







Idiomas
Páginas
Jurídico


Dr. Pablo Christian Barvosa
Médico Pediatra. Médico Principal del área ambulatoria del
Hospital Garrahan. Secretario del Grupo de enfermedades
poco frecuentes de la Sociedad Argentina de Pediatría.
Congreso Nacional de Pediatría 2015 - Mendoza

Detección de individuos PRESUNTAMENTE enfermos en una población
PRESUNTAMENTE sana.
• Objetivo: Detectar enfermedades o desórdenes en los recién nacidos:
cuyos síntomas clínicos no se hacen evidentes hasta que el daño irreversible ha ocurrido.
para las cuales están disponibles tratamientos.

• Métodos simples y prácticos para seleccionar individuos sospechosos.
• NO certifican diagnóstico.
• Pruebas diagnósticas confirmatorias.
• Si el RN presenta signos o síntomas de enfermedad requiere de una apropiada evaluación, aún cuando el resultado de la pesquisa fuera negativa.

Programa preventivo de gran importancia
en la Salud Pública
Según la AAP comprende: Educación de padres y pediatras sobre la pesquisa
Realización rápida y confiable del test
Recolección y trasporte confiable de la muestra
Pronta ubicación y seguimiento individual con test anormal
Diagnóstico de certeza con pruebas confirmatorias
Educación, consejo genético y apoyo psicológico de las familias de niños afectados
Manejo y tratamiento de los pacientes
Evaluación sistemática de la evolución

CRITERIOS DE SELECCIÓN DE LAS
ENFERMEDADES PARA REALIZAR PESQUISA
NEONATAL MASIVA
ENFERMEDAD
• Severidad y frecuencia
• Tratamiento satisfactorio
• Máxima efectividad de tratamiento en la etapa neonatal
• Posibilidad de control y tratamiento de los afectados
• Posibilidad de centralizar los datos
MÉTODO
• Obtención sencilla
• Fácil trasporte
• Fácil conservación y estabilidad de la muestra
• ALTA SENSIBILIDAD (baja tasa de falsos negativos)
• Costo razonable

¿Qué buscamos?
Fenilcetonuria Hipotiroidismo
Fibrosis quística
Galactosemia
Hiperplasia suprarrenal congénita
Déficit de biotinidasa

MARCO LEGAL • LEY 23.413 (1986) Obligatoriedad de realizar la pesquisa neonatal de
fenilcetonuria.
• LEY 23.874 (1990) Agrega la detección de hipotiroidismo congénito.
• DECRETO 1316 (1994) Reglamenta las leyes anteriores. Incorpora el plazo de
realización de las determinaciones y los responsables de la pesquisa.
• LEY 24438 (1994) Agrega la detección de fibrosis quística.

REGISTRO DE DATOS
IMPORTANTE: Para localizar a la familia en caso de resultados anormales, para
confirmar el diagnóstico e iniciar tratamiento rápidamente.

TOMA DE MUESTRA
A todos los RN vivos entre las 48 hs y el 5° día de vida (previo al alta
neonatal), 24 horas después de que el niño haya comenzado a alimentarse.
SITUACIONES ESPECIALES
Si el RN requiere plasmaféresis o transfusión, se debe tomar
la muestra previo al procedimiento o luego del 7° día de realizado.
PREMATUROS con EG < 35 S
• Se realiza la pesquisa y se repite a las 37 semanas
RN CON BAJO PESO < 1.500 G
• Se realiza la pesquisa y se repite cada 15 días hasta que alcance los 2.000 g
TRATAMIENTOS Clotrimozaxol y procaína benzilpenicilina interfieren con la determinación de biotinidasa.

TÉCNICA
Conservarse en lugar seco y protegido de la luz.
Dejar secar al aire 3 hs en posición horizontal.
Asegurarse que la sangre haya traspasado al reverso de la tarjeta. Colocar una sola gota de sangre por circulo.
Se deja que se forme espontáneamente la primer gota de sangre, que se retira con una gasa estéril, la segunda gota se coloca en el papel de filtro.
Punción con lanceta estéril.
Desinfección con alcohol, dejar secar.
La sangre se obtiene por punción del talón.


CONFIRMACIÓN DE RESULTADOS

HIPOTIROIDISMO CONGÉNITO HIPOTIROIDISMO CONGÉNITO PRIMARIO
evitar el retraso mental
El hipotiroidismo primario es la causa más frecuente de hipotiroidismo congénito, puede deberse a:
85% esporádicos vs 15% hereditarios
Mas frecuente en mujeres (2:1)
Incidencia: 1/2.500 a 3.000 RNV
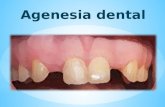
Agenesia Hipoplasia Ectopia Dishormonogénesis

MANIFESTACIONES CLÍNICAS
La mayoría asintomáticos.
5% síntomas clínicos: fontanela posterior amplia, ictericia
prolongada, hipotonía, constipación, somnolencia,
macroglosia.
MÉTODO DE PESQUISA: determinación de TSH.

FALSOS POSITIVOS FALSOS NEGATIVOS
Muestra precoz
Exposición materna a medicación
antitiroidea
RNPT < 35 Sem
Déficit de globulina ligadora de T4
Hipotiroidismo secundario o
terciario
Error de laboratorio

CONFIRMACIÓN DIAGNÓSTICA
• DOSAJE DE TSH Y T4.
• Acs antitiroideos y tiroglobulina.
• Ecografía tiroidea.
• Centellograma tiroideo.
• EVALUACION POR ENDOCRINÓLOGO.
INICIAR TRATAMIENTO INMEDIATAMENTE
luego de extraerse la muestra para la confirmación diagnóstica
(levotiroxina 10-15 ug/kg/dia)

FENILCETONURIA Deficiencia genética autosómica recesiva
Bloqueo de la conversión de la fenilalanina en tirosina.
Causas:
Déficit primario de la enzima fenilalanina hidroxilasa (98%)
Deficiencia en la síntesis o reciclaje de su cofactor
Incidencia de 1/10.000 RN (portadores sanos 1 cada 50 RN)

MANIFESTACIONES CLÍNICAS
(según el grado de actividad residual de la enzima)
Retraso mental severo
Convulsiones
Autismo
Hiperactividad
Microcefalia
MÉTODO DE PEQUISA:
detección de fenilalanina en sangre.

FALSOS POSITIVOS FALSOS NEGATIVOS
Alimentación parenteral
hipertirosinemia
Extracción precoz de la muestra
Prematuros
exsanguineotranfusión

CONFIRMACIÓN DIAGNÓSTICA
• Dosaje cuantitativo de fenilalanina y tirosina.
• Determinación genética de las mutaciones más frecuentes.
TRATAMIENTO
• Dieta restringida en fenilalanina.

FIBROSIS QUÍSTICA Enfermedad autosómica recesiva (7q31, mutación delta
F508)
Provoca insuficiencia pancreática exógena (y
posteriormente endógena) con síndrome malabsortivo,
infecciones respiratorias crónicas y una elevada
concentración de cloruros y sodio en el sudor.
Incidencia 1/7.600 RN (portadores sanos 1 cada 30)

MANIFESTACIONES CLÍNICAS
Ileo meconial, ictericia prolongada, síndrome malabsortivo
(por déficit de tripsina), prolapso rectal, enfermedad
hepática (colangitis esclerosante, cirrosis), diabetes.
Retraso del crecimiento.
Enfermedad respiratoria, infecciones frecuentes,
afectación de senos paranasales.
MÉTODO DE PESQUISA:
determinación de tripsina
inmunorreactiva.

Falsos positivos Falsos negativos
Obstruccion intestinal
Agenesia de conductos
pancreaticos
Hipoxia y mala perfusion del
pancreas
Ileo meconial

CONFIRMACIÓN DIAGNÓSTICA
• Segunda muestra para TIR.
• TEST DEL SUDOR Y ESTUDIO GENÉTICO.
TRATAMIENTO
• Prevención y tratamiento de la enfermedad respiratoria,
del déficit nutricional y otras manifestaciones o
complicaciones.

GALACTOSEMIA Error congénito del metabolismo de la galactosa
debido a la deficiencia de diferentes enzimas. Deficit de Galactosa 1-6 difosfato uridil(+ frec, Incidencia
1/17.000). Enfermedad grave de inicio temprano, autosómica recesiva. Se acumula galactosa 1-fosfato causando lesiones renales, hepáticas y en SNC (GALT).
Déficit de galactoepimerasa (condiciona cataratas sin enfermedad hepática) (GALE)
Déficit de galactoquinasa (GALK)
MÉTODO DE PESQUISA: determinación de galactosa en sangre.

Falso positivo Falso negativo
Muestra expuesta al calor o
humedad Transfusiones

CONFIRMACIÓN DIAGNÓSTICA
• Determinación de las diferentes enzimas.
• Estudio genético.
TRATAMIENTO
• Suspensión de la lactancia materna y dieta sin lactosa.

HIPERPLASIA SUPRARRENAL
CONGÉNITA (HSC)
• Enfermedad autosómica recesiva
• Prevalencia 1/12.000
• Causa
– 95% déficit de la enzima 21 hidroxilasa
• Disminución de aldosterona
• Disminución de cortisol aumento de ACTH
hiperplasia SR
• Exceso de 17-OH-progesterona y
androstenediona.


• FORMA CLÁSICA
• FORMA NO CLÁSICA (poco frecuente)
PRESENTACIÓN PRECOZ
• FORMA PERDEDORA DE SAL (75%) Crisis entre los 7 y 14 días de vida. Pérdida de peso, vómitos, letargia, deshidratación, hipoNa+, hiperK+.
• FORMA NO PERDEDORA DE SAL (25%) Virilización, sin alteración de los electrolitos.
PRESENTACIÓN TARDÍA
• Aumento de la velocidad de crecimiento con aceleración de la maduración ósea y signos de pubertad precoz.
MÉTODO DE PESQUISA:
determinación de 17-OH progesterona.

CONFIRMACIÓN DIAGNÓSTICA
• 17-OHP en suero.
• Ionograma.
• Estudio genético.
TRATAMIENTO
• Reemplazo hormonal (corticoide).
• Inhibidores de andrógenos.
• Corrección quirúrgica estética de niñas con virilización.
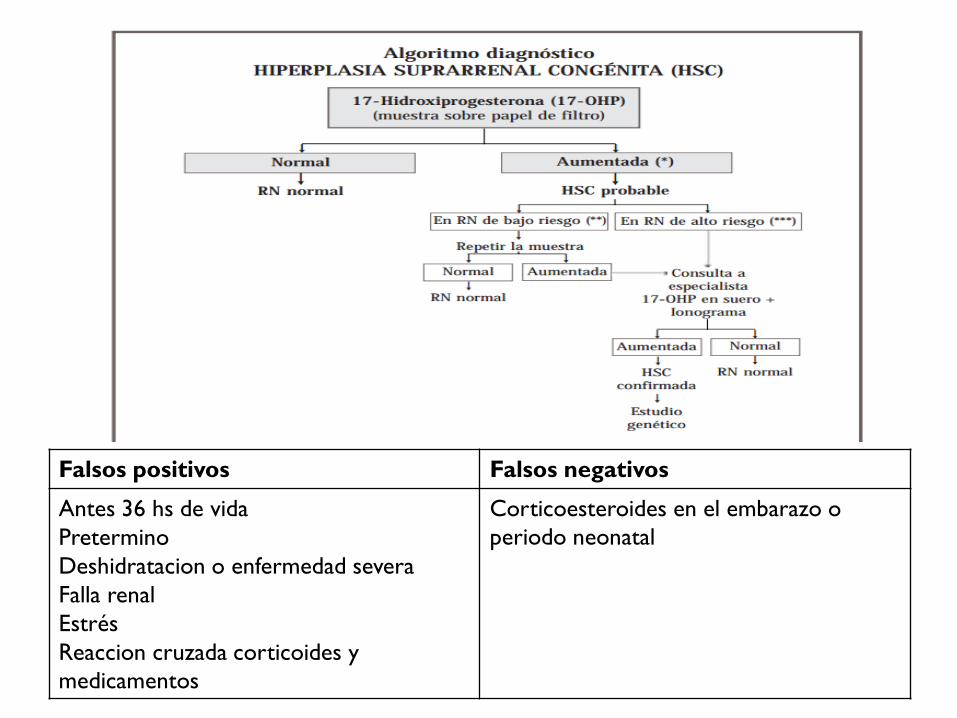
Falsos positivos Falsos negativos
Antes 36 hs de vida
Pretermino
Deshidratacion o enfermedad severa
Falla renal
Estrés
Reaccion cruzada corticoides y
medicamentos
Corticoesteroides en el embarazo o
periodo neonatal

DEFICIENCIA DE BIOTINIDASA Autosómico recesivo.
Deficiencia de la enzima biotinidasa encargada de la recuperación de la Biotina (cofactor de todas las carboxilasas del organismo)
Prevalencia 1/45.000-60.000

MANIFESTACIONES CLÍNICAS
Síntomas neurocutáneos: convulsiones, pérdida de
audición y visión, ataxia, hipotonía, dermatitis seborreica.
Retraso del crecimiento, hiperventilación , apneas,
alopecia, acidemia, cetoacidosis metabólica,
hiperamonemia.
MÉTODO DE PESQUISA:
determinación de biotinidasa en sangre.

Falsos positivos
Exposicion al calor o humedad
RN pretermino

CONFIRMACIÓN DIAGNÓSTICA
• Actividad enzimática.
• Análisis genético.
TRATAMIENTO • Biotina 5-20 mg.

EPF en cifras
La PARADOJA de las EPF
• Son entre 5.000 y 8.000 tipos de enfermedades representa 6-8% de la población total.
• En Europa se calculan 30 millones de personas afectadas.
• En Argentina según cálculos se estiman
3 millones de personas afectadas, que en la mayoría comienza en la infancia
(80% de etiología genética) Pocos Pacientes
Pocas Enfermedades
Poco impacto en Salud Pública PREGUNTAS COMUNES
¿Qué enfermedad tiene?
¿Dónde busco información?
¿Qué hago con este paciente?
¿Hay algún profesional que atienda esta enfermedad en mi provincia?
¿Requiere algún estudio de genética para confirmar la enfermedad?
¿Hay en el país algún grupo de profesionales que siga estos pacientes?
¿Cómo contacto a estos equipos?
DEFINICION según Ley Nacional 26689
Prevalencia en la población es igual o inferior a 1 en 2000 personas.


Enfermedades Poco Frecuentes
2011 – Promulgo la Ley de Enfermedades Poco Frecuentes: Ley 26. 689 de Asistencia Integral a Personas con enfermedades poco frecuentes.
2012 – Área de Enfermedades Poco Frecuentes
2014 – Creación del Programa Nacional de Enfermedades Poco Frecuentes
(Programa de Genética + Área de Hormona de Crecimiento + EPF)
2015 – Reglamentación de la Ley Nacional 26689

Enfermedades Poco Frecuentes
2015
- Creación de un Consejo Consultivo, incluyendo ONG
- Ciclo de Conferencias de EPF por Cibersalud, 270 lugares
- Armado de un recursero de profesionales y laboratorios que atienden EPF
- Genetistas itinerantes (La Pampa, Chubut y Santiago del Estero)
- Impulso de becas de Investigación
- Trabajo conjunto con el Servicio Nacional de Rehabilitacion para registro de
EPF.

Top Related