GUÍA INFLAMATORIAS NEUROLÓGICA JESÚS …acnweb.org/guia/g7cap10.pdf · GUÍA NEUROLÓGICA 7...
17
GUÍA NEUROLÓGICA 7 MIOPATÍAS INFLAMATORIAS JESÚS H. RODRÍGUEZ 71 MIOPATIAS INFLAMATORIAS JESÚS H. RODRÍGUEZ INTRODUCCI‹ N L as miopatía inflamatoria es una constela- ción de patologías con una incidencia de 1:100 000. Sus principales características son la debilidad muscular, un proceso histoló- gico inflamatorio, la elevación de enzimas musculares como la creatin kinasa (CK), y la aldolasa. No siempre es posible confirmar el proceso inflamatorio por patología, ni las enzimas musculares se elevan. La electromio- grafía cualitativa y cuantitativa respaldada por estudios como las neuroconducciones e incluso la electromiografía de fibra única pueden ayudar con una mejor aproximación diagnóstica, a tomar la decisión de instaurar un adecuado tratamiento y establecer un pronóstico. Los tipos más frecuentes de miopatías inflamatorias son la polimiositis, dermatomiositis y miositis de cuerpos de inclusión, aunque existen varias otras. CLASIFICACI‹ N Miopatías inmunes (polimiositis) Las características clínicas son un predo- minio de la debilidad de tipo proximal más que distal, y en ocasiones con debilidad selectiva por regiones como disfagia, dolor en la nuca o en el cuadriceps. Se presenta en mayores de, 20 años y progresa en meses. Cursa con dolor hasta en 30% de los casos y se relaciona mas con enfermedades del tejido conectivo. Se debe descartar la polimialgia por otras causas como la fascitis y la rabdomiolisis. Existen desórdenes asociados a la polimiositis que ocurren durante la evolución de la enfermedad. Entre ellas las arritmias y la cardiomiopatía inflamatoria, la debilidad de los músculos de la respiración y la enfermedad pulmonar intersticial. La paresia, generalmente el tercio superior del esófago está relacionada con debilidad muscular general o con escleroderma cuando el compromiso es del tercio inferior. Con la presencia de malignidad puede existir miopatía inflamatoria. Coexiste hasta en un 50% con casos de enfermedad autoinmune como lupus, Sjögren o con síndrome antifosfolípido en un 5-8%. En la tirotoxicosis es rara la elevación de la CK aun en un rango leve y hay una buena respuesta a la administración de antitiroideos. La elevación de las enzimas como la CK notoria entre 3-30 veces normal y la electromiografía (EMG) demuestra hiperexcitabilidad de la membrana (sarcolema) del músculo. Los anticuerpos relacionados con miopatías inflamatorias descritos en polimiositis se describen en las tablas 1 y 2. En la biopsia muscular se observa un patrón miopático con variación en el tamaño de las fibras musculares, necrosis con fagocitosis y regeneración de las fibras musculares e incremento moderado en parches del tejido conectivo endomisial. Hay inflamación endomisial y perivascular de tipo mononuclear como macrófagos y linfocitos CD8 con invasión de fibras musculares no necróticas. Las siguientes etiologías resaltan las claves diagnósticas que permiten distinguir las polimiositis: Idiopática. Se presenta con una debilidad más proximal que distal, moderada en la cara, simétrica, con mialgias. En solo el 20% hay elevación evidente de la CK. La EMG documenta un patrón miopático con actividad espontánea con fibrilaciones y ondas agudas. La biopsia muscular presenta características inflamatorias. Enfermedad vascular del colágeno. Se presenta en pacientes jóvenes con una edad promedio de 35 años y una razón 9:1 de
Transcript of GUÍA INFLAMATORIAS NEUROLÓGICA JESÚS …acnweb.org/guia/g7cap10.pdf · GUÍA NEUROLÓGICA 7...

GUÍA NEUROLÓGICA
7MIOPATÍAS INFLAMATORIAS
JESÚS H. RODRÍGUEZ
71
MIOPATIAS INFLAMATORIAS
JESÚS H. RODRÍGUEZ
INTRODUCCI‹ N
Las miopatía inflamatoria es una constela-ción de patologías con una incidencia de 1:100 000. Sus principales características
son la debilidad muscular, un proceso histoló-gico inflamatorio, la elevación de enzimas musculares como la creatin kinasa (CK), y la aldolasa. No siempre es posible confirmar el proceso inflamatorio por patología, ni las enzimas musculares se elevan. La electromio-grafía cualitativa y cuantitativa respaldada por estudios como las neuroconducciones e incluso la electromiografía de fibra única pueden ayudar con una mejor aproximación diagnóstica, a tomar la decisión de instaurar un adecuado tratamiento y establecer un pronóstico. Los tipos más frecuentes de miopatías inflamatorias son la polimiositis, dermatomiositis y miositis de cuerpos de inclusión, aunque existen varias otras.
CLASIFICACI‹ N Miopatías inmunes (polimiositis)
Las características clínicas son un predo-minio de la debilidad de tipo proximal más que distal, y en ocasiones con debilidad selectiva por regiones como disfagia, dolor en la nuca o en el cuadriceps. Se presenta en mayores de, 20 años y progresa en meses. Cursa con dolor hasta en 30% de los casos y se relaciona mas con enfermedades del tejido conectivo. Se debe descartar la polimialgia por otras causas como la fascitis y la rabdomiolisis. Existen desórdenes asociados a la polimiositis que ocurren durante la evolución de la enfermedad. Entre ellas las arritmias y la cardiomiopatía inflamatoria, la debilidad de los músculos de la respiración y la enfermedad pulmonar intersticial. La paresia, generalmente el tercio superior del esófago está relacionada con
debilidad muscular general o con escleroderma cuando el compromiso es del tercio inferior. Con la presencia de malignidad puede existir miopatía inflamatoria. Coexiste hasta en un 50% con casos de enfermedad autoinmune como lupus, Sjögren o con síndrome antifosfolípido en un 5-8%. En la tirotoxicosis es rara la elevación de la CK aun en un rango leve y hay una buena respuesta a la administración de antitiroideos. La elevación de las enzimas como la CK notoria entre 3-30 veces normal y la electromiografía (EMG) demuestra hiperexcitabilidad de la membrana (sarcolema) del músculo. Los anticuerpos relacionados con miopatías inflamatorias descritos en polimiositis se describen en las tablas 1 y 2.
En la biopsia muscular se observa un patrón miopático con variación en el tamaño de las fibras musculares, necrosis con fagocitosis y regeneración de las fibras musculares e incremento moderado en parches del tejido conectivo endomisial. Hay inflamación endomisial y perivascular de tipo mononuclear como macrófagos y linfocitos CD8 con invasión de fibras musculares no necróticas.
Las siguientes etiologías resaltan las claves diagnósticas que permiten distinguir las polimiositis:
Idiopática. Se presenta con una debilidad más proximal que distal, moderada en la cara, simétrica, con mialgias. En solo el 20% hay elevación evidente de la CK. La EMG documenta un patrón miopático con actividad espontánea con fibrilaciones y ondas agudas. La biopsia muscular presenta características inflamatorias.
Enfermedad vascular del colágeno. Se presenta en pacientes jóvenes con una edad promedio de 35 años y una razón 9:1 de
GUÍA NEUROLÓGICA
7MIOPATÍAS INFLAMATORIAS
JESÚS H. RODRÍGUEZ
72
mujeres a hombres. Frecuentemente cursa con artralgias, mialgias y anticuerpos de miositis de sobreposición (overlap) que responden satisfactoriamente a dosis bajas de corticoides. Se asocia a esclerodema y enfermedad del tejido conectivo mixto.
Polimiositis por anticuerpos especí-ficos. Anticuerpos Anti-T-ma sintetasa, Anti
Jo-1(Histidyl t-RNA sintetasa IgG). Es un cuadro clínico de frecuente presentación en mujeres, caucásicos(20-30%) y poco común en mestizos (5%). Inicia con debilidad de distribución proximal, simétrica, de moderada a severa y con mialgias. Fenómeno de Raynaud en 60%, queratosis en las manos o en el borde de los dedos y lesiones vasculíticas de tipo leucocitoclásticas. Una característica del síndrome anti-JO-1 es la neumonitis intersticial
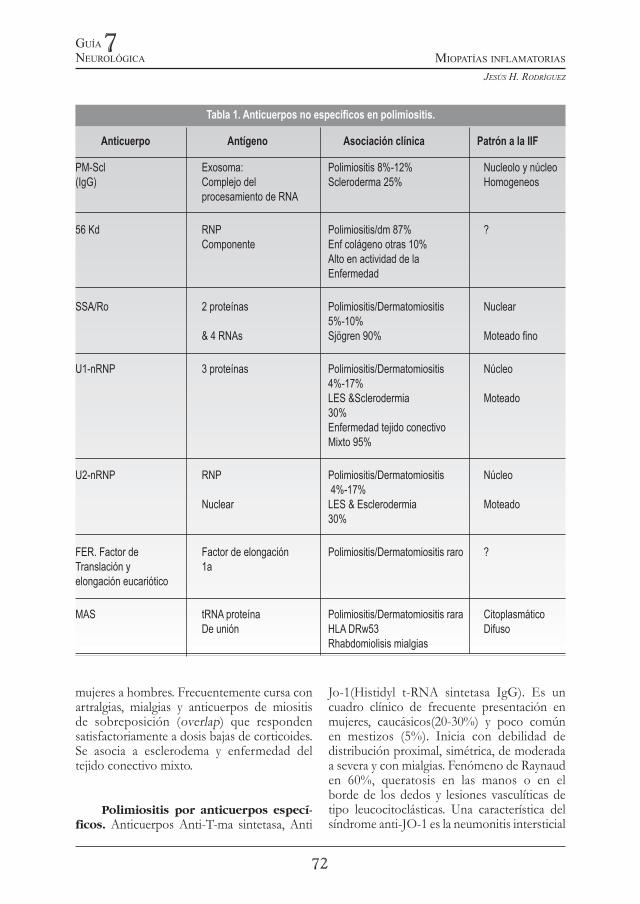
Tabla 1. Anticuerpos no especícos en polimiositis.
Anticuerpo Antígeno Asociación clínica Patrón a la IIF
PM-Scl Exosoma: Polimiositis 8%-12% Nucleolo y núcleo (IgG) Complejo del Scleroderma 25% Homogeneos procesamiento de RNA
56 Kd RNP Polimiositis/dm 87% ? Componente Enf colágeno otras 10% Alto en actividad de la Enfermedad
SSA/Ro 2 proteínas Polimiositis/Dermatomiositis Nuclear 5%-10% & 4 RNAs Sjögren 90% Moteado no
U1-nRNP 3 proteínas Polimiositis/Dermatomiositis Núcleo 4%-17% LES &Sclerodermia Moteado 30% Enfermedad tejido conectivo Mixto 95%
U2-nRNP RNP Polimiositis/Dermatomiositis Núcleo 4%-17% Nuclear LES & Esclerodermia Moteado 30%
FER. Factor de Factor de elongación Polimiositis/Dermatomiositis raro ? Translación y 1aelongación eucariótico
MAS tRNA proteína Polimiositis/Dermatomiositis rara Citoplasmático De unión HLA DRw53 Difuso Rhabdomiolisis mialgias
GUÍA NEUROLÓGICA
7MIOPATÍAS INFLAMATORIAS
JESÚS H. RODRÍGUEZ
73
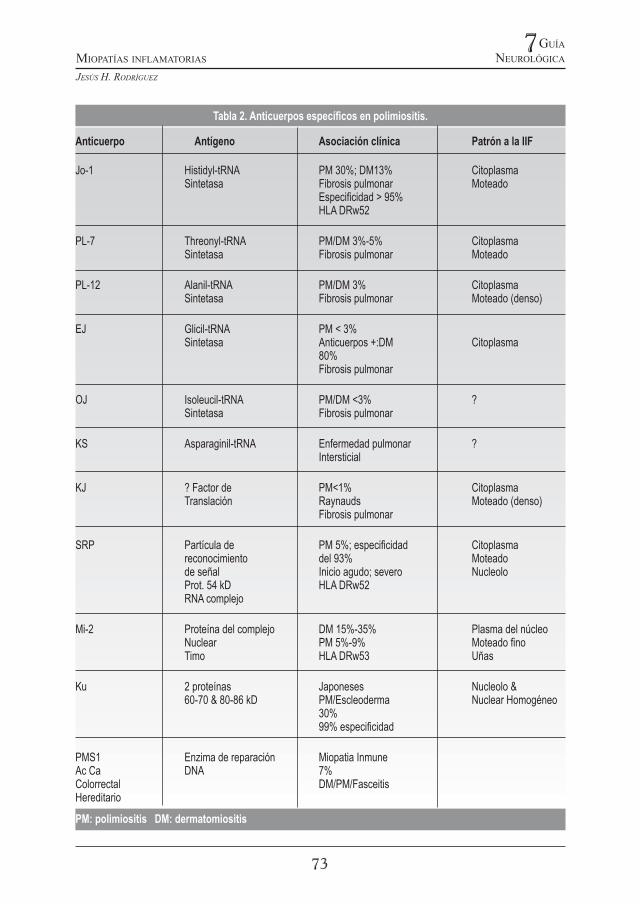
Tabla 2. Anticuerpos especícos en polimiositis.
Anticuerpo Antígeno Asociación clínica Patrón a la IIF
Jo-1 Histidyl-tRNA PM 30%; DM13% Citoplasma Sintetasa Fibrosis pulmonar Moteado Especicidad > 95% HLA DRw52
PL-7 Threonyl-tRNA PM/DM 3%-5% Citoplasma Sintetasa Fibrosis pulmonar Moteado
PL-12 Alanil-tRNA PM/DM 3% Citoplasma Sintetasa Fibrosis pulmonar Moteado (denso)
EJ Glicil-tRNA PM < 3% Sintetasa Anticuerpos +:DM Citoplasma 80% Fibrosis pulmonar
OJ Isoleucil-tRNA PM/DM <3% ? Sintetasa Fibrosis pulmonar
KS Asparaginil-tRNA Enfermedad pulmonar ? Intersticial
KJ ? Factor de PM<1% Citoplasma Translación Raynauds Moteado (denso) Fibrosis pulmonar
SRP Partícula de PM 5%; especicidad Citoplasma reconocimiento del 93% Moteado de señal Inicio agudo; severo Nucleolo Prot. 54 kD HLA DRw52 RNA complejo
Mi-2 Proteína del complejo DM 15%-35% Plasma del núcleo Nuclear PM 5%-9% Moteado no Timo HLA DRw53 Uñas
Ku 2 proteínas Japoneses Nucleolo & 60-70 & 80-86 kD PM/Escleoderma Nuclear Homogéneo 30% 99% especicidad
PMS1 Enzima de reparación Miopatia Inmune Ac Ca DNA 7%Colorrectal DM/PM/FasceitisHereditario
PM: polimiositis DM: dermatomiositis

GUÍA NEUROLÓGICA
7MIOPATÍAS INFLAMATORIAS
JESÚS H. RODRÍGUEZ
74
con un patrón radiológico linear predominante en las bases y adenopatía hiliar asociado a un cuadro de disnea, tos, inflamación, granulomas. Su respuesta a los corticoides es variable. Progresiva en 30% a artritis seronegativa no erosiva, con edema en las manos que afecta las pequeñas articulaciones, osteopenia y fiebre. Existe moderada a severa elevación de la CK (300-30000) anemia (24%) y calcinosis (24%). En la biopsia muscular se observa inflamación con predominio de macrófagos en regiones perivasculares y endomesiales, atrofia perifascicular, dege-neración y regeneración. Tiene una alta mortalidad.
Otro síndrome es el de anticuerpos de partícula de reconocimiento de señal 54kD y 72kD. Comienza entre los 40-50 años, más frecuente en mujeres que en hombres. La debilidad es severa en algunos músculos proximales, simétrica con mayor compromiso de piernas y brazos, mialgias, palpitaciones, disnea, sin lesiones en piel pero con fenómeno de Raynaud en 30%. Su curso es severo o fulminante con una alta mortalidad y recurrencias frecuentes en los casos que sobreviven. Existe elevación severa a muy severa de la CK. En la biopsia muscular se observa degeneración y regeneración con variedad en el tamaño de la fibras musculares con incremento en el tejido conectivo endomesial aún en el curso inicial de la enfermedad, las células inflamatorias son escasas o nulas. En su tratamiento se usan corticoides con resultado variable y mejoría en la iniciación de la enfermedad cuando la debilidad es moderada.
Síndrome de anticuerpos anti-MAS (Proteína de unión al tRNA). Su inicio es agudo a una edad promedio de 36 años. Se presenta con rabdomiolisis más frecuentemente en alcohólicos. Cursa con mialgias en 50% de los casos, palpitaciones y elevación alta de la CK tiene buena respuesta a los esteroides.
Miositis inducida por drogas
Con la d-penicilamina no existe una clara relación con la dosis. Tiene una frecuencia del 1% asociada a artritis reumatoidea, en la que inicia durante cualquier etapa del tratamiento. La debilidad es proximal y se puede asociar a disfagia (40%), mialgias, prurito en algunos pacientes y bloqueo en la conducción cardíaca. Su tratamiento consiste en la suspensión de la medicación, y corticoides con respuesta es variable aunque comúnmente se logra la recuperación. La elevación sérica de la CK es alta en el 75% o se mantiene en rangos normales. La d-penicilamina también puede inducir miastenia gravis, neuromiotonia y pénfigo.
La procainamida puede producir neuro-patía asociada que inicia entre el primer año de vida hasta los cuatro años. Produce parestesias y debilidad proximal simétrica.
Entre otras medicaciones que inducen miopatía se incluyen: la zidovudina (AZT), la hidralazina, el interferón alfa (polineuropatía axonal), y la fenitoina. Con ésta última, la miopatía inflamatoria cursa además con fiebre, rash, linfadenopatia y eosinofilia.
El síndrome del aceite toxico español se presentó por la ingesta de aceite adulterado en 1981 en España. Produce mialgias, que comienzan tres a cuatro semanas después de la exposición, de expresión difusa y severa asociado a falla respiratoria, cefalea y alta mortalidad en especial mujeres. Produce discapacidad a largo plazo en el 1-2% y neuropatía en el 10%.
Las estatinas: (Lovastatina, Pravastatina, Simvastatina) comprometen el músculo con una frecuencia de 1-6/10000. Afecta con más frecuencia en mujeres de edad mayor con enfermedad renal, hepática, diabetes, hipotiroidismo, estado de debilidad, cirugía, trauma o abuso de alcohol. También con el uso concomitante de gemfibrozilo cuando usualmente en los 2-3 meses de iniciada de terapia se producen mialgias y elevación de

GUÍA NEUROLÓGICA
7MIOPATÍAS INFLAMATORIAS
JESÚS H. RODRÍGUEZ
75
la CK. Cursa con calambres, incomodidad y dolor al reposo que se incrementa con el ejercicio. Aunque ocurre con CK normal, en el caso de mioglobinuria se recupera entre días a meses después de la suspensión. Se presenta asociado a neuropatía después de largo uso entre 1-7 años, generalmente más frecuente en edades entre los 41-68 años. La densidad de incidencia de asociación de uso de estatina con polineuropatía es de 1 en 2200 personas-año. Se descubre debilidad y parestesias en el 20% de estos casos.
Derivados del acido fibrico: (fibratos) clorfibrato, benzafibrato, ciprofibrato, clorfi-brato y gemfibrozil afectan el músculo con una frecuencia de 1 por 10000 pacientes, con un riesgo 42 veces mayor de miopatía en pacientes que usan fibratos versus los controles.
Así mismo aumenta el riesgo en falla renal, síndrome nefrótico también se asocia a drogas como probenecid, furosemida, después de los 2-3 meses de estar en tratamiento.
Otras drogas de precaución: niacina, ciclosporina, azoles, diltiazem, eritromicina con las que se presenta elevación alta de la CK, aún asintomáticas. Otras drogas posiblemente relacionadas son la penicilina, IPECA, emetina, sulfonamida, levodopa, cimetidina, leuprolide, propiltiouracil y carbimazole.
Familiar. Clínicamente similar a la miopatía idiopática, algunas son: distrofia fascioescapulohumeral, miositis por cuerpos de inclusión familiar, miopatía inflamatoria idiopática familiar, síndrome de Schmidt, síndrome de fiebre recurrente con miositis o fasceitis localizada (TRAPS).
Miopatía en enfermedad injerto vs. huésped. Es una reacción inmune mediada por una disparidad en el complejo mayor de histocompatibilidad HLA donante-receptor más frecuente en mujeres que se presenta con debilidad simétrica y mialgias en un 60%. La CK se eleva en el rango de 49-8400 Ul, la aldolasa alta en el 66% y en el rango de 5-88 UL y una elevación en el 90% de enzimas hepáticas en el rango moderado a alto. En la fase aguda en los primeros 100 días después
de transplante de médula ósea se manifesta en piel con exantema maculopapular, necrolisis epidérmica, cambios hepáticos similares a cirrosis biliar y microangiopatía intestinal. Después de 80 días en la fase crónica la expresión es variada con Liken plano en piel, esclerodermia-like, vitíligo, lesiones en boca y esófago liquenoides, queratoconjuntivitis y bronquiolitis que progresa a falla respiratoria. Aunque se complica rara vez con mielitis o neuritis óptica. Tiene mayor riesgo de presentarse en mayores de 20 años o cuando ha habido previamente rechazo (35%), cuando el donante es una mujer. Se maneja con ciclosporina, prednisona o azatioprina.
Miopatías por enfermedades granulo-matosas. (Tabla 3) algunas características clínicas son debilidad proximal que se puede asociar a disfagia, hipertrofia muscular ocasional con lenta progresión y dolor en la fase aguda, la CK puede estar alta o normal aunque la aldolasa se eleva más frecuentemente que la CK. En la patología los granulomas son no caseificantes con localización endomesial o perimesial. Generalmente, con el uso de esteroides se benefician entre el 40-80% de los casos.
Miopatía en la sarcoidosis. Presenta la mayoría de características de las miopatías por granulomatosis con una mayor presentación en mujeres de mediana edad. La debilidad es simetrica y puede ocurrir debilidad selectiva con falla respiratoria en los músculos ventilatorios. La CK se eleva moderadamente con pocos cambios a la resonancia magnética muscular. La patología demuestra signos miopáticos con granulomas no caseificantes con células gigantes. La presentación del cuadro clínico puede ser agudo con mialgias, con o sin debilidad proximal simétrica asociado a poliartritis, eritema nodoso con pocas o sin manifestaciones sistémicas. En la forma aguda la CK puede estar alta a medida que evoluciona la enfermedad, progresando de días hasta dos meses. Con tratamiento a base de corticoides, se pueden presentar nódulos en los músculos y crecimiento de la masa muscular con dolor que debe diferenciarse con una miositis nodular o artritis reumatoidea. La sarcoidosis muscular

GUÍA NEUROLÓGICA
7MIOPATÍAS INFLAMATORIAS
JESÚS H. RODRÍGUEZ
76
es asintomática hasta en un 50-80% de los casos y con CK normal.
Miopatías asociados a neoplasias (Miopatía Necrótica). Generalmente se presenta en pacientes ancianos y mayores de 40 años con debilidad proximal, simétrica de curso rápido que lleva a discapacidad severa en menos de dos a tres meses. La sobrevida esta relacionada con las posibilidades de respuesta al tratamiento de la neoplasia. Entre las neoplasias más frecuentemente asociadas con miositis se incluyen: pulmonar, tracto gastrointestinal, estómago, colon (adenocarcinoma), próstata y mama. La CK generalmente se elevada de 8-100 veces el valor normal de referencia. La patología demuestra fibras musculares necróticas e infiltración del músculo por macrófagos y fagocitosis en el endomisio con depósitos de complemento. Su tratamiento incluye la administración de corticoides.
Miosistis en enfermedades mitocon-driales. Ocurre a edades entre los 43-73 años con debilidad severa y temprana del cuadríceps (90%) y distribución proximal más habitualmente que distal en el 30% de los casos. Compromete la cara en el 50% y en el 80% de las veces es simétrica, progresa lentamente. Los niveles sericos de CK son normales o levemente elevados en el rango de 54-1200 UL. La patología muscular se encuentra en el citocromo oxidasa negativo, succinic dehidrogenasa positivo (SDH) o fibras rojas
rasgadas (ragged red fibers). En el 4%-27% de todas las fibras hay inflamación endomisial, invasión focal o deleciones mtDNA. El manejo es con metrotexate 10mgrs/K/semana. En los pacientes que no presenten respuesta debe descartarse la posibilidad de miositis de cuerpos de inclusión o repetir la biopsia. Por lo general no hay respuesta al tratamiento con corticoides.
Miosistis y otros desórdenes sistémicos
Miosistis asociada al SIDA. Se presenta tempranamente con polimiositis similar a la que ocurre en pacientes sin VIH. Mejora con corticoides y se ha establecido el beneficio del uso de pulsos cuando existen altos niveles de CK, necrosis de fibras musculares e infiltrado inflamatorio. También se presenta tardiamente y relacionado con el uso de zidovudina. Es frecuente en el inicio de la infección por VIH y con dosis altas del medicamento la ocurrencia de debilidad proximal, mialgia y niveles elevados de CK. Los hallazgos electromiográficos consisten en inestabilidad (hiperexcitabilidad) de membrana y actividad espontánea. Usualmente hay mejoría a los 3-4 meses de haber descontinuado la medicación.
Miopatías asociadas a enfermedades linfoproliferativas y gammapatia mono-clonal. El riesgo de transformación a neoplasia es de 1% a un año de seguimiento, 12% a los
Tabla 3. Miopatías por enfermedades granulomatosas.
Infecciosas: histoplasma, ccocidiodes, blastomices, Sporotrix, aspergillus, criptococcus, toxoplasma, lehismania, toxocara, schistosoma, Treponema pallidum, T pertunue, T carateum, M. tuberculosis, M lepra, M. Kansassi, M marinum, M avium-intracellulare, vacuna BCG, Brucilla, Yersinia, arañazo de gato, linfogranuloma
Neoplásicas: carcinoma, reticulosis, pinealoma, disgerminoma, seminoma, sarcoma de células reticulares, granuloma nasal linfoma
Químicas: aluminio, berilio, titanio, circonio, sílice, almidón. Inmunológicas sarcoidosis, miastenia gravis y timoma, enfermedad de Crohn, cirrosis 1ria biliar, granulomatosis de Wegener, arteritis de células gigantes, Churge Strauss, hipogammaglobulinemia, LES, granulomatisis linfoide, histiocitosis X, enfermedad granulomatosa hepática, enfermedades del complejo inmune, síndrome de Rosenthal-Melkersson, granulomatosis alérgica, enfermedad de Peyronie, Pneumonitis de hipersensibilidad
Otras: Whipple, radioterapia, panniculitis, chalazionpirexia, quiste cebáceo.

GUÍA NEUROLÓGICA
7MIOPATÍAS INFLAMATORIAS
JESÚS H. RODRÍGUEZ
77
10 años, 25% a los 20 años 30% a 25 años. Otros factores de riesgo son la concentración inicial de IgM, y la ausencia de cadenas ligeras en la orina.
Fascitis. Generalmente afecta más a cuadriceps. Aparece en síndromes específicos y entidades autoinmunes como eosinofilia-mialgia, intoxicación por aceite toxico español, eosinofilia-fasciitis, perimiositis..
Dermatomiositis
Se caracterizan por presentar concomi-tantemente alteraciones en la piel y miopatía. La frecuencia es mayor en meso-América y menor en el hemisferio norte. Representa mas del 90% de la miopatías inflamatorias en los niños. Las lesiones en piel se presentan con un rash heliotropo, edema de los parpados y cara, áreas expuestas al sol y superficies extensoras de las articulaciones. La debilidad es proximal y con disfagia en algunos casos. Otras característica son retinopatía, queratoconjuntivitis, iritis, uveítis, enfermedad cardíaca de pequeños vasos y angina de prinzmetal.
La dermatomiositis juvenil se presenta con una incidencia del 0,14-0,5 por 100 000 con un predominio en mujeres a hombres de 1.7-5.1. En algunos hay pródromos de infección del tracto respiratorio superior. Otros se han asociado a vacunación con HVB, suplencia con hormona del crecimiento, exposición excesiva al sol, medicamentos como D penicilamina, infección viral por coxsakie B, parvovirus, echovirus y algunos subtipos de HLA. La debilidad muscular es de predominio proximal con dolor muscular, rash característico que se puede exacerbar por la luz. La mortalidad es del 7% y es común una recuperación incompleta. Los niveles de CK pueden ser normales o altos en el momento de la presentación. Se puede presentar asociación con allotipos con cadena gamma de inmunoglobulina en niños mestizos.
Dermatomiositis del adulto anticuer-pos Mi-2(ANA de patrón nuclear moteado) negativo. Se asocia a arritmia cardíaca,
enfermedad intersticial del pulmón, un riesgo alto de malignidad, fenómeno de Raynaud y enfermedades del colágeno (CREST, AR, Scleroderma). Es más frecuente en mestizos aunque ocurre en caucásicos y el compromiso de piel es relativo en superficies expuestas a los rayos ultravioleta. El rash es característicamente unas en V con compromiso de las y crecimiento cuticular. La edad de inicio es desde los tres años hasta el adulto. Los ANA son positivos en el 100%.
Dermatomiositis asociada a maligni-dad. La frecuencia de presentación varía de un grupo étnico a otro y aumenta después de los 45 años de edad. La distribución es igual entre mujeres y hombres y se asocia más frecuentemente con dermatomiositis que con polimiositis, en blancos del norte de Europa y australianos. Hasta el 58% desarrolla neoplasia después del diagnóstico de dermatomiositis, en especial con cáncer de ovario (riesgo es 10,5 veces mayor), pulmón (5,9), páncreas (3,8), linfoma no Hodking (3,6), estómago (3,5) y colorectal (2,5).
Dermatomiositis con anticuerpos EJ. Se presenta también en polimisotis en 3% de los casos y es más frecuente con dermatomiositis en la que el 80% se asocia con fibrosis pulmonar.
Dermatomisitis con anticuerpos anti-pm-Scl (IgG). Anticuerpo contra exosoma y complejo proteico nuclear. Es común en polacos y no se ha escrito en japoneses. Se asocia en un 75%- 100% con el HLA-DR3. Con una presentación de sobre posición en un 24% con escleroderma. Cursa con debilidad proximal, lesiones en piel, manos de maquinistas (hiperqueratosis), fenómeno de Raynaud y telangiectasia. Presenta frecuen-temente respuesta a corticoides y es de buen pronóstico.
Dermatomiositis amiopática. Se pre-senta con una frecuencia del 8%-20% de las dermatomiositis en edad adulto media, aunque ocasionalmente es juvenil. Predomina en mujeres con excepción de la China. La fuerza puede conservarse normal. Característicamente

GUÍA NEUROLÓGICA
7MIOPATÍAS INFLAMATORIAS
JESÚS H. RODRÍGUEZ
78
se observa el rash en heliotropo, artralgias, nódulos de Gottron, lesiones periungueales, ocasionalmente prurito, eritema violáceo con máculas y pápulas. En la China se ha descrito asociación con malignidad. Los niveles de CK pueden ser normales. El manejo es conservador con inmunosupresión con corticoterpia sistémica o tópica.
Dermatomiositis Mi-2 anticuerpo positivo. Es más común en mestizos (Méjico y Guatemala), inicio desde los tres años hasta en el adulto y cursa con rash en V característico, crecimiento cuticular, rara vez asociado a enfermedades del tejido conectivo, con ANA positivos en el 100% y HLA característicos que varían según grupo étnico.
Miositis de cuerpos de inclusión
Es preponderante en hombres, con el 60-80% de forma esporádica. Aparece en mayores de 50 años con debilidad en las piernas y que produce en 70% dificultad para levantarse de sillas o subir escaleras. El compromiso predomina en el músculo cuadriceps aunque más distalmente en los brazos en un 15%. Una dificultad para la deglución ocurre en el 10% de los casos. Frecuentemente se respetan deltoides, pectorales, interóseos y músculos de la cara. La progresión es lenta en (5 a 20 años). Sin embargo, su progresión es más rápida cuando aparece en mayores de 60 años. La atrofia muscular es proporcional a la perdida de fuerza. Los reflejos osteotendinosos se disminuyen conforme progresa la enfermedad especialmente al comprometer la rodilla. La disfagia es subclínica en el 30% de los casos y compromete más en mujeres, el tercio superior del esófago. No existe asociación con enfermedades sistémicas. La CK se eleva moderadamente de 2-5 veces los valores de referencia. En la biopsia del nervio sural se observa pérdida de axones mielinizados. La electromiografía demuestran signos de hiperexcitabilidad de membrana y no se obtiene potencial sensorial en el nervio sural en mayores de 60 años. En la biopsia muscular se observa inflamación endomesial
con invasión focal de las fibras musculares, vacuolas redondeadas con material granular y filamentos. La inmunohistoquimica con tintura de rojo congo demuestra filamentos y muchas proteínas beta amiloide. Existen desórdenes mitocondriales en el 50% de los pacientes con miositis de cuerpos de inclusión como delecion en el mDNA. Clínicamente se puede observar hipertrofia de fibras musculares incluso más comúnmente que en la polimiositis. La resonancia magnética del músculo, en especial el gastronemius, demuestra infiltración grasa. No hay un tratamiento efectivo, excepto por los pocos reportes de mejoría con inmunoglobulina IgG intravenosa. En su variante hereditaria autosómica dominante, las características clínicas son similares a la variante esporádica y no existe un HLA asociado.
Otras miopatías inflamatorias
Miopatía asociada a la deficiencia de condroitin sulfato C en el músculo esquelético. Es un desorden raro descrito en pocos pacientes que, ocurre en adultos mayores de 42 años, con debilidad difusa, simétrica proximal, falla respiratoria y compromiso en meses de pares craneales con oftalmoplejia y disfagia. Los niveles de CK y aldolasa se man-tienen normales y se pueden presentar ANA elevados. La velocidad de neuroconduccion es normal y el EMG demuestra un patrón miopático. En la biopsia muscular no hay cambios miopáticos o existe atrofia de fibras musculares tipo II, con inflamación perimesial y perivascular. La tinción con condroitin sulfato indica ausencia en el endomisio. El tratamiento con corticoides produce mejoría en los meses siguiente de iniciados.
Miopatía con anticuerpos anti-decorin (antígeno BJ). Inicio generalmente en la 7ª década de la vida con debilidad proximal, simétrica, conservación de reflejos osteoten-dinsos y curso lentamente progresivo hasta una moderada discapacidad. Los niveles de CK son generalmente altos alrededor de las 1000 UL. Se presenta en asociación a macroglobulinemia de Waldenstrom con

GUÍA NEUROLÓGICA
7MIOPATÍAS INFLAMATORIAS
JESÚS H. RODRÍGUEZ
79
test positivo para el antígeno decorin, un proteoglicano localizado en la membrana basal. Hay actividad electromiográfica espontánea y patrón miopático. En la patología, las fibras musculares son de tamaño variable con fibrosis endomesial y depósitos de IgM.
Miositis aguda inflamatoria de la infancia. El 84% de los casos afecta niños. En promedio se inicio a los 8 años con rango de 3 - 13 años, 1 a 4 días después de un pródromos con fiebre (80%), tos (59%) y cefalea(37%) asociado a virus de influenza. Tiene relación con las estaciones de verano e invierno. Existe dolor en músculos, especialmente en pantorrilla o generalizado. La fuerza es normal en la mayoría. El curso de la enfermedad es generalmente durante una semana y existe recurrencia alrededor de un 8%. Los niveles de CK en la fase aguda son altos entre 250-14000 UL y después de un mes de la recuperación son normales. Se demuestran títulos virales positivos hasta en un 42% en especial para influenza (21%). En la biopsia muscular la fibras musculares son necróticas con algunos infiltrados inflamatorios.
Enfermedad celíaca. Es una reacción alérgica al gluten en los cereales. Con una respuesta inmune mediada por células T con predisposición genética y como blanco antigénico la transglutaminasa. Es más frecuente en mujeres que en hombres y geográficamente su distribución es mayor en Europa del este y rara en africanos, caribeños o asiáticos. Inicia en la niñez y hasta en la 7a década. Se presenta con diarrea, anemia, hemorragia, flatulencia, dermatitis herpetiforme, osteopenia y nefropatia mesangial IgA. Los hallazgos neurológicos son neuropatía de tipo axonal de predominio sensitivo, dolorosa, distal, simétrica y de lenta progresión. Otra forma de presentarse es como una neuropatía sensitivo motora o mononeuritis múltiple. La miopatía es poco frecuente y afecta el 5% de los casos de carácter inflamatoria y con tetania e hipocalcemia.
Miositis focales y fasceitis. (Ver polimiositis).
Linfohistiocitosis hemofagocítica. Se presenta como una actividad de los macrófagos con hemofagocitosis generalizada de las plaquetas y precursores. Sus caracte-rísticas clínicas son la fiebre persistente, pancitopenia, organomegalia, esplenomegalia, hepatomegalia, linfadenopatia, crisis convul-sivas, edema cerebral, rash, mialgias y niveles de enzimas hepáticas elevadas, especialmente la LDH. Su tratamiento es generalmente la administración de etoposido y prednisona o ciclofosfamida.
Miopatía necrotizante con capilares en boquilla de pipa (pipesteam). Esta miopatía aparece entre la 5a o 7ma edad con debilidad episódica subagudas. Después del ejercicio se inducen mialgias con edema en las siguientes 12 a 48 horas. La debilidad es proximal y llega ser severa, en ocasiones relacionada a dermatomiositis y rash característico. En el sistema nervioso central se presentan múltiples infartos con patrón de vasculitis. Se asocia a neoplasia de la vejiga. Así mismo, miocarditis y enfermedades no diferenciadas del tejido conectivo. La CK se eleva a rangos altos de 15 a 50 veces el valor de referencia. En la biopsia hay necrosis y fibras en rege-neración. Los característicos capilares en boquilla de pipa se presentan por depósitos amorfos de material del complemento en complejos de ataque de la membrana con reducción focal. Su tratamiento son los corticoides.
Miosistis idiopática orbitaria. Se define como uno o más músculos extraoculares infiltrados con células inflamatorias de etiología idiopática. Es mas frecuente en mujeres que en hombres con una razón de 2: 1. Inicia entre los 30-40 años con rango desde los 3-84 años. Su curso es agudo con recaídas y cronicidad. Se presenta visión doble con agudeza visual normal, proptosis, edema periorbitario, inyección conjuntival, quemosis y dolor. Usualmente los mismos músculos extrao-culares se comprometen bilateralmente. Los músculos más comúnmente involucrados

GUÍA NEUROLÓGICA
7MIOPATÍAS INFLAMATORIAS
JESÚS H. RODRÍGUEZ
80
son el recto medio (43%), recto superior (19%) y recto lateral (17%). El diagnóstico diferencial contempla el hipertiroidismo que no es dolorosa; las infecciones que con frecuencia reducen la agudeza visual y producen fiebre; la neoplasia orbitaria, fístula carótida-cavernosa y trombosis del seno cavernoso. En la resonancia magnética se observan músculos aumentados de tamaño y tendones engrosados. En la patología los músculos son edematosos con infiltrados inflamatorios y eosinofilia en algunos. Su tratamiento es con corticoides, inmunosupresores, manejo quirúrgico o con toxina botulínica de la diplopía crónica.
LABORATORIOS BÕSICOS EN MIOPAT◊AS (Tabla 4).
Hallazgos electrofisiológicos
Generalmente las miopatías no compro-meten las neuroconducciones sin embargo existen alguna patologías específicas, decritas anteriormente, que pueden afectarlas. Es recomendable limitar el estudio de EMG a un hemicuerpo ya que este puede elevar la CK y afectar la interpretación de la biopsia muscular. Otra recomendación es realizar la electromiografía con aguja bipolar. Los parámetros de morfología (fases) y duración cuyos valores se han descrito en varias tablas disponibles y que se discriminan por edad y músculos se han obtenido utilizanbdo este tipo de electrodo. El electrodo de aguja debe tener un área de análisis de aproximadamente 0,07 mm2. El análisis de los potenciales de unidad motora también se puede realizar con aguja monopolar, en cuyo caso los filtros deben modificarse. Al momento de decidir que músculos serán evaluados se deben tener en cuenta los músculos involucrados en la patología que se sospecha. Dado que existen diferentes estadíos en la enfermedad, así mismo el compromiso de los músculos puede variar. La selección se debe basar en los hallazgos clínicos como dolor, atrofia, debilidad a fin de
proceder a la exploración con aguja en aquellos músculos comprometidos. Es una buena práctica obviar la exploración de músculos con compromiso avanzado, atrofia e infiltración grasa o fibrosa.
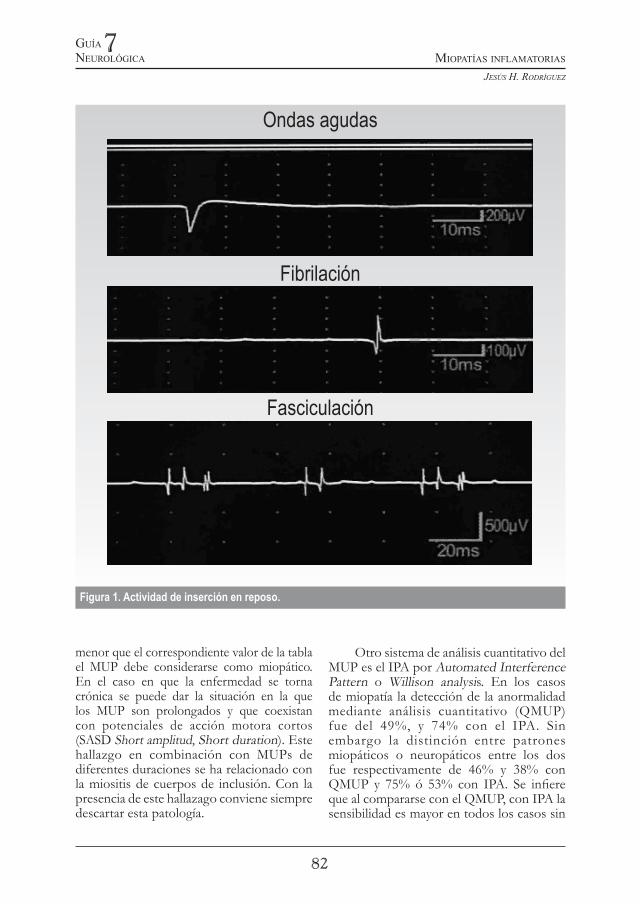
En el acto de insertar el electrodo el examinador experimentado aprecia la consistencia y resistencia del músculo al paso de la aguja. En el monitor y con el audio identifica y reconoce cualquier actividad espontánea que se genere con la inserción indicando la irritabilidad e hiperexcitabilidad de la membrana muscular del tipo descargas miotónicas repetitivas, ondas positivas, fibrilaciones o fasciculaciones (Figura 1).
Análisis cualitativo y cuantitativo del potencial de unidad motora a la actividad voluntaria. Generalmente se le pide al paciente que mantenga una contracción muscular mínima generando descargas a una frecuencia baja. Esto facilita la observación y determina-ción de las características del potencial de unidad motora (MUP). En ella se cuantificará el número de fases, su duración y amplitud. Los valores obtenidos se confrontan con los valores incluidas en las tablas mencionadas anteriormente (Tabla 5).
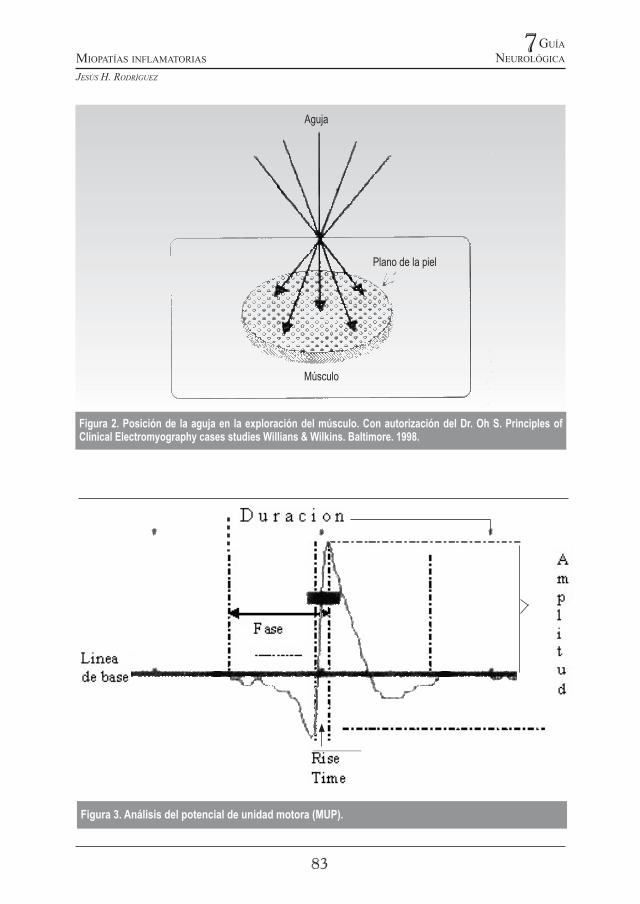
Existen requisitos técnicos también como que los filtros de alta frecuencia o high frequency filter (HFF) estén entre el rango estándar de de análisis de 3,2 Hz y 32 Khz en el que se garantiza no se falsee los parámetros por un recorte (cutoff) impuesto por una mala selección del filtro. Este error puede hacer variar el valor de la duración del MUP mostrándolo de corta duración. Cuando se utilice aguja monopolar se recomienda situar los filtros entre 2Hz y 10 Khz. La ganancia o gain, generalmente se utiliza en 100μv con una velocidad de 5 a 10 milisegundos/div. Al menos 20 potenciales de acción diferentes deben ser evaluados. Esta se logra con mínimos desplazamientos de la aguja en diferentes direcciones de la masa músculo, lo que permite explorar diferentes partes del músculo y por ende distintas unidades motoras (Figura 2).
GUÍA NEUROLÓGICA
7MIOPATÍAS INFLAMATORIAS
JESÚS H. RODRÍGUEZ
81
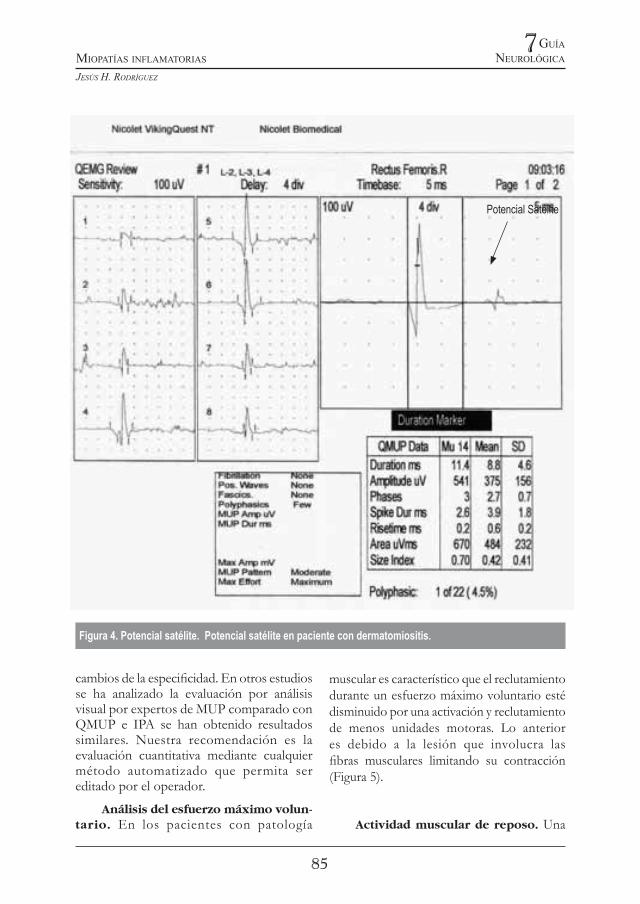
En algunos casos se debe remover la aguja e insertarla nuevamente en otro punto a poca distancia a fin de lograr otras MUP. En este punto se debe evaluar también que el MUP debe tener un “rise time” entre 0,2ms-0,5 ms. Este parámetro se mide en el registro como el tiempo desde el momento en que arranca la MUP hasta la máxima amplitud del primer pico (Figura 3). De esta manera se pueden utilizar los valores incluidos en tablas obtenidas en otras poblaciones. Se debe tener en cuenta también la posibilidad de existencia de potenciales satélites que se excluyen de la medición de la duración
(Figura 4). La medición de la duración del potencial de acción motora (MUP) comienza en la primera deflexión positiva y culmina en el punto en que se retorna a la línea de base original (Figura 3). Una vez se ha establecido la duración de los diferentes potenciales de unidad motora (mínimo 20) se realiza un promedio. Los equipos permiten calcular automáticamente este promedio (Tabla 4).
Para corregir por variabilidad a este resultado en la tabla se le debe restar un 20%. Si el resultado de la duración del MUP es
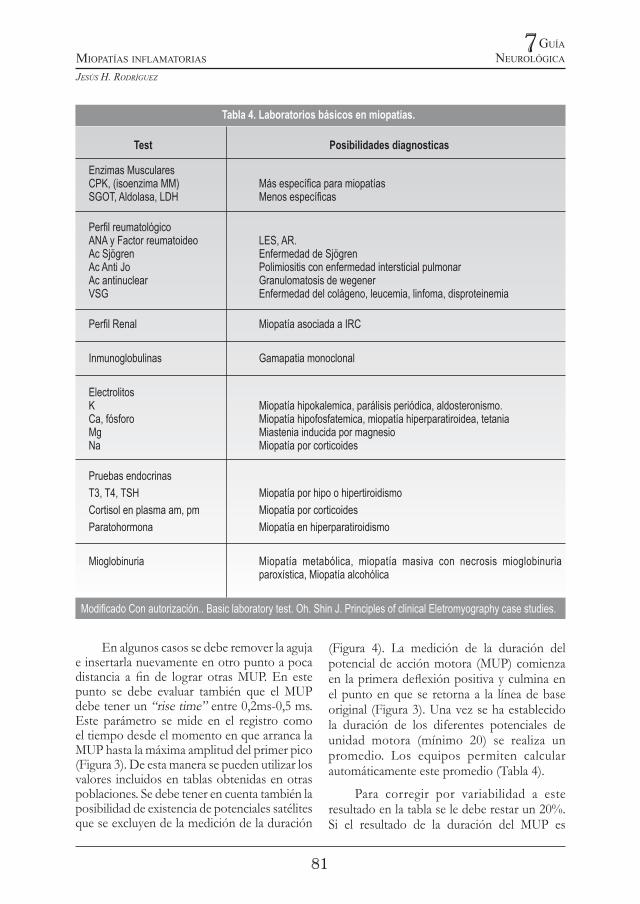
Tabla 4. Laboratorios básicos en miopatías.
Test Posibilidades diagnosticas
Enzimas MuscularesCPK, (isoenzima MM) Más especíca para miopatíasSGOT, Aldolasa, LDH Menos especícas
Perl reumatológicoANA y Factor reumatoideo LES, AR. Ac Sjögren Enfermedad de SjögrenAc Anti Jo Polimiositis con enfermedad intersticial pulmonarAc antinuclear Granulomatosis de wegenerVSG Enfermedad del colágeno, leucemia, linfoma, disproteinemia
Perl Renal Miopatía asociada a IRC
Inmunoglobulinas Gamapatia monoclonal
ElectrolitosK Miopatía hipokalemica, parálisis periódica, aldosteronismo.Ca, fósforo Miopatía hipofosfatemica, miopatía hiperparatiroidea, tetaniaMg Miastenia inducida por magnesioNa Miopatía por corticoides
Pruebas endocrinasT3, T4, TSH Miopatía por hipo o hipertiroidismoCortisol en plasma am, pm Miopatía por corticoidesParatohormona Miopatía en hiperparatiroidismo
Mioglobinuria Miopatía metabólica, miopatía masiva con necrosis mioglobinuria paroxística, Miopatía alcohólica
Modicado Con autorización.. Basic laboratory test. Oh. Shin J. Principles of clinical Eletromyography case studies.
GUÍA NEUROLÓGICA
7MIOPATÍAS INFLAMATORIAS
JESÚS H. RODRÍGUEZ
82
Figura 1. Actividad de inserción en reposo.
menor que el correspondiente valor de la tabla el MUP debe considerarse como miopático. En el caso en que la enfermedad se torna crónica se puede dar la situación en la que los MUP son prolongados y que coexistan con potenciales de acción motora cortos (SASD Short amplitud, Short duration). Este hallazgo en combinación con MUPs de diferentes duraciones se ha relacionado con la miositis de cuerpos de inclusión. Con la presencia de este hallazago conviene siempre descartar esta patología.
Otro sistema de análisis cuantitativo del MUP es el IPA por Automated Interference Pattern o Willison analysis. En los casos de miopatía la detección de la anormalidad mediante análisis cuantitativo (QMUP) fue del 49%, y 74% con el IPA. Sin embargo la distinción entre patrones miopáticos o neuropáticos entre los dos fue respectivamente de 46% y 38% con QMUP y 75% ó 53% con IPA. Se infiere que al compararse con el QMUP, con IPA la sensibilidad es mayor en todos los casos sin
GUÍA NEUROLÓGICA
7MIOPATÍAS INFLAMATORIAS
JESÚS H. RODRÍGUEZ
83
Figura 2. Posición de la aguja en la exploración del músculo. Con autorización del Dr. Oh S. Principles of Clinical Electromyography cases studies Willians & Wilkins. Baltimore. 1998.
Aguja
Plano de la piel
Músculo
Figura 3. Análisis del potencial de unidad motora (MUP).
GUÍA NEUROLÓGICA
7MIOPATÍAS INFLAMATORIAS
JESÚS H. RODRÍGUEZ
84
Tabla 5. Análisis cuantitativo de los potenciales de unidad motora (QMUP).
Del Laboratorio de neurosiología del Hospital de la University of Alabama At Birmingham. UAB. Con autorizacion del Dr. Shin J Oh S.
GUÍA NEUROLÓGICA
7MIOPATÍAS INFLAMATORIAS
JESÚS H. RODRÍGUEZ
85
cambios de la especificidad. En otros estudios se ha analizado la evaluación por análisis visual por expertos de MUP comparado con QMUP e IPA se han obtenido resultados similares. Nuestra recomendación es la evaluación cuantitativa mediante cualquier método automatizado que permita ser editado por el operador.
Análisis del esfuerzo máximo volun-tario. En los pacientes con patología
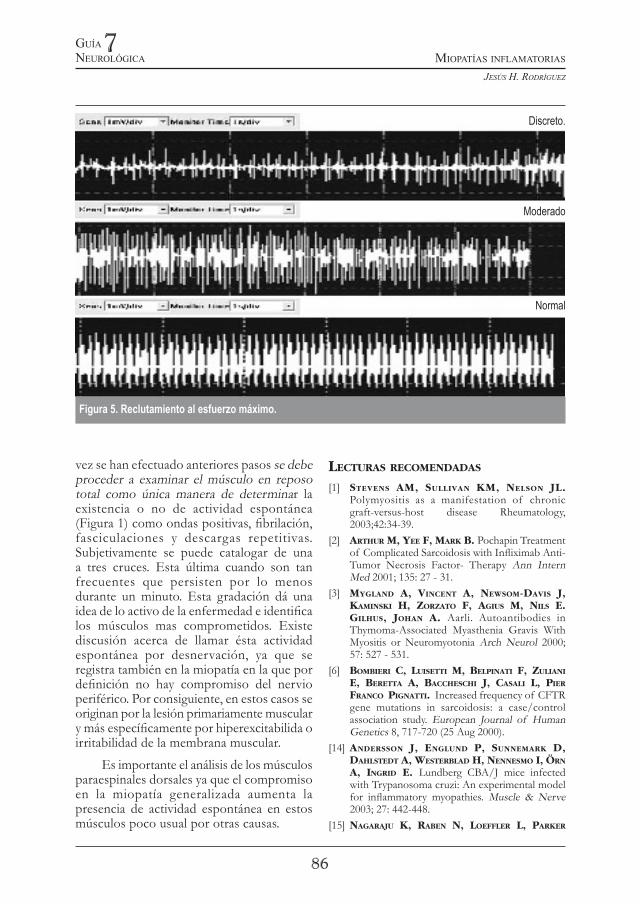
muscular es característico que el reclutamiento durante un esfuerzo máximo voluntario esté disminuido por una activación y reclutamiento de menos unidades motoras. Lo anterior es debido a la lesión que involucra las fibras musculares limitando su contracción (Figura 5).
Actividad muscular de reposo. Una
Figura 4. Potencial satélite. Potencial satélite en paciente con dermatomiositis.
Potencial Satélite
GUÍA NEUROLÓGICA
7MIOPATÍAS INFLAMATORIAS
JESÚS H. RODRÍGUEZ
86
vez se han efectuado anteriores pasos se debe proceder a examinar el músculo en reposo total como única manera de determinar la existencia o no de actividad espontánea (Figura 1) como ondas positivas, fibrilación, fasciculaciones y descargas repetitivas. Subjetivamente se puede catalogar de una a tres cruces. Esta última cuando son tan frecuentes que persisten por lo menos durante un minuto. Esta gradación dá una idea de lo activo de la enfermedad e identifica los músculos mas comprometidos. Existe discusión acerca de llamar ésta actividad espontánea por desnervación, ya que se registra también en la miopatía en la que por definición no hay compromiso del nervio periférico. Por consiguiente, en estos casos se originan por la lesión primariamente muscular y más específicamente por hiperexcitabilida o irritabilidad de la membrana muscular.
Es importante el análisis de los músculos paraespinales dorsales ya que el compromiso en la miopatía generalizada aumenta la presencia de actividad espontánea en estos músculos poco usual por otras causas.
Figura 5. Reclutamiento al esfuerzo máximo.
Discreto.
Moderado
Normal
LECTURAS RECOMENDADAS
[1] STEVENS AM, SULLIVAN K M, NELSON JL. Polymyositis as a manifestation of chronic graft-versus-host disease Rheumatology, 2003;42:34-39.
[2] ARTHUR M, YEE F, MARK B. Pochapin Treatment of Complicated Sarcoidosis with Infliximab Anti-Tumor Necrosis Factor- Therapy Ann Intern Med 2001; 135: 27 - 31.
[3] MYGLAND A, VINCENT A, NEWSOM-DAVIS J, KAMINSKI H, ZORZATO F, AGIUS M, NILS E. GILHUS, JOHAN A. Aarli. Autoantibodies in Thymoma-Associated Myasthenia Gravis With Myositis or Neuromyotonia Arch Neurol 2000; 57: 527 - 531.
[6] BOMBIERI C, LUISETTI M, BELPINATI F, ZULIANI E, BERETTA A, BACCHESCHI J, CASALI L, PIER FRANCO PIGNATTI. Increased frequency of CFTR gene mutations in sarcoidosis: a case/control association study. European Journal of Human Genetics 8, 717-720 (25 Aug 2000).
[14] ANDERSSON J, ENGLUND P, SUNNEMARK D, DAHLSTEDT A, WESTERBLAD H, NENNESMO I, fi RN A, INGRID E. Lundberg CBA/J mice infected with Trypanosoma cruzi: An experimental model for inflammatory myopathies. Muscle & Nerve 2003; 27: 442-448.
[15] NAGARAJU K, RABEN N, LOEFFLER L, PARKER

GUÍA NEUROLÓGICA
7MIOPATÍAS INFLAMATORIAS
JESÚS H. RODRÍGUEZ
87
T, ROCHON P, LEE E, DANNING C, WADA R, THOMPSON C, BAHTIYAR G, CRAFT J, HOOFT R VAN HUIJSDUIJNEN, PLOTZ P. Conditional up-regulation of MHC class I in skeletal muscle leads to self-sustaining autoimmune myositis and myositis-specific autoantibodies. PNAS 2000; 97: 9209-9214.
[19] OH SJ. Principles of Clinical Electro-myography. Baltimore:Williams & Wilkins 1998; 77: 391.
[20] OH SHIN J. Color atlas of Nerve biopsy pathology.
Boca Raton: CRC press 3th Ed. 2002: 110-111 Ilustraciones.
[21] OH SHIN J. Clinical Electromyography Nerve Conduction Studies. Third edition. Lippincott Philadelphia: Williams & Wilkins. 2003: 151.
[23] P.D.W.KIELY,C.W.HERON, ANDF.E.Bruckner Presentation and management of idiopathic inflammatory muscle disease: four case reports and commentary from a series of 78 patients Rheumatology, 2003;42:575-582.