Trastornos de las glándulas suprarrenales
55
-
Upload
leslie-pascua -
Category
Health & Medicine
-
view
321 -
download
0
Transcript of Trastornos de las glándulas suprarrenales


• Es un síndrome relacionado con la hipersecreción de aldosterona.
• Puede ser:
Primario:
Secreción excesiva de aldosterona, la causa se encuentra en la propia glándula suprarrenal.
Secundario:
Estímulo extrasuprarrenal.

• Generalmente se debe a un adenoma suprarrenal de aldosterona (Síndrome de Conn).
• Es unilateral y de tamaño pequeño.
• El hiperaldosteronismo es 2 veces más frecuente en las mujeres y suele aparecer entre los 30-50 años de edad.

• Estos pacientes tienen una hiperplasia nodular bilateral de la corteza suprarrenal. No se encuentra un adenoma solitario.
• Se denomina también hiperaldosteronismo idiopático o hiperplasia nodular.
• Su causa es desconocida y representa hasta el 80% de los casos de hiperaldosteronismo primario.

• Hipopotasiemia.
• HTA sistólica grave.
• Cefalea.
• Poliuria.
• Signos electrocardiográficos y radiográficos de HT ventricular izquierda.
• Signos electrocardiográficos de hipopotasiemia.
• Ausencia de edema.
• Alteraciones de la circulación cerebral, vasos retinianos y renales.
• Proteinuria (50%).
• Insuficiencia renal (15%).
• Daños en el aparato cardiovascular.

Criterios diagnósticos
1. Hipertensión diastólica sin edema.
2. Hiposecreción de renina.
3. Hipersecreción de aldosterona.
• Los adenomas productores de aldosterona deben ser localizados por medio de TC abdominal.
Datos de laboratorio
1. [] de la orina.
2. pH urinario neutro o alcalino.
3. Hipopotasiemia.
4. Hipernatriemia.
5. Alcalosis metabólica.
6. Disminución de los niveles séricos de magnesio.

• Extirpación quirúrgica del tumor (técnica laparoscópica).
• Restricción del Na en la dieta.
• Administración de antagonistas de la aldosterona:
• Espironolactona 25-100 mg/VO/c8hr.
• Eplerrenona 50-150 mg/VO/c8hr.

Se puede dar en casos de:
• Embarazo.
• Hipertensión.
• Hipersecreción secundaria de renina.
• Tumores raros que producen renina.
• Pacientes con edema.
• Insuficiencia cardíaca.
Se caracteriza por:
• Alcalosis hipopotasiémica.
• Aumento de la actividad de la renina.
• Elevación de la concentración de aldosterona.

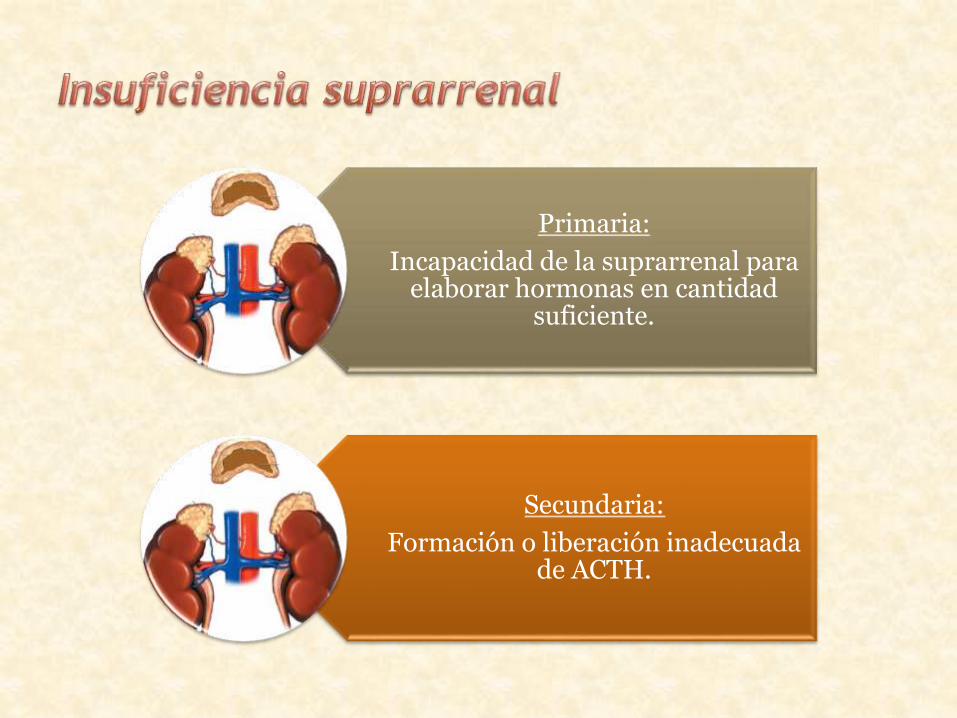
Primaria:
Incapacidad de la suprarrenal para elaborar hormonas en cantidad
suficiente.
Secundaria:
Formación o liberación inadecuada de ACTH.

Insuficiencia suprarrenalprimaria
Insuficiencia suprarrenalsecundaria
• Atrofia de la glándula.• Extirpación quirúrgica.• Infecciones.• Hemorragias.• Hiperplasia suprarrenal congénita.• Hipoplasia suprarrenal congénita.• Inhibidores enzimáticos.• Anticuerpos bloqueadores de la
ACTH.
• Hipopituitarismo debido a enfermedades hipotalamicohipofisarias.
• Inhibición del eje hipotálamo-hipófisis.


• La ISP es rara, puede aparecer a cualquier edad y afecta ambos sexos.
• Se debe a la destrucción progresiva de las suprarrenales (>90%).
• Actualmente la causa más frecuente es la atrofia suprarrenal idiopática, que tiene un mecanismo autoinmunitario.
• La destrucción autoinmunitaria de la glándula es secundaria a los linfocitos T citotóxicos.
• Varios procesos endócrinos autoinmunitarios en una misma persona, se conoce como síndrome autoinmunitario poliglandular de tipo II.

• Se debe a un gen mutante situado en el cromosoma 6 y se relaciona con los alelos B8 y DR3 del HLA.
• Suele manifestarse en la vida adulta.
• Se relaciona con:
» Anemia perniciosa.
» Vitíligo.
» Alopecia.
» Esprue no tropical.
» Miastenia gravis.

• Está integrado por la combinación de insuficiencia paratiroidea, suprarrenal y candidosis mucocutánea crónica.
• Se hereda con carácter autosómico recesivo y no está relacionado con el HLA.
• Suele aparecer en la niñez.
• Se relaciona con:
» Anemia perniciosa.
» Hepatitis crónica.
» Alopecia.
» Hipotiroidismo primario.
» Insuficiencia gonadal prematura.

• Fatiga, debilidad, síntomas gastrointestinales, pigmentación de la piel y mucosas, hipotensión e hipoglucemia.
• La astenia es el síntoma principal, el paciente está continuamente fatigado y necesita reposo en cama.
• La hiperpigmentación usualmente aparece como un oscurecimiento difuso de color moreno, pardo o bronceado en los codos, surcos de las manos y en las aréolas.
• En las mucosas pueden aparecer placas de color negro azulado.

• Fases tempranas:
» Disminución de los niveles de cortisol.
• Fases avanzadas:
» Hiponatriemia.
» Disminución de los niveles de cloruro y Bicarbonato.
» Hiperpotasiemia.
» Elevación de vasopresina y angiotensina II.
» Disminución del cortisol y aldosterona.
» Hipercalciemia leve a moderada.
» Cambios en el EKG y EEG.
» Anemia normocítica.
» Linfocitosis.
» Eosinofilia.

• Prueba de respuesta del cortisol:
– Respuesta del cortisol a los 60 min tras la inyección de 250 µg de cosintropinaIM o IV.
– Los niveles de cortisol deben superar los 495 nmol/L.
• Medición de la aldosterona:
– En la IS2, el incremento de la aldosterona será normal (5 ng/100ml).
– En la IS1, será anormal.
• Niveles de ACTH:
– Elevada: IS1.
– Normal- baja: IS2.

• Hormonoterapia restitutiva.
GLUCOCORTICOIDES
• Hidrocortisona (cortisol) 20-30 mg/día. (2/3 am y 1/3 pm.)
• RAM: Gastritis, insomnio, irritabilidad y excitación mental.
MINERALOCORTICOIDES
• Fludrocortisona 0.05-0.1 mg/VO/día.
• RAM: hipopotasiemia, HTA, cardiomegalia, ICC por retención de Na.
• Mujeres: 25-50 mg DHEA.
• Sal 3-4 gr/día.


• Puede ser:
» Déficit selectivo: aparece después de administrar prolongadamente glucocorticoides en exceso.
» Déficit múltiple: acompaña al déficit de muchas hormonas hipofisarias (panhipopituitarismo).
• Las concentraciones de ACTH sirven para distinguir la IS1 (ACTH elevada) de la IS2 (ACTH disminuída o ausente).
• Diagnóstico: prueba de la ACTH.
• Tratamiento: glucocorticoides, no es necesario administrar mineralocorticoides.


• Las crisis suprarrenales suponen una intensificación rápida y fulminante de una ISC que son desencadenadas por sepsis o estrés quirúrgico.
• Puede producirse una destrucción hemorrágica aguda de ambas glándulas. En niños, este acontecimiento suele ocurrir durante la septicemia por Pseudomonas o la meningococemia.
• También puede aparecer en condiciones de:
» Interrupción brusca de esteroides.
» Hiperplasia suprarrenal congénita.
» Fármacos que inhiben la síntesis o aceleran el metabolismo de los esteroides.

• En los pacientes sin tratamiento, los síntomas se acentúan y aparecen nauseas, vómitos, dolor abdominal intenso, deshidratación, fiebre, letargo, somnolencia seguido de colapso vascular por hipovolemia.
• La finalidad del tratamiento es reponer los glucocorticoides y el déficit de Na y agua.
• SSN IV + Infusión de hidrocortisona 100 mg/IV + Goteo de hidrocortisona 10 mg/hr.

• La fisiología del eje hipotálamo- hipófisis- suprarrenal se altera durante las enfermedades graves como traumatismos, intervenciones quirúrgicas, sepsis y choque.
• La producción inadecuada de cortisol durante las enfermedades graves puede causar hipotensión, disminución de la resistencia vascular generalizada, choque y muerte.


Hipoaldosteronismo aislado
Hipoaldosteronismo hiporreninémico
Seudohipoaldosteronismo

• El déficit aislado de aldosterona acompañado de una producción normal de cortisol ocurre en el hiporreninismo.
• La manifestación más común es la incapacidad para aumentar adecuadamente la secreción de aldosterona.
• Los pacientes tienen hiperpotasiemia y pérdida de Na por la orina.

• Hipoaldosteronismo aislado con déficit de la producción de renina.
• Se da sobre todo en pacientes con diabetes mellitus e insuficiencia renal leve.
• Mortalidad: 80%.
• En ella encontramos:
» Nefropatía.
» Neuropatía vegetativa.
» Expansión del volumen extracelular.
» Conversión defectuosa de los precursores de la renina en renina activa.
• Los niveles bajos de renina y aldosterona establecen el diagnóstico.

• Es un cuadro hereditario que se debe a una mutación del canal epitelial del sodio.
• Produce un hipoaldosteronismo con niveles elevados de renina y niveles elevados de aldosterona.

• Objetivo: corregir el déficit de mineralocorticoides.
• Fludrocortisona 0.05-0.15 mg.
• Reducción de la ingesta de sal.
• Furosemida.


Hiperplasia suprarrenal congénita
Hipoplasia suprarrenal congénita
Déficit aislado de glucocorticoides
Resistencia primaria al cortisol
AdrenoleucodistrofiaHiperplasia
suprarrenal lipoidea congénita

• Es consecuencia de mutaciones recesivas que provocan un defecto enzimático.
• Es el trastorno suprarrenal más frecuente de la lactancia y la niñez.
• La hiperplasia suprarrenal de comienzo tardío es la causa de 5-25% de los casos de hirsutismo y oligomenorrea en la mujer.
• Expresión clínica:
» Seudohermafroditismo femenino y masculino.
» Precocidad sexual.
» Edad ósea superior a la cronológica.
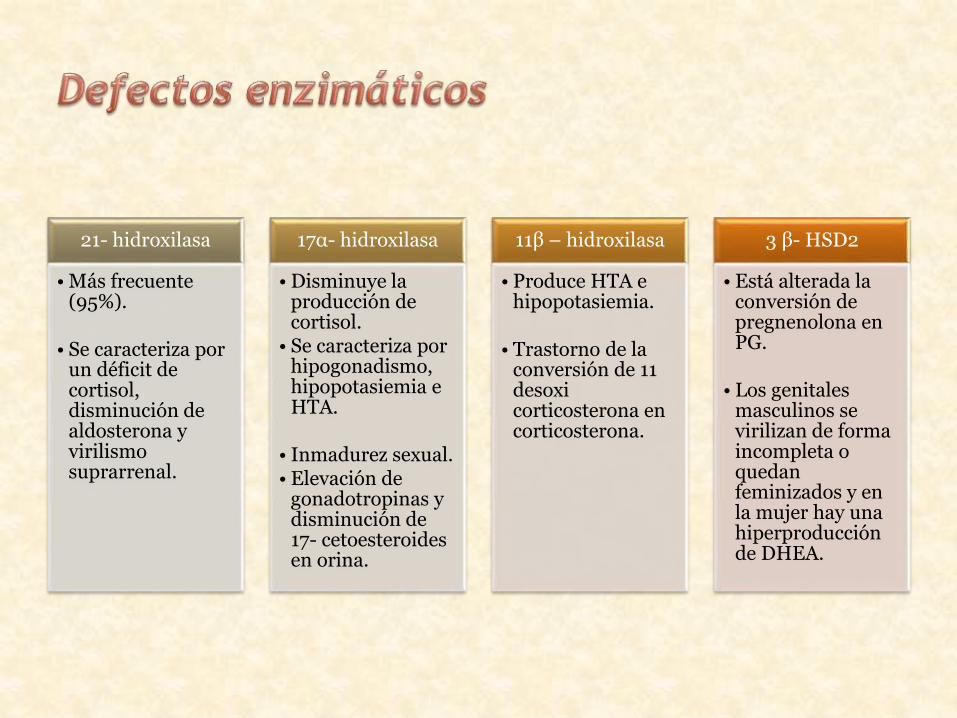
21- hidroxilasa
• Más frecuente (95%).
• Se caracteriza por un déficit de cortisol, disminución de aldosterona y virilismo suprarrenal.
17α- hidroxilasa
• Disminuye la producción de cortisol.
• Se caracteriza por hipogonadismo, hipopotasiemia e HTA.
• Inmadurez sexual.• Elevación de
gonadotropinas y disminución de 17- cetoesteroides en orina.
11β – hidroxilasa
• Produce HTA e hipopotasiemia.
• Trastorno de la conversión de 11 desoxi corticosterona en corticosterona.
3 β- HSD2
• Está alterada la conversión de pregnenolona en PG.
• Los genitales masculinos se virilizan de forma incompleta o quedan feminizados y en la mujer hay una hiperproducción de DHEA.

• El diagnóstico debe considerarse en lactantes que sufren episodios de ISA, HTA y pérdida de Na.
• Los hallazgos en la exploración física y en la excreta urinaria aumentan la sospecha.
• Diagnóstico prenatal en niñas afectadas: 14-16 SG.
• Tratamiento:
• Glucocorticoides (prednisona). 10-13 mg/BID o TID.
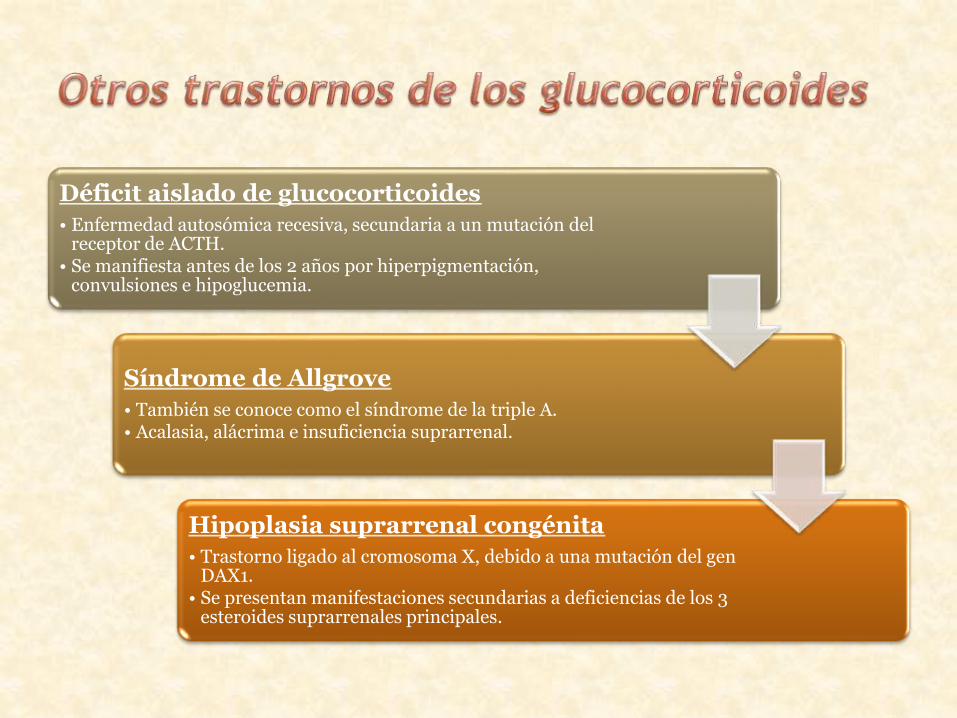
Déficit aislado de glucocorticoides
• Enfermedad autosómica recesiva, secundaria a un mutación del receptor de ACTH.
• Se manifiesta antes de los 2 años por hiperpigmentación, convulsiones e hipoglucemia.
Síndrome de Allgrove
• También se conoce como el síndrome de la triple A.• Acalasia, alácrima e insuficiencia suprarrenal.
Hipoplasia suprarrenal congénita
• Trastorno ligado al cromosoma X, debido a una mutación del gen DAX1.
• Se presentan manifestaciones secundarias a deficiencias de los 3 esteroides suprarrenales principales.
Resistencia primaria al cortisol
Mutaciones del receptor de glucocorticoides.
No se presentan síntomas de insuficiencia suprarrenal.
Adrenoleucodistrofia
Causa desmielinización y muerte prematura en los niños.
La adrenomieloneuropatía se acompaña de una neuropatía motora y sensitiva con paraplejía espástica en los adultos.
Hiperplasia suprarrenal lipoidea congénita
Se caracteriza por insuficiencia suprarrenal y esteroidogénesis gonadal defectuosa.

Hiperaldosteronismo corregible con
glucocorticoidesSíndrome de Bartter
Síndrome de Gitelmann
Síndrome de LiddleSíndrome de exceso
aparente de mineralocorticoides

• Trastorno heredado como rasgo autosómico dominante, se debe buscar en los sujetos con supresión de la actividad de la renina plasmática e HTA de comienzo juvenil.
• Los individuos afectados sufren tempranamente ACV de tipo hemorrágico.
• Tratamiento: administración de glucocorticoides o antimineralocorticoides (espironolactona, triamtereno o amilorida).

• Se caracteriza por hiperaldosteronismo grave (alcalosis hipopotasiémica), aumento de la actividad de la renina e hipercalciuria. P/A normal, no edema.
• Suele iniciar en la infancia.
• Biopsia renal muestra hiperplasia yuxtaglomerular.
• Implica un trastorno de la conservación renal de Na o Cl.
• La pérdida renal de Na, estimula la secreción de renina y la producción de aldosterona.

• Rasgo autosómico recesivo que se caracteriza por la pérdida de Na por los riñones.
• Produce la activación del sistema renina- angiotensina – aldosterona y los pacientes presentan:
» Hipotensión.
» Hipopotasiemia.
» Hipomagnesiemia.
» Bicarbonato sérico elevado.
» Disminución urinaria de calcio.

• Trastorno autosómico dominante que imita un hiperaldosteronismo.
• Los niveles de renina y aldosterona son bajos y hay un exceso de reabsorción de Na en el túbulo renal.
• Puede causar HTA e hipopotasiemia.
• La ingestión de dulces o la masticación de tabaco producen un síndrome que simula un hiperaldosteronismo primario.
• El componente de éstas sustancias que provoca la retención de Na es el ácido glicirrizínico.


• Los feocromocitomas y paragangliomas son tumores productores de catecolaminas provenientes del SNS o SNP.
• Pueden surgir en forma esporádica, heredarse o acompañar a otros trastornos.
• Constituye una causa de HTA reversible.
• 0.2% de las personas hipertensas tienen un feocromocitoma.
• Edad media del diagnóstico: 40 años.
• Regla de los dieces:
» 10% bilaterales.
» 10% extrasuprarrenales.
» 10% cancerosos.

• Los feocromocitomas y paragangliomas son neoplasias muy vascularizadas que provienen de células derivadas de paraganglios simpáticos o parasimpáticos.
• Los feocromocitomas tienen un parénquima oscuro ocasionado por la oxidación cromafínica de catecolaminas.
• Pueden estar situados en el plano retroperitoneal extrasuprarrenal, pelvis y tórax.
• El paraganglioma se usa para describir las neoplasias productoras de catecolaminas en cabeza y cuello y tumores que nacen del SNP.
• 25% de las personas que poseen un feocromocitoma son de carácter hereditario.

• Tríada clásica:
» Palpitaciones.
» Cefalea.
» Hiperhidrosis.
• Signo predominante: HTA.
• Las crisis catecolaminícas pueden causar:
» Insuficiencia cardiaca.
» Edema pulmonar.
» Arritmias.
» Hemorragia intracraneal.
• También pueden manifestarse episodios de ansiedad, taquicardia y palidez.

• Métodos bioquímicos: corroboración del exceso de catecolaminas.
• Hay un incremento de los niveles plasmáticos y urinarios de catecolaminas y metanefrinas (metabolitos metilados).
• Estudios de imágenes: localización del tumor.
• TC y RM.
• Especial utilidad para identificar feocromocitomas y paragangliomas extrasuprarrenales.
• Detecta 5% de las masas suprarrenales.
• Localización con radionúclidos: captación de los agentes es selectiva en los paragangliomas.

• HTA esencial.
• Ataques de ansiedad.
• Consumo de cocaína o anfetaminas.
• Lesiones intracraneales.
• Abstinencia de clonidina.
• Epilepsia.
• Crisis facticias.
• Diagnóstico diferencial del tumor:
• Adenoma suprarrenal no funcional.
• Aldosteronoma.
• Adenoma productor de cortisol.

• Extirpación quirúrgica:
• Laparotomía, laparoscopía transperitoneal o retroperitoneal y cirugía endoscópica atraumática.
• Bloqueadores α adrenérgicos:
• Fenoxibenzamina 5-10 mg/VO/TID/10-14 días.
• Propanolol 10 mg/VO/TID
• Para corregir los paroxismos: Prazosin VO o Fentolamina IV.
• Crisis hipertensivas transoperatorias: Nitroprusiato/goteo IV

• 5-10% de los feocromocitomas y paragangliomas son cancerosos.
• Son neoplasias que envían metástasis a distancia y a menudo aparecen en pulmones, hueso o hígado y su propagación es por vía vascular.
• Tratamiento:
• Citorreducción de la masa tumoral.
• Bloqueadores α contra los síntomas.
• Quimioterapia (Dacarbazina, ciclofosfamida, vincristina).
• Radioterapia con radionúclidos.
• Pronóstico de supervivencia: 30-60%.

Neurofibromatosis de tipo 1
Neoplasia endócrina múltiple
2 A o 2 B
Síndrome de Von Hippel-Lindau
Síndromes de paragangliomas

NF tipo 1 MEN 2 A y B
• MEN 2 A:
» Carcinoma medular tiroideo.
» Feocromocitoma.
» Hiperparatiroidismo.
• MEN 2 B:
» Carcinoma medular tiroideo.
» Feocromocitoma.
» Múltiples neuromas de la mucosa.
• Se caracteriza por:
» Múltiples neurofibromas.
» Manchas café con leche.
» Pecas en la piel de la axila.
» Nódulos de Lich del iris.
• 1% de los pacientes tienen feocromocitoma.

• Trastorno que predispone a hemangioblastoma de retina y cerebelo que afectan al tallo encefálico y la médula espinal.
• Produce también carcinomas de células claras renales, tumores de los islotes pancreáticos, tumores del oído interno, cistoadenomas del epidídimo y el ligamento ancho.
• Induce la angiogénesis; mayor expresión del factor endotelial vascular.
• 20-30%: feocromocitoma.