
HEMOFILIA ALCANZANDO LA TERCERA EDAD
43
HEMOFILIA ALCANZANDO LA TERCERA EDAD Lcda. Esp. María Salcedo
-
Upload
ministerio-del-poder-popular-para-la-salud-de-venezuela -
Category
Health & Medicine
-
view
34 -
download
6
Transcript of HEMOFILIA ALCANZANDO LA TERCERA EDAD

HEMOFILIA ALCANZANDO LA TERCERA EDAD
Lcda. Esp. María Salcedo


La hemofilia no distingue de clase social, religión o nacionalidad. se
extiende por todo el mundo y a todos los niveles socio-económicos.
HEMOFILIA

LA HEMOFILIA SE IDENTIFICO DESDE LOS TIEMPOS BÍBLICOS.

Leopoldo, fue el primer
descendiente de Victoria,
que padeció Hemofilia B
y dos de sus cinco hijas
La enfermedad afecto a gran parte de las familias Reales Europeas
descienden de la reina Victoria de Inglaterra (España, Alemania y Rusia)
Durante XIX Y XX

Una persona nacida con hemofilia la tendrá durante toda la vida.
El nivel de factor VIII ó IX en susangre por lo general permanece igual a lo largo de toda su vida.
HEMOFILIA

HEMOFILIA
La hemofilia es un trastorno de la coagulación, ligado al (cromosoma X), que afecta principalmente al sexo masculino.
El cromosoma X contiene muchos genes que son comunes a ambos sexos, como los genes para la producción del factor VIII y el factor IX, relacionados la hemofilia.
Afecta a más de 400.000 personas en el mundo.

Los factores de la coagulación son proteínas de la sangre que controlan el sangrado. Muchos factores trabajan en conjunto en una serie de reacciones químicas para detener una hemorragia. A esto se le llama el Proceso de coagulación
FACTORES DE LA COAGULACIÓN
Proceso de coagulación normal
Proceso de coagulación en la
hemofilia

Hemofilia A: Hemofilia Clásica, deficiencia del Factor VIII de la coagulación. Es el tipo más común de hemofilia que existe; la incidencia en la Hemofilia A se puede decir que es de 1 persona por cada 10.000 varones vivos
Hemofilia B: Enfermedad de Chistmas, deficiencia del Factor IX de la coagulación; La incidencia en la Hemofilia B de 1 por cada 100.000 varones vivos.
CLASIFICACIÓN DE LA HEMOFILIA

Es el trastorno de coagulación hereditario más común que afecta tanto a hombres como a mujeres. Se calcula que la EvW afecta a cerca del uno por ciento de la población.Estudios han demostrado que una cifra tan elevada como 9 de cada 10 personas con EvW no han sido diagnosticadas
Medir la adhesión de las plaquetas a los sitios de injuria vascular (heridas)
Estabilizar al F VIII.
ENFERMEDAD DE VON WILLEBRAND:

Hemofilia Leve
5% - 45%
Sangrado severo con trauma
o cirugía importante.
Hemofilia Moderada
1% - 5%
sangrado espontáneo ocasional.Sangrado severo con trauma y cirugía
Hemofilia Severa
por debajo del < 1%
Sangrados espontáneo. principalmente en articulaciones y músculos
SEVERIDAD DE LA HEMOFILIA
50% - 150%
NIVEL PORCENTAJE DE
ACTIVIDAD NORMAL DEL FACTOR EN SANGRE
EPISODIOS HEMORRÁGICOS
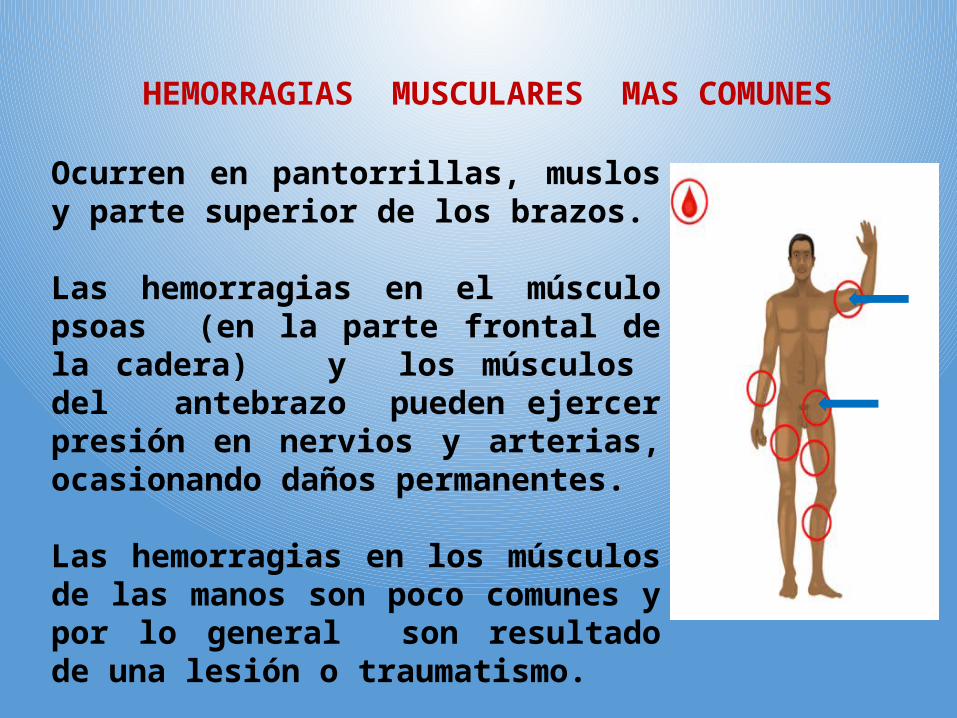
HEMORRAGIAS MUSCULARES MAS COMUNES
Ocurren en pantorrillas, muslos y parte superior de los brazos.
Las hemorragias en el músculo psoas (en la parte frontal de la cadera) y los músculos del antebrazo pueden ejercer presión en nervios y arterias, ocasionando daños permanentes.
Las hemorragias en los músculos de las manos son poco comunes y por lo general son resultado de una lesión o traumatismo.

HEMORRAGIAS ARTICULARES MAS COMUNES
Las hemorragias articulares más comunes ocurren en tobillos, rodillas y codos.
También pueden ocurrir en dedos de los pies, hombro y caderas
Por lo general, las articulaciones de las manos no se ven afectadas, excepto como resultado de una lesión
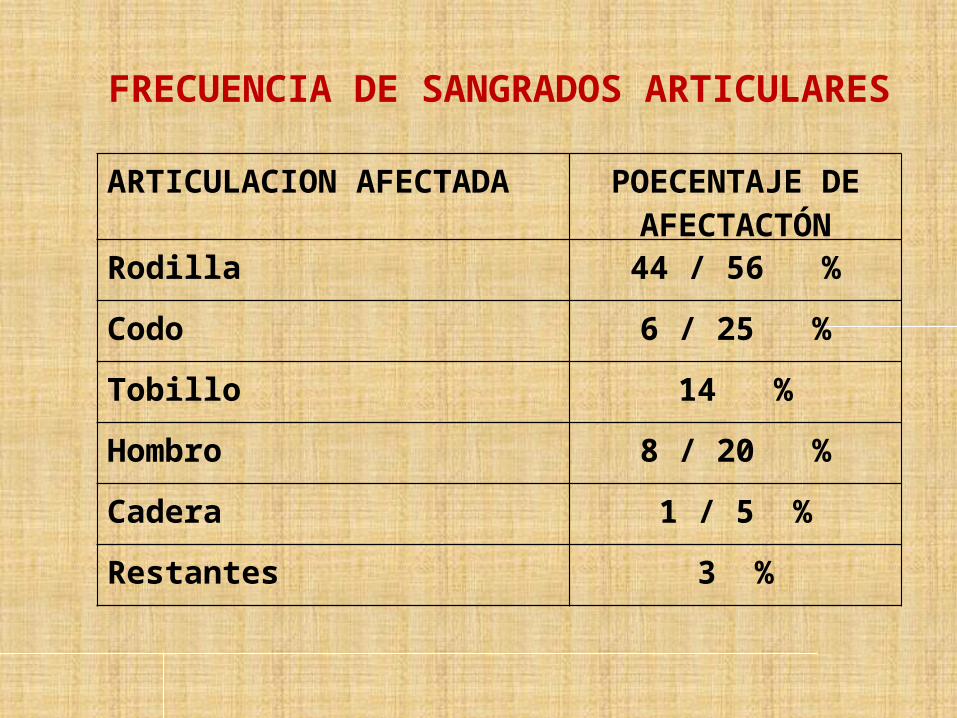
FRECUENCIA DE SANGRADOS ARTICULARES
ARTICULACION AFECTADA POECENTAJE DE AFECTACTÓN
Rodilla 44 / 56 %
Codo 6 / 25 %
Tobillo 14 %
Hombro 8 / 20 %
Cadera 1 / 5 %
Restantes 3 %

Sin un tratamiento adecuado y tras repetidas hemorragias en una misma articulación la membrana sinovial sangrará más fácilmente, los restos de sangre que se van depositando en la articulación van dañando los tejidos, se deja de producir el líquido sinovial y el roce de los huesos ocasiona el deterioro parcial o total de la articulación.
ATROPATIA HEMOLITICA
InflamadoDebilitamiento muscularRigidez matinalDolor crónicoMovimiento limitado
InflamadoDolorCalor
Burbujeo HormigueoCalor
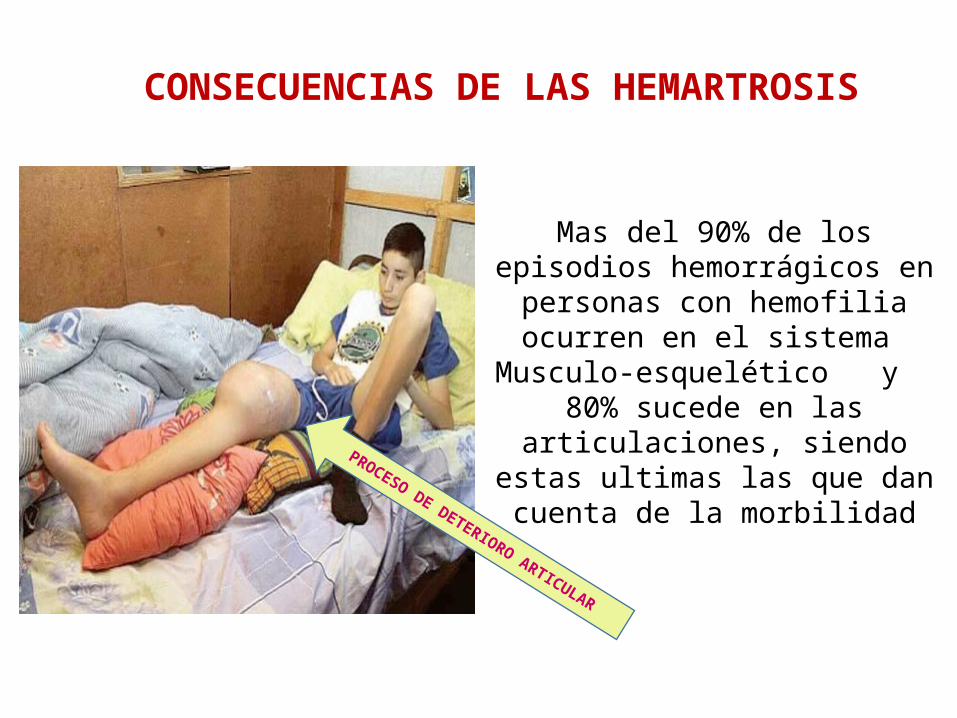
Mas del 90% de los episodios hemorrágicos en personas con
hemofilia ocurren en el sistema
Musculo-esquelético y 80% sucede en las articulaciones, siendo estas ultimas las que dan cuenta de la morbilidad
CONSECUENCIAS DE LAS HEMARTROSIS
PROCESO DE DETERIORO ARTICULAR
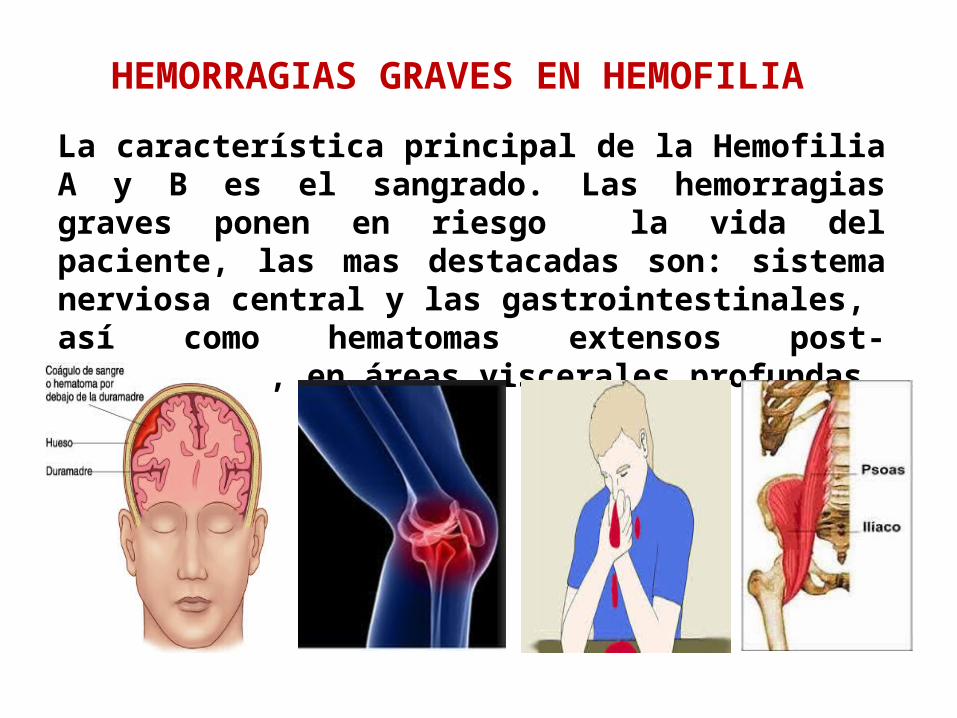
HEMORRAGIAS GRAVES EN HEMOFILIA
La característica principal de la Hemofilia A y B es el sangrado. Las hemorragias graves ponen en riesgo la vida del paciente, las mas destacadas son: sistema nerviosa central y las gastrointestinales, así como hematomas extensos post-traumáticos, en áreas viscerales profundas

Es hereditaria porque se
transmite a la descendencia
Es familiar porque distintos miembros de una misma familia
pueden heredar la misma alteración
HEMOFILIA

HERENCIA EN LA HEMOFILIA

En la década de los 50 Dra. Norma Blumenfel de Bosch, clasifica las primeras 9 familias atendidas en el Banco Municipal
de Sangre (Dto. Capital)
INICIO ATENCION P.C.H. EN VENEZUELA
Dra. En ciencias MédicasHematólogo
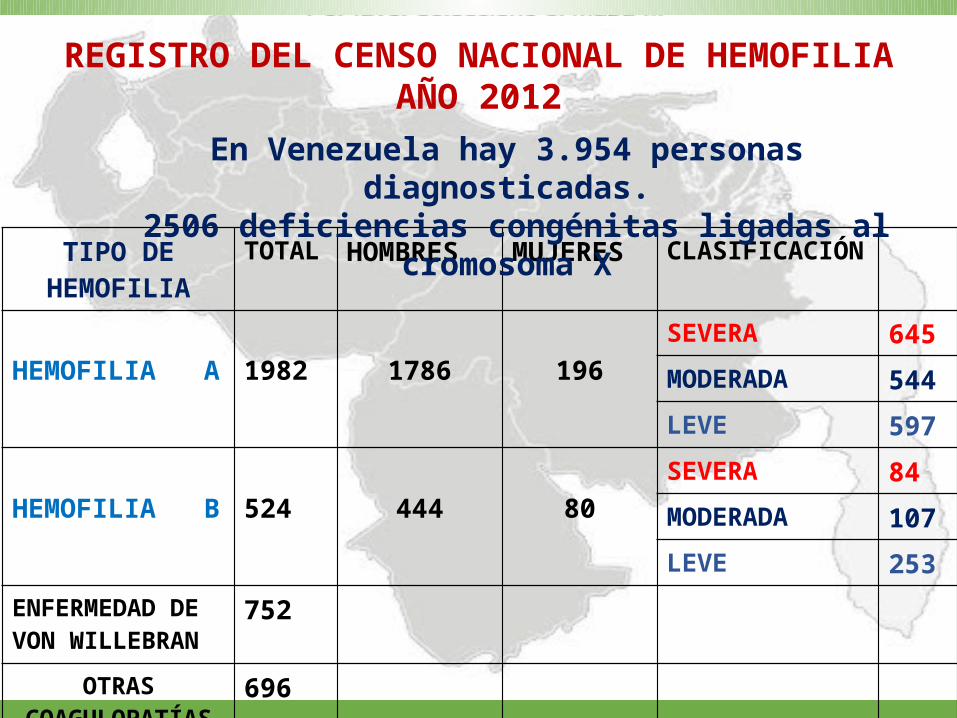
TIPO DE HEMOFILIA
TOTAL HOMBRES MUJERES CLASIFICACIÓN
HEMOFILIA A 1982 1786 196SEVERA 645
MODERADA 544
LEVE 597
HEMOFILIA B 524 444 80SEVERA 84
MODERADA 107
LEVE 253ENFERMEDAD DE VON WILLEBRAN
752
OTRAS COAGULOPATÍAS
696
REGISTRO DEL CENSO NACIONAL DE HEMOFILIA AÑO 2012
En Venezuela hay 3.954 personas diagnosticadas. 2506 deficiencias congénitas ligadas al cromosoma X

13 FILIALES
24 centros de atención
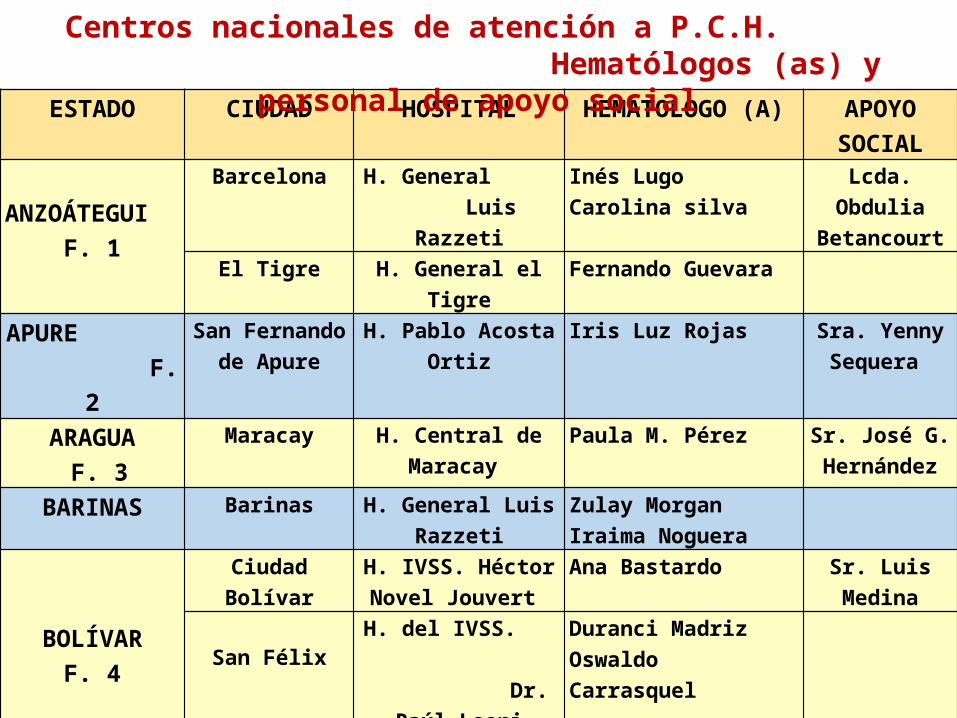
ESTADO CIUDAD HOSPITAL HEMATOLOGO (A) APOYO SOCIAL
ANZOÁTEGUI
F. 1
Barcelona H. General Luis Razzeti
Inés Lugo Carolina silva
Lcda. Obdulia Betancourt
El Tigre H. General el Tigre Fernando Guevara
APURE F. 2
San Fernando de Apure
H. Pablo Acosta Ortiz
Iris Luz Rojas Sra. Yenny Sequera
ARAGUA F. 3
Maracay H. Central de Maracay
Paula M. Pérez Sr. José G. Hernández
BARINAS Barinas H. General Luis Razzeti
Zulay Morgan Iraima Noguera
BOLÍVARF. 4
Ciudad Bolívar H. IVSS. Héctor Novel Jouvert
Ana Bastardo Sr. Luis Medina
San FélixH. del IVSS.
Dr. Raúl Leoni
Duranci MadrizOswaldo Carrasquel
Puerto Ordaz H. IVSS. Uyapar Yelitza Aguirre
CARABOBOF.5
Valencia H. Universitario Dr. Angel Laralde
Margorie RomeroSra. Miriam
GarcíaCOJEDES
San Carlos
H. General
Egor Nucete
Richard Figueredo
Centros nacionales de atención a P.C.H. Hematólogos (as) y personal de apoyo social
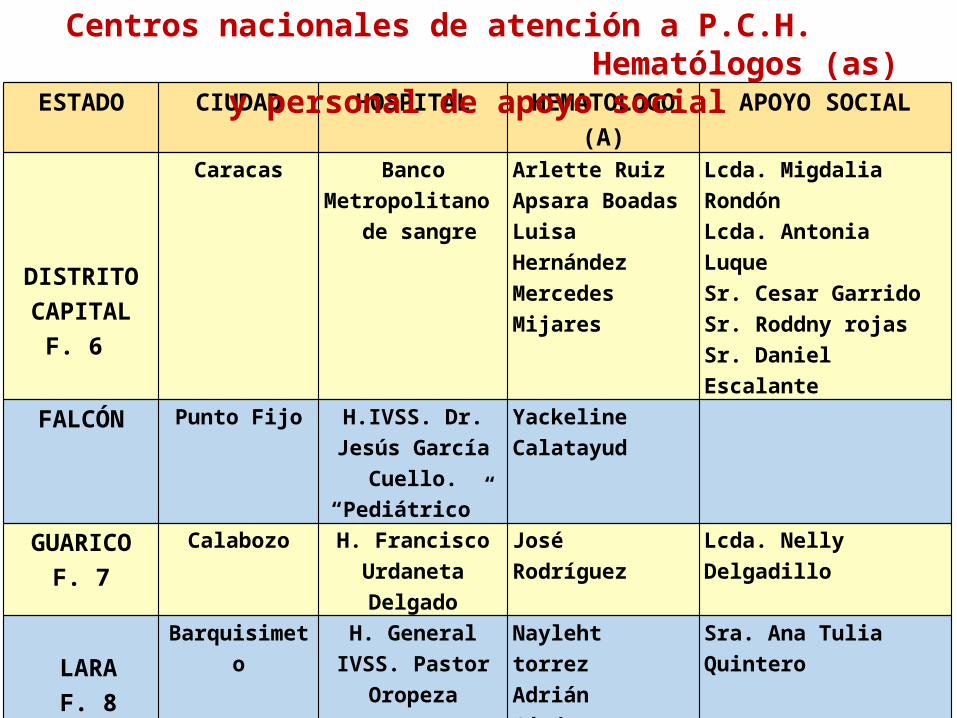
ESTADO CIUDAD HOSPITAL HEMATOLOGO (A)
APOYO SOCIAL
DISTRITO
CAPITALF. 6
Caracas Banco Metropolitano
de sangre
Arlette RuizApsara BoadasLuisa HernándezMercedes Mijares
Lcda. Migdalia RondónLcda. Antonia LuqueSr. Cesar GarridoSr. Roddny rojasSr. Daniel Escalante
FALCÓN Punto Fijo H.IVSS. Dr. Jesús García Cuello. “Pediátrico”
Yackeline Calatayud
GUARICOF. 7
Calabozo H. Francisco Urdaneta Delgado
José Rodríguez Lcda. Nelly Delgadillo
LARA F. 8
Barquisimeto H. General IVSS. Pastor Oropeza
Nayleht torrezAdrián Cárdenas
Sra. Ana Tulia Quintero
Carora H. Pastor Oropeza Riera
Carmen Mejías
MERIDAF. 9
Mérida H. IVSS. Dr. Tulio Carnevali
Salvatierra
Iris Luz Rojas Sra. Josefina Pico
MONAGAS Maturín H. Manuel Núñez Tovar
Edith Iguerey
Centros nacionales de atención a P.C.H. Hematólogos (as) y personal de apoyo social
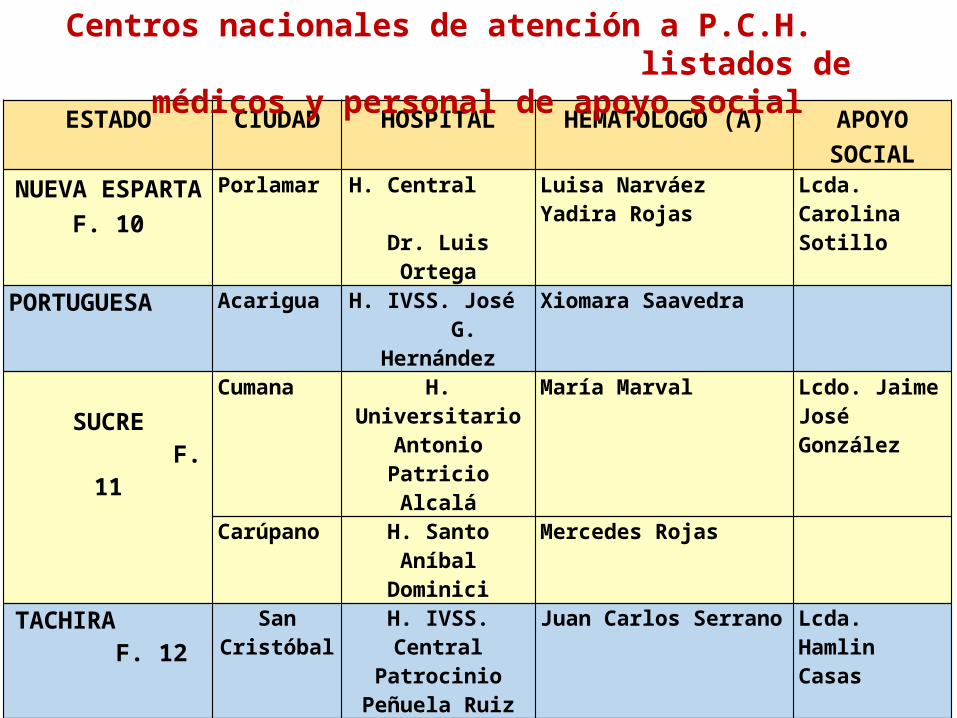
ESTADO CIUDAD HOSPITAL HEMATOLOGO (A) APOYO SOCIAL
NUEVA ESPARTA F. 10
Porlamar H. Central Dr. Luis Ortega
Luisa NarváezYadira Rojas
Lcda. Carolina Sotillo
PORTUGUESA Acarigua H. IVSS. José G. Hernández
Xiomara Saavedra
SUCRE
F. 11
Cumana H. Universitario Antonio Patricio
Alcalá
María Marval Lcdo. Jaime José González
Carúpano H. Santo Aníbal Dominici
Mercedes Rojas
TACHIRA F. 12
San Cristóbal
H. IVSS. Central Patrocinio
Peñuela Ruiz
Juan Carlos Serrano Lcda. Hamlin Casas
TRUJILLO Valera H. “Dr. José Gregorio
Hernández
Arturo PalomaresMaría Cristina Quevedo
YARACUY San Félix H. Placido Daniel Rodríguez
Iraida Tercek
ZULIA F 13
Maracaibo Banco de Sangre Edo. Zulia
Luis PazMariela Acosta
Lcda. Anny Cedeño
Centros nacionales de atención a P.C.H. listados de médicos y personal de apoyo social

EVOLUCIÓN DEL TRATAMIENTO P. C. H.
Antes de 1930. Inmovilización, hielo, reposo y analgesia
En 1930. Transfusiones de sangre
En 1936. Plasma fresco congelado
En 1937. globulina antihemofílica, ahora conocida como Factor VIII (8).

EVOLUCIÓN DEL TRATAMIENTO P. C. H.
1980. Concentrado de factor pureza elevada
1982. se dio una baja por el contagio del 80% de los que padecían hemofilia por el VIH/SIDA
En 1970. Concentrados por factores pureza intermediaria concentrados liofilizados de FVIII y FIX
En 1965. Crioprecipitados

EVOLUCIÓN DEL TRATAMIENTO P. C. H.
1990. Cada uno de los factores fueron sometidos a una inactivación viral, haciendo cada vez el proceso más seguro
1992. salió al mercado el primer factor que no fue derivado del plasma, obtenido por tecnología AND recombinante
1995. se introdujo el primer tratamiento profiláctico.

TRABAJO
EN
EQUIPO
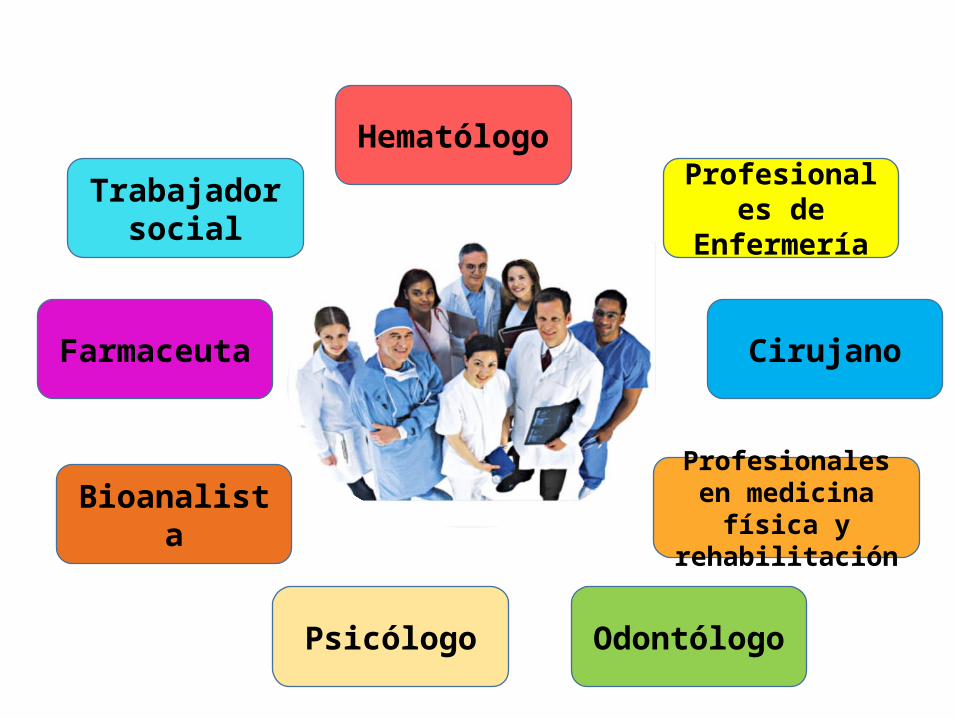
Trabajador social
Farmaceuta
Bioanalista
Psicólogo
Hematólogo
Profesionales de Enfermería
Cirujano
Profesionales en medicina física y
rehabilitación
Odontólogo

El tratamiento sustitutivo supuso un avance importantísimo
tanto para la calidad de vida como para la supervivencia
de los pacientes.
El tratamiento puede prevenir hemorragias o
minimizar sus efectos de manera que el paciente
permanezca libre de incapacidades y complicaciones
TRATAMIENTO

TERAPIA DE REEMPLAZO
Profilaxis En niños después de los dos
años luego de la primera hemartrosis, se administra el medicamento (factor) una a
tres veces por semana hasta que el hematólogo lo considere necesario
A DEMANDAEs la administración del factor liofilizado de manera manual,
posterior a un episodio hemorrágico, o cuando se va a
realizar una cirugía
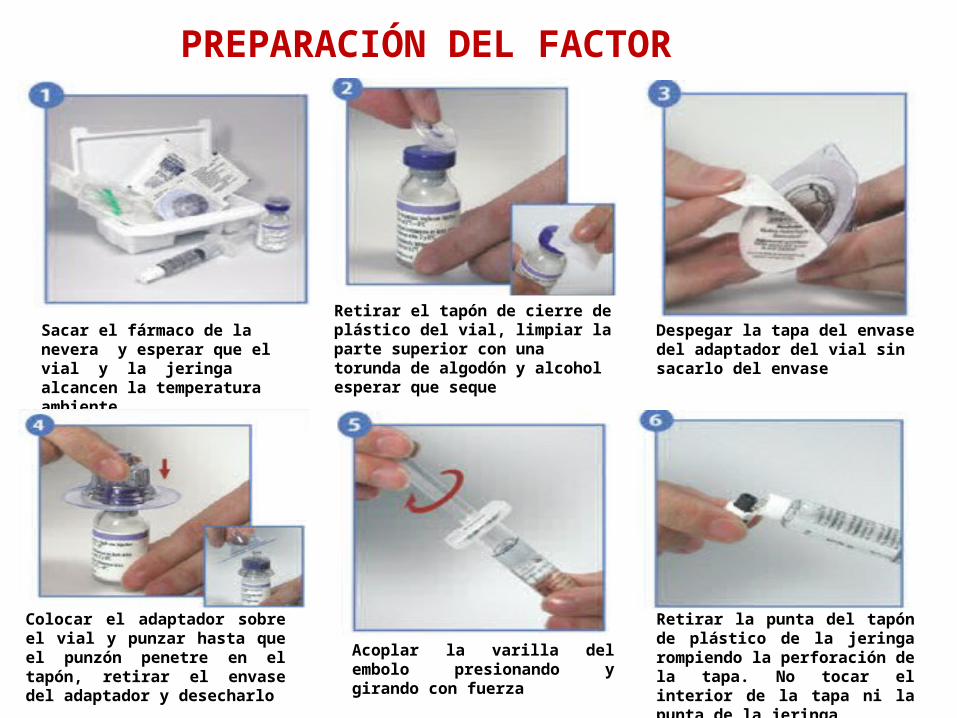
Sacar el fármaco de la nevera y esperar que el vial y la jeringa alcancen la temperatura ambiente
Retirar el tapón de cierre de plástico del vial, limpiar la parte superior con una torunda de algodón y alcohol esperar que seque
Despegar la tapa del envase del adaptador del vial sin sacarlo del envase
Retirar la punta del tapón de plástico de la jeringa rompiendo la perforación de la tapa. No tocar el interior de la tapa ni la punta de la jeringa
Acoplar la varilla del embolo presionando y girando con fuerza
Colocar el adaptador sobre el vial y punzar hasta que el punzón penetre en el tapón, retirar el envase del adaptador y desecharlo
PREPARACIÓN DEL FACTOR
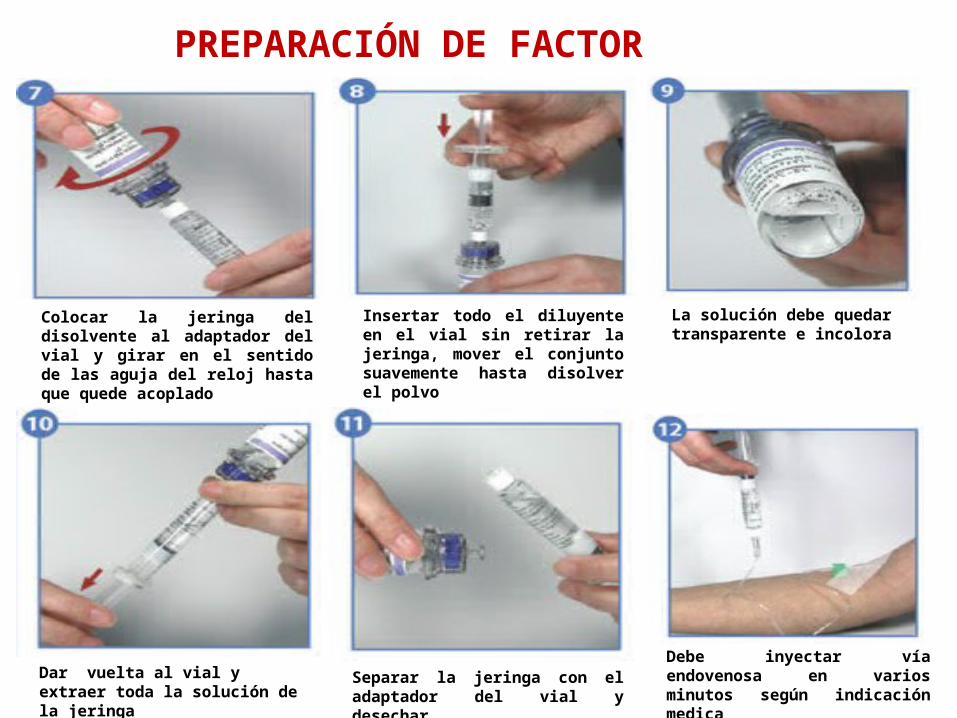
Colocar la jeringa del disolvente al adaptador del vial y girar en el sentido de las aguja del reloj hasta que quede acoplado
Insertar todo el diluyente en el vial sin retirar la jeringa, mover el conjunto suavemente hasta disolver el polvo
La solución debe quedar transparente e incolora
Debe inyectar vía endovenosa en varios minutos según indicación medica
Separar la jeringa con el adaptador del vial y desechar
Dar vuelta al vial y extraer toda la solución de la jeringa
PREPARACIÓN DE FACTOR

ADMINISTRACIÓN DE FACTORES
1. Lavado de manos
2. Si se rompe el envase del diluyente se debe prepara con agua destilada o solución fisiológica estéril. 3. Utilizar siempre el filtro para cada frasco de liofilizado.
4. Realizar asepsia en el área a colocar la vía periférica

5. El medicamento (factor) debe estar completamente disuelto ( no debe verse grumos) y evitar la espuma al momento de pasarlo a la jeringa
6. No llenar el área del scalp con el medicamento. (esperar fluido de sangre)
7. No golpear la zona (vena ) a cateterizar
8. Utilizar una banda ancha para fijar la vena a cateterizar.
PREPARACIÓN Y ADMINISTRACIÓN DE FACTORES

9. Colocar la banda por encima de la manga de la franela para evitar moretones y 5 a 10 cms. por encima a la vena a cateterizar.
10. Utilizar siempre el scalp 23 o 25.
11. Al intentar cateterizar la vena no lo logra, intentar nuevamente, si vuelve a fallar, retirar por completo el scalp y colocar una cura compresiva, buscar una nueva vena y utilizar un nueva scalp
ADMINISTRACIÓN DE FACTORES

Derivados plasmáticos obtenidos por pool de donantes
Factor IXFactor VIII
Factor VIII y Factor de von Willebrand
Productos Liofilizados Plasmáticos
Complejo protrombínico Factor II, V, VII, IX, X y los inhibidores
TRATAMIENTO

Productos Liofilizados Recombinantes
Por tecnología Clonación por cadena de ADN. Factor VIII (Factor VII bypasiante)
F VII, inhibidores y Otras coagulopatias según indicación del
hematólogo
TRATAMIENTO
Tiempo recomendado para la administración
Factor VIII 3 cc por minutoFactor VII, IX, X y complejo prothrombínico 1 cc x minuto.

Depende de la severidad, y el tratamiento adecuado
Antes los años 60; el promedio de vida llego a ser hasta de 11años
1980 el promedio de vida se aproxima de 50 a 60 años gracias al empleo de hemocomponentes. Década de los 80 insipiente pandemia de infección virus del (VIH/SIDA) hizo disminuir 10 años la esperanza de vida con respecto a varones sin hemofilia.
Hemofilia A 15% mas 45 años Hemofilia B 45% mas 45 añosEnfermedad de Von Willebrand 18% mas 45 años
PROMEDIO DE VIDA PCH

Hoy en día la supervivencia de un paciente con hemofilia es alta, gracias al
suministro por vía intravenosa del factor antihemofílico deficiente. Las personas que padecen esta enfermedad pueden
llevar una vida completamente normal con un tratamiento adecuado.



















