
Angio Displa Si As
47
UNIVERSIDAD CENTRAL DEL ECUADOR. FACULTAD DE CIENCIAS MÉDICAS. CARRERA DE MEDICINA. CIRUGÍA VASCULAR. ANGIODISPLASIAS. ALISON PAULINA MENA L. MELISSA DAYANA MENA C. Décimo semestre
-
Upload
carolina-salguero -
Category
Documents
-
view
34 -
download
1
Transcript of Angio Displa Si As

UNIVERSIDAD CENTRAL DEL ECUADOR.FACULTAD DE CIENCIAS MÉDICAS.
CARRERA DE MEDICINA.CIRUGÍA VASCULAR.
ANGIODISPLASIAS.
ALISON PAULINA MENA L.MELISSA DAYANA MENA C.
Décimo semestre
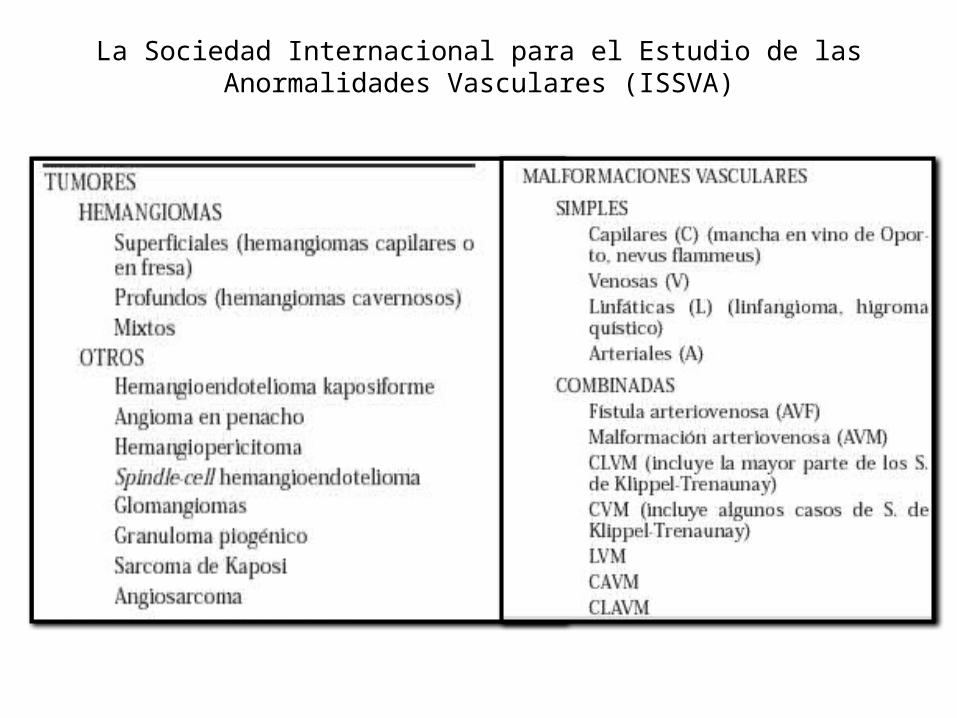
La Sociedad Internacional para el Estudio de las Anormalidades Vasculares (ISSVA)

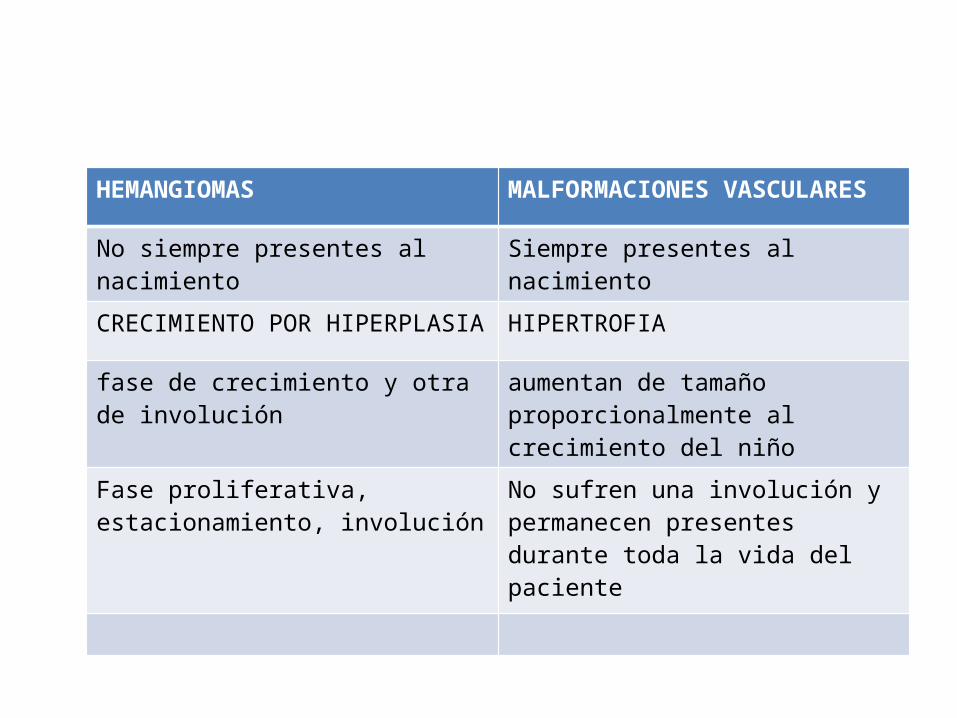
HEMANGIOMAS MALFORMACIONES VASCULARES
No siempre presentes al nacimiento Siempre presentes al nacimiento
CRECIMIENTO POR HIPERPLASIA HIPERTROFIA
fase de crecimiento y otra de involución aumentan de tamaño proporcionalmente al crecimiento del niño
Fase proliferativa, estacionamiento, involución
No sufren una involución y permanecen presentes durante toda la vida del paciente

HEMANGIOMAS
tumores benignos de las células endoteliales
Tumor vascular más común
Al nacer, las lesiones son a menudo pequeñas y discretas,60% ausente en el nacimiento
20% bilaterales
Localización : cabeza y cuello 60%Tórax:25 %
Extremidades:15% máculas telangiectáticas
rodeadas de un halo pálido, una mácula pálida, una mácula eritematosa o con menos frecuencia semejando una
contusión o una escoriación
1 -10nacimientos caucásicos, 1 -71 nacimientosafroamericanos y 1-125 nacimientosasiáticos

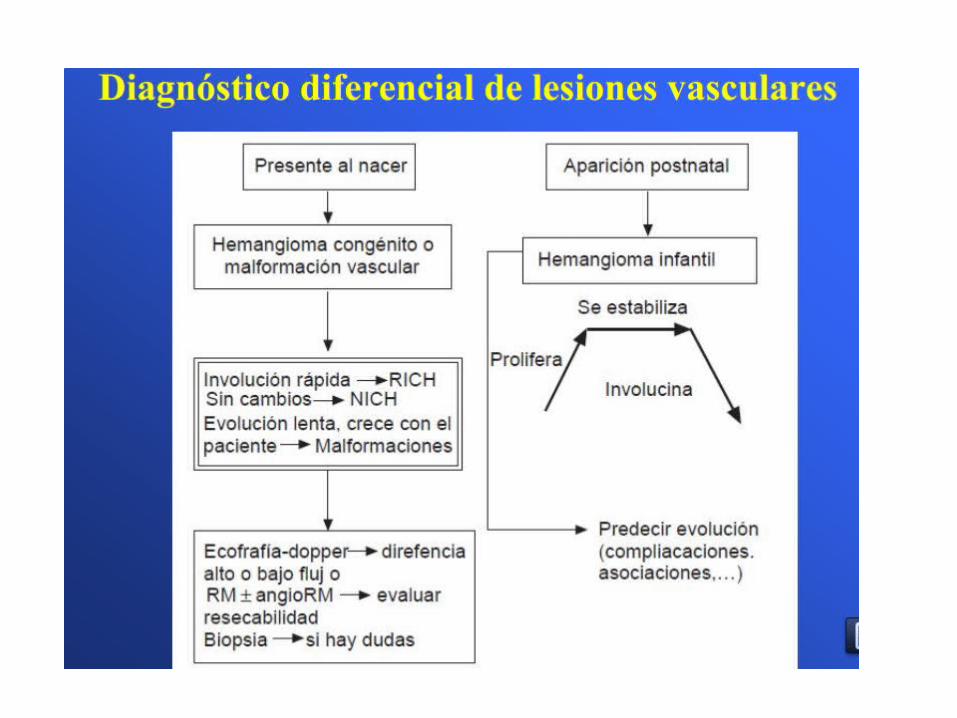
• Aparición por lo general días después del nacimiento
• FASE PROLIFERATIVA• Más intensa los primeros 6 meses de vida• En general: 1° año de vida, raro hasta los 2 años• Durante la fase proliferativa, los hemangiomas son lesiones de alto
flujo que a menudo son reveladas por bruit, pulsatilidad, y aumento de la temperatura
• FASE INVOLUTIVA• El hemangioma típico comenzará a involucionar aproximadamente 10 meses
después del nacimiento y 50% de las lesiones están completamente resuelto en 5 años
• Por lo general a partir del año
Historia Natural
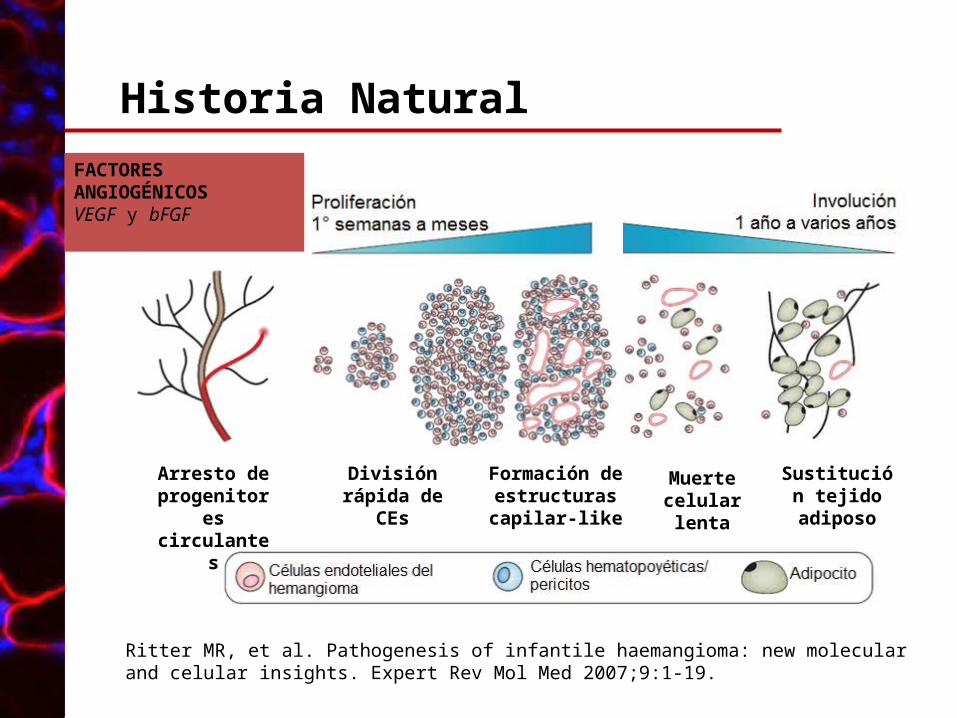
Historia Natural
Arresto de progenitores circulantes
División rápida de CEs
Formación de estructuras capilar-like
Muerte celular lenta
Sustitución tejido
adiposo
Ritter MR, et al. Pathogenesis of infantile haemangioma: new molecular and celular insights. Expert Rev Mol Med 2007;9:1-19.
FACTORES ANGIOGÉNICOSVEGF y bFGF
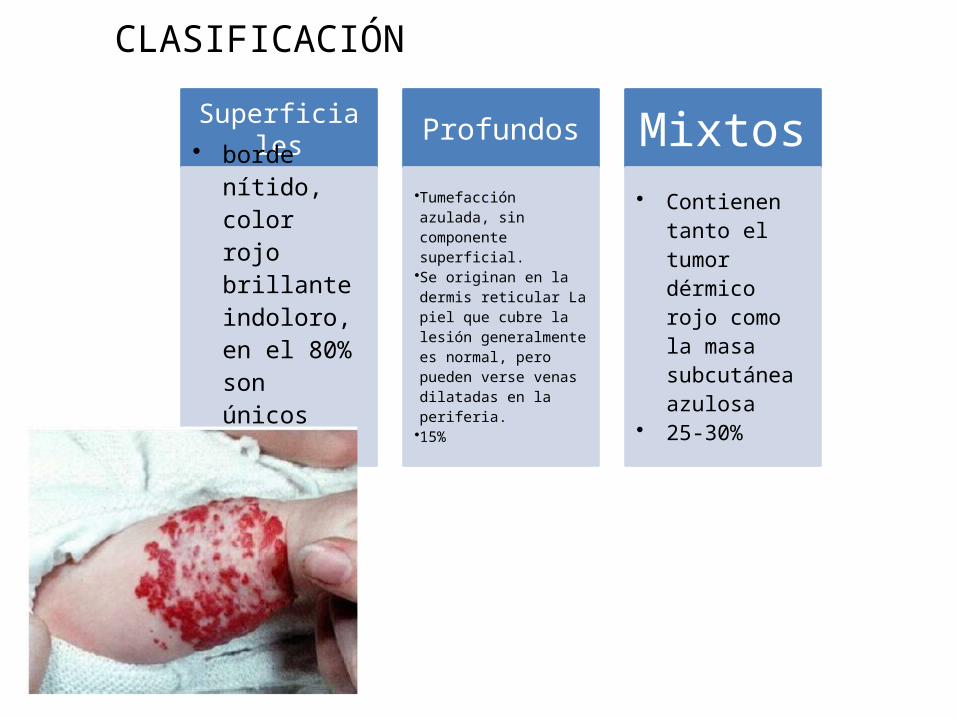
CLASIFICACIÓN
Superficiales
• borde nítido, color rojo brillante indoloro, en el 80% son únicos
• 50 y el 60%
Profundos •Tumefacción azulada, sin componente superficial.•Se originan en la dermis reticular La piel que cubre la lesión generalmente es normal, pero pueden verse venas dilatadas en la periferia.•15%
Mixtos
• Contienen tanto el tumor dérmico rojo como la masa subcutánea azulosa
• 25-30%
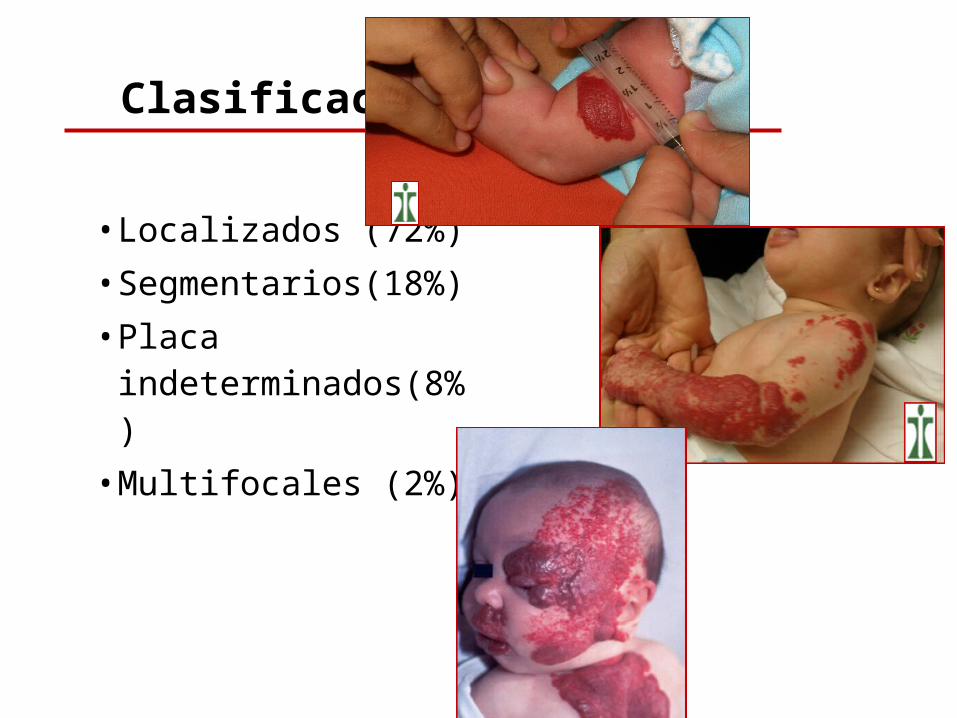
• Localizados (72%)• Segmentarios(18%)• Placa
indeterminados(8%)• Multifocales (2%)
Clasificación
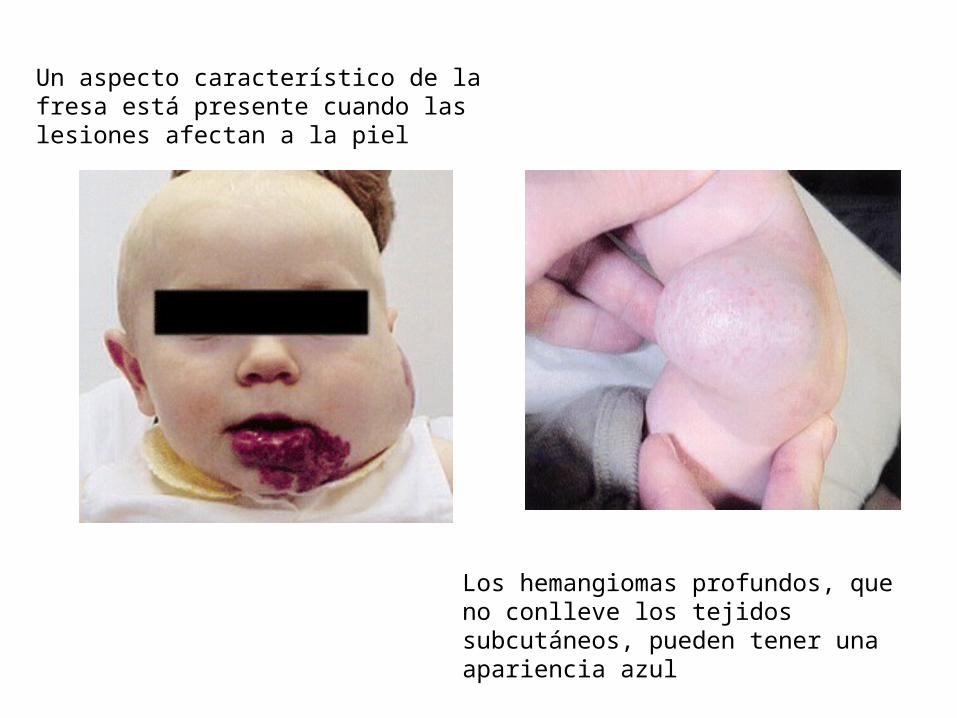
Un aspecto característico de la fresa está presente cuando las lesiones afectan a la piel
Los hemangiomas profundos, que no conlleve los tejidos subcutáneos, pueden tener una apariencia azul
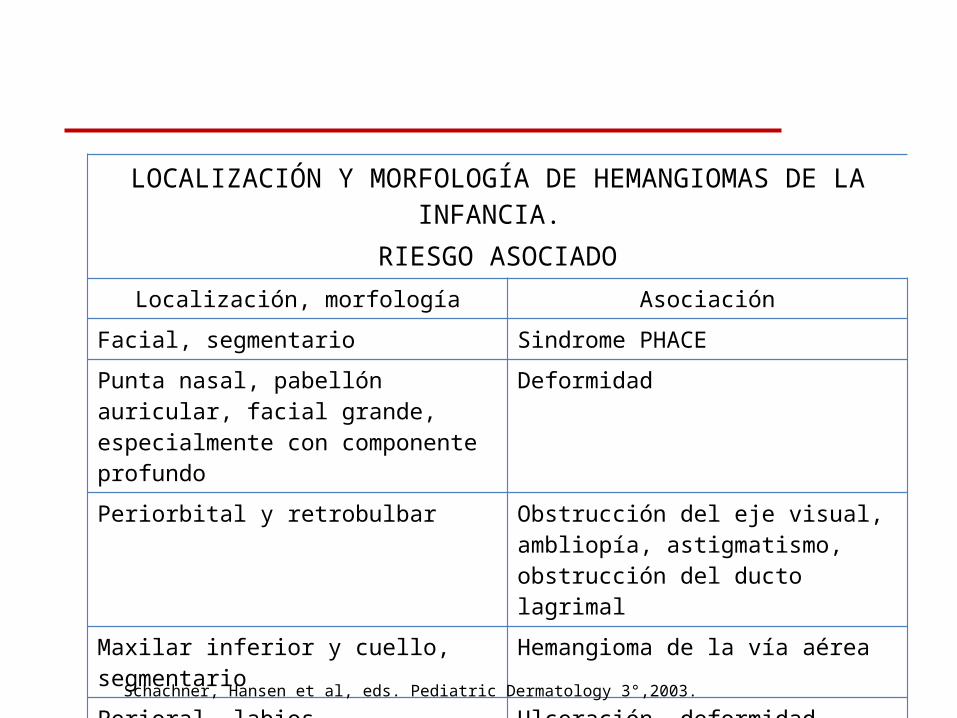
LOCALIZACIÓN Y MORFOLOGÍA DE HEMANGIOMAS DE LA INFANCIA. RIESGO ASOCIADO
Localización, morfología Asociación
Facial, segmentario Sindrome PHACE
Punta nasal, pabellón auricular, facial grande, especialmente con componente profundo
Deformidad
Periorbital y retrobulbar Obstrucción del eje visual, ambliopía, astigmatismo, obstrucción del ducto lagrimal
Maxilar inferior y cuello, segmentario Hemangioma de la vía aérea
Perioral, labios Ulceración, deformidad
Lumbo-sacro Espina bífida oculta, anomalías GU
Perianal, axila, cuello Ulceración
Hemangiomas múltiples (5) Compromiso visceral, ICC (HND)
Schachner, Hansen et al, eds. Pediatric Dermatology 3°,2003.
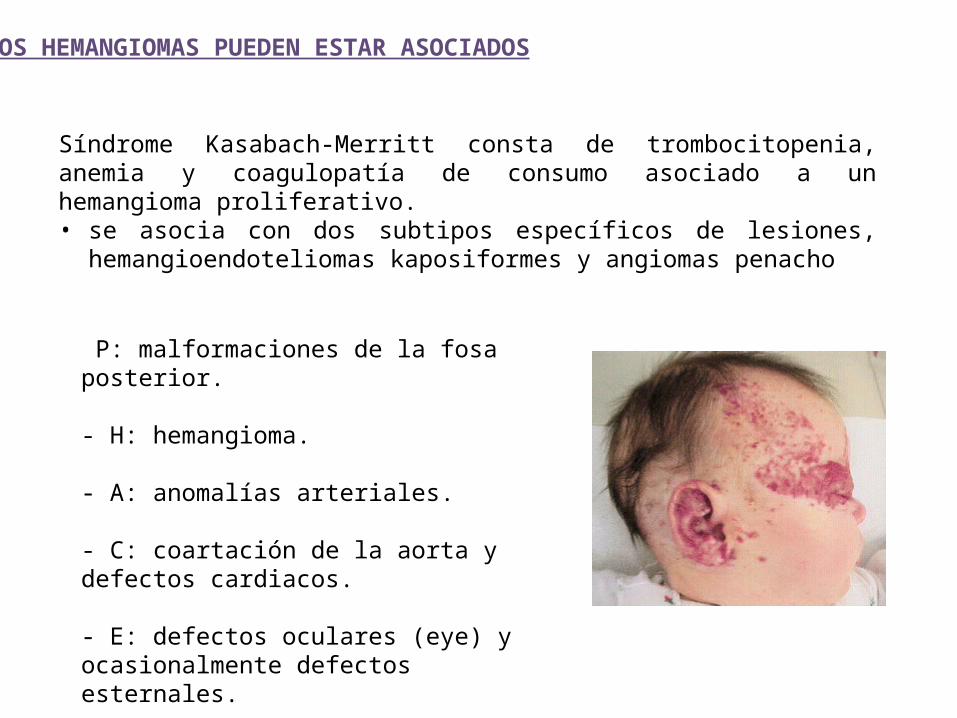
Síndrome Kasabach-Merritt consta de trombocitopenia, anemia y coagulopatía de consumo asociado a un hemangioma proliferativo.• se asocia con dos subtipos específicos de lesiones, hemangioendoteliomas
kaposiformes y angiomas penacho
P: malformaciones de la fosa posterior.
- H: hemangioma.
- A: anomalías arteriales.
- C: coartación de la aorta y defectos cardiacos.
- E: defectos oculares (eye) y ocasionalmente defectos esternales.
LOS HEMANGIOMAS PUEDEN ESTAR ASOCIADOS

DIAGNÓSTICO
Clínico: en 95% de los casos el diagnóstico puede establecerse por la historia y el examen físico.
Exámenes diagnósticos
1. Conteo de plaquetas.2. Ecografía.3. Ecografía Doppler.4. RMN: diferencia los hemangiomas de las malformaciones venosas o arteriovenosas, determina la extensión y el compromiso de las estructuras adyacentes.5. Tomografía axial computada.6. Biopsia.
Los hemangiomas aparecen como una lesión bien circunscrita; masa lobulada con irrigación y drenaje vascular. En la fase de involución se observa reemplazo por tejidos fibroso y adiposo

Indicaciones de tratamiento.- Hemangiomas que pueden comprometer la vida o la función
- Compromiso ocular.- Compromiso laríngeo.- Obstrucción nasal o auditiva.- Síndrome de Kasabach-Merritt.- Hemangiomatosis hepática.- Falla cardiaca.- Ulceración extensa de la piel.- Hemangiomas que pueden llevar a secuelas estéticas y consecuencias psicosociales adversas.- Hemorragia gastrointestinal

1. “Active, no intervention”- Hemangiomas pequeños, inocuos.
- Importante: hablar con los padres y explicarles la evolución natural de la lesión.
2. Intervención1. Hemangiomas de bajo riesgo: pequeño, sin compromiso funcional. 2. Hemangiomas de alto riesgo (grandes, localización de alto riesgo,
secuelas probables, compromiso funcional, compromiso de estructuras extracutáneas):
a. Médico a. Médico
1. Corticoides intralesionales: triamcinolona 10-40 mg/cc. Inyecciones múltiples en el tumor; se repiten a intervalos de 4 a 8 semanas. Las complicaciones son raras: oclusión de la arteria retiniana, necrosis palpebral, atrofia de la grasa subcutánea, hipopigmentación.
2. Corticoesteroides tópicos.
3. Oclusión.
1. Corticoesteroides sistémicos (indicados durante el periodo de crecimiento): prednisona 2-4 mg/kg/día; 3 mg/kg/día da una mejor respuesta.. Mecanismos de acción propuestos: incremento en la vasoconstricción, influencia hormonal e inhibición de la angiogénesis.
2. Acetonida de triamcinolona 10-40 mg/mL, algunas veces mezclada con dexametasona 4 mg/mL.
3. Interferon 2 alfa subcutáneo.
4. Terapia combinada.
b. Quirúrgico b. Quirúrgico1. Láser.
2. Criocirugía.
1. Láser pulsado.
2. Criocirugía c. Otros
1. Ciclofosfamida.
2. Embolización.
3. Radioterapia.
4. Acetato de leuprolide.
5. Ketotifeno.
6. Vincristina
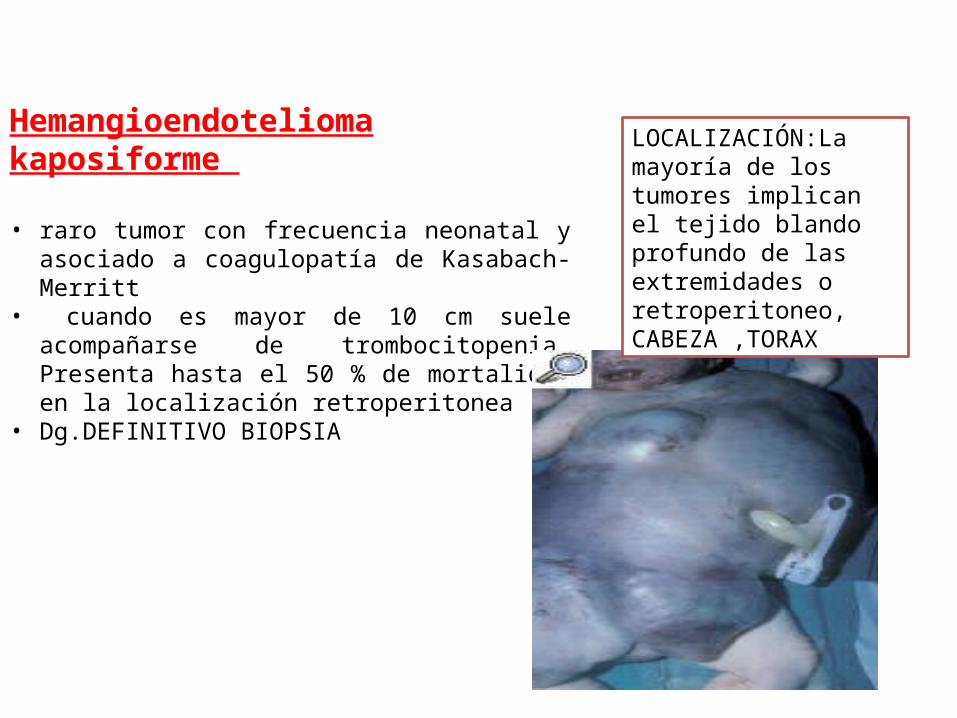
Hemangioendotelioma kaposiforme
• raro tumor con frecuencia neonatal y asociado a coagulopatía de Kasabach-Merritt
• cuando es mayor de 10 cm suele acompañarse de trombocitopenia. Presenta hasta el 50 % de mortalidad en la localización retroperitonea
• Dg.DEFINITIVO BIOPSIA
LOCALIZACIÓN:La mayoría de los tumores implican el tejido blando profundo de las extremidades o retroperitoneo, CABEZA ,TORAX
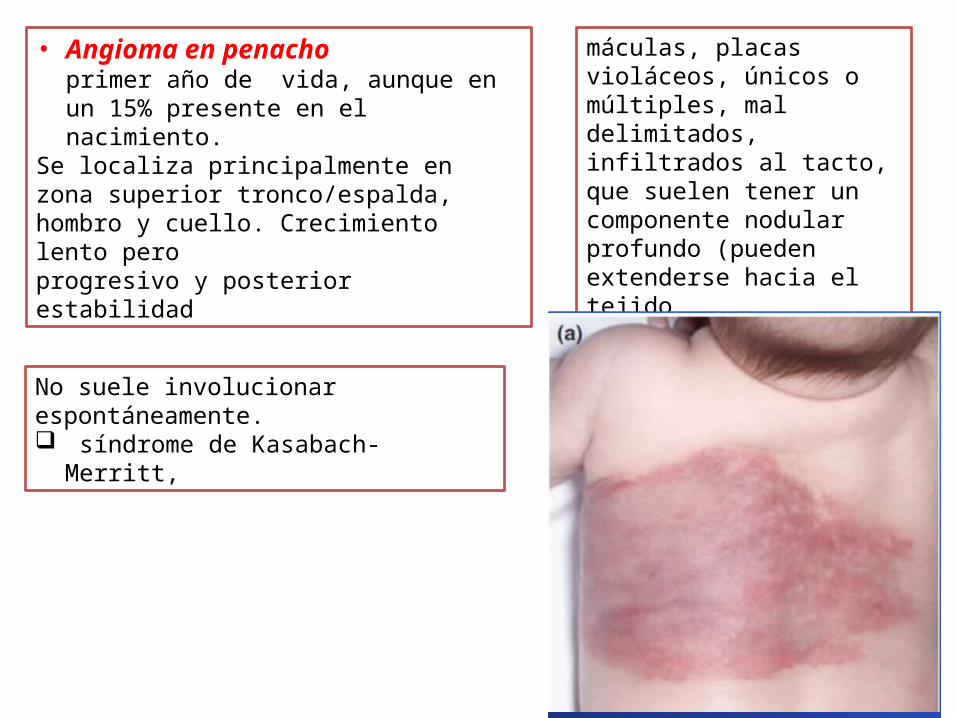
• Angioma en penacho primer año de vida, aunque en un 15% presente en el nacimiento.
Se localiza principalmente en zona superior tronco/espalda, hombro y cuello. Crecimiento lento pero progresivo y posterior estabilidad
No suele involucionar espontáneamente. síndrome de Kasabach-Merritt,
máculas, placas violáceos, únicos o múltiples, mal delimitados, infiltrados al tacto, que suelen tener un componente nodular profundo (pueden extenderse hacia el tejido celular subcutáneo, fascia y músculo)

Hemangiopericitoma
Hay dos formas clínica e histológicamente diferentes de hemangiopericitoma, el adulto y el congénito o infantil.
La forma infantil, a diferencia de los hemangiomas y a pesar de ser tumores muy vascularizados, raramente regresan; se dan más en niñas y se caracterizan por nódulos indurados únicos o múltiples de coloración rojiza recubiertos por piel en ocasiones necrótica o ulcerada. En caso de duda, la biopsia es necesaria para confirmar el diagnóstico
localización más frecuente son los tejidos blandos de extremidades, sobre todo muslo, fosa pélvica y retroperitoneo
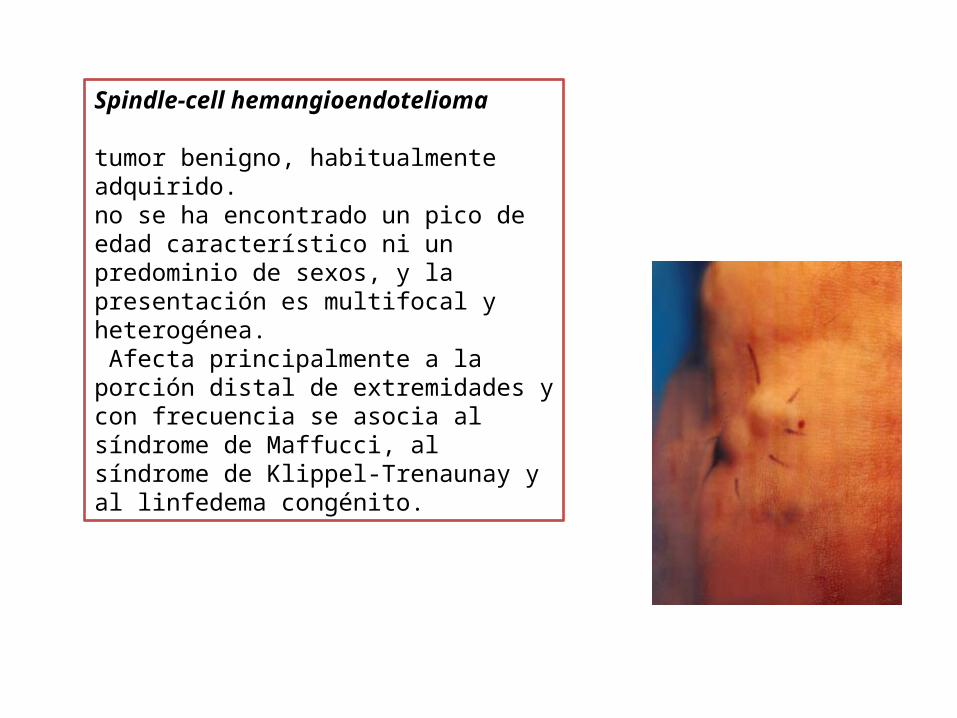
Spindle-cell hemangioendotelioma �
tumor benigno, habitualmente adquirido. no se ha encontrado un pico de edad característico ni un predominio de sexos, y la presentación es multifocal y heterogénea. Afecta principalmente a la porción distal de extremidades y con frecuencia se asocia al síndrome de Maffucci, al síndrome de Klippel-Trenaunay y al linfedema congénito.
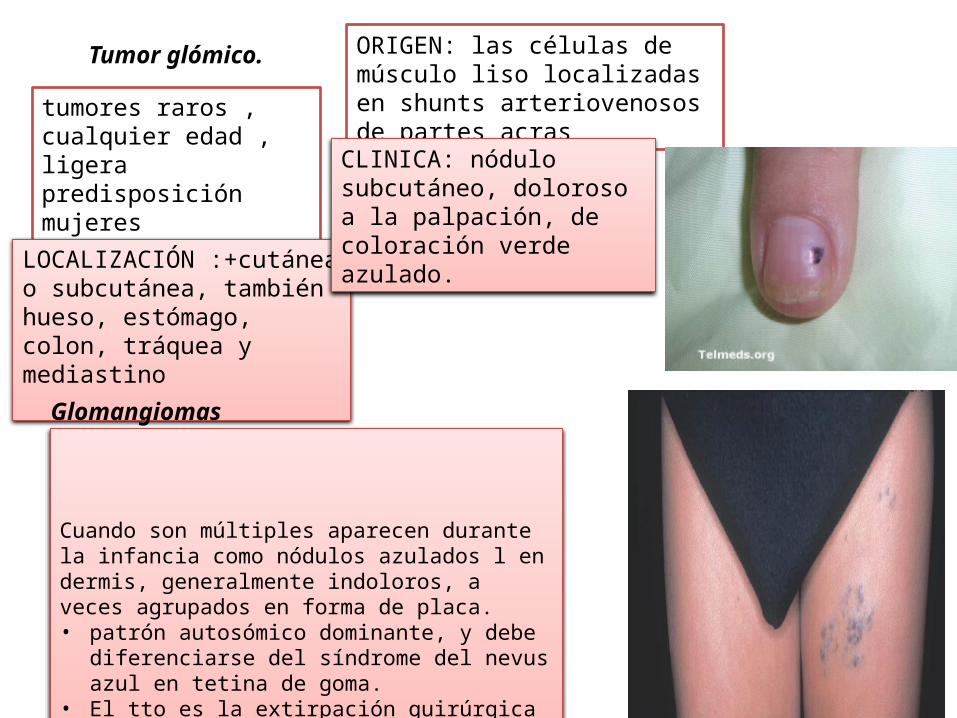
Cuando son múltiples aparecen durante la infancia como nódulos azulados l en dermis, generalmente indoloros, a veces agrupados en forma de placa.• patrón autosómico dominante, y debe diferenciarse
del síndrome del nevus azul en tetina de goma. • El tto es la extirpación quirúrgica
tumores raros , cualquier edad , ligera predisposición mujeres
ORIGEN: las células de músculo liso localizadas en shunts arteriovenosos de partes acras
LOCALIZACIÓN :+cutánea o subcutánea, también hueso, estómago, colon, tráquea y mediastino
CLINICA: nódulo subcutáneo, doloroso a la palpación, de coloración verde azulado.
Tumor glómico.
Glomangiomas

MALFORMACIONES DE ALTO FLUJO
Componentes Arteriales : MAV , Fístulas Arterio venosas, arterialesPoco frecuentes
• Las lesiones tienden a crecer con el niño, pero pueden agrandar :la trombosis, infección, o la estimulación hormonal
insuficiencia cardíaca congestiva, la embolia, el dolor, el sangrado y ulceración
• Exa.Físico : azul y pueden sentirse caliente con pulsaciones , puede thrill
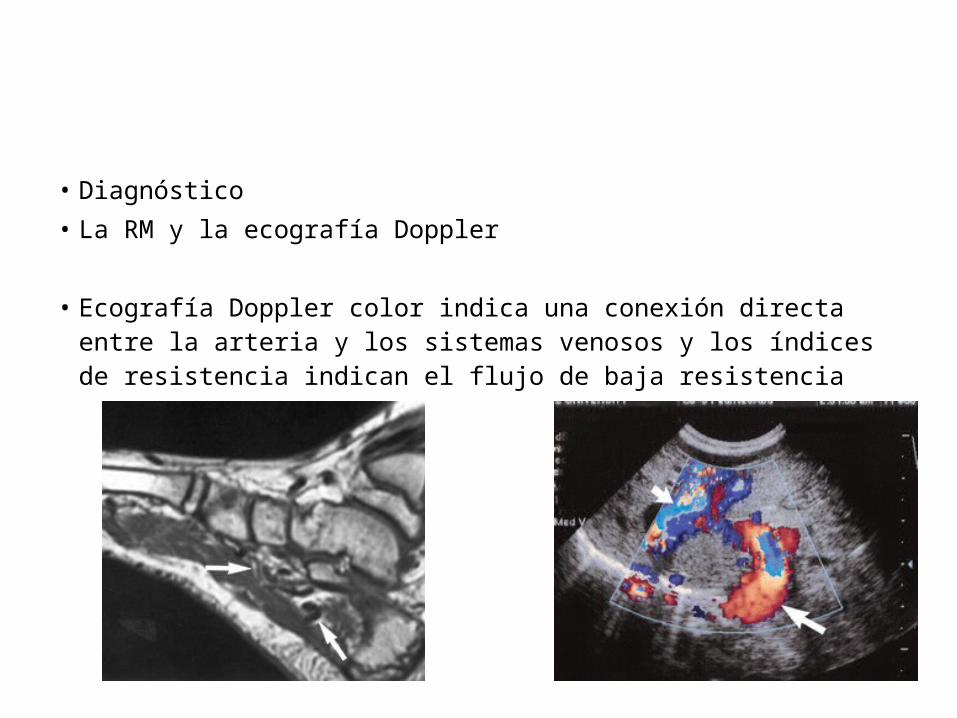
• Diagnóstico • La RM y la ecografía Doppler
• Ecografía Doppler color indica una conexión directa entre la arteria y los sistemas venosos y los índices de resistencia indican el flujo de baja resistencia
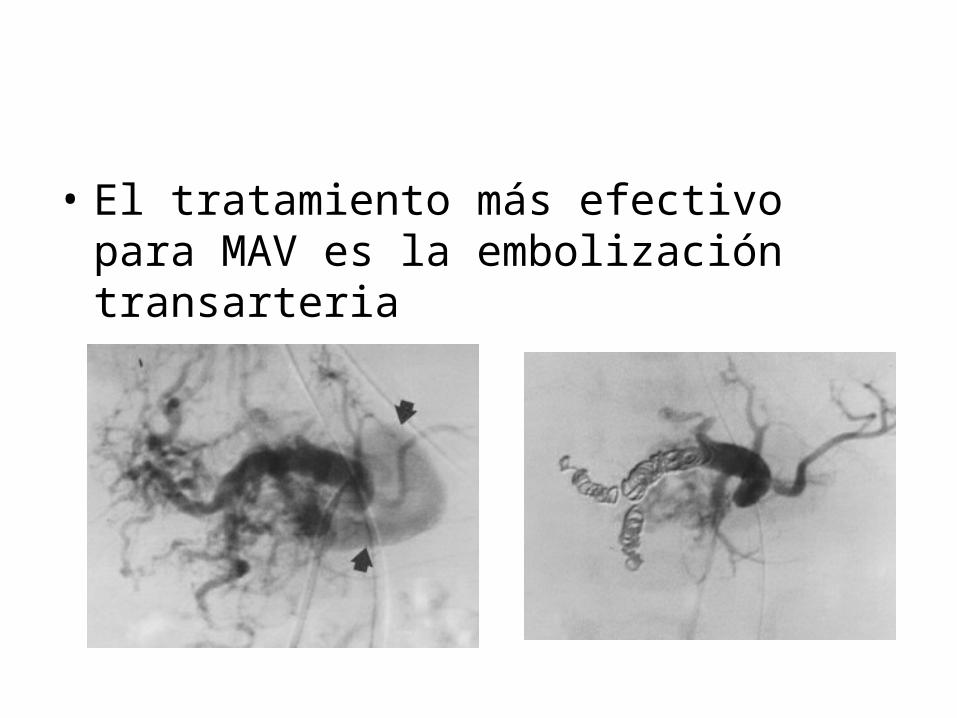
• El tratamiento más efectivo para MAV es la embolización transarteria
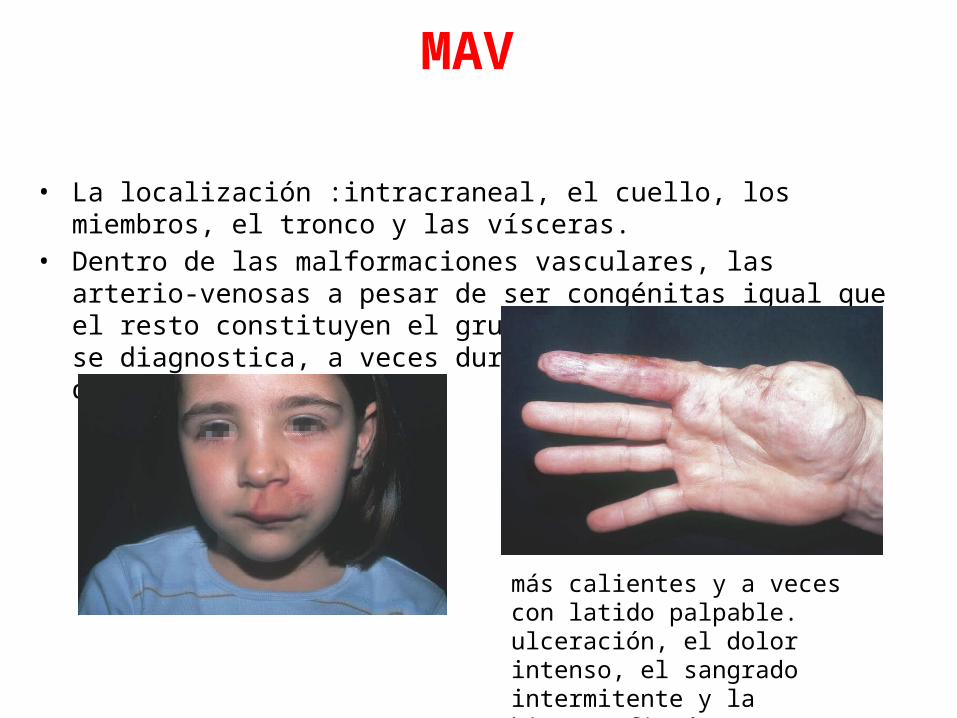
MAV
• La localización :intracraneal, el cuello, los miembros, el tronco y las vísceras.• Dentro de las malformaciones vasculares, las arterio-venosas a pesar de ser
congénitas igual que el resto constituyen el grupo que más tardíamente se diagnostica, a veces durante la cuarta-quinta década de la vida.
más calientes y a veces con latido palpable.ulceración, el dolor intenso, el sangrado intermitente y la hipertrofia ósea
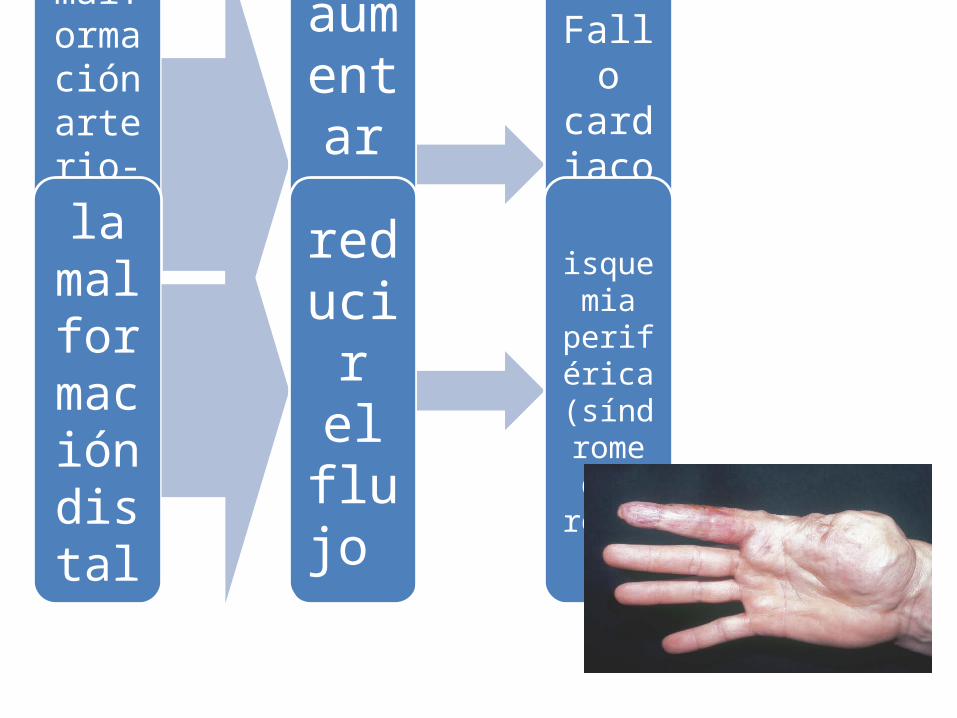
malformaci
ón arteri
o-venos
a proximal
aumentar el gasto
Fallo cardiaco congesti
vo
la malformació
n dista
l
reducir el flujo
isquemia periférica (síndrome del
robo)
QUIESCENTE
mancha rosada-violácea y la presencia de una derivación arterio-venosa detectable por eco-doppler
EXPANSIVO
+ clínicamente pulsátil, siendo evidente la presencia de vasos tensos tortuosos.
DESTRUCTIVO
+ junto a cambios cutáneos distróficos, ulceración, sangrado y dolor continuo
DES
COM
PEN
SAD
O
asociado a fallo cardiaco

Síndrome de Parkes Weber
• Malformación arterio-venosa.• Aparece en el nacimiento y afecta más al miembro
inferior
A diferencia del síndrome de Klippel-Trenaunay, la lesión vascular es de alto flujo con fístulas arterio-venosas, no suele presentar venas laterales anómalas, las malformaciones linfáticas y la afectación músculo-esquelética son raras
La principal complicación en el síndrome de Parkes Weber es el aumento del gasto que puede originar fallo cardiaco e isquemia cutánea
Se caracteriza por una mácula rosado-rojiza difusa de bordes geométricos o difusos que va aumentando de forma simétrica

MALFORMACIONES VENOSAS.
CLASIFICACIÓN.• Profundas o
superficiales.• Localizadas o difusas.
Localización más frecuente:1. Cara (lengua, paladar, labios).2. Cuello3. Extremidades
CARACTERÍSTICAS.• Blandas al tacto, a veces de aspecto nodular,
que se vacían con la compresión.• Color: más superficiales son moradas y las
profundas son azuladas o verdosas.• No son evidentes los cambios de
temperatura.
HISTOLOGÍA.Son canales venosos dilatados, de paredes irregulares y finas, con una anormal disposición de las fibras musculares lisas (responsable de la tendencia expansiva de estas lesiones.
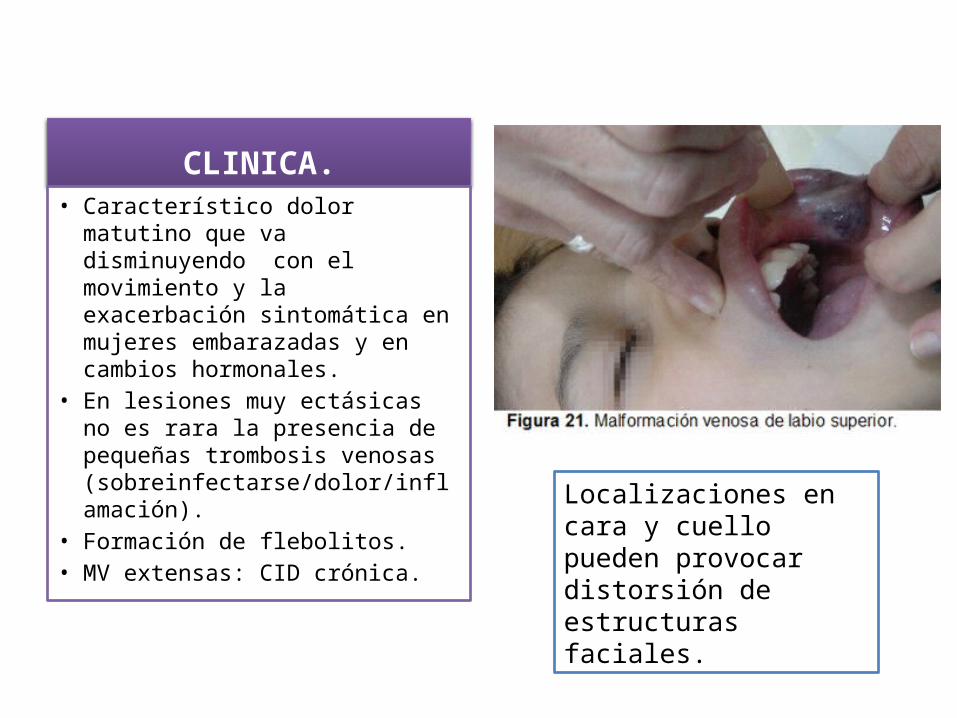
CLINICA.• Característico dolor matutino que
va disminuyendo con el movimiento y la exacerbación sintomática en mujeres embarazadas y en cambios hormonales.
• En lesiones muy ectásicas no es rara la presencia de pequeñas trombosis venosas (sobreinfectarse/dolor/inflamación).
• Formación de flebolitos.• MV extensas: CID crónica.
Localizaciones en cara y cuello pueden provocar distorsión de estructuras faciales.
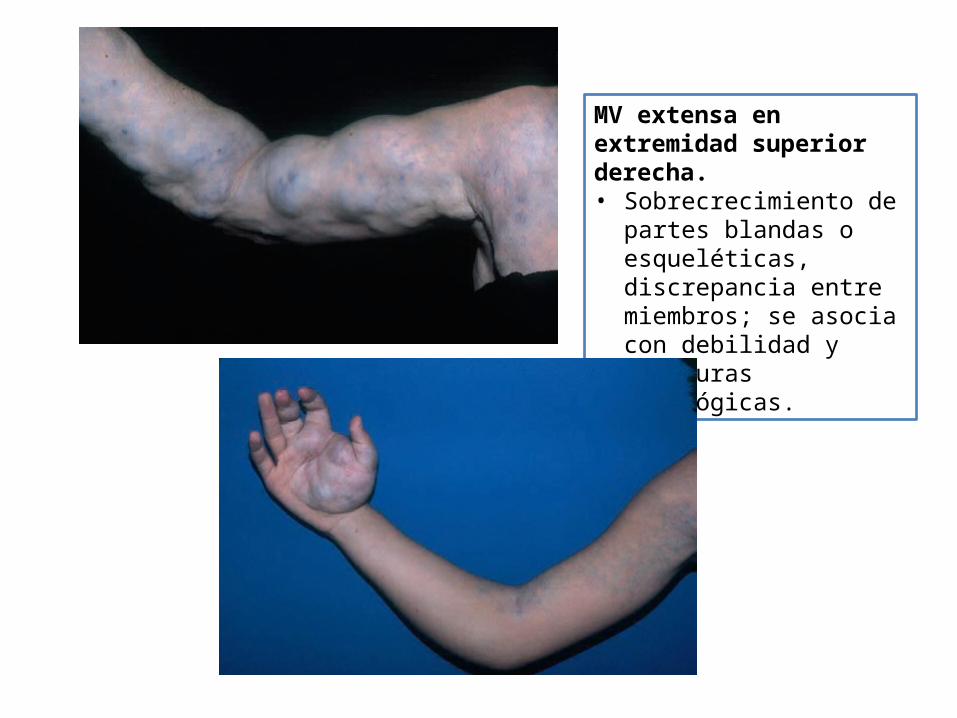
MV extensa en extremidad superior derecha.• Sobrecrecimiento de partes
blandas o esqueléticas, discrepancia entre miembros; se asocia con debilidad y fracturas patológicas.

Llamadas: • Mancha de vino
de oporto.• Nevus flammeus
• Son anomalías vasculares dérmicas.• RN 0.3% • Mayoría son esporádicos (patrón familiar
cromosoma 5).• Unilaterales y segmentarios.• Cualquier parte del cuerpo.• Más frecuente en la cara y cuello (83%).• Afecta más a la hemifacie derecha que a la
izquierda.
MALFORMACIONES CAPILARES.
HISTOLOGÍA.Vasos capilares-venulares ectásicos y dilatados localizados en la dermis superficial.
ESTUDIOS HISTOQUÍMICOS.• Morfología e índice mitóticos normales.• Disminución de fibras nerviosas = ectasia
vascular por falta de regulación nerviosa.• Vasos se dilatan y se oscurecen con un
aspecto empedrado.
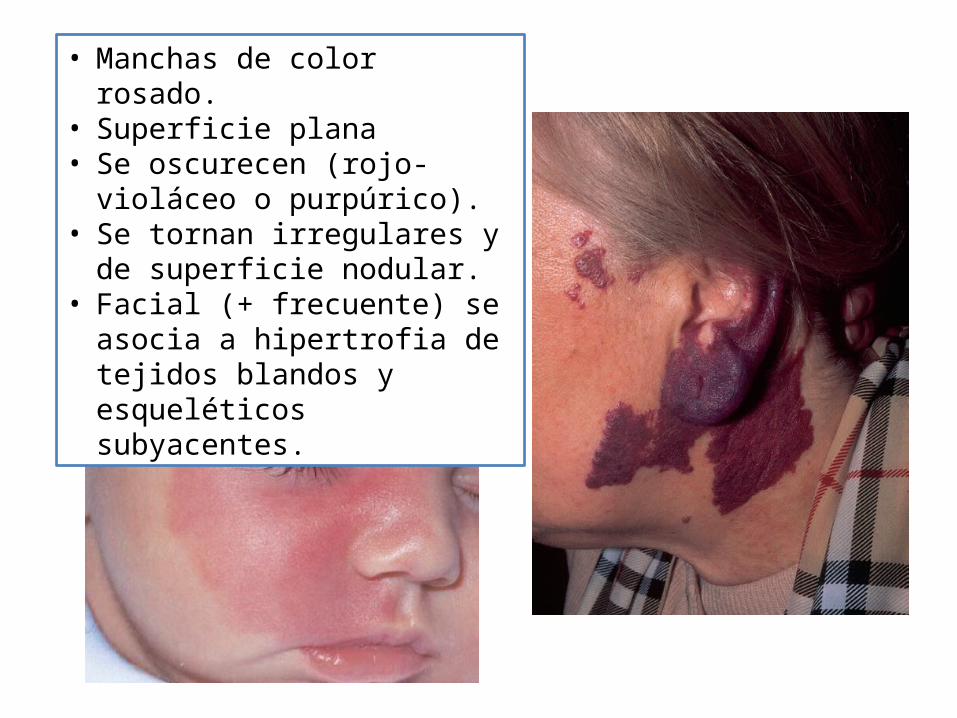
• Manchas de color rosado.• Superficie plana • Se oscurecen (rojo-violáceo o
purpúrico).• Se tornan irregulares y de
superficie nodular.• Facial (+ frecuente) se asocia a
hipertrofia de tejidos blandos y esqueléticos subyacentes.

DG diferencial.• Nevus flammeus neonatorum (RN
40%), es una mácula fluctuante de color rosado o rojo pálido.
• Frente, glabela, párpados superiores, región nasolabial, cuello o región lumbosacra.
• "Beso del ángel o piquete de cigüeña".
• No son verdaderas MC.• Dilataciones transitorias de los
vasos dérmico.• Tienden a desaparecer
espontáneamente antes 2 años.• No requiere tto.
TRATAMIENTO.• Fototermólisis selectiva con láser.
Bajo anestesia general, con intervalos de 2 a 3 meses.
• En hipertrofia de tejidos blandos y esqueléticos requiere tratamiento quirúrgico.

MALFORMACIONES LINFÁTICAS.
• Es la forma más común de malformación vascular.• No existe una única nomenclatura o clasificación de las ML.
CLASIFICACIÓN.• ML capilar (antiguamente
linfangioma capilar simple y circunscrito).
• ML microquística (antiguamente linfangioma cavernoso).
• ML macroquística (antiguamente linfangioma o higroma quístico)
• ML micro-macroquística
ML CAPILAR SIMPLE.• Pequeñas pápulas o vesículas
superficiales, ligeramente elevadas, únicas o múltiples.
• En cualquier parte del cuerpo (predilección: regiones oral, lengua y genital).
• Histológicamente presentan conductos linfáticos de paredes finas y dilatadas que comprometen dermis y epidermis.
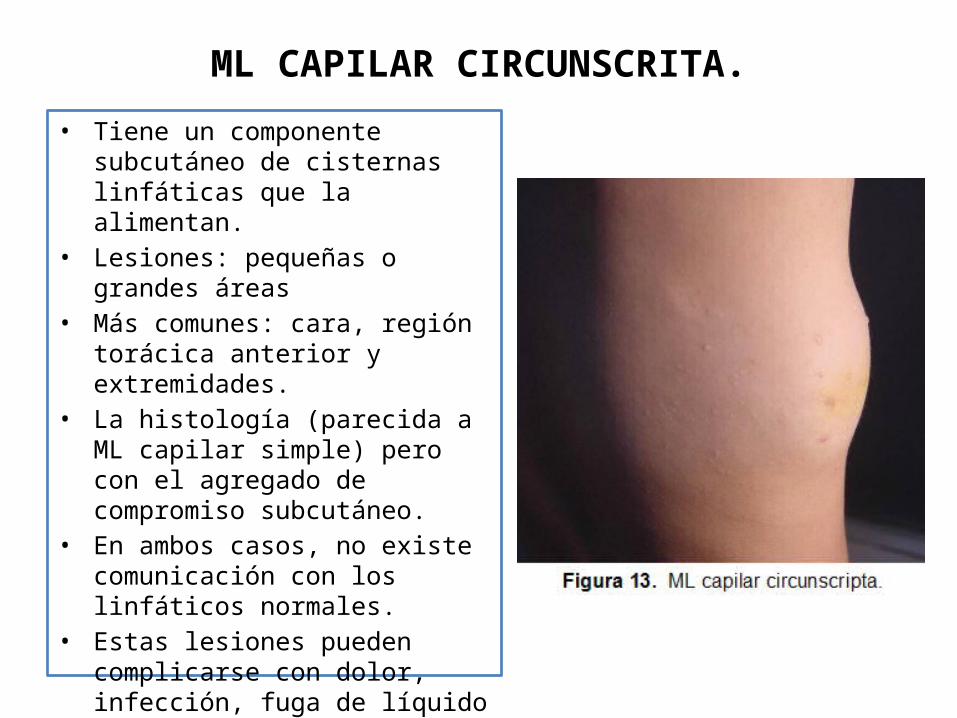
ML CAPILAR CIRCUNSCRITA.
• Tiene un componente subcutáneo de cisternas linfáticas que la alimentan.
• Lesiones: pequeñas o grandes áreas• Más comunes: cara, región torácica
anterior y extremidades. • La histología (parecida a ML capilar
simple) pero con el agregado de compromiso subcutáneo.
• En ambos casos, no existe comunicación con los linfáticos normales.
• Estas lesiones pueden complicarse con dolor, infección, fuga de líquido linfático o hemorrágico y problemas estéticos.
• El diagnóstico se establece por el examen físico.

ML microquística.
• Se presenta como una tumoración dura o duro-elástica, que compromete tejidos superficiales y profundos.
• Cualquier parte del cuerpo (más frecuente en la región cervicofacial 70-80%, la pared torácica, las extremidades y el retroperitoneo)
• Histológicamente presenta conductos linfáticos dilatados, revestidos de células endoteliales (a veces, con músculo liso) y múltiples espacios linfáticos pequeños que afectan epidermis, dermis, tejido subcutáneo y planos profundos.
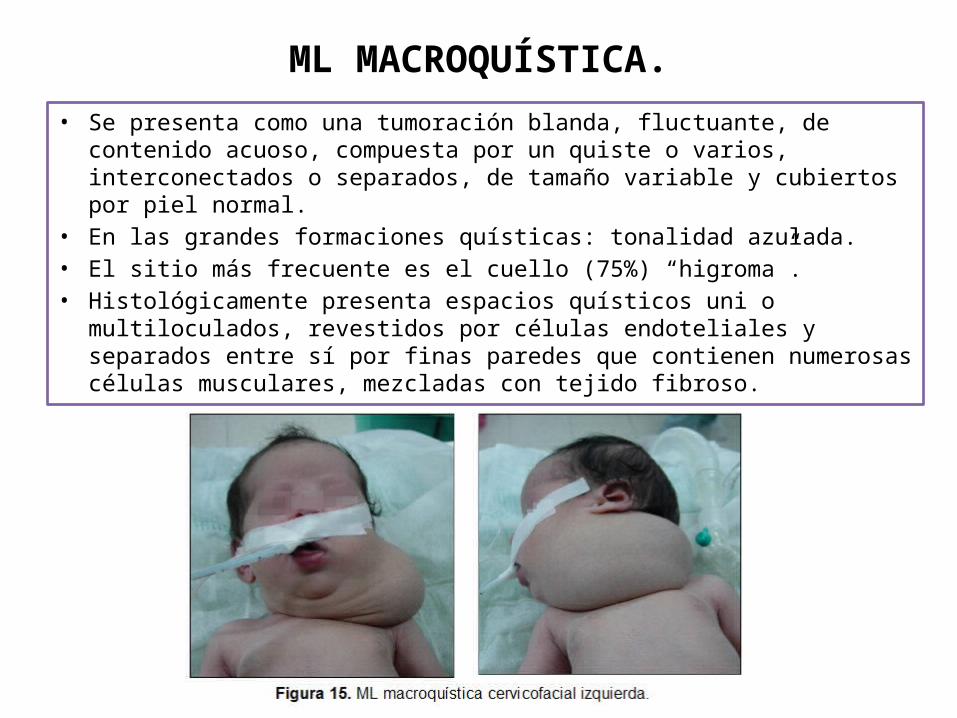
ML MACROQUÍSTICA.
• Se presenta como una tumoración blanda, fluctuante, de contenido acuoso, compuesta por un quiste o varios, interconectados o separados, de tamaño variable y cubiertos por piel normal.
• En las grandes formaciones quísticas: tonalidad azulada. • El sitio más frecuente es el cuello (75%) “higroma”.• Histológicamente presenta espacios quísticos uni o multiloculados,
revestidos por células endoteliales y separados entre sí por finas paredes que contienen numerosas células musculares, mezcladas con tejido fibroso.

COMPLEJOS SINDRÓMICOS ASOCIADOS A MALFORMACIONES VASCULARES.
MALFORMACIÓN VASCULARSimples.
SÍNDROMES.
Malformaciones venosas
• Síndrome de Bean• Síndrome de Maffucci• Glomangiomatosis familiar múltiple
Malformaciones capilares
• Síndrome de Sturge-Weber• Síndrome de Van Lohuizen• Enfermedad De Rendu- Osler- Weber• Ataxia telangectasia o síndrome de
Louis Bar• Síndrome de Divrye-Van Bogaert
Malformaciones linfáticas.
• Síndrome de Gorham

COMPLEJOS SINDRÓMICOS ASOCIADOS A MALFORMACIONES VASCULARES.
Complejas:
Venular-venosa-linfática
• Síndrome de Klippel-Trenaunay• Síndrome Proteus
Venular-venosa con fístula arterio-venosa
• Síndrome de Parkes Weber
Arterio-venosa • Síndrome de Rendú-Osler-Weber
Venosa o venosa-linfática
• Síndrome de Maffucci• Enfermedad de Gorham

SÍNDROME DE BEAN (malformación venosa).
Es una anomalía vascular rara (200 casos).
Etiología: desconoce (cromosoma 9p).
Caracterizada por MV multifocales en piel, tejidos blandos y tubo digestivo, aunque pueden afectar cualquier órgano o tejido.
Desde el punto de vista clínico, las lesiones del tubo digestivo son las más relevantes y se manifiestan por sangrado digestivo de tipo oculto, crónico y anemizante, desde temprana edad, que requiere terapia repetida con hierro y transfusiones frecuentes.
El diagnóstico se basa inicialmente en el hallazgo de las lesiones cutáneas características. El tratamiento de las lesiones cutáneas está indicado sólo con fines estéticos o cuando presentan un problema funcional.
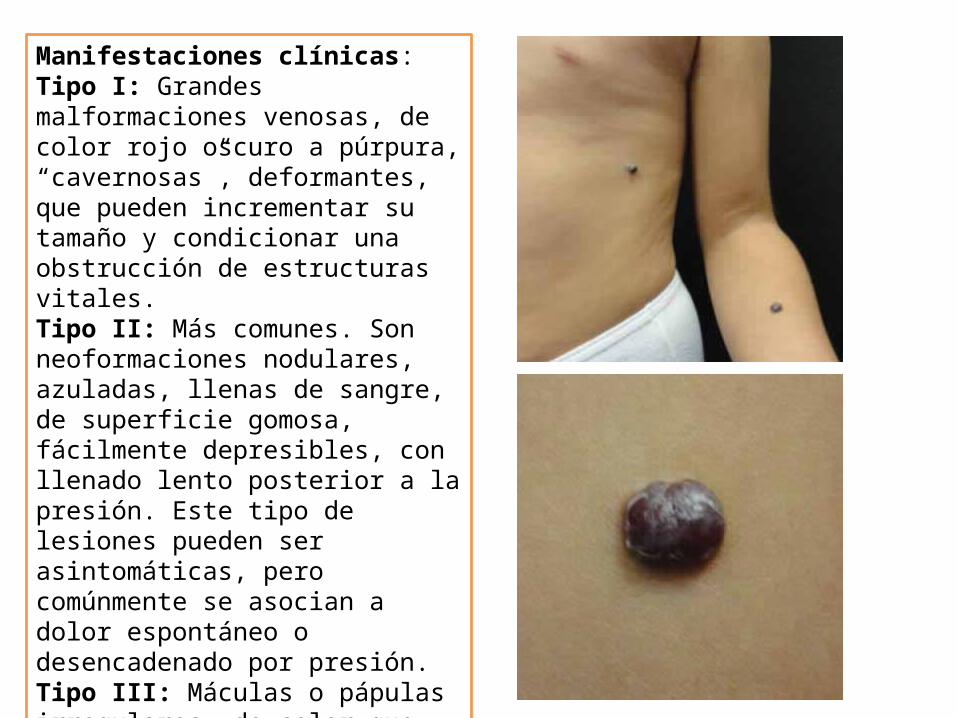
Manifestaciones clínicas:Tipo I: Grandes malformaciones venosas, de color rojo oscuro a púrpura, “cavernosas”, deformantes, que pueden incrementar su tamaño y condicionar una obstrucción de estructuras vitales. Tipo II: Más comunes. Son neoformaciones nodulares, azuladas, llenas de sangre, de superficie gomosa, fácilmente depresibles, con llenado lento posterior a la presión. Este tipo de lesiones pueden ser asintomáticas, pero comúnmente se asocian a dolor espontáneo o desencadenado por presión.Tipo III: Máculas o pápulas irregulares, de color que varía en tonos azul a negro, puntiformes, que pueden aparecer adyacentes o sobre una mácula azulada sin desaparecer a la presión.

Telangiectasia hemorrágica hereditaria o síndrome de Rendu-Osler-Weber (malformación capilar).
• Es un desorden hereditario autosómico dominante (incidencia 1-2 por 100.000).
• Caracterizado por telangiectasias en piel y mucosas, malformaciones arteriovenosas en cerebro y pulmones, y AV hepáticas.
• El síntoma de inicio suele ser la epistaxis, y es más frecuente en la pubertad. Las lesiones comienzan en los lechos capilares, donde delicados cortocircuitos capilares-venulares aparecen en piel y mucosas, pulmones, hígado y cerebro, generalmente durante la tercera o cuarta décadas de la vida.
• El diagnóstico se establece sobre la base de los antecedentes familiares y el cuadro clínico característico.

SÍNDROME DE GORHAM (malformación linfática).
• Entidad clínica extremadamente rara (no más de 200 casos descritos en la literatura) definida por osteolisis espontánea, masiva, idiopática y no seguida de producción de nuevo tejido óseo.
• Se caracteriza por una proliferación vascular de canales de origen linfático en el hueso y tejidos blandos circundantes.
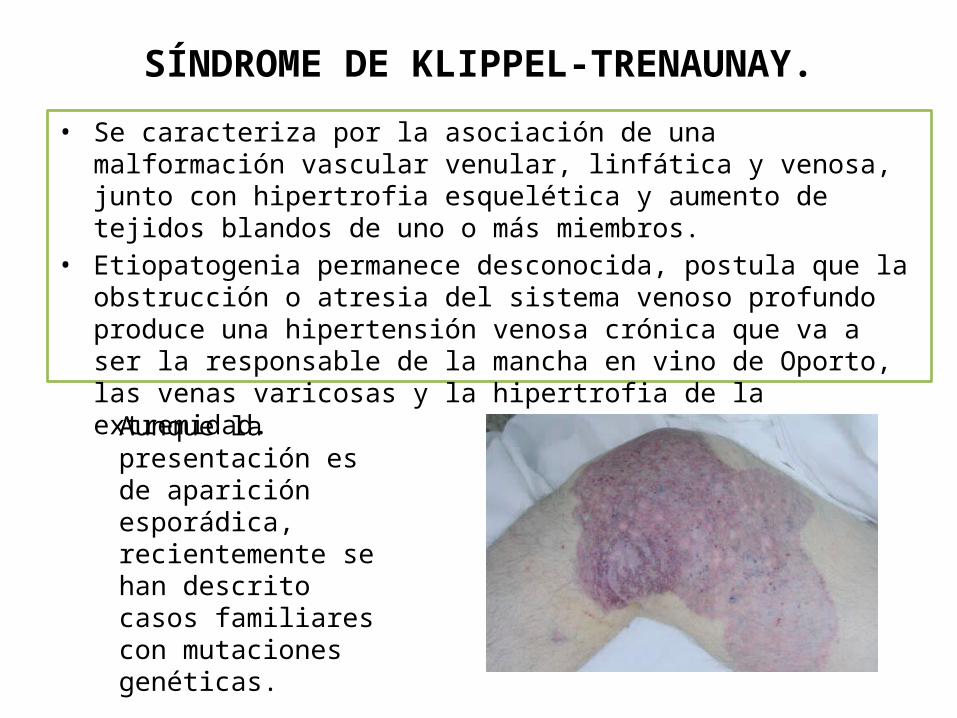
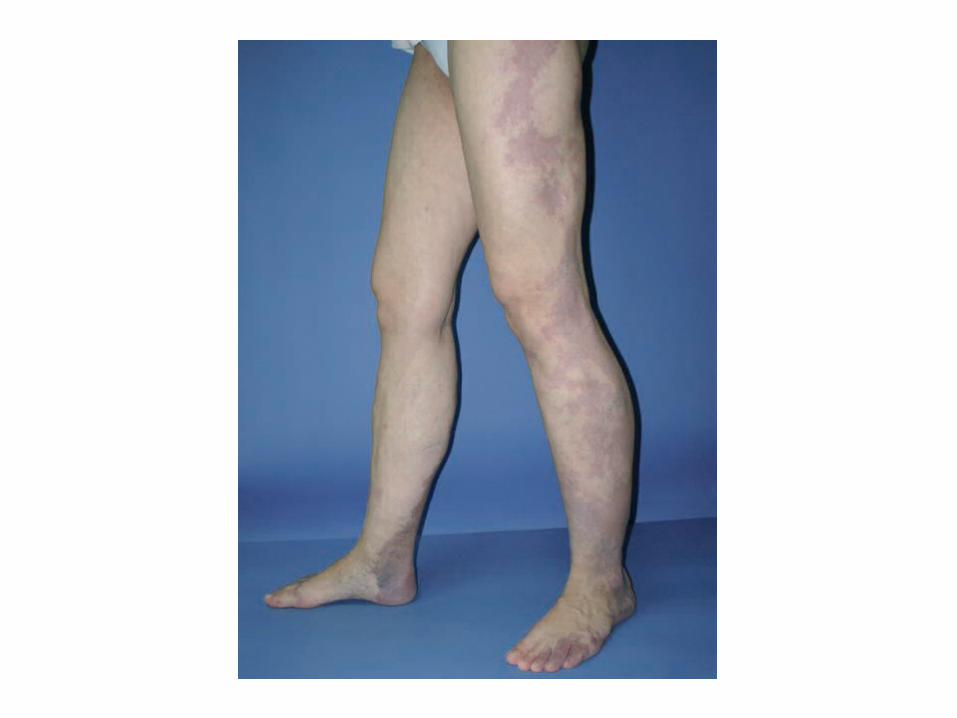
SÍNDROME DE KLIPPEL-TRENAUNAY.
• Se caracteriza por la asociación de una malformación vascular venular, linfática y venosa, junto con hipertrofia esquelética y aumento de tejidos blandos de uno o más miembros.
• Etiopatogenia permanece desconocida, postula que la obstrucción o atresia del sistema venoso profundo produce una hipertensión venosa crónica que va a ser la responsable de la mancha en vino de Oporto, las venas varicosas y la hipertrofia de la extremidad.
Aunque la presentación es de aparición esporádica, recientemente se han descrito casos familiares con mutaciones genéticas.

GRACIAS.


















