11):15-7.
31. Vlooswijk MCG, Majoie HJM, De Krom MCT, Tan IY, Aldenkamp AP. SUDEP in a dutch tertiary epilepsy setting: frequently feared but rarely seen. Epilepsia 2005; 46 (Suppl 11): 49.
32. Chapman D, Moss B, Panelli R. Pollard R. Sudden unexpected death in epilepsy. Epilepsy Australia; 2005. Australia. pp19.
33. Nashef L, Fish DR, Garner S, Sander JWAS, Shorvon SD. Sudden death in epilepsy: a study of incidence in a young cohort with epilepsy and learning difficulty. Epilepsia 1995; 36: 1187-4.
34. Beaussart-Defaye J, Beaussart M. Sudden unex-pected deaths in epilepsy. Epilepsia 2005; 46 (Suppl 11): 128.
35 Nilsson L, Bergman U, Diwan V, Farahmand, Person PG, Tomson T. Antiepileptic drug the-rapy and its management in sudden unexpected death in epilepsy: a case-control study. Epilepsia 2001; 42: 667-3
36. Nei M, Ho RT, Abou-Khalil BW, Drislane FW, Liporace J, et al. EEG and ECG in sudden unex-plained death in epilepsy. Epilepsia 2004; 45: 338-5.
37. Ansakorpi H, Korpelainen JT, Tanskanen P, Huikuri HV, Koivula A, et al. Cardiovascular regulation and hippocampal sclerosis. Epilepsia 2004; 45:933-9.
38. Devilat M, Gómez V, Muñoz P, Alarcón AM, Rivas M. Epilepsias catastróficas en la infancia: más que un concepto biológico. J Epilepsy Clin

��
Neurophysiol 2002; 8: 199-4.39. Walczak TS, Leppik LE, D´Amelio M, Rarick J,
So E, Ahman P, et al. Incidence and risk factors in sudden unexpected death in epilepsy. Neurology 2001; 56: 519-5.
40. Leestma JE, Walczal T, Hughes JR, Kalelkar MB, Teas SS. Ann Neurol 1989; 26: 195-3.
41. Sander JW, Bell GS. Reducing mortality : an im-portant aim of epilepsy management en J Neurol Neurosurg Psychiatry 2004; 75: 349-1.
42. Sperling MR, Harris A, Nei M, Liporace JD, O´Connor MJ. Mortality after epilepsy surgery. Epilepsia 2005; 46 (Suppl 11): 49-3.
Tabla 1Circunstancias y tipo de muerte súbita en 9 niños con epilepsia
Lugar Circunstancia Causa de muerte
1 Domicilio Encontrada sin vida al amanecer MSIEP definitiva
2 Domicilio Después de CTCG, con testigo MSIEP probable
3 Hospital Durante una CTCG, con testigo* MSIEP posible.
4 Domicilio Después de CPC, con testigo MSIEP probable
5 Domicilio Después de temblor, con testigo MSIEP probable
6 Domicilio Encontrada sin vida al amanecer MSIEP probable
7 Domicilio En sueño boca abajo MSIEP definitiva
8 Domicilio Encontrada sin vida al amanecer MSIEP probable
9 Domicilio Encontrada sin vida al amanecer MSIEP probable
*Cursaba con un shock séptico y escarlatina; CTCG: crisis tonicoclónica generalizada; CPC: crisis parcial compleja; MSIEP: muerte súbita inesperada.
Clínica y comorbilidades en niños con epilepsia fallecidos de muerte súbita Marcelo Devilat et al

12
Revista Chilena de Epilepsia Año 8, Nº 1, Diciembre de 2007
Tabla 2Características clínicas 9 niños con epilepsia fallecidos de muerte súbita
Tipo crisis Síndrome Evolución Tipo de la epilepsia etiológico
1 Mioclónicas+CTCG EMJ Inactiva Idiopática
2 CTCG Generalizado Resistente Sintomática
3 Parciales complejas Parcial Resistente Sintomática
4 Parciales complejas Parcial Resistente Sintomática
5 CTCG Generalizado Inactiva Criptogénica
6 CTCG Generalizado Catastrófica Sintomática
7 CPC + atónicas Parcial Activa Sintomática
8 Mioclónicas West Catastrófica Sintomática
9 Mioclónicas West Activa Sintomática
CTCG: crisis tonicoclónica generalizada; CPC: crisis parcial compleja; EMJ: epilepsia mioclónica juvenil.
Tabla 3EEG y tratamiento en 9 niños con epilepsia fallecidos de muerte súbita
Último EEG Nº de esquemas Último control Último Nivel de tratamiento de FAE de FAE
1 Normal 2 VPA Terapéutico 2 Puntas lentas 2 VPA Terapéutico 3 Específico 5 VPA-PB-CLB Terapéutico 4 Específico 2 PB-CBZ Terapéutico 5 Normal 2 VPA Terapéutico 6 Específico 8 VPA-CZP-TPM-DC Terapéutico 7 Específico 1 VPA Sin nivel 8 Específico 3 VPA-ACTH-Prednisona- VGB-FNT-PB Terapéutico9 Específico 1 VPA Terapéutico
FAE: antiepiléptico; VPA: ácido valproico; PB: fenobarbital; CLB: clobazam; VGB: vigabatrina; CBZ: carbamacepina; CZP: clona-zepam; TPM: topiramato; FNT: fenitoína; DC: dieta cetógena.

13
Tabla 4 Etiología y comorbilidades en 9 niños con epilepsia fallecidos de muerte súbita
Antecedentes Co-morbilidad Co-morbilidad patológicos neurológica no neurológica
1 no no Embarazo 21-22 semanas
2 EHI RM + PC no
3 Meningitis RM + AVC Bronconeumonías
4 no RM + Autismo Ehlers-Danlos
5 no no no
6 EHI RM + PC Desnutrición
7 no RM + PC Bronconeumonías
8 Agenesia cuerpo RM + PC Bronconeumonías, calloso Gastrostomía
9 Síndrome dismórfico RM + PC BronconeumoníasHoloprosencefalia
EHI: encefalopatía hipóxico isquémica; RM: retardo mental; PC: parálisis cerebral; AVC: accidente vascular cerebral
Correspondencia: Dr. Marcelo Devilat Barros. Silvina Hurtado 1713. Dpto 601. Santiago. Chile. Fax: + 02-235 2805.
Clínica y comorbilidades en niños con epilepsia fallecidos de muerte súbita Marcelo Devilat et al

14
Revista Chilena de Epilepsia Año 8, Nº 1, Diciembre de 2007
Cirugía de la epilepsia en niños. Hemisfere-tomía. Experiencia en el Instituto de Neuro-cirugía Asenjo.Dr. Manuel Loncomil Sánchez (1), Dra. Jovanka Pavlov Norambuena (1), Dra. Lilian Cuadra Olmos (2), Dr. Arturo Zuleta Ferreira (3), Dr. Sergio Valenzuela (3), Dr. Juan José Marengo (3)
(1) Médico-cirujano. Becado Neuropediatría. Hospital Roberto del Río(2) Médico-Cirujano. Neurólogo Infantil. Instituto Neurocirugía Asenjo(3) Médico-cirujano. Neurocirujano. Instituto Neurocirugía Asenjo
Trabajos Originales
ResumenLa epilepsia es una patología prevalente, asociada a morbilidad y mortalidad. Un alto porcentaje res-ponde al tratamiento con fármacos antiepilépticos, pero aproximadamente un 20 % de los casos, son refractarias al tratamiento. De este grupo, de acuerdo a una completa y correcta evaluación prequirúrgica, sólo un tercio será candidato a cirugía. La hemisfe-rectomía es una alternativa quirúrgica para ciertas condiciones patológicas.
En este artículo, se revisan las características de la Epilepsia refractaria en el niño, en cuanto a su etio-logía, evaluación prequirúrgica, decisión de cirugía y técnicas quirúrgicas, en especial, la Hemisferectomía, sus tipos, indicaciones y complicaciones asociadas descritas en la literatura. Posteriormente se hace una revisión de la casuística de este tipo de Cirugía en el Instituto de Neurocirugía Asenjo, desde 1992 a la fecha. Palabras claves: Epilepsia. Infantil. Tratamiento Quirúrgico. Hemisferectomía
AbstractEpilepsy is a prevalent pathology, associated with morbidity and mortality. Near 20% of the cases is unresponsive to pharmacological treatment and but only one third of this group – after an exhaustive presurgical evaluation – would finally be a candidate to surgical treatment. Hemispherectomy is suitable in some specific pathological situations.
In this article we review the following aspects of children refractory epilepsy: presurgical evaluation, surgical decision, surgical techniques, and parti-cularly the different modalities, indications, and complications of hemispherectomy described in the
literature. Then we present a review of the patients submited to hemispherectomy in the Asenjo Neuro-surgery Institute, since 1992 up to today.
Key words: Epilepsy. Children. Surgical treatment. Hemispherectomy.
EpidemiologíaLa Epilepsia es una enfermedad que afecta aproxima-damente al 1% de la población. Aproximadamente en el 70-80 % de los casos son controladas con fármacos antiepilépticos (FAE), pero existe un 20 -30 %de pacientes, con un diagnóstico correcto de epilepsia, que son resistentes a los Fármacos antiepilépticos.
Dado la Mortalidad asociada, mayor en pacientes epilépticos que en la población general, se hace aún más necesario, plantear en etapas tempranas de la en-fermedad, la posibilidad de un manejo quirúrgico.
Epilepsia RefractariaSe define como la persistencia de Crisis Epilépticas, en número y calidad suficiente, para provocar retraso en el desarrollo psicomotor, invalidez sociolaboral, retraso en la escolaridad, pese a un tratamiento far-macológico bien llevado (2 ensayos con Fármacos antiepilépticos de primera línea), y usando dosis máximas tolerables.
En el caso de Epilepsias Focales, la posibilidad de respuesta a un intento terapéutico con un 3º FAE, es menor al 5%.
Evaluación PrequirúrgicaResulta fundamental, pues sólo un tercio de los pacientes refractarios, serán en definitiva, candi-datos a cirugía. Su objetivo es determinar la Zona epileptógena, o zona cuya exéresis o desconexión

15
completa, determina quedar “libre de crisis”, sin producir secuelas secundarias a la cirugía.
Actualmente los métodos de evaluación se dividen en 2 fases:1. No invasivos: - Historia clínica - Examen neurológico - Video monitoreo EEG continuo - Neuroimágenes estructurales volumétricas - Neuroimágenes funcionales - Evaluación Neuropsicológica - Evaluación psiquiátrica
2. Invasivos: - Electrodos Intracraneanos (subdurales, foramen
oval) - Electrodos profundos
Mediante esta evaluación se pretende:1. Determinar si la epilepsia es concordante con una
naturaleza focal2. Localizar la zona epileptógena3. Identificar riesgos potenciales de cirugía al erra-
dicar la zona epileptógena4. Determinar la capacidad emocional, del paciente
y su familia, para enfrentar dificultades y resul-tados de la cirugía.
5. Descartar alto riesgo de dañar estructuras neuro-nales funcionales.
Decisión QuirúrgicaUna vez realizado el estudio prequirúrgico, eva-luando el marcado deterioro en la calidad de vida, conductual y desarrollo neuroevolutivo, asociado a las crisis epilépticas de difícil manejo y, si el tipo de resección propuesta supone un bajo riesgo morbilidad neurológica y de deterioro cognitivo, sólo entonces se plantea una indicación formal de cirugía.
Actualmente el manejo quirúrgico, debería indicarse en forma temprana, a fin de minimizar el deterioro ocasionado por las propias crisis epilépticas, así como por el uso prolongado de FAE.
Diferencias entre Adultos y Niños1. Las Crisis epilépticas son más frecuentes en
niños2. Las crisis epilépticas Recurrentes y el uso pro-
longado de FAE tienen efectos negativos en el neurodesarrollo
3. Niños presentan plasticidad cerebral
4. Etiología En Adultos la mayor causa de cirugía de la epi-
lepsia es la esclerosis temporal mesial (v/s 15% en niños)
En edad Pediátrica, los Tumores de Bajo grado y las Malformaciones Desarrrollo Cortical corres-ponden al 50-90%
5. Tipo de Cirugía, en directa relación a la etiolo-gía:
Adultos: Resección temporal Niños: Resecciones Extratemporales, Multiloba-
res, Hemisferectomías (HS)6. Riesgos Mayor Mortalidad Perioperatoria Pediátrica: 1.3
%. Asociado a resecciones de mayor amplitud y menores volúmenes sanguíneos
Desarrollo de nuevos déficits neurológicos. Aunque la edad confiere la ventaja de plasticidad cerebral
Etiología Fundamentalmente se divide en origen Temporal y Extratemporal; de acuerdo a la correlación de imá-genes y patología se clasifica en 4 grandes grupos:1. Patología del hipocampo: Esclerosis del Hipocampo2. Lesión circunscrita focal Tumoral: De bajo grado. Astrocitoma, Oligoden-
droglioma, Ganglioglioma, DNT. No tumoral: Malformativa: Hamartomas, Esclerosis Tubero-
sa Infecciosa / Inflamatoria: Neurocisticircosis,
Tuberculoma Vascular: Cavernomas3. Lesiones extensas glióticas o destructivas Patología secuelar: isquemia, trauma o hemorra-
gia4. Alteraciones del desarrollo cortical Microdisginesia, Displasia cortical, Paquigiria
Polimicrogiria, Hemimegalencefalia.
Rol de la cirugía en epilepsiaObjetivo: Resección completa del área epileptógena, evitando al máximo la resección de áreas de tejido sano y zonas elocuentes.
El éxito quirúrgico va a depender del grado de validez o concordancia de los datos localizatorios obtenidos a través del estudio prequirúrgico, y puede ser definido de acuerdo a distintas formas de evaluación:
Cirugía de la epilepsia en niños. Hemisferecetomía. Experiencia en el Instituto de Neurocirugía Asenjo Manuel Loncomil et al

16
Revista Chilena de Epilepsia Año 8, Nº 1, Diciembre de 2007
- Ausencia total de Crisis Epilépticas, en pacientes con epilepsia refractaria
- Disminuir la frecuencia y número de crisis, y con esto mejorar la calidad de vida del paciente y su familia
- Minimizar complicaciones psiquiátricas y cogni-tivas asociados a la epilepsia y el uso Fármacos antiepilépticos
- Que sea realizada en un momento oportuno, vale decir, precoz.
- La Tasa de mortalidad en pacientes epilépticos sin crisis, es similar a la de la población general
Clasificación de EngelEvaluación postoperatoria, según el control de las CE.I. Libre de crisis epilépticas (se excluyen las del 1º
mes post operatorio)II. Infrecuentes crisis epilépticas incapacitantesIII. Considerable mejoríaIV. Sin mejoría considerable
Técnicas Quirúrgicas.No siendo el objetivo de esta revisión, sólo se men-cionan las técnicas más utilizadas y se analiza en más detalle la Hemisferectomía.
1. Resecciones temporales y Corticectomías Se utiliza en epilepsias parciales y con foco úni-
co2. Lesionectomías Resección de lesión y área epileptógena, clara-
mente identificada y accesible a Cirugía3. Callosotomía Tratamiento paliativo. En casos de Epilepsia
Generalizada y de origen Bifrontal4. Radiocirugía5. Transección cortical Subpial múltiple6. Estimulación Nervio Vago7. Lesiones por estereotaxia8. Hemisferectomías y sus variantes Indicada para el manejo de las crisis epilépticas
cuyo origen está sólo en el hemisferio lesiona-do.
Sus indicaciones actuales quedan circunscritas a:
- Hemiplejia de origen vascular - Sindrome de Sturge Weber - Sindrome de Rasmussen - Hemimegalencefalia - Anormalidades desarrollo cortical, unilatera-
les
HEMISFERECTOMIAS. TECNICAS.
Anatómica: Resección en bloque o lobares. Presentan problemas en relación al tamaño de la cavidad residual, hidroce-falia secundaria y hemosiderosis cerebral superficial, motivo por el cual ya no se realizan.
Hemidecorticación: Resección cortical, con preservación de sustancia blanca y ganglios basales.
Funcional: Descrita por Rasmussen el año 1974. Corresponde a “Desconexión” de la zona epileotógena: incluye Lobectomía temporal con corticectomía central su-prasilviana, más callosotomía corporal y desconexión de lóbulos parieto-occipital y frontal.
Hemisferectomía funcional modificada:Permitió disminuir la invasividad quirúrgica, median-te el uso de una Craneotomía más pequeña, realizan-do una Desconexión de fibras, sin resección masiva, definiéndose como una “desconexión funcional total, con resección anatómica parcial”. Su uso determinó una disminución de los tiempos operatorios y de las pérdidas de sangre asociadas. Existen diversas técnicas, utilizadas de acuerdo al sustrato patológico.
Por convención se denominan Hemisferostomía.
Resultados Internacionales de la Hemisferecto-mía.De acuerdo a la revisión de las distintas series publi-cadas - Control de crisis: 70-80 %- Mejoría en la personalidad: 70 80 %- Aumento de coeficiente intelectual entre 5-10
pts.
Complicaciones asociadas.- Mayor deterioro motor y sensorial (campo vi-
sual)- Hidrocefalia 50 %. De las cuales un 10-20 %
requirió Válvula Derivativa.- Infección- Mortalidad 4-6 %

17
REVISION DE CASOS EN INSTITUTO DE NEU-ROCIRUGIA ASENJO
Desde 1992 hasta Noviembre 2007
Total: 28 niños operados H:18 M: 10
Edad promedio: 8 años H: 7a M: 9a 6m
Rango etario: 2a 2m – 21 años
Número de cirugías por año: 75 % en los últimos 4 años. El resto como casos aislados a partir del año 1992- 2007: 2 - 2006: 8- 2005: 8- 2004: 3
Etiología: Nº casos
Gliosis difusa 1 3,5 %Lesión focal tumoral 0 no tumoral 8 28,5 %Lesión destructiva extensa 8 28,5 %Malformaciones del desarrollocortical 11 39,5 %
Destacan como diagnóstico clínico: Nº casos
- Sd Sturge Weber 1- Sd Rasmussen 3- Hemimegalencefalia 4- Esclerosis tuberosa 1 - Secuela de infarto 2
Complicaciones Nº casos
Infecciosas Ventriculitis 2 ITU 2 Neumonia 4 Sepsis 1 Hidrocefalia 1Hemorragia ventricular 1Mayor compromiso motor 3 (en 2 casos fue sólo inicial)Anemia 1Colección extradural 1Mortalidad 0
SeguimientoSe logró en 26 de los 28 pacientes (19 pacientes son de regiones).
Promedio de tiempo de seguimiento: 33 meses- Menor tiempo: 3 meses. Vive en Coronel. Ciru-
gía:1998. Dg: Displasia cortical- Mayor tiempo: 15 años. Vive en Pichidehua. Ciru-
gía:1992. Dg: Displasia cortical. Actualmente sin crisis. Utilizando sólo 1 fármaco.
Clasificación Engel: Se logró actualizar en 15 pacientes, mediante el uso de llamado Telefónico (noviembre 2007).
24/28: Ia (15 actualizados) 85 % 2/28: II 7.5 % 2/28: Sin datos 7.5 %
Conclusiones
1. Hemisferectomía ofrece una excelente alternativa a la Epilepsia Refractaria
Cirugía de la epilepsia en niños. Hemisferecetomía. Experiencia en el Instituto de Neurocirugía Asenjo Manuel Loncomil et al

18
Revista Chilena de Epilepsia Año 8, Nº 1, Diciembre de 2007
2. Una Adecuada y PRONTA evaluación quirúrgica, mejora las crisis epilépticas, dejándolas en cero, y probablemente puede prevenir el deterioro psico-social, físico, cognitivo y económico asociados.
3. La mortalidad operatoria fue de 0 en el 100% de los niños operados.
4. La tasa de Mortalidad en pacientes epilépticos que controlan sus crisis es similar al de la pobla-ción general, meta que se alcanzó en al menos el 85 % de los pacientes (en 2 casos no hay datos) que fueron candidatos a cirugía de tipo Hemisfe-rectomía en el Instituto de Neurocirugía Asenjo, durante el periodo comprendido entre 1992 y Noviembre de 2007.
REFERENCIAS
1. Lüder HO, Ángel J, Munari C. General prin-ciples. Surgical Treatment of the epilepsy. 2ª ed.1993.
2. Engel Jr. J, Wieser HG, Spencer D. Overview: Surgical Therapy. A comprensive textbook, 1997.
3. Campos MG. Cirugía de la epilepsia. Rev Med Chile, 1994.
4. Kwan P., Brodie MJ. Early identification of refractory epilepsy. N Eng J of Med 2000; 342: 314-19.
5. Lüders HO, Awad I. Conceptual considerations. Epilepsy Surgery. 1991: 1063-70.
6. Paolicchi JM, JAyakar P, Dean P,y col. Predictors of outcome in pediatric epilepsy surgery. Neuro-logy 2000; 54: 642-47.
7. Neurocirugía Infantil Latinoamericana. Tomo
I. Selección del candidato para Cirugía de la epilepsia en pediatría. 2006; 507-54
8. Campos MG, Kanner AM, Epilepsia: diagnóstico y tratamiento. Evaluación prequirúrgica; 40: 574-96.
9. Fusco L., Vigevano F. Indications for surgical treatment of epilepsy in chilhood: a clinical and neurophysiological approach. Acta Pediatr Supp 2004; 93 (445): 28-31.
10. Shields WD. Defining medical intractability: the differences in children compared to adults. Paediatrics epilepsy syndromes and their surgical treatment, 1997: 93-98.
11. Campos MG, Kanner AM, Epilepsia: diagnóstico y tratamiento. Cirugía de la epilepsia en niños; 45: 646-64.
12. Willie E., Comair YG, Kotogal P. Seizure outcome after epilepsy surgery in children and adolescents. Ann Neurol 1998; 44: 740-48.
13. Lahl R, Villagrán R, Teixeira W. Neuropatologías de las epilepsias sintomáticas con consideración especial de las formas focales crónicas resistentes al tratamiento farmacológico. Neuropatología. Diagnóstico y clínica. Cruz-Sánchez. DIMSA 2000; 827-878.
14. Neurocirugía Infantil Latinoamericana. Tomo I. 2006. Patología de la Epilepsia; 469-506.
15. Campos MG, Kanner AM, Epilepsia: diagnóstico y tratamiento. Hemisferectomías. 46; 665-79.
16. Rasmussen T. Hemispherectomy for seizures revisited. Can J Neurol Sci 1983; 10: 71-78.
17. Vining EPG, Freeman JM, Pillas DJ. Why would you remove half a brain? The outcome of 58 chil-dren after hemispherectomy. The Johns Hopkins Experience: 1968-1996. Pediatric 1997; 100(2): 163-171.

19
Síndrome de West: Presentación clínica y pronóstico a largo plazoDra. Daniela Avila Smirnow, Dr. Marcelo Devilat BarrosCentro de Epilepsia Infantil. Servicio de Neurología y Psiquiatría Hospital Luis Calvo MackennaE-mail: [email protected]
Trabajos Originales
IntroducciónEl Síndrome de West se caracteriza por la combi-nación de espasmos infantiles, hipsarritmia en el electroencefalograma y detención del desarrollo psicomotor. Su inicio ocurre típicamente en el primer año de vida. El pronóstico a largo plazo del síndrome es en general pobre.
ObjetivosEste estudio tuvo como objetivos describir la presen-tación clínica de este cuadro, la evolución a corto y largo plazo, y pesquisar factores pronósticos.
MétodosEl presente estudio fue realizado en el Servicio de Neurología y Psiquiatría del Hospital Luis Calvo Mackenna. Se incluyeron 42 pacientes que cumplían con la tríada clínica característica. El tiempo de seguimiento promedio fue de 5 años 3 meses. Hubo 32 pacientes que tuvieron seguimiento de al menos 2 años, lo que se consideró como seguimiento a largo plazo.
ResultadosLos espasmos se iniciaron a los 5.9 meses de vida en promedio. Al inicio del cuadro hubo un 67% de pacientes con anormalidades neurológicas al exa-men físico, las que incluyeron retraso del desarrollo psicomotor, microcefalia y síndrome piramidal. El 74% de los casos fue sintomático y el 26% crip-togénico. A largo plazo todos los pacientes tenían retraso mental, el 75% permanecía con crisis, el 44% presentaba parálisis cerebral y el 41% microcefalia. Hubo una asociación estadísticamente significativa entre la presencia de microcefalia, RDSM y síndrome piramidal al inicio del cuadro y un peor pronóstico. La mortalidad fue de 17% y se asoció en forma estadísticamente significativa con microcefalia al inicio del cuadro.
ConclusionesEl pronóstico a largo plazo de estos pacientes fue desfavorable, todos evolucionaron con RDSM, la
mayoría persistieron con epilepsia, y la mortalidad fue elevada. Los hallazgos sugieren que el pronóstico parece estar determinado por el estado neurológico previo del paciente.
IntroducciónEl Síndrome de West o Espasmos Infantiles es una de las epilepsias generalizadas sintomáticas o crip-togénicas.� Se caracteriza por la tríada de espasmos masivos, hipsarritmia en el electroencefalograma (EEG) y retraso del desarrollo psicomotor (RDSM).2 Su inicio ocurre típicamente en el primer año de vida, con un peak a los 5 meses 3. La incidencia reportada es de 0.41 por mil nacidos vivos.4
Actualmente un 80% de los pacientes se clasifican dentro del grupo de los sintomáticos. Las causas pre-natales dan cuenta de casi el 50% de este grupo, inclu-yendo varias noxas intrauterinas, malformaciones del desarrollo cortical, síndromes neurocutáneos, desór-denes metabólicos y otros defectos cromosómicos y genéticos. El resto de los sintomáticos corresponden a los grupos perinatal y posnatal. Las etiologías pe-rinatales incluyen encefalopatía hipóxico isquémica (EHI), trauma obstétrico, y otras complicaciones del parto. Etiologías posnatales incluyen infección, trauma, noxas hipóxico isquémicos y tumores.3
El tratamiento más comúnmente usado en Estados Unidos es la corticotropina (ACTH). En Japón se inicia tratamiento con piridoxina, y de segunda y tercera línea se usa ácido valproico, y ACTH respec-tivamente. En el Reino Unido, el fármaco de primera línea es la vigabatina.5 La única revisión sistemática encontrada mostró que la ACTH era más eficaz que la prednisona, y que en esclerosis tuberosa la viga-batrina era más eficaz que el ACTH.6
El pronóstico a largo plazo se caracteriza por la frecuente aparición de otros tipos de crisis, epi-lepsia crónica intratable, evolución a Síndrome de Lennox-Gastaut, y retraso mental. La mortalidad varía entre 2 a 30 %.3

20
Revista Chilena de Epilepsia Año 8, Nº 1, Diciembre de 2007
ObjetivosLos objetivos de este estudio, fueron describir la pre-sentación clínica de este cuadro, la evolución a corto y largo plazo, y pesquisar factores pronósticos.
Métodos Los pacientes fueron seleccionados de la base de datos del Centro de Epilepsia del Servicio de Neuro-logía y Psiquiatría del Hospital Luis Calvo Macken-na. En ella se encontraron 73 pacientes que tenían el diagnóstico de Síndrome de West, ingresados entre enero de 1986 y diciembre de 2004.
Se definió como Síndrome de West, a la combinación de espasmos que ocurren en salvas, detención del desarrollo psicomotor (DSM), e hipsarritimia en el EEG.� La ocurrencia de RDSM, previo al inicio de los espasmos, no fue requisito para el diagnóstico.7
El término espasmos clínicos, se usó para definir movimientos breves y sincrónicos de la cabeza, tronco, y extremidades, o a veces sólo de la cabeza, o tronco, o extremidades.7 Estos movimientos podían ser en flexión, en extensión o ambos.7
Hipsarritmia se usó para describir un patrón electro-encefalográfico caracterizado por espigas y ondas lentas de alto voltaje, distribuidas al azar.7 Se optó por no diferenciar hipsarritmia clásica de hipsarritmia modificada, dado que en estudios randomizados, esta diferenciación tiene poca importancia práctica para el pronóstico.7
Se diseñó un protocolo para la revisión de las fichas de los pacientes. Este incluyó los antecedentes clíni-cos previos al inicio de los espasmos, y las caracterís-ticas clínicas y electroencefalográficas en el momento del inicio del cuadro, y durante la evolución.
Dentro de los antecedentes clínicos se incluyó el género del paciente y la presencia de crisis epilépticas
en el período neonatal.
Las variables incluidas en la presentación clínica fueron la edad de inicio de los espasmos, la presencia de RDSM, de microcefalia y de síndrome piramidal, el resultado del EEG y del estudio etiológico.
Se describió la evolución a corto y largo plazo en términos del RDSM, microcefalia, parálisis cerebral, persistencia de crisis epilépticas, EEG, y mortalidad.Se incluyeron sólo los pacientes que cumplieran con la definición de Síndrome de West previamente efec-tuada. Se excluyeron 6 pacientes cuyas fichas no se encontraron, 8 pacientes que no tenían la información requerida por el protocolo del estudio y 17 pacientes que no cumplieron con los criterios de inclusión. Se incluyeron los 42 pacientes restantes en el análisis de los datos y se realizó una revisión protocolizada de sus fichas (figura 1).
El tratamiento inicial consistió en ácido valproico asociado a ACTH sintética o a corticoesteroides en 30 pacientes (71%). En los restantes 12 pacien-tes (19%) se usaron otros fármacos antiepilépticos (FAE) en monoterapia o biterapia (tabla 1). La du-ración de la cura con ACTH y con prednisona varió de 10 días a 7 semanas, con un promedio de 20 días. Los pacientes tuvieron en promedio 4 esquemas de tratamiento durante la evolución. Cuatro pacientes fueron sometidos a dieta cetogénica. El inicio del tratamiento después de un mes del inicio de los es-pasmos, se denominó retraso de tratamiento.7
El estudio etiológico incluyó ultrasonografía ence-fálica (USG), tomografía axial computada (TAC) encefálica, resonancia nuclear magnética (RNM) encefálica, y estudio metabólico.
El Síndrome de West fue clasificado en los subgru-pos etiológicos de criptogénico y sintomático. Los
21
criptogénicos correspondieron a aquellos en los que se sospechó que tuvieran un trastorno subyacente, pero no se pudo identificar una causa estructural o bioquímica. Los sintomáticos fueron aquellos que tuvieron una causa identificada.7
La evaluación del (DSM) y del coeficiente intelectual fue realizada clínicamente por el médico tratante o utilizando escalas de desarrollo. La Escala de Bayley se usó en niños de 1 a 48 meses y la Escala de Inteli-gencia de Wechsler para Niños, se usó para niños de 6 a 16 años. Se registró el nivel de retraso del DSM en 25 pacientes a corto plazo y en 22 pacientes a largo plazo. En los demás pacientes sólo se consignó la presencia o ausencia de retraso de DSM, pero no su severidad.
La evolución fue descrita a corto y largo plazo. Se definió como evolución a corto plazo a aquella comprendida desde el inicio del cuadro hasta un año después. Se definió como evolución a largo plazo a la correspondiente a dos o más años desde el inicio de los espasmos.
Se definió persistencia de crisis a corto plazo, como la presencia de al menos una crisis durante el último mes. En la evolución a largo plazo denominó persis-tencia de crisis, a la presencia de una o más crisis durante los dos últimos años de seguimiento.
En el seguimiento a corto plazo 37 pacientes perma-necieron en control. Hubo 5 niños que no alcanzaron el seguimiento de un año, dos de ellos fallecieron, dos abandonaron controles y en uno el cuadro se inició menos de un año antes desde el punto de corte. En el seguimiento a largo plazo 32 pacientes permane-cieron en control. Hubo 10 que no alcanzaron los 2 años de seguimiento, los cinco recién nombrados y otros 5 más, de los cuales uno falleció, 3 abandonaron controles y uno se diagnosticó menos de dos años antes del punto de corte. (Tabla 2)
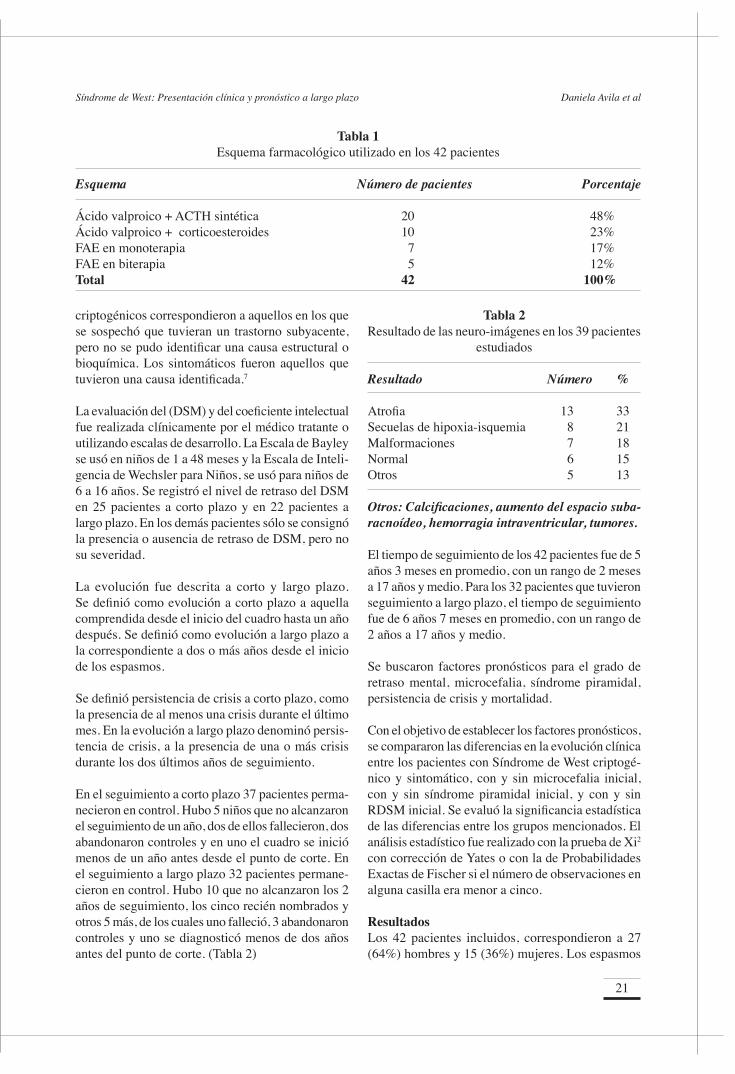
Tabla 2Resultado de las neuro-imágenes en los 39 pacientes
estudiados
Resultado Número %
Atrofia 13 33Secuelas de hipoxia-isquemia 8 21Malformaciones 7 18Normal 6 15Otros 5 13
Otros: Calcificaciones, aumento del espacio suba-racnoídeo, hemorragia intraventricular, tumores.
El tiempo de seguimiento de los 42 pacientes fue de 5 años 3 meses en promedio, con un rango de 2 meses a 17 años y medio. Para los 32 pacientes que tuvieron seguimiento a largo plazo, el tiempo de seguimiento fue de 6 años 7 meses en promedio, con un rango de 2 años a 17 años y medio.
Se buscaron factores pronósticos para el grado de retraso mental, microcefalia, síndrome piramidal, persistencia de crisis y mortalidad.
Con el objetivo de establecer los factores pronósticos, se compararon las diferencias en la evolución clínica entre los pacientes con Síndrome de West criptogé-nico y sintomático, con y sin microcefalia inicial, con y sin síndrome piramidal inicial, y con y sin RDSM inicial. Se evaluó la significancia estadística de las diferencias entre los grupos mencionados. El análisis estadístico fue realizado con la prueba de Xi2 con corrección de Yates o con la de Probabilidades Exactas de Fischer si el número de observaciones en alguna casilla era menor a cinco.
ResultadosLos 42 pacientes incluidos, correspondieron a 27 (64%) hombres y 15 (36%) mujeres. Los espasmos
Tabla 1Esquema farmacológico utilizado en los 42 pacientes
Esquema Número de pacientes Porcentaje
Ácido valproico + ACTH sintética 20 48%Ácido valproico + corticoesteroides 10 23%FAE en monoterapia 7 17%FAE en biterapia 5 12%Total 42 100%
Síndrome de West: Presentación clínica y pronóstico a largo plazo Daniela Avila et al

22
Revista Chilena de Epilepsia Año 8, Nº 1, Diciembre de 2007
se iniciaron en promedio a los 5.9 meses de vida, con un rango de 1 mes a un año y medio de vida.
Al inicio del Síndrome de West, se encontró un 67% de pacientes con RDSM, 29% con síndrome pira-midal y 29% con microcefalia (Figura 2). Hubo un 33% de pacientes que no tuvieron alteraciones al examen inicial, 29% que sólo tuvieron RDSM y el 38% restante tuvo una combinación de síntomas y signos (Figura 3).
Concomitante al inicio de los espasmos, hubo 5 pa-cientes que tuvieron asociadas crisis generalizadas y 6 que tuvieron crisis parciales. Todos los pacien-tes mostraron hipsarritmia en el EEG.
El estudio de neuroimágenes reveló anomalías en la RNM en todos los pacientes, en la TAC en el 80% y en la USG en el 50% de los niños (Figura 4). Las imágenes mostraron diversas anomalías, siendo las más frecuentes las secuelas de hipoxia isquemia, presentes en el 33% de los pacientes, y las malfor-maciones, encontradas en el 21% de los niños (tabla 2).
El estudio de enfermedades metabólicas mostró escasas alteraciones. La lactacidemia fue normal en los 13 pacientes estudiados. La amonemia re-sultó elevada en 1 de los 14 niños estudiados. El examen de aminoácidos en sangre, efectuado en 17 pacientes, mostró hiperglicinemia en un paciente,
Figura 3: Características de los 42 pacientes al inicio del Síndrome de West
Figura 2: Síntomas y signos de los 42 pacientes al inicio del Síndrome de West.
R: RDSM; P: Síndrome Piramidal; M: Microcefalia.

23
hiperalaninemia en otro, y en los restantes 15 ni-ños fue normal. El estudio de aminoácidos en orina, realizado en 17 pacientes, no mostró alteraciones significativas. El screening metabólico fue normal en los 6 pacientes estudiados. En el paciente con hiperglicinemia se midió aminoácidos en LCR, se encontró aumento de glicina, y se diagnosticó una hiperglicinemia no cetósica.
La etiología del Síndrome de West se identificó en el 74% de los pacientes, los que fueron catalogados como sintomáticos (figura 5).
La causa del cuadro se originó en el período prena-
tal en el 42%, en el perinatal en el 39% y en el pos-natal en el 19% de los niños. Las causas prenatales incluyeron enfermedades específicas, cromosómi-cas y malformativas. Estas últimas fueron las más frecuentes de este grupo y correspondieron al 23% de los pacientes. La asfixia intrauterina fue la más común de las etiologías perinatales, encontrándose en el 23% de los pacientes. Otras causas perinatales incluyeron hemorragia intracraneana no traumáti-ca, trastornos metabólicos, infecciones del sistema nervioso central (SNC) y septicemia neonatal.En-tre las causas pos natales, se encontró al accidente cerebro vascular, las enfermedades metabólicas, la EHI, y las infecciones del SNC. Estas últimas se
Figura 4: Resultado del estudio de neuroimágenes en 39 pacientes
Figura 5: Porcentajes de pacientes criptogénicos y sintomáticos
Síndrome de West: Presentación clínica y pronóstico a largo plazo Daniela Avila et al

24
Revista Chilena de Epilepsia Año 8, Nº 1, Diciembre de 2007
presentaron en el 9% de los casos y fueron las más frecuentes de este grupo. Las patologías específi-cas más comunes del total de los niños, fueron la EHI neonatal, que estuvo presente en el 23% y la esclerosis tuberosa que correspondió al 9 % de los pacientes (Tabla 3).
En la evolución a corto plazo, hubo un 97% de pa-cientes que tuvieron retraso del desarrollo psicomo-tor, un 49% que persistieron con crisis, un 43% que tuvieron parálisis cerebral y un 35% que tuvieron microcefalia. Las crisis fueron generalizadas en 12 pacientes, parciales en 5 y mixtas en 1 de ellos. El electroencefalograma se controló durante el primer año de seguimiento en 33 pacientes, observándose evolución a actividad epileptiforme en el 70% y a actividad inespecífica en el 30% de ellos. (Figura 6 y 7).
En la evolución a largo plazo, todos los pacientes
tuvieron retraso mental, el 75% tuvo persistencia de crisis, el 44% parálisis cerebral y el 41% microcefa-lia.. En 11 pacientes hubo crisis generalizadas, en 7 crisis parciales y en 6 mixtas. El EEG se realizó en 29 pacientes, en los que fue específico en el 76% e inespecífico en el 24% (Figura 6 y 8).
Al punto de corte, todos los pacientes tuvieron RDSM o RM. El 81% tuvo persistencia de crisis, el 43% tuvo microcefalia y el 40% tuvo parálisis cerebral. El 37% de ellos presentaron retraso men-tal y persistencia de crisis. El 29% tuvo persistencia de crisis, microcefalia, parálisis cerebral y retraso mental concomitantemente. El 34% restante tuvo diversas combinaciones de síntomas (Figura 9).
La mortalidad fue de un 17%. Las causas de muerte más frecuentes fueron la neumonía y bronconeumo-nía, que fue responsable del 72% de los fallecimien-tos (Tabla 4). Estas fueron muertes no relacionadas
Tabla 3Etiología del Síndrome de West en los 31 pacientes sintomáticos
Período Categoría diagnóstica Número de pacientes Etiología específica Número de pacientes
Prenatal Enfermedades específicas 4 (13%) Esclerosis tuberosa 3 (9%)(Congénitas) Síndrome de Seckel 1 (3%) N=13 (42%) Cromosómicas 2 (7%) Síndrome de Down 2 (7%) Malformaciones 7 (23%) Agenesia de cuerpo calloso 1 (3%) Encefalocele 1 (3%) Displasia Septo-Optica 1 (3%) Quiste aracnoidal 1 (3%) Paquigiria 1 (3%) Disgenesia cortical 1 (3%) Walker Warburg 1 (3%)Perinatal Asfixia intrauterina 7 (23%) EHI 7 (23%)N=12 (39%) Hemorragia intracraneana no traumática 1 (3%) HIV 1 (3%) Infecciones del SNC 2 (7%) Meningitis por estrepto- 2 (7%) coco beta hemolítico grupo B Trastornos Metabólicos 1 (3%) Hipoglicemia neonatal 1 (3%) Septicemia neonatal 1 (3%) 1 (3%)Posnatal Infecciones del SNCl 3 (9%) Meningitis 1 (3%)N=6 (19%) Encefalitis 2 (7%)
Accidente Cerebrovascular 1 (3%) Infarto cerebral 1 (3%) Metabólicas 1 (3%) Hiperglicinemia no cetósica 1 (3%) Otras 1 (3%) EHI 1 (3%)

25
Figura 6: Evolución clínica de los pacientes
Figura 7: Evolución a corto plazo en 37 pacientes (EEG realizado en 33 pacientes)
Síndrome de West: Presentación clínica y pronóstico a largo plazo Daniela Avila et al
Tabla 4Causas de muerte
Causa de muerte Número de pacientes (%)
Neumonía y Bronconeumonia5 (72)Muerte súbita1 (14)No consignada1 (14)
con epilepsia. Hubo un paciente en el que no se con-signó en la ficha su causa de muerte. Hubo un niño con muerte súbita inesperada (MSI). Este paciente murió en su casa durante el sueño y no se realizó autopsia. Todas las muertes por neumonía y bron-coneumonía recibieron tratamiento con corticoides
o ACTH. Sólo uno de estos pacientes no fue tratado con corticoides, ni ACTH y falleció de MSI. La di-ferencia en la incidencia de muertes por infecciones entre los pacientes tratados y no tratados con ACTH y corticoesteroides no fue estadísticamente signifi-cativa (p=0.1666).
Los pacientes fallecieron a los 3 años 8 meses en promedio, con rango de 1 año y 1 mes, a 8 años y 4 meses. El tiempo promedio entre el inicio del Síndrome de West y la muerte fue de 3 años 2 me-ses, con un rango de 6 meses a 8 años. El 57% por ciento falleció antes de los cuatro años de vida y de seguimiento. El 28% falleció antes de los dos años de vida y de seguimiento (Figura 10). El número

26
Revista Chilena de Epilepsia Año 8, Nº 1, Diciembre de 2007
de esquemas de tratamiento en estos pacientes fue de 5 en promedio, con un rango de 3 a 9 esquemas. Todos los pacientes tenían persistencia de crisis en el momento de su muerte.
En el seguimiento a corto plazo, fue más frecuen-te la parálisis cerebral y el RDSM severo y mo-derado en los niños que tenían RDSM al iniciarse los espasmos que en los que tenían DSM normal (p<0.05).A largo plazo fue más frecuente la micro-cefalia, la parálisis cerebral y el retraso mental se-vero y moderado en los pacientes con RDSM inicial
(p<0.05).(Tabla 5).
En la evolución a corto y a largo plazo fue más fre-cuente la parálisis cerebral y la microcefalia en los pacientes que al inicio tenían microcefalia que en los que tenían normo-cefalia (p<0.05). La morta-lidad fue mayor en los pacientes que inicialmente tenían microcefalia (p<0.05).(Tabla 6).
En el seguimiento a corto plazo los niños que ini-cialmente tenían un síndrome piramidal, tuvieron mayor frecuencia de microcefalia, de parálisis cere-
Figura 8: Evolución a largo plazo en 32 pacientes. (EEG realizado en 29 pacientes)
Figura 9: Características de los pacientes en el punto de corte.
Otros:M+P+R, P+R, R+MR: RDSM o retraso mental; M: microcefalia; P: parálisis cerebral; C: persistencia de crisis.
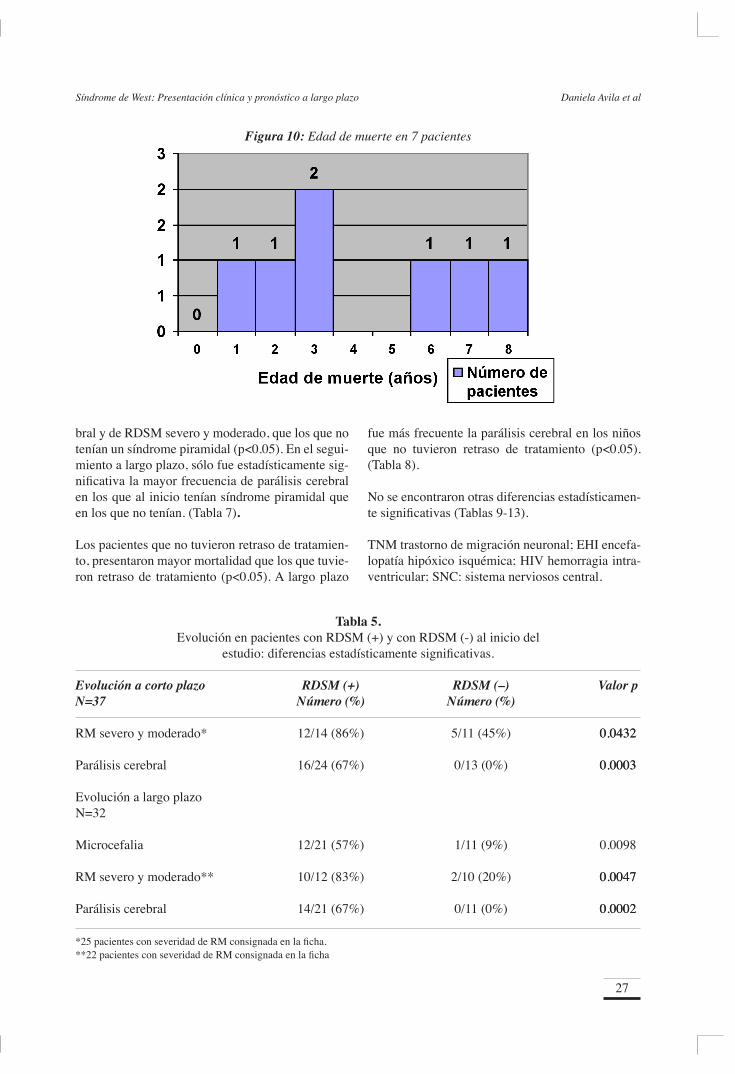
27
bral y de RDSM severo y moderado, que los que no tenían un síndrome piramidal (p<0.05). En el segui-miento a largo plazo, sólo fue estadísticamente sig-nificativa la mayor frecuencia de parálisis cerebral en los que al inicio tenían síndrome piramidal que en los que no tenían. (Tabla 7).
Los pacientes que no tuvieron retraso de tratamien-to, presentaron mayor mortalidad que los que tuvie-ron retraso de tratamiento (p<0.05). A largo plazo
fue más frecuente la parálisis cerebral en los niños que no tuvieron retraso de tratamiento (p<0.05). (Tabla 8).
No se encontraron otras diferencias estadísticamen-te significativas (Tablas 9-13).
TNM trastorno de migración neuronal; EHI encefa-lopatía hipóxico isquémica; HIV hemorragia intra-ventricular; SNC: sistema nerviosos central.
Figura 10: Edad de muerte en 7 pacientes
Tabla 5.Evolución en pacientes con RDSM (+) y con RDSM (-) al inicio del
estudio: diferencias estadísticamente significativas.
Evolución a corto plazo RDSM (+) RDSM (–) Valor pN=37 Número (%) Número (%) RM severo y moderado* 12/14 (86%) 5/11 (45%) 0.0432 0.0432
Parálisis cerebral 16/24 (67%) 0/13 (0%) 0.0003 0.0003
Evolución a largo plazoN=32
Microcefalia 12/21 (57%) 1/11 (9%) 0.0098
RM severo y moderado** 10/12 (83%) 2/10 (20%) 0.0047 0.0047
Parálisis cerebral 14/21 (67%) 0/11 (0%) 0.0002 0.0002
*25 pacientes con severidad de RM consignada en la ficha.**22 pacientes con severidad de RM consignada en la ficha
Síndrome de West: Presentación clínica y pronóstico a largo plazo Daniela Avila et al
28
Revista Chilena de Epilepsia Año 8, Nº 1, Diciembre de 2007
Tabla 6Evolución en pacientes con normocefalia y microcefalia al inicio del
estudio: diferencias estadísticamente significativas.
Evolución a corto plazo Microcefalia (+) Microcefalia (-) Valor pN=37 Número (%) Número (%)
Microcefalia 10/10 (100%) 3/27 (11%) 0.000008Parálisis cerebral 9/10 (90%) 7/27 (26%) 0.0007 0.0007
Evolución a largo plazo. N=32
Microcefalia 8/8 (100%) 5/24 (21%) 0.00018/8 (100%) 5/24 (21%) 0.0001Parálisis cerebral 7/8 (89%) 7/24 (29%) 0.00617/8 (89%) 7/24 (29%) 0.0061 0.0061Muerte 5/12 (42%) 2/30 (7%) 0.0138
Tabla 7Evolución en los grupos con y sin síndrome piramidal en el inicio del
estudio: diferencias estadísticamente significativas.
Evolución a Corto Plazo Síndrome Piramidal (+) Síndrome Piramidal (-) Valor pN=37 Número (%) Número (%)
Microcefalia 8/11 (73%) 5/26 (19%) 0.0032RM severo y moderado* 7/7 (100%) 10/18 (56%) 0.0404 0.0404Parálisis cerebral 11/11 (100%) 5/26 (19%) 0.000005 0.000005
Evolución a Largo plazo N=32
Parálisis cerebral 9/9 (100%) 5/23 (22%) 0.00007 0.00007
*25 pacientes con severidad del RM consignada en la ficha
Tabla 8Evolución en los grupos con y sin retraso de tratamiento: diferencias
estadísticamente significativas.
Retraso de tratamiento (+) Retraso de tratamiento (-) Valor p Número (%) Número (%)
Evolución a Largo plazo N=32
Parálisis cerebral 1/8 (14%) 13/24 (54%) 0.04565 0.04565
Muerte 0/12 (0%) 7/30 (23%) 0.00003 0.00003N=42

29
Tabla 9Diferencias no significativas entre pacientes criptogénicos y sintomáticos.
Criptogénicos Sintomáticos Valor p Número (%) Número (%)
Evolución a Corto plazoN=37
Parálisis cerebral 2/10 (20%) 14/27 (52%) 0.057Microcefalia 2/10 (20%) 11/27 (41%) 0.2462RDSM severo y moderado* 5/7 (71%) 12/18 (67%) 0.6068Persistencia de crisis 6/10 (60%) 12/27 (44%) 0.2637
Evolución a Largo plazoN=32
Parálisis cerebral 2/8 (25%) 12/24 (50%) 0.152Microcefalia 3/8 (38%) 10/24 (42%) 0.6306RDSM severo y moderado* 6/6 (100%) 12/16 (75%) 0.268Persistencia de crisis 5/8 (63%) 20/24 (83%) 0.235Muerte 0/11 (0%) 7/31 (23%)N=42 *25 pacientes con severidad de RM consignada en la ficha.**22 pacientes con severidad de RM consignada en la ficha
Tabla 10Diferencias no significativas entre pacientes con RDSM y DSM normal al inicio
RDSM (+) RDSM (-) Valor p Número (%) Número (%) Evolución a Corto plazoN=37
Microcefalia 11/24 (46%) 2/13 (15%) 0.065Persistencia de crisis 13/24 (34%) 5/13 (38%) 0.570
Evolución a Largo plazoN=32Persistencia de crisis 6/11 (55%) 17/21 (81%) 0.1230Muerte 6/28 (21%) 1/14 (7%) 0.02393N=42
Síndrome de West: Presentación clínica y pronóstico a largo plazo Daniela Avila et al

30
Revista Chilena de Epilepsia Año 8, Nº 1, Diciembre de 2007
Tabla 11Diferencias no significativas entre pacientes con microcefalia y
normocefalia al inicio.
Microcefalia (+) Microcefalia (-) Valor p Número (%) Número (%)
Evolución a Corto plazoN=37
RDSM severo y moderado* 5/17 (83%) 12/18 (63%) 0.3494Persistencia de crisis 6/10 (60%) 12/27 (44%) 0.3193
Evolución a Largo plazoN=32
RDSM severo y moderado** 4/5 (80%) 14/17 (82%) 0.6746Persistencia de crisis 7/8 (88%) 16/24 (67%) 0.0932
*25 pacientes con severidad de RM consignada en la ficha.**22 pacientes con severidad de RM consignada en la ficha
Tabla 12Diferencias no significativas entre pacientes con síndrome piramidal y sin síndrome piramidal al inicio.
Síndrome piramidal (+) Síndrome piramidal (-) Valor p Número (%) Número (%)
Evolución a Corto plazoN=37
Persistencia de crisis N=11 N=26 0.91485/11 (45%) 13/26 (50%)
Evolución a Largo plazoN=32
Microcefalia 6/9 (67%) 7/23 (30%) 0.0760RDSM severo y moderado 6/6 (100%) 12/16 (75%) 0.248Persistencia de crisis 8/9 (89%) 15/23 (65%) 0.1864Muerte 4/12 (33%) 3/30 (10%) 0.0883N=42

31
Tabla 13Diferencias no significativas entre con retraso de tratamiento y sin retraso de tratamiento.
Retraso Retraso Valor p de tratamiento (+) de tratamiento (-) Número (%) Número (%)
Evolución a Corto plazoN=37
Parálisis cerebral 2/10 (20%) 14/27 (52%) 0-0577Microcefalia 3/10 (30%) 10/27 (37%) 0.5345RDSM severo y moderado* 4/7 (57%) 13/18 (72%) 0.3931Persistencia de crisis 4/10 (40%) 13/27 (48%) 0.535
Evolución a Largo plazoN=32
Microcefalia 3/8 (38%) 10/24 (42%) 0.6306RDSM severo y moderado** 4/6 (67%) 14/16 (88%) 0.2704Persistencia de crisis 6/8 (75%) 17/24 (70%) 0.611
*25 pacientes con severidad de RM consignada en la ficha.**22 pacientes con severidad de RM consignada en la ficha
ConclusionesEl principal aporte de nuestro trabajo es reafirmar que la condición neurológica basal del paciente, es uno de los factores pronósticos de mayor importan-cia en la evolución a largo plazo, y que de ellos, el RDSM previo al inicio del cuadro resulta ser el más relevante.
Nuestro estudio evalúa y compara las evoluciones a
corto y largo plazo, y se revela que el pronóstico a corto plazo parece predecir lo que ocurrirá a largo plazo.
Respecto a la mortalidad, en este estudio encontra-mos un factor pronóstico específico, la presencia de microcefalia al inicio del cuadro, hallazgo que no había sido reportado previamente por ningún otro trabajo del que tuviéramos conocimiento.
Síndrome de West: Presentación clínica y pronóstico a largo plazo Daniela Avila et al

32
Revista Chilena de Epilepsia Año 8, Nº 1, Diciembre de 2007
Epilepsia, sueño y trastornos del sueñoDra. Julia Santín Martínez1, Dr. Jaime Godoy Fernández2
1 Profesor Auxiliar Asociado Departamento de Neurología, Escuela de Medicina, Pontificia Universidad Católica de Chile, Email: [email protected]
2 Profesor Titular Departamento de Neurología, Escuela de Medicina, Pontificia Universidad Católica de Chile, Email: [email protected]
Trabajos de Revisión
Introducción
La relación entre epilepsia y sueño es conocida desde la antigüedad. Es así como Aristóteles señalaba que “en muchos esta enfermedad comienza en el sueño, después las crisis son habituales durante el sueño” (1). Los vínculos posibles entre ambas condiciones son múltiples e incluyen la oscilación circadiana de algunos síndromes epilépticos (varios de ellos con manifestaciones restringidas al sueño), impacto en el curso de la epilepsia de la privación de sueño, coexistencia de afecciones como la apnea de sueño, efecto de los medicamentos antiepilépticos en el nivel de vigilancia diurna y en la calidad del sueño nocturno, fragmentación del sueño por crisis noctur-nas frecuentes, cambios en la expresión de las des-cargas epileptiformes durante el sueño, etc. Es fácil entonces comprender la importancia de tener presente esta información cuando se evalúa o trata un paciente con epilepsia. La anamnesis en estos casos siempre debería incluir aspectos como los hábitos de sueño, indagación sobre presencia de somnolencia diurna, ronquido, ubicación horaria de las crisis epilépticas, etc. A continuación revisaremos algunos aspectos relevantes de la relación entre sueño y epilepsia.
Trastornos del sueño y epilepsia: diagnóstico diferencial
Las epilepsias pueden tener manifestaciones clínicas extremadamente variadas; en el caso de las formas focales, puede haber tantas expresiones clínicas como funciones tiene la corteza. Sin embargo todas las crisis epilépticas presentan dos características consistentes que llevan a plantearse el diagnóstico: son eventos breves, generalmente de segundos a pocos minutos y simultáneamente estereotipados, siempre muy semejantes o idénticos en un mismo paciente. Por otra parte varios trastornos del sueño pueden manifestarse clínicamente como episodios breves y básicamente iguales, con las consiguientes dudas diagnósticas.
La cataplexia, síntoma cardinal de la narcolepsia (2), caracterizada por una súbita pérdida del tono postu-ral, completa (con caída del paciente) o parcial (caída de la cabeza o sensación de debilidad intermitente en las extremidades inferiores), puede confundirse con crisis epilépticas atónicas. La diferenciación clínica está dada por la preservación de la conciencia en la cataplexia -fenómeno que además tiene una duración que es variable- versus las crisis atónicas, siempre muy breves; a esto se debe agregar el característico desencadenamiento de la cataplexia por emociones y ciertamente su asociación con hipersomnolencia y otros elementos de la llamada “tétrada de la nar-colepsia”.
El síndrome de movimiento periódico de extremi-dades (3,4) constituye un diagnóstico diferencial de crisis epilépticas focales motoras. La diferencia clínica se establece por la bilateralidad de las mani-festaciones, la ocurrencia en salvas, su aparición en la primera mitad de la noche y la asociación con piernas inquietas, insomnio o hipersomnolencia.
El trastorno conductual del sueño REM (TCR) se caracteriza por episodios de descontrol conductual durante este estadio, que se manifiestan principal-mente durante la segunda mitad de la noche. Son eventos generalmente violentos y que sugieren a testigos la vivencia del correlato de actividad onírica, lo que efectivamente es corroborado por los propios pacientes si se les despierta durante estos eventos (5). Estos episodios pueden hacer sospechar crisis focales frontales nocturnas, las que pueden acom-pañarse de gran actividad motora, gesticulaciones y gritos (“crisis hipermotoras”). La diferencia clínica entre ambos eventos es la amnesia completa de lo ocurrido en el caso de las crisis epilépticas, que además ocasionalmente culminan en convulsiones generalizados tónico-clónicas. Crisis epilépticas de este tipo también se han descrito originadas en el ló-bulo temporal (6). La posible coexistencia de las dos condiciones aumenta la dificultad diagnóstica. Manni et al (7) describen un grupo de 6 pacientes de género

33
Epilepsia, sueño y trastornos del sueño Julia Santín et al
masculino, con edad promedio de 70.5 años, quienes tenían diagnóstico documentado de epilepsia y ade-más presentaban episodios de descontrol conductual nocturno, que correspondieron a TCR en estudios video-polisomnográficos. Los autores encontraron además actividad epileptiforme interictal en el 26.4 % de casos con TCR (9/34), cifra similar al porcenta-je de actividad epileptiforme interictal reportada por McBride y cols. (8) en pacientes mayores de 60 años de edad, con otros trastornos neurológicos.
Otras parasomnias, como el sonambulismo, terrores nocturnos y pesadillas, por su carácter episódico y en algunos de ellos la amnesia de lo ocurrido que conllevan, pueden confundirse con crisis epilépticas parciales complejas. La falta de estereotipia, mayor duración y, en el caso de las pesadillas y trastorno conductual del REM, la presencia de recuerdo frag-mentado, ayudan en el diagnóstico diferencial. El trastorno de pánico puede presentarse con eventos nocturnos. Estos tienen inicio brusco, se asocian a intenso miedo, manifestaciones autonómicas, temor a morir, etc. Aparecen principalmente en etapa II No-REM. Constituyen un diagnóstico diferencial de crisis epilépticas de las que se distinguen por no asociarse a alteración de conciencia, tener también una duración más prolongada y acompañarse de crisis de pánico diurnas (9).
En todos los casos antes mencionados los exámenes complementarios neurofisiológicos constituyen un elemento diferenciador. En los trastornos del sueño no habrá descargas epileptiformes intercríticas ni patrones de reclutamiento ictal, pero si podrán en-contrarse cambios específicos para narcolepsia en el test de latencias múltiples de sueño o alteraciones polisomnográficas en otras condiciones (movimiento periódico de extremidades, trastorno conductual del REM, etc). Cuando se plantea el diagnóstico diferencial con epilepsia, los registros poligráficos nocturnos deben incluir monitorización videográfica y suficiente número de canales para posibilitar una lectura electroencefalográfica confiable (10).
Actividad epileptiforme ictal e interictal y sueñoLa epilepsia del lóbulo frontal suele manifestarse por crisis casi exclusivamente nocturnas, algunas de ellas difíciles de identificar como de origen co-micial, especialmente si no hay eventos diurnos ni aparecen crisis tónico-clónicas nocturnas. Provini et al (11) presentaron una serie consecutiva de 100
pacientes con epilepsia frontal nocturna, estudiados poligráficamente. Establecieron la presencia de tres variedades semiológicamente identificables de crisis: a) despertares paroxísticos (que corresponden a crisis hipermotoras) en el 9%, b) distonías paroxísticas nocturnas (crisis del área motora suplementaria) en el 51% y c) “vagabundeo nocturno”, que corres-ponden a automatismos ambulatorios, en el 40%. Enfatizan entre las dificultades diagnósticas, el que sólo un tercio de los pacientes presentaba esporá-dicos eventos diurnos, más fácilmente reconocibles como epilépticos y cerca de un cuarto generalización secundaria en los eventos nocturnos, que también hacen más evidente el carácter epiléptico de las crisis. Una variedad particular de epilepsia frontal nocturna es la autosómica dominante, cuadro genético que se caracteriza por crisis de inicio en la infancia o adolescencia, generalmente sólo nocturnas y en las que en un 20 a 30% de los casos puede encontrarse mutación de genes que codifican receptores nicotí-nicos (12). Los “vagabundeos epilépticos nocturnos” fueron descritos por Pedley y Guilleminault en 1977 (13). Se caracterizan por deambulación y conductas bizarras paroxísticas manifestadas durante el sueño, que pueden interpretarse como sonambulismos. Estos eventos pueden también aparecer en epilepsias del lóbulo temporal (14). Nobili et al (15) describen el caso de un paciente que inició estos episodios a los 6 años de edad, siendo inicialmente diagnosticados como sonambulismo y finalmente como epilepsia. A los 39 años fue estudiado con monitoreo video-elec-troencefalográfico prolongado que mostró registros ictales con descargas bien localizadas de inicio en estructuras temporales derechas y ulterior dispersión al cíngulo.
Las crisis epilépticas nocturnas aparecen en distinta proporción en los estadios de sueño. Bazil et al. (16) estudiaron un total de 116 crisis en 188 pacientes, encontrando que de las crisis parciales complejas sólo un 3% se presentó durante REM, mientras que el 80% apareció en etapas I y II; ninguna crisis de inicio focal se generalizó durante REM. Estos mismos auto-res encontraron que la probabilidad de ocurrencia de crisis durante el sueño era signficativamente mayor en epilepsias frontales versus epilepsias temporales. La serie de Minecan et al (17), que incluyó 55 pacien-tes, encontró resultados semejantes, ya que el 95% de las crisis nocturnas se presentaron durante sueño No-REM, principalmente en estadios I y II. El electroencefalograma (EEG) es el examen com-plementario más útil en el estudio de las epilepsias,

34
Revista Chilena de Epilepsia Año 8, Nº 1, Diciembre de 2007
ya que puede mostrar alteraciones específicas de esta condición, tanto en los períodos intercríticos como durante crisis epilépticas clínicas. Dado que estas últimas son de ocurrencia relativamente infrecuente y por azar es menos probable obtener un registro de ellas en los estudios de rutina, el hallazgo más habi-tual que apoya la sospecha clínica de epilepsia son las descargas epileptiformes interictales (espigas o puntas). Estas alteraciones intercríticas se manifies-tan de manera distinta en vigilia y en las distintas etapas del sueño. Así existe en las etapas No-REM un incremento significativo del número de descargas epileptiformes, tanto en las epilepsias focales como en las generalizadas idiopáticas, con el consiguiente incremento de la positividad de los EEGs; de ahí la importancia de registrar períodos de sueño en los trazados intercríticos, especialmente cuando se practican para verificar el diagnóstico de epilepsia. Algunos síndromes epilépticos tienen como carac-terística distintiva un fuerte aumento de la actividad epileptiforme durante el sueño. Las epilepsias focales benignas de la infancia se caracterizan por crisis par-ciales motoras, que se presentan en niños, durante el sueño, comprometiendo principalmente cara y len-gua; las espigas y puntas focales característicamente muestran un acentuado incremento durante el sueño no-REM. Este síndrome parece corresponder a un extremo de un espectro que abarca otras variedades, que incluyen el Síndrome de Landau Kleffner, hasta otra variedad en la que durante el sueño se desenca-dena actividad epileptiforme continua, al modo de un status epiléptico y que desaparece en vigilia (estatus epiléptico eléctrico del sueño) (18,19,20,21,22).
En contraposición, en las etapas REM la frecuencia de aparición de elementos epileptiformes intercríti-cos se reduce significativamente, lo que también se da para las crisis epilépticas, como ya se mencionó. Estas diferentes expresiones pueden entenderse a la luz de la fisiología del sueño, ya que en el sueño No-REM, encontramos múltiples manifestaciones de sincronización neuronal (ondas del vértex, husos de sueño, transientes positivos occipitales, etc.), que fa-vorecerían de algún modo los fenómenos patológicos de tendencia sincronizante, como son los epilépticos. El sueño REM, en cambio, característicamente se asocia a desincronización, lo que dificultaría la acti-vación simultánea de grupos neuronales involucrados en las descargas epilépticas, produciendo además espigas o puntas de mayor focalización. En suma, los períodos no-REM aumentan la probabilidad de encontrar descargas epileptiformes pero son menos
útiles para definir carácter focal versus generaliza-do, mientras el REM tiene menos rendimiento en el diagnóstico pero aporta mayor precisión topográfica (23).
El sueño puede también modificar la morfología de las descargas, lo que incluso puede llevar a inter-pretaciones erradas. La ya mencionada tendencia a sincronizar poblaciones neuronales puede hacer que la actividad epileptiforme focal de la vigilia se exprese de un modo más generalizado. Ocurre tam-bién lo opuesto, que espigas y puntas perfectamente sincrónicas en vigilia se muestren fragmentadas y lateralizadas o focalizadas durante el sueño. Final-mente, otro punto que debe tenerse presente es que durante el sueño aparece no rara vez un sinnúmero de variantes normales, de aspecto epileptiformes que suelen llevar a confusión, entre las que pueden mencionarse espigas wicket, espigas positivas a 14-6 ciclos por segundo, descargas rítmicas temporales medias, espigas fantasmas y transientes epileptifor-mes benignos del sueño (24).
Una práctica habitual empleada para aumentar la po-sitividad del EEG en el diagnóstico de las epilepsias es realizarlo después de una privación de sueño. Este método incrementa las posibilidades de encontrar actividad epileptiforme interictal específica por al menos dos mecanismos, uno el simple aumento del tiempo de muestreo (estos registros son más largos que los llamados estándar) y porque en ellos au-menta la posibilidad de registrar sueño superficial, que, como se explicó, de suyo también produce un incremento de estas descargas.
Hipersomnolencia, insomnio y epilepsiaKhatami et al (25) estudiaron la prevalencia de trastornos específicos del sueño en un grupo de 100 pacientes consecutivos con epilepsia y 90 controles, mediante tres instrumentos validados (escala de som-nolencia de Epworth y escalas específicas para apnea del sueño y narcolepsia) encontrando que la frecuen-cia de apnea de sueño, piernas inquietas, bruxismo y pesadillas entre otros tenían una frecuencia semejante en ambos grupos, mientras el insomnio de manten-ción era más frecuente en epilépticos, aunque con una baja significación estadística.
Efecto de las crisis epilépticas y medicamentos antiepilépticos en el sueñoLa presencia de hipersomnolencia en pacientes con epilepsia es frecuente y se estima que alcanza a un

35
10-30%. Aunque en parte esto podría derivar del uso de fármacos antiepilépticos, el síntoma puede estar relacionado con la epilepsia per se. Drake et al (26) estudiaron con test de latencias múltiples de sueño un grupo de 30 pacientes con epilepsia con crisis parciales y generalizadas, de reciente diagnóstico y sin tratamiento farmacológico, a los que practicaron además EEG toda la noche anterior. Encontraron que el 10% tenía latencias promedio inferiores a 5 minutos y que el 27% tenía latencias inferiores a 8 minutos. De los hallazgos electroencefalográficos, la presencia de actividad epileptiforme en el lóbulo temporal derecho se correlacionó con somnolencia. Varios medicamentos antiepilépticos tienen efectos sedantes, transitorios o permanentes y en muchos casos con impacto en la arquitectura del sueño. Con relación a los antiepilépticos clásicos, la fenitoina se asocia a disminución de la eficiencia, latencia de sue-ño y aumento del sueño superficial (etapas I y II No-REM), sin afectar significativamente otros estadios. La carbamazepina en cambio mejora la eficiencia de sueño, disminuye su fragmentación y aumenta el sueño delta (etapas III/IV NoREM) pero disminuye la densidad de los elementos fásicos del REM. El ácido valproico genera aparentemente menos alteración del sueño; para algunos autores estabiliza los ciclos de sueño mientras otros encuentran escaso o nulo efecto sobre su arquitectura en dosis terapéuticas. El fenobarbital usado crónicamente acorta la latencia de sueño, aumenta las etapas profundas NoREN y disminuye el REM. Los benzodiazepínicos tiene un efecto semejante pero además disminuyen el sueño delta y ambos compuestos pueden causar sedación diurna (27). Sin embargo, algunos autores (6,28,29) no encuentran estos hallazgos.
Otros fármacos pueden inducir insomnio eventual-mente de tal magnitud que obliguen a su suspensión; entre estos están el felbamato (30) y la lamotrigina. Sadler et al (31) estudiaron retrospectivamente un grupo de 109 pacientes sin trastornos de sueño pre-vios que recibieron lamotrigina, encontrando que el 6.4% desarrolló un insomnio que obligó a cambios en la terapia. Foldvary et al (32) estudiaron con po-lisomnograma pre y post tratamiento a 10 pacientes que recibieron lamotrigina 400 mg/día como terapia añadida; demostraron que el fármaco producía una reducción del sueño delta, aumento de las etapas II, aumento del REM y disminución de los despertares; estos pacientes no tuvieron somnolencia diurna.
Bonnani et al (33) estudiaron con test de latencias múltiples de sueño 14 pacientes en monoterapia con topiramato, comparándose con controles pareados por sexo y edad, antes de inicio de la terapia y dos meses después, sin encontrar diferencias significa-tivas entre ambos grupos. El efecto de este fármaco, que ha sido usado en el trastorno nocturno del comer, sobre el sueño, no ha sido estudiado.
El levetiracetam parece no tener efectos deletéreos sobre el sueño. Un estudio con voluntarios normales tratados con este medicamento y estudiados antes y después de 3 semanas de terapia con PSG y test de latencias múltiples de sueño mostró consolidación del sueño (aumento de tiempo total de sueño y eficiencia de sueño) (34). Bell et al (35) muestran hallazgos semejantes en voluntarios sanos y pacientes con epilepsia, aunque otros (36) no encuentran efecto sig-nificativo alguno con una metodología muy similar. Respecto de la gabapentina, Foldvary-Schaefer et al (37) estudiaron un grupo de voluntarios normales con PSG y escala de Epworth basal y después de adminis-tración de 1800 mg/día, comparándolo con un grupo control. Encontraron aumento de la proporción de sueño delta, semejante a lo reportado por Legros et al (38) en un grupo de pacientes con epilepsia, sin cambios en la escala de somnolencia diurna.
Los pacientes con epilepsia pueden tener alteraciones en la arquitectura del sueño que incluyen aumento de la latencia de inicio del dormir, disminución y frag-mentación del REM, mayor frecuencia de cambios de etapa y despertares y disminución de la eficiencia de sueño (39). Estos cambios pueden ciertamente producirse en pacientes que experimentan crisis clí-nicas o subclínicas nocturnas, subgrupo de pacientes que muestra la mayor distorsión en la arquitectura de sueño, pero no se limitan a ellos ya que pueden apare-cer en pacientes con epilepsias con crisis sólo diurnas (16,39). El efecto de los fármacos tampoco explica estas alteraciones puesto que pueden presentarse en pacientes con epilepsias focales que no han recibido terapia (40). Estos cambios serían mas frecuentes e intensos en las epilepsias parciales (16).
Comorbilidad epilepsia y trastornos del sueñoLa alta prevalencia de trastornos del sueño que se manifiestan con hipersomnolencia, como la apnea de sueño y el síndrome de piernas inquietas hace probable su coexistencia con epilepsia y deben con-siderarse en el diagnóstico diferencial de este síntoma
Epilepsia, sueño y trastornos del sueño Julia Santín et al

36
Revista Chilena de Epilepsia Año 8, Nº 1, Diciembre de 2007
en pacientes con epilepsia (41). Particularmente relevante es el diagnóstico de apnea de sueño (42, 43,44,45), ya que se ha demostrado que este trastorno dificulta el control de las crisis y que el uso de CPAP nasal produce una mejoría al menos parcial en los eventos epilépticos; debe sospecharse la asociación especialmente en pacientes cuyas crisis epilépticas aparecen exclusiva o principalmente durante el sue-ño. (mas detalles sobre esta asociación se encuentran en el capítulo correspondiente).
ConclusiónLas relaciones entre epilepsia y sueño son múltiples, de gran interés y probablemente sólo parcialmente conocidas. Resultan de gran relevancia no sólo para mejor comprender la neurofisiología de las epilepsias sino por el impacto terapéutico que puede tener el diagnóstico de trastornos del sueño que simulan epilepsia, que coexisten con ella o incluso derivan de la terapia anticonvulsivante.
Bibliografía
1. On Sleep and Sleeplessness. By Aristotle. http://classics.mit.edu/Aristotle/sleep.html.
2. Mitler MM, Hajdukovic R, Erman M, Koziol JA. Narcolepsy. J Clin Neurophysiol. 1990;7:93-118.
3. Hornyak M, Feige B, Riemann D, Voderholzer U. Periodic leg movements in sleep and periodic limb movement disorder: prevalence, clinical significance and treatment. Sleep Med Rev. 2006;10:169-77. Review.
4. Mahowald MW. Restless Leg Syndrome and Periodic Limb Movements of Sleep. Curr Treat Options Neurol. 2003;5:251-260.
5. Schenck CH, Mahowald MW. REM sleep beha-vior disorder: clinical, developmental, and neu-roscience perspectives 16 years after its formal identification in SLEEP. Sleep. 2002;25:120-38. Review.
6. Bazil CW. Sleep, Sleep Apnea, and Epilepsy. Curr Treat Options Neurol. 2004 ;6:339-345.
7. Manni R, Terzaghi M, Zambrelli E. REM Sleep Behavior Disorder and Epileptic Phenomena: Clinical Aspects of the Comorbidity. Epilepsia. 2006;47:78-81.
8. McBride AE, Shih TT, Hirsch LJ. Video-EEG monitoring in the elderly: a review of 94 patients. Epilepsia. 2002;43:165-9.
9. Craske MG, Lang AJ, Mystkowski JL, Zucker BG, Bystritsky A, Yan-Go F. Does nocturnal
panic represent a more severe form of panic disorder? J Nerv Ment Dis. 2002;190:611-8.
10. Foldvary-Schaefer N, De Ocampo J, Mascha E, Burgess R, Dinner D, Morris H. Accuracy of seizure detection using abbreviated EEG during polysomnography. J Clin Neurophysiol. 2006;23:68-71.
11. Provini F, Plazzi G, Tinuper P, Vandi S, Lugaresi E, Montagna P. Nocturnal frontal lobe epilepsy. A clinical and polygraphic overview of 100 consecutive cases. Brain. 1999;122:1017-31.
12. Scheffer IE, Bhatia KP, Lopes-Cendes I, Fish DR, Marsden CD, Andermann F, Andermann E, Desbiens R, Cendes F, Manson JI, et al. Auto-somal dominant frontal epilepsy misdiagnosed as sleep disorder. Lancet. 1994;343:515-7. Re-view.
13. Pedley TA, Guilleminault C. Episodic nocturnal wanderings responsive to anticonvulsant drug therapy. Ann Neurol. 1977;2:30-5.
14. Nobili L, Cossu M, Mai R, Tassi L, Cardinale F, Castana L, Citterio A, Sartori I, Lo Russo G, Francione S. Sleep-related hyperkinetic seizures of temporal lobe origin. Neurology. 2004;62:482-5.
15. Nobili L, Francione S, Cardinale F, Lo Russo G. Epileptic nocturnal wanderings with a temporal lobe origin: a stereo-electroencephalographic study. Sleep. 2002;25:669-71.
16. Bazil CW, Castro LH, Walczak TS. Diurnal and nocturnal seizures reduce REM in patients with temporal lobe epilepsy. Arch Neurol. 2000;57:363-8.
17. Minecan D, Natarajan A, Marzec M, Malow B. Relationship of epileptic seizures to sleep stage and sleep depth. Sleep. 2002;25:899-904.
18. Blom S, Heijbel J. Benign epilepsy of children with centro-temporal EEG foci. Discharge rate during sleep. Epilepsia. 1975;16:133-40.
19. Luders H, Lesser R, Dinner DS, Morris HH. Benign focal epilepsy of childhood. En: Epilepsy electroclinical syndromes, Luders H y Lesser R eds, New York: Springer-Verlag, 1987, pp 303-346.
20. Hirsch E, Marescaux C, Maquet P, Metz-Lutz MN, Kiesmann M, Salmon E, Franck G, Kurtz D. Landau-Kleffner syndrome: a clinical and EEG study of five cases. Epilepsia. 1990;31:756-67.
21. Patry G, Lyagoubi S, Tassinari CA. Subclinical “electrical status epilepticus” induced by sleep in children. A clinical and electroencephalographic

37
study of six cases. Arch Neurol. 1971;24:242-52.
22. Gobbi G, Boni A, Filippini M. The spectrum of idiopathic rolandic epilepsy syndromes and idiopathic occipital epilepsies: from the benign to the disabling. Epilepsia. 2006;47:62-6.
23. Bazil CW, Walczak TS. Effects of sleep and sleep stage on epileptic and nonepileptic seizures. Epilepsia. 1997;38:56-62.
24. Santin J., Godoy J., Ríos L, Mesa T., Aranda L. Incidencia de variantes normales de aspecto epi-leptiforme en electroencefalogramas normales. Estudio prospectivo y revisión de la literatura. Rev. Chil. Neuropsiquiat. 2003;41: 281-290
25. Khatami R, Zutter D, Siegel A, Mathis J, Donati F, Bassetti CL. Sleep-wake habits and disorders in a series of 100 adult epilepsy patients a pros-pective study. Seizure. 2006;15:299-306.
26. Drake ME Jr, Weate SJ, Newell SA. Contin-gent negative variation in epilepsy. Seizure. 1997;6:297-301.
27. Sammaritano M, Sherwin A. Effect of anticon-vulsants on sleep. Neurology. 2000;54:S16-24.
28. Herman ST. Epilepsy and sleep. Curr Treat Op-tions Neurol. 2006;8:271-9.
29. Placidi F, Diomedi M, Scalise A, Marciani MG, Romigi A, Gigli GL. Effect of anticonvulsants on nocturnal sleep in epilepsy. Neurology. 2000;54:S25-32.
30. Ketter TA, Malow BA, Flamini R, Ko D, White SR, Post RM, Theodore WH. Felbamate mo-notherapy has stimulant-like effects in patients with epilepsy. Epilepsy Res. 1996;23:129-37.
31. Sadler M. Lamotrigine associated with insomnia. Epilepsia. 1999;40:322-5.
32. Foldvary N, Perry M, Lee J, Dinner D, Morris HH. The effects of lamotrigine on sleep in pa-tients with epilepsy. Epilepsia. 2001;42:1569-73.
33. Bonanni E, Galli R, Maestri M, Pizzanelli C, Fabbrini M, Manca ML, Iudice A, Murri L. Da-ytime sleepiness in epilepsy patients receiving
topiramate monotherapy.Epilepsia. 2004;45:333-7.
34. Cicolin A, Magliola U, Giordano A, Terreni A, Bucca C, Mutani R. Effects of levetiracetam on nocturnal sleep and daytime vigilance in healthy volunteers. Epilepsia. 2006;47:82-5.
35. Bell C, Vanderlinden H, Hiersemenzel R, Otoul C, Nutt D, Wilson S. The effects of levetiracetam on objective and subjective sleep parameters in healthy volunteers and patients with partial epilepsy. J Sleep Res. 2002;11:255-63.
36. Bazil CW, Battista J, Basner RC. Effects of levetiracetam on sleep in normal volunteers.Epilepsy Behav. 2005;7:539-42.
37. Foldvary-Schaefer N, De Leon Sanchez I, Karafa M, Mascha E, Dinner D, Morris HH. Gabapen-tin increases slow-wave sleep in normal adults. Epilepsia. 2002;43:1493-7.
38. Legros B, Bazil CW. Effects of antiepileptic drugs on sleep architecture: a pilot study. Sleep Med. 2003;4:51-5.
39. Wang WW, Xie QH, Wu X. Structure of the spontaneous all night sleep in epileptics with polysomnography. Clin EEG Neurosci. 2005;36:36-41.
40. Touchon J, Baldy-Moulinier M, Billiard M, Besset A, Cadilhac J. Sleep organization and epilepsy. Epilepsy Res Suppl. 1991;2:73-81.
41. Foldvary-Schaefer N. Sleep complaints and epilepsy: the role of seizures, antiepileptic drugs and sleep disorders. J Clin Neurophysiol. 2002;19:514-21.
42. Hollinger P, Khatami R, Gugger M, Hess CW, Bassetti CL. Epilepsy and obstructive sleep apnea. Eur Neurol. 2006;55:74-9.
43. Bazil CW. Sleep, Sleep Apnea, and Epilepsy. Curr Treat Options Neurol. 2004 ;6:339-345.
44. Foldvary-Schaefer N. Obstructive sleep apnea in patients with epilepsy: does treatment affect seizure control? Sleep Med. 2003;4:483-4.
45. Vaughn BV, D’Cruz OF. Obstructive sleep apnea in epilepsy. Clin Chest Med. 2003 ;24:239-48.
Epilepsia, sueño y trastornos del sueño Julia Santín et al

38
Revista Chilena de Epilepsia Año 8, Nº 1, Diciembre de 2007
Factores desencadenantes de crisis epilépticasErna Rauch AvilaUnidad de Neurología Infantil, Hospital Padre HurtadoE-mail: [email protected]
Trabajos de Revisión
INTRODUCCION
La epilepsia por definición, es la recurrencia de crisis espontáneas o no provocadas. En la mayoría de las veces, las crisis no parecen estar relacionadas direc-tamente a un evento desencadenante específico. Sin embargo, se acepta que no ocurren completamente al azar, pueden ser inducidas o inhibidas por estímulos externos o internos. La dependencia al ritmo circa-diano, la relación con estados endocrinos, fiebre, con factores emocionales, alcohol, privación de sueño y una serie de otras variables han sido descritas. Sólo un pequeño grupo sufre crisis inducidas por estímu-los específicos, conocidas como crisis reflejas. La prevalencia de las crisis reflejas en poblaciones de epilépticos varía según las series. Gastaut y Tassinari describen hasta un 6% de pacientes epilépticos con crisis reflejas. Las epilepsias reflejas representan una pequeña parte de todos los trastornos epilépticos. La detección de factores epileptogénicos es de suma importancia, especialmente en pacientes resistente a terapia farmacológica (1,2,3).
FACTORES DESENCADENANTES DE CRISIS EPILEPTICAS
Corresponde a cualquier factor endógeno o exógeno que promueve la ocurrencia de crisis epilépticas (4). Bardett los definió como “aquellas circunstancias que preceden al inicio de un ataque epiléptico y son con-sideradas tanto por el paciente como por el neurólogo una posible explicación por el cual la crisis sucedió” (5). Pueden ser inducidas por factores facilitadores no específicos como la privación de sueño, alcohol, fiebre, stress físico o psíquico, entre otros, que disminuyen transitoriamente el umbral convulsivo por mecanismos no bien dilucidados. También por mecanismos epilépticos específicos reflejos, en que estímulos sensoriales o cognitivos específicos activan áreas corticales localizadas o circuitos, los que debido a alguna “inestabilidad funcional” responden con una descarga epiléptica. Estos precipitantes de crisis reflejas pueden ser simples estímulos como luz inter-
mitente (fotosensibilidad), patrones lineales, cierre ocular, sobresalto y otros o estímulos complejos tales como lectura, praxias (juego de cartas o ajedrez), alimentación, música y otros. El origen puede ser genético (fotosensibilidad o epilepsia de la lectura) o lesiones corticales (epilepsia del sobresalto o ciertas crisis inducidas por movimiento)(3,6,7).
Tabla 1Factores inductores de crisis epilépticas
Endógenos Exógenos
- Stress emocional o físico - Privación de sueño- Fatiga - Alcohol (exceso o privación)- Infecciones o fiebre - Toxinas y drogas- Ciclo menstrual - Ayuno- Sueño - Luz intermitente- Metabólicos - Valor o humedad- Despertar súbito - Ejercicio físico- Hiperventilación - Incumplimiento de TTO
“Estos factores prueban la acce- sibilidad de la ictogénesis a in- fluencias exógenas”
PRECIPITANTES FRECUENTES DE CRISIS EPILEPTICAS
Privación de sueñoUn paciente con epilepsia debería “gastar el día despierto y la noche durmiendo. El disturbio de este hábito, no es bueno……em-peora todo si no duerme de día ni de noche”. Hipócrates.
La privación de sueño (PS) provoca efectos que van desde cambios en el apetito hasta alteraciones del aprendizaje y memoria (8).
¿La PS puede afectar el control de crisis en pa-cientes con epilepsia? La evidencia acumulada en los últimos 40 años, reconoce a la PS como un precipitante de crisis epi-lépticas. La PS puede promover crisis epilépticas y

39
facilitar las descargas epileptiformes interictales. Alteraciones electroencefalográficas después de una PS sugieren un importante rol activador de crisis generalizadas primarias. La pérdida de sueño puede promover crisis generalizadas en individuos sanos predispuestos. El riesgo a convulsionar aumenta después de 48 hrs de una PS (8,9).
¿La privación de sueño por sí sola o asociada a otros factores puede precipitar crisis epilépti-cas?Entre las décadas de 1960-70, varios estudios foca-lizados en pilotos y soldados privados de sueño por largos periodos, encontraron que las crisis epilépticas ocurrían efectivamente en el contexto de una PS, pero frecuentemente asociaban otros factores, como stress laboral, factores emocionales, somáticos, fatiga, consumo de alcohol, abuso de drogas. La mayoría registraba actividad espiga-onda generalizada en los EEGs. En 1962, Janz reportó que la PS, junto al consumo de alcohol, precipitaban CTCG, particular-mente crisis del despertar.
Resultados similares han sido publicados con poste-rioridad. Stress, fatiga y PS son factores comunes, a menudo coexistiendo en un mismo individuo. La PS frecuentemente se asocia a stress físico y emo-cional y abuso de sustancias, lo que dificulta evaluar su contribución individual. La PS aguda es usada en algunas unidades de monitorización para facilitar las crisis en el diagnóstico o evaluación prequirúrgica de la epilepsia. En un estudio que monitorizó a 84 adultos con epilepsia parcial refractaria, asignados algunos a PS y otros a sueño normal, no encontró que la PS afecte la frecuencia de las crisis. la mo-nitorización se hacía en un ambiente relativamente libre del stress de la vida diaria (como alcohol, drogas ilícitas y otros) lo que pudo influir en los resultados (10). Es un hecho reconocido que algunos pacientes sensibles a los efectos de la PS aumentan la fre-cuencia de sus convulsiones. La PS crónica puede aumentar la frecuencia, como sucede en la apnea obstructiva. En varias series de casos prospectivos y retrospectivos, el tratamiento de la apnea obstructiva mejoró el control de las crisis epilépticas. En una revisión retrospectiva en niños, el 56%, de un total de 9 niños, disminuyó la frecuencia de las crisis en los primeros 12 meses después del tratamiento de la apnea del sueño sin cambios en su medicación anticonvulsivante (11).
Privación de Sueño y Espigas interictalesEn paralelo a las series de casos que muestran que la PS puede activar las crisis, estudios EEGs también han implicado a la PS como promotor de descargas epileptiformes interictales (DEI). Estos registros pueden mostrar más descargas epileptiformes que un registro estándar, particularmente en pacientes con crisis generalizadas (Arné-Bes et al. 1982). Un EEG con PS, es un procedimiento activador de rutina, sim-ple y relativamente inocuo, su ventaja es que puede activar patrones epileptiformes más que crisis clíni-cas, así mejora la acuciosidad diagnóstica sin riesgo adicional al paciente (Rodin, 1984). Como el sueño no-REM es un potente activador de las DEI, la pre-gunta que se origina es si la PS facilita las descargas, simplemente por la facilidad de quedarse dormido durante el EEG o debido a que el estudio realmente demuestra descargas epileptiformes interictales no registradas anteriormente (8). La PS puede registrar anormalidad epileptiforme en el 35% de los pacientes con EEG inicial normal. Una revisión crítica de la literatura, con énfasis en el método, estableció que la PS facilita las DEI, independiente de sus efectos sobre el sueño. Otro gran estudio documentó el aumento de las DEI durante PS en pacientes cuyos EEGs estándar eran normales. La evidencia acumu-lada es suficiente para apoyar la hipótesis de que la PS activa las DEI. Se desconoce el mecanismo por el cual la PS puede activar regiones epilépticas.
¿La PS afecta la excitabilidad neuronal? ¿Los efectos de la PS y su presumible descenso del umbral convulsivo pueden ser explicados a nivel neuronal?La mayor parte de la literatura dedicada a los efectos de la PS sobre la función neuronal atañe a aspectos del aprendizaje y memoria. Sin embargo, los resul-tados no son extrapolables a la epileptogénesis. Los estudios sobre el efecto de la PS en epilepsia han sido principalmente observacionales, a nivel del organismo más que de la neurona. En gatos, la PS facilita las crisis inducidas por penicilina, tanto en sueño como en vigilia.
En un modelo genético de epilepsia ausencia en ratas que fueron privadas de sueño por 12 hrs., se registró un aumento en el número de descargas espiga-onda en las primeras 4 hrs de la PS. La limitación de este estudio es que la PS no fue separada de otros desenca-denantes. Otras investigaciones focalizadas en sueño
Factores desencadenantes de crisis epilépticas Erna Rauch

40
Revista Chilena de Epilepsia Año 8, Nº 1, Diciembre de 2007
indican que el sueño REM y sus componentes (ritmo theta hipocampal) suprimen la actividad convulsiva, mientras que la privación del sueño REM facilita las crisis. Se presume, que el aumento de la duración del sueño REM podría suprimir la actividad convulsiva. La disminución farmacológica del sueño REM en ratas disminuye el umbral convulsivo (8).
SUEÑO
A pesar del extenso número de estudios, la relación entre sueño y epilepsia no es completamente en-tendida. Es una situación reconocida que el sueño es proconvulsivante en pacientes con epilepsia, registrándose un incremento de la actividad epilép-tica ictal e interictal durante el sueño. Los estados de sueño afectan las crisis epilépticas. A su vez las crisis o la tendencia a convulsionar por sí misma pueden interrumpir el sueño y ocasionar diversos síntomas diurnos.
Al parecer, existen mecanismos similares compro-metidos en la propagación de las crisis y en la acti-vidad rítmica de ciertos estados de sueño. El sistema reticular activador del tronco cerebral y el tálamo están directamente relacionados tanto en la hiper-sincronía cerebral del sueño como en las descargas sincrónicas de las crisis generalizadas. Los cambios electroquímicos responsables de la hipersincronía cerebral facilitan también la propagación secundaria de las crisis parciales (9).
Aumento de la actividad epiléptica interictal e ictal durante el sueño. Durante el sueño no-REM aumentan las descargas epileptiformes generalizadas y parciales interictales, mientras que el sueño REM suprime las descargas epileptiformes generalizadas con un efecto variable sobre las descargas focales.
Durante el sueño, son más frecuentes las crisis epilép-ticas generalizadas (primaria o secundariamente). Las crisis parciales se inician más a menudo en vigilia. Janz encontró que el 55% de las crisis generalizadas secundariamente ocurrían durante el sueño.
El efecto del sueño en las crisis parciales es va-riable. Las CP complejas del lóbulo Frontal son más frecuentes durante el sueño que las del lóbulo Temporal (12).
Bazil et al. analizaron registros de Videomonitoreo-
EEG de 188 pacientes (1116 crisis), encontrando que el 20% de las crisis ocurrieron durante el sueño, más del 20% de los siguientes tipos de crisis ocurrió durante el sueño: subclínicas 60%, CTCG 45% y parciales complejas 31%. La distribución de las crisis parciales complejas según estados de sueño: 25% de crisis en estado I, 54% en estado 2; 13% en sueño onda-lenta, y 3% en REM comparado con 31, 45, 20 y 2% respectivamente por Terzano et al.
No ocurrieron crisis no epilépticas. Las crisis psicó-genas no epilépticas son consideradas raras durante el sueño.
En varias epilepsias y síndromes epilépticos, las crisis son más frecuentes en el sueño, en epilepsias generalizadas idiopáticas como el Gran mal del des-pertar y la Epilepsia Mioclónica Juvenil y epilepsias parciales como la Rolándica benigna, la epilepsia de la infancia con paroxismos occipitales, epilepsia autosómica dominante del lóbulo Frontal y ELF o ELT sintomáticas y criptogénicas y otras como el S. de Landau-Kleffner.
Las crisis ocurren durante ciertos estados de sueño.Especialmente en sueño no REM (particularmente onda-lenta) y disminuyen en el sueño REM. El estado 2 es un particular facilitador de epilepsias generalizadas, mientras el sueño de onda-lenta induce tanto crisis parciales como generalizadas. Algunos reportes sugieren que las crisis parciales incrementan en REM.
Las crisis pueden alterar la arquitectura del sueño, aunque la proporción total no es mayormente alte-rada. El sueño REM puede disminuir si ocurre una crisis nocturna, pero las noches libres de crisis el sueño REM es normal.
DESPERTAR SUBITO
Es un precipitante de importancia en Epilepsia Mioclónica Juvenil. El despertar provocado es más “peligroso” que el espontáneo, especialmente si se interrumpe en etapas inestables del sueño como sueño REM y fase 2 del sueño (13).
EJERCICIO Y FATIGA
Un escaso porcentaje de pacientes reporta crisis poco después del ejercicio. La actividad física intermitente

41
no parece aumentar la frecuencia de las crisis epilép-ticas. El ejercicio aeróbico puede aumentar el umbral convulsivo ¿efecto protector? La hiperventilación por ejercicio raramente provoca crisis. Probablemente aumenta el riesgo, un ejercicio sostenido más que intermitente y la hipoxia soste-nida o hipoglicemia después de un ejercicio físico vigoroso.
La acidosis metabólica post ejercicio, en la fase de recuperación provocaría anormalidades epileptifor-mes en el EEG.
La Fatiga es frecuentemente reportada como un fac-tor inductor de crisis, hasta un 13% y puede facilitar las crisis por video-juegos, entre otros (13,15).
ALCOHOL
Una ingesta moderada (social) no desencadena crisis epilépticas. Beber una o dos copas de vino, cerveza o licor: no induce crisis epilépticas (no incrementa la frecuencia de CTC ni parciales complejas), no interfiere con los fármacos antiepilépticos (no altera sus niveles plasmáticos) y no modifica el EEG.
La ingesta excesiva puede desencadenar crisis. Un alcohólico crónico, con o sin epilepsia o un paciente con epilepsia no alcohólico, pueden convulsionar durante una ingesta excesiva, pero más frecuente-mente lo harán durante la privación de alcohol o en el período de caída de los niveles de alcohol en la sangre (especialmente si está asociada con sueño insuficiente) (13, 14,15).
HIPERVENTILACION
La hiperventilación (HV) provoca alcalosis respi-ratoria y ésta a su vez vasoconstricción central que modifica el nivel de oxígeno y glucosa cerebral. Desencadena ausencias y menos acción en otros tipos de crisis.
La HV involuntaria puede ocurrir en el curso de acti-vidades de la vida diaria debido a ansiedad, sollozos o actividad sexual (13). FIEBRE
A cualquier edad, principalmente en el anciano, un episodio febril agudo puede provocar crisis si el
paciente es susceptible y en pacientes con epilepsia puede haber crisis durante episodios febriles (13).
CICLO MENSTRUAL- HORMONAS
Las crisis pueden agruparse en torno al ciclo mens-trual (epilepsia catamenial).
Su génesis se atribuye a las propiedades neuroactivas de esteroides y a la variación cíclica de sus niveles en el suero. La actividad epiléptica se relaciona con un incremento de la razón estrógeno/progesterona y la disminución de las crisis con un aumento de ni-veles de progesterona. El Estradiol inhibe al GABA y potencia la transmisión glutamaérgica. Aumenta el metabolismo neuronal y las descarga. En cambio, los metabolitos de Progesterona (allopregnanolona), son potentes pro Gabaérgicos, reducen el metabolismo neuronal y las descargas.
Otro fenómeno asociado es la disminución de fár-macos antiepilépticos (FAE) en sangre en los días previos a la menstruación. Los FAE y los esteroides son metabolizados en el mismo sistema microsomal. La caída premenstrual de esteroides gonadales per-mite el metabolismo de los FAE y esto explica su disminución.
Durante el embarazo, aumenta la frecuencia de las crisis en un 20% de las pacientes, un porcentaje similar las disminuye, no hay cambio en el resto (14,16).
STRESS Y EMOCIONES
El stress frecuentemente es mencionado como pre-cipitante de crisis. Su evaluación independiente es difícil, ya que a menudo coexiste con otros induc-tores, como la privación de sueño, fatiga y disturbios emocionales. En 177 pacientes (Mattson), 58% pade-cía crisis con mayor frecuencia en periodos de stress. Sin embargo, aquellos con epilepsias de larga data (más de 5años) parecían considerarlo menos. No hay diferencia de percepción entre pacientes con epilepsia refractaria y epilepsias bien controladas.
De los eventos vitales más significativos están la tensión familiar y muerte de la pareja.
Las crisis también son atribuidas a la acción de emociones; angustia, ansiedad, frustración e ira, que a su vez frecuentemente gatillan otros factores
Factores desencadenantes de crisis epilépticas Erna Rauch

42
Revista Chilena de Epilepsia Año 8, Nº 1, Diciembre de 2007
como pérdida de sueño, ingesta excesiva de alcohol e incluso hiperventilación.
Los pacientes con ELT parecen más vulnerables al stress y a factores emocionales.
Las terapias anti stress (yoga, acupuntura, medita-ción, psicoterapia) no están validadas por estudios rigurosos que apoyen su recomendación en pacientes con epilepsia.
Los mecanismos fisiopatológicos del stress no están aclarados. Neuroesteroides moduladores de recep-tores GABA derivados de la deoxicorticosterona (DOC) parecen jugar un rol en el control de las con-vulsiones. DOC es un esteroide sintetizado a nivel adrenal que aumenta durante el stress (17,18).
CRISIS EPILEPTICAS Y FASE LUNAR
Antiguas creencias han relacionado las crisis epilép-ticas con el ciclo lunar. En un trabajo retrospectivo, 859 pacientes que consultaron por convulsiones al servicio de emergencia, se relacionaron a la fase lunar. Una significativa acumulación de crisis fue observada en periodos de luna llena, apoyando la antigua creencia que éste periodo aumenta la frecuencia de convulsiones (19). Mientras en otro estudio, una revisión de las crisis ocurridas en un periodo de 3 años, reveló que las crisis no epilépticas incrementaban en periodo de luna llena, mientras que las crisis epilépticas aumentaban en cuarto menguan-te, no significativamente. Los autores concluyen que no hay efecto de luna llena en las crisis epilépticas como se cree, aunque es posible un efecto en crisis no epilépticas (20).
Varias son las encuestas publicadas sobre la per-cepción de factores desencadenantes. Frucht aplicó una encuesta a 400 pacientes con epilepsia, el 62% de ellos citó al menos un precipitante. En orden de frecuencia: - Stress 30%- Privación de sueño 18%- Sueño 14%- Fiebre y enfermedad 14%- Fatiga 13%- Stress, fatiga y privación de sueño se correlacio-
naron positivamente entre ellos.- No hubo influencia significativa de los diferentes
precipitantes al relacionarlos a Síndromes epilép-ticos. Pacientes con ELT citaron con menor fre-
cuencia el sueño como desencadenante comparado con otros síndromes. Efectos menstruales fueron citados mayormente por mujeres mayores de 12 años y especialmente con ELT (28%). El gráfico resume la frecuencia de factores (4).
FOTOSENSIBILIDAD
Es una respuesta clínica y electroencefalográfica anormal a la luz intermitente. Genéticamente de-terminada. El estímulo luminoso precipita las crisis (crisis fotosensibles). Las crisis fotosensibles son las más frecuentes de las crisis reflejas. La ILAE ha propuesto cambiar el término fotosensible por “visual-sensitive” (21).
¿Qué estímulos visuales provocan crisis fotosen-sibles?
1. Estimulación luminosa intermitente (simple flic-ker)
Destellos luminosos del ambiente (luz solar intermi-tente proyectada en una avenida de árboles cuando se viaja, luz solar reflejada sobre el mar o la nieve, relámpagos, faroles delanteros de autos, luz intermi-tente en películas, en galería de arcos, en discoteca o luces de Navidad).
2. Sensibilidad a Pattern Provocadas por patrones visuales (rayas en un papel mural, persianas Venecianas, cercos, peldaños de es-calera mecánica, radiadores, planchar ropa listada).
Alternar patrón blanco-negro, dibujos rayados sufi-cientemente contrastados con diferencias de tamaño y orientación de las rayas u oscilaciones, diferir

43
patrones en su brillo más que en color son altamente epileptogénicos.
Patern
3. Televisión y Video-juegoEn 1992 en Inglaterra una escena publicitaria pro-vocó crisis reflejas. El efecto se repitió en 1997 en Japón, donde 685 niños y jóvenes sufrieron una crisis epiléptica, la mayoría por primera vez, al ver una secuencia de dibujos animados de la serie Pó-kemon. El estímulo visual había sido especialmente epileptogénico al aparecer imágenes muy contrasta-das alternando el rojo y el azul con una frecuencia superior a 3 Hz.
Televisión (TV)La pantalla de TV parece ser el estímulo visual más frecuente. El primer reporte fue hecho por Livingston en 1952. Andermann y Charlton (70s) describieron crisis epilépticas al apagar la pantalla o cuando se ajustaba el canal.
La TV parece provocar crisis por varios mecanismos: (1)la fotosensibilidad del paciente, (2) los destellos de la pantalla, frecuencias de 50 Hz (Europa) y de 60 Hz (EEUU) aumentan el riesgo de convulsionar. Jeavons reportó 61% de anormalidades a 50Hz comparado con el 22% a 60 Hz,, lo que explicaba la mayor incidencia en Europa. Una pantalla a 100 Hz reduce el riesgo de crisis. (3)La sensibilidad a pattern (es un gatillante importante de crisis provocadas por video-juegos). (4)Algunos programas de TV evocan más crisis que otros. Escenas con brillo máximo (mayor a 10 seg), exceden los 100 lux son activantes, menos de 50 lux protegería. Imagen de alto contraste,
cambios de velocidad de la imagen (más de 3 por seg), Intermitencia en rojo o alternancia de rojo y azul (5). Distancia de separación menor de 60 cm. Aproximarse a ajustar la imagen.
Video-juegosEl primer reporte fue hecho por Rushton en 1981. La mayoría de los pacientes son fotosensibles. Varios factores involucrados: - Frecuencia de la imágenes del monitor- Número de líneas por segundo.- El efecto flash y la intermitencia.
Se suman procesos cognitivos, fatiga, movimiento de dedos, en conjunto parecen tener un rol desenca-denante de las crisis. En Gran Bretaña la incidencia anual de crisis gatilladas por VG entre 7 y 19 años, fue de 1.5/100000 (Quirk et al).
4. Autoinducción de crisisMovimiento de manos frente a sus ojos, cierre de ojos u observación de un pattern.
5. Otros estímulos visualesPérdida de fijación visual (fixation-Off seizures. FOS), se elimina la visión central y la fijación (al cerrar los ojos en una sala iluminada). Crisis es-cotosensibles ocurren frente a reducción de luz u oscuridad.
CRISIS REFLEJASDesencadenadas por estímulos sensoriales específi-cos. En las epilepsia reflejas sólo tienen crisis cuando se expone al estímulo. En figura 1, se presenta es-
Factores desencadenantes de crisis epilépticas Erna Rauch

44
Revista Chilena de Epilepsia Año 8, Nº 1, Diciembre de 2007
3. Salas-Puig J. Epilepsias reflejas. REV NEUROL 2000; 30 (Supl 1): S 85-S 89.
4. Frucht M.M. Distribution of Precipitants Among Epilepsy Syndromes. Epilepsia, 41 (12):
1534-39, 2000.5. Bardett et al. Factors that can exacerbate seizu-
res. The medical treatment of epilepsy. 1992: 79-89. Epilepsy and Behavior 2005: 85-89
6. Aird. The importante of seizure- inducing factors in the control of refractory forms of epilepsy. Epilepsia 1983; 24: 567-83)
7. Ferlazzo E. Cortical triggers in generalized reflex seizures and epilepsies. Brain (2005); 128: 700-710.
8. Beth A. Malow., M.S. Sleep Deprivation and Epilepsy. Epilepsy Currents, Vol. 4, Nº 5 (Sep-tiembre/Octubre) 2004 pp. 193-195
9. Bazil C. Effects of Sleep and Sleep Stage on Epileptic and Nonepileptic Seizures. Epilepsia 1997; 38: 56-62.
10. B.A.Malow et al Sleep deprivation does not affect seizure frecuency during inpatient video-EEG monitoring.. Neurology 2002; 59:1371-1374.
11. Koh et al Sleep Apnea Treatment improves seizu-res control in Children with Neurodevelopmental Disorders.. Pediatr Neurol 2000;22:36-39
12. Herman et al Neurology2001;56: 1453-913. Texto de Epilepsia. 1997. Engel 14. Manual de Epilepsia. Daniel Galdames. 2000 15. Denio et al. J Med 1989;20:171-7616. Herzog A. Three Patterns of Catamenial Epi-
lepsy. Epilepsia 1997; 38(10): 1082-8817. Neugebauer R. Stressful Life Events and Seizure
Frecuency in Patients with Epilepsy. Epilepsia 1994; 35 (2: 336-43)18. Doodipala S. Stress-Induced DOC…..The Jour-
nal of Neuroscience, May 1,2002, 22(9):3795-80519. Polychronopoulos P. Lunar phases and seizure
ocurrence. Just an ancient legend? Neurology 2006,96:1442-43 20. Benbadis S. The influence of the moon on seizure
frecuency: myth or reality? Epilepsy & Behavior 5(2004): 596-59721. Shashi S. Visual-Sesitive Epilepsies: Classifica-
tion and Review. Can. J. Neurol. Sci. 2005;32:298-305.
quema fisiopatológico. En las tablas 2 y 3 ejemplos de crisis reflejas generalizadas y parciales.
Tabla 2.Tipos de crisis reflejas en pacientes con actividad
epiléptica generalizada
Tabla 3.Tipos de crisis reflejas en pacientes con actividad
epiléptica focal
.
BIBLIOGRAFIA1. Spatt J.. Subjective perception of seizure preci-
pitants: results of a questionnaire study. Seizure 1998;7: 391-395.
2. Nakken K.O.Which seizure-precipitating factors do patients with epilepsy most frecuently report? Epilepsia and Behavior 6 (2005): 85-89.

45
Epilepsia Rolándica benigna de la infancia con puntas centro temporales: Un amplio espectro clínicoPerla DavidNeuropediatra. Profesor Agregado Facultad de Medicina Universidad de Chile.E-mail: [email protected]
Trabajos de Revisión
RESUMEN
Se revisan las características clínicas, exámenes de laboratorio, comorbilidad, evolución y pronóstico de la epilepsia Rolándica con espigas centro tempora-les, la cual permanece como un síndrome epiléptico parcial benigno, pero el cual tiene una evolución no tan benigna como se había planteado desde su descripción, sino dentro de un amplio espectro evolutivo desde escasas crisis que pueden ser no tratadas, trastornos transitorios del neurodesarrollo hasta secuelas neuropsicológicas severas con retardo mental, cuyo origen se plantea estaría relacionado con la precocidad de la presentación y la expresión de las descargas epileptiformes interictales, lo cual ha sido estudiado con múltiples métodos diagnósticos con resultados diversos en la ERB, la cual se encuen-tra en un espectro intermedio desde el síndrome de Landau-Kleffner hasta las descargas continuas de espiga ondas del sueño lento.
Palabras claves: Epilepsia Rolándica, compromiso neuropsicológico, evolución con amplio espectro clínico.
ABSTRACT
The clinics characteristic are revised, exams of la-boratory, comorbidity, evolution and outcome of the Rolandic epilepsy with centrotemporal spikes which remains like a benign partial epileptic syndrome but which has an evolution not as benign as had been pre-sented since their description but inside an extensive spectrum evolutionary since one seizure that can be not treaties, transitory with comorbidity to sequel’s comorbidity severe with mental retardation whose origin is presented would be related to the early of the presentation and the expression of the epileptic discharges which has been studied with multiple diagnostic methods with diverse results and evolu-tion since Landau-Kleffner syndrome to continuous
slow sleep wave spikes.
Key Words: Rolandic Epilepsy spectrum, comorbi-dity and neuropsychological sequel
INTRODUCCION
La descripción de este síndrome epiléptico parcial idiopático fue hecha por Loisseau en 1967. La epi-lepsia Rolándica benigna de la niñez (ERB) es el más frecuente síndrome epiléptico infantil (1-15). Entre los familiares de los niños que presentan el síndrome existen niños que presentan espigas centro temporales y no presentan crisis, otros con escasas crisis que son los portadores de la epilepsia benigna Rolándica típica (ERB) y se describe además los últimos años la Epilepsia Rolándica Atípica (ERBA), la cual tiene una evolución con compromiso neu-ropsicológico variable a pesar de la habitual buena evolución de las crisis.
MANIFESTACIONES CLINICAS
Los niños con ERB presentan crisis de caracterís-tica asociación al sueño y esto puede dificultar la descripción de las crisis. Es frecuente que los padres relaten ruidos guturales durante el sueño. En la mayo-ría de los casos las crisis son focales motoras simples, pueden presentar sacudidas clónicas de todo un hemi-cuerpo en 7% de los casos. En más de la mitad de los casos hay signos orofaríngeos como hipersalivación, ruido de deglución, disartria, anartria, movimientos de la lengua y atonía de un brazo o una pierna. En general presentan dos tipos de crisis las diurnas hemifaciales, breves sin compromiso de conciencia parciales simples y las complejas nocturnas en las que interviene la esfera bucofaríngea. Es posible tam-bién la asociación con las crisis de ausencia que son raras pero cada vez más descritas. Es más frecuente en el sexo masculino 61 a 65%, característicamente en niños con desarrollo psicomotor normal, examen

46
Revista Chilena de Epilepsia Año 8, Nº 1, Diciembre de 2007
neurológico normal y con antecedentes familiares de epilepsia. La duración de la crisis en 75% de los casos es no es más de 5 minutos. La asociación a comorbilidad como alteraciones del comportamiento, déficit atencional, hiperactividad agresividad y tics nerviosos son frecuentes (4).
La incorporación del Síndrome de Landau-Kleffner (SLK) dentro de los síndromes epilépticos cambia el concepto habitual de epilepsia pues incluye dentro de estos síndromes a alteraciones en el EEG sin crisis epilépticas y alteraciones neuropsicológicas, del mismo modo que el síndrome de espigas continuas del sueño lento (CSWS), ambos son reconocidos como síndromes epilépticos específicos con crisis no siempre presentes y asociados a alteraciones neuropsicológicas. Es decir los niños con SLK y con CSWS tienen manifestaciones fenotípicas similares a la ERBA por lo que varios autores su-gieren que son parte de un espectro de desórdenes fisiopatológicos comunes que varían en la severidad de sus manifestaciones clínicas dependiendo del grado y localización de la disfunción cerebral. (5). Clínicamente existe correlación entre ellos en la edad de presentación, el compromiso del lenguaje, la presencia de actividad epileptiforme intercrítica y CSWS con deterioro cognitivo conductual, con fluctuaciones clínicas y con alteraciones variables en el EEG en la mayoría de predominio temporal izquierdo con desaparición de estas alteraciones y de las crisis hacia la adolescencia (6)
La evolución atípica de la epilepsia Rolándica se ha publicado en forma progresiva durante los últimos años y los hechos más frecuentes dentro de los des-critos es su aparición en niños menores de 4 años que comprometen su coeficiente intelectual llegando a ser inferior a 80, la presencia de crisis de presentación diurna, el estado epiléptico, la parálisis post-ictal y el cuadro clínico dentro del espectro del SLK y del síndrome de CSWS (6-9).
EXAMENES DE LABORATORIO
Electroencefalograma (EEG): En la ERB la loca-lización de las espigas interictales es estereotipada y variable en el EEG el cual se caracteriza por un trazado de base normal sobre el cual se inscriben espigas interictales centro temporales con un dipolo antero-posterior en los montajes referenciales. Las espigas son de amplio voltaje con una onda lenta.
Aumentan en somnolencia y sueño, llegando a presentase en corridas. En un 60% son unilaterales. Pueden ser monofocales bifocales o multifocales. No hay correlación entre la intensidad de las descargas y la gravedad de las crisis. El foco puede variar con el tiempo. Como en un 30% de los casos el EEG se altera solo en sueño, por eso es necesario solicitar el examen con privación de sueño.
Magnetoencefalografía (MEG). La combinación EEG/MEG y los estudios neuropsicológicos pro-veen de mejor localización de las espigas que aquí muestran correlación entre localización y déficit lo que indica que las espigas interictales pueden inter-ferir con complejas funciones cognitivas en estudios con baterías neuropsicológicas paralelas. Por ser la ERB una epilepsia con signos de hiperexitabilidad cortical que varía en el tiempo en frecuencia, late-ralización y localización el patrón neuropsicológico puede ir cambiando en funciones cerebrales supe-riores y percepción espacial, incluyendo orientación y memoria espacial.
Resonancia Magnética cerebral (RM): Los exámenes de neuroimagen en la ERB son habitualmente nor-males, pero últimamente se describen anormalidades no solo comunes, sino también específicas para el lóbulo temporal y lateralización en el lado de las descargas y en estudios en que se han comparado con otras causas de consulta neurológica como cefaleas se encuentran anormalidades peri hipocampales, anormalidades por malformaciones congénitas como hiperintensidades subcorticales y periventriculares similares no específicas de ERB, por lo cual es ne-cesario solicitar un estudio de RM habitual en estos pacientes (9-11).
Estudios con RM funcional y EEG de MSA reve-lan un dipolo de origen central inicial en todos los pacientes estudiados con ERB, incluyendo el área de cara y mano. Un estudio muestra que la combinación de EEG/RMf y MSA puede ser una herramienta po-derosa para describir la zona irritativa de pacientes con epilepsias focales idiopáticas.
La ERB constituye un modelo in vivo para estudiar la función cortical. Los hechos clínicos dependen de dónde esté localizada la actividad paroxística y la dirección de propagación. El origen de este trastorno puede ser una disfunción de la fisiología neuronal con sincronía entre las neuronas vecinas.

47
TRATAMIENTO
El tratamiento habitual es la carbamazepina, oxcarba-zepina en la ERB y ácido valproico en ERBA. SLK, CSWS u otros como benzodiazepinas por ejemplo el Clobazam en algunos casos. En los casos de inicio precoz por su mal pronóstico y el empeoramiento con fármacos como carbamazepina y Fenobarbital, éstos no se deben utilizar. Por esto es importante definir en cada paciente que tipo de evolución de epilepsia es más probable que tenga de acuerdo a sus carac-terísticas clínicas y EEG prolongado de sueño de ser posible con monitoreo TV-EEG nocturno (12-14).
EVOLUCION PRONOSTICO Y COMORBILI-DAD
La evolución de ERB es muy variable y ha habido muchas discusiones en la literatura epileptológica sobre la ocurrencia de déficit cognitivos en ERB y ERBA con diferentes métodos y resultados. En lo que existe concordancia es en el amplio espectro clínico de estos cuadros de acuerdo a la menor edad de presentación con mayor gravedad y más pobre pronóstico y en su asociación a déficit atencional, trastornos específicos del aprendizaje, trastornos de conducta y tics nerviosos como comorbilidad. Todos los datos indican posible relación entre actividad eléctrica y trastorno maduracional con tendencia a performans cognitiva inferior en ERB de acuerdo a menor edad de presentación.
COMENTARIO
Un amplio espectro clínico de manifestaciones que se inician sólo desde manifestaciones electroence-falográficas de origen genético hasta el deterioro progresivo de las funciones cerebrales superiores comprende el espectro que va desde la ERB típica hasta la ERBA, SLK y CSWS lo cual no ha podido diferenciarse en este continuo clínico con múltiples investigaciones publicadas los últimos años utili-zando diversos métodos de estudio diagnóstico de la actividad epileptiforme, neuroimágenes y estudios neuropsicológicos con diversas baterías de test que intentan definir el impacto de la actividad epilepto-génica en el cerebro en diversas etapas de maduración y su efecto clínico a través del tiempo. Es necesario efectuar estudios evolutivos estandarizados y rando-mizados a largo plazo.
REFERENCIAS
1. Quijada C, David P, Espectro Autista Boletín So-ciedad de Psiquiatría y Neurología de la Infancia y Adolescencia 1998, año 9 N°1; 22-28.
2. David P, Quijada C. Síndrome de Landau-Kleff-ner. Boletín Sociedad de Psiquiatría y Neurología de la Infancia y Adolescencia 29-33.
3. David P, Quijada C. Epilepsia Rolándica Benigna con espigas Centro Temporales. Boletín Sociedad de Psiquiatría y Neurología de la Infancia y Ado-lescencia 1999, año 10 N° 2; 15-20.
4. Deltour L, Barathon M, QuaglinoV, Vernier MP, Despretz P et al. Children with benign epilepsy with centrotemporal spikes show impaired at-tentional control: evidence from an attentional capture paradigm. Epileptic Disord 2007 9 (1): 32-8.
5. Gobbi G, Boni A, Filippini M. The spectrum of idiopathic Rolandic epilepsy syndromes and idiopathic occipital epilepsies: from the benign to the disabling.
6. De Tiege X, Goldman S, Verheulpen D, Aeby, Poznanski N et al Coexistence of idiopathic rolandic epilepsy and CSWS in two families. Epilepsia 2006 47(10):1723-7.
7. Montenegro MA, Guerreiro MM. Coexistence of childhood absence and rolandic epilepsy J Child Neurol 2006 21(6):535-6.
8. You SJ, Kim DS, Ko TS. Benign Childhood epilepsy with centro temporal spikes: early onset of seizures is associated with poorer response to initial treatment Epileptic Disord 2006 8 (4)285-8.
9. Datta A, Sinclair DB. Benign epilepsy of child-hood with rolandic spikes: typical and atypical variants. Pediatr Neurol 2007; 36(3)141-5.
10. Boxerman JL, Hwash K, Bali B, Clarke T, Rogg J , Pal DK. Is Rolandic epilepsy associated with abnormal findings on cranial MRI? Epilepsy Res 2007; 75(2-3): 180-5.
11. Intelectual and language finding and their rela-tionship to EEG characteristics in benign child-hood epilepsy with centrotemporal spikes. Riva D, Vago C, Franceschetti S, Pantaleoni C, D Ar-rigo S et al Epilepsy Behav 2007 19(2):278-85.
12. Metz-Lutz mn, Filippini M. Neuropsychological findings in Rolandic epilepsy and Landau- Klef-fner syndrome. Epilepsia 2006 ; 47 Suppl 2:71-5.
Epilepsia rolándica benigna de la infancia con puntas centro temporales: Un amplio espectro clínico Perla David

48
Revista Chilena de Epilepsia Año 8, Nº 1, Diciembre de 2007
13. Stephani U, Carlsson G. The spectrum from BCECTS to LKS: The Rolandic EEG trait-impact on cognition. Epilepsia 2006;47 Suppl 2:67-70.
14. Andrade R, García-Espinoza A, De la Cruz-Turruelles A, Machado-Rojas A, Arteche-Prior M, Diaz- Pedraza A.Unilateral neglet transient cognitive impairment and intercritical activity in
rolandic epilepsy Rev Neurol 2007 1-15; 44(9); 537-40.
15. Northcott E, Connolly AM, Benoya A, Mc Intyre J, Christie J et al. Memory and phonological awareness in children with benign rolandic epilepsy compared to a matchet control group. Epilepsy Res 2007 75(1) 57-62.

49
¿Qué es la comorbilidad?Daniela Avila SmirnowNeurólogo PediátricoUnidad de Neurología, Servicio de Pediatría, Complejo Asistencial Dr. Sótero del RíoE-mail: [email protected]
Trabajos de Revisión
Comorbilidad v/s complicaciónUna de las primeras referencias al término comor-bilidad fue la que efectuó el fallecido epidemiólogo de la Universidad de Yale, Alvan Feinstein, en una publicación del Journal of Chronic Diseases en 1970. En este artículo, comorbilidad se definió como la ocurrencia de más de una patología en la misma persona. El objetivo de este término era usarlo en estudios clínicos para indicar patologías coexistentes (1,2). Posteriormente, diversos autores han redefinido el concepto de comorbilidad.
El Centro de Políticas de Salud de Manitoba (Manito-ba Centre for Health Policy), en el año 2003, definió comorbilidad como las condiciones médicas que aumentan el riesgo de muerte del paciente, además de la condición más significativa que causa su estadía en el hospital. Para ellos, el número de condiciones comórbidas se debería usar para proveer un indicador del estado de salud y del riesgo de muerte. En otras palabras, la comorbilidad sería un indicador de la utilización diferencial del cuidado hospitalario (3).
Un artículo del Journal of Gerontology: Medicial Sciences de 2004, define comorbilidad como la presencia concurrente de dos o más enfermedades diagnosticadas desde el punto de vista médico en el mismo individuo (4).
La conocida enciclopedia en línea, “Wikipedia”, agrega un nuevo concepto al término comorbilidad. Señala que en medicina y en psiquiatría, comorbili-dad se refiere a la presencia de uno o más trastornos o enfermedades, además del trastorno o enfermedad primaria de interés, y al efecto de dichos trastornos o enfermedades en el paciente (5).
Según la Organización mundial de la Salud (OMS), la comorbilidad es la ocurrencia simultánea de dos o más enfermedades en una misma persona, lo que se asemeja a la definición de Feinstein (6).El Centro de Políticas de Salud de Manitoba señala que es importante diferenciar una comorbilidad de
una complicación. Esta última se refiere a una condi-ción adquirida durante una estadía hospitalaria (3).
El Instituto para la Información de Salud Canadiense (Canadian Institute for Health Information), también efectuó su propia definición de Complicación en el año 1994. Esta se refiere a un diagnóstico de la Clasificación Internacional de Enfermedades (CIE) que describe una condición que se inicia después del comienzo de la observación hospitalaria y/o del tratamiento, que usualmente influye en la duración de la hospitalización del paciente, y/o influye significa-tivamente el manejo o tratamiento del paciente (7).
Pruebas de comorbilidadExisten pruebas de comorbilidad que intentan es-tandarizar el impacto o el valor de las condiciones comórbidas, y si es que éstas son enfermedades secundarias o terciarias. Estas pruebas intentan sintetizar las distintas condiciones comórbidas, en una única variable predictiva, que mida mortalidad u otros outcomes. Los investigadores han validado dichas pruebas por su valor predictivo, pero ninguna prueba es reconocida aún como un gold standard (3,5).
Hay algoritmos codificadores de comorbilidad, siendo los más usados los de Charlson y de Elixhauser. Estos algoritmos trabajan con la Clasificación Internacional de enfermedades (CIE) 9 y 10 (3,5,8,9,10). El CIE-9-MC se creó hace más de 30 años (1975) como un sistema moderno de clasificación de enfer-medades. El CIE-10 fue introducido en 1992 por la OMS, como una potencial mejora de al CIE-9-MC. Esta última versión contiene más códigos que la previa (11).
Charlson definió 17 comorbilidades basándose en condiciones clínicas registradas en fichas clínicas de pacientes. Posteriormente se propusieron algoritmos codificadores para las comorbilidades de Charlson a partir del CIE-9-MC. Estos algoritmos fueron la base

50
Revista Chilena de Epilepsia Año 8, Nº 1, Diciembre de 2007
para la creación del Indice de Charlson. El índice de comorbilidad de Charlson, predice la mortalidad a un año para un paciente que puede tener un rango de condiciones comórbidas como cardiopatía, SIDA o cáncer. Mientras más alto es el puntaje, mayor es la mortalidad. La importancia para el clínico radica en saber que tan agresivamente tratar una condición. Inicialmente eran las enfermeras quienes revisaban las fichas de los pacientes en estudio. Posteriormente se han adaptado cuestionarios que responden los mismos pacientes (3).
Elixhauser definió 30 comorbilidades basándose en el CIE-9-MC. Estas comorbilidades están aso-ciadas con aumentos sustanciales en la duración de las hospitalización, costos de la hospitalización y mortalidad (10). Varios estudios han traducido el índice de Charlson de los códigos del CIE-9-MC a los del CIE-10. Un estudio publicado en 2005 desarrolló algoritmos codificadores para las comorbilidades de Charlson y Elixhauser, con los diagnósticos del CIE-10. Con-cluyó que estos nuevos algoritmos codificadores producían estimaciones similares de la prevalencia de comorbilidad y que podrían mejorar a los algoritmos ya existentes (9).
Estos indicadores no incluyen a la epilepsia como una de las comorbilidades.
Comorbilidad en adultos con epilepsiaMuchas comorbilidades en personas adultas con epilepsia son más frecuentes que en aquellos que no tienen epilepsia (12).
Gaitatzis, el 2004 decía que una condición comórbida puede ser la causa de la epilepsia, estar asociada a la epilepsia vía factores etiológicos comunes, genéticos o ambientales, o estar asociada al tratamiento de la epilepsia. Como comorbilidades asociadas a la causa de la epilepsia, se encuentran el accidente vascular encefálico y los tumores cerebrales. Como factores etiológicos comunes, encontramos el traumatismo encéfalo craneano (cefalea y epilepsia) y el tabaco (bronquitis crónica y epilepsia) (12).
En adultos con epilepsia son comunes los trastornos psiquiátricos como los depresivos (18%), ansiosos (15%), y psicóticos (9%). Existen factores biológicos que pueden explicar la comorbilidad psiquiátrica en epilepsia, como la medicación, y también factores
psicológicos y sociales (12,13,14).
Desórdenes somáticos frecuentes en pacientes adul-tos con epilepsia son las fracturas (10%) y el asma (9%) (12).
Si se clasifican las comorbilidades según la CIE IX, las comorbilidades psiquiátricas más relevantes son las psicosis orgánicas, el abuso de alcohol, esqui-zofrenia y la psicosis no orgánica, las que son 4-6 veces más frecuentes que en la población general. Las enfermedades médicas más relevantes son los tumores cerebrales y tumores meníngeos, siendo 30 a 55 veces más frecuentes que en la población general. La enfermedad de Alzheimer y el AVE son 7 a 8 veces más frecuentes que en la población general (12).
Comorbilidad en niños con epilepsiaLas comorbilidades asociadas a epilepsia difieren en niños y adultos debido a la maduración del SNC y sistémica, y a las diferencias conductuales, cogniti-vas y estructurales. Las comorbilidades pueden ser específicas de cada edad, como el SDAH en niños y la enfermedad de Alzheimer en adultos. Algunas comorbilidades específicas de la niñez, son el SDAH, y el autismo.
Las comorbilidades son comunes en niños con epilepsia y algunas pueden incluso ensombrecer a la epilepsia misma. Los profesionales que tratan la epilepsia en niños, deben considerar el efecto de los fármacos antiepilépticos sobre las condiciones comórbidas.
Entre los trastornos del desarrollo comórbidos a la epilepsia, están el SDAH, el retraso del desarrollo psicomotor, el trastorno autista y el retraso mental.
Dunn, en 2003 encontró síntomas de SDAH en 38% de 175 niños con epilepsia. Estos fueron más frecuentes en los niños con crisis TCG y CG, que en los con crisis de ausencia o parciales.
La incidencia de epilepsia de inicio en la infancia asociada con retraso mental y parálisis cerebral es de 15-38%. Los niños con retraso mental y parálisis cerebral pueden tener el doble de probabilidad de pre-sentar epilepsia que con cada patología por sí sola.
La demencia es rara durante la niñez, pero se observa en enfermedades degenerativas. Con frecuencia la epilepsia es una comorbilidad.

51
En 8-28% de los pacientes con autismo hay una epi-lepsia asociada. Los fármacos antiepilépticos pueden exacerbar o mejorar algunas de sus conductas.
Ettinger et al. 1998 evaluó 44 niños con epilepsia, de 7 a 18 años de edad, encontrando 26% con scores de depresión significativamente elevados y 16% con criterios clínicos de ansiedad significativa. El tratamiento con agentes sedativos como el fenobar-bital, puede exacerbar los síntomas o a asociarse a su presentación.
Wirrell et al. encontró en un estudio retrospectivo un aumento significativo de lesiones accidentales, como lesiones relacionadas a la bicicleta, en niños con epilepsia de ausencia, comparado con otros niños con enfermedades crónicas.
ConclusionesLa Comorbilidad puede ser definida como la presen-cia de más de una patología en un mismo paciente.
Las comorbilidades son frecuentes en niños y en adultos con epilepsia.
Existen indicadores de comorbilidad basados en los Algoritmos de Charlson y Elixhauser, que permiten estimar la mortalidad de los pacientes.
Bibliografía1. Gaitatzis A., Carroll K, Majjed A., Sander J. The
Epidemiology of the Comorbidity of Epilepsy in the General Population. Epilepsia, 45 (12): 1613-1622, 2004.
2. Lilienfeld S. Comorbidity between and within childhood externalizing and internalizing dis-orders. Reflections and directions. Journal of Abnormal Child Psychology, Junio, 2003
3. http://www.umanitoba.ca/centres/mchp/concept/thesaurus/thesaurus_C.html
4. Fried L.P., Ferrucci L., Darer J., Williamson J.D., Anderson G. Untagling the concepsts of disbility, frailty and comorbidity:implications for improved targeting and care. Journal of Gerontol-ogy: Medical Sciences, 2004, 3 (59):255-263.
5. http://en.wikipedia.org/wiki/Comorbidity6. http://www.euro.who.int/ppt/mnh/Nilson.pdf7. http://secure.cihi.ca/cihiweb/en/downloads/diag-
nosis_typing_background_2007_e.pdf8. Coding Algorithms for Defining Comorbidities
in ICD-9-CM and ICD-10 Administrative Data. Quan Hude, et al. Medical Care. 43, 11, Noviem-bre 2005.
9. Roos10. Comorbidity Measures for Use with Administra-
tive Data. Elixhauser Anne, et al. CIE10.
¿Qué es la comorbilidad? Daniela Avila

52
Revista Chilena de Epilepsia Año 8, Nº 1, Diciembre de 2007
Programa “Coloquios en Epilepsia para mé-dicos”
Crónica
Directores: Dra. Perla David, Dr. Juan SalinasObjetivo: Difundir los conocimientos que permi-
tan al médico realizar un tratamiento seguro y actualizado a sus pacientes con epilepsia a un nivel de no especialista.
Inscripción: GratuitaLugar: Sede Sociedad de Epileptología de
Chile. Av. Providencia 2315 of. 215. Horario: 19.00 a 21.00 hrs.
Certificación de la Sociedad de Epileptología de Chile con el 80% de asistencia. PROGRAMA I. Miércoles 29 de agosto de 2007. “Introducción al estudio de las Epilepsias”. Dr. Marcelo Devilat - Crisis epilépticas y Epilepsia. ¿Una crisis o
dos? - Epidemiología: incidencia, prevalencia, mor-
talidad. - Pronóstico - Costos - Impactos sobre la persona y la Sociedad - Manejo del impacto
II. Miércoles 12 de septiembre de 2007 “Diagnóstico etiológico y clínico”. Dra. Leda Aguilera - Los factores de riesgo - Las causas a través del ciclo de vida “Electrofisiología clínica y neuroimagen” Dr. Darío Ramírez - Importancia del EEG. EEGst, EEG con priva-
ción, EEGvideo. - Síndromes electroclínicos especiales - El aporte de la tomografía axial computada y
de la resonancia nuclear magnética III. Miércoles 26 de septiembre de 2007. “Clasificación de crisis y de síndromes epilép-
ticos”. Dra. Daniela Triviño - Presentación de videos. - Juegos de video IV. Miércoles 10 de octubre de 2007. “Trastornos paroxísticos no epilépticos”. Dra. Mariana Weitzman - Diagnóstico diferencial de las epilepsias - Presentación de videos “Enfoque general del tratamiento de las epi-
lepsias”. Dr. Jorge Förster - Indicaciones psicosociales - Manejo de las restricciones - Manejo de las comorbilidades - La necesidad de los antiepilépticos (AE) y su
mecanismo de acción - Los AE según ciclo de vida. V. Miércoles 24 de octubre de 2007. “Tratamiento farmacológico”. Dra. Erna Rauch - Los AE convencionales y los de nueva gene-
ración - Indicaciones y tipo de crisis - Las interacciones más relevantes “Tratamiento no farmacológico”. Dr. Tomás Mesa - Dieta cetogénica - Estimulación vagal - Cirugía de la epilepsia - Indicaciones y sus resultados VI. Miércoles 07 de Noviembre de 2007. “Epilepsia y mujer”. Dr. Osvaldo Olivares - Efectos de los AE - La contracepción - El embarazo y sus riesgos - El feto “Aspectos psicosociales en epilepsia”.

53
Dr. Fernando Ivanovic - El trabajo y el colegio El estigma - La sexualidad - La calidad de vida Cada charla será 30 minutos y habrá un lapso de 15 minutos para discusión.Tendremos una pausa de 30 minutos.
INSCRIPCIONES
Las inscripciones se podrán realizar en la sede de la Sociedad o en [email protected]
Existe un cupo de 25 personas, por lo que se ruega cumplir con la solicitud de inscripción. Dr. Juan Salinas Dra. Perla David Director Directora
Programa “Coloquios en epilepsia para médicos” Crónica

54
Revista Chilena de Epilepsia Año 8, Nº 1, Diciembre de 2007
Acreditados 2007. Ingresados 2006-2007.Crónica
Acreditados 2007:
Dra. Daniela AvilaDra. Perla DavidDr. Marcelo DevilatDr. Jorge FörsterDr. Fernando IvanovicDr. Pedro JimenezDr. Cayetano NapolitanoDr. Darío RamírezDr. Enzo RiveraDr. Juan SalinasDra. Ledia TroncosoDra. Daniela TriviñoDra. Viviana Venegas
Ingresados 2006-2007:
Dr. Juan Pablo FigueroaDr. Jaime GonzálezDra. Eliana JeldresDra. Sonia Ponce de LeónDra. Andrea PérezDra. Scarlett WittingDra. Mariana WeitzmanDra. Macarena Oliver

55
Memoria anual correspondiente al año 200702 de diciembre del 2006 a 15 de diciembre del 2007
Crónica
REUNIONES DE TRABAJO DE EDUCACION CONTINUA
Realizadas los segundos sábados de cada mes durante el año 2007.
Diciembre 2006 Comorbilidad en Epilepsia en niños y adultos.
Dra. Eliana Jeldres (Trabajo de ingreso)
Dr. Marcelo Devilat. Asamblea General de Socios y
Elección de Directorio
Enero 2007 “Costo de la Epilepsia” Dr. Juan Pablo Figueroa
Marzo 2007 “Proposición De Clasificación De Epilepsia Refractaria”
Dr. Darío Ramírez C.
Abril 2007 “Utilidad del Video Monitoreo EEG en Niño”
Dra. Carolina Alvarez
Mayo 2007 “Metabolismo Oseo Y FAE” Dr. Jaime Godoy
Julio 2007 Encefalopatía Hipóxico Isquémica Neonatal y Epilepsia”
Dr. Manuel Arriaza
Agosto 2007 “Fundamentos éticos de la limita-ción de tratamiento en pacientes neurológicos”.
Dra. Beatriz Shand
Septiembre 2007 “Semiología de la epilepsia del ló-bulo temporal en niños y adultos”
Dr. Cayetano Napolitano Dra. Daniela Avila Conmemoración día latinoameri-
cano de la epilepsia”
Dr. Marcelo Devilat
Octubre 2007 Pérdida del efecto de los antiepi-lépticos. ¿Una forma de resisten-cia?”.
Dr. Marcelo Devilat
Noviembre 2007 “¿Se puede prevenir la Epilepsia refractaria?”.
Dr. Manuel Campos P.
COLOQUIOS EN EPILEPSIA PARA MEDICOS
Este año se realizó por segunda vez esta actividad de educación continua, que se llevó a cabo en la Sede de la Sociedad de Epileptología de Chile miércoles por medio, comenzando el 12 de septiembre y finalizando el 21 de noviembre. Los Directores fueron la Dra. Perla David y el Dr. Juan Salinas. Aquí expusieron destacados neurólogos y neuro-pediatras nacionales, tocando temas dirigidos especialmente a médicos generales y becados.
La asistencia promedio fue de 17 personas por se-sión y la actividad fue muy bien evaluada por los asistentes, por lo que existe el interés de repetirla el próximo año.
VII JORNADAS INVERNALES DE EPILEPSIA 2007
Los días 8 y 9 de Junio 2007, se realizaron en el Hotel Neruda las VII Jornadas Invernales de Epilepsia, bajo el título de “Epilepsia: Fotosensibilidad Pantallas y Sueño”. En ésta participaron como conferencistas varios especialistas nacionales, la mayoría de ellos, también miembros de nuestra Sociedad.
Especialistas y becados presentaron 19 trabajos de investigación originales, 15 en posters y 4 en plata-forma. Asistieron 120 personas. La evaluación de los asistentes arrojó una nota 6.6 promedio.

56
Revista Chilena de Epilepsia Año 8, Nº 1, Diciembre de 2007
REVISTA CHILENA DE EPILEPSIA
La Revista Chilena de Epilepsia, publicación oficial de la sociedad, cuenta con la Dra. Perla David como editora. Este año se está editando el 8º volumen de esta revista. En éste se presentarán trabajos originales y trabajos de revisión.
PAGINA WEB
Nuestra página web, www.epilepsiadechile.com, está a disposición de los socios y del público en general. En ella encontramos información sobre la Sociedad y temas relacionados a la Epilepsia, en las siguientes secciones:1) Directorio2) Socios 3) Congresos (Jornadas Invernales de Epilepsia)4) Referencias (referencias bibliográficas sobre
epilepsia)5) Publicaciones (Revista Chilena de Epilepsia y
Normas Técnicas)5) Galería de fotos6) Tribuna para el médico8) Tribuna el pacientes9) Vínculos con ILAE; IBE; WHO y MINSAL10) Día Latinoamericano de la Epilepsia.
CONTACTOS NACIONALES
La sociedad mantiene contactos con la Sociedad de Neurología, Psiquiatría y Neurocirugía, y con la Sociedad de Psiquiatría y Neurología de la Infancia y Adolescencia.
El delegado de ANLICHE, Dr. Manuel Campos, Presidente de la Liga Chilena Contra la Epilepsia, ha participado en el directorio de nuestra Sociedad.
Miembros de esta entidad, participan en el Grupo Normativo de Epilepsia, del Ministerio de Salud de Chile.
CELEBRACION DEL DIA LATINOAMERICANO DE LA EPILEPSIA El día 6 de septiembre se celebró el día Latinoameri-cano de la Epilepsia. Este evento fue organizado por el Dr. Manuel Campos, presidente de la Liga Chilena Contra la Epilepsia y contó con la presencia del Dr. Juan Salinas, presidente de la Sociedad de Epilepto-
logía de Chile, además de otras autoridades.
CONTACTOS INTERNACIONALES
Reunión de la Comisión de Asuntos Latinoame-ricanos de ILAE (CALE)Se realizó En Santa Mónica, Sao Paulo, Brasil, el 12 de Febrero de 2007 con asistencia de las autoridades máximas de ILAE.
Academia Latinoamericana de Epilepsia (ALA-DE)Con fecha de 12 de Febrero de 2007 se fundó en Santa Mónica, Sao Paulo, esta academia destinada a la enseñanza de la Epilepsia y a estimular la inves-tigación en Latinoamérica.
Primer Curso Latinoamericano de EpilepsiaSe realizó en Santa Mónica, Sao Paulo en Febrero pasado al que concurrieron 60 profesionales menores de 35 años.
XXVII Congreso Internacional de EpilepsiaSe realizó del 8 al 12 de Julio de 2007 en Singapur.
IV Congreso Latinoamericano de EpilepsiaSe realizará del 5 al 8 de Noviembre de 2008 en Montevideo.
REVISTA EPILEPSIA La Revista Epilepsia, publicación oficial de la ILAE, se recibe desde 1999 hasta la fecha, y se encuentra a disposición de los socios en la sede de la Sociedad de Epileptología de Chile.
SECRETARIA DE LA SOCIEDAD DE EPILEP-TOLOGIA DE CHILE La Sra. Luisa Esparza ejerce como secretaria desde el año 2003. Su horario de atención es de 10:00 a 14:00 hrs., los días lunes, miércoles y viernes.
NUEVOS SOCIOS El año 2007 se incorporaron como socios los doc-tores:1) Dra. Eliana Jeldres (reunión diciembre de
2006)2) Dr. Juan Pablo Figueroa (reunión enero 2007)

57
EQUIPAMIENTO PARA BIBLIOTECA DE LA SOCIEDAD DE EPILEPTOLOGIA DE CHILE La biblioteca, ubicada en la sede de la sociedad, cuenta con la Revista Epilepsia y con la Revista Chilena de Epilepsia.
ACREDITACION 2007 El directorio está recibiendo los antecedentes de los socios para la acreditación de la Sociedad de Epi-leptología de Chile, de acuerdo a su participación en actividades de la Sociedad, en libros y revistas cientí-ficas, en actividades internacionales y electrónicas.
ACTIVIDADES PARA EL 2008
VIII Jornadas Invernales de EpilepsiaLos días 6 y 7 de junio del 2008 se realizarán las VIII Jornadas Invernales de Epilepsia, con el tema “¿Por qué la persona sigue con crisis a pesar del tratamiento?”.
El Comité Organizador, invita a participar a los in-teresados, mediante trabajos originales, que pueden ser enviados al sitio Web de la Sociedad de Epilep-tología de Chile.
AGRADECIMIENTOS
El directorio agradece a la Industria Farmacéutica y Tecnológica la colaboración que han realizado a la Sociedad durante el año 2007, a Abott Laboratories de Chile, Glaxo-Smith-Kline Farmacéutica, Labora-torio Janssen-Cilag, Laboratorio Pfizer, Laboratorio Drugtech de la Corporación Farmacéutica Recalcine, Novartis Chile, Laboratorio Andrómaco, Tecnofarma y Sanofi-Aventis.
Dra. Daniela Avila S. Dr. Juan Salinas V. Secretaria Presidente Dr. Marcelo Devilat B.
Vicepresidente
Memoria anual correspondiente al año 2007 Crónica

58
Revista Chilena de Epilepsia Año 8, Nº 1, Diciembre de 2007
Programa VIII Jornadas Invernales de Epi-lepsiaHotel Neruda de Santiago, 6 y 7 de Junio de 2008
Crónica
Viernes 6 de Junio de 2008
¿POR QUE LA PERSONA SIGUE CON CRI-SIS A PESAR DEL TRATAMIENTO?
08:00-08:30 Inscripciones Bienvenida Presidente de la Sociedad.
Dr. Juan Salinas. Presidente de las VIII Jornadas.
Dr. Cayetano Napolitano
08.30-00:90 EL PACIENTE ES RESISTENTE A LOS ANTIEPILEPTICOS (AE)
Dr. Marcelo Devilat Se definirá la resistencia, los factores
que la condicionan, sus mecanismos, como evitarla y tratarla.
09.00-09:30 FALLAS EN LA ADHERENCIA O EN EL USO DE NIVELES DE AE Dr. Daniel Galdames
Se describirá la importancia del cumplimiento, del uso racional de los niveles de AE, detallando las interac-ciones de los AE entre sí y con otros medicamentos.
09.30-10.00 SE UTILIZA UN AE INADECUA-DO
Dra. Maritza Carvajal Se comentarán las indicaciones de
los AE de primera línea y los noveles según tipo de crisis y de epilepsia, detallando la escala de priorida-des.
10.00-10.30 EL AE ORIGINA O EXACERBA
CRISIS EPILEPTICAS Dra. Nelly Chiofalo Se mencionarán los AE que exacerban
las crisis o las originan, determinando los tipos de crisis y las repercusiones al EEG.
10.30-11.0 SE EXPONE A FACTORES PRECI-PITANTES
Dra. Viviana Venegas Se describirán los factores precipi-
tantes de las crisis, las consecuencias sobre el EEG y como evitarlos.
11.00-11:30 Pausa
11.30-12.0 NO SE INVESTIGA LA ETIOLO-GIA
Dr. Cayetano Napolitano Se hará un recorrido por las principales
causas de epilepsia (no progresivas) según grupo etario y de los proce-dimientos a emplear para llegar al diagnóstico.
12.00-12.30 ES UNA EPILEPSIA PROGRESI-VA
Dra. Scarlett Wittig Se describirán las epilepsias progresi-
vas, los métodos para su diagnóstico y su prevención.
12.30-13.00 Mesa redonda con los conferencistas
13.00-14.00 Almuerzo 14.00-14.30 EL PACIENTE PRESENTA COMOR-
BILIDAD Dra. Eliana Jeldres Se analizaran las enfermedades más
frecuentemente asociadas a la epi-lepsia que interfieren el tratamiento favoreciendo la resistencia.
14.30-15.0 NO SE UTILIZAN LOS NUEVOS AE
Dra. Erna Rauch Se señalará el aporte de los AE nove-
les como alternativa efectiva para el control de las crisis y para minimizar

59
los efectos adversos de los AE tradi-cionales.
15:00-15.30 ESTA EN ESTADO EPILÉPTICO NO CONVULSIVO
Dr. Osvaldo Olivares Se describirán los estados epilépticos
no convulsivos que pueden enmascarar el diagnóstico, destacando su detec-ción precoz y su tratamiento.
15:30-16:30 Mesa Redonda con los conferencis-tas
16:30-17:00 Pausa
17:00-18:30 Presentación Posters Dr. Enzo Rivera
18:30-19:30 Presentaciones en Plataforma Dr. Juan Salinas
19.30-20.00 Grupo Normativo Minsal
Dr. Darío Ramírez
20.00-20.15 Premiación
Sábado 7 de junio 2008.
¿POR QUE LA PERSONA SIGUE CON CRISIS A PESAR DEL TRATAMIENTO?
09.00-09.30 NO SE EMPLEA LA CIRUGIA Dra. Lilian Cuadra
Se expondrá la experiencia de la ci-rugía, destacando la relevancia de la precocidad del estudio prequirúrgico, las técnicas utilizadas, los resultados obtenidos y la conducta post cirugía.
09.30-10.00 NO SE USA DIETA CETOGENA NI ESTIMULADOR VAGAL
Dra. Keryma Acevedo Se describirán las indicaciones de estos procedimientos y su resultado, destacando cómo superar las dificul-
tades que ellos presentan. SIMPOSIO EPILEPSIA RESISTENTE. EXPE-RIENCIAS SOBRE SU MANEJO Y TRATA-MIENTO
Los Servicios invitados comunicaran las experiencias con sus pacientes resistentes, describiendo la meto-dología empleada para el manejo de los enfermos, las características de sus epilepsias, los tratamientos empleados y los resultados
10.00-10.10 Dr. Enzo Rivera Hospital van Buren. Valparaíso10.10-10.20 Dr. Emilio Brunie Hospital Grant. Concepción.
10.20-10.30 Dr. Darío Ramírez Hospital Salvador. Santiago 10.30 -10.40 Dr. Jaime Godoy Hospital Universidad Católica. Santia-
go 10.40 – 11.00 Pausa11.00 - 11.10 Dr. Marcelo Devilat Hospital Luis Calvo Mackenna11.10 - 11.20 Dra. Ledia Troncoso Hospital San Borja-Arriarán 11.20 - 11.30 Dra. Maritza Carvajal Hospital Exequiel González Cortés11.30 - 11.40 Dr. Leon Adlerstein Hospital Roberto del Río11.40 -12.40 Mesa redonda con los participantes en
el Simposio12.40 -13.00 Clausura y entrega de certificados
PATROCINAN
- Ministerio de Salud- Universidad de Chile- Universidad de los Andes- Universidad de Santiago- Universidad Mayor- Sociedad de Psiquiatría y Neurología de la Infancia
y Adolescencia- Liga Chilena contra la Epilepsia
Programa VIII Jornadas Invernales de Epilepsia Crónica

60
Revista Chilena de Epilepsia Año 8, Nº 1, Diciembre de 2007Comorbilidad psiquiátrica en epilepsia y su tratamiento Inés Lackington
Trabajos presentados en las VIII Jornadas Invernales de Epilepsia
Crónica
1. PACIENTES CON EPILEPSIA EN POLICLINI-CO DE NEUROLOGIA INFANTIL. CDT LOS ANGELES
Alvaro Wicki MonsalvesPoliclínico de Neurología Infantil. CDT Los Angeles Servicio de Salud Bio Bio.
Introducción. La mayoría de las Epilepsias co-mienza en la infancia o adolescencia, esperándose un buen control de crisis en 70% de los casos (Plan AUGE).
Objetivo. Determinar las características de los pa-cientes con Epilepsia confirmada controlados por Neurólogo Infantil en CDT de Los Angeles.
Material y Método. Se revisaron retrospectivamente 669 fichas de menores de 15 años que ingresaron a la Base de Datos Electrónica del CDT con el diagnósti-co de Epilepsia entre Enero 2006 y Marzo 2007.
Resultados: Se descartan 408 fichas (Eventos No Epilépticos, Error dígitico etc). De los 261 casos confirmados 146 (56%) son hombres, 115 (44%) mu-jeres, 136 (52%) Urbanos, 125 (48%) Rural, catalo-gándose como Epilepsia Idiopática (EI) 127 (48,6%), Sintomática (ES) 111 (42,5%) y Criptogénica (EC) 23 (8,9%). El 69% (180) en monoterapia, 23,3% (61) biterapia y 7,7% (20) triterapia. 165 casos (63,2%) con una comorbilidad a lo menos. Habían suspendido tratamiento 60 (23%). Ingresaron a PLAN AUGE 62 pacientes (23,7%), de ellos 4 EC, 3 ES y 55 EI correspondiendo a 43,3% del total de éI.
Conclusiones: La mitad de los pacientes en control tienen ES o EC, más del 60% de los casos presenta alguna comorbilidad. El 23,7% de los pacientes in-gresan al PLAN AUGE, cifra menor a la esperada.
2. EPILEPSIA Y COMORBILIDAD EN NIÑOS Eliana Jeldres, Santiago Peralta, Juan Figueroa,
Verónica Gómez, Marcelo Devilat.Centro de Epilepsia Infantil. Servicio de Neurolo-gía y Psiquiatría. Hospital Luis Calvo Mackenna, Santiago. Chile.
Introducción. En una presentación anterior presen-tamos nuestra experiencia en el tema con 198 niños, la actual contiene 239 pacientes de un universo no seleccionado y orientada al tipo de comorbilidad (CMB).
Objetivo. Determinar la CMB en niños con epilep-sia controlados en el Centro.
Material y Método. Se revisaron 239 historias clí-nicas, con un nivel de confianza de 92%, obtenidas aleatoriamente de la base datos. La edad de diag-nóstico fue de 43 meses (mediana) (0-176 meses). La edad de inicio del tratamiento fue de 47,5meses (mediana) (0-180meses). Hubo 35 (14.6%) niños con epilepsia criptogénica, 125 (52.3%) con epi-lepsia idiopática y 79 (33.1%) con epilepsia sinto-mática. Ciento treinta y ocho (57.7%) niños tenían crisis generalizadas, 89(37.2%), crisis parciales y 11 (4,6%) crisis mixtas.
Resultados. Ciento noventa y dos (80.3%) niños presentaron al menos una CMB y, 83 (43,2%) de ellos presentan tres o más. Cuarenta y siete (19.7%) niños no tuvieron CMB. De los 192 pacientes que presentan CMB, cada uno de ellos tiene en prome-dio 2,4 CMB. De las 464 CMB que se registraron en la muestra, 281 (60,6%) son de tipo neurológica y, 183 (39,4%) corresponden a etiologías no neu-rológicas.
Las CMB neurológicas más frecuentes fueron: Re-tardo mental (RM) y Retraso del desarrollo psico-motor (RDSM) en 71 (29,7%), Déficit atencional (SDA) en 51 (21,3%), Parálisis cerebral (PC) en 37 (15,5%) y Retraso del lenguaje en 21 (8,8%). Las comorbilidades no neurológicas afectan preferente-mente a los sistemas cardiovascular y respiratorio

61
en 32 (13,4%) niños, 31 (13,0%) niños con pato-logía digestiva y 27 (11,3%) niños con patología osteomuscular
Conclusiones. Alrededor del 80% de los niños pre-sentó al menos una CMB. Más de la mitad de estas CMB son de tipo neurológico, las más frecuentes fueron RM/RDSM, SDA, PC. Entre las CMB no neurológicas predominan las que afectan al sistema cardiovascular y respiratorio, sistema digestivo y sistema osteomuscular.
3. PROGRAMA DE EPILEPSIA EN LA UNIDAD DE NEUROLOGIA DEL HOSPITAL EXE-QUIEL GONZALEZ CORTES 2005 -2006
Marta Chang, Jessica Reyes, Vicky Mori, Juan Enrique González, Marisol Avendaño, Javier Es-cobari, Maritza Carvajal. Becadas de Psiquiatría Infanto-Juvenil. Universidad de Santiago. Equipo de Epilepsia.
Introducción. El Programa de Epilepsia se imple-mentó el año 2001, en nuestra unidad. Desde el pri-mer semestre del 2005 se implementa GES epilepsia. Desde junio se ingresan pacientes a SIGGES. Es de nuestro interés dar a conocer nuestra experiencia.
Objetivos. Dar a conocer las características de los pacientes adscritos a este Programa desde enero del 2005 a l 30 de abril del 2007.
Resultados. Se han ingresado hasta la fecha 1064 pacientes, de los cuales son pacientes activos 981 pacientes ( 92,19%) y se han dado de alta o trasla-dado 83 pacientes (7.8%). De estos 981 pacientes, 143 niños han sido catalogados como refractarios según criterios 4 y 5 de Schmidt, han sido ingresados al GES epilepsia, 255 niños y 583 niños tienen una epilepsia activa aun no catalogados en uno y otro grupo.
De los pacientes GES ingresaron 106 niños en año 2005, 117 niños el año 2006 y hasta el 30 de abril 32 casos.
Mas del 50% de los niños ingresados al GES lo hacen el primer semestre GES ( 63 /106 el año 2005 y 83 / 117 el año 2006). Se cerraron 13 casos el año 2005 y solo 1 caso el año 2006.
El 100% de nuestros pacientes tiene consentimiento
informado en la ficha clínica, y el 100% se cumplió la garantía de oportunidad de los 20 días.
Conclusiones. Estos resultados se lograron por un cambio en el funcionamiento de la Unidad de Neurología, asignando un equipo de Neurólogos a cargo del Programa, asignando EU en horario parcial, priorizando las horas para EEG a estos pacientes y conformando una red con los consultorios APS. Con capacitación continua de los encargados de programa en cada consultorio.
4. EPILEPSIA DEL SUEÑO EN NIÑOS. CLINICA Y TRATAMIENTO.
Karina Tirado, Katya Reinbach, Verónica Gómez, Marcelo Devilat.Centro de Epilepsia Infantil. Servicio de Neurolo-gía y Psiquiatría. Hospital Luis Calvo Mackenna. Santiago. Chile.
Introducción. Las epilepsias del sueño han sido bien descritas en el adulto desde hace muchos años. En los niños la experiencia es reducida.
Objetivo. Describir la clínica y tratamiento de niños con epilepsia del sueño.
Pacientes y Método. Se revisaron todas las historias clínicas, acerca del tema, de uno de nosotros (MD), de niños con epilepsia del sueño que resultaron ser 24.
Se revisaron las fichas clínica de 24 pacientes con epilepsia del sueño, definida como aquella en la cual las crisis epilépticas ocurren en >60% durante el sueño. La muestra consta de 10(41,7%) mujeres y 14(58,3%) hombres, con edad promedio actual de 11.4 años. Tiempo en control en el Centro fue en promedio de 7.3 años. El tiempo promedio de mono-terapia fue de 24 meses y 64 meses politerapia.
Resultados. No hubo diferencias de género. El diag-nóstico fue de epilepsia generalizada en 7(29,2%) pacientes y epilepsia parcial en 17(70,8%). Se regis-traron 13(54,1%) pacientes con epilepsia sintomática, 10(41,7%) idiopática y 1(4,2%) criptogénica. Veinte niños (83.3%) tenían un EEG específico para epilep-sia y en 4 (16 %) era normal o inespecífico.
Diez y ocho pacientes (75%) presentaban crisis sólo durante el sueño (Epilepsias puras del sueño (EPUS).
Trabajos presentados a las VIII Jornadas Invernales de Epilepsia Crónica

62
Revista Chilena de Epilepsia Año 8, Nº 1, Diciembre de 2007
Seis pacientes (25%) tenían crisis en sueño y en vigilia (Epilepsias predominantemente del sueño (EPRES). Tanto los niños con EPUS y EPRES tenían mayoritariamente crisis parciales.
Doce niños (50%) presentaron comorbilidad neu-rológica, 8(33,3%) mostraban comorbilidad no neurológica y sólo 4(17,7%) no tenían comorbilidad asociada. Al corte 8(33,3%) pacientes estaban en monoterapia, 15(62,5%) recibían politerapia y uno no ingería medicación. Se registraron 7(29,4%) pa-cientes con efectos adversos a los fármacos.
Conclusión. La mayoría de los niños tiene epilep-sias sintomáticas, parciales y con EEG específico. Así también, las comorbilidades y la politerapia son frecuentes. Dos tipos de epilepsia pueden describirse en relación a la exclusividad o no de crisis durante el sueño.
5. EPILEPSIA MIOCLONICA JUVENIL: CA-RACTERIZACION DE LOS PACIENTES EN CONTROL EN UN HOSPITAL PEDIATRICO
Andrea Schlatter, Keryma Acevedo, Leon Alderstein ,Andrea Godoy. Unidad Neurología Infantil. Hospital Roberto de Río.
Introducción. La epilepsia mioclónica juvenil (EMJ) representa el 5-10% de las epilepsias. La edad inicio tiene un máximo entre 12-14 años. Las ausencias y mioclonías son breves y suelen pasar desapercibidas, retrasando el diagnóstico de EMJ hasta la aparición de crisis tónico-clónicas generalizadas (TCG).
Objetivos. Establecer las características de pacientes con EMJ en control en el Hospital Roberto del Río, evaluando la respuesta al tratamiento, la evolución y la presencia de complicaciones secundarias al tratamiento.
Material y Método. Revisión retrospectiva de pacientes controlados en Policlínico de Neurología entre 1/01/05-31/12/06 codificados como Epilepsia Idiopática Generalizada (EIG).
Resultados. Revisión 264 fichas pacientes, encon-trándose 20 con EMJ (7,5%). 13 de sexo femenino con edad promedio 12 años (8–14). Motivo de con-sulta correspondió a crisis TCG en 60 % de los casos y sólo un 5% lo hizo por mioclonias. Un 75% había
presentado síntomas anteriores al diagnóstico como: ausencias (10), mioclonias (10) y crisis TCG (3). El primer EEG tomado fue compatible con EMJ en 80%. Ácido Valproico fue el tratamiento de elección en 19 pacientes. Sólo 4 pacientes continuaron con crisis. Como efectos secundarios al tratamiento destacan: aumento de peso y ovario poliquístico.
Conclusiones. Las características de nuestros pa-cientes se ajustan a lo descrito en la literatura en cuanto a edad, distribución por sexo y evolución. Llama la atención que la presencia de mioclonías o ausencias no motivaran la consulta espontánea de los pacientes.
6. ALTERACIONES DEL LENGUAJE EN PA-CIENTES CON EPILEPSIA DEL LOBULO TEMPORAL
Enzo Rivera, Carolina Bustos, Liliana Henríquez, Octavio Alarcón, Jorge Valdés. Facultad de Medicina, Carrera de Fonoaudiología, Universidad de Valparaíso, Chile
Introducción. Los efectos de las crisis y descargas eléctricas sobre la cognición y el lenguaje son un he-cho vislumbrado desde los inicios de la epileptología moderna. Al respecto, la mayoría de las investiga-ciones realizadas en el extranjero avalan la presencia de deterioro cognitivo en la Epilepsia del Lóbulo Temporal (ELT). La exploración del lenguaje a través de la ejecución de tareas lingüísticas, se plantea como un método útil para acercarse al problema. Objetivos. Demostrar posibles alteraciones lin-güísticas en una población de pacientes con ELT refractaria, en período interictal.
Pacientes y Método. Se evaluó a 13 pacientes per-tenecientes al policlínico de epilepsias refractarias del Hospital Carlos van Buren con diagnóstico de ELT (Grupo 1) y 13 adultos sin epilepsia (Grupo 2), a través de la pauta de evaluación de tareas lin-güísticas. Esta herramienta consta de 6 ítems que evalúan comprensión auditiva, memoria de trabajo, denominación visual y auditiva, así como fluidez fonológica y semántica. Se estableció un nivel de significación estadística de p < 0,05.
Resultados. Las personas con ELT obtuvieron rendi-mientos inferiores respecto a las personas sin ELT en todas las tareas aplicadas. Este rendimiento inferior

63
alcanzó niveles estadísticamente significativos para las tareas de comprensión auditiva, fluidez fonológica y semántica. Se observó asimismo un mayor tiempo de ejecución de las respuestas para las personas del Grupo 1.
Discusión. Las alteraciones del lenguaje encontradas concuerdan con lo descrito en la literatura. Sin em-bargo, debido a la naturaleza preliminar del trabajo, no es posible establecer conclusiones definitivas al respecto. Es necesario continuar esta línea de inves-tigación a fin de caracterizar mejor la población en riesgo y proponer programas dirigidos de rehabili-tación fonoaudiológica precoz.
7. MANEJO DE DEPRESION EN PACIENTES EPILEPTICOS. RESULTADOS DE UN ESTU-DIO PROSPECTIVO.
Elizabeth Guerrero, Silvia Guerrero.Consultorio San Antonio.
Introducción. La depresión es frecuente comorbi-lidad de la epilepsia. A pesar de ello escasamente diagnosticada, Neurology 2002.
Objetivos. Evaluar prevalencia de depresión en epilépticos del Consultorio.Detectar si existe algún efecto en las crisis convulsivas al manejar la depre-sión y efecto en el tiempo.
Material y Método. Incluye el 100 % de pacientes del Programa de Epilepsia, 52. Aplicándose anam-nesis y escala diagnóstica de Depresión CIE-10. Iniciándose tratamiento con ISRS, Fluoxetina .Se evalúa el número de crisis convulsivas al inicio, primer y segundo año de tratamiento.
Resultados. 17 pacientes presentan depresión, pre-valencia total del 32,69%, correspondiendo 9.6% varones y 23% mujeres. Distribuyéndose en leve, 3.84%; moderada: 21.15% y severa 7.69%. A 15 pacientes se logró aplicar tratamiento obteniéndose una reducción del número de crisis en 11 , 73.33% y reducción en el número de sus crisis de 61.89% promedio en más de la mitad de los tratados al primer año y 69.64% al segundo año.
Conclusiones. Obtuvimos alta prevalencia de de-presión, 32.69% asociada a las condiciones socio-culturales de San Antonio. Además el manejo de la depresión disminuyó las crisis, lo cual es constante
en el tiempo de tratamiento, probablemente a causa de la compensación de la depresión.
8. TRATAMIENTO CON LAMOTRIGINA EN NIÑOS CON EPILEPSIA
Katya Reinbach, Karina Tirado,Verónica Gómez, Marcelo DevilatCentro de Epilepsia Infantil. Servicio de Neurolo-gía y Psiquiatría. Hospital Luis Calvo Mackenna, Santiago. Chile
Introducción. La lamotrigina (LMT) es un antiepi-léptico (AE) de tercera generación que puede ser una ayuda eficaz en algunos niños con epilepsia
Objetivo. Evaluar la eficacia y los efectos adversos de LMT en niños con epilepsia.
Material y Método. Se revisaron las fichas clínicas de 27 niños tratados con LMT por uno de nosotros (MD) desde el año 1999, que recibieron LMT en algún momento de su evolución. Los pacientes fueron seguidos por 6 años 5 meses en promedio (0,2-18). Todos tenían calendario de crisis. Todos estaban en comedicación y la LMT se agregó como coadyuvante.
La edad promedio al iniciar LMT correspondió a 6 años 1 mes (1-18). La LMT fue usada en promedio por de 3 años 7 meses (0- 8,9). Todos los pacientes salvo uno, tenían epilepsia resistente.
Promedio de crisis fue de 185 al mes por paciente (3-999) al iniciar la LMT. Veinte y cuatro (89%) pacientes presentaron comorbilidad neurológica de los cuales 17 (70%) tenían retardo mental de diversos grados
Resultados. En 5(18,5%) enfermos hubo una reduc-ción de crisis del 100%. En 16(59,3%), la disminu-ción fue del 51 al 99% y en 6(22,2%), las crisis solo disminuyeron del 0 al 50%.
En 4 (22,2) niños con crisis parciales hubo una reduc-ción del crisis del 100%, en 9 (50%) fue de 51 a 99% y en 5(27,8%) la reducción fue de 0 a 50%.
Sólo un niño (11,1%) con crisis generalizadas tuvo una reducción del 100% de las crisis, 7 (77,8%) y en 1 (11,1) la reducción fue de 0 a 50%.
Trabajos presentados a las VIII Jornadas Invernales de Epilepsia Crónica

64
Revista Chilena de Epilepsia Año 8, Nº 1, Diciembre de 2007
En 4 pacientes (14.8%) hubo que retirar el AE, en uno por aumento de la frecuencia de crisis en otro por tolerancia y en 2 por reacciones alérgicas
Conclusión. La lamotrigina resultó ser beneficiosa para este grupo de pacientes con crisis parciales y generalizadas resistentes puesto que la mayoría disminuyó la frecuencia de crisis en más del 50%. Dos niños presentaron efectos adversos.
9. BROMURO DE POTASIO EN EPILEPSIA RE-FRACTARIA SINTOMATICA: EXPERIENCIA CLINICA
LoretoRíos, Christian Galleguillos, Scarlet Witting, Alejandra Hernández, Carla Rojas Carmen Qui-jada, Ledia Troncoso.Servicio Neuropsiquiatría Infantil, Hospital Clínico San Borja Arriarán, Santiago, Chile.
Introducción. El Bromuro de potasio es el más an-tiguo agente antiepiléptico descrito en la literatura. Su indicación actual va dirigida al tratamiento de epilepsias en veterinaria. La discontinuación en su uso se debió a su marcada sedación, pérdida de la libido, ausencia de monitoreo sanguíneo confiable y la aparición de nuevos antiepilépticos. Se desconoce su mecanismo de acción, aunque se cree actuaría similar al mecanismo de acción gabaérgica.
Objetivo. Conocer la efectividad de bromuro de potasio como alternativa terapéutica en pacientes con epilepsia refractaria a tratamientos antiepilépticos convencionales (fármacos antiepilépticos clásicos y de nueva generación, dieta cetogénica), tratados en el servicio de neurología Infantil del Hospital San Borja Arriarán.
Pacientes y Método. 8 pacientes con diagnóstico de Epilepsia refractaria sintomática tratados con bromuro de potasio fueron estudiados.
Resultados. Edad entre 1 y 11 años, sexo 5 hom-bres y 3 mujeres. 2 pacientes con diagnóstico de malformaciones desarrollo cortical, 2 con parálisis cerebral secundaria a meningitis y asfixia, 4 con encefalopatía epiléptica de causa no precisada. El rango de dosis varió entre 40 a 90 mgrs/Kg/día. 3 pacientes resultaron con reducción cercana al 90% y 1 con disminución del 50%. La presencia de reacciones adversas fue infrecuente siendo sólo la somnolencia el efecto registrado en 2 de ellos e
hiperkalemia leve en otro.
Conclusiones. El bromuro de potasio puede ser una alternativa efectiva y de muy bajo costo a conside-rar en algunos pacientes con epilepsia refractaria cuando la terapéutica antiepiléptica habitual no tiene respuesta.
10. COSTO DIRECTO MEDICO DEL TRATA-MIENTO DE LAS EPILEPSIAS PARCIALES CON CRISIS PARCIALES COMPLEJAS EN NIÑOS
Juan Pablo Figueroa, Marcelo Devilat, Bolívar Va-lenzuela, Verónica Burón, Ricardo Erazo, Ximena Carrasco, Daniela Triviño, Verónica Gómez.Centro de Epilepsia Infantil. Servicio de Neurolo-gía y Psiquiatría. Hospital Luis Calvo Mackenna, Santiago. Chile.
Introducción. Existe escasa literatura sobre costos en epilepsia, a pesar de la importancia para la plani-ficación de acciones costo-efectivas
Objetivo. Cuantificar costos directos médicos en la epilepsia parcial con crisis parciales complejas en niños.
Pacientes y Método. 23 niños, 12 varones y 11 mujeres, se seleccionaron por muestreo aleatorio sencillo de un total de 210 pacientes atendidos entre 1997 y 2006 con el diagnóstico de epilepsia parcial con crisis parciales complejas (3 sintomáticas y 20 idiopáticas o criptogénicas), se excluyeron pacientes con epilepsia resistente a tratamiento con AE.
Los valores de las unidades de costos los proporcionó el centro de costos del hospital.
Los pacientes estuvieron bajo control en promedio 4,3 años (0,8 a 8,8), con 139,1 años acumulados. El tiempo promedio con antiepilépticos (AE) fue 3,7 años (0,8 a 8,75) y sin AE fue 0,62 años (0 a 3,17). Resultados. Consulta neuropediatra 6.778.460 pesos, consulta enfermera 32.000 pesos, consulta psicóloga 6.000 pesos, EEG 866.026 pesos, TAC cerebro 995.998 pesos, RM cerebro 1.120.000 pesos, hemograma 39.353 pesos, pruebas hepáticas 137.150 pesos, niveles plasmáticos AE 70.600 pesos, otros exámenes 44.000, costo AE 4.399.556 pesos.

65
Conclusiones. El costo directo médico total fue 14.489.143 pesos, el costo directo médico promedio por paciente fue 629.962 pesos y el costo médico directo mensual por paciente fue 12.352 pesos.
11. PROPUESTA DE ESCALA DE EVALUACION PARA PREVENCION DE OSTEOPOROSIS EN PACIENTES CON USO CRONICO DE ANTIEPILEPTICOS
Dra. Loreto Ríos, Dra. Loreto Llanos, Dr. Jorge Orellana*, Dra. Scarlet Witting, Dra. Alejandra Hernández, Dra. Carla Rojas, Dra. Carmen Qui-jada, Dra. Ledia TroncosoServicio de Neuropsiquiatría infantil Hospital San Borja Arriarán- Santiago*Servicio de Pediatría Hospital San Borja Arriarán-Santiago
Introducción.El uso crónico de antiepilépticos produce alteraciones del metabolismo óseo no considerado en la práctica habitual del manejo de la epilepsia.
Objetivos. Establecer protocolo de factores de riesgo a fin de prevenir osteoporosis en los niños en trata-miento antiepiléptico.
Material y Método. Con la finalidad de construir escala de evaluación del riesgo de osteoporosis en pacientes con uso crónico de antiepilépticos se recolecta información de 100 pacientes de nuestro servicio, analizando edad; sexo; peso; número, tipo y tiempo de uso de antiepilépticos; características de las crisis; actividad motora diaria; lugar de origen (exposición solar) y exámenes realizados para estudio de metabolismo óseo.
Resultados. Menos del 30% de los pacientes tenían estudio dirigido de la función ósea. Ninguno de los pacientes tenía realizado estudio con densitometría ósea.
Discusión. El tratamiento de la osteoporosis en pacientes con factores de riesgo como uso crónico de antiepilépticos y postración es difícil y poco efectivo. El uso de un protocolo de seguimiento en edad pediátrica es fundamental para la prevención de secuelas irreversibles.
Conclusiones. El monitoreo de metabolismo óseo no se realiza en pacientes en tratamiento crónico con antiepilépticos. Se propone protocolo de riesgo acce-
sible al sistema público a fin de iniciar profilaxis para alteraciones del metabolismo óseo. Considerando que la densitometría ósea es un examen no accesible económicamente al sistema público, se propone es-tudio prospectivo utilizando escala de puntuación de factores de riesgo que permitan seleccionar aquellos pacientes que deben iniciar tratamiento profiláctico para osteoporosis.
12. PRESENTACION DEL SINDROME CON-VULSIVO EN EL SERVICIO DE URGENCIA PEDIATRICO EN EL HOSPITAL SAN MAR-TIN DE QUILLOTA 2004-2005
Diego Barros, Daisy Pezoa, Dr. Yébenes, Juan Pezoa, Javier Arellano, Yessenia Orellana.Hospital San Martín de Quillota. Chile
Introducción. La epilepsia es una enfermedad de alta prevalencia, y su presentación en la edad pediátrica involucra no sólo el seguimiento y diagnóstico de la patología en sí, sino también el manejo multidisci-plinario del “impacto social” tanto en la familia del paciente como en el círculo social inserto.
Objetivos. Determinar la frecuencia y realizar una descripción de la presentación del Síndrome Con-vulsivo en el Servicio de Urgencia Pediátrico en el Hospital San Martín de Quillota durante el período 2004-2005.
Pacientes y Método. Revisión de todos los datos de urgencia pediátrica presentados durante el periodo descrito (67487) y el análisis de Ficha Médica. Se realiza base de datos en Microsoft Office Access 2003 para tabulación y análisis de datos para estudio descriptivo retrospectivo de estos pacientes.
Resultados. Del total de consultas N= 67487, un 3,27% (2212 pacientes) presentó dentro de sus diagnósticos el Síndrome Convulsivo. De los cuales un 41,9% (927) correspondía al sexo femenino y un 58,1% a varones. Las edades fluctuaron entre 4 meses y 12 años (ver tabla de distribución etárea). Sólo un 2,17% requirió de hospitalización (48) y el prome-dio de días de hospitalización de estos niños fue de 5,6. Cabe destacar que de la población estudiada un 31,4% (695) correspondía a población rural y esto es una sobreestimación del porcentaje de población rural atendida en este Servicio de Urgencia. Del tipo de Síndrome Convulsivo un 77,9% (1724) fueron Convulsión Febril (CF), un 10% (222) ECPC, un
Trabajos presentados a las VIII Jornadas Invernales de Epilepsia Crónica

66
Revista Chilena de Epilepsia Año 8, Nº 1, Diciembre de 2007
8,5% (189) no fue clasificado. Del tratamiento utili-zado el más frecuente fue el uso de Diazepam Rectal (DZR) con un 93,6% (2072).
Conclusiones. El Síndrome Convulsivo presenta una alta frecuencia en las consultas de Urgencia Pediátricas del HSMQ (3,27%), siendo su principal presentación la CF (77,9%), con una distribución etárea homogénea y el uso de DZR fue el tratamiento más frecuente, con un bajo porcentaje de hospitali-zación 2,17%.
13. EVIDENCIA DE RESPUESTA EEG ANOR-MALA FOTOESTIMULACION EN PACIEN-TES CON COMPROMISO DE CONCIEN-CIA.
Lucila Andrade, Gisela Kuester, Debora Pollak, Tomás Mesa, Julia Santin, Jaime Godoy. Departamento de Enfermedades Neurológicas y Departamento de Pediatría, Pontificia Universidad Católica de Chile. Santiago, Chile.
Objetivo. Reportar la existencia de respuestas EEG anormales a fotoestimulación (FE) en pacientes no seleccionados, niños y adultos, con compromiso de conciencia, referidos para EEG portátil.
Material y Método. Se revisaron retrospectiva-mente los EEG portátiles consecutivos efectuados en pacientes del Hospital Clínico Pontificia Univer-sidad Católica de Chile entre Enero 2004-Diciembre 2006. Analizamos demografía; distribución/morfo-logía/clínica de las respuestas a FE; rasgos del EEG basal y presencia de actividad epileptiforme interic-tal/ictal no asociada a FE.
Resultados. Se incluyeron siete pacientes y 11 EEG. La edad promedio fue 42 años (rango: 2-88) y hubo cinco mujeres. Siete EEG mostraron RFP no asociada a cambio clínico, la que fue focal, bifocal o multifocal, todos ellos con patrón autolimitado y autosostenido en un mismo registro. En seis EEG hubo respuesta fótica anormal electroclínica, todas de origen focal, pero sólo dos con inicio occipital.
Conclusiones. La fotosensibilidad se asocia tradi-cionalmente a epilepsias idiopáticas generalizadas y occipitales. En la literatura hay reportes aislados de actividad epileptiforme inducida por FE en pacientes con alteración de conciencia. A nuestro entender, esta es la primera descripción de una serie de pa-
cientes con compromiso de conciencia cuyo estudio EEG mostró respuesta anormal a fotoestimulación, hallazgo que podría tener eventuales implicancias fisiopatológicas y terapéuticas.
14. CARACTERISTICAS DE LOS EEG DE LA UNIDAD DE ELECTROENCEFALOGRA-FIA. SERVICIO DE PEDIATRIA HOSPITAL EXEQUIEL GONZALEZ CORTES. AÑO 2006.
Maritza Carvajal, Juan Enrique González, Cristina Hidalgo.
Introducción. Con la implementación del Programa de Epilepsia el año 2001, se crea la necesidad de priorizar la solicitud del EEG en nuestro hospital por los médicos tratantes neurólogos, contando con la adquisición del equipo Digital con Video-Monitoreo de 32 canales a fines del año 2005.
Objetivos. Dar a conocer las características de los EEG realizados desde Enero hasta Diciembre del año 2006, en virtud del tipo de examen, procedencia, distribución etaria y resultados del examen.
Material y Método. Estudio Descriptivo Observa-cional Prospectivo. Se obtuvieron los datos de tabla EXCEL previamente diseñada, donde se registraron los datos durante el año 2006. Se analizaron estadís-ticamente los resultados.
Los registros EEG se realizaron en 1 equipo Digital de 32 canales y dos equipos análogos de 8 canales uno de ellos en UCI y otro en unidad de EEG. Fueron realizados en sistema internacional 10-20, con electrodos en cuero cabelludo, entre Enero y Diciembre del 2006 e informados en su mayoría por un solo médico.
Resultados. Durante el año 2006 se realizaron 1874 Electroencefalogramas. El 63.6% fueron realizados en formato digital y el 36.6% se realizó en formato análogo. El 78.4% se realizó en forma ambulatoria y el 21.6% a pacientes hospitalizados. Se realizaron 35 Video-Monitoreo para diagnóstico de crisis. Sólo el 4.4% de los exámenes fueron realizados con inducción de sueño. Se observó igual distribución por sexos. La distribución etaria correspondió a: Lactantes (28d a 2ª) 37.4%, Escolares (5ª a 11 a) 24.5%, Preescolares 15.7%, Adolescentes (>12ª)

67
17.6% y Recién Nacidos a 4.9%. Los exámenes fueron solicitados en su mayoría (75.4%) por el departamento de Neuropediatría, seguidos por UCI (7.1%), Intermedio (4%), Lactantes (3.6%), Neo-natología Hospital Barros Luco (2.4%), Segunda Infancia (2%), Psiquiatría CDT Hospital Barros Luco (1%), Servicio de Urgencia (0.6%) y Onco-logía (0.4%).Del total de EEG realizaron un 52.2% fue informado como normal y un 47.8% presentó alguna alteración, de las cuales destaca en orden de frecuencia: Actividad Epileptiforme Focal (17.4%), Actividad Epileptiforme Generalizada (17%), Alte-ración difusa (5.2%), Actividad Multifocal (1.2%), Alteración inespecífica (2.8%), Muerte Cerebral (0.7%) e Hipsarritmia (0.5%).
Discusión. En la actualidad la gran mayoría de los exámenes se está realizando en formato digital, por sus ventajas al momento del análisis en relación al formato análogo. El 47.8% presentó anormalidades, lo cual es concordante con la información existente en la literatura. Cabe destacar que sólo un 4.4% de nuestros pacientes recibieron inducción de sueño con Hidrato de Cloral. En el 75% de nuestros pacientes el examen fue indicado por un médico Neurólogo o una Enfermera altamente capacitada en Epilepsia. Nuestra casuística es concordante con lo publicado en la literatura.
15. ANALISIS DE LA RESPUESTA FOTOPA-ROXISTICA.
Mariana Weitzman, Erna Rauch , Mª Eugenia López, Jorge Lasso.Hospital Padre Hurtado
Introducción. La respuesta fotoparoxística (RFP) se manifiesta con ondas cerebrales de tipo punta o espiga onda, o como lentitudes en rango theta generalizada de predominio anterior o posterior, o puede ser focal. Su relación con epilepsia está muy determinada en ciertos síndromes.
Objetivos. Describir la evolución clínica de pacien-tes con resultado en electroencefalograma de RFP.
Material y Método. Estudio descriptivo, revisión de ficha clínica y registro EEG.
Resultados. De 6.500 electroencefalogramas, se encontraron 53 exámenes con RFP, el promedio de edad fue 23 años (4 a 57 a). Un 73% (38 pacientes)
tuvo diagnóstico de epilepsia (70% de tipo genera-lizado, 22% focal y un 8% indeterminado). De los síndromes generalizados la mayoría era EMJ 51%, seguido de epilepsia ausencia 13%, en cambio en los focales, la mayoría se presentó en la infancia, la epilepsia rolándica fue la más frecuente. Al analizar el tipo de RFP la mayoría fue limitada al estímulo 72%, seguida de la respuesta autosostenida 25%. El grupo sin epilepsia (27%) tuvo diagnóstico de cefalea 32% y crisis convulsiva febril 22%.
Conclusión. La RFP fue un hallazgo poco frecuente en el registro EEG, pero se asoció en alto porcentaje con epilepsia, principalmente formas generalizadas idiopáticas.
16. CLASIFICACION DE LOS PATRONES DE RESPUESTA EEG A FOTOESTIMULACION EN PACIENTES AMBULATORIOS.
Lucila Andrade, Debora Pollak, Gisela Kuester, Jaime Godoy, Julia Santin, Tomás Mesa. Departamento de Enfermedades Neurológicas y Departamento de Pediatría, Pontificia Universidad Católica de Chile. Santiago, Chile.
Objetivos. Describir tipos de respuesta fotopa-roxística (RFP) y crisis epilépticas (CE) por fo-toestimulación (FE) en pacientes no seleccionados, niños/adultos, ambulatorios, referidos para estudio EEG. Correlacionar hallazgos con clasificaciones propuestas en la literatura.
Material y Método. Se revisaron retrospectivamen-te los EEG consecutivos efectuados en Laboratorio EEG Pontificia Universidad Católica de Chile entre Enero 2004-Abril 2007. Analizamos demografía, distribución/morfología de descargas epilépticas (DE) por FE, características del EEG basal vigilia/sueño y de las DE interictales/ictales independien-tes de FE. Se intentó agrupar los distintos tipos de RFP según clasificaciones de “Jeavons y Harding”,” Waltz” y “Kasteleijn-Nolst Trenité et. al”.
Resultados. Se incluyeron 33 pacientes y 39 EEG. Edad promedio: 13.6 años (rango: 3-42), 61% mu-jeres. En 62% de los EEG la RFP tuvo morfología variable en un mismo registro, incluyendo DE focales y generalizadas. Sólo en 21% de los EEG se pudo definir RFP como estrictamente focal, focal con ge-neralización o generalizada. Las CE por FE fueron mayoritariamente de origen focal. Las tres clasifica-
Trabajos presentados a las VIII Jornadas Invernales de Epilepsia Crónica

68
Revista Chilena de Epilepsia Año 8, Nº 1, Diciembre de 2007
ciones de respuestas EEG a FE fueron insuficientes a la luz de nuestros hallazgos.
Conclusiones. Existen variados modelos de RFP en-tre pacientes referidos para EEG ambulatorio que van más allá de los propuestos en la literatura, impidiendo adecuadas descripción y correlación clínica.
17. UTILIDAD DEL REGISTRO POLIGRAFICO EN EPILEPSIA.
Dra. Loreto Ríos, Dra. Francisca López, Dra. Scarlet Witting, Dra. Carla Rojas, Dra. Carmen Quijada, Dra. Ledia Troncoso.Servicio de Neuropsiquiatría. Hospital Clínico San Borja Arriarán (HCSBA), Santiago, Chile.
Introducción. En los últimos años, el videomo-nitoreoEEG ha sido un aporte valioso para lograr una aproximación diagnóstica más precisa de los pacientes con epilepsia. Sin embargo , recientemente la incorporación del registro simultáneo de EEG y EMG a través de la poligrafía, ha permitido preci-sar el origen cortical o extracortical de la actividad motora observada y diferenciar el tipo de paroxismo observado por ejemplo, mioclonía, clonía, ausencia mioclónica, espasmo.
Material y Método. Presentamos tres pacientes con-trolados en el Servicio de Neuropsiquiatría Infantil del HCSBA, en quienes el registro poligráfico fue de importancia precisión diagnóstica.
Resultados. Se describen tres pacientes, dos con sospecha de Epilepsia Parcial Continua y uno con Epilepsia Frontal de difícil manejo. En los dos primeros se pudo descartar el diagnóstico en base a los hallazgos del registro. En el tercer caso, se pudo determinar con certeza ausencia de epilepsia y presencia de pseudocrisis.
Conclusión. El registro poligráfico simultáneo EEG-EMG puede ser de suma utilidad en el diagnóstico y por lo tanto manejo de los pacientes con epilepsia.
18. EPILEPSIA PARCIAL COMPLEJA Y ALTERA-CION METABOLISMO ACIDOS GRASOS DE CADENA MEDIA ¿EFECTO SECUNDARIO DE ACIDO VALPROICO?
Camilo Zapata, Jorge Díaz. Hospital Las Higueras de Talcahuano
Introducción. El Acido Valproico puede producir excreción de Octanoil-carnitina (C8) simulando el perfil de acilcarnitina encontrado en el déficit de MCAD, patología que habitualmente debuta como hipoglicemia hipocetósica en lactantes. Presentamos un caso en que se planteó déficit de MCAD atípico el cual se descarta por estudio genético pero plantea el diagnóstico diferencial de esta patología y el uso de dosis altas de Valproato.
Caso Clínico. Se presenta caso de escolar de 13 años sin antecedentes de asfixia, hijo de padres sa-nos no consanguíneos, que presenta Crisis parciales complejas repetidas desde los 7 años, EEG muestra actividad irritativa epileptiforme temporal izquierda, TAC y RNM cerebral normal, se inicia Acido Val-proico en dosis crecientes hasta dosis máxima de 1500 mg /d, por el difícil control de crisis (permanece con 2 a 3 crisis mensuales), se agrega Carbamazepi-na 30 mg/k/día y Clonazepam 1 mg/día con lo que actualmente lleva 6 meses sin crisis.
Desde los 10 años paciente presenta episodios de letargia, debilidad y al menos un episodio de hipo-glicemia (el que requirió uso de solución Glucosada ev) en relación al ayuno, por lo que se le indica alimentación fraccionada e ingesta de Hidratos de carbono nocturna y L-Carnitina. Dentro de su estudio se realiza Espectrometría de masa en Tándem (EMT) la que muestra categórica elevación de Octanoil carnitina (0,44 mmol/l) resultado que se confirma con EMT realizada en USA (IVX), lo que plantea alteración del metabolismo de Acidos Grasos de cadena media atípico.
Se realiza Estudio de Mutación de enzima deshidro-genada de AGCM, no encontrándose mutaciones 985A>G y 199T>C. Otros exámenes muestran Amonio y Acido Láctico Normal, carnitina total disminuida con aumento de carnitina esterificada, función hepática normal, estudio aminoacidopatías (-). Se evalúa exámenes por expertos en enfermeda-des metabólicas de Pediatrix (USA) quienes sugieren que las alteraciones del metabolismo de los AGCM son consecuencia del uso de altas dosis de Acido Valproico.
Se presenta este caso por presentar alteración del metabolismo de los ácidos Grasos en forma sinto-mática y adquirida (no hay síntomas sugerentes de esta patología en el período de lactante y preescolar) y sería un efecto adverso poco conocido del Acido

69
Valproico.
19. CASO CLINICO: DESCRIPCION DEL SIN-DROME CONVULSIVO EN PACIENTE CON SINDROME DE HUNTER
Diego Barros, Daisy Pezoa, Teresa Román, Juan Pezoa, Javier Arellano y Yessenia Orellana.Hospital El Pino. Santiago, Chile
Introducción. La Mucopolisacaridosis tipo II o el síndrome de Hunter es heredado como enfermedad ligada al cromosoma X. La anomalía metabólica que causa el síndrome es la falta de la enzima sulfatasa de sulfoiodurodinato y al estar esta enzima ausente los mucopolisacáridos se acumulan en varios tejidos causando el daño.
Objetivos. Describir los episodios convulsivos en una patología poco frecuente como el Síndrome de Hunter.
Pacientes y Método. Análisis de la Ficha Médica. Estudio Caso-Control.
Resultados. El paciente corresponde a un varón de 10 años con familiares portadores de la misma patología controlados en este Servicio. El Síndrome de Hunter
es una patología que se puede presentar con la si-guiente clínica: rasgos faciales toscos, cabeza grande, nariz corta y ancha (presentando una rinorrea mucosa crónica, afecciones de oído y senos paranasales en forma crónica, cara plana con pómulos rojos, cuello corto, macroglosia, rigidez de las articulaciones, sordera, hipertricosis, hepato y esplenomegalia que empuja al diafragma hacia arriba disminuyendo su expansibilidad torácica y aumentando los problemas respiratorios, retina anormal, manos cortas anchas y con los dedos gordos, síndrome del túnel carpia-no, vientre muy abultado y herniopatía frecuente, además de una piel muy gruesa. El paciente había presentado durante su vida 6 episodios convulsivos (3 durante el último año de control) de los cuales, el primero de estos fue una Convulsión Febril a 39,5° C por una Pansinusitis, el segundo interpretado como tónico-clónico generalizado y el último como focalizado. El paciente actualmente se encuentra en estudio de Epilepsia, sin medicación específica y con deterioro de su condición neurológica por su patología de base.
Conclusión: En relación al Síndrome de Hunter no existe descripción en la literatura sobre una relación directa, incidencia y/o tipo de presentación de la Epilepsia, razón por la cual se decide la presentación del Caso Clínico.
Trabajos presentados a las VIII Jornadas Invernales de Epilepsia / Congresos y Cursos 2008 Crónica
Congresos y Cursos 2008Crónica
CONGRESO SOPNIAXXVI Congreso Anual de SOPNIA15 al 18 de Octubre de 2008Pucón, Chilewww.sopnia.com
CONGRESO SONEPSYN63º Congreso Anual Chileno 2008Neurología, Psiquiatría y NeurocirugiaHotel del Mar, Viña del Mar30, 31 de Octubre y 1 de Noviembre de 2008www.sonepsyn.com

70
Revista Chilena de Epilepsia Año 8, Nº 1, Diciembre de 2007
Sugerencias para las contribuciones a los autores
Las contribuciones podrán tener la forma de traba-jos originales de investigación clínica o experimen-tal, de medicina social y salud pública relacionadas con las epilepsias, revisiones de temas, casos clíni-cos, crónica y cartas al editor. Las colaboraciones deberán ser enviadas a la secre-taría de la Sociedad Chilena de Epilepsia y revisa-das por el Comité Editorial. Los artículos se entregarán mecanografiados en pa-pel tamaño carta con doble espacio, con un máximo de 26 líneas por página, con un margen de 2.5 cm en todos sus bordes, escritos con letra Arial Nivel 12. La extensión máxima para los artículos originales y de revisión será de 16 páginas, de 8 para los casos clínicos y de 3 para los artículos de crónica y cartas al editor. Se incluirá un original con dos fotocopias y un impreso en disquete de 3.5 (90 mm) utilizando programa Word Perfect o Word para PC. Se aceptarán figuras (dibujos y gráficos) enviados en forma de copia fotográfica en papel satinado blanco y negro de 10 x 15 cm. La lectura de las fi-guras se hará en hoja separada. En el dorso de cada figura se marcará el número que la identifica y una flecha con su orientación con lápiz de carbón. En el texto se indicará dónde debe ser intercalada. Las tablas (cuadros o tablas) se enviarán mecano-grafiados y numerados según orden de aparición en el texto, en el cual se señalará su ubicación. Se aceptará un máximo de 5 elementos (figuras o tablas) por artículo. El título deberá ser claro y conciso. Se incluirá el nombre de los autores con el primer apellido, el títu-lo profesional de cada uno de ellos y el lugar donde se realizó el trabajo. Las referencias bibliográficas deben limitarse a un máximo de 15. Se sugiere refe-rir y citar bibliografía latinoamericana y chilena y al terminar mencionar el e-mail del autor principal.
Clasificación de las contribuciones:1. Trabajo original. Realizado según el siguiente
esquema: a) Introducción, donde se plantea la si-tuación general del problema b) Objetivos, don-de se plantean los antecedentes y los problemas que se quiere resolver. c) Material o Pacientes y Métodos, en el que se hacen explícitas las carac-terísticas del universo y cómo se instrumentali-zó. d) Resultados, donde se expone la situación obtenida. e)Discusión, en la que se comentan los resultados con relación a los problemas plantea-dos o a la información proporcionada por otros autores. f) Resumen de 200 palabras en español o inglés.
2. Trabajos de revisión. Se trata de una revisión bibliográfica acerca de un tema específico, pre-sentado según las instrucciones de longitud y referencias bibliográficas ya señaladas.
3. Casos clínicos. Presentación de casos de interés práctico, según el esquema de trabajo original.
4. Actualidades, revisión de capítulos de interés especial, realizadas por profesionales que ten-gan experiencia en el tema y contribuyan a cla-rificar conceptos.
5. Crónica. Espacio destinado a noticias de interés en el campo de la clínica, neurofisiología, imá-genes, Salud Pública o administración. Presen-tación según instrucciones detalladas más arri-ba.
6. Cartas al editor, cuyo objetivo es ser una tribuna abierta de la Revista a sus lectores.
Presentación de las referencias bibliográficasDeben enumerarse en el texto en forma consecuti-va, en el mismo orden en que aparecen citadas por primera vez y acompañarse la lista total de ellas. En caso de haber más de 5 autores, se colocará la pala-bra “et al” para incluir los restantes. Cada referen-cia de revista debe anotarse en el orden siguiente: Apellido paterno del autor con la primera inicial del nombre; título del trabajo; revista en que aparece el artículo según “Index Medicus”, año, volumen, página inicial y final del texto. Las referencias de libros se anotarán así: título del libro, ciudad en que fue publicado, editorial, año. Se usarán comas para separar a los autores entre si.Ejemplos:Pérez J, Santos G. Serotonina humana. Rev Med Chile 1967; 45:12-14.
Crónica


















